

Abstract
Small molecules targeting the enzymes responsible for human immunodeficiency virus (HIV) maturation, DNA synthesis and its subsequent chromosomal integration as ribonucleotide-free double-stranded DNA remain the mainstay of combination antiretroviral therapy. For infected individuals harboring drug-susceptible virus, this approach has afforded complete or near-complete viral suppression. However, in the absence of a curative strategy, the predictable emergence of drug-resistant variants requires continued development of improved antiviral strategies, inherent to which is the necessity of identifying novel targets. Regulatory elements that mediate transcription, translation, nucleocytoplasmic transport, dimerization, packaging and reverse transcription of the (+) strand RNA genome should now be considered viable targets for small molecule, peptide- and oligonucleotide-based therapeutics. Where target specificity and cellular penetration and toxicity have been the primary obstacle to successful “macromolecule therapeutics”, this chapter summarizes (a) novel approaches targeting RNA motifs whose three-dimensional structure is critical for biological function and consequently may be less prone to resistance-conferring mutations and (b) improved methods for delivery.
Keywords: Human Immunodeficiency Virus, Human Immunodeficiency Virus Replication, Frameshift Site, External Guide Sequence, Dimer Initiation Sequence
Introduction
Seminal studies by Rosen et al. in 1985 designated a region at the immediate 5′ terminus of the human immunodeficiency virus type 1 (HIV-1) RNA genome the trans-acting response, or TAR element. This cis-acting motif, in conjunction with the viral trans-activator of transcription (Tat) protein, significantly enhanced transcription of the HIV-1 genome via modification of the transcription complex (Karn 1999; Peterlin and Price 2006). Shortly thereafter, a second HIV accessory protein, the regulator of expression of virion proteins (Rev) whose interaction with another cis-acting RNA (the Rev responsive element or RRE) regulated nucleocytoplasmic export of full-length and singly-spliced viral RNAs was reported (Feinberg et al. 1986). Surprisingly, more than 25 years after the crucial contribution of the Tat/TAR and Rev/RRE axes to HIV-1 replication was demonstrated, these RNAs, the viral and host proteins with which they interact, and the resulting nucleoprotein complexes have not proven therapeutically accessible, based primarily on a lack of selectivity of small molecules and inefficient delivery of larger therapeutic oligonucleotides. The discovery of numerous new classes of RNAs and their function in a variety of biological processes has revolutionized molecular biology, which undoubtedly will have profound implications for clinical sciences. The application of “ macromolecule therapeutics” in other fields, while still their in fledgling stage, is exemplified by:
Splice-Switching Oligonucleotides (SSOs). Alternative splicing enables a single pre-messenger RNA to support multiple protein isoforms, thereby increasing the diversity of the proteome. While essential for normal development, aberrant splicing underlies a significant number of human diseases, and methods for manipulating alternative splicing could thus have significant therapeutic value. As an example, modified antisense oligonucleotides that re-direct splice site selection of Bcl-x pre-mRNA from Bcl-xL to -xS were demonstrated to induce apoptosis in breast and prostate cancer cells, while a second nanoparticle-delivered SSO reduced tumor load in lung metastases (Bauman et al. 2010).
Steric Blocking Oligonucleotides. Myotonic dystrophy is characterized by expansion of triplet repeats of the mRNA (typically CAG and CUG) from 5 to 35 copies, in healthy individuals, to as many as 2500 in the disease state. In myotonia, the expanded CUG repeat-containing transcript binds and sequesters the splicing factor muscleblind-like protein 1, rendering it unavailable for splicing of several pre-mRNAs, including the chloride channel protein 1 (CLCN1), resulting in muscle hyperexcitability (Lee and Cooper 2009). In a mouse model, Wheeler et al. (2009) demonstrated that injecting a phosphorodiamidate morpholino oligonucleotide (PMO) complementary to the CUG repeat displaced muscleblind-like protein 1, corrected CNCL1 pre-mRNA splicing, resulting in restoration of transmembrane chloride channel conductance and a marked reduction in myotonia.
External Guide Sequence (EGS) Oligonucleotides. EGS oligonucleotides are designed to bind their mRNA target and assume a structure that is recognized and degraded by RNase P (a ubiquitous endonuclease that matures the termini of tRNAs), thereby impairing translation. Jiang et al. have used the EGS strategy to block human cytomegalovirus (HCMV) gene expression by targeting the viral protease-specifying mRNA for degradation (Jiang et al. 2011), while the clinical implications of an EGS comprised of a (PMO) backbone have been suggested by effectiveness against antibiotic-resistant gram negative bacteria (Wesolowski et al. 2011).
RNA Interacting Polynucleotides (RIPtides). Microarrays allowing multiple candidate ligand sequences to be evaluated in parallel should provide an invaluable platform for oligonucleotide-based RNA targeting. Early studies of small libraries (300–400) combining 2′-O-methyl ribonucleotides and locked nucleic acids (Kierzek 2009) have been superseded by the development of 2′-O-methyl RIPtide arrays bearing the four natural nucleobases, varying from 4- to 8-m in length (Gude et al. 2012), which provide almost 90,000 individual probes. The RIPtide approach has been successfully used to identify oligonucleotides that inhibit human telomerase function both in vitro and in cultured cells (Gude et al. 2012).
Translation Suppressing Oligonucleotides (TSOs). PMOs described above represent a class of 2′-O-substituted ribonucleotides that are not recognized by host RNasesH or the RNA-induced silencing complex, and thus do not promote target degradation. These attributes can be exploited by TSOs in that their binding at or near the initiation codon will have the consequence of antagonizing ribosome binding and translation of the target protein. A more detailed description of these approaches can be found in excellent reviews on RNA therapeutics by Kole et al. (2012), Burnett and Rossi (2012).
Knowledge accumulated for over the last three decades for regulatory HIV-1 RNA motifs would therefore be expected to create new modalities for therapeutic intervention, comprising small molecules, polypeptides, inhibitory RNAs, and possibly combinations thereof. Examples of “genome targets” are briefly reviewed in the following section.
RNA Control of HIV Replication
As our understanding of the HIV (+) strand RNA genome organization has progressed, multiple regulatory elements dispersed throughout the viral genome (Fig. 1) that have been identified and subject to extensive examination can now be considered as therapeutically accessible.
Primer binding site (pbs). Complementarity between this 18-nt sequence preceding the unique 5′ (U5) region and the 3′ terminal nucleotides of the tRNA replication primer (tRNALys,3 in the case of HIV-1 and HIV-2), mediate primer binding, thereby defining the initiation site for (−) strand, RNA-dependent DNA synthesis (Abbink and Berkhout 2008a; Le Grice 2003).
Primer activation signal (PAS). Located upstream of the pbs, an interaction of the PAS and “anti-PAS” TΨC nucleotides of the tRNA primer have been shown to play an important role in regulating tRNA-primed initiation of reverse transcription (Beerens et al. 2001; Huthoff et al. 2003).
Dimer initiation site (DIS). Together with the dimerization linkage sequence (DLS) the DIS promotes dimerization of the viral RNA genome prior to its packaging (Laughrea and Jette 1994; Skripkin et al. 1994).
Packaging signal (Ψ). Interaction of Ψ with the Gag-encoded nucleocapsid protein (NC) is critical to incorporation of the dimeric RNA genome into assembling virions (L’Hernault et al. 2007; Lever 2007).
Major splice donor site (MSD), used to generate all subgenomic spliced transcripts (Abbink and Berkhout 2008b).
Gag/Pol frameshift sites. This heptanucleotide “slippery” sequence, in combination with an adjacent RNA hairpin, promote the −1 ribosomal frameshifting event required for low-frequency translation of the Gag/Pol polyprotein (Brakier-Gingras et al. 2012).
Polypurine tracts (PPTs). Located in the center (cPPT) and 3′ region of the genome (3′ PPT), these all-purine ribonucleotide tracts, in the context of the RNA/DNA replication intermediate, are resistant to reverse transcriptase (RT)-associated ribonuclease H (RNase H) hydrolysis and critical for priming (+) strand, DNA-dependent DNA synthesis (Rausch and Le Grice 2004).
Splice acceptor sites, present at several positions on the genome, participate in the production of several spliced viral transcripts (Purcell and Martin 1993).
Polyadenylation signal, used to generate the 3′ end of the viral RNA (Wilusz 2013).
Fig. 1.
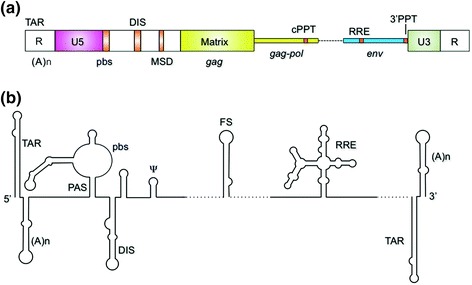
a Schematic representation of the HIV-1 RNA genome. Cis-acting signals: TAR, transactivation response element; (A)n, poly A; R, repeat sequence, U5, unique 5′ sequence; pbs, primer binding site; DIS, dimer initiation sequence; MSD, major splice donor; cPPT, central polypurine tract; RRE, Rev response element; 3′ PPT, 3′ polypurine tract; U3, unique 3′ sequence. b Proposed secondary structure of cis-acting signals. PAS, primer activation sequence; Ψ, psi, or encapsidation sequence; FS, frameshift sequence
The following sections will outline recent advances that have been made in small molecule, polypeptide and oligonucleotide-based targeting of these cis-acting elements, while a later section will deal with the general issues of their cellular uptake, specificity and stability.
Transcriptional Regulation and More: The Transactivation Response (TAR) Element
Nucleotides 1–59 at the 5′ terminus of the viral genome define the TAR hairpin (Fig. 2a), whose apical loop and nearby trinucleotide bulge (Fig. 2b) serve as a binding site for the virus-coded Tat protein. This interaction, in conjunction with the cellular co-factor transcription elongation factor-b (P-TEFb), significantly enhances transcription elongation (Karn 1999). As such, targeting the Tat/TAR axis to interrupt the virus life cycle at the level of transcription can be considered. However, additional roles of TAR suggest it could also be targeted to affect multiple steps of reverse transcription. Since the TAR and poly(A) hairpins collectively constitute the repeat (R) region, they are represented at the 5′ and 3′ termini of the (+) strand viral RNA, and likely play a role in (−) strand DNA transfer, which exploits R homology. Mutagenesis studies (Berkhout et al. 2001) have shown that structural features of the TAR hairpin facilitate (−) strand DNA transfer, while Huthoff and Berkhout (2001) have demonstrated that mutations in the same region affect the equilibrium between the alternate configurations of the 5′ UTR (designated the long distance base pairing and branched conformations) that regulate tRNA-primed initiation of reverse transcription. A role for TAR in initiation of reverse transcription has also been proposed (Harrich et al. 2000). Thus, while the mechanistic basis for these observations requires further experimentation, they highlight a potentially pleiotropic role for the Tat/TAR axis that should be exploited therapeutically.
Fig. 2.
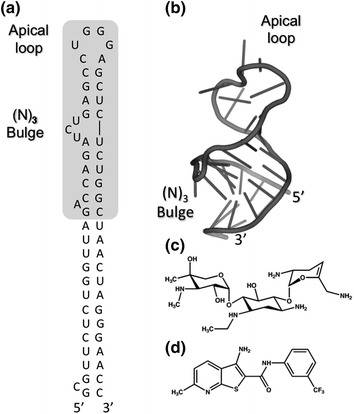
a, b Secondary and tertiary structures of the HIV-1 TAR hairpin, respectively. The full-length hairpin is depicted in (a), within which shorter version used for NMR spectroscopy is shaded. Nucleotides of the apical loop and trinucleotide (N3) bulge constitute the primary Tat binding site. c, d TAR binding small molecules identified by computational molecular dynamics (c, Stelzer et al. 2011) and high throughput screening of small molecule microarrays (d, Sztuba-Solinska et al. 2014), respectively
In view of the role of the highly conserved TAR apical loop in transactivation and stimulation of transcription, the notion of disrupting the Tat/TAR interaction with steric blocking oligonucleotides has been investigated by Arzumanov et al. who found that several classes of modified, dodecameric, nucleotides efficiently antagonized Tat-dependent transcription in vitro in HeLa cell nuclear extracts (Arzumanov et al. 2001). Despite this promising result, in vivo efficacy was not observed. However, subsequent studies have been encouraging, showing that polyamide nucleic acid analogs (PNAs) designed to interact with the TAR apical loop and adjacent bulged nucleotides substantially inhibited Tat-mediated transactivation in vitro system and, when transfected into CEM cells, lead to a significant drop in virus infectivity (Kaushik et al. 2002).
Polypeptide analogs, or peptidomimetics, that antagonize the Tat/TAR interaction represent an alternative therapeutic strategy. Although Lee et al. have elegantly demonstrated inhibition of HIV-1 replication in MT-4 cells at non-toxic concentrations (Lee et al. 2005), the conformational flexibility of linear polypeptides had the drawback of promoting off-target interactions and sensitivity to proteolysis. These drawbacks can, however, be overcome by the construction of “stapled” or conformationally-constrained peptides. First described in 2004 as a means of activating apoptosis to target leukemia (Walensky et al. 2004), stapling of native peptides represents a novel therapeutic strategy by modulating protein-protein interactions. Using a similar strategy, Varani and colleagues (Lalonde et al. 2011) have designed a series of cyclic, cell-permeable peptides that competitively inhibit the Tat/TAR interaction in vivo at sub-micromolar concentrations with minimal cytotoxicity. Surprisingly, but in keeping with the pleiotropic nature of the Tat/TAR axis, time-of-addition experiments indicated that, in addition to viral transcription, these cyclic peptides also acted at the level of reverse transcription. Resistance of branched peptide (BP) ligands to proteases and peptidases has promoted their use as a novel class of multivalent non-toxic, cell-permeable therapeutics. These features have been exploited to create a class of peptides that bound TAR RNA with low to sub-micromolar affinity (Bryson et al. 2012), most likely spanning nucleotides of both the apical loop and neighboring trinucleotide bulge.
Although requiring experimental validation, the potential of such therapeutic peptides might be enhanced by designing bifunctional “catalytic” inhibitors linking a targeting and effector moiety. Amino-terminal Cu(II)/Ni(II) binding motifs, or ATCUNs, are small, high affinity metal binding sites located at the N-terminus of many naturally occurring proteins, and have been shown to cleave DNA, modify RNA and inactivate proteins via release of reactive oxygen species (Jin et al. 2007). The ATCUN motif features a three amino acid sequence, X1-X2-His, of which X1 and X2 can be any amino acid and X1 must have a free amino terminal group. This approach would require simple modification of linear peptides and, while the cyclic peptides afford no free N-terminus, appending the tripeptide to a lysine side chain is feasible, thereby creating a “metallo-peptide” capable of targeting TAR and promoting its nucleolytic degradation. A later section addresses application of the metal chelate-peptide strategy as a means of targeting the HIV-1 RRE.
Finally, small molecules targeting the TAR hairpin have for the larger part been aminoglycosides that, while active, have generally suffered from a lack of specificity, binding to 16S and 18S rRNAs, tRNA and several catalytic RNAs (Luedtke et al. 2003; Blount et al. 2005). However, two recent studies have been encouraging. By combining NMR spectroscopy and computational molecular dynamics, Stelzer et al. have taken advantage of virtual screening to identify several compounds that antagonize the Tat/TAR interaction by interacting with nucleotides of both the apical loop and trinucleotide bulge (Fig. 2c) (Stelzer et al. 2011). Our own work has involved the construction of small molecule microarrays that can be probed with fluorescently-labeled RNA structural motifs, such as the TAR apical loop. Using this approach (Sztuba-Solinska et al. 2014), we have identified a novel chemotype (Fig. 2d) that binds the TAR hairpin with micromolar affinity, inhibits HIV-1 replication in culture and is not cytotoxic. While in a developmental stage, small molecule microarrays hold immense potential for targeting additional structural motifs in the HIV-1 genome.
Reverse Transcription: Initiation of (−) Strand DNA Synthesis
Retroviral minus (−) strand RNA-dependent DNA synthesis initiates from a host-coded tRNA hybridized to the primer binding site (pbs) located immediately downstream of the unique sequence of the 5′ UTR (U5). Beyond pbs-mediated base pairing, additional intermolecular tRNA-viral RNA interactions have been proposed as critical mediators of the HIV-1 initiation process. These include interactions between (i) an the A-rich loop of the U5-IR hairpin and the U-rich tRNALys,3 anticodon domain (ii), nucleotides of the U5-IR stem, designated the primer activation signal or PAS, with those of the tRNA TΨC stem and (iii) nucleotides of the unique 3′ region (U3) with those of the tRNA anticodon stem (Abbink and Berkhout 2008a). Thus, from a steric interference perspective, the tRNA/viral RNA duplex offers several potential therapeutic targets, an example of which was the ability of a short 2′-O-methyl labeled oligonucleotide directed to the pbs to competitively inhibit tRNALys,3 binding in vitro and HIV-1 replication in HeLa cells (Freund et al. 2001).
Curiously, although a considerable body of literature is available for the structure of the tRNA/viral RNA complex (Lanchy et al. 2000) kinetic characterization of tRNA-primed DNA synthesis (Lanchy et al. 1996) and the conformational dynamics of HIV-1 RT associated with these events (Liu et al. 2010), the mechanism whereby the tRNA primer is incorporated into the budding virion and subsequently hybridized to the pbs has, until only recently, received little attention. The interaction of human lysyl tRNA synthetase (LysRS) with the capsid (CA) domain of the Gag precursor (via its C-terminal domain or CTD) was identified by Kleiman and colleagues, whose related studies elegantly showed that disrupting LysRS synthesis reduced tRNALys,3 incorporation into virions its and annealing to the viral RNA genome (Guo et al. 2003). With the goal of disrupting LysRS/CA interactions, combinatorial library screening, Dewan et al. (2012) have subsequently identified two cyclic peptides (whose clinical application takes advantage of their enhanced resistance to proteolytic degradation) that are active in vitro in the low micromolar range. While the in vivo selectivity of this first-in-class LysRS/CA antagonists has not be demonstrated, it paves the way for development of modified cyclic peptides with improved cell penetration and pharmacokinetic properties.
Genome Dimerization: The Dimer Initiation Sequence (DIS)
Dimerization of the retroviral genome is mediated through the DIS palindromic sequence located between the primer binding and encapsidation sites (Fig. 3), disruption of which induces pleiotropic effects that severely impair infectivity (Paillart et al. 2004). The absolute requirement for this cis-acting signal, for which high resolution crystallographic data for the DIS “kissing” and extended duplex are available, promoted construction of a neamine dimer (Fig. 3d) as a novel small molecule class that targeted the DIS interface by interacting with both RNA strands (Bodlenner et al. 2007). In vitro analysis has confirmed that such compounds indeed targeted the DIS loop and stabilized the kissing complex, although their cellular toxicity and ability to inhibit virus replication remains to be established.
Fig. 3.
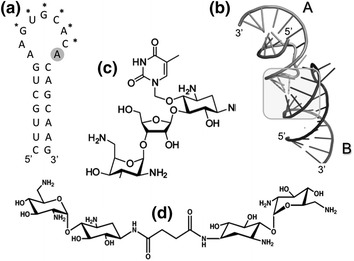
a Sequence and secondary structure of the DIS hairpin of HIV-1 subtypes A and F. Asterisks indicate nucleotides of the DIS palindrome. b Tertiary structure of the DIS-induced dimer. The region of dimerization is indicated by the shaded box. c, d structures of small moelcules targeting the DLS (c) Thymine-neomycin conjugate (Ennifar et al. 2013) whose targeting to the DIS is mediated through Watson-Crick base-pairing with A280 (shaded in a). d Linked neamine dimer (Bodlenner et al. 2007)
Crystallographic data of kissing loop/aminoglycoside complexes has illustrated a critical interaction of DIS residue A280 with loop 1 of the aminoglycoside neomycin (Fig. 3a). Ennifar and colleagues have taken advantage of this information to targeted the RNA dimer with a thymine-containing aminoglycoside chimera (Fig. 3c) capable of hydrogen bonding with A280 (Ennifar et al. 2013). Binding to the DIS kissing loop was confirmed by isothermal titration calorimetry and supported by the inability of their analog to interact with an A280U mutant predicted to disrupt Watson-Crick base pairing. Although the affinity for this thymine-neomycin chimera for the DIS was moderate (K d = 5.3 μM), an encouraging observation was its ability, in contrast to previous aminoglycosides, to interact with the DIS of multiple HIV-1 subtypes without any significant change in affinity. Although antiviral activity remains to be established, this work provides an encouraging example of the potential for structure-based drug design to direct the development of dimerization antagonists with improved potency and selectivity.
Virion Packaging and Assembly: The Encapsidation Site
Despite extensive mutagenesis and structural probing efforts, there has been little consensus on structures within the 5′ UTR responsible for interacting with the dimeric viral genome and its subsequent trafficking to the plasma membrane for encapsidation into the budding virion. However, a landmark NMR study of a dimeric RNA containing the entire 5′ UTR and the immediate 5′ region of gag has provided an elegant model that proposes the -G-G-A-G- tetraloop of the packaging signal (Ψ, defined by stem-loop (SL) 3 of the core HIV-1 packaging domain) is recognized by the NC domain of the Gag precursor through an RNA structural switch linking translation, dimerization and packaging (Lu et al. 2011) (Fig. 4a, b). A subsequent study (Stephenson et al. 2013), has coupled chemical probing data of the 5′ UTR with through-space distances derived from single molecule fluorescence resonance energy transfer experiments. This analysis, while supporting models derived by NMR, highlighted a kink-turn motif whose mutagenesis was previously shown to impair packaging, suggesting it may provide a Gag binding site. Together, these complementary, high-resolution techniques have provided a much-needed structural basis for antiviral strategies designed to interrupt genome packaging, possibly antagonizing the proposed Gag-mediated structural switch from translation of the viral genome to its incorporation into the budding virion.
Fig. 4.
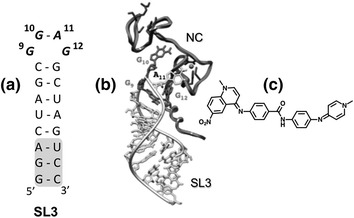
a Secondary structure of the SL3 hairpin defining the HIV-1 encapsidation signal. Nucleotides enclosed within the grey box were added to stabilize SL3 for NMR spectroscopy. G9, G10, and G12 indicate guanines of the SL3 tetraloop (designating the 5′ G as nucleotide 1). b 3-D model of the HIV-1 NC−SL3 complex. The NC protein is depicted in dark grey and the RNA hairpin in light grey. Guanines 9, 10 and 12, which are essential for NC binding, are indicated. c Structure of NSC260594, which antagonizes the SL3-NC interaction. Modified from Bell et al. (2013)
Using a combination of computational docking and high throughput screening to identify ligands selective for GNRA tetraloops, Warui et al. identified several small molecules that bound SL-3 RNA (Warui and Baranger 2012). Two of these ligands bound with micromolar affinity, displayed enhanced selectivity for Ψ-containing RNA over non-specific single- and double-stranded RNA, and disrupted the interaction with HIV-1 NC protein as measured by gel electrophoretic mobility shift analysis. Bell et al. have designed a high-throughput fluorogenic assay that monitors Gag-mediated disruption of the Ψ-containing SL-3 RNA and used this to screen two small molecule libraries totaling ~2600 compounds (Bell et al. 2013). In addition to chemotypes previously shown to interact with DNA, these authors identified the quinolinium derivative NSC260594 (Fig. 4c) that in culture significantly reduced virus infectivity in the low micromolar range. 1H-NMR spectroscopy of SL-3 in the presence of NSC260594 indicated little to no influence on stem nucleotides, while in contrast, significant perturbation of nucleotides comprising the -G-G-A-G- tetraloop was observed in the presence of a molar equivalent of the ligand, indicating a 1:1 stoichiometry. An equivalent analysis demonstrated that the non-discriminatory intercalator ellipticine interacted exclusively with nucleotides of the SL-3 stem, providing a promising measure of NSC260594 specificity. The notion that an important interaction of Gag with viral RNA might be the targeted by this small molecule can be inferred from 1H-NMR studies (De Guzman et al. 1998), which have shown that the SL-3 -G-G-A-G- tetraloop provides a tight binding site for HIV-1 NC. Finally, targeting Ψ RNA may not be restricted to small molecules, evidenced by 1H-NMR analysis demonstrating that a short, tryptophan-rich polypeptide (proposed as an NC mimic) also interacts with the -G-G-A-G- tetraloop (Dietz et al. 2008).
Protein Synthesis and Ribosomal Frameshifting: A Delicate Balance
Ribosomal frameshifting provides a mechanism in most retroviruses through which a −1 frameshift during translation controls the transition between high level synthesis of structural proteins, encoded on the Gag open reading frame, to low-level synthesis of enzyme-containing proteins of the Gag-Pol precursor polyprotein (Jacks et al. 1988). The frameshift signal of HIV-1 group M is illustrated in Fig. 5a, b, comprising a heptanucleotide ‘slippery’ sequence followed by the frameshift stimulatory signal, an irregular helix whose upper and lower stem are separated by a 3-nucleotide purine bulge (a feature maintained within an individual subtype, while varying between subtypes). Although the precise mechanism underlying frameshifting remains open to debate, maintaining the correct Gag:Gag-Pol ratio appears critical to virion assembly and maturation. Small molecules or nucleic acids that interact with the frameshift signal and alter this critical balance through its disruption or stabilization would therefore seem an attractive therapeutic strategy.
Fig. 5.
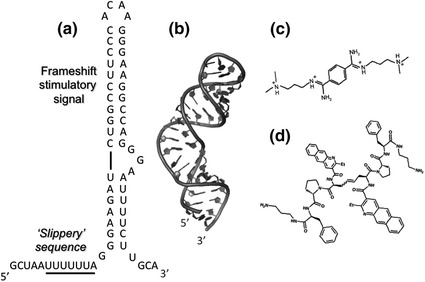
Secondary (a) and tertiary structure (b) of the HIV-1 frameshift sequence that maintains the ~20:1 ratio of Gag:Gag/pol proteins, comprising the slippery sequence and frameshift stimulatory signal. c Structure of the frameshift stimulator Bis[N-(3 dimethylaminopropyl)amidino]benzene tetrahydrochloride (RG501). Modified from Marcheschi et al. (2011). d Compound 4, a frameshift stimulator that inhibits HIV-1 replication (Ofori et al. 2014)
High throughput screening of a library of ~50,000 chemical entities by Hung et al. (1998) indicated that the benzene derivative RG501 (Fig. 5c) was capable of increasing HIV-1 frameshift efficiency approximately two-fold and impairing virus spread in culture. However, RG501 was shown to interact with a variety of RNA helices, suggesting toxicity issues would arise as a consequence of preferential targeting of ribosomal RNA. Despite the negative outcome, Marscheschi et al. (2011) have demonstrated by NMR spectroscopy that RG501 binds in the major groove of the frameshift signal and, by hydrogen bonding to phosphate groups on opposite sides, alters the conformation of the -G-G-A- trinucleotide bulge, providing important structural data for the rational design of derivatives with increased potency and selectivity. Although modification of the basic screening strategy (Dulude et al. 2008) yielded a series of arginine-rich peptides capable of reducing frameshift efficiency, these appeared to be equally non-selective. More encouraging results have been reported by Ofori et al. (2014), who have investigated the effect of nucleic acid-intercalating peptide antibiotics on ribosomal frameshifting. Among these, a benzo [g] quinolone-containing analog (Fig. 5d) bound the frameshift site with an affinity of ~100 nM, and with increased selectivity over non-specific RNA. This analog was cell permeable, exhibited minimal cytotoxicity, and decreased virus infectivity, which correlated with increased levels of frameshifting. In parallel with the search for improved small molecule ligands, the potential of oligonucleotide-based inhibition, via steric blocking, should not be overlooked, based on studies by Ahn et al. (2011a) who demonstrated that PNAs designed to hybridize to the frameshift site suppressed replication of the severe acute respiratory syndrome coronavirus, while a second class that targets the three major stem-loop (SL) domains of X-RNA inhibited RNA synthesis initiation in hepatitis C virus (Ahn et al. 2011b).
Nucleocytoplasmic RNA Transport: The Rev Response Element (RRE)
Early attempts to target the HIV-1 RRE, which is responsible for nucleocytoplasmic transport of unspliced and singly-spliced viral RNAs, have involved small molecules, such as neomycin B and related aminoglycosides which, as outlined earlier, have suffered from lack of specificity and poor cellular uptake (Ahn et al. 2011b). Likewise, trans-dominant negative Rev mutants, while a promising alternative, led to rapid acquisition of mutations conferring resistance (Legiewicz et al. 2008). Thus, new modalities of targeting the Rev/RRE interaction are warranted. Both structural and mutagenesis analyses have shown that the α-helical arginine-rich motif (ARM) of Rev mediates a critical interaction with the RRE by inserting into the major groove of an asymmetric bulge in stem-loop IIB. Based on these observations, Mills et al. (2006) synthesized a series of ARM-like conformationally-constrained peptidomimetics to antagonize the Rev/RRE interaction. Of 15 candidate peptidomimetics examined, one bound the RRE with equivalent affinity as the ARM (K d ~50 nM), while control experiments indicated that its unconstrained counterpart showed little specificity.
Targeting the RRE and its subsequent irreversible inactivation with reactive metal chelates appended to the Rev ARM has been proposed by Cowan and colleagues as a strategy of developing “catalytic” metallo-inhibitors that theoretically would not be required in saturating amounts to achieve maximum potency. Such bifunctional metallo-inhibitors combine a high affinity targeting motif (the Rev ARM) with a metal chelate complex capable of damaging RNA in the vicinity of the ARM binding site via a variety of oxidative chemistries. Proof of this strategy was demonstrated by the ability of Cu++-Gly-Gly-His-ARM complexes to selectively cleave the RRE both in vitro (Jin and Cowan 2006) and in vivo (Jin and Cowan 2007). This approach has more recently been extended to investigate the oxidative properties of the Rev ARM when linked to the metal chelators 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), diethylenetriamineepentaacetic acid (DTPA), ethylenediaminetetraacetic acid (EDTA) and nitrilotriacetic acid (NTA) (Joyner and Cowan 2013) (Fig. 6). For each of these complexes, high affinity binding to RRE stem-loop IIB was retained (varying from 0.2 to 16 nM versus 1.6 nM for the unmodified peptide). The Cu++-bound metal chelates of each were found to be most efficient in RRE cleavage, with activity varying 15-fold in the order Cu-NTA-Rev > Cu-DOTA-Rev > Cu-DTPA-Rev > Cu-EDTA-Rev, representing a ~50-fold improvement over the Cu++-Gly-Gly-His-ARM complex. In a biological setting, oxidative damage of the RRE would result in dissociation of the metal chelate-ARM complex, which can be “reactivated” by reducing agents such as ascorbic acid or glutathione, the concentration of which is sufficiently high in vivo. Also, since the K d for the bound metal is of the order of 10−15 M, toxicity resulting from its leaching from the complex is likely to be minimal.
Fig. 6.
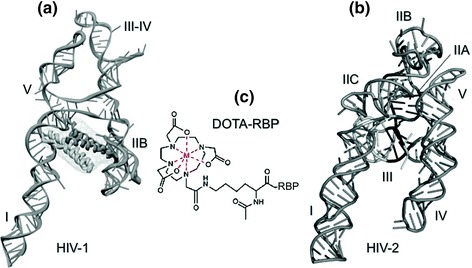
a, b 3D representations of the A-like topology assumed by the RREs of HIV-1 and HIV-2, respectively. In a, the proposed binding site for the Rev arginine-rich motif (ARM) is indicated. Designations I–V represent the individual stem-loops. c Schematic representation of DOTA-RBP, a bifunctional metallopeptide combining the Rev binding peptide (RBP) with a metal-chelating center (DOTA) that can be induced to release reactive oxygen species
Branched peptides, comprising either natural or unnatural amino acids (or combinations thereof) are gaining attention as therapeutic agents, based on their potential for multivalent targeting of RNA, as well as their resistance to proteolysis (Bryson et al. 2012). High throughput screening of an ~50,000 branched peptide boronic acid (BPBA) library, (whose boronic acid substituent was designed to mimic an acceptor for RNA 2′ OH groups as well as improve selectivity for RNA over DNA) identified a candidate peptide (BPBA1) that bound RRE stem-loop IIB with micromolar affinity (Zhang et al. 2013) and a 1:1 stoichiometry. Significant losses in binding affinity with linear peptides (~50-fold) suggested that all “arms” of this branched peptide participated in its interaction with stem-loop IIB. A complementary analysis with stem-loop IIB variants provided strong support for the notion that the correct tertiary structure of the target RNA was a prerequisite to high affinity BPBA1 binding. Consistent with these findings, enzymatic footprinting highlighted several regions of stem-loop IIB that were rendered nuclease-insensitive in the presence of the branched peptide. Finally, studies with fluorescent derivative suggest that BPBA1 can be taken up by cells, although its ability to suppress HIV-1 replication remains to be established.
Lastly, the availability of a high resolution structure of the RRE, which at a two-dimensional level comprises a collection of hairpins, loops and bulges, should promote novel strategies that target additional, unique pockets as sites for either small molecule or macromolecule therapeutics. In this respect, our recent efforts have provided working models for the RREs of HIV-2 (Fig. 6b) (Lusvarghi et al. 2013) and subsequently HIV-1 (Fig. 6a) (Fang et al. 2013). As illustrated in Fig. 6, although the structural organization of individual domains may differ, both RREs appear to assume an unusual “A-like” topology. For the HIV-1 RRE, the legs of this structure, whose ~55A separation has been proposed as necessary for RRE function, are suggested to constitute binding tracks for cooperative binding of Rev. These structures will hopefully serve as targets for new generations of molecular scaffolds.
Polypurine Tract Primers of Plus Strand DNA Synthesis
HIV (+)-strand DNA synthesis initiates from the 3′ terminus of polypurine tracts (PPTs) located adjacent to the U3 region of the 3′ LTR and in the center of the genome (cPPT). While the role for the cPPT has been somewhat controversial, mutational studies (Hu et al. 2010) have raised the intriguing notion that, perhaps in conjunction with the central termination sequence (CTS), the cPPT is less relevant to nuclear import than to minimize exposure of unpaired (−) strand DNA to host restriction factors such as the family of APOBEC proteins. These issues notwithstanding, accurate selection of the 3′ and cPPT primers from the RNA/DNA replication intermediate and their removal from nascent (+) strand DNA represent critical steps in the viral replication cycle.
Despite extensive mutagenesis analysis (Rausch and Le Grice 2007), the structural basis for resistance of PPT-containing RNA/DNA hybrids to RNase H-mediated cleavage remains unclear. However, structural studies that combined mass spectrometry with NMR (Turner et al. 2008), raise the possibility that the PPT could be targeted by small molecules designed to antagonize its selection or removal from nascent (+) DNA. Electrospray ionization-Fourier transfer ion cyclotron resonance mass spectrometry has the particular advantage of allowing multiplexing, i.e. the interaction of nucleic acid ligands with several PPT variants could be simultaneously investigated, based on their mass signatures. Using this approach, Turner et al. showed that short PPT-containing RNA/DNA hybrids contained two binding sites for the aminoglycoside neomycin B (Turner et al. 2008). Tandem mass spectrometry (which essentially provided a “footprint” of the PPT/neomycin B complex) demonstrated that, at a 1:1 neomycin B:PPT ratio, the aminoglycoside bound at the PPT/U3 junction. Increasing this ratio to 2:1 identified the second, upstream aminoglycoside binding site at the 5′ extremity of the PPT (Fig. 7). Consistent with these observations, 1H-NMR of a 1:1 neomycin B:PPT hybrid showed shifts in imino proton signals at positions corresponding to the PPT/U3 junction and evidence for a second binding site as the ratio was increased. Subsequent NMR analysis in the presence of HIV-1 RT indicated contact between protein and nucleic acid in the same two regions, while equivalent findings with RT from the LTR-retrotransposon Ty3 and its cognate PPT (Brinson et al. 2009) suggested that structural features at either extremity of the PPT may be a more general feature contributing to its recognition. Such data are not intended to imply the therapeutic use of neomycin B, which is known to bind to a variety of RNA structures, but rather to illustrate that more selective nucleic acid ligands could be developed to disrupt (+) strand DNA synthesis.
Fig. 7.
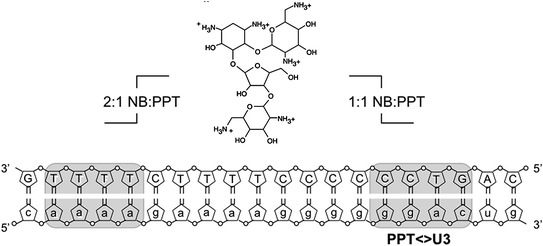
Aminoglycoside targeting of the HIV-1 PPT RNA/DNA hybrid. Nucleotides of the PPT RNA and its DNA complement are represented in small and capital letters, respectively. Shaded portions of the RNA/DNA hybrid indicate the binding sites for neomycin B (NB, Center) at a 1:1 and 2:1 NB-RNA/DNA ratio. Modified from Turner et al. (2008)
An intriguing means by which the HIV PPT might be targeted in the context of the single-stranded RNA genome, the RNA/DNA intermediate, or the integrated double-stranded DNA involves the use of triplex forming oligonucleotides (TFOs), which bind to the oligopurine strand of the duplex through Hoogsteen or reverse Hoogsteen hydrogen bonds. Studies by Volkmann and Moelling have demonstrated that, in vitro, a TFO targeted against the PPT rendered the PPT/U3 junction refractory to RNase H-mediated hydrolysis and, as a consequence, inhibited initiation of (+) strand DNA synthesis (Volkmann et al. 1995). In order to determine whether the PPT sequence of the single-stranded viral RNA genome and not the RNA/DNA replication intermediate might also respond to triplex formation, these authors targeted the single-stranded PPT with a TFO wherein the Watson-Crick and Hoogsteen base pairing sequence were on the same strand and separated by a short linker sequence. This “sandwich” TFO, which was suggested to be thermodynamically more efficient than the three-stranded system, also rendered the PPT refractory to RNase H-mediated cleavage in vitro, and inhibited de novo HIV infection in culture. Finally, although the HIV RNA genome is the focus of this review, it is worth noting that Giovannangeli et al. have used the TFO approach to target the HIV-1 PPT in the context of the integrated proviral DNA. Using a TFO-psoralen conjugate, these authors successfully converted the non-covalent three-stranded structure into a localized covalent lesion on genomic DNA following UV irradiation of cells (Giovannangeli et al. 1997).
Delivery Strategies
As the concept of targeting the HIV RNA genome with small molecules, peptides and oligonucleotides is gaining popularity, their ability to penetrate the cell membrane and, for peptides and nucleic acids, access their target without initiating an immune response presents a significant challenge. A promising advance in this area has been the development of short cell-penetrating peptides (CPPs), comprising combinations of natural or unnatural amino acids, the basis of which was the original observation that HIV-1 Tat could enter cells when added to the culture medium. In addition to the arginine-rich Tat peptide, other natural CPPs have been identified, including penetratin, herpesvirus tegument protein VP22 and inv3 of Mycobacterium tuberculosis, each of which has the capacity to promote intracellular uptake of conjugated cargoes (Copolovici et al. 2014). As proof-of-principle, Roisin et al. (2004) have shown that a Tat-derived CPP, linked to peptide ligands, antagonized the interaction of Rev with cellular factors critical to nucleocytoplasmic RNA transport, inhibiting HIV-1 replication in both primary lymphocytes and macrophages.
Although the application of steric blocking and splice-switching oligonucleotides as antagonists of HIV replication has been limited, CPP-conjugated oligonucleotides have retained their attraction as therapeutic agents. Oligonucleotides containing peptide nucleic acid (PNA) locked nucleic acid (LNA) or phosphorodiamidate morpholino (PMO) analogs are most commonly used in order to avoid complications arising through interactions between negatively charged nucleic acid and the positively charged CPP. Following their uptake, endosomal release of CPP-oligonucleotide complexes presents the next challenge. Inhibitory CPP-PNA conjugates targeted to the TAR hairpin could be released from endosomes by chloroquine treatment which, while clinically impractical, demonstrated endosomal release as a rate limiting step (Turner et al. 2005). Alternative strategies to promote endosomal release have included the identification of CPPs that themselves elicit endosomolytic activity and CPP modification via stearylation, myristoylation or incorporation of chloroquine-functionalized dendrons on the backbone. A more comprehensive review of CPP-directed oligonucleotide delivery, its challenges and future potential can be found in Lee et al. (2013).
Finally, while the efficacy small molecules targeting cis-acting sequences on the RNA genome might currently be limited due to poor cellular uptake, CPP-mediated delivery of doxorubicin, methotrexate and paclitaxel (Stewart et al. 2008) provides encouraging evidence for further exploitation of this novel technology.
Conclusions and Perspectives
Although development of a cure for HIV is a desirable goal, until this is achieved, antiviral therapies will remain the mainstay for inhibiting virus replication and spread of AIDS. With respect to the viral RNA genome, technological advances have provided the first glimpses the complete HIV-1 genome structure, highlighting a complex collection of inter- and intramolecular interactions that are critical mediators of its replication, as well as its nucleocytoplasmic transport, translation, dimerization and packaging. Many of these complexes are now being dissected at the atomic level by a variety of biophysical approaches, including X-ray crystallography, NMR spectroscopy, small angle X-ray scattering and cryo-EM tomography. Such successes have collectively provided us with a wealth of RNA–RNA and RNA–protein interactions that can be antagonized to disrupt HIV replication, in the form of small molecules, peptides and therapeutic RNAs. The development of novel, cost-effective fluorescence-based high-throughput screening methodologies recapitulating these interactions should be an encouragement to accelerate our “assault” on the HIV-1 RNA genome and complement ongoing efforts to target RNA for the treatment of a multitude of disease states.
Acknowledgments
I would like to thank Jennifer T. Miller for critical reading of the manuscript. This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Social Services.
Abbreviations
- ARM
Arginine-rich motif
- CA
Capsid
- DIS
Dimerization initiation site
- HIV
Human immunodeficiency virus
- PAS
Primer activation signal
- ppt
Polypurine tract
- Ψ
Packaging element
- Rev
Regulator of expression of virion proteins
- RRE
Rev response element
- RT
Reverse transcriptase
- SL
Stem-loop
- TAR
Trans-activation response element
- Tat
Trans-activator of transcription
- U3
Unique 3′ sequence
- U5
Unique 5′ sequence
- UTR
Untranslated region
Footnotes

The HIV gag polyprotein (A, shown in red) is translated from the HIV RNA genome (in yellow) by cellular ribosomes (B). A stem-loop structure in the genome (C) induces a frame shift roughly 5 % of the time, producing the longer gag-pol protein (D)
Contributor Information
Bruce E. Torbett, Email: betorbet@scripps.edu
David S. Goodsell, Phone: +1858784-2839, Email: goodsell@scripps.edu
Douglas D. Richman, Phone: 858552-8585-7439, Email: drichman@ucsd.edu
Stuart F. J. Le Grice, Email: legrices@mail.nih.gov
References
- Abbink TE, Berkhout B. HIV-1 reverse transcription initiation: a potential target for novel antivirals? Virus Res. 2008;134(1–2):4–18. doi: 10.1016/j.virusres.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Abbink TE, Berkhout B. RNA structure modulates splicing efficiency at the human immunodeficiency virus type 1 major splice donor. J Virol. 2008;82(6):3090–3098. doi: 10.1128/JVI.01479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn DG, Lee W, Choi JK, Kim SJ, Plant EP, Almazan F, Taylor DR, Enjuanes L, Oh JW. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res. 2011;91(1):1–10. doi: 10.1016/j.antiviral.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn DG, Shim SB, Moon JE, Kim JH, Kim SJ, Oh JW. Interference of hepatitis C virus replication in cell culture by antisense peptide nucleic acids targeting the X-RNA. J Viral Hepatitis. 2011;18(7):e298–e306. doi: 10.1111/j.1365-2893.2010.01416.x. [DOI] [PubMed] [Google Scholar]
- Arzumanov A, Walsh AP, Rajwanshi VK, Kumar R, Wengel J, Gait MJ. Inhibition of HIV-1 Tat-dependent trans activation by steric block chimeric 2′-O-methyl/LNA oligoribonucleotides. Biochemistry. 2001;40(48):14645–14654. doi: 10.1021/bi011279e. [DOI] [PubMed] [Google Scholar]
- Bauman JA, Li SD, Yang A, Huang L, Kole R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010;38(22):8348–8356. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Groot F, Berkhout B. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J Biol Chem. 2001;276(33):31247–31256. doi: 10.1074/jbc.M102441200. [DOI] [PubMed] [Google Scholar]
- Bell NM, L’Hernault A, Murat P, Richards JE, Lever AM, Balasubramanian S. Targeting RNA-protein interactions within the human immunodeficiency virus type 1 lifecycle. Biochemistry. 2013;52(51):9269–9274. doi: 10.1021/bi401270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Vastenhouw NL, Klasens BI, Huthoff H. Structural features in the HIV-1 repeat region facilitate strand transfer during reverse transcription. RNA. 2001;7(8):1097–1114. doi: 10.1017/S1355838201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount KF, Zhao F, Hermann T, Tor Y. Conformational constraint as a means for understanding RNA-aminoglycoside specificity. J Am Chem Soc. 2005;127(27):9818–9829. doi: 10.1021/ja050918w. [DOI] [PubMed] [Google Scholar]
- Bodlenner A, Alix A, Weibel JM, Pale P, Ennifar E, Paillart JC, Walter P, Marquet R, Dumas P. Synthesis of a neamine dimer targeting the dimerization initiation site of HIV-1 RNA. Org Lett. 2007;9(22):4415–4418. doi: 10.1021/ol701760k. [DOI] [PubMed] [Google Scholar]
- Brakier-Gingras L, Charbonneau J, Butcher SE. Targeting frameshifting in the human immunodeficiency virus. Expert Opin Ther Targets. 2012;16(3):249–258. doi: 10.1517/14728222.2012.665879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinson RG, Turner KB, Yi-Brunozzi HY, Le Grice SF, Fabris D, Marino JP. Probing anomalous structural features in polypurine tract-containing RNA-DNA hybrids with neomycin B. Biochemistry. 2009;48(29):6988–6997. doi: 10.1021/bi900357j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson DI, Zhang W, McLendon PM, Reineke TM, Santos WL. Toward targeting RNA structure: branched peptides as cell-permeable ligands to TAR RNA. ACS Chem Biol. 2012;7(1):210–217. doi: 10.1021/cb200181v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copolovici DM, Langel K, Eriste E, Langel U (2014) Cell-penetrating peptides: design. ACS Nano Synth Appl. doi:10.1021/nn4057269 [DOI] [PubMed]
- De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279(5349):384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- Dewan V, Liu T, Chen KM, Qian Z, Xiao Y, Kleiman L, Mahasenan KV, Li C, Matsuo H, Pei D, Musier-Forsyth K. Cyclic peptide inhibitors of HIV-1 capsid-human lysyl-tRNA synthetase interaction. ACS Chem Biol. 2012;7(4):761–769. doi: 10.1021/cb200450w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz J, Koch J, Kaur A, Raja C, Stein S, Grez M, Pustowka A, Mensch S, Ferner J, Moller L, Bannert N, Tampe R, Divita G, Mely Y, Schwalbe H, Dietrich U. Inhibition of HIV-1 by a peptide ligand of the genomic RNA packaging signal Psi. ChemMedChem. 2008;3(5):749–755. doi: 10.1002/cmdc.200700194. [DOI] [PubMed] [Google Scholar]
- Dulude D, Theberge-Julien G, Brakier-Gingras L, Heveker N. Selection of peptides interfering with a ribosomal frameshift in the human immunodeficiency virus type 1. RNA. 2008;14(5):981–991. doi: 10.1261/rna.887008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennifar E, Aslam MW, Strasser P, Hoffmann G, Dumas P, van Delft FL. Structure-guided discovery of a novel aminoglycoside conjugate targeting HIV-1 RNA viral genome. ACS Chem Biol. 2013;8(11):2509–2517. doi: 10.1021/cb400498n. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang J, O’Carroll IP, Mitchell M, Zuo X, Wang Y, Yu P, Liu Y, Rausch JW, Dyba MA, Kjems J, Schwieters CD, Seifert S, Winans RE, Watts NR, Stahl SJ, Wingfield PT, Byrd RA, Le Grice SF, Rein A, Wang YX. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155(3):594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg MB, Jarrett RF, Aldovini A, Gallo RC, Wong-Staal F. HTLV-III expression and production involve complex regulation at the levels of splicing and translation of viral RNA. Cell. 1986;46(6):807–817. doi: 10.1016/0092-8674(86)90062-0. [DOI] [PubMed] [Google Scholar]
- Freund F, Boulme F, Michel J, Ventura M, Moreau S, Litvak S. Inhibition of HIV-1 replication in vitro and in human infected cells by modified antisense oligonucleotides targeting the tRNALys3/RNA initiation complex. Antisense Nucleic Acid Drug Dev. 2001;11(5):301–315. doi: 10.1089/108729001753231687. [DOI] [PubMed] [Google Scholar]
- Giovannangeli C, Diviacco S, Labrousse V, Gryaznov S, Charneau P, Helene C. Accessibility of nuclear DNA to triplex-forming oligonucleotides: the integrated HIV-1 provirus as a target. Proc Natl Acad Sci USA. 1997;94(1):79–84. doi: 10.1073/pnas.94.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude L, Berkovitch SS, Santos WL, Kutchukian PS, Pawloski AR, Kuimelis R, McGall G, Verdine GL. Mapping targetable sites on human telomerase RNA pseudoknot/template domain using 2′-OMe RNA-interacting polynucleotide (RIPtide) microarrays. J Biol Chem. 2012;287(22):18843–18853. doi: 10.1074/jbc.M111.316596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Javanbakht H, Kleiman L. Specific inhibition of the synthesis of human lysyl-tRNA synthetase results in decreases in tRNA(Lys) incorporation, tRNA(3)(Lys) annealing to viral RNA, and viral infectivity in human immunodeficiency virus type 1. J Virol. 2003;77(18):9817–9822. doi: 10.1128/JVI.77.18.9817-9822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrich D, Hooker CW, Parry E. The human immunodeficiency virus type 1 TAR RNA upper stem-loop plays distinct roles in reverse transcription and RNA packaging. J Virol. 2000;74(12):5639–5646. doi: 10.1128/JVI.74.12.5639-5646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Saenz DT, Fadel HJ, Walker W, Peretz M, Poeschla EM. The HIV-1 central polypurine tract functions as a second line of defense against APOBEC3G/F. J Virol. 2010;84(22):11981–11993. doi: 10.1128/JVI.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung M, Patel P, Davis S, Green SR. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol. 1998;72(6):4819–4824. doi: 10.1128/jvi.72.6.4819-4824.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Berkhout B. Mutations in the TAR hairpin affect the equilibrium between alternative conformations of the HIV-1 leader RNA. Nucleic Acids Res. 2001;29(12):2594–2600. doi: 10.1093/nar/29.12.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Bugala K, Barciszewski J, Berkhout B. On the importance of the primer activation signal for initiation of tRNA(lys3)-primed reverse transcription of the HIV-1 RNA genome. Nucleic Acids Res. 2003;31(17):5186–5194. doi: 10.1093/nar/gkg714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331(6153):280–283. doi: 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]
- Jiang X, Bai Y, Rider P, Kim K, Zhang CY, Lu S, Liu F. Engineered external guide sequences effectively block viral gene expression and replication in cultured cells. J Biol Chem. 2011;286(1):322–330. doi: 10.1074/jbc.M110.158857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Cowan JA. Targeted cleavage of HIV rev response element RNA by metallopeptide complexes. J Am Chem Soc. 2006;128(2):410–411. doi: 10.1021/ja055272m. [DOI] [PubMed] [Google Scholar]
- Jin Y, Cowan JA. Cellular activity of Rev response element RNA targeting metallopeptides. J Biol Inorg Chem [JBIC] (a publication of the Society of Biological Inorganic Chemistry) 2007;12(5):637–644. doi: 10.1007/s00775-007-0221-2. [DOI] [PubMed] [Google Scholar]
- Jin Y, Lewis MA, Gokhale NH, Long EC, Cowan JA. Influence of stereochemistry and redox potentials on the single- and double-strand DNA cleavage efficiency of Cu(II) and Ni(II) Lys-Gly-His-derived ATCUN metallopeptides. J Am Chem Soc. 2007;129(26):8353–8361. doi: 10.1021/ja0705083. [DOI] [PubMed] [Google Scholar]
- Joyner JC, Cowan JA (2013) Target-directed catalytic metallodrugs. Braz J Med Biol Res (Revista brasileira de pesquisas medicas e biologicas/Sociedade Brasileira de Biofisica [et al]) 46(6):465–485. doi:10.1590/1414-431X20133086 [DOI] [PMC free article] [PubMed]
- Karn J. Tackling Tat. J Mol Biol. 1999;293(2):235–254. doi: 10.1006/jmbi.1999.3060. [DOI] [PubMed] [Google Scholar]
- Kaushik N, Basu A, Pandey VN. Inhibition of HIV-1 replication by anti-trans-activation responsive polyamide nucleotide analog. Antiviral Res. 2002;56(1):13–27. doi: 10.1016/S0166-3542(02)00024-4. [DOI] [PubMed] [Google Scholar]
- Kierzek E. Binding of short oligonucleotides to RNA: studies of the binding of common RNA structural motifs to isoenergetic microarrays. Biochemistry. 2009;48(48):11344–11356. doi: 10.1021/bi901264v. [DOI] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11(2):125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hernault A, Greatorex JS, Crowther RA, Lever AM. Dimerisation of HIV-2 genomic RNA is linked to efficient RNA packaging, normal particle maturation and viral infectivity. Retrovirology. 2007;4:90. doi: 10.1186/1742-4690-4-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde MS, Lobritz MA, Ratcliff A, Chamanian M, Athanassiou Z, Tyagi M, Wong J, Robinson JA, Karn J, Varani G, Arts EJ. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLoS Pathog. 2011;7(5):e1002038. doi: 10.1371/journal.ppat.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanchy JM, Ehresmann C, Le Grice SF, Ehresmann B, Marquet R. Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J. 1996;15(24):7178–7187. [PMC free article] [PubMed] [Google Scholar]
- Lanchy JM, Isel C, Keith G, Le Grice SF, Ehresmann C, Ehresmann B, Marquet R. Dynamics of the HIV-1 reverse transcription complex during initiation of DNA synthesis. J Biol Chem. 2000;275(16):12306–12312. doi: 10.1074/jbc.275.16.12306. [DOI] [PubMed] [Google Scholar]
- Laughrea M, Jette L. A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry. 1994;33(45):13464–13474. doi: 10.1021/bi00249a035. [DOI] [PubMed] [Google Scholar]
- Le Grice SF. “In the beginning”: initiation of minus strand DNA synthesis in retroviruses and LTR-containing retrotransposons. Biochemistry. 2003;42(49):14349–14355. doi: 10.1021/bi030201q. [DOI] [PubMed] [Google Scholar]
- Lee CW, Cao H, Ichiyama K, Rana TM. Design and synthesis of a novel peptidomimetic inhibitor of HIV-1 Tat-TAR interactions: squaryldiamide as a new potential bioisostere of unsubstituted guanidine. Bioorg Med Chem Lett. 2005;15(19):4243–4246. doi: 10.1016/j.bmcl.2005.06.077. [DOI] [PubMed] [Google Scholar]
- Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009;37(Pt 6):1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Castagner B, Leroux JC. Is there a future for cell-penetrating peptides in oligonucleotide delivery? Eur J Pharm Biopharm (official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV) 2013;85(1):5–11. doi: 10.1016/j.ejpb.2013.03.021. [DOI] [PubMed] [Google Scholar]
- Legiewicz M, Badorrek CS, Turner KB, Fabris D, Hamm TE, Rekosh D, Hammarskjold ML, Le Grice SF. Resistance to RevM10 inhibition reflects a conformational switch in the HIV-1 Rev response element. Proc Natl Acad Sci USA. 2008;105(38):14365–14370. doi: 10.1073/pnas.0804461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever AM. HIV-1 RNA packaging. Adv Pharmacol. 2007;55:1–32. doi: 10.1016/S1054-3589(07)55001-5. [DOI] [PubMed] [Google Scholar]
- Liu S, Harada BT, Miller JT, Le Grice SF, Zhuang X. Initiation complex dynamics direct the transitions between distinct phases of early HIV reverse transcription. Nat Struct Mol Biol. 2010;17(12):1453–1460. doi: 10.1038/nsmb.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, Dorjsuren B, Kulandaivel G, Jones S, Hiremath A, Divakaruni SS, LaCotti C, Barton S, Tummillo D, Hosic A, Edme K, Albrecht S, Telesnitsky A, Summers MF. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science. 2011;334(6053):242–245. doi: 10.1126/science.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedtke NW, Liu Q, Tor Y. RNA-ligand interactions: affinity and specificity of aminoglycoside dimers and acridine conjugates to the HIV-1 Rev response element. Biochemistry. 2003;42(39):11391–11403. doi: 10.1021/bi034766y. [DOI] [PubMed] [Google Scholar]
- Lusvarghi S, Sztuba-Solinska J, Purzycka KJ, Pauly GT, Rausch JW, Grice SF. The HIV-2 Rev-response element: determining secondary structure and defining folding intermediates. Nucleic Acids Res. 2013;41(13):6637–6649. doi: 10.1093/nar/gkt353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheschi RJ, Tonelli M, Kumar A, Butcher SE. Structure of the HIV-1 frameshift site RNA bound to a small molecule inhibitor of viral replication. ACS Chem Biol. 2011;6(8):857–864. doi: 10.1021/cb200082d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills NL, Daugherty MD, Frankel AD, Guy RK. An alpha-helical peptidomimetic inhibitor of the HIV-1 Rev-RRE interaction. J Am Chem Soc. 2006;128(11):3496–3497. doi: 10.1021/ja0582051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofori LO, Hilimire TA, Bennett RP, Brown NW, Jr, Smith HC, Miller BL. High-affinity recognition of HIV-1 frameshift-stimulating RNA alters frameshifting in vitro and interferes with HIV-1 infectivity. J Med Chem. 2014;57(3):723–732. doi: 10.1021/jm401438g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J. Dimerization of retroviral RNA genomes: an inseparable pair. Nat Rev Microbiol. 2004;2(6):461–472. doi: 10.1038/nrmicro903. [DOI] [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23(3):297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Purcell DF, Martin MA. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J Virol. 1993;67(11):6365–6378. doi: 10.1128/jvi.67.11.6365-6378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch JW, Le Grice SF. ‘Binding, bending and bonding’: polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int J Biochem Cell Biol. 2004;36(9):1752–1766. doi: 10.1016/j.biocel.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Rausch JW, Le Grice SF. Purine analog substitution of the HIV-1 polypurine tract primer defines regions controlling initiation of plus-strand DNA synthesis. Nucleic Acids Res. 2007;35(1):256–268. doi: 10.1093/nar/gkl909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roisin A, Robin JP, Dereuddre-Bosquet N, Vitte AL, Dormont D, Clayette P, Jalinot P. Inhibition of HIV-1 replication by cell-penetrating peptides binding Rev. J Biol Chem. 2004;279(10):9208–9214. doi: 10.1074/jbc.M311594200. [DOI] [PubMed] [Google Scholar]
- Rosen CA, Sodroski JG, Haseltine WA. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell. 1985;41(3):813–823. doi: 10.1016/S0092-8674(85)80062-3. [DOI] [PubMed] [Google Scholar]
- Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci USA. 1994;91(11):4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7(8):553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson JD, Li H, Kenyon JC, Symmons M, Klenerman D, Lever AM. Three-dimensional RNA structure of the major HIV-1 packaging signal region. Structure. 2013;21(6):951–962. doi: 10.1016/j.str.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart KM, Horton KL, Kelley SO. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org Biomol Chem. 2008;6(13):2242–2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- Sztuba-Solinska J, Shenoy SR, Gareiss P, Krumpe LR, Le Grice SF, O’Keefe BR, Schneekloth JS., Jr Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J Am Chem Soc. 2014;136(23):8402–8410. doi: 10.1021/ja502754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Arzumanov AA, Gait MJ. Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res. 2005;33(1):27–42. doi: 10.1093/nar/gki142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KB, Brinson RG, Yi-Brunozzi HY, Rausch JW, Miller JT, Le Grice SF, Marino JP, Fabris D. Structural probing of the HIV-1 polypurine tract RNA:DNA hybrid using classic nucleic acid ligands. Nucleic Acids Res. 2008;36(8):2799–2810. doi: 10.1093/nar/gkn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann S, Jendis J, Frauendorf A, Moelling K. Inhibition of HIV-1 reverse transcription by triple-helix forming oligonucleotides with viral RNA. Nucleic Acids Res. 1995;23(7):1204–1212. doi: 10.1093/nar/23.7.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305(5689):1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warui DM, Baranger AM. Identification of small molecule inhibitors of the HIV-1 nucleocapsid-stem-loop 3 RNA complex. J Med Chem. 2012;55(9):4132–4141. doi: 10.1021/jm2007694. [DOI] [PubMed] [Google Scholar]
- Wesolowski D, Tae HS, Gandotra N, Llopis P, Shen N, Altman S. Basic peptide-morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc Natl Acad Sci USA. 2011;108(40):16582–16587. doi: 10.1073/pnas.1112561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325(5938):336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz J. Putting an ‘End’ to HIV mRNAs: capping and polyadenylation as potential therapeutic targets. AIDS Res Ther. 2013;10(1):31. doi: 10.1186/1742-6405-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bryson DI, Crumpton JB, Wynn J, Santos WL. Targeting folded RNA: a branched peptide boronic acid that binds to a large surface area of HIV-1 RRE RNA. Org Biomol Chem. 2013;11(37):6263–6271. doi: 10.1039/C3OB41053F. [DOI] [PMC free article] [PubMed] [Google Scholar]