

Abstract
The yeast two-hybrid system (Y2H) is a powerful method to identify binary protein–protein interactions in vivo. Here we describe Y2H screening strategies that use defined libraries of open reading frames (ORFs) and cDNA libraries. The array-based Y2H system is well suited for interactome studies of small genomes with an existing ORFeome clones preferentially in a recombination based cloning system. For large genomes, pooled library screening followed by Y2H pairwise retests may be more efficient in terms of time and resources, but multiple sampling is necessary to ensure comprehensive screening. While the Y2H false positives can be efficiently reduced by using built-in controls, retesting, and evaluation of background activation; implementing the multiple variants of the Y2H vector systems is essential to reduce the false negatives and ensure comprehensive coverage of an interactome.
Keywords: Yeast two-hybrid system, Protein–protein interactions, Pooled library screening, Two-hybrid array
Introduction
Specific interactions between proteins form the basis of most biological processes, thus the knowledge of an organism’s protein interaction network provides insights into the function(s) of individual proteins, the structure of functional complexes, and eventually, the organization of the entire cell. The Protein-protein interactions (PPIs) can be identified by a multitude of experimental methods. However, a vast majority of the PPIs available today are generated by yeast two-hybrid (Y2H) method and affinity purification or co-fractionation coupled to mass spectrometry (AP-MS) (Kerrien et al. 2012). It is important to note that these methods yield different types of information; Y2H analyses reveal binary interactions, including transient interactions, whereas the AP-MS approaches report multiple interactions connecting all of the proteins in fairly stable complexes. Protein interactome analysis on a genome scale was first achieved by using yeast two-hybrid (Y2H) screens (Ito et al. 2001; Uetz et al. 2000) and next by large-scale mass spectrometric analysis of affinity-purified protein complexes (Gavin et al. 2002; Ho et al. 2002). Here we describe the high-throughput Y2H screening strategies, applied to map high-quality proteome-scale interactome networks of model organisms and pathogenic infectious agents.
Yeast Two-Hybrid System
The Y2H system is a genetic screening extensively used to identify binary protein–protein interactions in vivo (in yeast cells). The system was originally developed by Stanley Fields in 1989 (Fields and Song 1989). The principle of the assay relied on major discoveries on transcription initiation (Brent and Ptashne 1985). In general the protein domains can be separated and recombined and can retain their properties. In particular, transcription factors can frequently be split into the DNA-binding domain (DBD) and activation domains (ADs). In the two-hybrid system, a DNA-binding domain (in this case, from the yeast Gal4 protein) is fused to a protein generally called bait (“B”) for which one wants to find interacting partners. A transcriptional activation domain (from the yeast Gal4 protein) is then fused to one or more ORFs (preys) (Fig. 11.1). The bait and prey fusion proteins are then co-expressed in the same yeast cell. If, the two proteins bait and prey interact, a transcription factor is reconstituted which in turn activates the reporter gene(s) (Fig. 11.1). The expression of the reporter gene allows the yeast cell to grow under certain conditions. For example, the HIS3 reporter encodes imidazoleglycerolphosphate (IGP) dehydratase, a critical enzyme in histidine biosynthesis. In a screening yeast strain lacking an endogenous copy of HIS3, expression of a HIS3 reporter gene is driven by a promoter that contains a Gal4p-binding site, so the bait protein fusion can bind to it. However, the bait fusion does not contain a transcriptional activation domain it remains inactive. If, a prey protein with an attached activation domain binds to the bait protein, this activation domain can recruit the basal transcription machinery, and expression of the reporter gene ensues. Thus, these cells can now grow in the absence of histidine in the media because they can synthesize their own.
Fig. 11.1.
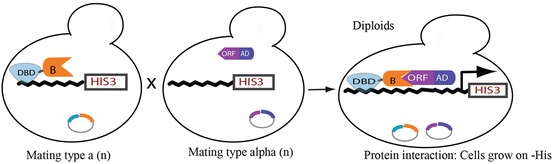
Yeast two-hybrid principle: A protein of interest ‘B’ is expressed in yeast as a fusion to a Gal4p DNA-binding domain (DBD, “bait”; circles denote expression plasmids). Another protein or library of proteins of interest ‘ORF’ is fused to Gal4p transcriptional activation domain (AD, “prey”). The two yeast strains are mated to combine the two plasmids expressing bait and prey fusion proteins in the same cell (diploid). If, proteins ‘B’ and ‘ORF’ interact in the resulting diploids cells, they reconstitute a transcription factor which activates a reporter gene (HIS3) and therefore allows the cell to grow on selective synthetic media (media lacking histidine)
Y2H Applications
Initially, the two-hybrid system was invented to demonstrate the association of two proteins (Fields and Song 1989). Later, it was demonstrated that completely new protein interactions can be identified with this system. Over time, it has become clear that the ability to perform unbiased large-scale library screens is the most powerful application of the system. In recent years, Y2H method has been extensively applied to map high-quality proteome-scale binary interactome networks of server model organism, including human proteome and may pathogenic infectious agents. In a recent study, Rolland et al., published a human interactome map, which is based on a systematic Y2H screening of 13,000 human proteins that uncovered 14,000 PPIs (Rolland et al. 2014). Similarly, several large-scale Y2H projects have been successful in systematically mapping binary interactome landscape of Escherichia coli (Rajagopala et al. 2014), Saccharomyces cerevisiae (Uetz et al. 2000; Yu et al. 2008), Caenorhabditis elegans (Gong et al. 2004), Drosophila melanogaster (Giot et al. 2003); these studies have shown that most of the proteins in a cell are actually connected to each other. The Y2H screening has also been implemented on many pathogenic infectious agents, to name a few, the Kaposi sarcoma-associated herpesvirus (Uetz et al. 2006), varicella-zoster (Uetz et al. 2006; Stellberger et al. 2010), Epstein–Barr (Calderwood et al. 2007), SARS (von Brunn et al. 2007), influenza (Shapira et al. 2009) viruses, the Campylobacter jejuni (Parrish et al. 2007), Helicobacter pylori (Hauser et al. 2014; Rain et al. 2001) and Treponema pallidum (Titz et al. 2008) bacteria, and Trypanosoma brucei (Lacomble et al. 2009) parasite. These interactome maps enhance our knowledge on these infectious agents and suggest potential therapeutic targets. Another potential of the Y2H method is to map host–pathogen protein interactions, which has been achieved for Epstein–Barr (Calderwood et al. 2007), hepatitis C (de Chassey et al. 2008), influenza (Shapira et al. 2009) and dengue (Khadka et al. 2011) viruses as well as mapping the interactions of bacterial effectors proteins with the it’s host (Memisevic et al. 2013). These studies have the potential to both fundamentally change our understanding of how pathogens (virus/bacteria) modulate the host proteome and aid the development of countermeasures to control infections/diseases. Likewise, the two-hybrid screens, can also be adapted to a variety of related questions, such as the identification of mutants that avert or advance interactions (Schwartz et al. 1998; Wang et al. 2012), the screening for drugs that affect protein interactions (Vidal and Endoh 1999; Vidal and Legrain 1999), the identification of RNA-binding proteins (SenGupta et al. 1996), or the semiquantitative determination of binding affinities (Estojak et al. 1995). The system can also be exploited to map binding domains (Rain et al. 2001; Ester and Uetz 2008), to study protein folding (Raquet et al. 2001), or to map interactions within a protein complex, for example, spliceosome (Hegele et al. 2012), proteasome (Cagney et al. 2001), flagellum (Rajagopala et al. 2007).
High-Throughput Yeast Two-Hybrid Screens
Array-Based Screening
In an array screening, a number of pre-defined prey proteins are tested for interactions with a bait protein. Typically the bait protein is expressed in one haploid yeast strain and the prey is expressed in another haploid yeast strain of different mating type (Fig. 11.1). The two strains are then mated so that the two proteins are expressed in the resulting diploid cell (Fig. 11.2). The screenings are done side-by-side under identical conditions with several prey proteins, and negative controls, so they can be well controlled, i.e., compared with control assays. In an array, usually each element (prey) is sequence validated and therefore it is immediately clear which two proteins are interacting when positives are selected. Most importantly, all the assays are done in an ordered array, so that background signals can be easily distinguished from true signals (Fig. 11.2, step 3). However, to perform the array screens the prey library need to be made upfront. This can be done for a subset of genes or for a whole genome (i.e., all ORFs of a genome). The array-based Y2H screenings are ideal for small genomes, for example mapping the interactions of phages (Rajagopala et al. 2011), virus (Uetz et al. 2006; Shapira et al. 2009; Khadka et al. 2011) and mapping the interactions within a protein complex (Rajagopala et al. 2007, 2012). Hands-on time and the amount of used resources grow exponentially with the number of tested proteins; this is a disadvantage for large genome sizes. However, cloned ORFeome collections of whole genomes become increasingly available for several organisms and modern cloning systems also allow direct transfer of entry clones into many specialized vectors (Walhout et al. 2000). One of the first applications of such clone collections is often a high-throughput protein interaction screening.
Fig. 11.2.
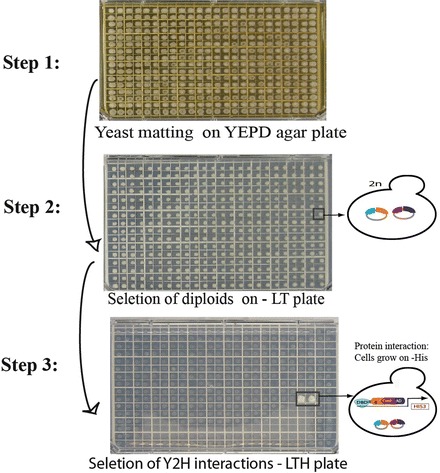
Array-based yeast two-hybrid screens. Step 1: Yeast mating combines the bait and prey plasmids. The bait (DNA-binding domain (DBD) fusion) liquid culture is pinned onto YEPDA agar plates using a 384-pin pinning tool, and then the prey array (activation-domain (AD) fusion) is pinned on top of the baits using the sterile pinning tool. The mating plates are incubated at 30 °C for 16 h. Step 2: The cells from the yeast mating plates are transferred onto –Leu –Trp medium plates using a sterile 384-pin pinning tool. Only diploid cells will grow on the media lacking leucine and tryptophan agar plates and ensures that both the prey and bait plasmids are combined in the diploid yeast cells. Step 3: The diploid cells are transferred onto –Leu –Trp –His medium plates for protein interaction detection. If the bait and prey proteins interact, and an active transcription factor is reconstituted and transcription of a reporter gene is activated. Thus, the cells can grow on selective media plates
Pooled Array Screening
The pooling strategy has the potential to accelerate screening but require sequencing capacity and/or extensive pairwise Y2H screening. In the pooled array screening, preys of known identity (systematically cloned or sequenced cDNA library clones) are combined and tested as pools against bait strains. The identification of the interacting protein pair commonly requires either sequencing preys in the positive yeast colonies or retesting of all members of the respective pool clones. Prof. Vidal lab at the Dana-Farber Cancer Institute, Boston MA, employed a pooling strategy for several large-scale interactome mapping projects (Rolland et al. 2014; Yu et al. 2008; Rual et al. 2005). Often, they tested each bait against pools of ∼188 preys (mini-libraries) and the identity of the interacting prey in the mini-libraries was identified by sequencing the prey PCR amplicons by end-read sequencing (Rual et al. 2005), or ‘stitched’ the interacting bait and preys together into a single amplicon and sequenced using next-generation sequencing technology (Rolland et al. 2014; Yu et al. 2008, 2011). Stelzl et al. tested pools of 8 baits against a systematic library of individual preys and identified interactions by a 2nd interaction mating (Stelzl et al. 2005). Likewise, Jin et al. proposed an “smart-pool-array” system, which allows the deconvolution of the interacting pairs through the definition of overlapping bait pools (Jin et al. 2007), and thus usually does not depend on sequencing or a 2nd pairwise retest procedure. Preferably, the preys are pooled rather than baits, because the former do not generally result in self-activation of transcription.
Pooled Library Screening
The pooled library screening strategy significantly accelerates screening but might also have the disadvantage of increasing the number of false negatives and multiple sampling is essential to achieve a reasonable sampling sensitivity rate (Rajagopala et al. 2014; Yu et al. 2008) and they require significant sequencing capacity. Similar, to pooled array screening the prey library is constructed by systematically cloning each of the sequenced ORFeome or cDNA library clones into Y2H prey vector(s). In a recent study we implemented this approach to map the E. coli interactome network (Rajagopala et al. 2014). In this study all the prey yeast strains (∼4000) were combined into a single pool and tested against each bait strains. After Y2H screening the identity of the interacting preys are identified by sequencing (Fig. 11.3).
Fig. 11.3.

Pooled-library yeast two-hybrid screens: A haploid yeast strain expressing a single protein as a DBD fusion is mixed with the yeast haploid strains expressing a prey library (systematical cloned). The bait and prey (1:1 ratio) culture is plated on YEPDA agar plate and the plates are incubated at 30 °C for 6 h or overnight at room temperature. During this process (yeast mating) both the prey and bait plasmids are combined in the diploid yeast cells. The cells from the mating plates are collected and transferred onto –Leu –Trp –His medium plates (supplemented with different concentration of 3-AT) for protein interaction detection, and plates are incubated at 30 °C for 4–6 days. The identity of interacting prey is identified by yeast colony PCR of positive yeast colonies, followed by DNA sequencing of the PCR product. The Y2H interactions obtained from the pool screening are subjected to pairwise retest (Phenotyping II) using fresh archival yeast stock, the screening was performed as quadruplet. Interactions which are not reproduced or showed signal in the auto-activation test (marked in red) should be removed from the interaction list
Random Library Screening
Random library screens do not require systematic cloning of all prey constructs, however, the prey library must be created. Therefore, the complete DNA sequence of the genome of interest is no prerequisite. Random prey libraries can be made using genomic DNA or cDNA based libraries. For genomic libraries, the genomic DNA of interest is randomly cut, size-selected, and the resulting fragments ligated into one or more two-hybrid prey vector(s). Previous yeast two-hybrid and bacterial two-hybrid screening projects used random genomic DNA libraries (Rain et al. 2001; Joung et al. 2000). A cDNA library is made through reverse transcription of mRNA collected from specific cell types or whole organisms. To simplify the task even more, many cDNA libraries are commercially available. For example, Clontech has a collection of human and tissue-specific cDNA libraries. However the bait clones that need to be screened with a random library need to be made independently.
Similar to pooled library screens, in a random library screen a library of prey proteins is tested for interactions with a bait protein. Similar to pooled library screens the bait protein is expressed in one yeast strain and the prey is expressed in another yeast strain of different mating type. The two strains are then mated so that the two proteins are expressed in the resulting diploid cell. The diploids are plated on interaction selective medium where only yeast cells having bait and its interacting prey will grow. The prey is identified by isolating the prey plasmids, PCR amplification of the insert, and sequencing (Sect. 11.6.11). The major limitation of the random library screening is unavailability of the indusial prey clones to perform pairwise Y2H retest or other validation assays, for example, validate a subset of interactions using orthogonal assays. Thus, evaluating the quality of PPIs relies on computation methods.
Adapting Next-generation Sequencing
The major shortfall of the high-throughput protein-protein interactome datasets is low coverage (Rajagopala et al. 2014; Yu et al. 2008). Even for the well-studied bacterium E. coli, more than 50 % of the interactome remains to be mapped (Rajagopala et al. 2014). An impotent step for high-throughput interactome-mapping methods using Y2H is determining the identities of the interacting proteins. Adapting the next-generation DNA sequencing technologies (Bennett et al. 2005; Margulies et al. 2005) as opposed to Sanger technology, would substantially increase throughput and decrease cost. However, next-generation DNA sequencing technologies are not readily applicable for identification of interacting pairs. Yu et al. describe a massively parallel interactome-mapping strategy that incorporates next-generation DNA sequencing and test the strategy in a high-throughput Y2H system (Yu et al. 2011). The methodology, termed Stitch-seq, which used PCR approach to amplify and stitch the bait and prey ORF or cDNA inserts in to a single amplican. Then the PCR products are pooled and sequenced by next-generation DNA sequencing to produce stitched interacting sequence tags. The sequencing produced an average read length of 207 bases, which are 125 bases longer than the 82-bp linker sequence between bait and preys. To identify the ORFs encoding pairs of interacting proteins, they selected reads that contained the linker sequence and also covered at least 15 bases of ORF-specific sequences on both ends of the linker. These reads could unambiguously identify pairs of unique bait and prey ORFs, after matching these sequences to human ORFeome collection used for the study. This general scheme can be readily extended to increase throughput and decrease cost for other interactome-mapping methods, particularly for binary protein-protein interaction assays
Analysis of Y2H Data
Analysis of raw results significantly improves the data quality of the protein interaction set. It is important to consider at least the following three parameters to obtain a high-quality Y2H data. Auto-activation: Is the background self-activation strength of the tested bait. The protein interaction strength of interacting pairs must be significantly higher than with all other (background) pairs. Ideally, no activation (i.e. no colony growth) should be observed in non-interacting pairs or vector control. Reproducibility: The protein interactions that are not reproduced in a pairwise retest experiment should be discarded. Sticky preys: For each prey the number of different interacting baits (prey count) is counted; preys interacting with a large number of baits are non-specific (“sticky” preys) and thus may have no biological relevance. The cut-off number depends also on the nature of baits and the number of baits screened: if a large family of related proteins are screened it is expected that many of them find the same prey. As a general guideline the number of baits interacting with a certain prey should not be larger than 5–10 % of the bait number, in an unbiased set of baits or genome-wide screenings. Furthermore, more sophisticated statistical evaluations of the raw can be adapted, i.e., using logistic regression approach that uses statistical and topological descriptors to predict the biological relevance of protein-protein interactions obtained from high-throughput screens as well as integrating known and predicted interactions from a variety of sources (Bader et al. 2004; von Mering et al. 2007).
False Negatives and Multiple-Variants of Y2H System
Although Y2H screens have been among the most powerful methods to detect binary protein-protein interactions, a limitation of the technology is the high incidence of false negative interactions (true interactions that are not detected) which is on the order of 70–90 % (Rajagopala et al. 2014; Yu et al. 2008). The interactome studies that have implemented proteome-scale Y2H screening in E. coli and yeast are shown to have identified 20–25 % of the PPIs (Rajagopala et al. 2014; Yu et al. 2008). In a previous studies, Rajagopala et al, have investigated underlying causes for this high degree of false negatives and uncovered that the structural constrains and expression levels of recombinant proteins play a major role (Rajagopala et al. 2009). Traditionally, Y2H screens have been performed using N-terminal fusion proteins of DNA-binding and activation domains. To mitigate the structural constrains Stellberger et al. constructed two new vectors that allows to make both C-terminal fusion proteins of DNA-binding and activation domains and showed that permutations of C- and N-terminal Y2H vectors detect different subsets of interactions (Stellberger et al. 2010). A study by Chen et al. benchmarked several variants of two-hybrid vectors (i.e., pGBGT7g-pGADCg, pGBGT7g-pGADT7g, pDEST32-pDEST22, pGBKCg-pGADT7g and pGBKCg-pGADCg) using a human positive reference set and a random reference set of protein-protein interaction pairs (Braun et al. 2009; Chen et al. 2010). In addition to each vector pair, they tested each protein both as activation (prey) and DNA-binding domain fusion (bait), including C-terminal fusions in pGBKCg and pGADCg. This way, they tested each protein pair in ten different configurations (Chen et al. 2010). This study clearly demonstrates that different Y2H variants (multiple-variants) detect markedly different subsets of interactions in the same interactome. All ten different configurations of bait-prey fusions were required to detect 73 of 92 interactions (79.3 %), whereas individual vector pairs detected only 23.3 out of 92 interactions (25.3 %) on average (Chen et al. 2010). Furthermore, recent studies demonstrate the general effectiveness of the multiple-variants of Y2H system in detecting true direct binary interactions and topology among the protein complex subunits (Rajagopala et al. 2012). Having multiple-variants of Y2H vectors that detect different subsets of interactions will be of great value to generate more comprehensive protein interactions data set, thus future interactome projects must adopt multiple Y2H vector systems with proper controls and adequate stringency.
Quality of Y2H Interaction Data
Like any other assay system, the two-hybrid system has the potential to produce false positives. The false positives may be of technical or biological nature. A “technical” false positive is an apparent two-hybrid interaction that is not based on the assembly of two hybrid proteins (that is, the reporter gene(s) gets activated without a protein–protein interaction between bait and prey). Frequently, such false positives are associated with bait proteins that act as transcriptional activators. Some bait or prey proteins may affect general colony viability and hence enhance the ability of a cell to grow under selective conditions and activate the reporter gene. Mutations or other random events of unknown nature may be invoked as potential explanations as well. A number of procedures have been developed to identify or avoid false positives, including the utilization of multiple reporters, independent methods of specificity testing, or simply retesting the interactions in a pairwise Y2H assays to make sure that the interaction is reproducible (Rajagopala et al. 2014; Yu et al. 2008; Koegl and Uetz 2007).
A biological false positive involves a true two-hybrid interaction with no physiological relevance. Those include the partners that can physically interact but that are never in close proximity to one another in the cell because of distinct subcellular localization or expression at different times during the life cycle. Examples may include paralogs that are expressed in different tissues or at different developmental stages. The problem is that the “false positive” nature can rarely be proven, as there may be unknown conditions under which these proteins do interact with a biological purpose. Overall, few technical false positives can be explained mechanistically, although many may simply do interact non-physiologically. While it often remains difficult to prove the biological significance of an interaction, many studies have attempted to validate them by independent methods. Validating an interaction by other methods certainly increases the probability that it is biologically significant. In a recent study, Rajagopala et al. assess the quality of Y2H interaction by evaluating 114 randomly selected Y2H interactions in two different methods, i.e., coimmunoprecipitation and luminescence-based mammalian interactome mapping (LUMIER) assays and confirmed ∼86 % of the Y2H interactions by at least one of these biochemical methods (Rajagopala et al. 2014). Similarly, when subsets of the large-scale human Y2H interactomes were evaluated about 65 % of them could be verified by independent orthogonal methods (Rual et al. 2005; Stelzl et al. 2005).
Integration of AP-MS and Y2H Data
It is important to note that affinity-purification followed by mass spectrometry (AP-MS) derived information about protein complexes does not provide information about the internal topology of multiprotein assemblies. Protein complexes are often interpreted as if the proteins that co-purify are interacting in a particular manner, consistent with either a spoke or matrix model (Goll and Uetz 2006). The yeast two-hybrid and other orthogonal assays detect direct binary interactions. Combination of both methods will give a better picture of protein complex topology and an experimentally derived confidence score for each interaction. In a recent study Rajagopala et al. compiled a list of 227 E. coli protein complexes that have three or more components as identified by AP-MS studies (Rajagopala et al. 2014). They identified the binary interactions between subunits of these complexes using proteome-scale Y2H data set and literature-curated binary interactions. Integrating these two data sets were able to map 745 binary interactions in 203 complexes, which deduce a putative complete internal topology for 15 multiprotein complexes. For another 45 complexes they determined the putative internal topology of a sub-complex with at least three subunits. However, even the combination of both methods is usually not sufficient to establish accurate topology as some interactions may be too weak to be detected individually.
Proteome-Scale Y2H Screening
Making an entire proteome library of an organism that can be screened in vivo under uniform conditions is a challenge. When proteins are screened on a genome scale, automated robotic procedures are necessary. The Y2H screening protocols described here have been extensively tested with human, yeast, bacterial, and viral proteins, but they can be applied to any other genome. Different high-throughput methods used to generate Y2H clones, i.e., proteins with AD fusions (preys) and the DBD fusions (baits), proteome-scale Y2H screening are included below. Usually, the processes starts with construction of the prey and bait libraries (Protocol 11.6.1–11.6.6); bait auto-activation tests (Protocol 11.6.7) followed by high-throughput array-based Y2H screening (Protocol 11.6.8) or pooled library screening (Protocol 11.6.9) including the selection of positives and identifying the interaction proteins by sequence (Protocol 11.6.11). Finlay, conducting the pairwise Y2H retests (Protocol 11.6.10) to make sure that the interactions are reproducible.
Materials
Yeast Media
YEPD liquid medium: 10 g yeast extract, 20 g peptone, 20 g glucose, dissolve in 1 L sterile water, and autoclave.
YEPDA liquid medium: 10 g yeast extract, 20 g peptone, 20 g glucose, dissolve in 1 L sterile water, and autoclave. After autoclaving, cool the medium to 60–70 °C, and then add 4 ml of 1 % adenine solution (see below).
YEPDA agar medium: 10 g yeast extract, 20 g peptone, 20 g glucose, 16 g agar, dissolve in 1 L sterile water and autoclave. After autoclaving, cool the medium to 60–70 °C, and then add 4 ml of 1 % adenine solution. Pour 40 ml into each sterile Omnitray plate (Nunc) under sterile hood, and let them solidify.
Medium concentrate: 8.5 g yeast nitrogen base, 25 g ammonium sulfate, 100 g glucose, 7 g dropout mix (see below). Make up to 1 L with sterile water, and filter-sterilize (Millipore).
Yeast Minimal Media
For 1 L of selective medium, autoclave 16 g agar in 800 ml water, cool the medium to 60–70 ° C, and then add 200 ml medium concentrate. Depending on the minimal media plates, the missing amino acids and/or 3AT (3-amino-1,2,4-triazole) solution should be added.
For media lacking tryptophan plates (-Trp): Add 8.3 ml leucine and 8.3 ml histidine from the amino acid stock solution (see below).
For media lacking leucine plates (-Leu): Add 8.3 ml tryptophan and 8.3 ml histidine solution from the amino acid stock solution (see below).
For media lacking tryptophan and leucine plates (-Leu –Trp): Add 8.3 ml histidine from the stock solution (see below).
For media lacking tryptophan, leucine, and histidine plates (-Leu –Trp –His): Nothing needs to be added.
For –Leu –Trp -His + 3 mM 3AT plates: Add 6 ml of 3AT (3-amino-1,2, 4-triazole, 0.5 M) to a final concentration of 3 mM.
Dropout mix (-His, -Leu, -Trp): Mix 1 g methionine, 1 g arginine, 2.5 g phenylalanine, 3 g lysine, 3 g tyrosine, 4 g isoleucine, 5 g glutamic acid, 5 g aspartic acid, 7.5 g valine, 10 g threonine, 20 g serine, 1 g adenine, and 1 g uracil and store under dry, sterile conditions.
- Amino acid stock solutions
- Adenine solution (1 %): Dissolve 10 g of adenine in 1 L, 0.1 M NaOH solution and sterile filter.
- Histidine solution (His): Dissolve 4 g of histidine in 1 L sterile water and sterile filter.
- Leucine solution (Leu): Dissolve 7.2 g of leucine in 1 L sterile water and sterile filter.
- Tryptophan solution (Trp): Dissolve 4.8 g of tryptophan in 1 L sterile water and sterile filter
Reagents for Yeast Transformation
Salmon sperm DNA (Carrier DNA): Dissolve 7.75 mg/ml salmon sperm DNA (Sigma) in sterile water, autoclave for 15 min at 121 °C, and store at −20 °C.
Dimethylsulfoxide (DMSO, Sigma).
Competent yeast strains, e.g., AH109 (for baits), and Y187 (for preys).
0.1 M Lithium acetate (LiOAc).
Yeast minimal media plates (depending on the selective markers on the yeast expression plasmid).
96PEG solution: Mix 45.6 g PEG (Sigma), 6.1 ml of 2 M LiOAc, 1.14 ml of 1 M Tris, pH 7.5, and 232 μl 0.5 M EDTA. Make up to 100 ml with sterile water and autoclave.
Plasmid clones (i.e., bait and prey clones)
Reagents for Bait Auto-Activation Test
YEPDA liquid medium.
–Trp –Leu (“–LT”) selective media agar plates (see Sect. 11.5.2).
Selective media agar plates without Trp, Leu, and His (“–LTH”), but with different concentrations of 3-AT, e.g., 0 mM, 1 mM, 3 mM, 10 mM, and 50 mM (–LTH/3-AT plates).
Bait strains that need to be tested and prey strains carrying the prey vector (empty vector), e.g., Y187 strain with pGADT7g plasmid.
Array-Based Y2H Screening (Work Station)
20 % (v/v) bleach (1 % sodium hypochlorite).
95 % (v/v) ethanol.
Single-well microtiter plate (e.g., OmniTray; Nalge Nunc) containing solid YEPD + adenine medium (see Sect. 11.5.1), –Leu –Trp, –His –Leu –Trp, and –His –Leu –Trp + different concentrations of 3AT.
384-Pin replicator for Beckman Biomek FX work station.
Bait liquid culture (DBD fusion-expression yeast strain).
Yeast prey array on solid YEPDA plates.
Reagents for Pairwise Y2H Retests
96-well microtiter plates (U- or V-shaped).
YEPDA medium and YEPDA agar in Omnitrays (Nunc).
Selective agar media plates (–LT, –LTH with 3-AT).
Prey yeast strain carrying empty prey plasmid, e.g., pGADT7g in Y187 strain.
Bait and prey strains to be retested.
Protocols
Construction of Y2H Libraries for Proteome-Scale Screening
After the set of proteins or entire ORFeome to be included in the systematic array or library is defined, the coding genes need to be cloned into several Y2H bait and prey expression vectors. In order to facilitate the cloning of a large number to proteins, site-specific recombination-based systems are commonly used (e.g., Gateway cloning, Fig. 11.4) (Walhout et al. 2000).
Fig. 11.4.

Generating Y2H baits and preys using Gateway cloning. The Gateway-based Y2H expression clones are made by sub-cloning the ORFs of interest from a Gateway entry vector (pDONR/zeo or pDONR201) into the Y2H expression vectors (Table 11.1) by Gateway LR reaction (Walhout et al. 2000). It is possible to simultaneously transfer a single ORF from an entry vector to four target vectors using a single LR reaction, as long as the resulting expression plasmids can be separated using prototrophic markers specific to each vector (Stanyon et al. 2003). We have used up to three Y2H expression vectors (i.e., pGADT7g, pGBGT7g, and pGBKCg, they carry Ampicillin, Gentamycin and Kanamycin resistance respectively) in a LR reaction
Gateway Cloning
Adapting Gateway (Life Technologies) technology provides a fast and efficient way of cloning the ORFs (Walhout et al. 2000). It is based on the site-specific recombination properties of bacteriophage lambda (Landy 1989); recombination is mediated between so-called attachment sites (att) of DNA molecules: between attB and attP sites or between attL and attR sites. In the first step of cloning the gene of interest is inserted into a specific Gateway entry vector by recombining a PCR product of the ORF flanked by attB sites with the attP sites of a pDONR vector (Life Technologies). The resulting entry clone plasmid contains the gene of interest flanked by attL recombination sites. These attL sites can be recombined with attR sites on a destination vector, resulting in a plasmid for functional protein expression in a specific host. For example, a Gateway entry clone (pDONR vector) can be subsequently cloned into multiple Y2H vectors (Table 11.1) and other Gateway compatible expression vectors as required.
Table 11.1.
Reagents for a yeast two-hybrid screen
| Bait and prey vectors | |||||||
|---|---|---|---|---|---|---|---|
| Gal4-fusion | Selection | ||||||
| Vector | Promoter | DBD | AD | Yeast | Bacterial | Ori | Source |
| pDEST22 | fl-ADH1 | – | N-term | Trp1 | Amp. | CEN | Life Technologies |
| pDEST32 | fl-ADH1 | N-term | – | Leu2 | Gent. | CEN | Life Technologies |
| pGBKT7g | t-ADH1 | N-term | – | Trp1 | Kan. | 2 μ | Uetz et al. (2006) |
| pGBGT7g | t-ADH1 | N-term | – | Trp1 | Gent. | 2 μ | Rajagopala et al. (2014) |
| pGBKCatg | t-ADH1 | C-term | Trp1 | Kan. | 2 μ | Rajagopala et al. (2014) | |
| pGADT7g | fl-ADH1 | – | N-term | Leu2 | Amp. | 2 μ | Uetz et al. (2006) |
| pGBKCg | t-ADH1 | C-term | – | Trp1 | Kan. | 2 μ | Stellberger et al. (2010) |
| pGADCg | fl-ADH1 | – | C-term | Leu2 | Amp. | 2 μ | Stellberger et al. (2010) |
| Yeast strains | |||||||
| Bait yeast strain | AH109 (Clontech) | ||||||
| Prey yeast strain | Y187 (Clontech) | ||||||
| Media and instruments | |||||||
| Yeast media | YEPDA, selective liquid media and agar plates | ||||||
| Pin tool | Optional but necessary when large number are tested | ||||||
Fl full-length, N/C-term. N/C-terminal (fusion), Amp. Ampicillin, Kan. Kanamycin, Gen. Gentamicin
ORFeome Collections
The starting point of a systematic proteome-scale Y2H screening is the construction of an ORFeome. An ORFeome represents all ORFs of a genome; in some cases selected gene set is individually cloned into Gateway entry vector. More and more ORFeomes are available and can be directly used for generating the Y2H bait and prey constructs. These ORFeome range from small viral genomes, e.g., KSHV and VZV (Uetz et al. 2006), Phages (Rajagopala et al. 2011), to several bacterial genomes such as E. coli, Helicobacter pylori, Bacillus anthracis or Yersinia pestis (Rajagopala et al. 2010). Clone sets of multicellular eukaryotes, e.g. C. elegans (Lamesch et al. 2004), human (Rual et al. 2004), or plant (Gong et al. 2004), have also been described. However, not all genes of interest are already available in entry vectors.
The Prey Array
The Y2H array is set up from an ordered set of AD-containing strains (preys). The prey constructs are assembled by transfer of the ORFs from entry vectors into specific prey vectors by recombination. Several prey vectors for the Gateway system are available. In our lab we primarily use the Gateway-compatible pGADT7g, pGADCg vectors, a derivative of pGADT7 (Clontech), and pDEST22 (Life Technologies) (Table 11.1). These prey constructs are transformed into haploid yeast cells using yeast transformation protocol (described in Protocol 11.6.6), e.g. the Y187 strain (mating type alpha) (Table 11.2). Finally, individual yeast colonies, each carrying one specific prey construct, are arrayed on agar plates in a 96- or 384-format usually as duplicates or quadruplicates.
Table 11.2.
Yeast strains and their genotypes
| Yeast strains | Genotypes |
|---|---|
| Y187 | MATα, ura3- 52, his3- 200, ade2- 101, trp1- 901, leu2- 3, 112, gal4Δ, met–, gal80Δ,URA3::GAL1UAS -GAL1TATA -lacZ (after Harper et al. 1993) |
| AH109 | MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1UAS-MEL1 TATA-lacZ (after James et al. 1996) |
The Bait Strains
Similar to prey construction, the bait clones are also constructed by recombination-based transfer of the ORFs into specific bait vectors. Bait vectors used in our lab are the Gateway technology compatible pGBGT7g, pGBKCg vectors, a derivative of pGADT7 vector (Clontech) and pDEST32 (Life Technologies) (Table 11.1). The bait constructs are also transformed into haploid yeast cells (Protocol 11.6.6), e.g. the AH109 strain (mating type a) (Table 11.2). After auto-activation testing, the baits can be tested for interactions against the Y2H prey array or pooled prey library. It is important to note that bait and prey must be transformed into yeast strains of opposite mating types to combine bait and prey plasmids by yeast mating and to co-express the recombinant proteins in diploids. Bait and prey plasmids can go into either mating type. However, this decision also depends on existing bait or prey libraries to which the new library may be screened later. Moreover, at least one of the haploid strains must contain a two-hybrid reporter gene (here, HIS3 gene under GAL4 promoter).
Yeast Transformation
This method is recommended for the high-throughput transformation of the bait or prey plasmid clones into corresponding yeast strains. This protocol is suitable for ∼1000 transformations; it can be scaled up and down as required. Selection of the transformed yeast cells requires synthetic media plates (leucine- or tryptophan-free agar media depending on the selective marker on the Y2H plasmid).
Prepare Competent Yeast Cells
Inoculate 250 ml YEPD liquid medium with freshly grown yeast strains on YEPD agar medium in a 1 L flask and grow in a shaker (shaking at 200 rpm) at 30 °C. Remove the yeast culture from the shaker when the cell density reaches OD 0.8–1. This usually takes 12–16 h.
Spin the cells at 2000 × g for 5 min at room temperature; pour off the supernatant.
Dissolve the cell pellet in 30 ml of LiOAc (0.1 M); make sure pellet is completely dissolved and there are no cell clumps.
Transfer the cells into a 50-ml Falcon tube and spin the cells at 2000 × g for 5 min at room temperature
-
Pour off the supernatant, and dissolve the cell pellet in 10 ml LiOAc (0.1 M).
Prepare the yeast transformation mix: Mixing the following components in a 200-ml sterile bottle:Component For 1000 reactions 96PEG 100 ml Salmon sperm DNA 3.2 ml DMSO 3.4 ml Add the competent yeast cells prepared above (steps 1–5) to the yeast transformation mix; shake the bottle vigorously by hand, or vortex for 1 min.
Pipette 100 μl of the yeast transformation mix into a 96-well plate (we generally use Costar 3596 plates) by using a robotic liquid handler (e.g., Biomek FX) or a multistep pipette.
Now add 25–50 ng of plasmid; keep one negative control (i.e., only yeast transformation mix).
Seal the 96-well plates with plastic or aluminum tape and vortex for 2–3 min. Care should be taken to seal the plates properly; vigorous vortexing might cause cross-contamination.
Incubate the plates at 42 °C for 30 min.
Spin the 96-well plate for 5 min at 2000 × g; discard the supernatant and aspirate by tapping on a cotton napkin a couple of times.
Wash the cell pellet with 150 μl sterile H2O
Spin the 96-well plate for 5 min at 2000 × g; discard the supernatant
Add 25 μl sterile H2O
Transfer 10 μl the cells to selective agar plate to select yeast with transformed plasmid (single-well Omnitrays from Nunc are well suited for robotic automation). As an alternative to the robotic automation, one can use a multichannel pipette to transfer the cells. Allow the yeast spots to dry on the plates.
Incubate at 30 °C for 2–3 days. Colonies start appearing after 24 h.
Bait Auto-activation Tests
Prior to the Y2H screening, the bait yeast strains should be examined for auto-activation (self-activation). Auto-activation is defined as detectable bait-dependent reporter gene activation in the absence of any prey interaction partner. Weak to intermediate strength auto-activator baits can be used in two-hybrid array screens because the corresponding bait–prey interactions confer stronger signals than the auto-activation background. In case of the HIS3 reporter gene, the self-activation background can be suppressed by titrating with 3-Amino-1,2,4-triazole(3-AT), a competitive inhibitor of HIS3. Auto-activation of all the baits is examined on selective plates containing different concentrations of 3-AT. The lowest concentration of 3-AT that suppresses growth in this test is used for the interaction screen (see below), because it avoids background growth while still detecting true interactions.
The aim of this test is to measure the background reporter activity of bait proteins in absence of an interacting prey protein. This measurement is used for choosing the selection conditions for the Y2H screening.
Bait strains are arrayed on a single-well Omnitray agar plate; usually standard 96-spot format.
The arrayed bait strains are mated with a prey strain carrying the empty prey plasmid, e.g., Y187 strain with pGADT7g. Mating is conducted according to the standard screening protocol as described in Protocol 11.6.8. Note that here an array of baits is tested whereas in a “real” screen (Protocol 11.6.8) an array of preys is tested.
After selecting for diploid yeast cells (on –LT agar), the cells are transferred to media selecting for the HIS3p reporter gene activity as described in Protocol 11.6.8. The -LTH transfer may be done to multiple plates with increasing concentrations of 3-AT. Recommended 3-AT concentrations for the –LTH plates are 0, 1, 3, 10, 25, and 50 mM.
These –LTH + 3-AT plates are incubated for 4–6 days at 30 °C. The auto-activation level of each of the bait is assessed and the lowest 3-AT concentration that completely prevents colony growth is noted. As this concentration of 3-AT suppresses reporter activation in the absence of an interacting prey, this 3-AT concentration is added to –LTH plates in the actual interaction screens as described in Protocol 11.6.8.
Screening for Protein Interactions Using a Protein Array
The Y2H prey array can be screened for protein interactions by a mating procedure that can be carried out using robotics (Biomek FX work station). A yeast strain expressing a single candidate protein as a DBD fusion is mated to all the colonies in the prey array (Fig. 11.2, step 1). After mating, the colonies are transferred to a diploid-specific medium, and then to the two-hybrid interaction selective medium.
A 384-pin replicating tool (e.g., High-Density Replication Tool; V&P Scientific) can be used to transfer the colonies form one agar plate to another and between the transfer steps, the pinning tool must be sterilized (described below).
Note that not all plastic ware is compatible with robotic devices, although most robots can be reprogrammed to accept different consumables. In the procedure described here, the prey array is gridded on 86 × 128 mm single-well microtiter plates (e.g., OmniTray, Nalge Nunc International) in a 384-colony format (see Fig. 11.2).
-
Sterilization: Sterilize a 384-pin replicator by dipping the pins in the sequential order into 20 % bleach for 20 s, sterile water for 1 s, 95 % ethanol for 20 s, and sterile water again for 1 s. Repeat this sterilization after each transfer.
Note 1: Immersion of the pins into these solutions must be sufficient to ensure complete sterilization. When automatic pinning devices are used, the solutions need to be checked and refilled occasionally (especially ethanol which evaporates faster than the others).
Day 1:
-
Preparing prey array for screening: Use the sterile replicator to transfer the yeast prey array from selective plates to single-well microtiter plates containing solid YEPD medium and grow the array overnight in a 30 °C incubator.
Note 2: Usually in a systematic array-based Y2H screening, duplicate or quadruplicate prey arrays are used. Ideally, the master prey array should be kept on selective agar plates. The master array should only be used to make “working” copies on YEPDA agar plates for mating. The template can be used for 1–2 weeks; after 2 weeks it is recommended to copy the array onto fresh selective agar plates. Preys and bait clones tend to lose the plasmid if stored on YEPDA for longer periods, which may reduce the mating and screening efficiency.
-
Preparing bait liquid culture (DBD fusion-expressing yeast strain): Inoculate 20 ml of liquid YEPD medium in a 250-ml conical flask with a bait strain and grow overnight in a 30 °C shaker
Note 3: If the bait strains are frozen, they are grown on selective agar medium plates and grown for 2–3 days at 30 °C. Baits from this plate are then used to inoculate the liquid YEPD medium. It is important to make a fresh bait culture for Y2H mating, as keeping the bait culture on reach medium (YEPD) for a long time may cause loss of plasmids. Usually we grow baits overnight for the screening.
Day 2:
-
3.
Mating procedure: Pour the overnight liquid bait culture into a sterile Omnitray plate. Dip the sterilized pins of the pin replicator (thick pins of ∼1.5 mm diameter should be used to pin baits) into the bait liquid culture and place directly onto a fresh single-well microtiter plate containing YEPDA agar media. Repeat with the required number of plates and allow the yeast spots to dry onto the plates.
-
4.
Pick up the fresh prey array yeast colonies with sterilized pins (thin pins of ∼1 mm diameter should be used to pin the preys) and transfer them directly onto the baits on the YEPDA plate, so that each of the 384 bait spots per plate receives different prey yeast cells (i.e., a different AD fusion protein). Incubate overnight at 30 °C to allow mating (Fig. 11.2, step 1).
Note 4: Mating usually take place in <15 h, but a longer period is recommended because some bait strains show poor mating efficiency. Adding adenine into the bait culture before mating increases the mating efficiency of some baits.
-
5.
Selection of Diploids: For the selection of diploids, transfer the colonies from YEPDA mating plates to diploids selection minimal media agar plates (–Leu –Trp plates) using the sterilized pinning tool (thick pins should be used in this step). Grow the plates for 2–3 days at 30 °C until the colonies are >1 mm in diameter (Fig. 11.2, step 2).
Note 5: This step is an essential control step to ensure successful mating because only diploid cells containing the Leu2 and Trp1 markers on the prey and bait vectors, respectively, will grow in this medium. This step also helps the recovery of the colonies and increases the efficiency of the next interaction selection step.
-
6.
Interaction selection: Transfer the diploid yeast cells from –Leu –Trp plates to interaction selection minimal media agar plates (–His –Leu –Trp plates), using the sterilized pinning tool. If the baits are auto-activating, they have to be transferred onto –His –Leu –Trp supplemented with a specific concentration of 3-AT plates (Protocol 11.6.1). Incubate the plates at 30 °C for 4–6 days.
-
7.
Score the interactions by looking for growing colonies that are significantly above background by size and are present as duplicate or quadruplicate colonies.
-
8.
Most two-hybrid-positive colonies appear within 3–5 days, but occasionally positive interactions can be observed later. Very small colonies are usually designated as background; however, the real positives signal should be compared with background vector control.
-
9.
Scoring can be done manually or using automated image analysis procedures. When using image analysis, care must be taken not to score contaminated colonies as positives.
Screening for Protein Interactions Using Pooled Libraries
Although the Y2H array screening ensures each pairwise combination in the library will be tested, it may be not feasible of large genomes, as the screening throughput increases exponentially with the genome size. Pooled library screening is an alternative strategy to significantly accelerate the screening. The prey clones are made by systematically cloning the ORFeome into Y2H vectors. The interacting preys in the library are identified by sequencing the Y2H positive yeast colonies (Protocol 11.6.11). Furthermore, to ensure the reproducibility of the interaction, all the interaction pairs will be subjected to Y2H pairwise retest (Protocol 11.6.10)
Day 1:
Preparing pooled prey libraries: The prey library strains (yeast) expressing each ORF is grown on a selective liquid medium (in 2 ml deep well plate) for 48 h in a 30 °C shaker. Equal amount of each of the freshly grown preys are combined into to a single pool. Ideally preys should be grown freshly for each batch of screening.
Inoculate the empty prey vector in 200 ml selective medium (Y2H negative control)
Preparing bait liquid culture (DBD fusion-expressing yeast strain): Inoculate 10 ml of selective medium (medium lacking tryptophan, depending on the selective marker on the bait plasmid) with bait fusion-expressing yeast strain and grow the yeast overnight in a 30 °C incubator.
Day 2:
-
4.
Mating procedure: Mix bait and prey at a 1:1 ratio, for example 4 OD bait (4 ml of OD = 1) and 4 OD prey (4 ml of OD = 1) culture in 15 ml Falcon tubes.
Note 6: The amount of (OD units) bait and prey used for the screening depends on the complexity of the prey library, in case of E. coli which contains about 4000 ORFs; we use 4 OD units of baits and preys. In case of human cDNA library screening we recommend using 12 OD units of baits and preys each
-
5.
For each of the bait include one negative control, i.e., mix bait and empty prey vector at a 1:1 ratio.
-
6.
Centrifuge the bait and prey solution for 2 min, at 3000 rpm at room temperature, discard supernatant
-
7.
Suspend the yeast pellet in 500 μl YPDA liquid medium, and plate on YEPDA agar plate (60 × 15 mm), and air dry the plates.
-
8.
Incubate the plates at 30 °C for 6 h or overnight at room temperature.
-
9.
After incubation, collect the cells by washing the plate with 2 ml of sterile water.
-
10.
Spin down the cells, remove the supernatant and wash the cells by adding 2 ml of sterile water
-
11.
Suspend the cells in 2 ml of selective medium (media lacking tryptophan, and leucine).
-
12.
Plate 500–1000 μl on the interaction selective agar plates –Leu –Trp -His supplemented with predefined concentration of 3-AT, based on auto-activation test of the bait. The remaining sample can be stored at 4 °C for 4–6 days for further use.
Note 7: This step is an essential control step to ensure successful Y2H mating procedure, because only diploid cells containing the Leu2 and Trp1 markers on the prey and bait vectors, respectively, will grow on media lacking tryptophan, and leucine medium. This step also helps the recovery of the colonies and increases the efficiency of the next interaction selection step. To measure the diploids make an aliquot of 1:100 dilution of the sample (step 11) and plate the cells on –Leu –Trp plates, the screening depth in millions, should be > 0.1 million, up to 1 million diploids in case of E. coli library screening. This is at least twenty times the number of library size.
-
13.
Interaction selection: Incubate the –Leu –Trp -His + 3-AT for 4–6 days at 30 °C until the colonies are ∼1 mm in diameter.
-
14.
Two-hybrid positives: The interaction selection plates that show colony growth but no colonies on control plates (bait mated to empty prey vector) are the two-hybrid positive yeast clones. If the control plates show even few colonies the diploids should be plated on selective plates with higher concentration of 3-AT.
-
15.
Identity of interacting preys: The positive yeast colonies are picked either manually or using robotics and subjected to yeast colony PCR (Protocol 11.6.11), followed by DNA sequencing to identify the preys.
Pairwise Y2H Retesting
A major consideration when using the Y2H system is the number of false positives, particularly in the pooled library screening. The major sources for false positives are non-reproducible signals that arise through little-understood mechanisms. Thus, pairwise retesting can identify most of the false positives. We routinely use at least duplicate tests, although quadruplicates should be used if possible (Fig. 11.2). Retesting is done by mating the interaction pair to be tested and by comparing the activation strength of this pair with the activation strength of a control, usually the bait mated with the strain that contains the empty prey vector. Testing the reproducibility of an interaction greatly increases the reliability of the Y2H data.
Re-array bait and prey strains of each interaction pair to be tested into 96-well microtiter plates. Use separate 96-well plates for baits and preys. For each retested interaction, fill one well of the bait plate and one corresponding well of the prey plate with 150 μl selective liquid medium (media lacking Leucine or Tryptophan).
For each retested interaction, inoculate the bait strain into a well of the 96-well plate and the prey strain at the corresponding position of the 96-well prey plate, for example, bait at position B2 of the bait plate and prey at position B2 of the prey plate. In addition, inoculate the prey strain with the empty prey vector (e.g., strain Y187 with plasmid pGADT7g) into 20 ml selective liquid medium.
Incubate the plates overnight at 30 °C.
-
Mate the baits grown in the bait plate with their corresponding preys in the prey plate. In addition, mate each bait with the prey strain carrying an empty prey vector as a background activation control. The mating is done as described in Protocol 11.6.8, using the bait and prey 96-well plates directly as the source plates.
Note 8: First the baits are transferred from their 96-well plate to two YEPDA plates (interaction test and control plate) using a 96-well replication tool. Let the plate dry for 10–20 min. Then transfer the prey’s from their 96-well plate onto the first YEPDA plate and the empty prey vector control strain onto the second YEPDA plate
The transfers to selective plates and incubations are done as described in Protocol 11.6.8. As before, test different baits with different activation strengths on a single plate and pin the diploid cells onto –LTH plates with different concentrations of 3-AT. For choosing the 3-AT range, the activation strengths (Protocol 11.6.7) serve as a guideline.
After incubating for 4–6 days at 30 °C on –LTH/3-AT plates, the interactions are scored; positive interactions show a clear colony growth at a certain level of 3-AT, whereas no growth should be seen in the control (bait mated with empty vector strain).
Yeast Colony PCR and Sequencing Sample Preparation
Yeast Colony PCR
This protocol is designed to amplify the insert of the preys or baits in the two-hybrid positive yeast clones, using primers that bind to the upstream and downstream region of the insert. The PCR is optimized for 30 μl reaction; the total volume of the reaction can be scaled up and down as required.
Pick the yeast colony from interaction selective plate into 100 μl of sterile H2O, in 96 well plate; store the plate at −80 freezer for longer storage.
Take a new 96 well PCR plate and pipette 5 unit of zymolyase (1 μl) enzyme to each well.
Add 9 μl of above yeast (step 1), and incubate at 30 °C for 60 min.
After incubation, add 20 μl PCR master mix with forward and reverse primers specific to prey or bait vector used in the Y2H screening.
Run PCR cycles as recommended by the enzyme (polymerase) provider.
After PCR, load 5 μl of PCR reaction into agarose gel to check PCR amplicons.
Purification of the PCR Amplicons for Sequencing
To clean up PCR products before sequencing, the PCR reaction is subjected to the exonuclease I, which removes leftover primers while the Shrimp Alkaline Phosphatase (SAP) removes the dNTPs
Spin the Yeast colony PCR plate at 2000 rpm for 3 min (to sediment yeast debris).
Pipette 8 μl of PCR sample without touching the bottom yeast debris, into new PCR plate.
- Make the SAP master mix by mixing the following reagents
SAP master mix Components 100 samples 10× SAP buffer 50 μl Water 890 μl SAP (1 U/μl) 50 μl Exonuclease I (10 U/μl) 10 μl Add 10 μl of SAP master mix to 8 μl of PCR sample.
Incubate in the thermocycler as follows: 37 °C for 60 min, 72 °C for 15 min, then put on hold at 4 °C.
Use the sample for DNA sequencing using primers specific to prey or bait vector.
Acknowledgements
Work on this chapter was funded by NIH grant RO1GM79710.
Contributor Information
Nevan J. Krogan, PhD, Email: Nevan.Krogan@ucsf.edu
Mohan Babu, PhD, Email: mohanbabu_r@yahoo.com.
Seesandra Venkatappa Rajagopala, Email: rajgsv@gmail.com.
References
- Bader JS, Chaudhuri A, Rothberg JM, Chant J. Gaining confidence in high-throughput protein interaction networks. Nat Biotechnol. 2004;22(1):78–85. doi: 10.1038/nbt924. [DOI] [PubMed] [Google Scholar]
- Bennett ST, Barnes C, Cox A, Davies L, Brown C. Toward the 1,000 dollars human genome. Pharmacogenomics. 2005;6(4):373–382. doi: 10.1517/14622416.6.4.373. [DOI] [PubMed] [Google Scholar]
- Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6(1):91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent R, Ptashne M. A eukaryotic transcriptional activator bearing the DNA specificity of a prokaryotic repressor. Cell. 1985;43(3 Pt 2):729–736. doi: 10.1016/0092-8674(85)90246-6. [DOI] [PubMed] [Google Scholar]
- Cagney G, Uetz P, Fields S. Two-hybrid analysis of the Saccharomyces cerevisiae 26S proteasome. Physiol Genomics. 2001;7(1):27–34. doi: 10.1152/physiolgenomics.2001.7.1.27. [DOI] [PubMed] [Google Scholar]
- Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, Vidal M, Kieff E, Johannsen E. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A. 2007;104(18):7606–7611. doi: 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Rajagopala SV, Stellberger T, Uetz P. Exhaustive benchmarking of the yeast two-hybrid system. Nat Methods. 2010;7(9):667–668. doi: 10.1038/nmeth0910-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chassey B, Navratil V, Tafforeau L, Hiet MS, Aublin-Gex A, Agaugue S, Meiffren G, Pradezynski F, Faria BF, Chantier T, Le Breton M, Pellet J, Davoust N, Mangeot PE, Chaboud A, Penin F, Jacob Y, Vidalain PO, Vidal M, Andre P, Rabourdin-Combe C, Lotteau V. Hepatitis C virus infection protein network. Mol Syst Biol. 2008;4:230. doi: 10.1038/msb.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester C, Uetz P. The FF domains of yeast U1 snRNP protein Prp40 mediate interactions with Luc7 and Snu71. BMC Biochem. 2008;9:29. doi: 10.1186/1471-2091-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estojak J, Brent R, Golemis EA. Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol. 1995;15(10):5820–5829. doi: 10.1128/MCB.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340(6230):245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415(6868):141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, Vijayadamodar G, Pochart P, Machineni H, Welsh M, Kong Y, Zerhusen B, Malcolm R, Varrone Z, Collis A, Minto M, Burgess S, McDaniel L, Stimpson E, Spriggs F, Williams J, Neurath K, Ioime N, Agee M, Voss E, Furtak K, Renzulli R, Aanensen N, Carrolla S, Bickelhaupt E, Lazovatsky Y, DaSilva A, Zhong J, Stanyon CA, Finley RL, Jr, White KP, Braverman M, Jarvie T, Gold S, Leach M, Knight J, Shimkets RA, McKenna MP, Chant J, Rothberg JM. A protein interaction map of Drosophila melanogaster. Science. 2003;302(5651):1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Goll J, Uetz P. The elusive yeast interactome. Genome Biol. 2006;7(6):223. doi: 10.1186/gb-2006-7-6-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Shen YP, Ma LG, Pan Y, Du YL, Wang DH, Yang JY, Hu LD, Liu XF, Dong CX, Ma L, Chen YH, Yang XY, Gao Y, Zhu D, Tan X, Mu JY, Zhang DB, Liu YL, Dinesh-Kumar SP, Li Y, Wang XP, Gu HY, Qu LJ, Bai SN, Lu YT, Li JY, Zhao JD, Zuo J, Huang H, Deng XW, Zhu YX. Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol. 2004;135(2):773–782. doi: 10.1104/pp.104.042176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805–816. doi: 10.1016/0092-8674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- Hauser R, Ceol A, Rajagopala SV, Mosca R, Siszler G, Wermke N, Sikorski P, Schwarz F, Schick M, Wuchty S, Aloy P, Uetz P. A second-generation protein-protein interaction network of Helicobacter pylori. Mol Cell Proteomics. 2014;13(5):1318–1329. doi: 10.1074/mcp.O113.033571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, Stelzl U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45(4):567–580. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415(6868):180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98(8):4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144(4):1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Avramova L, Huang J, Hazbun T. A yeast two-hybrid smart-pool-array system for protein-interaction mapping. Nat Methods. 2007;4(5):405–407. doi: 10.1038/nmeth1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Ramm EI, Pabo CO. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci U S A. 2000;97(13):7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, Pfeiffenberger E, Porras P, Raghunath A, Roechert B, Orchard S, Hermjakob H (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res 40(Database issue):D841–D846. doi: 10.1093/nar/gkr1088 [DOI] [PMC free article] [PubMed]
- Khadka S, Vangeloff AD, Zhang C, Siddavatam P, Heaton NS, Wang L, Sengupta R, Sahasrabudhe S, Randall G, Gribskov M, Kuhn RJ, Perera R, LaCount DJ (2011) A physical interaction network of dengue virus and human proteins. Mol Cell Proteomics 10(12):M111 012187. doi: 10.1074/mcp.M111.012187 [DOI] [PMC free article] [PubMed]
- Koegl M, Uetz P. Improving yeast two-hybrid screening systems. Brief Funct Genomic Proteomic. 2007;6(4):302–312. doi: 10.1093/bfgp/elm035. [DOI] [PubMed] [Google Scholar]
- Lacomble S, Portman N, Gull K. A protein-protein interaction map of the Trypanosoma brucei paraflagellar rod. PLoS One. 2009;4(11):e7685. doi: 10.1371/journal.pone.0007685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamesch P, Milstein S, Hao T, Rosenberg J, Li N, Sequerra R, Bosak S, Doucette-Stamm L, Vandenhaute J, Hill DE, Vidal M. C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions. Genome Res. 2004;14(10B):2064–2069. doi: 10.1101/gr.2496804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landy A. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem. 1989;58:913–949. doi: 10.1146/annurev.bi.58.070189.004405. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memisevic V, Zavaljevski N, Pieper R, Rajagopala SV, Kwon K, Townsend K, Yu C, Yu X, DeShazer D, Reifman J, Wallqvist A. Novel Burkholderia mallei virulence factors linked to specific host-pathogen protein interactions. Mol Cell Proteomics. 2013;12(11):3036–3051. doi: 10.1074/mcp.M113.029041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JR, Yu J, Liu G, Hines JA, Chan JE, Mangiola BA, Zhang H, Pacifico S, Fotouhi F, DiRita VJ, Ideker T, Andrews P, Finley RL., Jr A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007;8(7):R130. doi: 10.1186/gb-2007-8-7-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, Chemama Y, Labigne A, Legrain P. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409(6817):211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- Rajagopala SV, Titz B, Goll J, Parrish JR, Wohlbold K, McKevitt MT, Palzkill T, Mori H, Finley RL, Jr, Uetz P. The protein network of bacterial motility. Mol Syst Biol. 2007;3:128. doi: 10.1038/msb4100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Hughes KT, Uetz P. Benchmarking yeast two-hybrid systems using the interactions of bacterial motility proteins. Proteomics. 2009;9(23):5296–5302. doi: 10.1002/pmic.200900282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Yamamoto N, Zweifel AE, Nakamichi T, Huang HK, Mendez-Rios JD, Franca-Koh J, Boorgula MP, Fujita K, Suzuki K, Hu JC, Wanner BL, Mori H, Uetz P. The Escherichia coli K-12 ORFeome: a resource for comparative molecular microbiology. BMC Genomics. 2010;11:470. doi: 10.1186/1471-2164-11-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Casjens S, Uetz P. The protein interaction map of bacteriophage lambda. BMC Microbiol. 2011;11:213. doi: 10.1186/1471-2180-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Sikorski P, Caufield JH, Tovchigrechko A, Uetz P. Studying protein complexes by the yeast two-hybrid system. Methods. 2012;58(4):392–399. doi: 10.1016/j.ymeth.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Sikorski P, Kumar A, Mosca R, Vlasblom J, Arnold R, Franca-Koh J, Pakala SB, Phanse S, Ceol A, Hauser R, Siszler G, Wuchty S, Emili A, Babu M, Aloy P, Pieper R, Uetz P. The binary protein-protein interaction landscape of Escherichia coli. Nat Biotechnol. 2014;32(3):285–290. doi: 10.1038/nbt.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raquet X, Eckert JH, Muller S, Johnsson N. Detection of altered protein conformations in living cells. J Mol Biol. 2001;305(4):927–938. doi: 10.1006/jmbi.2000.4239. [DOI] [PubMed] [Google Scholar]
- Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, Kamburov A, Ghiassian SD, Yang X, Ghamsari L, Balcha D, Begg BE, Braun P, Brehme M, Broly MP, Carvunis AR, Convery-Zupan D, Corominas R, Coulombe-Huntington J, Dann E, Dreze M, Dricot A, Fan C, Franzosa E, Gebreab F, Gutierrez BJ, Hardy MF, Jin M, Kang S, Kiros R, Lin GN, Luck K, MacWilliams A, Menche J, Murray RR, Palagi A, Poulin MM, Rambout X, Rasla J, Reichert P, Romero V, Ruyssinck E, Sahalie JM, Scholz A, Shah AA, Sharma A, Shen Y, Spirohn K, Tam S, Tejeda AO, Trigg SA, Twizere JC, Vega K, Walsh J, Cusick ME, Xia Y, Barabasi AL, Iakoucheva LM, Aloy P, De Las RJ, Tavernier J, Calderwood MA, Hill DE, Hao T, Roth FP, Vidal M. A proteome-scale map of the human interactome network. Cell. 2014;159(5):1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A, Li N, Rosenberg J, Lamesch P, Vidalain PO, Clingingsmith TR, Hartley JL, Esposito D, Cheo D, Moore T, Simmons B, Sequerra R, Bosak S, Doucette-Stamm L, Le Peuch C, Vandenhaute J, Cusick ME, Albala JS, Hill DE, Vidal M. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 2004;14(10B):2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437(7062):1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Schwartz H, Alvares CP, White MB, Fields S. Mutation detection by a two-hybrid assay. Hum Mol Genet. 1998;7(6):1029–1032. doi: 10.1093/hmg/7.6.1029. [DOI] [PubMed] [Google Scholar]
- SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci U S A. 1996;93(16):8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139(7):1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanyon CA, Limjindaporn T, Finley RL., Jr Simultaneous cloning of open reading frames into several different expression vectors. Biotechniques. 2003;35(3):520–522. doi: 10.2144/03353st05. [DOI] [PubMed] [Google Scholar]
- Stellberger T, Hauser R, Baiker A, Pothineni VR, Haas J, Uetz P. Improving the yeast two-hybrid system with permutated fusions proteins: the Varicella Zoster Virus interactome. Proteome Sci. 2010;8:8. doi: 10.1186/1477-5956-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksoz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122(6):957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Titz B, Rajagopala SV, Goll J, Hauser R, McKevitt MT, Palzkill T, Uetz P (2008) The binary protein interactome of Treponema pallidum--the syphilis spirochete. PLoS One 3(5):e2292. doi: 10.1371/journal.pone.0002292 [DOI] [PMC free article] [PubMed]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403(6770):623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, Haas J. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311(5758):239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- Vidal M, Endoh H. Prospects for drug screening using the reverse two-hybrid system. Trends Biotechnol. 1999;17(9):374–381. doi: 10.1016/S0167-7799(99)01338-4. [DOI] [PubMed] [Google Scholar]
- Vidal M, Legrain P. Yeast forward and reverse ‘n’-hybrid systems. Nucleic Acids Res. 1999;27(4):919–929. doi: 10.1093/nar/27.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Brunn A, Teepe C, Simpson JC, Pepperkok R, Friedel CC, Zimmer R, Roberts R, Baric R, Haas J. Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS One. 2007;2(5):e459. doi: 10.1371/journal.pone.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7--recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35(Database issue):D358–D362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/S0076-6879(00)28419-X. [DOI] [PubMed] [Google Scholar]
- Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol. 2012;30(2):159–164. doi: 10.1038/nbt.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M (2008) High-quality binary protein interaction map of the yeast interactome network. Science (New York, NY) 322(5898):104–110. doi: 10.1126/science.1158684 [DOI] [PMC free article] [PubMed]
- Yu H, Tardivo L, Tam S, Weiner E, Gebreab F, Fan C, Svrzikapa N, Hirozane-Kishikawa T, Rietman E, Yang X, Sahalie J, Salehi-Ashtiani K, Hao T, Cusick ME, Hill DE, Roth FP, Braun P, Vidal M. Next-generation sequencing to generate interactome datasets. Nat Methods. 2011;8(6):478–480. doi: 10.1038/nmeth.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]