

Abstract
Sialic acids have a pivotal functional impact in many biological interactions such as virus attachment, cellular adhesion, regulation of proliferation, and apoptosis. A common modification of sialic acids is O-acetylation. O-Acetylated sialic acids occur in bacteria and parasites and are also receptor determinants for a number of viruses. Moreover, they have important functions in embryogenesis, development, and immunological processes. O-Acetylated sialic acids represent cancer markers, as shown for acute lymphoblastic leukemia, and they are known to play significant roles in the regulation of ganglioside-mediated apoptosis. Expression of O-acetylated sialoglycans is regulated by sialic acid-specific O-acetyltransferases and O-acetylesterases. Recent developments in the identification of the enigmatic sialic acid-specific O-acetyltransferase are discussed.
Keywords: 9-O-Acetylated sialic acids, Anemia, Anti-9-O-acetylated sialoglycoprotein antibody, Apoptosis, Erythrocytes, Indian visceral leishmaniasis, Leukemia, O-Acetylated disialoganglioside, O-Acetylated sialoglycoproteins, Sialate O-acetylesterase, Sialate O-acetyltranferase, Sialic acids
Introduction
The generic term sialic acid defines a large family of 9-carbon monosaccharides which commonly occur as terminal residues of the oligosaccharide moiety of glycoconjugates. The diversity of sialic acids results from differential N- and O-substitutions of the two basic molecules, i.e., N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). The most common modification of sialic acids is O-acetylation preferentially at the hydroxyl groups of carbon C4, C7, C8, and C9, whereas that at C9 seems to be the most frequent one.
The complexity of sialic acids is further augmented by their different glycosidic linkage types (for example, α2,3, α2,6, α2,8, and α2,9) to the oligosaccharide chain. Both modification of the terminal sialic acid and the anomericity of linkage determine their functional impact in many biological interactions, such as, e.g., virus attachment, cellular adhesion, and regulation of proliferation and apoptosis.
Occurrence and Functions of O-Acetylated Sialic Acids
Analysis of O-Acetylated Sialic Acids
Detection and analysis of O-acetylated sialic acids (O-Ac-Sias) requires several precautions in sample preparation. Even by taking special care to avoid the loss of O-acetyl groups during the purification and analysis process of sialoglycans, results may lead to an underestimation of their O-acetylation. Exposure of O-Ac-Sias to moderate alkaline conditions results in migration of acetyl groups from carbon C7 to carbons C8 or C9 [1, 2]. Procedures for isolation of gangliosides often involve incubation with NaOH to remove phospholipids, which results in a complete loss of O-acetyl groups due to saponification.
Histochemical staining with mild periodic acid-Schiff reaction (mPAS) is a general method to detect sialic acids. O-Ac-Sias with acetyl groups at C-8 or C-9 do not react under these conditions, but will stain after saponification with NaOH [3]. Mild oxidation in combination with detection of liberated formaldehyde with acetyl-acetone and ammonium acetate, yielding a fluorogen, has been described as a sensitive method to determine sialic acid concentration. After removal of O-acetyl groups at C-8 and C-9, an increase in formaldehyde production can be observed, which allows an indirect quantitative determination of O-Ac-Sias [4]. Lectins from the crab Cancer antennarius [5] and Achatinin-H derived from the snail Achatina fulica [6, 7] were shown to bind to 9-O-Ac-Sias. Viral lectins have also been used to detect this sialic acid derivative. The influenza C virus surface hemagglutinin-esterase glycoprotein termed HEF specifically binds to 9-O-Ac-Sias [8–10]. Influenza C virus preparations were used to stain cells and tissues of different sources [11–18]. To avoid the use of infectious virus, the influenza C virus surface glycoprotein was expressed as a chimeric protein fused to an Fc immunoglobulin domain [19] or to enhanced green fluorescent protein [20]. The chimeric proteins are useful to remove specifically 9-O-acetyl groups from sialic acids [10, 19–27]. The chimeric protein was also useful as lectin: by inhibition of the esterase domain with diisopropyl fluorophosphate, a general serine hydrolase inhibitor, the lectin function of the CHE-Fc protein was employed to detect 9-O-Ac-Sias [19, 25, 28, 29].
In addition to lectins, monoclonal antibodies with specificity for O-acetylated gangliosides are available, which can be used for immunodetection and fluorescence activated cell sorting. Examples are MAb Jones [30], MAb UM4D4 [31], MAb U5 [32], and MAb 7H2 [33]. These antibodies can also be used for immunodetection of O-Ac gangliosides on HPTLC plates using a method originally described by Saito et al. [34]. Most recently, a method to determine the distribution of O-Ac gangliosides in rat brain by matrix assisted laser desorption/ionization (MALDI) mass spectrometry imaging was published [35]. This procedure combines high resolution mass spectrometry with imaging software to map and image gangliosides with detailed structural information and histological accuracy.
A highly sensitive method to analyze O-Ac-Sias relies on fluorescence detection of sialic acids derivatized with 1,2-diamino-4,5-methylenedioxybenzene (DMB), followed by fluorometric high-performance liquid chromatography (HPLC) [36]. For analysis of O-Ac-Sias the acidic hydrolysis of glycosidically bound sialic acids should be performed with propionic instead of acetic acid [37]. The presence of O-Ac-Sias can be determined by their respective Rf values [38]. In biological samples carboxyl groups of other compounds may result in additional peaks in chromatograms. Therefore, the presence of O-Ac-Sias in HPLC peaks should be confirmed by saponification, specific de-O-acetylation with influenza C virus esterase (for Neu5,9Ac2) or rat coronavirus esterase for Neu4,5Ac2, or by mass spectrometry. The position of side groups of sialic acids can be determined by coupled gas chromatography/mass spectrometry (GC/MS) [39–43]. Very recently another highly sensitive method to identify different forms of O-acetylated sialic acids by electrospray ionization travelling wave ion mobility mass spectrometry coupled with low-energy collision-induced dissociation was described, allowing unambiguous assignment of the position of O-acetylation [44].
O-Acetylated Sialic Acids in Bacteria
Sialic acids are constituents of different opportunistic human pathogens. Bacteria can obtain sialic acids by either de novo biosynthesis or by acquisition from the environment. Different Gram negative bacteria like Escherichia coli K1, Neisseria meningitidis, and Campylobacter jejuni, and Gram positive bacteria like Group B Streptococcus (GBS) can synthesize sialic acids [45]. Sialic acid uptake in other Gram negative bacteria like Haemophilus Influenza, Pasteurella multocida, Haemophilus ducreyi, and Pseudomonas aeruginosa from exogenous sources has recently been established [45, 46]. These bacteria use their sialic acids for a variety of different purposes that play important roles in their ability to colonize and persist in organs of their hosts. For instance, sialic acids were shown to subvert immune clearance mechanisms by restricting complement C3b deposition on its surface [47]. Once synthesized, sialic acid residues in the capsular polysaccharide or oligosaccharide of E. coli K1, GBS serogroup III, N. meningitidis, one or more of the hydroxyl groups in positions 4, 7, 8, and 9 are substituted by acetyl groups [48]. Sia O-acetylation and de-O-acetylation is regulated by the gene neuD and neuA, respectively, to reach a final level of the surface expressed O-Ac-Sia modification [47, 49, 50]. In addition to neuD, which O-acetylates monomeric sialic acid, E. coli K1 strains harboring the prophage CUS-3 express neuO, a polysialic acid-specific O-acetyltransferase [51, 52]. Expression of neuO is regulated by phase variation [53]. In E. coli K1 strains O-acetylation increases immunogenicity of the K1 capsule and correlates with increased virulence in patients [54]. O-Acetylation of polySia increases desiccation resistance, which may favor survival in the environment [55]. In N. meningitides the genes for O-acetyltransferases are located immediately downstream of the capsule synthesis genes siaA – siaD [48] and termed oatC and oatWY [48, 56, 57].
Most bacterial O-acetyltransferases are characterized by a hexapeptide repeat sequence folding into a left-handed β-helix [48, 57, 58]. OatC, transferring acetyl groups exclusively onto polysialic acid joined by α2,9-linkages, apparently evolved separately and is characterized by an α/β hydrolase fold topology [56]. The presence of the sialic acids and Neu5,9Ac2α2-6GalNAc sialoglycotope has been demonstrated on P. aeruginosa [46]. Molecular analysis of GBS serogroup III strain indicates that high levels of Sia O-acetylation disrupt interactions with human Siglec-9 present on neutrophils and block removal of capsular polysaccharide Sia by bacterial sialidase, but do not alter deposition of complement on its surface [47, 59]. Blocking the interaction of GBS with neutrophils increases their activation followed by increasing bacterial killing.
O-Acetylated Sialic Acids as Virus Receptors
Many viruses use sialic acids as receptors for binding to target cells, which is the critical first step of infection. Influenza C viruses bind to cells via their surface glycoprotein termed hemagglutinin-esterase-fusion (HEF) protein. These viruses bind to Neu5,9Ac2 via the hemagglutinin function of the HEF protein [9, 10]. In addition, the HEF protein has a sialate-9-O-acetylesterase activity [8, 10]. Infectious salmon anemia viruses, another genus of the Orthomyxoviridae, encode a hemagglutinin-esterase (HE) surface glycoprotein which binds to Neu4,5Ac2 [60]. The esterase of these viruses is also specific for Neu4,5Ac2 [24]. Several coronaviruses and toroviruses also express HE proteins which interact with O-Ac-Sias (Table 1). A more comprehensive review of “Sialovirology” will be published in another chapter of this issue.
Table 1.
Viruses recognizing O-acetylated sialic acids
| Virus | Viral sialic acid binding protein | Sialic acid recognized | Reference |
|---|---|---|---|
| Influenza C virus | HEF | Neu5,9Ac2 | [8–10, 61] |
| Infectious salmon anemia virus | HE | Neu4,5Ac2 | [24, 60] |
| Human coronavirus OC43 | HE, S | Neu5,9Ac2 | [62–64] |
| Bovine coronavirus | HE, S | Neu5,9Ac2 | [64–69] |
| Hemagglutinating encephalomyelitis virus | HE | Neu5,9Ac2 | [70, 71] |
| Murine coronavirus | HE | Neu4,5Ac2 | [72, 73] |
| Murine coronavirus strain DVIM | HE | Neu5,9Ac2 | [74] |
| Puffin coronavirus | HE | Neu4,5Ac2 | [73, 75] |
| Sialodacryo-adenitis virus | HE | Neu4,5Ac2 | [20, 73] |
| Porcine torovirus | HE | Neu5,9Ac2 | [67, 76] |
| Bovine torovirus | HE | Neu5,7(8),9Ac3 | [67] |
Functions and Biosynthesis of O-Acetylated Sialic Acids in Parasites
Extensive research for the past decade has associated 9-O-acetylated sialic acids with promastigotes and amastigotes kinetoplastid parasites Leishmania sp. [77–83]. During the disease manifestation of visceral, cutaneous, and mucocutaneous leishmaniasis, increased presence of 9-O-acetylated sialic acids is observed in virulent strains of Leishmania sp., indicating their probable relevance in pathogenesis. In contrast minimal or undetectable presence of 9-O-acetylated sialic acids on a virulent strain of UR6 also signifies the role of 9-O-acetylated sialoglycotope as markers of virulence [78, 81]. The function of parasite-associated 9-O-acetylated sialic acids for entry of promastigotes into macrophages has been demonstrated in comparison to the minimal internalization of de-O-acetylated promastigotes. Analysis of the sialylation during the differentiation of internalized virulent promastigotes into amastigotes indicates that 9-O-acetylated sialic acids not only facilitate promastigote-entry but also play a probable role in differentiation and persistence of infection [81]. Additionally, increased presence of 9-O-acetylated sialic acids during metacyclic stages of virulent promastigotes has pointed to the direct correlation between the association of 9-O-acetylated sialic acids and virulence. Apart from being a marker of virulence, 9-O-acetylated sialic acids also play an important role in conferring nitric oxide resistance in virulent Leishmania sp. having enhanced distribution of the sialoglycotope. Furthermore they also influence the intracellular survival of the parasite within macrophages and modulate the host responses in their favor as evidenced by decreased level of IL-12 and IFN-γ, the signature TH1 cytokines [81]. In contrast, macrophages show increased levels of these cytokines, when they are infected with de-O-acetylated promastigotes. This observation suggests that the parasite is capable of modulating the host responses via 9-O-acetylated sialic acids for the successful infection. Apparently they act as effective ligands whose expression supports parasite internalization, intracellular differentiation.
De novo synthesis of sialic acids usually occurs as a result of the fine-tuning of four enzymes, namely sialidase, trans-sialidase, esterase, and O-acetyltransferase. Extensive work from the author’s group (CM) has convincingly demonstrated that these parasites lack an active machinery for the biosynthesis of this unique sialoglycotope as corroborated by the absence of activity of UDP-GlcNAc 2-epimerase which catalyzes the first step of sialic acid synthesis [77, 79]. The presence of N-acetyltransferase in L. amazonensis has indicated the presence of enzymes for acetylation [84]. However, any such claim for the presence of O-acetyltransferase requires the identification of the respective genes that, at present, is lacking. Direct transfer of sialoglycoproteins from the serum demands extensive study of proteomic characterization of surface proteins on promastigotes and is a subject of future research.
O-Acetylated Sialic Acids During Development and Differentiation
As there are multiple changes known to occur during the development of organs and tissues, glycosylation patterns are also subject to change [85]. As an example, aberrant expression of sialyltransferases can result in displacement of cells, as shown for sialyltransferase ST6GalNAc5, which is normally expressed only in brain. Expression of this sialyltransferase in breast cancer cells alleviates their migration into the brain by mediating cell passage through the blood–brain barrier [86].
In order to identify the functional impact of sialic acids, knockout mice with deleted sialyltransferase genes were created which exhibited a number of developmental changes. Knockout of sialyltransferase ST8Sia1, also known as GD3 synthase, resulted in a complete absence of b and c series gangliosides. These mice appeared to undergo normal development and had a normal life span. Double knockout mice with an additional disruption of the GalNAcT gene encoding β1,4-N-acetyl-galactosaminyltransferase, thereby expressing GM3 as the sole ganglioside, were extremely sensitive to sound stimuli even leading to sudden death [87]. Mice with a knockout of the ST6Gal1 gene exhibited tissue specific alterations in sialylation, concomitant with highly selective losses of 9-O-acetylation of sialic acid residues [88]. Knockout of polysialyltransferase ST8Sia4 allowed for the first time a discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity [89]. Addition of polysialic acid is an important modification of the neural cell adhesion molecule NCAM, directing migration and differentiation of neuronal cells within the central nervous system [90]. Recent investigations point to a role of polysialic acids in the development of social interactions and aggression in mice [91].
Due to the fact that the understanding of the molecular functions of sialic acids in development and differentiation are just emerging, it is not surprising that the functions of O-acetylation of sialic acids are even less well understood. However, some examples pointing to specific roles of O-AcSias are available.
By expression of the influenza C virus sialate-9-O-acetylesterase in transgenic mice, it was shown that O-acetylation is a prerequisite for normal development. Mouse embryos constitutively expressing the esterase were arrested as early as in the two cell stage [92]. In the rat nervous system the 9-O-acetylated ganglioside GD3 (9-O-AcGD3) was shown to exhibit discrete patterns during neuronal development [30]. In the fetal mouse cortex 9-O-acetylated ganglioside GT3 is strongly expressed and decreases to undetectable levels after birth [93]. In another study glycolipid-bound Neu5,9Ac2 was highest in embryonic mouse brain E13, and gradually decreased until birth. Significant amounts of Neu5,9Ac2 were found in adult mouse brain in the glycolipid fractions of the olfactory bulb, hippocampus, and telencephalon [40]. In pig brain, an increase of Neu5,9Ac2 was observed during the maturation of the cortex and cerebellum [94]. In patients with Guillain-Barré and Fisher´s syndromes, which are manifested as acute inflammatory demyelinating polyneuropathies, antibodies against O-acetylated gangliosides were found [95]. For 9-O-Ac-GD3, roles in neuronal motility were suggested [96, 97]. Antibodies to 9-O-Ac-GD3 induce microtubule depolymerization in growing neurits [98], and 9-O-Ac-GD3 was found in point contacts of neuronal growth cones [99]. Cerebellar granule neuron migration was blocked by the 9-O-Ac-GD3 mAB Jones in live animals [100]. This block was also observed in GD3 synthase knockout mice [101]. Results from the latter study indicated that the inhibitory effect of mAB Jones may be caused by binding to β1-integrin.
In chicken erythrocytes O-Ac-Sias represent a differentiation marker, which appears in 6-day-old birds and is fully developed in 20-day-old chickens [102]. During the early stages of human development, 9-O-AcGD3 is present in different tissues. In contrast, during the erythropoiesis, 9-O-AcGD3 level is decreased during maturation in the erythroid progenitor cells in bone marrow. Mature erythrocytes show lower 9-O-AcGD3 levels than immature cells.
Alterations of membrane characteristics and morphology occur in mature erythrocytes via 9-O-AcGD3 mediated signaling [103]. Such signaling via 9-O-AcGD3 also induces membrane alterations, vesicularization, phosphatidyl serine exposure, and activation of cysteine proteases like caspase 3, suggesting a programmed cell death like pathway in mature erythrocytes. In contrast, enhanced level of 9-O-AcGD3 is observed in lymphoblasts whereas GD3 expression is insignificant compared to normal lymphocytes. The anti-apoptotic role of 9-O-AcGD3 in lymphoblasts in contrast to mature erythrocytes suggests a cell specific role of 9-O-AcGD3 [104].
Role of O-Acetylated Sialic Acids in Immunological Processes
The major task of the immune system is to fight against invading microorganisms, to differentiate between self and non-self, i.e., to control autoimmune reactivity, and to eliminate defective or mutated cells such as tumor cells. The immune detection of tumor cells is often hampered by the fact that they are able, by many mechanisms, to disguise themselves as being normal. In a wider sense repair mechanisms such as wound healing and angiogenesis as part of the inflammatory process should also be included within the range of immune reactions. In general, immune reactions can be divided into the branches of innate immunity as first line of defense and adaptive immunity for highly specific reactions including immunological memory. However, there are many molecular structures which bridge these two systems.
During recent years it has become increasingly apparent that carbohydrate– lectin interactions play a vital role in many of these immune reactions regulated by the innate and the adaptive branch of the immune system [105]. This includes both the recognition of microbial structures by immune cells and the intricate crosstalk between immune cells. It was hypothesized that attachment to and invasion of microorganisms into host cells taking advantage of carbohydrate–lectin interactions had induced a selective pressure to modulate their carbohydrate structures and the respective lectin receptors in order to prevent infection or on the side of the microorganisms to counteract these alterations on the host’s side [106]. By means of this putative evolutionary process the complexity of carbohydrate structures expressed on the cell surface might have developed. Interestingly, a majority of these protein-carbohydrate interactions depends on the presence of terminal sialic acids as decisive recognition elements of the oligosaccharide ligand. Accordingly there are at least two lectin families known, the selectins and the Siglecs, which are specialized in the recognition of sialoglycans in various anomeric linkage patterns [107, 108]. Within the evolutionary development of carbohydrate complexity, O-acetylation of sialic acids seems to play a major role in infections and possibly also in defense mechanisms of immune cells. For example, coronaviruses have adapted specialized hemagglutinins to attach to cell surface expressed O-acetylated sialoglycans and to destroy further the linkage using sialic acid specific O-acetylesterases by removing the respective O-acetyl group [64, 67–69, 109].
When analyzing O-acetylation of lymphocytes it became evident that these immune cells preferentially synthesize 7-O-acetyl- and 9-O-acetyl sialoglycans [22, 32, 110–112].
O-Acetylated sialic acids resulted in increased susceptibility to alternate complement pathway-mediated lysis of murine erythrocytes. Progressive loss of Neu5,9Ac2-GPs with differentiation is concomitantly associated with an increasing resistance to alternate complement pathway activation. Increased presence of Neu5,9Ac2-GPs on erythrocytes of patients with visceral leishmaniasis may be responsible for ~ two- to threefold greater susceptibility to alternate complement-mediated hemolysis as compared to healthy individuals [80, 113].
O-Acetylation of Ganglioside GD3
Although theoretically O-acetyl sialoglycans can occur on glycoproteins and glycosphingolipids, the majority of studies focused on the disialo-ganglioside GD3 as the major carrier of terminal O-acetylated sialic acid. GD3 itself has been described to be involved in several immune reactions. These results were obtained to a large extent by application of specific monoclonal antibodies against GD3 and its 9-O- and 7-O-acetylated variants (designated as CD60a (GD3), CD60b (9-O-acetyl GD3), and CD60c (7-O-acetyl-GD3) [114]. By means of these CD60 antibodies the expression of these gangliosides can be observed in the sterical context of the cell surface of live cells. In addition these anti-ganglioside antibodies are useful tools to study the content of GD3 and its O-acetylated variants in distinct cellular compartments.
In earlier in vitro studies it was shown that gangliosides such as GD3 shed by tumors can inhibit the activity of natural killer (NK) cells [115, 116]. It was proposed that anti-tumor cytotoxicity of NK cells was blocked, thereby alleviating undisturbed tumor growth. While at that time the mechanism of this possible inhibition was not clear, consecutive work may explain the mechanism. Nicoll et al. described that GD3 expressed on target cells is able to modulate NK cell cytotoxicity by interaction with Siglec-7 expressed on NK cells. Siglec-7 has a preference for binding of α2,8 linked disialo glycans and seems to be one of the inhibitory NK cell receptors [117]. In the case of melanoma cells the inhibitory function of GD3 could not be verified since GD3 expressed on melanoma cells can induce another class of cytotoxic cells, the NKT cells [118]. It was further elucidated that the fine specificity of GD3 reactive NKT cells is mediated by binding to CD1d, a surface molecule structurally related to the major histocompatibility antigen (MHC) which is expressed on T cells. CD1d has a preference for carbohydrate ligands. It is not clear at the moment whether in the in vivo situation there may be a balance between anti-melanoma NK and NKT cell-mediated immunity which affects the outcome of an effective immune surveillance in melanoma patients. Another mechanism to explain immune escape of melanoma cells may be that GD3 seems to be able to impair dendritic cell differentiation from monocytes and may induce their apoptosis [119]. On the other hand it is known that melanoma cells not only express GD3 but also its O-acetylated variants in various degrees [16]. One may speculate that O-acetylation of GD3 can provide another protective mechanism of melanoma cells against cytotoxic attack of immune cells.
Role of O-Acetylated Sialic Acids on Glycoproteins
O-Acetylated sialoglycans may also be part of glycoproteins as shown for the mucin family of O-glycosylated glycoproteins. In the colon, sialyl Lewisx (CD15s) moieties of MUC1 and MUC2 were found to be differentially O-acetylated [120, 121]. It may be that O-acetylated CD15s is a negative regulator of the metastasis process because it may block the recognition of CD15s by E-selectin (CD62E) expressed on vascular endothelial cells and thereby inhibit the attachment of metastasizing cells to the vessel wall as a prerequisite to invade the host tissue. Krishna and Varki described the presence of 9-O-acetylated sialomucins on murine CD4 T lymphocytes [28].
Whether sialic acid O-acetylation has an effect on the binding capacity of Siglec proteins is still not unequivocally resolved. In an earlier report Sjoberg et al. stated that semisynthetic N-linked oligosaccharides with terminal O-acetylation have reduced binding to CD22 (Siglec2) and, further, treatment of murine lymphocytes with a chimeric influenza C esterase (CHE-Fc) clipping off O-acetyl residues increased binding of CD22 to various murine lymphocyte subsets [27]. CD22 is a B lymphocyte specific lectin which recognizes preferentially terminally α2,6 sialylated lactosaminyl oligosaccharides. It does not react with α2,3 sialylated structures. In the murine system CD22 has a preference for Neu5Gc whereas in humans CD22 solely recognizes Neu5Ac. Whether Neu5Gc is synthesized and expressed in the human system is most unlikely although some reports described its presence though in small quantities [122].
The conceptional problem with the above-mentioned results of Sjoberg et al. is that natural O-acetylated α2,6 sialylated lactosaminyl ligands have not yet been identified. Effects of the influenza esterase on CD22 binding as described may be a result of steric alterations in the composition of surface oligosaccharides.
To clarify the influence of 9-O-acetylated sialoglycans on the intracellular functions of CD22 as a negative regulator of B cell activation, Cariappa et al. used murine mutants with a defect in the cellular sialate O-acetylesterase and investigated the function of CD22 in B cell signaling [123]. Indeed, they found increased 9-O-acetylation in B cells of these mouse mutants and subsequently also enhanced B cell receptor signaling, pointing to a possible suspension of CD22 control. Although these results prove a regulatory role of the sialate O-esterase towards B cell activation, the direct effect of 9-O-acetylation of CD22 ligands is still to be shown. Interestingly, mutations in the sialate O-esterase gene can be linked to certain human autoimmune disorders [124].
The overall expression of O-acetylated sialoglycoconjugates at a given stage of lymphocyte differentiation depends on the intricate balance of enzymes involved both in synthesis and degradation of these oligosaccharides. Wipfler et al. recently measured the transcription of GD3 synthase ST8SIA1, the putative sialic acid-specific O-acetyltransferase CASD1, the human sialidases NEU1 and NEU3, and the sialic acid O-esterase SIAE in various human lymphocyte subsets representing various differentiation and activation stages in comparison to the expression of GD3 and its 7-O-acetylated and 9-O-acetylated variants (CD60a,b,c) [112]. It became apparent that the transcription of anabolic and catabolic enzymes was different in lymphocytes of various stages which had an impact on the intracellular and surface expression of CD60 structures.
Reduced O-acetylation may help tumor cells to escape from complement-mediated lysis because recognition of carbohydrates by elements of the alternative complement pathway may be inhibited by O-acetylation [26].
O-Acetylated Sialic Acids in Cancer
In malignant cells glycosylation is often altered in different ways. Examples are changes in glycosaminoglycans, altered branching on N-glycans, changes in mucin O-glycans, and in many instances elevated expression of sialic acids [125]. Changes in O-acetylation are also observed regularly in cancer cells.
9-O-Acetyl GD3 and Cancer
The identification of the disialo ganglioside 9-O-acetyl GD3, considered as an oncofetal marker, has been achieved using a lectin derived from the crab Cancer antennarius that recognizes sialic acids which are O-acetylated at both C4 and C9 positions and have been shown to be a biomarker in human melanoma [5]. Enhanced presence of O-acetylated GD3 has been reported in breast cancer, basaliomas, tumors of neuroectodermal origin [17, 126], childhood lymphoblastic leukemia (ALL) [104], and glioblastoma [127].
Ravindranath et al. [128] found that in one melanoma patient only metastatic lesions expressed the O-acetylated forms of GD2 and GD3 whereas the primary tumor expressed exclusively the non-O-acetylated gangliosides. The same was observed in basalioma. In basalioma the expression of this antigen was generally up to 60-fold higher than in surrounding normal skin [17]. However, in breast carcinomas the situation seems to be more complex. In normal ducts and in benign lesions 9-O-acetylated sialoglycans were present in Golgi regions and at the plasma membrane as detected by reaction with a CD60b antibody. This is in contrast to carcinomas of the breast where 9-O-acetylated sialoglycans were distributed in the cytoplasm in a disorderly fashion [129]. Cell surface expression of CD60b structures was only observed in well-differentiated carcinomas and overall expression decreased with progression of malignancy. Similar changes in distribution of sialoglycans and the sialyltransferase ST6Gal1 responsible for α2,6 sialylation have been found in hepatocellular carcinomas in which the disorder of sialoglycan distribution was correlated to the grade of malignancy [130]. It is still unclear whether loss of O-acetylation in malignant colon carcinomas is an advantage for tumor progression.
Surface-expressed 9-O- and 7-O-acetylated GD3 are abundantly expressed on human T lymphocytes [110]. Expression of both CD60b and CD60c (7-O-acetylated GD3) has also been detected on small resting lymphocytes of peripheral blood and on mature, activated T lymphocytes in lymph nodes [22, 112]. Tonsillar B lymphocytes, though to a smaller extent than T lymphocytes, express CD60b and c [22, 111]. Additionally the occurrence of CD60c was described on CD16+ NK cells, monocytes, and granulocytes to various extents [32] and on human CD34 hematopoietic progenitor cells derived from bone marrow (Schwartz-Albiez, unpublished). Cell surface-expressed CD60b and c on T and B lymphocytes may have a functional role in the lymphocytic activation process because anti-CD60b and c monoclonal antibodies can influence lymphocytic proliferation [22, 32, 131]. An interesting observation was that T and B lymphocytes react towards stimulation with CD60b and c antibodies in a different way. While in T lymphocytes CD60c antibodies alone, like a mitogen, can stimulate proliferation in B cells, additional signals such as addition of the cytokine Il-4 and triggering of the B cell receptor (surface expressed immunoglobulin) are required. For stimulation with antibodies against CD60b in T and B lymphocytes, additional signals are required [22]. This differential behavior may have a basis in a different surface distribution of the antigens. While CD60b structures are found in dot-like formations, possibly as components of rafts, both in T and B cells, CD60c on T cells showed a more homogenous distribution on the cell surface [22]. It is most likely that CD60b is a cno-stimulatory signal for raft-concentrated receptors while CD60c in T cells acts as a mitogen. It is rather unlikely that gangliosides themselves confer transmembrane signaling because they do have neither a transmembrane nor an intracellular domain. An explanation may be that distinct glycosphingolipids cross-linked by antibodies can contribute to raft formation by pulling together a certain array of receptors.
Anti-apoptotic Role of O-AcGD3
The disialoganglioside GD3 is a well-known inducer of the apoptotic-program and its proapoptotic-effects can be counteracted by O-acetylation. Exogenous addition of GD3 to lymphoblasts promotes the apoptotic program whereas 9-O-acetyl-GD3 has anti-apoptotic effects. Unlike GD3, 9-O-acetyl-GD3 fails to depolarize mitochondrial membranes followed by the release of cytochrome c and caspase 9 and 3. The removal of O-acetyl groups by sodium salicylate in lymphoblasts re-establishes the GD3-responsiveness to apoptotic signals. Thus, the balance of de novo synthesized GD3 and 9-O-acetyl-GD3 plays important roles in the survival of lymphoblasts in leukemia [104].
Recently, differential expression and possible function of 9-O- and 7-O-acetylated GD3 during apoptosis of human tonsillar B and T lymphocytes has also been reported [22]. Malisan et al. [132] also demonstrated that acetylation suppresses the proapoptotic activity of ganglioside GD3.
Interestingly, this ganglioside and its O-acetylated variants also have a function inside the cell, breaking the dogma that the final destiny for gangliosides is the cell surface. Intracellular GD3 is involved in CD95- and ceramide-mediated apoptosis [133]. Upon receptor-triggering, de novo synthesized GD3 accumulates intracellularly which can be demonstrated by increased activity of GD3 synthase. GD3 as a rule is restricted to be present in the Golgi network and at the plasma membrane it is transferred to mitochondria via endosomal transport [134] to raft-like mitochondrial membrane domains [135]. The mechanisms of GD3-induced apoptosis can be traced back to a GD3-mediated change in the mitochondrial membrane potential and consequently increased production of reactive oxygen species [136, 137]. Oxidation of GD3 to GD3-7-aldehyde was shown to increase the apoptotic effect by targeting adenine nucleotide translocase [138]. O-Acetylation of GD3 suppresses this pro-apoptotic function of non-acetylated GD3 and does not have the deleterious effects of GD3 on mitochondria membranes [132]. Cells which are resistant to over-expression of GD3 convert existing GD3 more readily to 9-O-acetylated GD3 [132]. In further confirmation of the anti-apoptotic effects of O-acetylated GD3, Kniep et al. found that exogenous O-acetylated GD3 given to cells in vitro is internalized and can prevent apoptosis [139]. It was also observed that a T leukemia cell line resistant to apoptosis induced by N-acetyl-sphingosine or daunorubicin transferred GD3 more readily into 9-O-acetyl GD3 [139]. Targetting 9-O-acetylated GD3 with sialate-9-O-acetylesterase results in apoptosis of biopsy-derived human glioblastoma cells. Compared to treatment of cells with exogenous O-acetylesterase, the effect of de-O-acetylation is more pronounced when the esterase is expressed within the cells from a recombinant baculovirus vector [127]. Thus, these data strongly point to an anti-apoptototic function of intracellular 9-O-acetyl GD3 that may protect tumor cells from apoptosis. While data have been gathered on the anti-apoptotic effect of 9-O-acetyl GD3, no data are available on possible effects of 7-O-acetyl GD3. We have observed that in T and B cells 7-O-acetyl GD3 followed in its expression intensity by 9-O-acetyl GD3 is present in an intracellular pool [112]. Given that intracellular 9-O-acetyl GD3 confers anti-apoptotic capacity, can this protective effect possibly be accelerated by conversion of 7-O-acetyl- into 9-O-acetyl GD3?
We also have no knowledge of what regulates the apparently differential transport of both acetylated forms of GD3 to the cell surface and what mechanisms with regard to the balance between the intracellular pool and cell surface expression of CD60b and c are decisive for regulation of either anti-apoptotic or proliferative effects. It was shown that human tonsillar B lymphocytes undergoing in vitro either spontaneous or staurosporine-induced apoptosis are characterized by surface expression of CD60b but not CD60c [22]. It may be that O-acetylated gangliosides fulfil different tasks at the cell surface and in intracellular compartments.
Neu5,9Ac2-GPs and Cancer
In the gastrointestinal tract, the concentration of O-acetylated sialic acids of colonic mucin decreases in colorectal carcinomas, colonic adenomas, ulcerative colitis, and Hirschsprung’s disease, suggesting its reversal to the embryonic form. However, human skin contains very little O-acetylated sialic acids. This decrease in O-acetylated sialic acids is associated with a concomitant increase in expression of the sialylated antigens, sialyl Tn, sialyl Lewis (a), and sialyl Lewis (x), which are considered to be adverse prognostic indicators.
An increased amount of O-acetylated sialic acids (Neu5,9Ac2-GPsALL) on erythrocytes [140] and peripheral blood mononuclear cells (PBMC) of patients suffering from childhood acute lymphoblastic leukemia (ALL) [141–145] has been demonstrated using the preferential specificity of a lectin, Achatinin-H, towards Neu5,9Ac2-α2,6-GalNAc [6, 7]. The absence of Neu5,9Ac2-GPs and corresponding anti-Neu5,9Ac2-GPs antibodies in corresponding cells of healthy children or in patients with other cross-reactive hematological disorders such as acute myelogenous leukemia, chronic myeloid leukemia, chronic lymphocytic leukemia, non-Hodgkin’s lymphoma, thalassemia, and aplastic anemia confirmed the specificity of these biomarkers [141–145]. The binding of Neu5,9Ac2-GPsALL with Achatinin-H in the presence of several synthetic sialic acid analogs further confirmed the presence of this sialoglycotope on lymphoblasts in leukemia [144].
Function of Neu5,9Ac2 for Detection of Minimal Residual Disease
In spite of successful treatment, patients may retain small numbers of malignant cells which is referred to as minimal residual disease (MRD) responsible for relapse. MRD is the main cause of a relapse of the disease. Neu5,9Ac2-GPs are strongly expressed in childhood ALL at the onset of disease, then they decrease with chemotherapy and reappear with relapse. This observation makes Neu5,9Ac2-GPs a potential biomarker for diagnosis and monitoring the disease status in childhood ALL (Fig. 1) [146, 151–155].
Fig. 1.
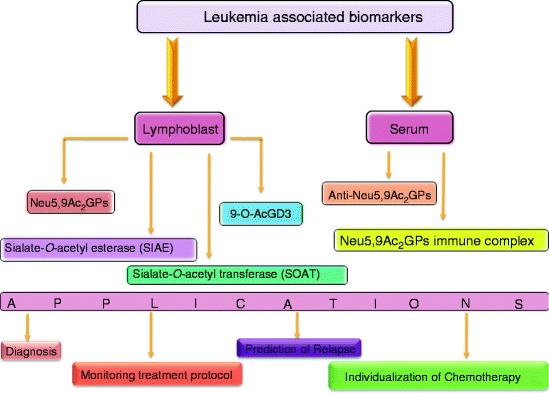
Schematic overview of enhanced sialylation, Neu5,9Ac2GPs [141–145], 9-OAcGD3 [104] on lymphoblasts, SOAT in microsomes [146], and anti-Neu5,9Ac2GPs antibodies [147–150] in serum as signature molecules useful for diagnosis and monitoring childhood ALL
A 6-year longitudinal follow-up study reveals that the expression of three newly induced leukemia-associated Neu5,9Ac2-GPsALL (90, 120, and 135 kDa) disappears after treatment in patients who have disease free survival [143, 144]. The 90-kDa band persists in a few patients who subsequently relapse with the re-expression of the 120-kDa band. Early clearance of Neu5,9Ac2-GPs+ALL cells, during 4–8 weeks of treatment, shows a good correlation with low risk of relapse [143]. Therefore, close monitoring of 90- and 120-kDa 9-O-AcSGs may serve as a reliable index for long-term management of these children and merits therapeutic consideration.
Subsequently, a suitable template has been established by using the differential expression of Neu5,9Ac2-GPsALL along with other known CD antigens to monitor MRD [151]. A 2-year longitudinal follow-up study of 89 patients [B- (n = 75) or T- (n = 14) ALL], from the onset of the disease until the end of chemotherapy, reveals the sensitivity of MRD detection reaching 0.01% for a patient in clinical remission using flow cytometry. Presence of enhanced MRD due to failure in early clearance of lymphoblasts is implicated in an elevated risk of relapse. Elevated MRD during the chemotherapeutic regime predicts clinical relapse, at least 2 weeks before clinical manifestation. Therefore, these templates can function for MRD detection, during and post-chemotherapy for proper patient management strategies, thereby helping in designing tailor-made chemotherapy.
Function of Neu5,9Ac2 for the Survival of Lymphoblasts
The role of O-acetylated Sias for the survival of lymphoblasts has been reported (Fig. 2) [23, 104, 156–158, 160].
Fig. 2.
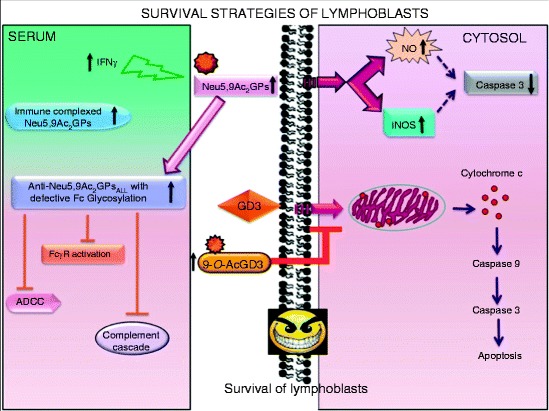
Immune escape of lymphoblasts possibly due to enhanced sialylation, O-acetylation of glycoproteins or disialo gangliosides GD3 [156–158], SOAT [146], and reduced membrane bound sialidase (Neu 3) [159] in ALL. Subclass switching of anti-Neu5,9Ac2GPs antibodies from IgG1 to IgG2, modulation of Fc-glycosylation, and weakening a few Fc-glycosylation-sensitive effector functions seem to be responsible for evading the host’s immune response [160]
Anti-Neu5,9Ac2-GPsALL Antibodies
Enhanced levels of antibodies against Neu5,9Ac2-GPs in leukemia patients as compared to normal individuals have also been used for monitoring disease status [147–150, 152, 160].
An enhanced amount of Neu5,9Ac2-GPs specific IgG2 in leukemia was unable to trigger the complement cascade, activation of FcγR and cell-mediated cytotoxicity, although its sialoglycotope binding ability remains unaffected [160]. Interestingly, only Neu5,9Ac2-GPs specific IgG1N purified from normal human serum emerged as the potent mediator of cell-mediated cytotoxicity, complement fixation, and activator of effector cells through FcγR. Therefore, the generation of customized O-acetylated sialic acid specific chimeric anti-9-Neu5,9Ac2-GPs-IgG1 antibody constructs bearing functional normal Fc domain, having a homogenous glycoform and a pre-determined profile of functional potential would be ideal for therapeutic applications. Such customized antibodies might lead to their proper functioning and therefore possibly being useful along with cytokine therapy to activate in vivo anti-cancer pathways for proper immune-surveillance in leukemia.
Subclass switching of anti-Neu5,9Ac2-GPs specific to IgG2, alteration in their Fc-glycosylation profile, along with impairment of a few Fc-glycosylation-sensitive effector functions hint towards an unbalanced homeostasis helpful for evading the host’s immune defense, suggesting a possible mechanism for functional unresponsiveness of tumor antibodies in general [160].
An interesting phenomenon is that patients suffering from certain tumors, for instance medullablastomas, a neural tumor disease, carry antibodies of the IgM subtype against O-acetylated GD3 in their serum [161].
Biosynthesis and Degradation of O-Acetylated Sialic Acids
Appearance of O-acetylated sialoglycoproteins or glycosphingolipids is cell type specific and developmentally regulated. Their synthesis and turnover is a finely tuned phenomenon. Following the translocation of cytidine monophosphate (CMP)-sialic acid residues into the Golgi apparatus, sialyltransferases catalyze the transfer of sialic acid onto an acceptor like galactose or N-acetylgalactosamine or less commonly N-acetylglucosamine or 5-N-acetyl neuraminic acid of an appropriate oligosaccharide chain as part of a nascent glycoconjugate in α2,3, α2,6, α2,8, or α2,9 linkages. Subsequently, sialate O-acetyltransferases (SOAT) transfer the acetyl group from acetyl-CoA onto sialoglycoconjugates at the C-7/8/9 positions, generating O-acetylated sialoglycoconjugates. The primary insertion site for the O-acetyl group may well be the C7–OH group, from where it can non-enzymatically migrate to the C-9 position, presumably via the C8–OH group, leaving the C7–OH group available for a new transfer [1, 162].
These O-acetyl esters are removed by a family of other important enzymes in sialic acid metabolism, the sialate-O-acetylesterases (SIAE). Both SOAT and SIAE are the two main enzymes responsible for the quantity of the O-acetyl ester groups on sialic acids. Therefore the activities of sialyltransferases and SOAT at one end of the spectrum, and the SIAE and a group of another key catabolic enzyme (sialidases), responsible for cleaving sialic acid residues from glycoproteins and glycolipids, at the other end of the spectrum, regulate the expression of O-acetylated sialoglycoconjugates.
Sialate-O-Acetyltransferase
Cancer cells frequently alter the regulation of sialylation processes leading to the appearance of characteristic sialoglycoproteins and sialoglycosphingolipids. A reduced SOAT enzyme activity in human colon and colorectal carcinoma is corroborated with decreased O-acetylation in the course of tumor development [120, 163, 164]. In contrast, enhanced SOAT in microsomes of lymphoblasts from bone marrow of children with leukemia, irrespective of their lineage, is corroborated with increased Neu5,9Ac2-GPs [146]. The O-acetylation of exogenously added GD3 by ALL-microsomes extends the specificity of this SOAT towards gangliosides. Enhanced activity of SOAT with higher V max in leukemia is one of the few descriptions of an enzyme of this type in human. However, a higher acetylation rate may also be partly due to differences in transporters and natural acceptors.
Besides endogenous acceptors, exogenous substrates like different sialoglycoproteins, CMP-Neu5Ac and GD3, are substrates for the enzyme. However, it is difficult to discover the selectivity of the SOAT in vivo. The reaction products are mainly Neu5,7Ac2 and Neu5,8Ac2, suggesting the primary insertion site of the O-acetyl group to be at C-7, followed by C-8 of Neu5Ac. The acetyl group possibly migrates from the seven position to the primary alcohol group of sialic acid at C-9 presumably via C-8. This is corroborated by enhanced Neu5,9Ac2 exclusively in isolated microsomes of these lymphoblasts. Accordingly, the leukemia SOAT was denoted as sialate-7(9)-O-acetyltransferase [146].
The possibility that a number of distinct SOATs are controlling O-acetylation of sialic acids attached to glycans via different linkages cannot be ruled out, suggesting another level at which O-acetylation is possibly controlled in cancer and normal tissue. This has been supported by the observations wherein SOAT activity with high specificity for terminal α2,8-linked sialic acid residues and no detectable activity for α2,3-linked sialic acids is reported [165].
Interestingly, expression of 9-O-AcGD3 is higher in leukemic cells, which gives the plausible answer that increased amounts of GD3 might be converted to 9-O-AcGD3 by means of enhanced SOAT and reduced membrane-bound sialidase (Neu3) [159], thereby reducing the GD3 content. Complex regulations of the overall metabolism of sphingolipids through different activation of other enzymes are involved in association with sialyltransferases in leukemia. Augmented lactosylceramide might contribute to the increased resistance of malignant lymphocytes towards apoptosis.
The SOAT activities increase rapidly with the onset of disease, decrease with clinical remission, and increase sharply again with clinical relapse and correlate well with high levels of cell surface Neu5,9Ac2-GPs and 9-O-AcGD3 on lymphoblasts. Thus understanding the mechanisms of O-acetylation of sialic acids will enhance our knowledge of the functions of sialic acids in animals and humans in general and not only in cancer. Analysis of SOAT may provide insight into the pathogenesis of disease and its progression, and may even provide clues for designing new drugs. Clearly, further studies are needed to unravel the sialic acid linkage specificity of SOAT in cancer.
As already indicated, O-acetylation depends on multiple factors, including the origin of tissues or the type of cell lines used for SOAT assays. Attempts to isolate the enzyme by biochemical procedures [2, 166–169] led to the identification of at least two different SOAT activities: partially purified SOAT from bovine submandibular glands transfers acetyl groups to C7 of Sias. It was proposed that migration of acetyl groups from C7 to C9 might be enzymatically catalyzed [2]. In a later publication the existence of a “migrase” could not be substantiated [167]. A second type of SOAT was found in Golgi-enriched fractions of guinea pig liver, which transfers acetyl groups to C4 [170]. SOAT activity isolated from guinea pig liver preferred gangliosides as substrate. In addition, a heat-stable low molecular weight cofactor, which could be separated from SOAT activity by ultrafiltration, was proposed to enhance 4-O-SOAT activity [166]. In vitro several substrates, including free sialic acid, CMP-Sia, gangliosides, and glycoprotein-bound Sias could be acetylated by SOAT derived from different sources. In vivo free Sias most likely do not represent natural substrates for SOAT, because Sia is transferred into the Golgi as CMP-Sia by a nucleotide-sugar transporter [171, 172].
Higa and Paulson isolated CMP-Sia synthase and used this enzyme to prepare 9-O-Ac-CMP-Sia [173]. Interestingly, 4-O-Ac-CMP-Sia could not be synthesized with this enzyme. Sialyltransferases were able to transfer Neu5,9Ac2 from the donor 9-O-Ac-CMP-Sia to glycoproteins, but at lower rates than Neu5Ac or Neu5Gc. It was concluded that a direct transfer of O-Ac-Sias to glycoproteins like bovine mucin from 9-O-Ac-CMP-Sia could account only for a fraction of the total O-Ac-Sias found. Moreover, it was proposed that 4-O-acetylation would result from the action of an O-acetyltransferase on the glycosidically-bound Sia [173]. Later it was found that both the CMP-Sia and acetyl-CoA transporters are critical components for the O-acetylation of CMP-Sia in the Golgi lumen. In addition, it was also suggested that a sialyltransferase exists that preferentially utilizes CMP-Neu5,9Ac2 as the donor substrate to sialylate Galβ1,3(4)R- residues [164]. This finding was in contrast to earlier observations that acetyl-CoA does not enter isolated rat liver Golgi vesicles which are able to perform the acetylation of α2,6 linked sialic acids on N-glycans [174]. Additional data had led to the proposal that O-acetylation is the product of a trans-membrane reaction, involving a membrane protein with essential histidine and lysine residues [175]. In summary, it has not yet been possible to obtain a purified eukaryotic SOAT preparation suitable to determine its amino acid sequence and the gene(s) encoding SOAT.
Other laboratories have tried to identify SOAT by expression cloning. Expression of different cDNAs was found to stimulate O-acetylation of sialic acids. With such experiments, Ogura and coworkers reported the cloning of an O-acetyl ganglioside synthase with a significant homology to milk fat globule membrane protein [176]. Kanamori et al. isolated a trans-membrane protein that most likely represents an acetyl-CoA transporter. Interestingly, they found that expression of this transporter induced the formation of O-Ac-GD3 [177]. During the search for the SOAT gene Shi et al. also isolated cDNAs which most likely are not directly involved in transfer of O-acetyl groups: a cDNA clone encoding a chimeric protein composed of a bacterial tetracycline resistance gene repressor and a plasmid sequence was found to enhance O-acetylation. Also, a clone encoding a truncated form of vitamin D binding protein was isolated. In both cases, expression of the recombinant proteins was required to observe increased O-acetylation [178]. This finding may indicate that the expressed mRNAs or proteins are recognized by pattern recognition receptors that then induce an “alarm” pathway, finally resulting in O-acetylation of sialic acids. Binding to the bacterial tetracycline resistance gene repressor may have triggered the initiation of cells to become pre-apoptotic by inducing the expression of 9-O-Ac-GD3. To speculate further, induction of apoptosis would then just require activation of the cellular SIAE to generate the pro-apoptotic ganglioside GD3. Another molecule possibly involved in O-acetylation of GD3 was identified as Tis21, a cell cycle regulator and cell death molecule [179]. It is possible that Tis21 may be a mediator of O-acetylation, but it appears unlikely that Tis21 represents the elusive SOAT. In GM2/GD2 knockout mice high amounts of 9-O-Ac-GD3 were found to accumulate in nerve tissue [180]. In this publication expression levels of the previously reported inducers of O-acetylation were also examined. No up-regulation was found for vitamin D binding protein, acetyl-CoA transporter, or the putative O-acetyl ganglioside synthase, while Tis21 was partially down regulated.
Recently, a new player in the field was identified. A screening of the human genome database revealed the CASD1 gene as a candidate to encode a key enzyme involved in sialic acid O-acetylation [181]. When the Cas1 protein was expressed in COS cells together with ST8Sia1, a substantial increase of synthesis of 7-O-Ac-GD3 was observed. Human Cas1p was shown to co-localize with the Golgi marker ST6Gal1. At the mRNA level, elevated CASD1 expression was concomitant with increased levels of O-Ac-GD3 in primary human cells and in cell lines derived from human melanoma and liver cancers. Transfection of CASD1 specific siRNA resulted in a reduction of O-Ac-GD3 expression. On the other hand, expression of Cas1p in COS cells is not sufficient to direct O-acetylation of sialic acids on N- or O-linked glycans on the model glycoprotein erythropoietin (manuscript in preparation).
Cas1p is encoded by the CASD1 gene located on chromosome 7q21.3. The gene is homologous to the CAS1 gene of Cryptococcus neoformans. The designation is derived from the function of its encoded protein in the formation of the fungal Capsule Structure. Genetic evidence strongly indicates that the fungal Cas1 protein is involved in the O-acetylation of glucorono-xylomannans [182]. Deletion of the CAS1 gene resulted in a loss of O-acetylation on C-6 of either Man or ManGlcA residues. O-Acetylation could be restored by expression of CAS1 from a plasmid.
The human and fungal Cas1 proteins are composed of an N-terminal serine–glycine–asparagine–histidine (SGNH) domain and a C-terminal trans-membrane domain with 8–12 trans-membrane regions (Fig. 1). Homologs of CAS1 are also present in plants. They were shown to be directly involved in O-acetylation of plant cell walls. This modification is present in high amounts in different plant polysaccharides. O-Acetylation is a hurdle in the processing of plant material into biofuels, because glycosidases used for degradation of plant polysaccharides are negatively affected by O-acetylation [183]. On top of that, acetate and its conversion products are inhibitory to microorganisms used for fermentation [184].
During the screening of Arabidopsis mutants, plants were identified with insertional mutations in the plant CAS1 genes, which were termed RWA (reduced wall acetylation) [185]. In Arabidopsis four RWA genes are present, and their expression in different plant tissues partially overlaps. The rwa2 mutant exhibited a 15–30% reduction in cell wall acetylation. Different polysaccharides were affected at similar rates. Therefore, it was speculated that rwa proteins act immediately upstream of the transfer of acetyl groups to different acceptors. Interestingly, the plant Cas1 proteins lack the N-terminal SGNH domain. Instead, a large number of proteins with similarity to the N-terminal domain of Cas1p was identified in a bioinformatics approach [186]. In plants, the DUF231 (domain of unknown function) family of proteins may be involved in the transfer of O-acetyl groups to specific acceptors, possibly by a trans-esterase mechanism [185]. In mammals, other proteins, including members of the FAM55 and FAM113 families, and C7orf58, exhibit similarities to the SGNH domain of Cas1p [186]. Thus, it may be speculated that these proteins are candidates to direct O-acetyl groups to sialic acids in different glycosidic linkages and/or to different carbons of sialic acids (Fig. 3).
Fig. 3.
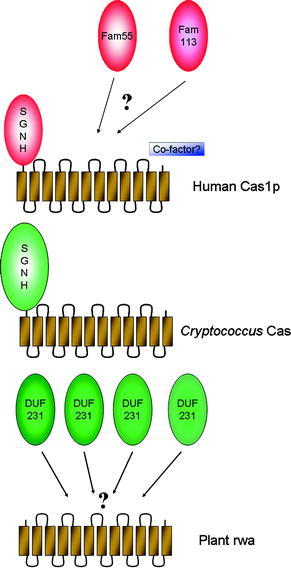
Proposed schematic structures of Cas1p and rwa proteins and possible recruitment of FAM55, FAM113, or DUF231 proteins for catalytic activity of O-acetyltransferases. The human and cryptococcal Cas1 proteins consist of N-terminal serine–glycine–asparagine–histidine (SGNH) and C-terminal transmembrane domains. Plants express similar proteins termed reduced wall acetylation (rwa) lacking the N-terminal SGNH domain. Rwa may recruit DUF231 proteins to deliver acetyl groups from acetyl-CoA to different polysaccharides of the plant cell wall. In mammalian systems members of the FAM55 or FAM113 proteins may interact with Cas1p and a cofactor to transfer acetyl groups to sialic acids in different linkages
In addition, other yet unidentified components of the cellular SOAT activities may be required for the fine specificity of O-acetylation. Moreover, it is also unclear how SOAT and SIAE work in concert to regulate acetylation. This regulation may be at transcriptional level, but interactions at the protein level may also play a role.
Sialate-O-Acetylesterase
SIAE hydrolyses either C4- or C9-O-acetyl groups of glycosidically-linked or free sialic acid released from glycoconjugates by sialidases [162, 187–190]. At least two forms of cellular 9-O-acetyl-SIAE, one in the cytoplasm and the other in the lysosomal compartment (i.e., membrane-bound), exist in mammals. The secreted form, originally termed luminal sialic acid esterase [191, 192], was later termed lysosomal sialic acid O-acetylesterase (Lse) [193], because it was shown by immuno-electron microscopy to co-localize with acid hydrolases and lysosomal membrane glycoproteins [191]. This SIAE contains a cleavable N-terminal signal sequence and is secreted from COS cells as a glycoprotein with an apparent molecular mass of 62 kDa [193], whereas in rat liver it is found predominantly as a heterodimer composed of a small (28-kDa) and large (38-kDa) subunit connected by disulfide bridges. The small subunit could be labeled with the serine hydrolase inhibitor diisopropyl fluorophosphate, indicating that SIAE is a serine esterase. No sequence similarities to other known serine esterases were found [192]. Amino acid residues, important for catalytic activity and secretion, were determined by expression of mutated cDNA. The mutations tested were found in the SIAE gene of patients with autoimmune disease [124]. Both subunits are encoded by the same mRNA in the order signal sequence – small subunit – large subunit. The precursor is presumably cleaved in lysosomal compartments [192, 194]. SIAE was identified independently by another research team as cDNA derived from a gene that is upregulated during B cell maturation. Several cDNAs were isolated which apparently are derived from differentially spliced SIAE mRNA [195]. One of the spliced mRNAs is lacking the exon for the signal sequence and encodes the cytosolic SIAE. While the mRNA for the secreted SIAE is widely expressed in different adult tissues, the expression of the cytosolic SIAE is restricted. Elevated expression of the latter was found mainly in liver, ovary, and brain [196]. SIAE regulates B cell antigen receptor signal strength and peripheral B cell development in mice [123], presumably by regulating recognition of α2,6-linked sialic acids by CD22 [27], a negative regulator of the B cell receptor [197–200] and Toll-like receptors [201]. Mutations in the SIAE gene resulting in a loss of function are strongly linked to autoimmune disease [124], while overexpression of SIAE is linked to pre-eclampsia [202], a condition of pregnant women who exhibit high blood pressure and proteinurea.
Concerning the function of SIAE, many open questions remain. While it can be envisaged that cytosolic SIAE is involved in de-O-acetylation of O-Ac-Sias delivered from lysosomes, the function of the “lysosomal” SIAE remains controversial due to the unfavorable pH in lysosomes. The pH optimum of SIAE is in the neutral to alkaline range. Data indicate that intracellular vesicles exist which contain mannose-6-phosphate positive glycoproteins [191]. Such entities possibly represent transport vesicles on the way from the Golgi to lysosomes. In any case, precise models of how SIAE is involved in tuning the balance of O-acetylation of sialic acids are currently not available. Consequently, a large field is open for future investigations. Another question concerns the enzymatic activity of secreted SIAE, which was shown to be increased upon proteolytic cleavage [193]. The protease required for cleavage activation was proposed to be a lysosomal enzyme, but the nature of this protease remains to be determined. Moreover, it is apparently not a common protease. COS cells were shown to express and secrete the uncleaved SIAE [193].
Another puzzle remains concerning the distribution of the secreted SIAE. Available data indicate that it is intracellularly concentrated in lysosomal compartments [191]. On the other hand it is efficiently secreted into the culture supernatant of cells over-expressing SIAE, and therefore it has access to O-Ac-Sias at the cell surface [193, 195]. Furthermore, the substrate specificity was shown for O-acetyl groups on free sialic acids [193]. In Siae knockout mice, an increase in α2,6-linked O-Ac-Sias was observed by using the influenza C virus lectin [123]. Strictly speaking, the influenza C virus lectin binds to any accessible O-Ac-Sia regardless of the underlying linkage. Thus, it remains an open question whether α2,3- or α2,8- linked sialic acids are also de-O-acetylated by SIAE. Furthermore, no SIAE hydrolyzing Neu4,5Ac2 has been purified to homogeneity allowing the determination of its relationship to SIAE specific for Neu5,9Ac2. In summary, the expression of 9-O-acetylated sialoglycoproteins seems to be controlled by the relative activities of SOAT and SIAE. Many details on O-acetylation and de-O-acetylation still require clarification.
Future Perspectives
The existence of diverse O-acetylated Sias together with the enormous variations in sialoglycotopes having O-acetylated Sias in different linkages, with different subterminal sugars and their regulative functions in proliferation, and their controlled expression may be explored in view of possible applications in cancer therapy. Despite huge efforts to elucidate the factors mediating the escape of cancer cells from immunological surveillance, our knowledge in this regard is still rather limited. Exploring the function of O-acetylated sialic acids on the immune cells may contribute to our understanding of this problem.
In this direction, much emphasis of current research is invested in development of target-oriented anti-cancer drugs. Future investigation using both enzymological and molecular biology approaches of some key enzymes like SOAT, SIAE, sialyltranferases, and sialidases as drug targets may also lead into the direction of therapeutic interventions. For instance, deeper understanding of their functioning could be explored in a more practical direction for pharmacological manipulation of the apoptotic pathways.
Acknowledgments
The Council of Scientific and Industrial Research (CSIR) under IAP-0001, Systems Biology (HCP004), New Millennium Indian Technology leadership Initiative (NMITLI, TLP-004) projects, CSIR-I.I.C.B. and Department of Biotechnology under cancer Biology (GAP 235), Govt. of India supported this work. CM also acknowledges support from JC Bose Fellowship, Department of Science and Technology, Govt. of India. The work of RSA was supported by a grant of the Deutsche José Carreras Leukämie Stiftung (DJCLS, project No DJCLS R08/13). CM and RSA are also grateful to financial support by a mutual grant from the Indian Medical Research Council and the German Cancer Research Center. RV was supported by grants from the Austrian Science Fund (Projects P20080 and L608-B03) and by Salzburg Research Fellowships P147200-06 and P144001-01.
Abbreviations
- 9-O-AcGD3
9-O-Acetylated ganglioside GD3
- ALL
Acute lymphoblastic leukemia
- GC/MS
Gas chromatography/mass spectrometry
- GP
Glycoprotein
- HPLC
High performance liquid chromatography
- MAb
Monoclonal antibody
- Neu4,5Ac2
5-N-Acetyl-4-O-acetyl neuraminic acid
- Neu5,7(8),9Ac3
5-N-Acetyl-7(8),9-O-acetyl neuraminic acid
- Neu5,9Ac2
5-N-Acetyl-9-O-acetyl neuraminic acid
- Neu5,9Ac2-GPs
Glycoproteins with 5-N-acetyl-9-O-acetyl neuraminic acid
- Neu5Ac
5-N-Acetyl neuraminic acid
- Neu5Gc
5-N-Glycolyl neuraminic acid
- O-Ac
O-Acetyl
- O-Ac-Sia
O-Acetylated sialic acid
- SIAE
Sialate-O-acetylesterase, sialic acid O-acetylesterase
- SOAT
Sialate-O-acetyltransferase, sialic acid O-acetyltransferase
References
- 1.Kamerling JP, Schauer R, Shukla AK, Stoll S, Van Halbeek H, Vliegenthart JF. Eur J Biochem. 1987;162:601. doi: 10.1111/j.1432-1033.1987.tb10681.x. [DOI] [PubMed] [Google Scholar]
- 2.Vandamme-Feldhaus V, Schauer R. J Biochem (Tokyo) 1998;124:111. doi: 10.1093/oxfordjournals.jbchem.a022069. [DOI] [PubMed] [Google Scholar]
- 3.Haverkamp J, Schauer R, Wember M, Kamerling JP, Vliegenthart JF. Hoppe Seylers Z Physiol Chem. 1975;356:1575. doi: 10.1515/bchm2.1975.356.2.1575. [DOI] [PubMed] [Google Scholar]
- 4.Shukla AK, Schauer R. Hoppe Seylers Z Physiol Chem. 1982;363:255. doi: 10.1515/bchm2.1982.363.1.255. [DOI] [PubMed] [Google Scholar]
- 5.Ravindranaths MH, Paulson JC, Irie RF. J Biol Chem. 1988;263:2079. [PubMed] [Google Scholar]
- 6.Mandal C, Basu S. Biochem Biophys Res Commun. 1987;148:795. doi: 10.1016/0006-291x(87)90946-6. [DOI] [PubMed] [Google Scholar]
- 7.Sen G, Mandal C. Carbohydr Res. 1995;268:115. doi: 10.1016/0008-6215(94)00311-3. [DOI] [PubMed] [Google Scholar]
- 8.Herrler G, Rott R, Klenk HD, Muller HP, Shukla AK, Schauer R. EMBO J. 1985;4:1503. doi: 10.1002/j.1460-2075.1985.tb03809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogers GN, Herrler G, Paulson JC, Klenk HD. J Biol Chem. 1986;261:5947. [PubMed] [Google Scholar]
- 10.Vlasak R, Krystal M, Nacht M, Palese P. Virology. 1987;160:419. doi: 10.1016/0042-6822(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 11.Muchmore EA, Varki A. Science. 1987;236:1293. doi: 10.1126/science.3589663. [DOI] [PubMed] [Google Scholar]
- 12.Zimmer G, Reuter G, Schauer R. Eur J Biochem. 1992;204:209. doi: 10.1111/j.1432-1033.1992.tb16626.x. [DOI] [PubMed] [Google Scholar]
- 13.Zimmer G, Suguri T, Reuter G, Yu RK, Schauer R, Herrler G. Glycobiology. 1994;4:343. doi: 10.1093/glycob/4.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harms G, Reuter G, Corfield AP, Schauer R. Glycoconj J. 1996;13:621. doi: 10.1007/BF00731450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.do Valle Matta MA, Sales Alviano D, dos Santos Silva Couceiro JN, Nazareth M, Meirelles L, Sales Alviano C, Angluster J (1999) Parasitol Res 85:293 [DOI] [PubMed]
- 16.Hubl U, Ishida H, Kiso M, Hasegawa A, Schauer R. J Biochem (Tokyo) 2000;127:1021. doi: 10.1093/oxfordjournals.jbchem.a022693. [DOI] [PubMed] [Google Scholar]
- 17.Fahr C, Schauer R. J Invest Dermatol. 2001;116:254. doi: 10.1046/j.1523-1747.2001.01237.x. [DOI] [PubMed] [Google Scholar]
- 18.Alviano DS, Rodrigues ML, Almeida CA, Santos AL, Couceiro JN, Soares RM, Travassos LR, Alviano CS. Arch Microbiol. 2004;181:278. doi: 10.1007/s00203-004-0653-9. [DOI] [PubMed] [Google Scholar]
- 19.Klein A, Krishna M, Varki NM, Varki A. Proc Natl Acad Sci USA. 1994;91:7782. doi: 10.1073/pnas.91.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strasser P, Unger U, Strobl B, Vilas U, Vlasak R. Glycoconj J. 2004;20:551. doi: 10.1023/B:GLYC.0000043292.64358.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Argueso P, Sumiyoshi M. Glycobiology. 2006;16:1219. doi: 10.1093/glycob/cwl041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erdmann M, Wipfler D, Merling A, Cao Y, Claus C, Kniep B, Sadick H, Bergler W, Vlasak R, Schwartz-Albiez R. Glycoconj J. 2006;23:627. doi: 10.1007/s10719-006-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, Bandyopadhyay S, Mallick A, Pal S, Vlasak R, Bhattacharya DK, Mandal C. J Cell Biochem. 2005;95:206. doi: 10.1002/jcb.20382. [DOI] [PubMed] [Google Scholar]
- 24.Hellebo A, Vilas U, Falk K, Vlasak R. J Virol. 2004;78:3055. doi: 10.1128/JVI.78.6.3055-3062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi WX, Chammas R, Varki A. J Biol Chem. 1996;271:31517. doi: 10.1074/jbc.271.49.31517. [DOI] [PubMed] [Google Scholar]
- 26.Shi WX, Chammas R, Varki NM, Powell L, Varki A. J Biol Chem. 1996;271:31526. doi: 10.1074/jbc.271.49.31526. [DOI] [PubMed] [Google Scholar]
- 27.Sjoberg ER, Powell LD, Klein A, Varki A. J Cell Biol. 1994;126:549. doi: 10.1083/jcb.126.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishna M, Varki A. J Exp Med. 1997;185:1997. doi: 10.1084/jem.185.11.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumermuth E, Beuret N, Spiess M, Crottet P. J Biol Chem. 2002;277:18687. doi: 10.1074/jbc.M109408200. [DOI] [PubMed] [Google Scholar]
- 30.Blum AS, Barnstable CJ. Proc Natl Acad Sci USA. 1987;84:8716. doi: 10.1073/pnas.84.23.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kniep B, Peter-Katalinic J, Flegel W, Northoff H, Rieber EP. Biochem Biophys Res Commun. 1992;187:1343. doi: 10.1016/0006-291x(92)90450-y. [DOI] [PubMed] [Google Scholar]
- 32.Kniep B, Claus C, Peter-Katalinic J, Monner DA, Dippold W, Nimtz M. J Biol Chem. 1995;270:30173. doi: 10.1074/jbc.270.50.30173. [DOI] [PubMed] [Google Scholar]
- 33.Cerato E, Birkle S, Portoukalian J, Mezazigh A, Chatal JF, Aubry J. Hybridoma. 1997;16:307. doi: 10.1089/hyb.1997.16.307. [DOI] [PubMed] [Google Scholar]
- 34.Saito M, Kasai N, Yu RK. Anal Biochem. 1985;148:54. doi: 10.1016/0003-2697(85)90627-x. [DOI] [PubMed] [Google Scholar]
- 35.Colsch B, Jackson SN, Dutta S, Woods AS. ACS Chem Neurosci. 2011;2:213. doi: 10.1021/cn100096h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hara S, Yamaguchi M, Takemori Y, Furuhata K, Ogura H, Nakamura M. Anal Biochem. 1989;179:162. doi: 10.1016/0003-2697(89)90218-2. [DOI] [PubMed] [Google Scholar]
- 37.Mawhinney TP, Chance DL. Anal Biochem. 1994;223:164. doi: 10.1006/abio.1994.1564. [DOI] [PubMed] [Google Scholar]
- 38.Schauer R, Kamerling JP. In: Glycoproteins II. Montreuil J, Vliegenthart JFG, Schachter H, editors. Amsterdam: Elsevier; 1997. p. 243. [Google Scholar]
- 39.Bulai T, Bratosin D, Pons A, Montreuil J, Zanetta JP. FEBS Lett. 2003;534:185. doi: 10.1016/s0014-5793(02)03838-3. [DOI] [PubMed] [Google Scholar]
- 40.Rinninger A, Richet C, Pons A, Kohla G, Schauer R, Bauer HC, Zanetta JP, Vlasak R. Glycoconj J. 2006;23:73. doi: 10.1007/s10719-006-5439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robbe C, Capon C, Maes E, Rousset M, Zweibaum A, Zanetta JP, Michalski JC. J Biol Chem. 2003;278:46337. doi: 10.1074/jbc.M302529200. [DOI] [PubMed] [Google Scholar]
- 42.Zanetta JP, Pons A, Iwersen M, Mariller C, Leroy Y, Timmerman P, Schauer R. Glycobiology. 2001;11:663. doi: 10.1093/glycob/11.8.663. [DOI] [PubMed] [Google Scholar]
- 43.Zanetta JP, Srinivasan V, Schauer R. Biochimie. 2006;88:171. doi: 10.1016/j.biochi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 44.Winkler W, Huber W, Vlasak R, Allmaier G. Rapid Commun Mass Spectrom. 2011;25:3235. doi: 10.1002/rcm.5217. [DOI] [PubMed] [Google Scholar]
- 45.Severi E, Hood DW, Thomas GH. Microbiology. 2007;153:2817. doi: 10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- 46.Khatua B, Ghoshal A, Bhattacharya K, Mandal C, Saha B, Crocker PR. FEBS Lett. 2010;584:555. doi: 10.1016/j.febslet.2009.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiman S, Dahesh S, Carlin AF, Varki A, Nizet V, Lewis AL. Glycobiology. 2009;19:1204. doi: 10.1093/glycob/cwp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claus H, Borrow R, Achtman M, Morelli G, Kantelberg C, Longworth E, Frosch M, Vogel U. Mol Microbiol. 2004;51:227. doi: 10.1046/j.1365-2958.2003.03819.x. [DOI] [PubMed] [Google Scholar]
- 49.Lewis AL, Cao H, Patel SK, Diaz S, Ryan W, Carlin AF, Thon V, Lewis WG, Varki A, Chen X, Nizet V. J Biol Chem. 2007;282:27562. doi: 10.1074/jbc.M700340200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis AL, Nizet V, Varki A. Proc Natl Acad Sci USA. 2004;101:11123. doi: 10.1073/pnas.0403010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bergfeld AK, Claus H, Vogel U, Muhlenhoff M. J Biol Chem. 2007;282:22217. doi: 10.1074/jbc.M703044200. [DOI] [PubMed] [Google Scholar]
- 52.Deszo EL, Steenbergen SM, Freedberg DI, Vimr ER. Proc Natl Acad Sci USA. 2005;102:5564. doi: 10.1073/pnas.0407428102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vimr ER, Steenbergen SM. Mol Microbiol. 2006;60:828. doi: 10.1111/j.1365-2958.2006.05158.x. [DOI] [PubMed] [Google Scholar]
- 54.Frasa H, Procee J, Torensma R, Verbruggen A, Algra A, Rozenberg-Arska M, Kraaijeveld K, Verhoef J. J Clin Microbiol. 1993;31:3174. doi: 10.1128/jcm.31.12.3174-3178.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mordhorst IL, Claus H, Ewers C, Lappann M, Schoen C, Elias J, Batzilla J, Dobrindt U, Wieler LH, Bergfeld AK, Muhlenhoff M, Vogel U. Environ Microbiol. 2009;11:3154. doi: 10.1111/j.1462-2920.2009.02019.x. [DOI] [PubMed] [Google Scholar]
- 56.Bergfeld AK, Claus H, Lorenzen NK, Spielmann F, Vogel U, Muhlenhoff M. J Biol Chem. 2009;284:6. doi: 10.1074/jbc.M807518200. [DOI] [PubMed] [Google Scholar]
- 57.Lee HJ, Rakic B, Gilbert M, Wakarchuk WW, Withers SG, Strynadka NC. J Biol Chem. 2009;284:24501. doi: 10.1074/jbc.M109.006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulz EC, Bergfeld AK, Ficner R, Muhlenhoff M. PLoS One. 2011;6:e17403. doi: 10.1371/journal.pone.0017403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weiman S, Uchiyama S, Lin FY, Chaffin D, Varki A, Nizet V, Lewis AL. Biochem J. 2010;428:163. doi: 10.1042/BJ20100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Falk K, Aspehaug V, Vlasak R, Endresen C. J Virol. 2004;78:3063. doi: 10.1128/JVI.78.6.3063-3071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenthal PB, Zhang X, Formanowski F, Fitz W, Wong CH, Meier-Ewert H, Skehel JJ, Wiley DC. Nature. 1998;396:92. doi: 10.1038/23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krempl C, Schultze B, Herrler G. Adv Exp Med Biol. 1995;380:371. doi: 10.1007/978-1-4615-1899-0_60. [DOI] [PubMed] [Google Scholar]
- 63.Kunkel F, Herrler G. Virology. 1993;195:195. doi: 10.1006/viro.1993.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vlasak R, Luytjes W, Spaan W, Palese P. Proc Natl Acad Sci USA. 1988;85:4526. doi: 10.1073/pnas.85.12.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schultze B, Gross HJ, Brossmer R, Herrler G. J Virol. 1991;65:6232. doi: 10.1128/jvi.65.11.6232-6237.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schultze B, Herrler G. J Gen Virol. 1992;73:901. doi: 10.1099/0022-1317-73-4-901. [DOI] [PubMed] [Google Scholar]
- 67.Smits SL, Gerwig GJ, van Vliet AL, Lissenberg A, Briza P, Kamerling JP, Vlasak R, de Groot RJ. J Biol Chem. 2005;280:6933. doi: 10.1074/jbc.M409683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vlasak R, Luytjes W, Leider J, Spaan W, Palese P. J Virol. 1988;62:4686. doi: 10.1128/jvi.62.12.4686-4690.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng Q, Langereis MA, van Vliet AL, Huizinga EG, de Groot RJ. Proc Natl Acad Sci USA. 2008;105:9065. doi: 10.1073/pnas.0800502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schultze B, Gross HJ, Brossmer R, Klenk HD, Herrler G. Virus Res. 1990;16:185. doi: 10.1016/0168-1702(90)90022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schultze B, Wahn K, Klenk HD, Herrler G. Virology. 1991;180:221. doi: 10.1016/0042-6822(91)90026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Regl G, Kaser A, Iwersen M, Schmid H, Kohla G, Strobl B, Vilas U, Schauer R, Vlasak R. J Virol. 1999;73:4721. doi: 10.1128/jvi.73.6.4721-4727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wurzer WJ, Obojes K, Vlasak R. J Gen Virol. 2002;83:395. doi: 10.1099/0022-1317-83-2-395. [DOI] [PubMed] [Google Scholar]
- 74.Langereis MA, van Vliet AL, Boot W, de Groot RJ. J Virol. 2010;84:8970. doi: 10.1128/JVI.00566-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klausegger A, Strobl B, Regl G, Kaser A, Luytjes W, Vlasak R. J Virol. 1999;73:3737. doi: 10.1128/jvi.73.5.3737-3743.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langereis MA, Zeng Q, Gerwig GJ, Frey B, von Itzstein M, Kamerling JP, de Groot RJ, Huizinga EG. Proc Natl Acad Sci USA. 2009;106:15897. doi: 10.1073/pnas.0904266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatterjee M, Chava AK, Kohla G, Pal S, Merling A, Hinderlich S, Unger U, Strasser P, Gerwig GJ, Kamerling JP, Vlasak R, Crocker PR, Schauer R, Schwartz-Albiez R, Mandal C. Glycobiology. 2003;13:351. doi: 10.1093/glycob/cwg027. [DOI] [PubMed] [Google Scholar]
- 78.Chava AK, Bandyopadhyay S, Chatterjee M, Mandal C. Glycoconj J. 2004;20:199. doi: 10.1023/B:GLYC.0000024251.30100.08. [DOI] [PubMed] [Google Scholar]
- 79.Chava AK, Chatterjee M, Gerwig GJ, Kamerling JP, Mandal C. Biol Chem. 2004;385:59. doi: 10.1515/BC.2004.008. [DOI] [PubMed] [Google Scholar]
- 80.Chava AK, Chatterjee M, Sharma V, Sundar S, Mandal C. J Infect Dis. 2004;189:1257. doi: 10.1086/382752. [DOI] [PubMed] [Google Scholar]
- 81.Ghoshal A, Gerwig GJ, Kamerling JP, Mandal C. Glycobiology. 2010;20:553. doi: 10.1093/glycob/cwp207. [DOI] [PubMed] [Google Scholar]
- 82.Ghoshal A, Mukhopadhyay S, Chava AK, Gerwig GJ, Kamerling JP, Chatterjee M, Mandal C. Parasitology. 2009;136:159. doi: 10.1017/S0031182008005180. [DOI] [PubMed] [Google Scholar]
- 83.Mukhopadhyay S, Mandal C. Indian J Med Res. 2006;123:203. [PubMed] [Google Scholar]
- 84.Rojas-Chaves M, Hellmund C, Walter RD. Parasitol Res. 1996;82:435. doi: 10.1007/s004360050141. [DOI] [PubMed] [Google Scholar]
- 85.Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 86.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Nature. 2009;459:1005. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL. J Biol Chem. 2001;276:6885. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- 88.Martin LT, Marth JD, Varki A, Varki NM. J Biol Chem. 2002;277:32930. doi: 10.1074/jbc.M203362200. [DOI] [PubMed] [Google Scholar]
- 89.Eckhardt M, Bukalo O, Chazal G, Wang L, Goridis C, Schachner M, Gerardy-Schahn R, Cremer H, Dityatev A. J Neurosci. 2000;20:5234. doi: 10.1523/JNEUROSCI.20-14-05234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Angata K, Huckaby V, Ranscht B, Terskikh A, Marth JD, Fukuda M. Mol Cell Biol. 2007;27:6659. doi: 10.1128/MCB.00205-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calandreau L, Marquez C, Bisaz R, Fantin M, Sandi C. Genes Brain Behav. 2010;9:958. doi: 10.1111/j.1601-183X.2010.00635.x. [DOI] [PubMed] [Google Scholar]
- 92.Varki A, Hooshmand F, Diaz S, Varki NM, Hedrick SM. Cell. 1991;65:65. doi: 10.1016/0092-8674(91)90408-q. [DOI] [PubMed] [Google Scholar]
- 93.Zhang G, Ji L, Kurono S, Fujita SC, Furuya S, Hirabayashi Y. Glycoconj J. 1997;14:847. doi: 10.1023/A:1018542105832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vallejo V, Reyes-Leyva J, Hernandez J, Ramirez H, Delannoy P, Zenteno E. Comp Biochem Physiol B Biochem Mol Biol. 2000;126:415. doi: 10.1016/S0305-0491(00)00213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koga M, Yuki N, Ariga T, Hirata K. J Neurol Sci. 1999;164:50. doi: 10.1016/s0022-510x(98)00331-1. [DOI] [PubMed] [Google Scholar]
- 96.Mendez-Otero R, Santiago MF. Braz J Med Biol Res. 2003;36:1003. doi: 10.1590/s0100-879x2003000800006. [DOI] [PubMed] [Google Scholar]
- 97.Miyakoshi LM, Mendez-Otero R, Hedin-Pereira C. Braz J Med Biol Res. 2001;34:669. doi: 10.1590/s0100-879x2001000500016. [DOI] [PubMed] [Google Scholar]
- 98.Araujo H, Menezes M, Mendez-Otero R. Eur J Cell Biol. 1997;72:202. [PubMed] [Google Scholar]
- 99.Negreiros EM, Leao AC, Santiago MF, Mendez-Otero R. J Neurobiol. 2003;57:31. doi: 10.1002/neu.10248. [DOI] [PubMed] [Google Scholar]
- 100.Santiago MF, Costa MR, Mendez-Otero R. J Neurosci. 2004;24:474. doi: 10.1523/JNEUROSCI.0116-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang CR, Liour SS, Dasgupta S, Yu RK. J Neurosci Res. 2007;85:1381. doi: 10.1002/jnr.21264. [DOI] [PubMed] [Google Scholar]
- 102.Herrler G, Reuter G, Rott R, Klenk HD, Schauer R. Biol Chem Hoppe Seyler. 1987;368:451. doi: 10.1515/bchm3.1987.368.1.451. [DOI] [PubMed] [Google Scholar]
- 103.Mukherjee K, Chowdhury S, Mondal S, Mandal C, Chandra S, Bhadra RK. Biochem Biophys Res Commun. 2007;362:651. doi: 10.1016/j.bbrc.2007.08.048. [DOI] [PubMed] [Google Scholar]
- 104.Mukherjee K, Chava AK, Mandal C, Dey SN, Kniep B, Chandra S. J Cell Biochem. 2008;105:724. doi: 10.1002/jcb.21867. [DOI] [PubMed] [Google Scholar]
- 105.van Kooyk Y, Rabinovich GA. Nat Immunol. 2008;9:593. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 106.Varki A. Glycoconj J. 2009;26:231. doi: 10.1007/s10719-008-9183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crocker PR, Redelinghuys P. Biochem Soc Trans. 2008;36:1467. doi: 10.1042/BST0361467. [DOI] [PubMed] [Google Scholar]
- 108.Schwartz-Albiez R. In: The sugar code. Fundamentals of glycosciences. Gabius HJ, editor. Weinheim: Wiley-Blackwell; 2009. p. 447. [Google Scholar]
- 109.de Groot RJ. Glycoconj J. 2006;23:59. doi: 10.1007/s10719-006-5438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kniep B, Flegel WA, Northoff H, Rieber EP. Blood. 1993;82:1776. [PubMed] [Google Scholar]
- 111.Vater M, Kniep B, Gross HJ, Claus C, Dippold W, Schwartz-Albiez R. Immunol Lett. 1997;59:151. doi: 10.1016/s0165-2478(97)00116-8. [DOI] [PubMed] [Google Scholar]
- 112.Wipfler D, Srinivasan GV, Sadick H, Kniep B, Arming S, Willhauck-Fleckenstein M, Vlasak R, Schauer R, Schwartz-Albiez R. Glycobiology. 2011;21:1161. doi: 10.1093/glycob/cwr050. [DOI] [PubMed] [Google Scholar]
- 113.Sharma V, Chatterjee M, Mandal C, Sen S, Basu D. Am J Trop Med Hyg. 1998;58:551. doi: 10.4269/ajtmh.1998.58.551. [DOI] [PubMed] [Google Scholar]
- 114.Schwarz-Albiez R, Claus C, Kniep B. In: Leukocyte Typing VII. Mason D, Simmons D, Buckley C, Schwartz-Albiez R, Hadam M, Saalmuller A, Clark E, Malavasi F, Morrissey JA, Vivier E, Horejsi V, De Haas M, Van der Schoot E, Uguccioni M, Hart D, Hadam M, Goyert S, Zola H, Civin C, Meuer S, Shaw S, Turni L, Andre P, Bensussan A, editors. Oxford: Oxford University Press; 2002. p. 185. [Google Scholar]
- 115.Bergelson LD, Dyatlovitskaya EV, Klyuchareva TE, Kryukova EV, Lemenovskaya AF, Matveeva VA, Sinitsyna EV. Eur J Immunol. 1989;19:1979. doi: 10.1002/eji.1830191102. [DOI] [PubMed] [Google Scholar]
- 116.Grayson G, Ladisch S. Cell Immunol. 1992;139:18. doi: 10.1016/0008-8749(92)90096-8. [DOI] [PubMed] [Google Scholar]
- 117.Nicoll G, Avril T, Lock K, Furukawa K, Bovin N, Crocker PR. Eur J Immunol. 2003;33:1642. doi: 10.1002/eji.200323693. [DOI] [PubMed] [Google Scholar]
- 118.Park JE, Wu DY, Prendes M, Lu SX, Ragupathi G, Schrantz N, Chapman PB. Immunology. 2008;123:145. doi: 10.1111/j.1365-2567.2007.02760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peguet-Navarro J, Sportouch M, Popa I, Berthier O, Schmitt D, Portoukalian J. J Immunol. 2003;170:3488. doi: 10.4049/jimmunol.170.7.3488. [DOI] [PubMed] [Google Scholar]
- 120.Corfield AP, Myerscough N, Warren BF, Durdey P, Paraskeva C, Schauer R. Glycoconj J. 1999;16:307. doi: 10.1023/a:1007026314792. [DOI] [PubMed] [Google Scholar]
- 121.Mann B, Klussmann E, Vandamme-Feldhaus V, Iwersen M, Hanski ML, Riecken EO, Buhr HJ, Schauer R, Kim YS, Hanski C. Int J Cancer. 1997;72:258. doi: 10.1002/(sici)1097-0215(19970717)72:2<258::aid-ijc10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 122.Muchmore EA, Diaz S, Varki A. Am J Phys Anthropol. 1998;107:187. doi: 10.1002/(SICI)1096-8644(199810)107:2<187::AID-AJPA5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 123.Cariappa A, Takematsu H, Liu H, Diaz S, Haider K, Boboila C, Kalloo G, Connole M, Shi HN, Varki N, Varki A, Pillai S. J Exp Med. 2009;206:125. doi: 10.1084/jem.20081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Surolia I, Pirnie SP, Chellappa V, Taylor KN, Cariappa A, Moya J, Liu H, Bell DW, Driscoll DR, Diederichs S, Haider K, Netravali I, Le S, Elia R, Dow E, Lee A, Freudenberg J, De Jager PL, Chretien Y, Varki A, Macdonald ME, Gillis T, Behrens TW, Bloch D, Collier D, Korzenik J, Podolsky DK, Hafler D, Murali M, Sands B, Stone JH, Gregersen PK, Pillai S. Nature. 2010;466:243. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Varki A, Kannagi R, Toole BP. In: Essentials in glycobiology. 2. Varki A, Cummings RD, Esko JD, Freeze HD, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 126.Kohla G, Stockfleth E, Schauer R. Neurochem Res. 2002;27:583. doi: 10.1023/a:1020211714104. [DOI] [PubMed] [Google Scholar]
- 127.Birks SM, Danquah JO, King L, Vlasak R, Gorecki DC, Pilkington GJ. Neuro Oncol. 2011;13:950. doi: 10.1093/neuonc/nor108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ravindranath MH, Muthugounder S, Presser N. Melanoma Res. 2008;18:47. doi: 10.1097/CMR.0b013e3282f43acf. [DOI] [PubMed] [Google Scholar]
- 129.Gocht A, Rutter G, Kniep B. Histochem Cell Biol. 1998;110:217. doi: 10.1007/s004180050284. [DOI] [PubMed] [Google Scholar]
- 130.Cao Y, Merling A, Crocker PR, Keller R, Schwartz-Albiez R. Lab Invest. 2002;82:1515. doi: 10.1097/01.lab.0000038503.34655.98. [DOI] [PubMed] [Google Scholar]
- 131.Claus C, Schlaak J, Dittmayer M, Meyer zum Buschenfelde K, Dippold W (1994) Eur J Immunol 24:1208 [DOI] [PubMed]
- 132.Malisan F, Franchi L, Tomassini B, Ventura N, Condo I, Rippo MR, Rufini A, Liberati L, Nachtigall C, Kniep B, Testi R. J Exp Med. 2002;196:1535. doi: 10.1084/jem.20020960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Maria R, Lenti L, Malisan F, d’Agostino F, Tomassini B, Zeuner A, Rippo MR, Testi R. Science. 1997;277:1652. doi: 10.1126/science.277.5332.1652. [DOI] [PubMed] [Google Scholar]
- 134.Garcia-Ruiz C, Colell A, Morales A, Calvo M, Enrich C, Fernandez-Checa JC. J Biol Chem. 2002;277:36443. doi: 10.1074/jbc.M206021200. [DOI] [PubMed] [Google Scholar]
- 135.Garofalo T, Tinari A, Matarrese P, Giammarioli AM, Manganelli V, Ciarlo L, Misasi R, Sorice M, Malorni W. FEBS Lett. 2007;581:3899. doi: 10.1016/j.febslet.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 136.Garcia-Ruiz C, Colell A, Paris R, Fernandez-Checa JC. FASEB J. 2000;14:847. doi: 10.1096/fasebj.14.7.847. [DOI] [PubMed] [Google Scholar]
- 137.Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G, Testi R. FASEB J. 2000;14:2047. doi: 10.1096/fj.99-1028com. [DOI] [PubMed] [Google Scholar]
- 138.Brenner C, Kniep B, Maillier E, Martel C, Franke C, Rober N, Bachmann M, Rieber EP, Sandhoff R. Biochem Biophys Res Commun. 2010;391:248. doi: 10.1016/j.bbrc.2009.11.044. [DOI] [PubMed] [Google Scholar]
- 139.Kniep B, Kniep E, Ozkucur N, Barz S, Bachmann M, Malisan F, Testi R, Rieber EP. Int J Cancer. 2006;119:67. doi: 10.1002/ijc.21788. [DOI] [PubMed] [Google Scholar]
- 140.Ghosh S, Bandyopadhyay S, Bhattacharya DK, Mandal C. Ann Hematol. 2005;84:76. doi: 10.1007/s00277-004-0933-0. [DOI] [PubMed] [Google Scholar]
- 141.Bandyopadhyay S, Mukherjee K, Chatterjee M, Bhattacharya DK, Mandal C. J Immunol Methods. 2005;297:13. doi: 10.1016/j.jim.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 142.Mandal C, Chatterjee M, Sinha D. Br J Haematol. 2000;110:801. doi: 10.1046/j.1365-2141.2000.02105.x. [DOI] [PubMed] [Google Scholar]
- 143.Pal S, Ghosh S, Bandyopadhyay S, Mandal C, Bandhyopadhyay S, Kumar Bhattacharya D. Int J Cancer. 2004;111:270. doi: 10.1002/ijc.20246. [DOI] [PubMed] [Google Scholar]
- 144.Pal S, Ghosh S, Mandal C, Kohla G, Brossmer R, Isecke R, Merling A, Schauer R, Schwartz-Albiez R, Bhattacharya DK. Glycobiology. 2004;14:859. doi: 10.1093/glycob/cwh111. [DOI] [PubMed] [Google Scholar]
- 145.Sinha D, Mandal C, Bhattacharya DK. Leukemia. 1999;13:119. doi: 10.1038/sj.leu.2401239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mandal C, Srinivasan GV, Chowdhury S, Chandra S, Schauer R. Glycoconj J. 2009;26:57. doi: 10.1007/s10719-008-9163-3. [DOI] [PubMed] [Google Scholar]
- 147.Bandyopadhyay S, Chatterjee M, Banavali SD, Pal S, Nair CN, Advani SH, Mandal C. Indian J Biochem Biophys. 2006;43:7. [PubMed] [Google Scholar]
- 148.Pal S, Bandyopadhyay S, Chatterjee M, Bhattacharya DK, Minto L, Hall AG, Mandal C. Clin Biochem. 2004;37:395. doi: 10.1016/j.clinbiochem.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 149.Pal S, Chatterjee M, Bhattacharya DK, Bandhyopadhyay S, Mandal C. Glycobiology. 2000;10:539. doi: 10.1093/glycob/10.6.539. [DOI] [PubMed] [Google Scholar]
- 150.Pal S, Chatterjee M, Bhattacharya DK, Bandhyopadhyay S, Mandal C. Glycoconj J. 2001;18:529. doi: 10.1023/a:1019692329568. [DOI] [PubMed] [Google Scholar]
- 151.Chowdhury S, Bandyopadhyay S, Mandal C, Chandra S. BMC Cancer. 2008;8:40. doi: 10.1186/1471-2407-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chowdhury S, Mandal C. Biotechnol J. 2009;4:361. doi: 10.1002/biot.200800253. [DOI] [PubMed] [Google Scholar]
- 153.Sinha D, Bhattacharya DK, Mandal C. Leuk Res. 1999;23:803. doi: 10.1016/s0145-2126(99)00093-4. [DOI] [PubMed] [Google Scholar]
- 154.Sinha D, Mandal C, Bhattacharya DK. Leuk Res. 1999;23:433. doi: 10.1016/s0145-2126(98)00184-2. [DOI] [PubMed] [Google Scholar]
- 155.Sinha D, Mandal C, Bhattacharya DK. Leukemia. 1999;13:309. doi: 10.1038/sj.leu.2401312. [DOI] [PubMed] [Google Scholar]
- 156.Ghosh S, Bandyopadhyay S, Mukherjee K, Mallick A, Pal S, Mandal C, Bhattacharya DK. Glycoconj J. 2007;24:17. doi: 10.1007/s10719-006-9007-y. [DOI] [PubMed] [Google Scholar]
- 157.Ghosh S, Bandyopadhyay S, Pal S, Das B, Bhattacharya DK, Mandal C. Br J Haematol. 2005;128:35. doi: 10.1111/j.1365-2141.2004.05256.x. [DOI] [PubMed] [Google Scholar]
- 158.Mukherjee K, Chava AK, Bandyopadhyay S, Mallick A, Chandra S, Mandal C. Biol Chem. 2009;390:325. doi: 10.1515/BC.2009.036. [DOI] [PubMed] [Google Scholar]
- 159.Mandal C, Tringali C, Mondal S, Anastasia L, Chandra S, Venerando B. Int J Cancer. 2010;126:337. doi: 10.1002/ijc.24733. [DOI] [PubMed] [Google Scholar]
- 160.Bandyopadhyay S, Bhattacharyya A, Mallick A, Sen AK, Tripathi G, Das T, Sa G, Bhattacharya DK, Mandal C. Int Immunol. 2005;17:177. doi: 10.1093/intimm/dxh198. [DOI] [PubMed] [Google Scholar]
- 161.Ariga T, Suetake K, Nakane M, Kubota M, Usuki S, Kawashima I, Yu RK. Neurosignals. 2008;16:226. doi: 10.1159/000111565. [DOI] [PubMed] [Google Scholar]
- 162.Schauer R. Zoology (Jena) 2004;107:49. doi: 10.1016/j.zool.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 163.Shen Y, Kohla G, Lrhorfi AL, Sipos B, Kalthoff H, Gerwig GJ, Kamerling JP, Schauer R, Tiralongo J. Eur J Biochem. 2004;271:281. doi: 10.1046/j.1432-1033.2003.03927.x. [DOI] [PubMed] [Google Scholar]
- 164.Shen Y, Tiralongo J, Kohla G, Schauer R. Biol Chem. 2004;385:145. doi: 10.1515/BC.2004.033. [DOI] [PubMed] [Google Scholar]
- 165.Chen HY, Challa AK, Varki A. J Biol Chem. 2006;281:7825. doi: 10.1074/jbc.M512379200. [DOI] [PubMed] [Google Scholar]
- 166.Iwersen M, Dora H, Kohla G, Gasa S, Schauer R. Biol Chem. 2003;384:1035. doi: 10.1515/BC.2003.116. [DOI] [PubMed] [Google Scholar]
- 167.Lrhorfi LA, Srinivasan GV, Schauer R. Biol Chem. 2007;388:297. doi: 10.1515/BC.2007.033. [DOI] [PubMed] [Google Scholar]
- 168.Tiralongo J, Schauer R. Trends Glycosci Glycotechnol. 2004;16:1. [Google Scholar]
- 169.Tiralongo J, Schmid H, Thun R, Iwersen M, Schauer R. Glycoconj J. 2000;17:849. doi: 10.1023/a:1010965128335. [DOI] [PubMed] [Google Scholar]
- 170.Iwersen M, Vandamme-Feldhaus V, Schauer R. Glycoconj J. 1998;15:895. doi: 10.1023/a:1006911100081. [DOI] [PubMed] [Google Scholar]
- 171.Eckhardt M, Gotza B, Gerardy-Schahn R. J Biol Chem. 1998;273:20189. doi: 10.1074/jbc.273.32.20189. [DOI] [PubMed] [Google Scholar]
- 172.Eckhardt M, Muhlenhoff M, Bethe A, Gerardy-Schahn R. Proc Natl Acad Sci USA. 1996;93:7572. doi: 10.1073/pnas.93.15.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Higa HH, Paulson JC. J Biol Chem. 1985;260:8838. [PubMed] [Google Scholar]
- 174.Diaz S, Higa HH, Hayes BK, Varki A. J Biol Chem. 1989;264:19416. [PubMed] [Google Scholar]
- 175.Higa HH, Butor C, Diaz S, Varki A. J Biol Chem. 1989;264:19427. [PubMed] [Google Scholar]
- 176.Ogura K, Nara K, Watanabe Y, Kohno K, Tai T, Sanai Y. Biochem Biophys Res Commun. 1996;225:932. doi: 10.1006/bbrc.1996.1274. [DOI] [PubMed] [Google Scholar]
- 177.Kanamori A, Nakayama J, Fukuda MN, Stallcup WB, Sasaki K, Fukuda M, Hirabayashi Y. Proc Natl Acad Sci USA. 1997;94:2897. doi: 10.1073/pnas.94.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shi WX, Chammas R, Varki A. Glycobiology. 1998;8:199. doi: 10.1093/glycob/8.2.199. [DOI] [PubMed] [Google Scholar]
- 179.Satake H, Chen HY, Varki A. J Biol Chem. 2003;278:7942. doi: 10.1074/jbc.M210565200. [DOI] [PubMed] [Google Scholar]
- 180.Furukawa K, Aixinjueluo W, Kasama T, Ohkawa Y, Yoshihara M, Ohmi Y, Tajima O, Suzumura A, Kittaka D. J Neurochem. 2008;105:1057. doi: 10.1111/j.1471-4159.2008.05232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Arming S, Wipfler D, Mayr J, Merling A, Vilas U, Schauer R, Schwartz-Albiez R, Vlasak R. Glycobiology. 2011;21:553. doi: 10.1093/glycob/cwq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Janbon G, Himmelreich U, Moyrand F, Improvisi L, Dromer F. Mol Microbiol. 2001;42:453. doi: 10.1046/j.1365-2958.2001.02651.x. [DOI] [PubMed] [Google Scholar]
- 183.Selig M, Adney W, Himmel M, Decker S. Cellulose. 2009;16:711. [Google Scholar]
- 184.Carroll A, Somerville C. Annu Rev Plant Biol. 2009;60:165. doi: 10.1146/annurev.arplant.043008.092125. [DOI] [PubMed] [Google Scholar]
- 185.Manabe Y, Nafisi M, Verhertbruggen Y, Orfila C, Gille S, Rautengarten C, Cherk C, Marcus SE, Somerville S, Pauly M, Knox JP, Sakuragi Y, Scheller HV. Plant Physiol. 2011;155:1068. doi: 10.1104/pp.110.168989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Anantharaman V, Aravind L. Biol Direct. 2010;5:1. doi: 10.1186/1745-6150-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schauer R, Shukla AK. Glycoconj J. 2008;25:625. doi: 10.1007/s10719-008-9109-9. [DOI] [PubMed] [Google Scholar]
- 188.Schauer R, Reuter G, Stoll S. Biochimie. 1988;70:1511. doi: 10.1016/0300-9084(88)90288-x. [DOI] [PubMed] [Google Scholar]
- 189.Angata T, Varki A. Chem Rev. 2002;102:439. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 190.Schauer R. Glycoconj J. 2000;17:485. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Butor C, Griffiths G, Aronson NN, Jr, Varki A. J Cell Sci. 1995;108(Pt 6):2213. doi: 10.1242/jcs.108.6.2213. [DOI] [PubMed] [Google Scholar]
- 192.Higa HH, Manzi A, Varki A. J Biol Chem. 1989;264:19435. [PubMed] [Google Scholar]
- 193.Guimaraes MJ, Bazan JF, Castagnola J, Diaz S, Copeland NG, Gilbert DJ, Jenkins NA, Varki A, Zlotnik A. J Biol Chem. 1996;271:13697. doi: 10.1074/jbc.271.23.13697. [DOI] [PubMed] [Google Scholar]
- 194.Butor C, Diaz S, Varki A. J Biol Chem. 1993;268:10197. [PubMed] [Google Scholar]
- 195.Stoddart A, Zhang Y, Paige CJ. Nucleic Acids Res. 1996;24:4003. doi: 10.1093/nar/24.20.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Takematsu H, Diaz S, Stoddart A, Zhang Y, Varki A. J Biol Chem. 1999;274:25623. doi: 10.1074/jbc.274.36.25623. [DOI] [PubMed] [Google Scholar]
- 197.Courtney AH, Puffer EB, Pontrello JK, Yang ZQ, Kiessling LL. Proc Natl Acad Sci USA. 2009;106:2500. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ghosh S, Bandulet C, Nitschke L. Int Immunol. 2006;18:603. doi: 10.1093/intimm/dxh402. [DOI] [PubMed] [Google Scholar]
- 199.Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. J Immunol. 2010;184:3618. doi: 10.4049/jimmunol.0902711. [DOI] [PubMed] [Google Scholar]
- 200.Kelm S, Gerlach J, Brossmer R, Danzer CP, Nitschke L. J Exp Med. 2002;195:1207. doi: 10.1084/jem.20011783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kawasaki N, Rademacher C, Paulson JC. J Innate Immun. 2011;3:411. doi: 10.1159/000322375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tsai S, Hardison NE, James AH, Motsinger-Reif AA, Bischoff SR, Thames BH, Piedrahita JA. Placenta. 2011;32:175. doi: 10.1016/j.placenta.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]