

Abstract
Diffuse alveolar hemorrhage (DAH) is a clinical syndrome characterized by generalized intra-alveolar bleeding originating from the pulmonary microcirculation. The finding of DAH carries an extended differential diagnosis and may be associated with a number of histopathologic patterns. The prompt recognition and diagnosis of DAH is of critical importance to the practicing clinician as accurate diagnosis and prompt initiation of therapy may dramatically improve patient outcomes. This chapter reviews the diagnosis and management of diffuse alveolar hemorrhage.
Keywords: Alveolar Hemorrhage, Vasculitis, Capillaritis, Granulomatosis with Polyangiitis, Microscopic Polyangiitis, Systemic Lupus Erythematosus, Anti-Phospholipid Antibody Syndrome, Anti-Basement Membrane Antibody Disease, Idiopathic Pulmonary Hemosiderosis
Introduction
Diffuse alveolar hemorrhage (DAH) is a clinical syndrome defined by generalized intra-alveolar bleeding originating from the pulmonary microcirculation. Patients commonly present with dyspnea, hemoptysis, anemia, diffuse radiographic pulmonary infiltrates and hypoxemia. The severity can range from mild dyspnea to severe hypoxemic respiratory failure requiring mechanical ventilation. Diagnosis is frequently made at the time of bronchoscopy, when serial aliquots of bronchoalveolar lavage (BAL) fluid reveal a progressively hemorrhagic return. However, the presence of DAH carries a broad differential diagnosis (Table 10.1) and is associated with a number of histopathologic patterns. This chapter will review the approach to the diagnosis and the management of DAH.
Table 10.1.
Differential diagnosis of DAH based on pathology
| Histology | Etiologies |
|---|---|
| Pulmonary Capillaritis | Granulomatosis with polyangitis |
| Microscopic polyangitis | |
| Isolated pulmonary capillaritis | |
| Systemic lupus erythematosus | |
| Primary antiphospholipid antibody syndrome | |
| Other collagen vascular disorders/connective tissue diseases | |
| Henoch-Schönlein purpura | |
| Behçet Syndrome | |
| Goodpasture syndrome | |
| Acute lung transplant rejection | |
| Hematopoietic stem cell transplantation | |
| Cryoglobulinemia | |
| Drugs and medications (e.g. propylthiouracil) | |
| Bland Pulmonary Hemorrhage | Idiopathic pulmonary hemosiderosis |
| Goodpasture syndrome | |
| Systemic lupus erythematosus | |
| Coagulation disorders | |
| Inhalational exposures (e.g. trimellitic anhydride, isocyanates) | |
| Drugs and medications (e.g. penicillamine, amiodarone, nitrofurantoin) | |
| Mitral stenosis/valvular heart disease | |
| Left ventricular dysfunction | |
| Obstructive sleep apnea | |
| Pulmonary veno-occlusive disease | |
| Diffuse Alveolar Damage | Acute respiratory distress syndrome |
| Acute idiopathic pneumonia | |
| Hematopoietic stem cell transplantation | |
| Drugs and medications (e.g. cocaine inhalation) | |
| Acute exacerbation of interstitial lung disease | |
| Miscellaneous | Lymphangioleiomyomatosis |
| Human immunodeficiency virus infection | |
| Pulmonary capillary hemoangiomatosis |
Clinical Vignettes
Case One
A 19 year old man presented to the emergency room complaining of 1 week of progressive dyspnea on exertion and non-productive cough, initially thought to be a respiratory infection. On further history, he revealed a 1 month history of a non-pruritic rash on his legs and ankles. He denied any fever, chills, chest pain, sputum production, recent inhalational injury, cocaine or other drug use, or any human immunodeficiency virus (HIV) risk factors. Review of systems was positive for fatigue, malaise, abdominal pain that was worse after meals, and diffuse arthralgias, particularly of the large joints. He also endorsed multiple episodes of hematochezia (passing bright red blood per rectum) over the past month. He denied any sinus disease, gross hematuria, focal weakness, or paresthesias. His only medications were non-steroid anti-inflammatory agents on an as needed basis.
Physical exam revealed tachycardia, tachypnea, increased respiratory effort with accessory muscle use and significant hypoxemia with an oxygen saturation of 92 % while on high flow oxygen through a non-rebreather mask. He was anxious and speaking only in short sentences. His pulmonary exam revealed diffuse bilateral crackles. His abdominal examination revealed diffuse tenderness and a positive guaiac test for occult blood. His skin exam was notable for irregular, palpable, slightly raised, purpuric lesions with surrounding petechiae on his lower extremities.
The patient’s respiratory status deteriorated over the ensuing 4–5 h, ultimately requiring intubation and mechanical ventilation. Laboratory testing was notable for an elevated white blood cell count of 17,000 cells/mm3 and an elevated erythrocyte sedimentation rate of 87 mm. His laboratory testing also indicated acute renal failure with a creatinine of 1.8 mg/dl, and his urinalysis revealed both granular casts and microscopic hematuria, but no red blood cell casts. Chest imaging revealed patchy, heterogenous, diffuse bilateral infiltrates and bronchoscopy revealed an increasingly bloody return on serial aliquots. Skin biopsy confirmed a leukocytoclastic vasculitis and IgA positive immunofluorescence. The patient was diagnosed with Henoch-Schönlein purpura.
The patient was treated aggressively with intravenous corticosteroids, cyclophosphamide and plasmapharesis. He had resolution of his respiratory failure and was liberated from mechanical ventilation on hospital day #5. His renal function also subsequently returned to normal, and he was discharged to home on hospital day #16.
Case Two
A 28 year old man presented to clinic for progressive dyspnea and fatigue. The patient has a complex past medical history notable for multiple episodes of deep venous thrombosis and a known diagnosis of anti-phospholipid antibody syndrome. Further work-up for systemic lupus erythematosus and other collagen vascular diseases was negative. The patient has been maintained on chronic oral anti-coagulation for the past 4–5 years.
Approximately, 1 year ago the patient had a “flare” of his disease that began with a non-productive cough, fatigue and dyspnea, similar to his current presentation. However, with the earlier episode, he went on to develop hemoptysis and respiratory distress. Surgical lung biopsy at an outside hospital revealed alveolar hemorrhage and an underlying fibrotic non-specific interstitial pneumonitis. He was treated with intravenous corticosteroids and improved. Since that time, his oral corticosteroids have slowly been weaned, and at the time of the current presentation, he was down to 10 mg of oral Prednisone every other day. Of note, the patient also reported that he had recently resumed smoking 1/4–1/2 pack of cigarettes per day.
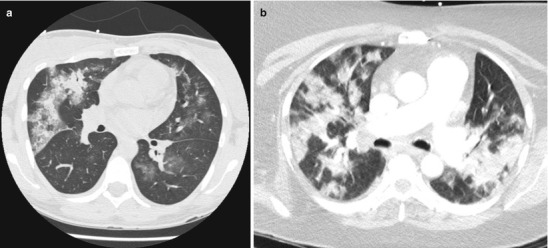
Physical examination was notable for a mildly elevated heart rate of 100 beats per minute and a mildly elevated respiratory rate of 20 breaths per minute. Auscultation revealed crackles at the right base, but breathing was otherwise easy, symmetric and unlabored. Pulmonary function testing revealed a forced vital capacity that was 65 % predicted and FEV1 that was 70 % predicted, but a normal diffusing capacity of carbon monoxide (DLCO) at 90 % predicted that corrected to 108 % predicted when adjusted for alveolar volume. High resolution computed tomography (HRCT) of the chest demonstrated patchy ground glass opacities (Fig. 10.1a, b). Bronchoscopy revealed diffuse alveolar hemorrhage on lavage.
Fig. 10.1.
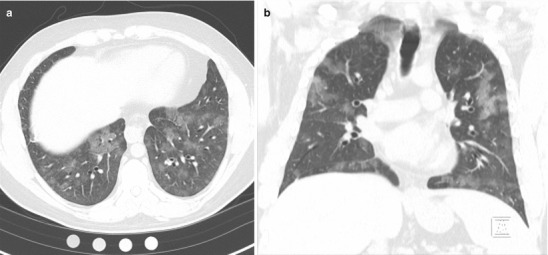
(a, b) High resolution computed tomography images demonstrating patchy ground glass opacities consistent with alveolar hemorrhage
The patient was diagnosed with DAH secondary to recurrent anti-phospholipid antibody syndrome and was successfully treated with increased doses of oral corticosteroids and the addition of a steroid-sparing, cytotoxic agent. Upon achieving a goal maintenance dose of cytotoxic agent, the corticosteroids were successfully tapered to 5 mg of oral Prednisone daily.
Case Three
The patient is a 75 year old gentleman who was in good health and quite active until 6–8 months prior to presentation. At that time, he was noted to develop dyspnea on exertion by family members and was encouraged to seek medical attention. Pulmonary evaluation revealed significant functional impairment, and HRCT demonstrated a basilar predominant, reticular pattern of interstitial lung disease. Autoimmune serologies including anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, rheumatoid factor, anti-Scl-70, anti-SS-A and anti-SS-B antibodies were all negative. Surgical lung biopsy revealed usual interstitial pneumonitis, and the patient was diagnosed with idiopathic pulmonary fibrosis.
Over the first few months following the diagnosis, the patient noticed a slow, steady decline in function, but over the 2–3 weeks prior to presentation, he became dramatically worse with markedly increased oxygen requirements and dyspnea that occurred with ambulating room to room. Upon presenting to clinic, he was found to be in respiratory distress with a respiratory rate of 32 breaths per minute, accessory muscle use and increased work of breathing. The patient was admitted to the Intensive Care Unit.
Further evaluation included HRCT of the chest which revealed diffuse ground glass infiltrates superimposed on an underlying fibrosing interstitial pneumonia consistent with his known diagnosis of idiopathic pulmonary fibrosis (IPF) (Fig. 10.2a, b). White blood cell count was normal, but his hematocrit was reduced at 35 %. Bronchoscopy reveal diffuse alveolar hemorrhage (Fig. 10.2c). Differential cell counts from the BAL revealed a 60 % neutrophilia, but no infectious organisms were isolated. Echocardiography confirmed normal left ventricular function and filling pressures and was otherwise unremarkable. No evidence of pulmonary embolus or other precipitant of respiratory decline could be identified. Given that no specific precipitant for the patient’s acute respiratory decline could be identified and that infection, heart failure, thromboembolic disease and other potential causes of acute lung injury were all excluded, the patient was diagnosed with an acute-exacerbation of IPF. Furthermore, the bronchoscopic finding of alveolar hemorrhage did not prove to represent clinically-significant hemorrhage and repeat serologies, ANCA testing, anti-basement membrane antibodies were all negative. Following a prolonged ICU course, he died of his respiratory failure.
Fig. 10.2.
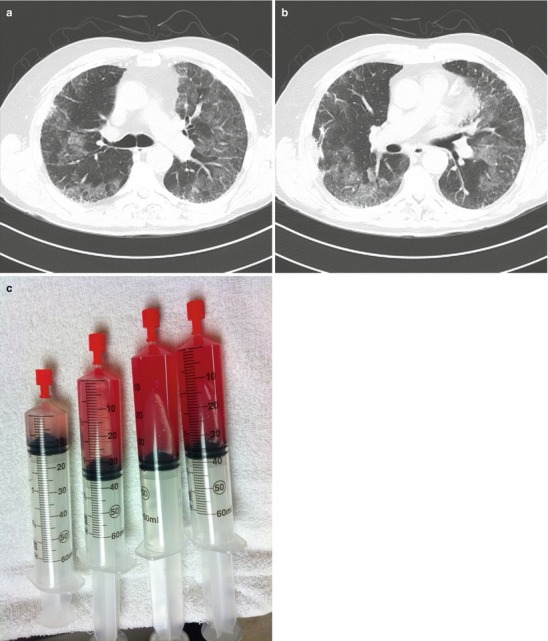
(a, b)High resolution computed tomography images demonstrating patchy ground glass opacities superimposed on peripheral-predominant reticular infiltrates and early honeycomb changes. (c), Serial aliquots of bronchoalveolar lavage fluid demonstrating an increasingly hemorrhagic return diagnostic of alveolar hemorrhage
Clinical Presentation
Patients who present with DAH can present at any age. Patients may have a known predisposing condition such as a systemic vasculitis, collagen vascular disease, or mitral stenosis, or the DAH may represent the initial manifestation of their disease state. DAH may occur as an isolated event or with repeated episodes of bleeding. Hemoptysis, the most characteristic sign of DAH, may evolve slowly over a period of weeks (i.e. anti-phospholipid antibody syndrome [1]) or more dramatically over a period of days or hours (i.e. crack cocaine inhalation [2]). However, it has also been reported that up to one-third of cases of DAH will present without evidence of hemoptysis [3]. Additional pulmonary symptoms may include dyspnea, non-productive cough, exercise intolerance, and/or vague chest discomfort or heaviness. As mentioned, patients may present earlier in a disease course with more mild symptoms of dyspnea or hemoptysis, or they may present with fulminant disease including profound hypoxemia or respiratory failure. Constitutional symptoms may also commonly be seen including fatigue, malaise, anorexia, fever, and myalgias.
In evaluating any patient with DAH, a comprehensive and detailed history is very important. Areas to consider include: (1) Does the patient have any elements to suggest a systemic autoimmune or collagen vascular disorder? The identification of extra-pulmonary signs and symptoms may be helpful in revealing a potential underlying etiology for the DAH. For example, the identification of skin lesions consistent with a cutaneous leukocytoclastic vasculitis, the presence of destructive upper airway lesions, or the finding of inflammatory ocular disease may point the clinician towards the diagnosis of a primary small vessel vasculitis. Similarly, does the patient have a malar rash or synovitis to suggest possible systemic lupus erythematosus? (2) Does the patient have any underlying cardiac disease? Specifically, does the patient have valvular heart disease (i.e. mitral stenosis or rheumatic heart disease) or disease that might result in elevated left-sided filling pressures? (3) Does the patient take any potentially causal medications such as penicillamine or propythiouracil? Or engage in illicit drug use, such as crack cocaine? (4) Does the patient have a coagulation disorder or take any anti-coagulants that might contribute to hemorrhage?
Physical examination findings in DAH are nonspecific. Objective findings may include fever, tachypnea, tachycardia, hypoxemia, diffuse crackles/rales, bronchial breath sounds or other findings consistent with alveolar consolidation on chest auscultation. The search for extra-pulmonary findings though may be extremely fruitful as regards identifying an inciting underlying systemic disease. Such findings may include palpable purpura, conjunctivitis, septal perforation, iridocyclitis, synovitis, or focal neurologic deficits/mononeuritis multiplex.
On laboratory testing patients will be noted to have a low and/or falling hemoglobin. However, the presence of a normochromic, normocytic anemia in acutely-ill patients tends to be a non-specific finding. In the case of subclinical bleeding or recurrent bouts of DAH, iron deficiency anemia may develop as well. Generally speaking, elevations of the white blood cell counts and platelets will be noted, although thrombocytopenia may be seen in conjunction with DAH in entities such as idiopathic thrombocytopenic purpura, thrombotic thrombocytopenia purpura, hemolytic uremic syndrome, or disseminated intravascular coagulation [4, 5]. Of note, these conditions are generally associated with bland pulmonary hemorrhage rather than a capillaritis lesion. Additionally, the presence of thrombocytopenia with DAH should also raise suspicion for possible systemic lupus erythematosus (SLE) [6] or primary anti-phospholipid antibody syndrome (APLAS) [7].
Coagulation studies are critical to excluding coagulopathy as the inciting etiology of DAH (bland hemorrhage). Elevated inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein are commonly elevated, but are non-specific findings. Serologic testing for specific autoimmune disorders and immune complex mediated diseases is a necessary part of the evaluation of DAH and extremely helpful when positive. Urinalysis should be obtained in all patients with DAH to evaluate for the presence of a “pulmonary-renal syndrome” which is defined as the presence of DAH plus glomerulonephritis. Glomerulonephritis in turn is characterized by the presence of (i) proteinuria, (ii) microscopic (or gross) hematuria, ideally with dysmorphic, crenulated red blood cells, and (iii) red blood cell casts. Renal insufficiency and renal failure will commonly ensue such that the presence of a pulmonary renal syndrome calls for rapid treatment to prevent permanent renal failure.
Chest radiography is extremely informative and is characterized by diffuse alveolar infiltrates, but is difficult to distinguish from other diseases characterized by an alveolar filling pattern (i.e acute respiratory distress syndrome, congestive heart failure or pneumonia.) The alveolar opacities themselves may vary from a patchy focal process to confluent, diffuse alveolar filling. Still, those cases that initially present with unilateral or lobar infiltrates will usually rapidly progress to diffuse alveolar filling if unrecognized or untreated. HRCT of the chest can confirm the presence of air space filling. While DAH will typically be characterized by patchy, bilateral ground glass opacities ± consolidation with a central and lower lobe predominance, the higher resolution images tend to add only a marginal amount of information over a standard chest radiograph. Finally, although not commonly recognized, repeated bouts of DAH may lead to findings of fibrosis or even obstructive lung disease on chest radiography [8, 9].
Pulmonary function testing may be performed in patients in whom the disease onset is less acute, and in these cases, the diffusing capacity for carbon monoxide (DLCO) may be elevated, or if measured sequentially, may be noted to increase. This increase in DLCO is secondary to the presence of carbon monoxide-avid hemoglobin in the airspaces. However, in patients who present with more acute disease, pulmonary function testing is rarely feasible. Longitudinally, pulmonary function testing can be useful in cases of DAH in which the bleeding may be chronic or recur frequently, such as idiopathic pulmonary hemosiderosis or anti-phospholipid antibody syndrome as these patients may go on to develop obstructive and/or restrictive physiology.
Diagnosis (Table 10.1)
While the presence of DAH may be strongly suggested in a patient with marked hemoptysis, bilateral alveolar infiltrates, anemia and respiratory distress, this classic presentation appears to represent a minority of cases. Ultimately, DAH remains on the differential diagnosis of any patient with bilateral or diffuse alveolar infiltrates, hypoxemia and dyspnea, and in point of fact, autopsy studies have shown that 2–4 % of patients with clinical acute respiratory distress syndrome (ARDS) who died of their disease will be found to have unsuspected DAH [10, 11]. Thus, although pneumonia, heart failure and ARDS are all much more common than DAH, in those cases where the diagnosis is less than certain, bronchoscopy with BAL should be considered.
When performing the procedure, the BAL should always be performed prior to any concomitant procedure such as biopsy to avoid precipitating any confounding bleeding or even bronchoscope trauma. When choosing the anatomic location for the BAL, the operator should choose the areas most involved by chest radiograph, or in diffuse disease, may choose the right middle lobe or lingula so as to optimize the return volumes. The bronchoscope should be advanced until “wedged” or impacted in a segmental or subsegmental bronchus. Once a position has been secured, four to five standard saline aliquots of between 30 and 60 ml should be serially instilled and removed via the bronchoscope up to a total lavage volume of no less than 100 ml and no more than 300 ml (and ideally >30 % of the total instilled volume should be obtained on return to assure the accuracy of differential cell counts) [12]. In DAH, the recovered fluid will become increasingly hemorrhagic from aliquot to aliquot, or at a minimum, will not clear with serial lavage. This bronchoscopic finding is diagnostic of DAH (Box 10.1). Nevertheless, the finding of DAH is not a final diagnosis in and of itself as the general presence of DAH carries an extended differential diagnosis and cannot by itself define the underlying etiology for the DAH.
Box 10.1
Diagnosis of Diffuse Alveolar Hemorrhage
Entities
Diffuse alveolar hemorrhage is diagnosed at the time of bronchoscopy. With the bronchoscope in “wedge position” in a segmental or subsegmental bronchus, four to five standard saline aliquots of between 30 and 60 ml are serially instilled and removed for a total lavage volume of no less than 100 ml and no more than 300 ml. A diagnosis of diffuse alveolar hemorrhage is made when the recovered fluid is identified to be increasingly hemorrhagic from aliquot to aliquot, or at a minimum, does not clear with serial lavage. Alternatively, a diagnosis of DAH may also be made at time of surgical lung biopsy when a pathologic finding of diffuse alveolar hemorrhage is made (red blood cells filling the alveolar spaces.) If a diagnosis of DAH is made at the time of surgical lung biopsy, a concurrent pathologic diagnosis of capillaritis or bland hemorrhage should also be identified
As mentioned above, serologic testing is central to the evaluation of DAH, and in specific cases, serologic studies can confirm a diagnosis without the need for surgical biopsy. In Goodpasture’s syndrome, diagnosis may be confirmed by the presence of serum anti-basement membrane antibodies (ABMAs.) [13] Similarly, serum anti-cardiolipin antibodies (and Russell Viper Venom Time) should be measured to assess for primary anti-phospholipid antibody syndrome [1]. The presence of serum anti-neutrophil cytoplasmic antibodies (ANCA) and/or a positive anti-proteinase-3 or anti-myeloperoxidase enzyme-linked immuosorbant assay (ELISA) will assist with the diagnosis of a primary, small vessel, ANCA-associated vasculitis (AAV) such as granulomatosis with polyangiitis (the entity formerly known as Wegener’s granulomatosis), microscopic polyangiitis, pauci-immune idiopathic pulmonary capillaritis, or Eosinophilic Granulomatosis with Polyangiitis (EGPA), the entity formerly known as Churg Strauss Syndrome [14]. In cases of DAH complicating SLE, the diagnosis of SLE is usually established [3]. However, in cases where DAH is the presenting manifestation, serum testing for low serum complement (specifically C3 and C4), serum antinuclear antibodies, and the presence of anti-double-stranded deoxyribonucleic acid antibodies will help point to the diagnosis. Anti-SS-A (Ro) and SS-B (La) antibodies are less specific, but may be associated with SLE as well as primary Sjogren’s syndrome and scleroderma. Anti-streptolysin O testing is helpful in the identification of post-streptococcal disease, and cryoglobulins and hepatitis serologies are helpful in the assessment of cryoglobulinemia.
Additional testing that is less specific but may be helpful in diagnosis includes a complete blood count, liver function testing, renal function testing, inflammatory markers, urinalysis with sediment examination, and coagulation studies. Furthermore, echocardiography is often required to evaluate for mitral stenosis, severe diastolic dysfunction and other causes of elevated left-sided filling pressures that potentially may cause bland hemorrhage. Additional imaging studies, beyond chest radiography and HRCT, that may yield diagnostic information depending upon the clinical scenario include CT of the sinuses (i.e. to assess for evidence of granulomatosis with polyangiitis), CT/MRI of the brain, and CT of the abdomen and pelvis.
In some cases, surgical lung biopsy may be required to establish the underlying cause if serologic testing or history are unrevealing. The decision to proceed to surgical lung biopsy should not be taken lightly as the procedure, whether done as a less invasive video assisted thoracoscopic procedure or a more invasive thoracotomy, requires general anesthesia and is an invasive surgical thoracic procedure in a moderately or severely ill patient. On the other hand, surgical lung biopsy may be safely accomplished in the hands of an experienced surgeon in the vast majority of cases. Ultimately, the decision to proceed or not to proceed to surgical lung biopsy must take into account a careful weighing of the risks, benefits and alternatives. It should be clear that the biopsy is revealing critical diagnostic information that cannot be obtained in other ways and that this information will affect treatment decisions.
Three broad categories of pulmonary histopathology are associated with DAH, namely (i) capillaritis, (ii) bland hemorrhage and (iii) diffuse alveolar damage with hemorrhage, and the identification of the underlying histopathologic pattern can often be used to focus the differential diagnosis.
Pulmonary Capillaritis
Histology (Fig. 10.3)
Fig. 10.3.
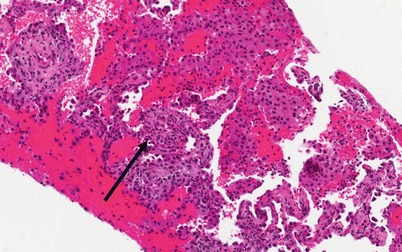
Photomicrograph (20× magnification) of an H&E stained section showing lung parenchyma with diffuse airspace filling by hemorrhage and scattered macrophages. The arrow points to a region of capillaritis in which the alveolar septa are expanded by necrotic neutrophils and karyorrhexitic nuclear debris (Courtesy of Dr. Steven Groshong, Division of Pathology, Department of Medicine, National Jewish Health, Denver, Colorado, USA)
Pulmonary capillaritis, also known as alveolar capillaritis, necrotizing alveolar capillaritis, and neutrophilic capillaritis, is one of the three core histologic pattern that may be associated with DAH. Pulmonary capillaritis is characterized by neutrophils infiltrating the alveolar septa along the pulmonary capillaries with associated nuclear debris and hemorrhage. Capillaritis may, in some cases, also involve other small vessels such as the venules and arterioles. There is associated disruption of the alveolar-capillary basement membrane, and red blood cells, edema fluid, fragmented neutrophils, debris and fibrin leak into the alveolar spaces [15]. The alveolar interstitium itself is broadened by the presence of edema, fibrinoid necrosis, inflammatory cells, and red blood cells. The neutrophils in the interstitium and vessel walls degranulate and undergo apoptosis, and as such, often appear pyknotic and undergo karyorrhexis leaving behind characteristic basophilic nuclear debris. Other features that may occur or be identified on this background pattern of lung injury include: small-vessel thrombosis, organizing pneumonia, and type II alveolar epithelial cell hyperplasia. Lastly, it should be noted that pulmonary capillaritis is a subset of pulmonary vasculitis in which the microcirculation of the lung (alveolar capillaries, arterioles, and venules) is predominantly affected and the larger pulmonary vessels are spared [16].
Etiologies
ANCA-Associated Small Vessel Vasculitis: Microscopic Polyangiitis
Microscopic polyangiitis (MPA) is another of the small vessel ANCA associated vasculitides and has a predilection for the microvasculature of the kidney and the lung. MPA is universally associated with a focal segmental necrotizing glomerulonephritis and is characterized by marked constitutional symptoms. MPA can be differentiated from classic polyarteritis nodosa by the lack of involvement of medium sized blood vessels and the absence of systemic hypertension. Distinguishing MPA from GPA can be difficult, but MPA lacks the granulomatous inflammation seen in GPA, only affects the upper airway in <15 % of patients, and is commonly associated with a peri-nuclear ANCA staining pattern (p-ANCA) rather than a c-ANCA pattern. DAH due to pulmonary capillaritis occurs in up to one-third of patients with MPA and represents by far the most common pulmonary manifestation of the disease. In patients with recurrent bouts of DAH related to MPA, both obstructive lung disease and pulmonary fibrosis have been reported [20, 26].
As mentioned above, a diagnosis of MPA essentially requires that the patient have a focal segmental necrotizing glomerulonephritis. Other common clinical manifestations include arthralgias/arthritis, myalgias/myositis, gastrointestinal disease, peripheral nervous system involvement and cardiac disease. As seen in other AAV, non-specific inflammatory markers are elevated (erythrocyte sedimentation rate and C-reactive protein), and non-specific increases in both serum antinuclear antibodies and rheumatoid factor may be present. Circulating immune complexes may be found in a significant minority of patients with MPA, but tissue localization of circulating immune complexes is rarely seen. ANCA are frequently present in patients with MPA, but less so than with GPA, on the order of 50–75 %. Additionally, 85 % of the ANCAs will have a p-ANCA staining pattern that in turn is more commonly associated with anti-myeloperoxidase (MPO) antibodies [17, 26, 27].
As with GPA, DAH in MPA represents life-threatening disease and is associated with an increased mortality. Indeed, an episode of DAH secondary to MPA is associated with a 30 % mortality. For those patients who do survive, the 1-year and 5-year survival is reduced to 82 and 68 %, respectively [26, 28].
Treatment of DAH secondary to MPA is very similar to GPA and consists of supportive care elements plus glucocorticoids and plasmaphresis followed by cyclophosphamide or rituximab. Additionally, recombinant factor VIIa has been tried at the case report level as a modality by which to control refractory alveolar hemorrhage and unremitting respiratory failure in severe cases of DAH due to MPA [29].
ANCA-Associated Small Vessel Vasculitis: Granulomatosis with Polyangiitis (GPA)
Granulomatosis with polyangiitis is the entity formerly known as Wegener granulomatosis and represents one of the more common etiologies associated with pulmonary capillaritis as well as pulmonary renal syndrome. GPA is one of the AAVand is characterized by granulomatous inflammation of the upper and lower respiratory tract and a necrotizing small vessel vasculitis. While the American College of Rheumatology and Chapel Hill Consensus Conference have developed criteria for the classification of the AAV, these criteria perform poorly when used to diagnose an individual patient. The diagnosis of GPA and the other AAV rests upon the clinician integrating clinical, laboratory, radiographic and pathologic data and making an informed clinical judgment that the data do or do not support a diagnosis of GPA.
DAH is estimated to occur in 5–15 % of patients with GPA. Indeed, the presence of DAH alone should raise the possibility of GPA and the other AAV within the differential diagnosis [17]. DAH can occur as an initial manifestation of the disease or it may occur during an exacerbation of a previously established case. DAH may occur as an isolated finding or in conjunction with other pulmonary manifestations of GPA. In a patient series published by Cordier and colleagues, pulmonary capillaritis was identified in 31 % of open lung biopsies obtained in patients with GPA [18]. The presence of DAH, by definition, represents severe, life-threatening disease and correlates with a considerably increased mortality [19].
As mentioned above, GPA commonly presents with upper airway involvement (>80 %) and may manifest with epistaxis, nasal discharge or crusting, septal perforation, otitis, hearing loss, or subglottic or tracheal stenosis. Similarly, the lower respiratory tract is also frequently involved (>80 %) and patients may manifest with cough, dyspnea, chest discomfort or hemoptysis. Radiographically, patients may have infiltrates, consolidation, nodules, cavities, and/or effusion(s). Extra-pulmonary manifestations will commonly include renal involvement/glomerulonephritis, constitutional symptoms, myalgias, arthralgias/arthritis, cutaneous involvement, ocular involvement, and cardiac manifestations [20].
Anti-neutrophil cytoplasmic antibodies (ANCA) are a hallmark of GPA and contribute to the pathogenesis of AAV. Three distinct ANCA staining patterns have been identified, namely cytoplasmic, peri-nuclear and atypical, and it is the cytoplasmic or c-ANCA that have been most closely associated with GPA. c-ANCA in turn, have been shown to recognize the proteinase-3 (PR3) antigen in the vast majority of cases. 85–90 % of patients with generalized active GPA will be c-ANCA and/or anti-PR3 positive [17]. While ANCA titers correlate with disease activity, a rise in ANCA titers needs to be considered within the context of the full clinical assessment, as a change in ANCA titers alone lacks sufficient sensitivity and specificity for predicting disease relapse to be used as such [21]. Also, it should be noted that while a positive c-ANCA or PR3 is very helpful in diagnosing AAV, a negative test does not exclude GPA or AAV in an individual patient.
Treatment of DAH begins with basic supportive care elements such as a secure airway, oxygen therapy and ventilatory support. Once the patient has been stabilized and the “A, B, Cs” of airway, breathing and circulation have been addressed, and any potential coagulopathic state or bleeding diasthesis similarly addressed, treatment directed towards the underlying precipitating disease may begin.
Treatment of GPA requires the use of immunosuppressive agents (cytotoxic medications and systemic corticosteroids) that carry the risk of serious adverse side effects. As such, the intensity of the immunosuppresion must carefully be titrated to disease activity, and disease activity must be careful assessed in each patient. DAH clearly represents organ and life threatening disease and as such qualifies as “severe” disease that necessitates the use of more aggressive immunosuppressive regimens to control the disease activity.
The initial regimen of choice for both generalized active and severe life-threatening disease had been oral cyclophosphamide plus oral corticosteroids based upon the original National Institutes of Health studies demonstrating the efficacy of this regimen for the induction of disease remission [22]. In 2007, Jayne and colleagues published the MEPEX trial (Randomized Trial of Plasma Exchange or High-Dosage Methylprednisolone as Adjunctive Therapy for Severe Renal Vasculitis) in which patients with severe renal disease were treated with corticosteroids and oral cyclophosphamide and additionally randomized to plasma exchange or high dose intravenous methylprednisolone [23]. Dialysis-independent survival was greater in the plasma exchange group than the intravenous corticosteroid group such that the addition of plasma exchange to corticosteroids and cyclophosphamide has since been recommended for the management of patients with severe renal disease. Whether this same strategy may be applied to patients with DAH was tested in a 20 patient case series, and indeed, this strategy appears to be effective in diffuse alveolar hemorrhage as well [24].
In 2010, the Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis (RAVE) trial evaluated the anti-CD-20 monoclonal biologic rituximab for the management of generalized active and severe AAV and found that rituximab was non-inferior when compared with cyclophosphamide [25]. No significant differences in total or serious adverse events were noted between the treatment groups. Subgroup analysis further showed that rituximab was equally effective with cyclophosphamide for the management of alveolar hemorrhage. Thus, rituximab may be used as a potential alternative to cyclophosphamide in severely ill patients, including those with DAH.
Isolated Pulmonary Capillaritis
Isolated pulmonary capillaritis or idiopathic pauci-immune pulmonary capillaritis refers to a small vessel vasculitis that is confined to the lungs. Some experts liken this entity to a lung-limited MPA. DAH in isolated pulmonary capillaritis may or may not be p-ANCA positive, and while no differences can be discerned between those patients who are ANCA positive and ANCA negative, this may be due to inadequate longitudinal follow-up. In one case series of 29 patients, isolated pulmonary capillaritis was the most common cause of DAH with biopsy proven pulmonary capillaritis, followed by GPA and MPA [30]. In this study, isolated pulmonary capillaritis accounted for 28 % of the cases, and there were no clinical, serologic, or histologic features of an alternative systemic disorder. Clinically, three quarters of patients presented with respiratory distress and half required mechanical ventilation. Despite this, there was an 88 % in hospital survival and an overall favorable prognosis for the group. Isolated pulmonary capillaritis is treated along the same lines as AAV and responds well to standard therapy with corticosteroids and cytotoxic medications [27, 31].
Systemic Lupus Erythematosus
DAH affects only 4 % of patients with SLE, but along with acute lupus pneumonitis represents one of the most devastating pulmonary complications of SLE with a mortality rate approaching 50 %. Histopathologically, DAH due to SLE is associated with pulmonary capillaritis in the vast majority of cases, but bland pulmonary hemorrhage and DAH secondary to diffuse alveolar damage may also be seen. Co-morbid and/or precipitating infectious complications should excluded as a contributing factor to the DAH [3, 32, 33].
As with SLE itself, there is a strong female preponderance in DAH secondary to SLE, and patient are on average in their third to fourth decade. In the majority of cases of DAH associated with SLE, glomerulonephritis is also present at the time of presentation. As with other cases of DAH, patients present with dyspnea, hemoptysis, hypoxemia and respiratory distress/respiratory failure; however, this clinical presentation is common to both DAH and acute lupus pneumonitis (ALP) and distinguishing between these entities can be exceedingly difficult. In point of fact, 20 % of cases of ALP may present with hemoptysis. Still, the majority of DAH cases occur in patients with a known diagnosis of SLE and will frequently have concomitant glomerulonephritis, whereas 50 % of cases of ALP are an initial presentation of SLE. Ultimately, as with most cases of DAH, diagnosis is made at time of BAL. In those patients who undergo biopsy, ALP is characterized by diffuse alveolar damage complicated by hemorrhage and may also have features of organizing pneumonia, but should not demonstrate frank capillaritis [3, 6].
As mentioned previously, mortality rates associated with DAH in SLE are high and have traditionally ranged from 50–90 %, although more recent data suggests that the use of aggressive immunosuppressive treatment, increased recognition of concomitant infections, and advances in the management of critically-ill patients, survival is far better. Negative prognostic factors include the need for mechanical ventilation, the presence of infection, and the requirement for cyclophosphamide therapy [3, 6, 33].
Treatment of DAH secondary to SLE includes the use of intravenous, high-dose methylprednisolone and cyclophosphamide. While plasmapharesis is also used for DAH complicating SLE, it is unclear whether or not this intervention provides additional benefit [27, 33].
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome, along with GPA, MPA, idiopathic pauci-immune capillaritis, and SLE, represents one of the more common etiologies of capillaritis. As with the other entities, DAH may be an initial presentation or later complication of the disease. Symptoms again include cough, dyspnea, fatigue, malaise, fever, hemoptysis, hypoxemia, and acute respiratory failure. Thrombocytopenia may be present at the time of the DAH episode helping to focus the differential diagnosis on APLAS along with SLE, disseminated intravascular coagulation, and thrombotic thrombocytopenic purpura. On histology, there is evidence of pulmonary capillaritis with or without concomitant microvascular thrombosis [7, 34].
The management of APLAS is commonly complicated thromboemoblic disease and the need for anti-coagulation. When an episode of DAH occurs in a patient with an established diagnosis of APLAS, the presence of capillaritis and diffuse hemorrhage is further complicated by the presence of therapeutic anti-coagulation (as well as the possibility of concomitant pulmonary thromboemboli.) Nevertheless, it must be recognized that more often than not, it is the capillaritis driving the DAH and controlling the vasculitis is key to achieving therapeutic success. At the same time, diagnosing, treating and/or preventing the thromboembolic manifestations of the disease, as well as controlling the DAH, cannot be ignored. Thus, even in centers experienced in the management of complex autoimmune diseases, the management of these patients is extremely challenging and referral to a center of expertise is recommended when feasible. Nevertheless, first line therapy for DAH associated with antiphospholipid antibody syndrome is intravenous corticosteroids combined with optimal supportive care. Rapid resolution of most cases of DAH is typically seen after treatment with corticosteroids. In cases of catastrophic antiphospholipid syndrome, IVIG or plasma exchange may be added to the intravenous corticosteroids and supportive care. Most recently, case reports describing the use of rituximab for refractory APLAS, including APLAS complicated by refractory DAH, have suggested that this agent may ultimately proven to have a role when more conventional therapies are unsuccessful [1, 34–36].
As with other cases of chronic and/or recurrent DAH, fibrosis and/or obstructive disease may evolve over time. Lastly, it should be noted that in catastrophic cases of APLAS (Asherson’s syndrome), ARDS and multi-system organ dysfunction may develop. In these cases, the pathology will demonstrate diffuse alveolar damage, capillaritis and diffuse small vessel occlusion and obliteration [14, 35].
Goodpasture’s Syndrome (Anti-basement Membrane Antibody Disease)
Goodpasture’s Syndrome, or anti-basement membrane antibody disease, is an autoimmune disorder mediated by antibodies directed against the non-collagenous domain (NC1) of the alpha-3 chain of type IV collagen (and to a lesser degree the alpha-5 chain) found in basement membranes [37]. The majority of patients, approximately 60–80 %, present with a pulmonary-renal syndrome of diffuse alveolar hemorrhage and glomerulonephritis. Indeed, the presence of a true pulmonary renal syndrome helps focus the differential diagnosis upon ABMA disease, GPA, MPA, and SLE. Still, 15–30 % of cases may present with isolated glomerulonephritis, and conversely up to 10 % of patients may present with DAH alone without renal involvement. Interestingly, although type IV collagen is found elsewhere in the body, including skin, eye, and gastrointestinal tract, end organ damage in ABMA disease is limited to the kidneys and lung. DAH represents a major cause of mortality in patients with ABMA disease, and among the competing causes of DAH, ABMA disease has a relatively poorer prognosis. On the other hand, in those cases of ABMA disease in which the pulmonary manifestations are dominant, renal outcomes are better when compared to patients who present with renal disease alone [13, 37].
The clinical presentation of DAH due to ABMA disease is very similar to other cases of DAH of differing etiologies of DAH with the caveat that glomerulonephritis is present in most albeit not all cases. Again, patients will complain of dyspnea, cough, hemoptysis, hypoxemia, and/or constitutional symptoms (fatigue, fever, anorexia, weight loss, arthralgias and myalgias). ABMA disease preferentially affects men more than women (approximately 2:1) and has a predilection for young adults (the average patient age reported ranges between 20 and 30). In those patients with ABMA who develop DAH, a history of smoking is extremely common (50–90 %), although recent viral infection or other inhalation exposures (e.g. hydrocarbons, marijuana, fire smoke, cocaine) may also be seen immediately antecedent to the onset of DAH. In point of fact, it is hypothesized that cigarette smoking (or alternatively infection or other inhalational exposure) plays a pathophysiologic role in the development of DAH either through a secondary injury to the alveolar-capillary unit, facilitating antigen presentation, or allowing ABMAs entry into the lung [13]. On laboratory testing, anemia will commonly be present, and frequently will be accompanied by an elevated blood urea nitrogen or serum creatinine consistent with renal insufficiency. Urinalysis frequently reveals microscopic hematuria, proteinuria, and red blood cell casts diagnostic of glomerulonephritis. Interestingly, 3–7 % of patients will be p-ANCA positive, suggesting the possibility of an overlap syndrome with MPA. ABMA disease has also been shown to have an association with specific human leukocyte antigen (HLA)-DR alleles, specifically HLA-DRB1*1501 [37]. Chest imaging studies, as with other cases of DAH, will show patchy diffuse alveolar infiltrates that appears as ground glass infiltrates or consolidation on HRCT. While it is rare to obtain pulmonary function testing in acutely-ill patients, in more chronic cases or more slowly evolving cases, an increased diffusing capacity of carbon monoxide may be identified.
Diagnosis may be made by identifying the presence of serum anti-basement membrane antibodies (ABMAs) in a patient with a compatible clinical presentation and circulating antibodies may be identified in two thirds to three quarters of patients at time of diagnosis [37]. At the bedside, it may be necessary to make a tentative clinical diagnosis and initiate therapy while awaiting the results of the serologic testing which frequently must be sent to a referral laboratory. Of note, while antibody titers appear to correlate with the severity of the renal disease, no such correlation has been identified with the pulmonary manifestations of the disease. Alternatively, patients may be diagnosed via lung or kidney biopsy and immunofluorescence studies. On light microscopy, the histopathology of the lung in ABMA disease may demonstrate either bland hemorrhage or capillaritis (although the appearance of the capillaritis in ABMA disease tends to lack some of the more aggressive and destructive features seen in MPA or GPA). Similarly, the renal biopsy will demonstrate a focal, segmental, rapidly progressive glomerulonephritis with crescent formation that is indistinguishable from other etiologies of rapidly progressive glomerulonephritis. However, the frozen sections should demonstrate a positive immunofluorescence pattern with a linear, continuous, “ribbon-like” appearance, reflecting antibody that has bound to the basement membrane. This immunofluorescence pattern is distinct from the punctate, patchy staining pattern seen in SLE and the negative or “pauci-immune” pattern associated with the AAV, and hence, is diagnostic for ABMA disease [38].
DAH associated with Goodpasture’s is managed very similarly to DAH (or severe renal failure) secondary to GPA or MPA. Plasmapharesis combined with corticosteroids and a cytotoxic agent (i.e. cyclophosphamide) has been shown to be effective in both of these clinical contexts and is associated with improved mortality and renal recovery [39]. Early diagnosis and prompt institution of therapy is crucial to optimizing outcomes, and as such, it is sometimes necessary to initiate therapy pending a conclusive diagnosis. Ultimately, the similarities in therapeutic recommendations for pulmonary renal syndrome, whether it is due to ANCA associated vasculitis, SLE or Goodpasture’s syndrome combined with the adverse effects associated with delays in therapy makes this approach judicious. Steroids alone, or steroids combined with cytotoxic agents without the use of plasmaphresis do not achieve equivalent results. While there is no definitive data informing the optimal duration of plasmaphresis, the duration of therapy for Goodpasture’s tends to be longer than in AAV, on the order of 10–14 exchanges, or until ABMAs become undetectable. With regard to choice of cytotoxic agent, cyclophosphamide is the most common agent utilized for life threatening alveolar hemorrhage. In more mild cases or as the patient’s conditions improves, azathioprine (and more recently mycophenolate mofetil) have been used [20]. Lastly, rituximab has been proposed by some experts as a potential therapeutic agent for Goodpasture’s syndrome based upon its mechanism of action and the pathogenetic role of ABMAs in the disease; however, beyond a handful of anecdotal reports, there is currently no evidence to support its use and its role in managing ABMA disease awaits further study.
In terms of prognosis, patients with more severe renal disease have a worse outcome. A retrospective review of patients with ABMA disease by Levy and colleagues, patients with a creatinine concentration of <5.7 mg/dl had a 1 year survival of 100 % and a renal survival of 95 %, those with a creatinine ≥5.7 mg/dl but who did not require immediate hemodialysis had a 1 year survival of 83 % and renal survival of 82 %, and those who required immediate hemodialysis had a 1 year survival of 65 % and renal survival of only 8 % [40]. A similar pattern is seen if one assesses renal involvement via biopsy in that patients with ≥70 % crescentic glomeruli and renal insufficiency may be expected to have persistent renal failure whereas patients with less than 30 % of their glomeruli having undergone crescent formation have improved survival and renal function. In a 28 patient case series of patients with ABMA disease and DAH reported by Lazor and colleagues, patients with pulmonary predominant disease (a paucity or absence of renal involvement) had a lower requirement for immunosuppressive therapy and plasma exchange than patients with a combined pulmonary renal syndrome. Interestingly, this cohort was characterized by frequent worsening of their pulmonary or renal disease but 100 % survival [13]. Unlike ANCA associated vasculitis, ABMA disease has generally been characterized more as a monophasic process without multiple recurrences, and those cases characterized by recurrence have tended to relapse in close proximity to disease onset [37].
Lung Allograft Rejection
Pulmonary capillaritis as a manifestation of acute lung transplant rejection was first noted in a case series of five patients in 1998, four of case of which were confirmed histopathologically on surgical lung biopsy [41]. Interestingly, immunofluorescence studies identified septal capillary deposition of antibodies specific for complement factors and immunoglobulin subtypes. To differentiate between acute cellular rejection and post-transplant capillaritis, a biopsy is required; however, these conditions can be found concomitantly in greater than 50 % of cases. When post-tranplantation capillaritis is identified, intensification of immunosuppressive regimen is recommended. Plasmapharesis has also been tried on a compassionate use basis, but remains unproven [42].
Others
In addition to SLE and antiphospholipid syndromes, DAH has been documented in other collagen vascular diseases. While exceedingly rare, there have been case reports of pulmonary capillaritis in rheumatoid arthritis, scleroderma, mixed connective tissue disease, polymyositis/anti-synthestase syndromes, and undifferentiated connective tissue diseases [31, 43]. Distinguishing DAH from other pulmonary manifestations and complications of the underlying autoimmune disease may be difficult, especially given the rarity of DAH in these other entities, and the fact that hemoptysis need not be present. Competing considerations include diffuse alveolar damage (i.e. acute interstitial pneumonitis or lupus pneumonitis), organizing pneumonia, infection, pulmonary edema/heart failure and drug toxicity. Treatment for DAH associated with these other connective tissue disease entities is similar to that recommended for DAH in SLE. Ultimately, these entities are all considered to be a secondary, autoimmune-mediated, small vessel vasculitis secondary to an underlying collagen vascular disease and therapy must be directed at the underlying process.
Henoch-Schönlein purpura is an immune-complex mediated autoimmune disorder most commonly seen in pediatric populations which may also occur, albeit less frequently, in young adults. Patients typically present with a palpable, purpuric rash, most prominently over the lower extremities, and a focal segmental glomerulonephritis. Constitutional symptoms, arthalgias with synovitis, and gastrointestinal tract manifestations are common. Pulmonary capillaritis has been reported in patients with Henoch-Schönlein purpura, but it is exceedingly rare. Indeed, a large case series of 37 adult patients revealed no cases of DAH [44]. In those cases where DAH is found to complicate Henoch-Schönlein purpura, IgA immune complexes may be demonstrated in the serum, lung and kidney. Once again, management centers upon best supportive care combined with immunosuppressive therapy (corticosteroids and cytotoxic agents).
Behçet’s disease or Behçet’s syndrome, is a clinical syndrome of unclear pathophysiology, characterized by mucocutaneous oral and genital ulcers, skin lesions and pathergy, ocular disease (pan-uveitis, iridocyclitis, retinal vasculitis), arthritis, and vascular disease, generally manifested as thrombophlebitis, venous thrombosis and/or arterial aneurysms and/or occlusions. The syndrome preferentially affects individuals of Middle Eastern origin, but is also found with increased incidence in Japanese populations. The most common pulmonary manifestation of Behçet’s disease is pulmonary artery aneurysms, and these occur in 1–8 % of all individuals with Behçet’s. The presence of pulmonary artery aneurysms, however, is associated with a 50 % mortality. Pulmonary hemorrhage may occur either due to involvement of the microvasculature that in turn leads to DAH, or alternatively, patient may present with massive hemorrhage secondary to the erosion of an aneurysm into the airway [45–47]. As with the other entities, management centers upon best supportive care combined with immunosuppressive therapy (corticosteroids and cytotoxic agents) [48].
DAH from pulmonary capillaritis has also been documented in mixed cryoglobulinemia. Mixed cryoglobulinemia is a small- to medium-sized vessel, immune complex and complement mediated vasculitis, and is frequently seen in association with hepatitis B and C infection. Patients often present with cutaneous vasculitis and glomerulonephritis, but rare cases of DAH have been reported [49].
Finally, some rare causes of pulmonary capillaritis include inflammatory bowel disease, idiopathic glomerulonephritis, IgA nephropathy, EGPA, myasthenia gravis, and drug-sensitivities due to diphenylhydantoin, retinoic acid, and propylthiouracil. With regards to inflammatory bowel disease, a number of pulmonary complications have been associated with both ulcerative colitis and Crohn’s disease including bronchiolitis (panbronchiolits and bronchiolitis obliterans) bronchiectasis and interstitial lung diseases [50]. There are at least two case reports of DAH with a capillaritis lesion associated with ulcerative colitis. Both cases responded to corticosteroids and cytotoxic therapy. A number of cases of propylthiouracil-associated p-ANCA-positive vasculitis with DAH have also been reported, and these patients in general responded to therapy with corticosteroids and the discontinuation of propylthiouracil [51].
Bland Pulmonary Hemorrhage (Fig. 10.4)
Fig. 10.4.
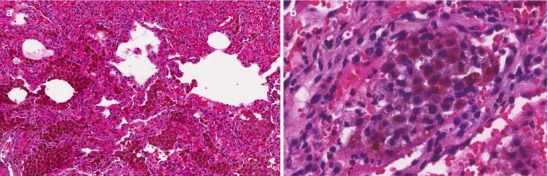
(a) Photomicrograph (20× magnification) of a histopathologic section of lung demonstrating diffuse alveolar hemorrhage without features of capillaritis (bland hemorrhage). (b), Photomicrograph (60× magnification) of hemosiderin-laden macrophages filling the alveolar space (Courtesy of Dr. Steven Groshong, Division of Pathology, Department of Medicine, National Jewish Health, Denver, Colorado, USA)
Histology
While both bland pulmonary hemorrhage and capillaritis show airspaces intra-alveolar filling by red blood cells, fibrin and hemosiderin-laden macrophages along with septal expansion be edema and reactive type II cell hyperplasia, the vessel walls of bland pulmonary hemorrhage lack the inflammatory cell infiltrates and necrotic features seen in capillaritis. After repeated episodes of hemorrhage from any cause, fibrotic changes and/or microvascular “drop out” may evolve. In some cases of bland hemorrhage due to idiopathic pulmonary hemosiderosis, electron micrographs have demonstrated abnormalities in the integrity of the alveolar-capillary membrane suggesting a possible etiology. Finally, cases of capillaritis that have undergone a course of treatment or partial treatment may histopathologically appear as bland hemorrhage confounding the diagnostic algorithm [15, 52].
Etiologies
Idiopathic Pulmonary Hemosiderosis
The diagnosis of idiopathic pulmonary hemosiderosis (IPH) is a diagnosis of exclusion, and by definition, is the presence of bland diffuse alveolar hemorrhage in the absence of an identifiable etiology. Patients who present with IPH typically are children (80 %) and young adults (20 %). Males are preferentially affected relative to females (2:1). Familial cases have been reported. Clinically, the disorder is characterized by recurrent episodes of DAH. As such patients present with cough, dyspnea, hemoptysis, constitutional symptoms (fever, fatigue, malaise, anorexia), anemia (especially iron deficiency anemia), hypoxia, recurrent pulmonary infiltrates and/or exercise intolerance. The severity of individual episodes of hemorrhage may vary from asymptomatic to fulminant respiratory failure requiring mechanical ventilation. Given the recurrent nature of the episodes of DAH, pulmonary fibrosis and restrictive lung disease will develop in up to a quarter of patients. Alternatively, patients with more chronic and refractory courses may also develop obstructive lung disease. Impaired gas exchanged with a reduced DLCO has similarly been reported in patients with chronic and relapsing disease. An isolated elevation of serum IgA may be seen in up to half of pediatric patients with IPH and may help raise the possibility of IPH in the differential diagnosis of a younger patient with DAH [14, 53].
By definition, in IPH there should be no evidence of a systemic disorder (vasculitis, immune complex mediated disease, collagen vascular disease, etc.), no potentially inciting drugs or exposures, no significant cardiac lesions, no coagulopathy and no appreciable pathophysiologic explanation for the disease [53]. As such, a detailed history (including occupational, exposure, drug and medication history) full serologic evaluation for autoantibodies and markers of autoimmune disease, urinalysis with sediment examination, urine toxicology screening (e.g. cocaine) ECG, and echocardiography should all be within normal limits. Even then, given that this is a diagnosis of exclusion, as cases of IPH are followed longitudinally, they may later be re-classified as a disease of known etiology as additional disease manifestations develop or objective data support an alternative diagnosis. It is believed that a number of cases in the published literature would have been classified differently had serum anti-basement membrane antibody testing, ANCA testing and PR3/MPO ELISA testing been widely available at the time of publication. One caveat to this would be an observation that IPH may be associated with celiac disease or jejunal villous atrophy in some patients, and at least at present, would still be considered IPH [54]. Similarly, subtle findings of capillaritis may easily be missed and cases of idiopathic pulmonary capillaritis may be mis-classified as IPH. This is especially true in the patient who has started corticosteroid therapy or other disease modifying therapy prior to surgical lung biopsy. Moreover, this may explain why some patients with IPH are observed to respond to immunosuppression with corticosteroids and cytotoxic agents. Nevertheless, to diagnose IPH, the biopsy must demonstrate bland hemorrhage and an absence of any features of vasculitis/capillaritis [53]. Indeed, to make a definitive diagnosis of IPH a lung biopsy is essentially required.
As mentioned above, histologic evaluation reveals bland alveolar hemorrhage with filling of the alveolar spaces with red blood cells, fibrin and hemosiderin-laden macrophages. Alveolar epithelial type II cell hyperplasia may be noted, and the vessels of the microvasculature may appear dilated and/or tortuous. Electron microscopy studies have further revealed subtle alveolar epithelial type I cell injury, basement membrane thickening, excessive collagen deposition and an absence of immune complexes [53].
As with other cases of DAH, treatment of IPH begins with supportive care elements of oxygen, reversing any bleeding diasthesis, and when indicated, ventilatory support and red blood cell transfusion. Pharmacologically, corticosteroids and cytotoxic agents again represent the mainstays of therapy, although their effectiveness specifically in IPH is unproven. Plasmapharesis has been used in severe, refractory episodes of DAH at the case report level [14]. The prognosis is variable–25 % of patients will have limited disease characterized by a single episode of hemorrhage without recurrence, 25 % will have recurrent hemorrhage but remain free of fibrosis or other major structural lung disease, 25 % will have progressive, chronic lung disease as a result of chronic, recurrent hemorrhage, and 25 % of patients will die of massive hemorrhage or other major complication of their disease [8, 53].
Drugs and Medications
A number of drugs and chemicals have been associated with the development of DAH and a pathologic correlate of bland hemorrhage including penicillamine, amiodarone, nitrofurantion, and isocyanates. The reader is directed to the website www.pneumotox.com maintained by Drs. Foucher and Camus and the Groupe d’Etudes de la Pathologie Pulmonaire Iatrogène for up-to-date information regarding medication associated pulmonary toxicity.
DAH and pulmonary renal syndrome are rare but reported complication of penicillamine therapy regardless of indication (rheumatoid arthritis, Wilson’s disease, or primary biliary cirrhosis) [55, 56]. As with other drug-induced pulmonary complications, the key to diagnosis is eliciting a truly complete list of current medications as well as past medication history. On average, patients will have been taking penicillamine for a year prior to the onset of DAH, but the duration of therapy prior to the development of toxicity is highly variable. In patients who have undergone biopsy, the histopathology will demonstrate bland hemorrhage and immunofluorescence studies will show granular deposition of IgG similar to patients with SLE [57, 58]. Pulmonary capillaritis has not been associated with pencillamine therapy. Treatment includes cessation of pencillamine plus corticosteroids, cytotoxic therapy and plasmapharesis [55].
While the majority of patients with amiodarone pulmonary toxicity will demonstrate “classic” histopathologic features, namely the presence of interstitial edema and fibrosis, copious vacuolated histiocytes, and foamy alveolar macrophages with or without elements of organizing pneumonia and/or diffuse alveolar damage, cases of diffuse alveolar hemorrhage associated with amiodarone therapy have been reported and histopathologically will demonstrate bland hemorrhage [57, 58]. Similarly, nitrofurantoin therapy is most commonly associated with a subacute or chronic cellular interstitial pneumonitis and/or pulmonary fibrosis, but in rare cases, an acute nitrofurantoin toxicity may develop and present with an acute-onset diffuse alveolar hemorrhage [58]. Therapy requires discontinuation of the drug with or without concomitant corticosteroids.
Coagulopathy
Coagulation disorders are among the most common etiologies of DAH associated with bland pulmonary hemorrhage. Thrombocytopenia of a variety of etiologies including idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, drug-induced thrombocytopenia (i.e. chemotherapy), hemolytic uremic syndrome and disseminated intravascular coagulation may all lead to bland hemorrhage [4, 5]. Similarly, pharmacologic anticoagulation with vitamin K antagonists, fractionated or unfractionated heparin, direct thrombin inhibitors, IIb/IIIa inhibitors and fibrinolytic therapy may also lead to alveolar hemorrhage [59]. Less obvious causes may include vitamin K deficiency and advanced liver disease.
As with other patients with DAH, patients may present with dyspnea, exercise intolerance, hypoxemia, anemia, and pulmonary infiltrates. Hemoptysis appears to be less common than with other etiologies of DAH, but bronchoscopy and lavage are generally diagnostic. Therapy includes supportive care and reversal of the coagulopathy.
Valvular Heart Disease and Left Ventricular Dysfunction
Mitral stenosis may produce DAH in those instances in which the disease is severe enough to produce severe pulmonary venous hypertension, and histopathologically appears as bland hemorrhage. Although patients may have a known history of mitral stenosis, a history of rheumatic heart disease or the development of insidious exercise intolerance and dyspnea may herald a diagnosis of valvular heart disease, or patients may have an initial presentation of pulmonary infiltrates or intermittent hemoptysis [60]. DAH has also been reported in patients with markedly elevated left ventricular filling pressures of other etiologies such as severe aortic stenosis, severe diastolic dysfunction, and cardiomyopathy. Treatment in these cases is directed at the underlying cardiovascular pathology.
Other
Inhalation of acid anhydrides and isocyanates have been associated with alveolar hemorrhage. These reactive organic chemicals are used in the manufacturing of plastics, paints, varnishes, and other resins. Hence, obtaining an occupational and exposure history is important in the evaluation of the patient with DAH. In general, the disease process is lung-limited, and interestingly, cases related to acid anhydride exposure appear to have a latency period between initial exposure and the development of hemorrhage of 1–3 months suggesting an immunologic mechanism. As with other exposure related processes, treatment requires elimination of the exposure [61, 62].
Extremely severe cases of obstructive sleep apneaand obesity hypoventilation syndrome have also been associated with alveolar hemorrhage. These cases are generally associated with marked pulmonary hypertension, chronic hypoxemia, pulmonary capillary network proliferation, and biventricular heart failure. Histology in these cases demonstrates bland hemorrhage, non-specific injury, and capillary proliferation [63].
Lastly, there are rare cases of pulmonary veno-occlusive disease associated with DAH. Pulmonary veno-occlusive disease may occur in the setting of bone marrow transplantation, chemotherapy-induced lung injury, radiation, collagen vascular disease, HIV, a familial disorder or as an idiopathic process. Patients most commonly present with signs and symptoms of severe pulmonary hypertension, including exercise intolerance, syncope, lightheadedness and dyspnea, but may report hemoptysis and in rare cases may be demonstrated to have DAH. Pulmonary function testing will demonstrate normal lung volumes and spirometry but a reduced diffusing capacity of carbon monoxide. Right heart catheterization will reveal pulmonary hypertension but a normal pulmonary capillary wedge pressure. Histology in these cases demonstrates obliteration, thrombosis, and fibrosis in and around the pulmonary venules and bland hemorrhage. The prognosis in pulmonary veno-occlusive disease is poor, and lung transplantation is the only definitive therapy, although immunosuppressive therapy (cytotoxics and corticsteroids), vasodilator therapy, and anti-coagulation have all been attempted [64, 65] (Fig. 10.5).
Fig. 10.5.
(a) High resolution computed tomography image and (b), CT angiography image demonstrating a spectrum of disease associated with alveolar hemorrhage. Note that while both CT images demonstrate patchy alveolar filling patterns, the density of the infiltrates may range from ground glass to frank consolidation, and the extent of the proportion of involved lung may similarly vary (Courtesy of Dr Gregory P. Cosgrove, Division of Pulmonary and Critical Care Medicine, National Jewish Health, Denver, Colorado, USA)
Diffuse Alveolar Damage
Histology
Diffuse alveolar damage (DAD) is a histopathologic pattern associated with acute lung injury. In those cases in which patients present with an acute illness, such as septic shock, pneumonia, trauma, aspiration or pancreatitis, severe hypoxemia, bilateral alveolar radiographic infiltrates, and a histopathologic correlate of DAD, they are diagnosed with ARDS. Other etiologies of DAD include acute exacerbations of fibrosing interstitial lung diseases, acute lupus pneumonitis, bone marrow transplantation, and acute interstitial pneumonitis. While DAH is not considered “characteristic” of underlying DAD, DAH is clearly associated with underlying DAD. Histology in these cases will show a dominant lesion of DAD characterized by non-cardiogenic pulmonary edema, alveolar type I cell injury and necrosis, denudation of the basement membrane, hyaline membrane formation in the alveolar spaces, thrombi in the microvasculature, and an influx of plasma cells, histiocytes, lymphocytes and scattered neutrophils, combined with evidence of focal hemorrhage in the alveolar spaces. As the entity evolves over days, the alveolar spaces and interstitium will fill with loose fibromyxoid tissue, the type II alveolar epithelial cells will proliferate and appear hyperplastic and cuboidal and elements of organizing pneumonia may develop as airspace fibrin begins to organize. While gross examination of the aliquots of serial bronchoalveolar lavage will be largely indistinguishable from cases of bland hemorrhage or capillaritis, in general, the absolute red blood cell counts identified by formal differential cell counts seem to be lower in DAD with hemorrhage, and there is a concurrent pronounced neutrophilia related to the lung injury [15, 66]. In many ways, the presence of DAH in the setting of DAD is more confounder than clinically significant, and should not be mis-interpreted as being a major manifestation of DAD (Box 10.2).
Box 10.2
Entities Characterized by Prominent and Severe Diffuse Alveolar Hemorrhage
Entities
Granulomatosis with polyangitis
Microscopic polyangitis
Isolated pulmonary capillaritis
Systemic lupus erythematosus
Primary antiphospholipid antibody syndrome
Goodpasture syndrome
Hematopoietic stem cell transplantation
Idiopathic pulmonary hemosiderosis
Coagulation disorders
Etiologies
Hematopoietic Stem Cell Transplantation (HSCT)
Both infectious and non-infectious pulmonary complications are extremely common following bone marrow transplantation or hematopoietic stem cell transplantation (HSCT). In 2011, the American Thoracic Society published an official research statement on the spectrum of noninfectious lung injury after HSCT or the idiopathic pneumonia syndrome (IPS) [67]. Approximately 3–15 % of patients who undergo allogenic HSCT will develop a non-infectious lung injury within 120 days of transplantation. A subset of these patients will have DAH. As with other patients with DAH, patients will present with dyspnea, hypoxemia, cough, constitutional symptoms and bilateral radiographic infiltrates. Hemoptysis occurs less frequently than expected and may be found in perhaps 15 % of patients. Respiratory failure requiring mechanical ventilation is common. The onset of DAH usually occurs within 1–5 weeks of the HSCT. Risk factors associated with the development of DAH in these patients include advanced age, total body radiation, type of myeloablative conditioning regimens, and presence of severe acute graft-vs-host disease. While most cases of clinical DAH will be a subset of IPS, infection as a contributing feature to the development of DAH must be excluded, as hemorrhage in this subset of patients may also be due to infection. Mortality for DAH in this setting is 60–100 %. Furthermore, even for the minority of patients that survive an episode of DAH, the follow-up 6-month mortality is on the order of 40 %. Treatment for DAH associated with HSCT is high-dose corticosteroids and supportive care, but given the exceedingly high mortality, the efficacy of this strategy is clearly limited. Attempts at protective strategies, including reductions in the intensity of the conditioning regimen, have not been proven to decrease the risk of disease [68–71].
Cocaine Inhalation
Cocaine is an illicit drug derived from leaves of the Erythroxylon coca plant. While cocaine may have legitimate medicinal properties as a local anesthetic, its stimulant properties make it an attractive drug of abuse. Cocaine may be inhaled nasally, smoked and inhaled in its free base form (“crack” cocaine) or injected intravenously. Inhaled crack cocaine is commonly associated with an array of pulmonary complications including thermal injuries to the upper airways, cough and carbonaceous sputum, barotrauma (pneumothorax and pneumomediastinum), cardiogenic and noncardiogenic pulmonary edema, bronchospasm and asthma, eosinophilic lung reactions, organizing pneumonia, “crack lung,” hemoptysis and pulmonary hemorrhage. Acute respiratory symptoms usually develop with several hours of use but may develop in a matter of minutes or may evolve over several days. Presenting complaints will include cough, chest pain (usually pleuritic), shortness of breath, hemoptysis, and wheezing. Hypoxic, tachycardia, tachypnea, and abnormalities on auscultation are common. Imaging findings depend upon the manifestation of cocaine pulmonary toxicity, but in the case of “crack lung” or alveolar hemorrhage, bilateral pulmonary infiltrates are identified. Toxicology screening should reveal the presence of cocaine metabolites in the urine [2].
Diffuse alveolar hemorrhage as an isolated complication or as part of the more heterogenous “crack lung” pattern of acute parenchymal injury is believed to be relatively common but data is limited. Histology in “crack lung” is characterized by DAD, edema, inflammatory infiltrates, and hemorrhage. The mechanism of injury is believed to relate to profound vasocontriction of the pulmonary vascular bed and its resulting cellular damage. Additionally, direct toxic effect of the inhaled substances is also believed to play a role [2].
Management is supportive in nature and in most cases, the injury will spontaneously resolve. At present, there appears to be no role for corticosteroids or other immunosuppressive therapy, but substance abuse counseling is critical to the longitudinal management of these patients.
Acute Exacerbation of Interstitial Lung Disease
Acute exacerbation of interstitial lung disease (AE-ILD) is a relatively rare cause of DAD associated DAH, but conversely is a common cause of death among patients with fibrosing interstitial lung diseases. While the occurrence of AE-ILD is now well recognized in IPF collagen vascular disease associated ILD, hypersensitivity pneumonitis, drug-associated ILD, and fibrotic non-specific interstitial pneumonitis, the incidence and pathophysiology remain largely unknown, and the diagnosis remains one of exclusion. By definition, AE-ILD is the presence of an acute respiratory decline (≤30 days) in a patient with a pre-existing fibrosing interstitial lung disease accompanied by new radiographic infiltrates (ground glass or consolidation superimposed on pre-existing fibrotic changes) in whom no infection and no alternative cause of the decline can be identified [72].
Recently, two groups of investigators have reported their experience with this entity improving our understanding of the clinical features of AE-ILD. In one large retrospective study focusing on idiopathic pulmonary fibrosis, the incidence of AE-ILD among an at risk population was 14.2 % at 1-year and 20.7 % at 3-year follow-up. In the study, in hospital mortality was approximately 50 % with a 5-year survival of only 18.4 %. Diffuse alveolar hemorrhage was noted as the cause of death in 2.2 % of patients. Predictors of poor outcome included older age, low FVC and DLCO, and immunosuppressive therapy [73].
In another smaller study of patients hospitalized with suspected AE-ILD, 100 % of patients were noted to present with worsening dyspnea and increased oxygen requirements. Most patients were found to have a cough and constitutional symptoms. Bronchoscopy was performed routinely to exclude infectious etiologies for the patients’ decline, and surprisingly, diffuse alveolar hemorrhage was identified on serial lavage in 21.7 % of patients. On chest imaging, the majority of patients had diffuse ground glass opacities superimposed on their underlying lung disease. In the patients that underwent lung biopsy or autopsy, nine out of ten had evidence of DAD superimposed on underlying fibrosis and one patient demonstrated significant acute and chronic alveolar hemorrhage with no significant acute lung injury. Hospital survival was only 37 % and 1-year survival 14.8 % in this patient cohort [74].
Treatment of AE-ILD is largely empiric and focused on supportive care. Most experts recommend the administration of broad-spectrum antimicrobial therapy and high dose corticosteroids, but no significant controlled trials have been performed to confirm their efficacy. Additionally, the ACE-IPF study of Anti-Coagulant Effectiveness in Idiopathic Pulmonary Fibrosis was stopped early due to an excess of mortality in the warfarin treatment arm. In patient who require mechanical ventilation, the mortality approaches 100 %, thus while a lung-protective strategy is recommended given the similarities to ARDS, counseling the patient and family regarding the dismal prognosis is strongly recommended.
Acute Interstitial Pneumonia
Acute interstitial pneumonia (AIP) is unique clinical-histopathologic entity characterized by a rapidly progressive course of respiratory decline over days to weeks that is associated with diffuse bilateral radiographic infiltrates, and the pathologic correlation of organizing DAD [75]. By definition, AIP is an idiopathic interstitial pneumonia, and the clinician must eliminate known causes of DAD in order to make a diagnosis. This can often be difficult, and indeed, some experts have coined AIP to be “idiopathic ARDS.”
AIP is very rare with only about 250 cases in the literature. On average patients are in their fifth through seventh decades, and there appears to be a slight male preference. Unlike ARDS, symptoms tend to evolve over days to weeks (and in some reports, months) and include cough, dyspnea, fatigue, malaise, fever, and “flu-like” illness. Symptoms typically progress to include more marked dyspnea, hypoxemia and respiratory distress [76]. Imaging studies universally reveal bilateral infiltrates, and on more refined HRCT imaging, appear as ground-glass abnormalities and consolidation [77]. The pathologic pattern of AIP is that of fibroproliferative or organizing DAD. Proliferative hyperplastic type II alveolar epithelial cells are seen lining airspaces filled with fibromyxoid tissue, edema, fibrinous exudates and remnants of hyaline membranes. Subacute and chronic inflammatory infiltrates are noted as is fibroblast proliferation, interstitial widening and collagen deposition. Occasionally, features of alveolar hemorrhage are also identified on histology [75].
Given the very low numbers of patients reported in the literature, it is difficult to prognosticate in AIP, but overall it appears to be better than AE-ILD or HSCT-IPS, but worse than ARDS [78]. As with DAD of other etiologies, patients receive supportive care, including lung protective ventilatory strategies (6 ml/kg tidal volumes) and restrictive fluid strategies if they require mechanical ventilation. High dose corticosteroids are commonly deployed, but again, objective data supporting their use is lacking and is largely based upon extrapolation from other entities.
Acute Respiratory Distress Syndrome
The acute respiratory distress syndrome (ARDS) is a major cause of respiratory failure and ICU admission. The 1992 American European Consensus Conference criteria defined ARDS by the presence of (i) acute onset (≤7 days) respiratory failure, (ii) bilateral lung infiltrates, (iii) severe hypoxemia (PaO2/FiO2 ratio of <200) and (iv) the absence of evidence of left atrial hypertension (pulmonary capillary wedge pressure <18 mmHg) [79]. The most common etiologies or precipitants of ARDS are septic shock, pneumonia, aspiration, trauma, and pancreatitis. Other previously described precipitants include fat emboli, thermal burns, transfusion-associated lung injury, and inhalational injuries. Risk factors for the development of ARDS include advanced age and alcohol consumption. Initial evaluation of patients with ARDS is focused on (i) determining and treating the underlying cause of the lung injury and (ii) optimally supporting the patient’s ventilation and gas exchange. Hence, patients typically are broadly cultured for the presence of infection and cardiogenic causes of pulmonary edema are excluded (echocardiography, serial electrocardiography, cardiac enzymes). Bronchoscopy may be performed in patients in whom no obvious inciting etiology is identified. Mechanical ventilatory support is required in virtually all patients, and a lung protective ventilatory strategy is recommended in all ARDS patients (see below) [80, 81].
The histology of ARDS is DAD, but super-imposed intra-alveolar hemorrhage may be seen, especially during the acute phase of DAD [10].
Mortality in ARDS has improved over the past 10–15 years with advances in care informed by large multicenter clinical trials, in particular the National Heart, Lung and Blood Institute ARDS Clinical Network trials. The landmark ARMA trial published in 2000 of patient with ARDS maintained on mechanical ventilation, demonstrated that a “lung-protective” tidal volume of 6 ml/kg was associated with a 22 % reduction in mortality when compared with a tidal volume of 12/ml/kg [82]. Further investigation into ventilatory strategies using differing levels of positive end expiratory pressure were unable to achieve clinically significant differences, but re-inforced the desirability of keeping the end inspiratory plateau pressure below 30 mmHg [83]. The Fluid and Catheter Treatment Trial compared a liberal versus a conservative fluid management strategy in ARDS as well as methodology for monitoring fluid management, and the investigators found that a conservative strategy of fluid management improved lung function and shortened the duration of mechanical ventilation [84]. Additionally, investigation into the optimal management of analgesia and sedation, liberation strategies from mechanical ventilation, nutritional management, the prevention of hospital acquired conditions (catheter related blood stream infections, ventilator associated pneumonia, pressure ulcers, venous thromboembolic disease, etc.) and restrictive transfusion strategies have all contributed to the improvements seen in the management of the critically-ill patient including those with ARDS. Hence, the 28-day mortality has declined from 35 to 40 % to approximately 20–25 % [81].
As the mortality in ARDS has improved, a new appreciation has developed for the long-term morbidity of survivors. Problems including persistent dyspnea, fatigue and exercise intolerance, restrictive lung disease, cognitive dysfunction, post-traumatic stress disorder, and neuromuscular weakness are increasingly recognized as important long term sequellae following an episode of ARDS [80].
Miscellaneous Causes
Etiologies
Human Immunodeficiency Virus (HIV)
Infection with HIV predisposes patients not only to its all to well known infectious complications, but also a broad array of non-infectious pulmonary complications such as Kaposi’s sarcoma, interstitial lung disease (nonspecific interstitial pneumonitis, lymphocytic interstitial pneumonitis, organizing pneumonia), pulmonary hypertension and alveolar hemorrhage. Vincent and colleagues prospectively analyzed the BAL results of a cohort of HIV-infected patients undergoing bronchoscopy as part of their evaluation for new respiratory symptoms and/or fever and found that approximately one third of patients had evidence of DAH [85]. Independent risk factors for DAH in this study were (i) pulmonary Kaposi’s sarcoma, (ii) hydrostatic pulmonary edema, (iii) cytomegalovirus pneumonia, and (iv) thrombocytopenia (platelets <60,000 cell per microliter). However, the absolute degree of hemorrhage was relatively mild and did not appear to affect survival. Treatment is largely supportive care combined with treating reversible, underlying pulmonary processes (i.e. pneumonia or Kaposi’s).
Pulmonary Capillary Hemoangiomatosis
Pulmonary capillary hemangiomatosis is an exceedingly rare entity characterized by diffuse proliferation of the pulmonary capillary network. Patients present with signs and symptoms of both alveolar hemorrhage (secondary to capillary rupture into the airspaces) and pulmonary hypertension (secondary to obstruction to flow at the capillary/post-capillary level.) [86] Given its rarity, no effective therapies have been identified. There is one case report of clinical improvement following treatment with interferon alpha-2a [87].
Treatment
When approaching a patient with hemoptysis, bilateral infiltrates with hypoxemia, or known lower respiratory tract bleeding, the differential diagnosis remains quite broad, and yet, the bedside clinician must support the patient’s vital physiologic functions, and correctly diagnosis and reverse the disease process in an efficient and timely fashion. Moreover, as has been discussed, the degree and severity of dyspnea, hypoxia and/or respiratory distress can vary dramatically between patients from the asymptomatic to profound hypoxemic respiratory failure. Nevertheless, the initial steps of management revolve around supportive care and distinguishing between a focal source of hemorrhage, a non-hemorrhagic diagnosis, and diffuse alveolar hemorrhage.
From a supportive care standpoint, oxygen therapy titrated to a saturation of arterial hemoglobulin ≥90 % is recommended for all patients. Similarly, reversing any contributing coagulopathy is recommended unless firm contraindications or mitigating circumstances exist (i.e. anti-phospholipid antibody syndrome with active or life-threatening thromboembolic disease). In those patients who are unable to protect their airway or who require ventilatory support, endotracheal intubation and initiation of mechanical ventilation is recommended. In general, non-invasive positive pressure ventilation would be contraindicated in any patient suffering respiratory failure secondary to active DAH. Still, to date, no randomized trials have been performed to determine the most effective mode of mechanical ventilation in DAH. As has been discussed previously, extrapolating from the extensive data informing the ARDS literature, most experts would recommend a lung-protective, low tidal volume strategy (tidal volumes of 6 ml/kg ideal body weight). Given the clinical similarities between DAH complicated by respiratory failure and ARDS, it is reasonable to extrapolate this data to the care of patients with DAH.
Similarly, volume resuscitation is recommended for patients with clinical evidence of hypovolemia or shock. Following definitive reversal of the hypoperfused state, adopting a conservative fluid strategy, again based on data from the ARDS literature, would also be advisable. For those patients with clinically significant anemia, transfusion may be necessary, but there is no data to specifically inform transfusion thresholds in DAH. Recent data in more general populations of critically-ill patients have found that blood transfusions increase the risk of adverse outcomes, particularly in younger and less severely-ill patients, and many critical care units will now uniformly deploy restrictive transfusion strategies that transfuse to a goal hemogloblin of no higher than 7.0–9.0 g/dl [88]. Mitigating against this, would be the rate of active bleeding and whether or not the hemorrhage is controlled at the time of decision-making.
As outlined earlier, the diagnosis of DAH rests upon bronchoscopy and bronchoalveolar lavage demonstrating an absence of clearing, or paradoxical worsening of hemorrhage during serial lavage. Competing considerations that may be identified on bronchoscopy include focal hemorrhage (malignancy, arterio-venous malformation, bronchiectasis, necrotizing pneumonia, pulmonary embolus, tuberculosis/Rasmussen’s aneurysms, etc.), aspirated blood (gastrointestinal or upper airway bleeding), or non-hemorrhagic disease (ARDS, pneumonia, pulmonary edema, etc). Once a diagnosis of DAH has been established, further distinguishing between capillaritis and non-capillaritis lesions determines whether or not additional targeted therapies may provide benefit. Generally, the specific underlying etiology is not known even when a diagnosis of DAH is established, but rapid intervention may be required depending upon disease severity.
For patient’s with suspected pulmonary capillaritis, in whom a diagnosis of GPA, MPA, SLE or Goodpasture’s syndrome is being seriously entertained and who demonstrate respiratory distress, respiratory failure requiring mechanical ventilation and/or demonstrate a clinically-significant pulmonary renal syndrome of DAH and glomerulonephritis, initiating therapy with IV corticosteroids and plasmaphresis is recommended along with the introduction of a cytotoxic agent (e.g. cyclophosphamide). The data informing these treatment recommendations are derived primarily from the AAV and Goodpasture’s syndrome literature and have been discussed earlier. The optimal timing of the introduction of the cyclophosphamide or other cytotoxic agent in the critical care setting, especially in patients with concomitant infection and/or requiring mechanical ventilation remains subject to debate. For patients with lesser degrees of disease severity, not requiring mechanical ventilation and without significant end-organ impairment, clinicians may elect to treat with corticosteroids alone pending the results of the definitive evaluation (which in turn may then drive additional treatment decisions such as the introduction of a cytotoxic agent). However, these cases can often evolve rapidly and close serial observation is required. If patients demonstrate clinical deterioration, therapy may need to be escalated.
As was discussed earlier, two recently published randomized control trials were performed in generalized, active and severe AAV comparing cyclophosphamide to rituximab for the induction of remission, and based upon the finding of non-inferiority, rituximab may now be considered an alternative first line agent for the management of AAV [25, 89]. Furthermore, specifically with the RAVE study, rituximab was found to be as effective as cyclophosphamide in patients with alveolar hemorrhage. Thus, one may consider substituting rituximab for cyclophosphamide in patients with AAV and alveolar hemorrhage. However, recognizing that rituximab is a monoclonal antibody, the timing of its administration relative to any use of plasmapharesis must be carefully considered, and in point of fact, most experts would recommend beginning administration after completion of any plasma exchange (or limiting its use to those patients who do not require plasma exchange).
Additional therapies that have been considered in these severe cases of DAH include activated human factor VII and extracorporeal membrane oxygenation (ECMO). Exogenous activated factor VII has been used on a compassionate use basis and reported at the case report level to aid in hemostasis of cases of refractory hemorrhage. Similarly, ECMO has been utilized at the case report level to prolong survival while allowing the above therapies to have an effect, but pragmatically, the pro-inflammatory cascade triggered by the ECMO circuit and the need for anti-thrombotic therapies during ECMO administration complicates its use in DAH.
Conclusions
Diffuse alveolar hemorrhage is a complex and life-threatening clinical-pathologic syndrome associated with a broad differential diagnosis and a number of distinct histopathologic patterns. Accurate and timely diagnosis of DAH and its specific underlying cause permits the bedside clinician to effectively treat the majority of patients such that a detailed knowledge of its diagnosis and management is critical for the Pulmonary/Critical Care physician.
Contributor Information
Vincent Cottin, Email: vincent.cottin@chu-lyon.fr.
Jean-Francois Cordier, Email: jean-francois.cordier@chu-lyon.fr.
Luca Richeldi, Email: L.Richeldi@soton.ac.uk.
Jason Wells, Email: Jaw2014@gmail.com.
Stephen K. Frankel, Email: Frankels@NJHealth.org.
References
- 1.Gertner E. Diffuse alveolar hemorrhage in the antiphospholipid syndrome: spectrum of disease and treatment. J Rheumatol. 1999;26(4):805–7. [PubMed] [Google Scholar]
- 2.Walkenstein M. The pulmonary complications of crack cocaine a comprehensive review. Chest. 1995;107(1):233–40. doi: 10.1378/chest.107.1.233. [DOI] [PubMed] [Google Scholar]
- 3.Zamora MR, Warner ML, Tuder R, Schwarz MI. Diffuse alveolar hemorrhage and systemic lupus erythematosus. Clinical presentation, histology, survival, and outcome. Medicine. 1997;76(3):192–202. doi: 10.1097/00005792-199705000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Martinez AJ, Maltby JD, Hurst DJ. Thrombotic thrombocytopenic purpura seen as pulmonary hemorrhage. Archives of internal medicine. Am Med Assoc. 1983;143(9):1818–20. [PubMed] [Google Scholar]
- 5.Robboy SJ, Minna JD, Colman RW, Birndorf NI, Lopas H. Pulmonary hemorrhage syndrome as a manifestation of disseminated intravascular coagulation: analysis of ten cases. Chest. 1973;63(5):718–21. doi: 10.1378/chest.63.5.718. [DOI] [PubMed] [Google Scholar]
- 6.Santos-Ocampo AS, Mandell BF, Fessler BJ. Alveolar hemorrhage in systemic lupus erythematosus: presentation and management. Chest. 2000;118(4):1083–90. doi: 10.1378/chest.118.4.1083. [DOI] [PubMed] [Google Scholar]
- 7.Ford HJ, Roubey RAS. Pulmonary manifestations of the antiphospholipid antibody syndrome. Clin Chest Med. 2010;31(3):537–45. doi: 10.1016/j.ccm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Buschman DL, Ballard R. Progressive massive fibrosis associated with idiopathic pulmonary hemosiderosis. Chest. 1993;104(1):293–5. doi: 10.1378/chest.104.1.293. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz M, Mortenson R, Colby T. Pulmonary capillaritis: the association with progressive irreversible airflow limitation and hyperinflation. Am Rev Respir Dis. 1993;148(2):507–11. doi: 10.1164/ajrccm/148.2.507. [DOI] [PubMed] [Google Scholar]
- 10.de Hemptinne Q, Remmelink M, Brimioulle S, Salmon I, Vincent J-L. ARDS: a clinicopathological confrontation. Chest. 2009;135(4):944–9. doi: 10.1378/chest.08-1741. [DOI] [PubMed] [Google Scholar]
- 11.Esteban A, Fernández-Segoviano P, Frutos-Vivar F, Aramburu JA, Nájera L, Ferguson ND, et al. Comparison of clinical criteria for the acute respiratory distress syndrome with autopsy findings. Ann Intern Med. 2004;141(6):440–5. doi: 10.7326/0003-4819-141-6-200409210-00009. [DOI] [PubMed] [Google Scholar]
- 12.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–14. doi: 10.1164/rccm.201202-0320ST. [DOI] [PubMed] [Google Scholar]
- 13.Lazor R, Bigay-Gamé L, Cottin V, Cadranel J, Decaux O, Fellrath J-M, et al. Alveolar hemorrhage in anti-basement membrane antibody disease: a series of 28 cases. Medicine. 2007;86(3):181–93. doi: 10.1097/md.0b013e318067da56. [DOI] [PubMed] [Google Scholar]
- 14.Lara AR, Schwarz MI. Diffuse alveolar hemorrhage. Chest. 2010;137(5):1164–71. doi: 10.1378/chest.08-2084. [DOI] [PubMed] [Google Scholar]
- 15.Colby TV, Fukuoka J, Ewaskow SP, Helmers R, Leslie KO. Pathologic approach to pulmonary hemorrhage. Ann Diagn Pathol. 2001;5(5):309–19. doi: 10.1053/adpa.2001.27923. [DOI] [PubMed] [Google Scholar]
- 16.Fishbein GA, Fishbein MC. Lung vasculitis and alveolar hemorrhage: pathology. Semin Respir Crit Care Med. 2011;32(3):254–63. doi: 10.1055/s-0031-1279823. [DOI] [PubMed] [Google Scholar]
- 17.Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int. 1998;53(3):743–53. PMID: 9507222 [DOI] [PubMed]
- 18.Cordier J, Valeyre D, Guillevin L, Loire R. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906–12. doi: 10.1378/chest.97.4.906. [DOI] [PubMed] [Google Scholar]
- 19.Bligny D, Mahr A, Toumelin PL, Mouthon L, Guillevin LC. Predicting mortality in systemic Wegener’s granulomatosis: a survival analysis based on 93 patients. Arthritis Rheum. 2004;51(1):83–91. doi: 10.1002/art.20082. [DOI] [PubMed] [Google Scholar]
- 20.Frankel SK, Jayne D. The pulmonary vasculitides. Clin Chest Med. 2010;31(3):519–36. doi: 10.1016/j.ccm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Boomsma M, Stegeman C. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum. 2000;43(9):2025–33. doi: 10.1002/1529-0131(200009)43:9<2025::AID-ANR13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983;98(1):76–85. doi: 10.7326/0003-4819-98-1-76. [DOI] [PubMed] [Google Scholar]
- 23.Jayne DRW, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18(7):2180–8. doi: 10.1681/ASN.2007010090. [DOI] [PubMed] [Google Scholar]
- 24.Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, Henke DC, Falk RJ. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis. Am J Kidney Dis. 2003;42(6):1149–53. doi: 10.1053/j.ajkd.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 25.Stone J, Merkel P, Spiera R, Seo P. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillevin L, Durand-Gasselin B, Cevallos R. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999;42(3):421–30. doi: 10.1002/1529-0131(199904)42:3<421::AID-ANR5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 27.Frankel SK. Update in the diagnosis and management of pulmonary vasculitis. Chest. 2006;129(2):452–65. doi: 10.1378/chest.129.2.452. [DOI] [PubMed] [Google Scholar]
- 28.Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax. 2000;55(6):502–10. doi: 10.1136/thorax.55.6.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heslet L, Nielsen J, Levi M, Sengeløv H. Successful pulmonary administration of activated recombinant factor VII in diffuse alveolar hemorrhage. Crit Care. 2006;10(6):R177. doi: 10.1186/cc5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jennings C, King JT, Tuder R. Diffuse alveolar hemorrhage with underlying isolated, pauciimmune pulmonary capillaritis. Am J Respir Crit Care Med. 1997;155(3):1101–9. doi: 10.1164/ajrccm.155.3.9116994. [DOI] [PubMed] [Google Scholar]
- 31.Schwarz MI, Zamora MR, Hodges TN, Chan ED, Bowler RP, Tuder RM. Isolated pulmonary capillaritis and diffuse alveolar hemorrhage in rheumatoid arthritis and mixed connective tissue disease. Chest. 1998;113(6):1609–15. doi: 10.1378/chest.113.6.1609. [DOI] [PubMed] [Google Scholar]
- 32.Rojas-Serrano J, Pedroza J, Regalado J, Robledo J, Reyes E, Sifuentes-Osornio J, et al. High prevalence of infections in patients with systemic lupus erythematosus and pulmonary haemorrhage. Lupus. 2008;17(4):295–9. doi: 10.1177/0961203307086930. [DOI] [PubMed] [Google Scholar]
- 33.Kamen DL, Strange C. Pulmonary manifestations of systemic lupus erythematosus. Clin Chest Med. 2010;31(3):479–88. doi: 10.1016/j.ccm.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Gertner E, Lie JT. Pulmonary capillaritis, alveolar hemorrhage, and recurrent microvascular thrombosis in primary antiphospholipid syndrome. J Rheumatol. 1993;20(7):1224–8. [PubMed] [Google Scholar]
- 35.Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, Pons-Estel G, et al. Catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the “CAPS Registry”. J Autoimmun. 2009;32(3–4):240–5. doi: 10.1016/j.jaut.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Elazary AS, Klahr P, Hershko A, Dranitzki Z, Rubinow A, Naparstek Y. Rituximab induces resolution of recurrent diffuse alveolar hemorrhage in a patient with primary antiphospholipid antibody syndrome. Lupus. 2012;21(4):438–40. doi: 10.1177/0961203311422713. [DOI] [PubMed] [Google Scholar]
- 37.Kluth DC, Rees AJ. Anti-glomerular basement membrane disease. J Am Soc Nephrol. 1999;10(11):2446–53. doi: 10.1681/ASN.V10112446. [DOI] [PubMed] [Google Scholar]
- 38.Magro CM, Morrison C, Pope-Harman A, Rothrauff SK, Ross P. Direct and indirect immunofluorescence as a diagnostic adjunct in the interpretation of nonneoplastic medical lung disease. Am J Clin Pathol. 2003;119(2):279–89. doi: 10.1309/EHUQHW6K0G41E0DX. [DOI] [PubMed] [Google Scholar]
- 39.Cui Z, Zhao J, Jia X-Y, Zhu S-N, Jin Q-Z, Cheng X-Y, et al. Anti-glomerular basement membrane disease. Medicine. 2011;90(5):1. doi: 10.1097/MD.0b013e31822f6f68. [DOI] [PubMed] [Google Scholar]
- 40.Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001;134(11):1033–42. doi: 10.7326/0003-4819-134-11-200106050-00009. [DOI] [PubMed] [Google Scholar]
- 41.Badesch DB, Zamora M, Fullerton D, Weill D, Tuder R, Grover F, et al. Pulmonary capillaritis: a possible histologic form of acute pulmonary allograft rejection. J Heart Lung Transplant. 1998;17(4):415–22. [PubMed] [Google Scholar]
- 42.Astor TL, Weill D, Cool C, Teitelbaum I, Schwarz MI, Zamora MR. Pulmonary capillaritis in lung transplant recipients: treatment and effect on allograft function. J Heart Lung Transplant. 2005;24(12):2091–7. doi: 10.1016/j.healun.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Cordier J-F, Cottin V. Alveolar hemorrhage in vasculitis: primary and secondary. Semin Respir Crit Care Med. 2011;32(3):310–21. doi: 10.1055/s-0031-1279827. [DOI] [PubMed] [Google Scholar]
- 44.Shrestha S, Sumingan N, Tan J, Alhous H, Althous H, McWilliam L, et al. Henoch Schönlein purpura with nephritis in adults: adverse prognostic indicators in a UK population. QJM. 2006;99(4):253–65. doi: 10.1093/qjmed/hcl034. [DOI] [PubMed] [Google Scholar]
- 45.Erkan F. Pulmonary involvement in Behçet disease. Curr Opin Pulm Med. 1999;5(5):314–8. doi: 10.1097/00063198-199909000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Raz I, Okon E, Chajek-Shaul T. Pulmonary manifestations in Behçet’s syndrome. Chest. 1989;95(3):585–9. doi: 10.1378/chest.95.3.585. [DOI] [PubMed] [Google Scholar]
- 47.Erkan F, Çavdar T. Pulmonary vasculitis in Behçet’s disease. Am Rev Respir Dis. 1992;146(1):232–9. doi: 10.1164/ajrccm/146.1.232. [DOI] [PubMed] [Google Scholar]
- 48.Yazici H, Pazarli H, Barnes C. A controlled trial of azathioprine in Behçet’s syndrome. N Engl J Med. 1990;322(5):281–5. doi: 10.1056/NEJM199002013220501. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez-Vidigal F, Roig F. Alveolar hemorrhage in mixed cryoglobulinemia associated with hepatitis C virus infection. An Med Interna. 1998;15(12):661–3. [PubMed] [Google Scholar]
- 50.Black H, Mendoza M. Thoracic manifestations of inflammatory bowel disease. Chest. 2007;131(2):524–32. doi: 10.1378/chest.06-1074. [DOI] [PubMed] [Google Scholar]
- 51.Hadjiangelis NP, Harkin TJ. Propylthiouracil-related diffuse alveolar hemorrhage with negative serologies and without capillaritis. Respir Med. 2007;101(4):865–7. doi: 10.1016/j.rmed.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 52.Travis WD, Colby TV, Lombard C, Carpenter HA. A clinicopathologic study of 34 cases of diffuse pulmonary hemorrhage with lung biopsy confirmation. Am J Surg Pathol. 1990;14(12):1112–25. doi: 10.1097/00000478-199012000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. Eur Respir J. 2004;24(1):162–70. doi: 10.1183/09031936.04.00116302. [DOI] [PubMed] [Google Scholar]
- 54.Pacheco A, Casanova C, Fogue L, Sueiro A. Long-term clinical follow-up of adult idiopathic pulmonary hemosiderosis and celiac disease. Chest. 1991;99(6):1525–6. doi: 10.1378/chest.99.6.1525. [DOI] [PubMed] [Google Scholar]
- 55.Gavaghan T, McNaught P, Ralston M. Penicillamine-induced “Goodpasture’s syndrome”: successful treatment of a fulminant case. Aust N Z J Med. 1981;11(3):261–5. [PubMed] [Google Scholar]
- 56.Louie S, Gamble CN, Cross CE. Penicillamine associated pulmonary hemorrhage. J Rheumatol. 1986;13(5):963–6. [PubMed] [Google Scholar]
- 57.Israel-Biet D, Labrune S, Huchon GJ. Drug-induced lung disease: 1990 review. Eur Respir J. 1991;4(4):465–78. [PubMed] [Google Scholar]
- 58.Camus P, Foucher P, Bonniaud P. Drug-induced infiltrative lung disease. Eur Respir J Suppl. 2001;32:93s–100. [PubMed] [Google Scholar]
- 59.Barnett V, Bergmann F, Humphrey H, Chediak J. Diffuse alveolar hemorrhage secondary to superwarfarin ingestion. Chest. 1992;102(4):1301–2. doi: 10.1378/chest.102.4.1301. [DOI] [PubMed] [Google Scholar]
- 60.Spence TH, Connors JC. Diffuse alveolar hemorrhage syndrome due to “silent” mitral valve regurgitation. South Med J. 2000;93(1):65–7. doi: 10.1097/00007611-200093010-00013. [DOI] [PubMed] [Google Scholar]
- 61.Zeiss CR, Leach CL, Smith LJ, Levitz D, Hatoum NS, Garvin PJ, et al. A serial immunologic and histopathologic study of lung injury induced by trimellitic anhydride. Am Rev Respir Dis. 1988;137(1):191–6. doi: 10.1164/ajrccm/137.1.191. [DOI] [PubMed] [Google Scholar]
- 62.Patterson R, Nugent KM, Harris KE, Eberle ME. Immunologic hemorrhagic pneumonia caused by isocyanates. Am Rev Respir Dis. 1990;141(1):226–30. doi: 10.1164/ajrccm/141.1.226. [DOI] [PubMed] [Google Scholar]
- 63.Ahmed Q, Chung-Park M, Tomashefski JF. Cardiopulmonary pathology in patients with sleep apnea/obesity hypoventilation syndrome. Hum Pathol. 1997;28(3):264–9. doi: 10.1016/S0046-8177(97)90122-2. [DOI] [PubMed] [Google Scholar]
- 64.Mandel J, Mark EJ, Hales CA. Pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2000;162(5):1964–73. doi: 10.1164/ajrccm.162.5.9912045. [DOI] [PubMed] [Google Scholar]
- 65.Rabiller A, Jaïs X, Hamid A, Resten A, Parent F, Haque R, et al. Occult alveolar haemorrhage in pulmonary veno-occlusive disease. Eur Respir J. 2006;27(1):108–13. doi: 10.1183/09031936.06.00054105. [DOI] [PubMed] [Google Scholar]
- 66.Beasley MB. The pathologist’s approach to acute lung injury. Arch Pathol Lab Med. 2010;134(5):719–27. doi: 10.5858/134.5.719. [DOI] [PubMed] [Google Scholar]
- 67.Panoskaltsis-Mortari A, Griese M, Madtes DK, Belperio JA, Haddad IY, Folz RJ, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med. 2011;183(9):1262–79. doi: 10.1164/rccm.2007-413ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Afessa B, Peters SG. Chronic lung disease after hematopoietic stem cell transplantation. Clin Chest Med. 2005;26(4):571–86. doi: 10.1016/j.ccm.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 69.Afessa B, Tefferi A, Litzow M. Diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med. 2002;166(5):641–5. doi: 10.1164/rccm.200112-141CC. [DOI] [PubMed] [Google Scholar]
- 70.Afessa B. Outcome of diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med. 2002;166(10):1364–8. doi: 10.1164/rccm.200208-792OC. [DOI] [PubMed] [Google Scholar]
- 71.Lewis ID, DeFor T, Weisdorf DJ. Increasing incidence of diffuse alveolar hemorrhage following allogeneic bone marrow transplantation: cryptic etiology and uncertain therapy. Bone Marrow Transplant. 2000;26(5):539–43. doi: 10.1038/sj.bmt.1702546. [DOI] [PubMed] [Google Scholar]
- 72.Kim D, Park J, Park B, Lee J. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27(1):143–50. doi: 10.1183/09031936.06.00114004. [DOI] [PubMed] [Google Scholar]
- 73.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37(2):356–63. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 74.Huie TJ, Olson AL, Cosgrove GP, Janssen WJ, Lara AR, Lynch DA, et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: etiology and outcomes. Respirology. 2010;15(6):909–17. doi: 10.1111/j.1440-1843.2010.01774.x. [DOI] [PubMed] [Google Scholar]
- 75.American Thoracic Society, European Respiratory Society American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 76.Vourlekis J. Acute interstitial pneumonia. Clin Chest Med. 2004;25(4):739–47. doi: 10.1016/j.ccm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Ichikado K, Johkoh T, Ikezoe J. Acute interstitial pneumonia: high-resolution CT findings correlated with pathology. Am J Roentgenol. 1997;168(2):333–8. doi: 10.2214/ajr.168.2.9016201. [DOI] [PubMed] [Google Scholar]
- 78.Quefatieh A. Low hospital mortality in patients with acute interstitial pneumonia. Chest. 2003;124(2):554–9. doi: 10.1378/chest.124.2.554. [DOI] [PubMed] [Google Scholar]
- 79.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt1):818–24. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 80.Bernard GR. Acute respiratory distress syndrome: a historical perspective. Am J Respir Crit Care Med. 2005;172(7):798–806. doi: 10.1164/rccm.200504-663OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Susannah K, Leaver TWE. Acute respiratory distress syndrome. BMJ. 2007;335:389. doi: 10.1136/bmj.39293.624699.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342(18):1301–8. [DOI] [PubMed]
- 83.Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351(4):327–36. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 84.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354(24):2564–75. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 85.Vincent B. AIDS-related alveolar hemorrhage: a prospective study of 273 BAL procedures. Chest. 2001;120(4):1078–84. doi: 10.1378/chest.120.4.1078. [DOI] [PubMed] [Google Scholar]
- 86.Faber CN, Yousem SA, Dauber JH, Griffith BP, Hardesty RL, Paradis IL. Pulmonary capillary hemangiomatosis: a report of three cases and a review of the literature. Am Rev Respir Dis. 1989;140(3):808–13. doi: 10.1164/ajrccm/140.3.808. [DOI] [PubMed] [Google Scholar]
- 87.White C, Sondheimer H, Crouch E. Treatment of pulmonary hemangiomatosis with recombinant interferon alfa-2a. N Engl J Med. 1989;320(18):1197–200. doi: 10.1056/NEJM198905043201807. [DOI] [PubMed] [Google Scholar]
- 88.Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340(6):409–17. doi: 10.1056/NEJM199902113400601. [DOI] [PubMed] [Google Scholar]
- 89.Jones R, Tervaert JC. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211–20. doi: 10.1056/NEJMoa0909169. [DOI] [PubMed] [Google Scholar]