

Abstract
The recombinant antigen obtained by cloning and expressing two IBV nucleocapsid protein fragments (143-414 aa, 281-414 aa) in Escherichia coli was used for the detection of avian infectious bronchitis virus (IBV) specific antibodies in chicken sera by the indirect ELISA (rNpIBV-ELISA). As a result of testing 1524 serum samples the diagnostic sensitivity and specificity of rNpIBV-ELISA when comparing those of the routine whole IBV ELISA have been shown to be 93.81% and 87.36%, respectively. The agreement value was 91.5%.
Keywords: Avian IBV, Nucleoprotein, Recombinant antigen, ELISA
Infectious bronchitis (IB) is an important disease economically to the poultry industry because it affects chickens of all ages and causes respiratory and nephrotic syndromes in broilers, reduced egg production in layers and breeders (Cavanagh and Naqi, 1997). This disease is caused by infectious bronchitis virus (IBV), a member of the family Coronaviridae (order Nidovirales) and genus Coronavirus (Cavanagh, 1997, Ziebuhr et al., 2000). IBV is an enveloped virus containing an unsegmented, single-stranded, positive-sense RNA genome. The virion includes four major structural proteins: the surface spike glycoprotein (S) consisting of two subunits S1 and S2, the membrane (M) glycoprotein, the phosphorylated nucleocapsid (N) protein and the envelope (E) protein. N protein of IBV is highly conserved, highly immunogenic. It carries epitopes inducing cross-reactive antibodies and is the most abundant virus-derived protein produced throughout infection (Seah et al., 2000). N protein may also induce cross-protective immunity (Boots et al., 1992, Seo et al., 1997, Yu et al., 2001).
Currently, indirect enzyme-linked immunosorbent assay (ELISA) using whole virus IBV antigen is carried out worldwide for measuring the level of IBV specific antibodies.
However, the production of IBV in SPF-chicken embryo eggs or tissue cultures, the inactivation of viral suspension, the concentration and the purification of IBV antigen for ELISA are very expensive and laborious procedures. In contrast, the use of recombinant full-length N protein or fragments of IBV N protein cloned and expressed into Escherichia coli or yeast as ELISA antigens for IBV-specific antibody makes testing serum samples a much cheaper and more convenient process (Chen et al., 2003, Gibertoni et al., 2005, Ndifuna et al., 1998).
In the study, two recombinant proteins, analogues of the IBV nucleoprotein fragments, were used as antigen for an IBV-specific antibody ELISA (rNpIBV-ELISA).
IBV vaccine strain H52 Massachusetts type was passaged initially in 9–11-days chicken SPF-embryos of to extract viral RNA as described by Gribanov et al. (1997).
Two fragments of N gene were chosen for cloning. One clone coded the fragment of N protein (143-414 aa) with four linear immunodominant epitopes, and the other coded the fragment of N protein (281-414 aa) with two epitopes (Seah et al., 2000). Three primers for sequencing the H52 reference strain were used to amplify two overlapping fragments of IBV N gene by PCR: N1IBV–N3IBV, fragment 1; N2IBV–N3IBV, fragment 2 (Table 1 , Fig. 1c). Restriction sites BamHI were added to 5′-ends of the two upstream primers N1IBV and N2IBV or HindIII to 3′-end of the downstream primer N3IBV. The open reading frame (ORF) was between 433 and 1242 bp. RT-PCR amplification of two truncated fragments of the N-gene was performed as follows: reverse transcription (50 °C, 15 min), denaturation of RNA and inactivation of reverse transcriptase (94 °C, 3 min), standard PCR steps including denaturation (94 °C, 0.5 min), annealing (50 °C, 0.5 min) and extension (72 °C, 1 min) for 35 cycles (Lugovskaya et al., 2002). The PCR products of expected sizes are shown in Fig. 1a. The sizes of the two truncated fragments were 809 and 398 bp, respectively. Amplified fragments were sequenced using a “fmol DNA Cycle Sequencing System” (Promega Corp., USA) and both sequences corresponded to the nucleocapsid IBV H52 gene.
Table 1.
List of primers used to amplify cDNA fragments coding for targeted regions of the rNp2IBV and rNp4IBV proteins
| Primer | Nucleotide sequence | Position (nucleotides) |
|---|---|---|
| N1IBV | 433 to 458 | |
| N2IBV | 844 to 868 | |
| N3IBV | 1242 to 1216 |
Fig. 1.
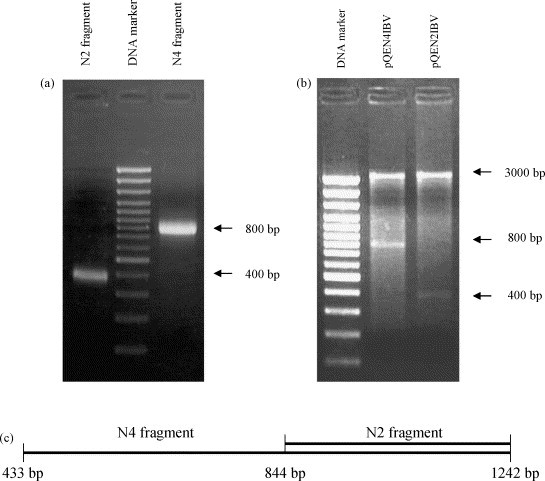
IBV nucleocapsid gene fragments. (a) Electrophoresis in 1.5% agarose gel. Amplified PCR products of the nucleocapsid gene fragments. (b) Electrophoresis in 1.5% agarose gel. The nucleocapsid gene fragments isolated from pQEN4IBV and pQEN2IBV (N4 fragment and N2 fragment, respectively) by the restriction with BamHI and HindIII. N4 fragment, 809 bp; N2 fragment, 398 bp. (c) Locations of overlapping N gene fragments of IBV encoding four (N4 fragment) and two (N2 fragment) antigen sites.
The amplified PCR products were purified using the “Total RNA Isolation System” (Promega Corp., USA), digested using BamHI and HindIII enzymes and ligated into a pQE vector (QIAGEN GmbH, Hilden, Germany) followed by transformation into E. coli strain M15 according to the manufacturer's protocol. The constructed recombinant plasmids designated pQEN2IBV and pQEN4IBV were sequenced confirming that they were both in frame. The size of insertions was confirmed by BamHI and HindIII restriction as shown in Fig. 1b.
Positive ampicillin- and kanamicin-resistant clones were identified by PCR screening and were confirmed further by enzymatic cutting. Bacteria from a single colony were grown to an optical density at 600 nm of 0.5–0.7 in Luria-Bertani medium supplemented with ampicillin and kanamicin up to final concentrations of 100 and 25 μg/ml, respectively, and induced with 2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). In the course of IPTG induction for 5 h at 37 °C, the inserted with a six–histidine tag (His6) genes were expressed as fusion proteins. Cells were pelleted at 5000 rpm for 20 min and lysed with 6 M guanidine-HCl (Dia M, Russia) for 1 h. Proteins were purified using nickel-nitrilotriacetic acid metal affinity chromatography (Ni-NTA) agarose binding the His6 tag according to the manufacture's recommendation (QIAGEN GmbH, Hilden, Germany). The concentration of purified recombinant N proteins designated rNp2IBV and rNp4IBV was determined by the quantitative Bradford's method (Bradford, 1976) using bovine serum albumin (BSA) standards.
The protein concentration of each of the proteins was found to be approximately 2.3–6 mg/ml.
Expression of pQEN2IBV and pQEN4IBV in E. coli system and purification of proteins from cell lysates were analyzed by SDS-PAGE according to the Laemmli method (Laemmli, 1970) (Fig. 2a). Recombinant protein specificity was tested using Western blot with chicken antisera (Fig. 2c). Bacterial whole-cell lysates and purified recombinant proteins were applied to 12.5% polyacrylamide gels and separated by electrophoresis at constant voltage 200 V. The gels were stained with Coomassie blue R-250 to detect proteins. The protein band of approximately 20 kDa was clearly visualized following the induction of fusion protein from pQEN2IBV with IPTG. At the same time, partial SDS-PAGE proteolysis was shown to proceed in the course of expression of the fusion protein from pQEN4IBV; two protein bands of approximately 35 and 30 kDa were seen (Fig. 2a and b). However, the proteolytic products did not have any effect on the specificity or sensitivity of an indirect ELISA based on the recombinant protein as antigen (rNpIBV-ELISA) as seen below. For Western blots, the proteins were transferred to nitrocellulose membranes 0.45 μm pore size (Millipore Corp., USA) at 15 V and 200 mA for 1.5 h. The membranes were then treated with blocking buffer including 1% BSA before being incubated with chicken serum samples diluted 1:50 in TBST buffer (pH 7.4), containing 0.02 M Tris–HCl, 0.15 M NaCl, 0.05% Tween-20, at room temperature for 1 h, followed by incubation with a horseradish peroxidase-conjugated secondary anti-chicken immunoglobulin G (Synbiotics Corp., USA), washed three times with TBST each time, and finally 4-chloro-1-naphtol (Sigma Chemical Company, USA) was added to visualize protein bands.
Fig. 2.
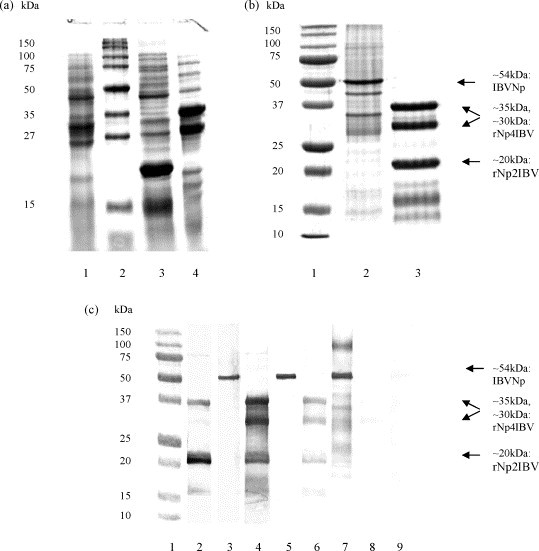
SDS-PAGE on a 12.5% gel and Western blotting. (a) SDS-PAGE of bacterial lysates. Lane 1, lysate of E. coli cells in the absence recombinant plasmid; lane 2, MW marker; lane 3, lysate of E. coli cells with pQEN2IBV after IPTG expression; lane 4, lysate of E. coli cells with pQEN2IBV after IPTG expression. (b) SDS-PAGE of rNp2IBV and rNp4IBV mixture and purified and concentrated antigen of IBV. Lane 1, MW marker; lane 2, antigen of IBV; lane 3, mixture of recombinant proteins. (c) Western blot analysis. Lane 1, MW marker. The transferred mixture of rNp2IBV and rNp4IBV and proteins of IBV antigen were reacted with rNp2IBV-specific chicken serum (lanes 2 and 3, respectively), with rNp4IBV-specific chicken serum (lanes 4 and 5, respectively), with IBV-specific chicken serum (lanes 6 and 7, respectively), with normal chicken serum (lanes 8 and 9, respectively).
In order to detect IBV-specific antibodies in chicken sera an rNpIBV-ELISA was developed. The commercial IBV antibody test kit contained IBV antigen of Massachusetts type (The Federal Center for Animal Health, Russia) is certified for Russia and used as the standard routine IBV-ELISA. The reaction was performed according to the instructions.
Flat-bottomed plate wells (NUNC, Immunoplate, Denmark) were coated with 0.5–1 μg of the mixture of two recombinant proteins rNp2IBV and rNp4IBV in sodium carbonate–bicarbonate buffer pH 9.6 at 4 °C overnight and unbound sites were blocked with a 1% (v/v) BSA in TBST (BSA-TBST) for 1 h. Using the mixture of proteins by an ELISA detected more positive samples than ELISAs using each of the proteins separately (data not shown). The control serum samples and the samples to be tested were diluted in BSA-TBST and incubated for 1 h at room temperature. Thereafter, goat anti-chicken horseradish peroxidase labeled antibodies (Synbiotics Corp., USA) were added at a working dilution and held for 1 h at room temperature. Washing of wells in TBST four times ended each stage. The antigen–antibody complexes were detected with ABTS (40 mM 2.2′-azino-di-3-ethyl-benzthiazoline sulfonic acid) as substrate and 4 mM hydrogen peroxide in 100 mM citrate buffer pH 4.5. The color development was stopped with 1% sodium dodecyl sulphate (SDS) after 15-min incubation and the intensity was measured at 405 nm (SLT-spectra, Austria).
Sera from vaccinated chicken (n = 216) were tested by a serial two-fold dilution method to determine the respective IBV-specific antibody rNpIBV-ELISA end-point. Simultaneously the same serum samples were analyzed at a 1:100 dilution. The reactivity of each test sample was determined by subtracting the mean optical density (OD) of the negative control from the mean OD of duplicate test samples. The resulting corrected OD value for a test sample was divided by the mean OD value of the positive control from which the mean negative OD value was subtracted, and the results were expressed as the S/P ratio. The regression line to correlate the results of single and serial two-fold dilution methods was plotted with the aid of the computer program “Statistica, correlation matrices” (Stat. Soft. Inc., USA). A regression line was plotted to correlate the rNpIBV-ELISA end-point and the S/P value of 216 chicken serum samples at a 1:100 dilution. The equation of regression line was log10 T = 3.612 + 1.755 × log10 S/P. The correlation coefficient r = 0.9615 was obtained. The coefficient was statistically significant (p < 0.05), indicating a good linearity and a strong association between the rNpIBV-ELISA titre calculated by the above formula or determined by the serial two-fold dilution method.
Sera from unvaccinated chicken (n = 130) were examined to determine the serum cutoff of rNpIBV-ELISA. From these results the mean and the sample standard deviation were calculated. The negative threshold was then determined as the lower 99% confidence limit of the mean plus 3 S.D. The mean (±S.D.) A405 and S/P ratio of all negative sera by rNpIBV-ELISA were 0.163 (±0.026) and 0.076 (±0.039), respectively, using the mean ± 3 S.D. as cutoff value (S/P ratio, 0.193).
Field serum samples from vaccinated and unvaccinated chicken (n = 1524) from 28 farms of the Russian Federation were tested by both rNpIBV-ELISA and the routine specific IBV ELISA (IBV-ELISA). The obtained data were compared (Table 2 ). The diagnostic specificity, sensitivity of both assays and agreement value between two ELISAs were determined as described by Jacobson (1998). As a result the sensitivity and specificity of the developed rNpIBV-ELISA was found to be not inferior (93.81% and 87.36%, respectively) to those of the routine IBV-ELISA (92.86% and 88.97%, respectively). The agreement value was 91.5%. Heterologous sera to the viruses of infectious bursal disease, Newcastle disease, infectious laryngotracheitis, egg drop syndrome—76, encephalomyelitis, to adeno- and reoviruses and to Mycoplasma gallisepticum were negative by both rNpIBV-ELISA and IBV-ELISA (data not shown).
Table 2.
Comparison of rNpIBV-ELISA and IBV-ELISA for detection of IBV-specific antibodies in chicken sera
| IBV-ELISA result | rNpIBV-ELISA result (no. of analyzed serum samples) |
||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 910 | 60 | 970 |
| Negative | 70 | 484 | 554 |
| Total | 980 | 544 | 1524 |
The rNpIBV-ELISA using the mixture of the two recombinant N proteins as an antigen demonstrated high sensitivity and specificity for the detection of IBV-specific antibodies in 1524 chicken serum samples. This test-system can be use for routine investigations.
Acknowledgement
Assistance of Mrs. L.K. Frymovich in preparing the manuscript for publication is greatly appreciated.
References
- Boots A.M., Benaissa-Trouw B.J., Hesselink W., Rijke E., Schrier C., Hensen E.J. Induction of anti-viral immune responses by immunization with recombinant-DNA encoded avian coronavirus nucleocapsid protein. Vaccine. 1992;10:119–124. doi: 10.1016/0264-410X(92)90028-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- Cavanagh D., Naqi S.A. Infectious bronchitis virus. In: Calnek B.W., Barnes Hj., Beard C.W., McDougald L.R., Saif Y.M., editors. Disease of Poultry. 9th ed. Iowa State University Press; Ames, IA: 1997. pp. 511–526. [Google Scholar]
- Chen H., Coote B., Attree S., Hiscox J.A. Evaluation of a nucleoprotein-based enzyme-linked immunosorbent assay for the detection of antibodies against infectious bronchitis virus. Avian Pathol. 2003;32:519–526. doi: 10.1080/0307945031000154125. [DOI] [PubMed] [Google Scholar]
- Gibertoni A.M., Montassier Mde.F., Sena J.A., Givisiez P.E., Furuyama C.R., Montassier H.J. Development and application of Saccharomyces cerevisiae-expressed nucleocapsid protein-based enzyme-linked immunosorbent assay for detection of antibodies against infectious bronchitis virus. J. Clin. Microbiol. 2005;43:1982–1984. doi: 10.1128/JCM.43.4.1982-1984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribanov O.G., Shcherbakov A.V., Perevozchikova N.A., Drygin V.V., Gusev A.A. A simple method for RNA isolation and purification. Bioorg. Khim. 1997;23:692–694. (Russian) [PubMed] [Google Scholar]
- Jacobson R.H. Validation of serological assays for diagnosis of infectious diseases. Rev. Sci. Tech. 1998;17(2):469–526. doi: 10.20506/rst.17.2.1119. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lugovskaya N.N., Lugovskoy A.A., Bochkov Yu.A., Mudrak N.S., Drygin V.V., Borisov A.V., Borisov V.V., Gusev A.A. Detection and estimation of avian infectious bronchitis virus antigen by novel indirect liquid-phase blocking enzyme-linked immunosorbent assay using chicken and rabbit affinity purified immunoglobulins. Avian Pathol. 2002;31:549–557. doi: 10.1080/0307945021000024571. [DOI] [PubMed] [Google Scholar]
- Ndifuna A., Waters A.K., Minglong Z., Collinsson E.W. Recombinant nucleocapsid protein is potentially an inexpensive, effective serodiagnostic reagent for infectious bronchitis virus. J. Virol. Methods. 1998;70:37–44. doi: 10.1016/S0166-0934(97)00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seah J.N., Yu L., Kwang J. Localization of linear B-cell epitopes on infectious bronchitis virus nucleocapsid protein. Vet. Microbiol. 2000;75:11–16. doi: 10.1016/s0378-1135(00)00202-9. [DOI] [PubMed] [Google Scholar]
- Seo S.H., Wang L., Smith R., Collisson E.W. The carboxyl-terminal 120-residue polypeptide of infectious bronchitis virus nucleocapsid induces cytotoxic T lymphocytes and protects chickens from acute infection. J. Virol. 1997;71:7889–7894. doi: 10.1128/jvi.71.10.7889-7894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Liu W., Schnitzlein W.M., Tripathy D.N., Kwang J. Study of protection by recombinant fowl poxvirus expressing C-terminal nucleocapsid protein of infectious bronchitis virus against challenge. Avian Dis. 2001;45:340–348. [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]