

Abstract
Recently, polyomaviruses KI and WU were identified in the airways of patients with acute respiratory symptoms. The epidemiology and pathogenesis of these two viruses are not fully understood, and the development of molecular assays, such as Real Time PCR, was useful for examining their biology and role in different clinical syndromes. The evaluation of different target regions for the amplification of polyomaviruses KI and WU, comparing published primer/probe sets and sets designed in the laboratory is described and was used for testing 175 clinical specimens (84 stools and 91 tonsils). The results showed that the laboratory designs were more sensitive for the detection of polyomaviruses KI and WU DNA in clinical samples. The choice of the primer/probe set, and primarily of the region for amplification, may be relevant for understanding the pathogenic role of viruses such as polyomaviruses KI and WU.
Keywords: Polyomavirus KI, Polyomavirus WU, Real Time polymerase chain reaction, Primer/probe set
1. Introduction
The Polyomaviridae family is constituted by small double-stranded DNA viruses which are widespread in the human population with a seroprevalence of 60–80% in adults. Two polyomaviruses are known to infect humans, JC virus (JCV) and BK virus (BKV), both discovered in 1971 (Gardner et al., 1971, Padgett et al., 1971). SV40, another polyomavirus which infects naturally the rhesus monkey, was introduced accidentally into the human population as a contaminant during the early use of poliomyelitis vaccines (Butel and Lednicky, 1999, Bergallo et al., 2007). Following primary infection, which occurs usually through the respiratory tract and is often asymptomatic, polyomaviruses can establish latency in the urinary tract, brain, and in the B cells (Jiang et al., 2009). Reactivation is frequent in immunocompromised patients, such as transplant recipients, with severe consequences (Costa et al., 2008). Two novel polyomaviruses have been described recently and named Karolinska Institutet virus (KIV) and Washington University virus (WUV) (Allander et al., 2007, Gaynor et al., 2007). KIV and WUV have been isolated first in respiratory secretions from human patients with acute respiratory tract symptoms and, in the case of KIV, also in faecal samples (Allander et al., 2007, Gaynor et al., 2007, Babakir-Mina et al., 2008, Babakir-Mina et al., 2009, Jiang et al., 2009). WUV shares only 15–49% identity with BKV and JCV and is most similar to KIV. KIV and WUV nonstructural proteins display amino acid sequence similar to those of the other primate polyomaviruses, while the structural proteins exhibit a very low degree of similarity to those of other known polyomaviruses, suggesting that WUV and KIV define a novel branch within the Polyomaviridae family with unexplored biology and controversial clinical significance (Allander et al., 2007, Gaynor et al., 2007, Norja et al., 2007). The development of molecular assays, such as Real Time PCR, for KIV and WUV detection could be useful to determine the epidemiology, pathogenesis and pathogenic role of the two viruses in different clinical syndromes. In a Real Time PCR protocol design different parameters, such as the target region and the conditions of amplification, have to be considered in order to obtain enhanced sensitivity and specificity. In a high-throughput routine laboratory, the use of “in-house” protocols requires optimization, standardization, and validation processes, consisting in the assessment of sensitivity, specificity, efficiency, precision and linearity. Furthermore, the choice of the sample material, also referred to as the matrix, could have a strong influence on the results and has to be considered in the validation process (Dimech et al., 2004, Rabenau et al., 2007).
The aim of this study was to evaluate different target regions for the amplification of KIV and WUV, comparing published primer/probe sets and sets designed in the laboratory on different clinical specimens and to optimize a quantitative assay using the most suitable primer/probe set.
2. Materials and methods
2.1. Clinical specimens
A total of 175 clinical specimens (84 stools and 91 tonsils provided by the Mediterranean Hematology Institute of Rome and the Cytodiagnostic and Histopathology Unit of the Maggiore Hospital of Trieste, respectively) were studied.
2.2. Nucleic acid extraction
Twenty-five milligrams of paraffin-embedded tonsillar tissue were deparaffinized with xylene and ethanol 100–70% and digested subsequently overnight at 56 °C with proteinase K. In the case of faecal samples, 200 mg of stool were extracted. Total DNA extraction was carried out using QIAamp DNA mini kit (Qiagen, Milan, Italy), according to the manufacturer's specification.
2.3. Real Time PCR assay on clinical specimens
The following target regions were evaluated: for KIV, the regulatory region (primer/probe set KI-A), the small t antigen (KI-B), both described previously (Bialasiewicz et al., 2007), and the VP1 gene (laboratory designed primer/probe set KI-C); for WUV, the regulatory region (WU-A and WU-C), the large T antigen (WU-B), as described previously (Bialasiewicz et al., 2007), and the VP2 gene (laboratory designed primer/probe set WU-D)(Table 1 ). The laboratory design of the Real Time PCR primers and TaqMan probe was performed using the Primer Express 3.0 software (Applied Biosystems, Cheshire, UK). The reaction mixture contained 1× Master Mix (Platinum qPCR supermix—UDG with ROX, Invitrogen), 0.1 mM primers and 0.09 mM VIC probe for the housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), as internal control. Five microliters of DNA were added to 15 μl of the reaction mix, giving a final reaction volume of 20 μl. The amplification profile was optimized for the 7300 Real Time PCR System (Applied Biosystems) as follows: one cycle of decontamination at 50 °C for 2 min, one cycle of denaturation at 95 °C for 10 min followed by 45 cycles of amplification at: 95 °C for 15 s, 60 °C for 60 s. The cycle number during which the fluorescence signal is above the background (Ct) is proportional to the initial log concentration of the target DNA.
Table 1.
Primer/probe sets for Real Time PCR.
| Oligonucleotide (virus-gene) | Sequence (5′ → 3′) | Amplicon size (bp) | Reference |
|---|---|---|---|
| KI-C-F (KI-C-VP1) | GAGCCCACCCCTCATTACTG | 61 | This study |
| KI-C-R | CTTGAACCGCTTTCCTTGTCA | ||
| KI-C-probe | FAM-TCAATTAGCTCTGCCATTG-MGB | ||
| WU-D-F (WU-D-VP2) | AGTCAACCCACAAGAGTGCAAA | 63 | This study |
| WU-D-R | CAGCACGTCTACCCCTCCTTT | ||
| WU-D-probe | FAM-CCTTCCAAAACAAGTCAG-MGB | ||
| GAPDHF (GAPDH) | GCCAAAAGGGTCATCATCTC | 512 | This study |
| GAPDHR | GGGGCCATCCACAGTCTTCT | ||
| GAPDH probe | VIC-TGGTATCGTGGAAGGA-MGB | ||
| KI-A-F (KIV-regulatory region) | ACCTGATACCGGCGGAACT | 95 | Bialasiewicz et al. (2007) |
| KI-A-R | CGCAGGAAGCTGGCTCAC | ||
| KI-A-TM | FAM-CCACACAATAGCTTTCACTCTTGGCGTGA-TAMRA | ||
| KI-B-F (KIV-small T antigen) | GAATGCATTGGCATTCGTGA | 114 | Bialasiewicz et al. (2007) |
| KI-B-R | GCTGCAATAAGTTTAGATTAGTTGGTGC | ||
| KI-B-TM | FAM-TGTAGCCATGAATGCATACATCCCACTGC-TAMRA | ||
| WU-A-F (WUV-regulatory region) | GGCCTACAACAGGGCTTATTTG | 97 | Bialasiewicz et al. (2007) |
| WU-A-R | GAACCCAAGGACGTCTCTGTTAA | ||
| WU-A-TM | FAM-CTTTGTAGTCCAGCGGAAAGTGAAGGGT-TAMRA | ||
| WU-B-F (WUV-large T antigen) | CTACTGTAAATTGATCTATTGCAACTCCTA | 136 | Bialasiewicz et al. (2007) |
| WU-B-R | GGGCCTATAAACAGTGGTAAAACAACT | ||
| WU-B-TM | FAM-CCTTTCCTCCACAAAGGTCAAGTAAA-TAMRA | ||
| WU-C-F (WUV-regulatory region) | GGCACGGCGCCAACT | 115 | Bialasiewicz et al. (2007) |
| WU-C-R | CCTGTTGTAGGCCTTACTTACCTGTA | ||
| WU-C-TM | FAM-TGCCATACCAACACAGCTGCTGAGC-TAMRA | ||
| POLVP1-39F (KIV-VP1)a | AAGGCCAAGAAG TCAAGT TC | 325 | Allander et al. (2007) |
| POLVP1-363Ra | ACA CTC ACT AAC TTG ATT TGG | ||
| AG0044 (WUV-VP2)a | TGTTACAAATAGCTGCAGGTCAA | 490 | Modified from Gaynor et al. (2007) |
| WUVclonRa | TTAAACTCTGTTTCTTCTGACAG | ||
Primers for plasmid construction.
2.4. Standard plasmid construction, optimization, standardization, and validation of the “in-house” Real Time PCR assays
For standard plasmid construction, primers external to KI-C and WU-D were used (Table 1). Plasmids were obtained using the pGEM-T-easy vector system (Promega, Italy). For Real Time PCR quantification a standard curve was created in a 4–log range by serial 10-fold dilutions of the KIV and WUV standard plasmids. Quantitation was determined on the basis of spectrophotometric analysis at 260 nm. The two laboratory designed Real Time PCR assays KI-C and WU-D were optimized by testing different concentrations of the target primers and probe: 0.9/0.25, 0.5/0.15 and 0.2 mM/0.1 mM primer/probe.
The sensitivity of Real Time PCR assays was defined by the lowest concentration of target quantified at a frequency of 100%. The dynamic range, which is defined as the range of dilutions in which a linear regression curve can be constructed, was evaluated using 10-fold dilutions (from 1010 to 100 copies/reaction) of KIV and WUV plasmids. The Ct values outside the measurements comprised within the dynamic range are not quantifiable and a Ct > 40 is considered negative. The precision or intra- and inter-assay variability (CV) was evaluated using different concentrations of standard plasmids (ranging from 105 to 102 copies/reaction) within a single run (n = 10) or different run experiments (n = 10). The efficiency value, defined as 10(−1/slope) and ranging usually from 1.7 to 2.2, was evaluated.
In order to ascertain the ability of detecting the target, a cycle sequencing was performed. The PCR amplification products were used as templates for DNA sequencing using the BigDye Terminator chemistry, following the manufacturer's instructions (BigDye Terminator v3.1 Cycle Sequencing Kit [Applied Biosystems, Warrington, UK]). Subsequently, the nucleotide sequence alignment was performed using the ClustalX 2.0.10 software.
The in vitro specificity of the two laboratory designed Real Time PCR assays was determined by testing positive samples to BKV, JCV and SV40, and viral and microbial strains (exclusivity testing), including human polyomavirus BK (ATCC VR-837), JC (ATCC VR-1583), SV-40 (ATCC VR-820), coxsackievirus types A18 (ATCC VR-176), A20 (ATCC VR-178), B1 (ATCC VR-28), B2 (ATCC VR-29) and B3 (ATCC VR-30), echovirus types 1 (ATCC VR-31), 6 (ATCC VR-36) and 9 (ATCC VR-39), enterovirus type 68 (ATCC VR-561), 69 (ATCC VR-785), 70 (ATCC VR-836) and 71 (ATCC VR-1432), human rhinovirus 15 (ATCC VR-285) and 43 (ATCC VR-336), human parainfluenza viruses (PIV 1 [ATCC VR-94], PIV 2 [ATCC VR-92], PIV 3 [ATCC VR-93]), influenza viruses (influenza A virus H1N1 [ATCC VR-95], H3N2 [ATCC VR-547], influenza B virus [ATCC VR-101]), human respiratory syncytial virus (RSV-A [ATCC VR-26], human adenovirus 3 (ATCC VR-3), 5 (ATCC VR-5), 7 (ATCC VR-7), 40 (ATCC VR-931D) and 41 (ATCC VR-930D), human cytomegalovirus (ATCC VR-538), human coronavirus types 229E (ATCC VR-740) and OC43 (ATCC VR-1558), human rotavirus (ATCC VR-2018), Streptococcus pneumoniae (ATCC 6301), Streptococcus viridans (ATCC 19950), Streptococcus oralis (ATCC 10557), Legionella pneumophila (ATCC 33152), Mycoplasma pneumoniae (ATCC 15377), Chlamydia pneumoniae (ATCC 53592), Clostridium difficile (ATCC 9689), Proteus vulgaris (ATCC 6059), Proteus mirabilis (ATCC 4630), and Escherichia coli (ATCC 4157).
The following formula was used for the quantitation of KIV- and WUV-DNA (100 ng of sample × quantity indicated by the PCR software)/(amplification volume × spectrophotometer reading).
2.5. Prevention of PCR contamination
Precautions must be taken to prevent contamination of the reaction tubes with a product previously amplified, or target DNA from other specimens and controls. The preparation of reagents and processing of samples were carried out in safety cabinets located in separate laboratories, all distant from the area where amplified products were analyzed. Each cabinet was equipped with an independent batch of reagents, micropipette sets, sterile reagent tubes, and filtered pipette tips. Uracil-DNA glycosylase was used to eliminate PCR ‘carry-over’ contamination from previous PCRs.
3. Results
3.1. Clinical samples
A total of 84 stool samples and 91 tonsils were tested to determine the sensitivity of the Real Time PCR assays. The results are summarized in Table 2, Table 3 . Considering stool specimens, KIV was found positive in 17/84 (20.2%), 18/84 (21.4%), and 26/84 (31%) with KI-A, KI-B, and KI-C, respectively; while WUV was positive in 6/84 (7.1%), 6/84 (7.1%), 7/84 (8.3%), and 21/84 (25%) with WU-A, WU-B, WU-C, and WU-D, respectively. In the case of tonsil specimens, KIV was positive in none (0/91), 12/91 (13.2%), and 12/91 (13.2%) with KI-A, KI-B, and KI-C, respectively; while WUV was found in none (0/91), 0/91, 0/91, and 4/91 (4.4%) with WU-A, WU-B, WU-C, and WU-D, respectively.
Table 2.
Results of the Real Time PCR assays.
| N sample | KI-A | KI-B | KI-C | WU-A | WU-B | WU-C | WU-D |
|---|---|---|---|---|---|---|---|
| Stool | |||||||
| 1 | 31.9 | 32.4 | 28.0 | N | N | N | 34.0 |
| 2 | 33.2 | 34.1 | 29.6 | 32.1 | 33.5 | 32.7 | 33.1 |
| 3 | 33.0 | 33.6 | 28.5 | 30.9 | 32.3 | 31.8 | 31.9 |
| 4 | 28.6 | 28.5 | 28.3 | N | N | N | 32.6 |
| 5 | 28.6 | 28.5 | 29.4 | N | N | N | N |
| 6 | 30.7 | 30.7 | 30.9 | N | N | N | N |
| 7 | 26.2 | 26.0 | 27.1 | N | N | N | 33.9 |
| 8 | 32.5 | 32.2 | 32.3 | N | N | N | N |
| 9 | 30.1 | 30.8 | 28.8 | N | N | N | 31.5 |
| 10 | 28.4 | 28.8 | 29.5 | N | N | N | 36.7 |
| 11 | 29.8 | 30.1 | 30.6 | N | N | N | 36.0 |
| 12 | 23.4 | 23.2 | 23.8 | N | N | N | N |
| 13 | 37.4 | N | 33.8 | N | N | N | 37.1 |
| 14 | 33.1 | 33.9 | 31.7 | 31.3 | 32.1 | 31.7 | 32.1 |
| 15 | 35.8 | 36.7 | 28.1 | N | N | N | N |
| 16 | N | 37.4 | 23.7 | N | N | N | 38.2 |
| 17 | 22.1 | 21.6 | 23.1 | N | N | N | 37.3 |
| 18 | N | 37.4 | 29.3 | 21.9 | 22.5 | 22.2 | 22.6 |
| 19 | N | N | 26.6 | 29.8 | 31.5 | 30.3 | 30.8 |
| 20 | N | N | 34.3 | N | N | 37.5 | 36.43 |
| 21 | N | N | 29.0 | N | N | N | 32.4 |
| 22 | N | N | 28.4 | N | N | N | 32.5 |
| 23 | N | N | 29.4 | 29.8 | 31.3 | 30.2 | 30.6 |
| 24 | 24.1 | 24.0 | 24.3 | N | N | N | 35.2 |
| 25 | N | N | 30.0 | N | N | N | 31.6 |
| 26 | N | N | 28.1 | N | N | N | 31.8 |
| Tonsils | |||||||
| 8 | N | 31.8 | 36.0 | N | N | N | 37.8 |
| 15 | N | 33.1 | 38.0 | N | N | N | N |
| 16 | N | 29.7 | 36.7 | N | N | N | 37.2 |
| 19 | N | 33.7 | 36.8 | N | N | N | 36.4 |
| 25 | N | 35.4 | 36.0 | N | N | N | N |
| 39 | N | 39.2 | 38.6 | N | N | N | N |
| 59 | N | 34.5 | 35.2 | N | N | N | N |
| 70 | N | 34.4 | 36.9 | N | N | N | 36.5 |
| 71 | N | 35.4 | 33.3 | N | N | N | N |
| 77 | N | 38.9 | 39.5 | N | N | N | N |
| 80 | N | 37.1 | 36.9 | N | N | N | N |
| 93 | N | 35.8 | 36.8 | N | N | N | N |
Cycle threshold (Ct) values are indicated. N = negative. Reported values of Ct represent the mean of measurements obtained in triplicate.
Table 3.
Prevalence of KIV and WUV by the different PCR assays evaluated in this study.
| KI-A |
KI-B |
KI-C |
WU-A |
WU-B |
WU-C |
WU-D |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | Positive samples (%) | Negative samples (%) | |
| Stool (N = 84) | 17 (20.2%) | 67 (79.8%) | 18 (21.4%) | 66 (78.6%) | 26 (31.0%) | 58 (69.0%) | 6 (7.1%) | 78 (92.9%) | 6 (7.1%) | 78 (92.9%) | 7 (8.3%) | 77 (91.7%) | 21 (25.0%) | 63 (75.0%) |
| Tonsils (N = 91) | 0 (0%) | 91 (100%) | 12 (13.2%) | 79 (86.8%) | 12 (13.2%) | 79 (86.8%) | 0 (0%) | 91 (100%) | 0 (0%) | 91 (100%) | 0 (0%) | 91 (100%) | 4 (4.4%) | 87 (95.6%) |
3.2. Optimization, standardization, and validation of the “in-house” Real Time PCR assays
The following primer/probe concentrations were chosen: 0.5/0.15 and 0.9 mM/0.25 mM for KIV and WUV, respectively. The dynamic range of the two laboratory designed Real Time PCRs was evaluated by carrying out in triplicate serial dilutions of standard plasmids (from 1010 to 100 copies/reaction) and was as follows: from 1010 to 102 for KIV (Fig. 1 ) and from 1010 to 101 for WUV (Fig. 2 ). The sensitivity of the assays was evaluated in triplicate and resulted 102 and 101 for KIV and WUV, respectively. The precision, defined as the level of concordance of the individual test results within a single round (intra-assay variability) and in different experiments (inter-assay variability), was evaluated using different concentrations of plasmid standards, ranging from 105 to 102 copies/reaction, and is reported in Table 4 . The efficiency value was 2.06 and 1.92 for KIV and WUV, respectively. Individual assays were compared to the respective performances in duplex (with GAPDH) and no differences, in terms of Ct, were recorded.
Fig. 1.
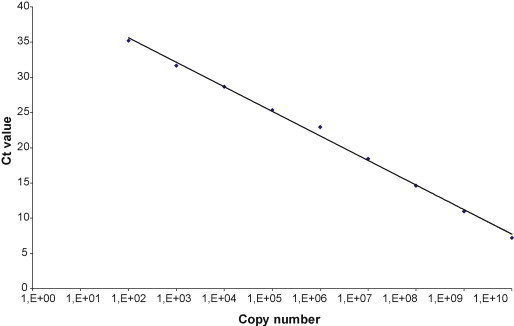
KIV (KI-C) dynamic range, from 1010 to 102 copies/reaction (y = −3.5x + 39.1; R2 = 0.99).
Fig. 2.
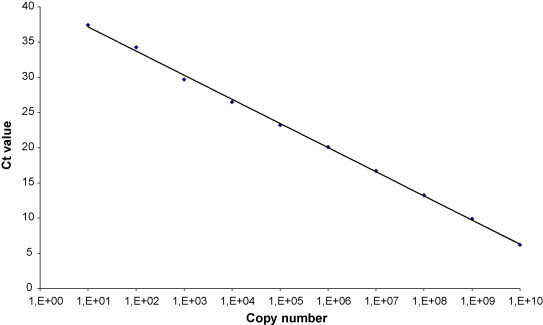
WUV (WU-D) dynamic range, from 1010 to 101 copies/reaction (y = −3.4x + 40.6; R2 = 0.99).
Table 4.
Intra- and inter-assay variability of KIV and WUV standard curve by Real Time PCR.
| Standard dilution (plasmid copies/reaction) | Coefficient of variability (%) |
|||
|---|---|---|---|---|
| Intra-assay (%) |
Inter-assay (%) |
|||
| KIV | WUV | KIV | WUV | |
| 102 | 0.83 | 0.18 | 1.68 | 3.29 |
| 103 | 0.35 | 0.93 | 0.83 | 0.62 |
| 104 | 0.14 | 0.70 | 1.94 | 0.80 |
| 105 | 1.48 | 0.68 | 2.42 | 1.53 |
The sensitivity of assay was determined by the lowest standard dilution detectable consistently in triplicate at a frequency of 100% and it was found to be of 102 and 101 copies/reaction for KIV and WUV, respectively.
The cycle sequencing showed that the two PCR assays have a complete homology with all the eight KIV isolates and 12/14 WUV isolates described to date (www.ncbi.nlm.nih.gov web site search performed on March 31, 2009), as forward primer for WUV recognized 95% of the sequence of WU/Wuerzburg/01/06 and WU/Wuerzburg 03/07 (GenBank accession nos. EU711055.1 and EU711058.1), due to the presence of a puntiform mutation at the nucleotide 1739. No significant homology to any other sequence was found. No positive result was demonstrated for the other viral and microbial strains indicating that these molecular assays are specific for KIV and WUV.
Primers and probes were tested in silico for possible cross-reactivity with human sequences, unrelated viral and other microbial sequences based on the data available at the BLAST alignment software (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Viral load was determined, based on standard curve from the two laboratory designed Real Time PCRs, and resulted as follows: for the faecal samples the average load was 3508 genome equivalents/100 ng of extracted DNA (median 785) and 10733 (median 90) for KIV and WUV, respectively; viral load quantification in tonsil specimens was not possible due to Ct values behind linearity range (i.e. Ct values corresponding to 102 and 101 copies/reaction for KIV and WUV, respectively).
4. Discussion
Currently, the presence of the new polyomaviruses KI and WU has not been investigated extensively (Allander et al., 2007, Gaynor et al., 2007, Norja et al., 2007, Payungporn et al., 2008, Jiang et al., 2009, Lindau et al., 2009), thus the availability of optimized and clinically validated Real Time PCR assays could be useful for investigating the epidemiological and clinical impact of these viruses, as well as their tissue tropism.
Real Time PCR assays are described as ‘closed’ systems, since no post-amplification manipulation of the amplicon is required. The advantages of these systems, in comparison to conventional PCR, include a reduced turnaround time, minimising of the potential for carry-over contamination and the ability to scrutinise closely the assay performance, thus representing a suitable tool for routine diagnosis in virology. Real Time PCR offers significant improvements to viral load quantitation because of its wide dynamic range which can accommodate at least eight log 10 copies of nucleic acid template. This is made possible because the data are chosen from the linear phase of amplification where conditions are optimal, rather than the end-point where the final amount of amplicon may have been affected by inhibitors, poorly optimized reaction conditions or saturation by inhibitory PCR by-products and double-stranded amplicon. In regards to Real Time PCR, the design of primer/probe sets, although relatively simple from a computational point of view, has to take into account several factors, especially the choice of target region.
In this study, the assessment of two laboratory designed Real Time PCR TaqMan assays for the detection of polyomaviruses KI and WU, targeting the VP1 and VP2 genes, respectively, is described. These methods have been compared to those described previously (Bialasiewicz et al., 2007) and targeting different regions, in the light of the potential impact of its choice on the detection of KIV- and WUV-DNA, and the interpretation of prevalence results in different clinical syndromes.
Considering the detection rates of KIV and WUV in clinical samples using the different assays, the primer/probe sets designed in this study appeared more sensitive in comparison to those published. This could be attributed to the choice of the target region. Among the 26 faecal specimens positive by the KI-C assay, only 17/26 (65.4%) and 18/26 (69.2%) were positive by KI-A and KI-B, respectively; while, of the 12 tonsil samples positive with the KI-C, all were positive by KI-B and none by KI-A. However, KI-B exhibited lower Ct values in comparison to KI-C. Despite that, in a high-throughput diagnostic laboratory is essential to enhance an assay to make it suitable for use on a wide range of matrices. In this regard, KI-C seems to be more reliable for the detection of KIV in the two matrices analyzed. Considering the WU-D assay, this appeared to be superior to the other assays as additional stool and tonsil specimens were found to be positive.
As the epidemiological and clinical impact of these new viruses is still elusive, the availability of highly sensitive and specific methods is mandatory to obtain reliable and relevant data. The assessment of a PCR assay for virus detection is based on knowledge of a representative number of genome sequences to allow for primer/probe set design. At present, the complete genome sequence of only eight and 14 clinical isolates for KIV and WUV, respectively, is known; while a larger number of partial gene sequences have been characterized. The assays developed in this study show complete sequence homology with all the KIV and 12 WUV isolates; this is due to the presence of a puntiform mutation in the target region VP2. The choice of this region, instead of the large T used commonly for human polyomaviruses, was considered unavoidable in a diagnostic laboratory working on BKV and JCV. Therefore, the comparison of methods targeting different genome regions can represent a useful tool and avoid false-negative findings.
The optimization of the two laboratory designed Real Time PCRs allowed the identification of amplification conditions which are more suitable in terms of primer/probe concentrations, efficiency, linearity, sensitivity, specificity, and precision.
5. Conclusions
In conclusion, the development of two laboratory designed Real Time PCR TaqMan assays for quantitative detection of polyomaviruses KI and WU and the optimization and standardization of these assays using different clinical samples (in particular stool and tonsils) are described. Among the various parameters that have to be considered in the development of a Real Time PCR assay, the choice of the primer/probe set, and primarily of the region to amplify, could represent a useful tool to understand the pathogenic role of viruses such as KIV and WUV.
Source of funding
This work was supported by a research grant of Regione Piemonte, Ricerca Sanitaria Finalizzata 2008bis to Cristina Costa.
References
- Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M.A., Dalianis T., Ramqvist T., Andersson B. Identification of a third human polyomavirus. J. Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babakir-Mina M., Ciccozzi M., Dimonte S., Farchi F., Valdarchi C., Rezza G., Perno C.F., Ciotti M. Identification of the novel KI polyomavirus in the respiratory tract of an Italian patient. J. Med. Virol. 2008;80:2012–2014. doi: 10.1002/jmv.21303. [DOI] [PubMed] [Google Scholar]
- Babakir-Mina M., Ciccozzi M., Campitelli L., Aquaro S., Lo Coco A., Perno C.F., Ciotti M. Identification of the novel KI polyomavirus in paranasal and lung tissues. J. Med. Virol. 2009;81:558–561. doi: 10.1002/jmv.21417. [DOI] [PubMed] [Google Scholar]
- Bergallo M., Costa C., Margio S., Sidoti F., Segoloni G.P., Ponzi A.N., Cavallo R. Detection and typing of BKV, JCV, and SV40 by multiplex nested polymerase chain reaction. Mol. Biotechnol. 2007;35:243–252. doi: 10.1007/BF02686010. [DOI] [PubMed] [Google Scholar]
- Bialasiewicz S., Whiley D.M., Lambert S.B., Gould A., Nissen M.D., Sloots T.P. Development and evaluation of real-time PCR assays for the detection of the newly identified KI and WU polyomaviruses. J. Clin. Virol. 2007;40:9–14. doi: 10.1016/j.jcv.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butel J.S., Lednicky J.A. Cell and molecular biology of simian virus 40: implications for human infections and disease. J. Natl. Cancer Inst. 1999;91:119–134. doi: 10.1093/jnci/91.2.119. [DOI] [PubMed] [Google Scholar]
- Costa C., Bergallo M., Astegiano S., Terlizzi M.E., Sidoti F., Segoloni G.P., Cavallo R. Monitoring of BK virus replication in the first year following renal transplantation. Nephrol. Dial. Transplant. 2008;23:3333–3336. doi: 10.1093/ndt/gfn289. [DOI] [PubMed] [Google Scholar]
- Dimech W., Bowden D.S., Brestovac B., Byron K., James G., Jardine D., Sloots T., Dax E.M. Validation of assembled nucleic acid-based tests in diagnostic microbiology laboratories. Pathology. 2004;36:45–50. doi: 10.1080/0031302032000174941. [DOI] [PubMed] [Google Scholar]
- Gardner S.D., Field A.M., Coleman D.V., Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;1:1253–1257. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- Gaynor A.M., Nissen M.D., Whiley D.M., Mackay I.M., Lambert S.B., Wu G., Brennan D.C., Storch G.A., Sloots T.P., Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M., Abend J.R., Johnson S.F., Imperiale M.J. The role of polyomaviruses in human disease. Virology. 2009;384:266–273. doi: 10.1016/j.virol.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau C., Tiveljung-Lindell A., Goh S., Ramqvist T., Allander T. A single-tube, real-time PCR assay for detection of two newly characterized human KI and WU polyomaviruses. J. Clin. Virol. 2009;44:24–26. doi: 10.1016/j.jcv.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Norja P., Ubillos I., Templeton K., Simmonds P. No evidence for an association between infections with WU and KI polyomaviruses and respiratory disease. J. Clin. Virol. 2007;40:307–311. doi: 10.1016/j.jcv.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett B.L., Walker D.L., ZuRhein G.M., Eckroade R.J., Dessel B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–1260. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- Payungporn S., Chieochansin T., Thongmee C., Panjaworayan N., Samransamruajkit R., Theamboolers A., Poovorawan Y. Detection and discrimination of WU/KI polyomaviruses by real-time PCR with melting curve analysis. J. Virol. Methods. 2008;153:70–73. doi: 10.1016/j.jviromet.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Rabenau H.F., Kessler H.H., Kortenbusch M., Steinhorst A., Raggam R.B., Berger A. Verification and validation of diagnostic laboratory tests in clinical virology. J. Clin. Virol. 2007;40:93–98. doi: 10.1016/j.jcv.2007.07.009. [DOI] [PubMed] [Google Scholar]