

Abstract
A powerful approach utilizing polyproteins for balancing stoichiometry of recombinant multiprotein complexes overproduced in baculovirus expression vector systems (BEVS) is described. This procedure has been implemented here in the MultiBac system but can also be directly adapted to all commonly used BEVS. The protocol details the design principles of polyprotein-expressing DNA constructs, the generation of composite baculovirus for polyprotein production, and the expression and in vivo processing of polyproteins in baculovirus infected insect cells.
Key words: Polyprotein, Multiprotein complexes, Subunit stoichiometry, Baculovirus-insect cell expression system, Tobacco etch virus (TEV) NIa protease, In vivo proteolysis, Multigene delivery, MultiBac system, Cre recombinase, Tn7 transposition
Introduction
Multiprotein complexes catalyze essential cellular activities. Studying their structure and function is an emerging focus of biological research. In most cases, recombinant production is required for obtaining sufficient amounts of homogenous material for detailed analyses [1]. MultiBac is an advanced expression system that has been designed for overexpressing multiprotein complexes in insect cells infected by a single composite multigene baculovirus. MultiBac has enabled production of many challenging multiprotein complexes, setting the stage to unlock their mechanism [2–4]. A bottleneck which can be encountered when many heterologous proteins are co-produced from individual expression cassettes derives from imbalanced expression levels of the individual proteins prohibiting proper complex assembly [3, 5]. Some subunits may be expressed stronger, others weaker, and occasionally one subunit is expressed at such a low level that it becomes detrimental to overall yield and a complex containing all desired subunits cannot be obtained.
In order to balance expression levels and achieve properly assembled complexes with correct subunit stoichiometry, we have implemented a novel strategy based on polyproteins that are processed in vivo into individual subunits by a highly specific protease [3, 5] (Fig. 1). This approach derives from the strategy used by certain viruses such as Coronavirus to realize their proteome [6]. To facilitate polyprotein production with the MultiBac BEVS, new transfer plasmids have been created that rely on Tn7 transposon-mediated gene integration into the MultiBac baculovirus (Fig. 2). Other baculoviruses that are in use rely on homologous recombination for composite baculovirus generation (flashBAC from OET, BacVector series from Novagen, others). These baculoviruses can likewise be accessed for polyprotein expression by using the pOmni-PBac plasmid [7].
Fig. 1.
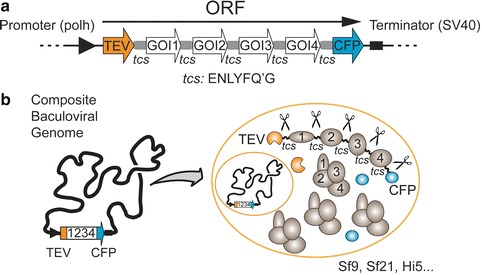
Design and in vivo processing of polyproteins. (a) Genes of interest (GOIs) are assembled into a single open reading frame (ORF), giving rise to a polyprotein. In this polyprotein, individual genes of interest are spaced apart by cleavage sites (tcs) for tobacco etch virus (TEV) NIa protease which is also encoded by the ORF. The C-terminal CFP serves to monitor heterologous expression level. (b) Composite MultiBac baculoviral genome DNA containing the polyprotein expression cassette (left ) is used to transfect cultured insect cells for multiprotein complex production (right)
Fig. 2.
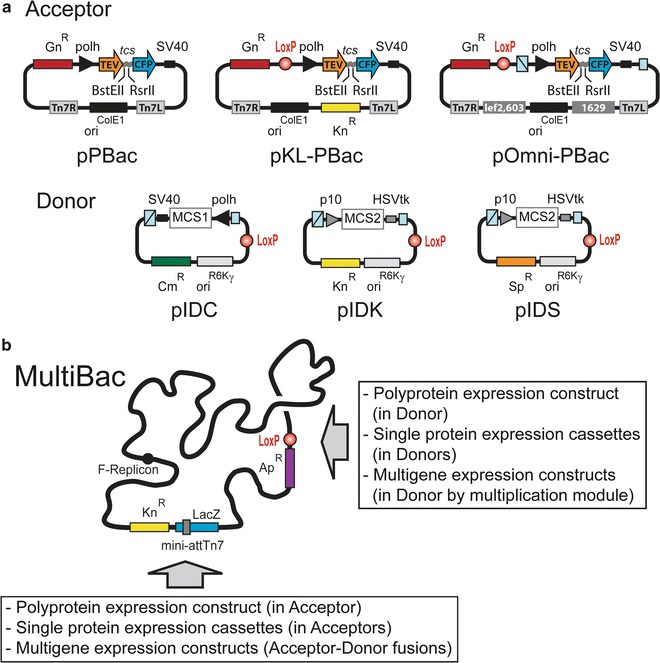
Integration of polyprotein expression cassettes into MultiBac baculovirus. (a) MultiBac Acceptor vectors pPBac, pKL-PBac, and pOmni-PBac, tailored for polyprotein production, are shown schematically (top). They contain a regular ColE1 origin of replication and a polyprotein expression cassette, which encodes an N-terminal TEV protease and a C-terminal CFP spaced by a TEV cleavage site (tcs). BstEII and RsrII sites are used for inserting the polyprotein encoding ORF of interest. Donor vectors pIDC, pIDK, and pIDS contain a conditional origin of replication derived from the R6Kγ phage [13]. The multiplication module flanking the expression cassettes contain a homing endonuclease site and a complementary BstXI site (boxes in light blue). Polh and p10 are baculoviral very late promoters; SV40 and HSVtk are polyadenylation signals. MCS1 and MCS2 stand for multiple cloning sites. Tn7L and Tn7R are specific DNA sequences for Tn7 transposition; the lef2/603 and Ori1629 homology regions are shown as gray boxes. LoxP sites are shown as red balls. Cm stands for chloramphenicol, Gn for gentamicin, Kn for kanamycin, Sp for spectinomycin. (b) Besides polyprotein expression cassettes, single protein and multigene expression cassettes can also be integrated into the Tn7 attachment site (mini-attTn7) harbored by the LacZ (lacZα) gene, or the LoxP site of the MultiBac baculovirus. Ap stands for ampicillin. The F-Replicon is a single-copy bacterial origin of replication. For reagents contact: iberger@embl.fr
All polyprotein transfer plasmids contain the same expression cassette which encompasses a very late viral promoter (polyhedrin) followed by the gene encoding for NIa protease from tobacco etch virus (TEV) for subunit liberation, a short oligonucleotide sequence presenting a BstEII and an RsrII restriction endonuclease site, and finally a gene encoding for cyan fluorescent protein (CFP) for direct read-out of polyprotein expression. A TEV NIa protease cleavage site (tcs) is placed upstream of the CFP encoding gene (Fig. 2a). Heterologous genes of interest (GOIs) can be inserted into this polyprotein expression cassette by using the BstEII and RsrII sites and restriction–ligation cloning (see Note 1). Transfer plasmids are then used to integrate the resulting polyprotein expression cassette into baculovirus genomes of choice to generate composite baculovirus for protein expression (Fig. 2b). With this strategy, a number of complexes have been successfully produced with balanced subunit expression levels, including a ~700 kDa physiological core complex of human general transcription factor TFIID [4] (Fig. 3).
Fig. 3.
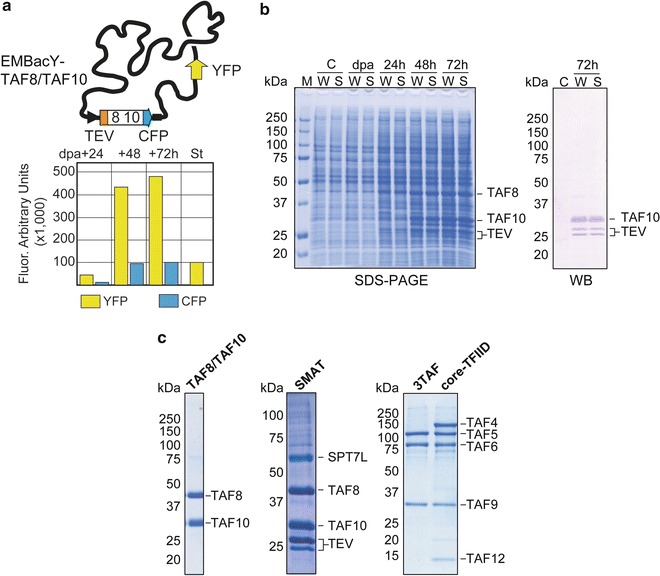
Multiprotein complexes produced from polyproteins. (a) The TAF8/TAF10 dimer (inserted into the Tn7 attachment site) was co-expressed as a polyprotein with the yellow fluorescent protein (YFP) inserted into the viral LoxP site from a composite baculovirus (EMBacY-TAF8/TAF10). YFP and CFP expression per one million cells were tracked for evaluating the viral infection and polyprotein production. St stands for a defined fluorescence standard (used to calibrate for 100,000 arbitrary units), dpa stands for day of proliferation arrest in the infected culture [5]. (b) SDS-PAGE (left ) shows balanced expressions of TAF8 and TAF10. Complete proteolysis of the TAF8/TAF10 polyprotein was confirmed by Western blot (right ) using antibody specific for the hexa histidine-tags of TAF10 and TEV protease (doublet ). M stands for molecular weight marker, C stands for cell control (uninfected insect cells), W stands for whole cell extract, S stands for supernatant. (c) Sections from SDS-PAGE are shown for TAF8/TAF10 dimer from size exclusion chromatography purification, SMAT complex from IMAC batch purification [5], 3TAF and core-TFIID complexes from size exclusion chromatography purification [4]
Multiprotein complexes can be expressed from a single polyprotein, or alternatively, from several polyproteins that are co-expressed, or a combination of single protein expression cassettes and one or several polyproteins, depending on the complex of choice. We recommend combining a maximum of four to five genes (in addition to the genes encoding for TEV protease and the fluorescent protein) into a single open reading frame (ORF). Otherwise, any later work to modify the genes of interest may become complicated.
We observed that while it is sufficient to provide one TEV NIa protease gene in a co-expression experiment using several polyproteins, it appears that “tagging” all polyproteins with TEV NIa protease at the N-terminus balances overexpression levels between polyproteins (IB, unpublished data).
Materials
We strongly recommend carrying out the design of polyproteins in silico using a DNA cloning software of choice (i.e., VectorNTI, ApE, others). Gene synthesis may be preferred for generating the individual genes of interest, in which internal BstEII and RsrII sites must be eliminated. If synthetic genes are used in conjunction with other plasmids of the MultiBac system, we recommend to further eliminate also any restriction sites that are part of the so-called multiplication modules (AvrII, ClaI, SpeI, BstZ17I, NruI, PmeI, BstXI) in the MultiBac plasmids [2, 3, 9]. Thereby, maximum flexibility of gene assembly is achieved for co-expressing proteins.
The modular concept of the MultiBac system allows transferring expression cassettes between various plasmids [2, 9]. This option can be used if several polyproteins are to be co-expressed, for example by inserting a polyprotein expression cassette into a Donor and accessing the LoxP site present on the MultiBac baculoviral backbone (Fig. 2b). Alternatively, Acceptor–Donor fusions can be generated by Cre-LoxP reaction of Donors of choice with pKL-PBac or pOmni-PBac, following published protocols [9, 10]. When co-expressing several polyproteins, we recommend using different fluorescent markers (CFP, YFP, mCherry, others) to monitor polyprotein expression instead of tagging each polyprotein with the same fluorescent protein.
All reagents are prepared using ultrapure water (Millipore Milli-Q system or equivalent; conductivity of 18.2 MΩ cm at 25 °C) and analytical grade reagents. Buffers, antibiotics, and enzymes are stored at −20 °C.
Materials for Inserting Polyprotein Constructs into Transfer Vectors via Restriction–Ligation Cloning
Restriction endonucleases BstEII and RsrII and reaction buffers (New England Biolabs, NEB).
T4 DNA ligase and buffer (NEB).
Gel extraction kit (i.e., Qiagen, Germany).
Plasmid purification kit (i.e., Qiagen, Germany).
Regular E. coli competent cells (TOP10, HB101, or comparable).
E. coli competent cells containing pir gene (if Donor plasmids are used, see Note 2).
Antibiotics: chloramphenicol, gentamicin, kanamycin, spectinomycin (for concentrations see ref. 8).
Agar for pouring plates.
Media (LB, TB, SOC) for growing minicultures.
Materials for Integrating Polyprotein Expression Cassettes into Baculovirus Genome
Methods
The genes encoding for the polyproteins are designed in silico, and then inserted into the transfer plasmid of choice. Once designed, polyprotein encoding genes can be created by a variety of means including DNA synthesis, restriction–ligation cloning, or sequence and ligation independent cloning (SLIC) [10, 11] or other methods, according to the preferences of the user. We recommend custom DNA synthesis to facilitate polyprotein construction.
Polyprotein In Silico Design
Group genes into polyproteins based on a set of chosen criteria (such as putative interaction partners, physiological (sub)assemblies, subunits with the same copy number within a complex).
Decide on the number of polyproteins that should be co-expressed (we recommend not to catenate more than four to five genes in addition to the protease and fluorescent marker encoding genes in each polyprotein ORF).
Decide on placement of tags. Note that cleavage sites other than TEV protease cleavage sites have to be used if tags are to be removed at a later stage by a specific protease (i.e., PreScission protease, thrombin, enterokinase, others).
Remove stop codons from individual genes, except for the last gene of interest if the option to monitor polyprotein expression via the plasmid-encoded fluorescent marker protein is not desired. If fluorescence read-out is desired, delete stop codons of all genes that are to be inserted.
Decide on TEV protease cleavage site containing linker in between individual protein entities in the polyprotein. In particular if long unstructured tails are already predicted for example at the C-terminus of a given protein, we recommend adjoining the TEV NIa protease cleavage site (typically ENLYFQ’G) directly. The glycine residue replaces the starting methionine of the following protein.
Generate the DNA sequence. Add BstEII site to 5′ end and RsrII site to 3′ end.
Create complete polyprotein expression construct in silico, predict translation, verify reading frame through the TEV NIa protease and the fluorescent marker.
Decide on DNA assembly strategy (SLIC, restriction–ligation, PCR assembly, others).
Create all DNA sequences in silico and validate by simulating the reading frame.
Preparation of Transfer Plasmid DNA
Choose from pOmni-PBac, pPBac, or pKL-PBac to generate the polyprotein expressing construct for expression with the baculovirus of choice (see Note 4). All polyprotein expression cassettes have the same design with BstEII and RsrII sites for DNA insertion between the gene encoding for TEV NIa protease and the gene encoding for CFP. pKL-PBac contains a LoxP site for integrating Donor plasmids with further genes of interest; pOmni-PBac contains elements for homologous recombination in addition to elements for Tn7 transposition.
Digest several micrograms transfer plasmid by BstEII and RsrII enzymes according to manufacturers’ recommendation. Sequential digestion is recommended as BstEII cuts optimally at 60 °C, while RsrII prefers 37 °C.
Analyze the digestions by agarose gel electrophoresis to confirm that the digestions are complete.
Purify digested plasmid by using commercial gel extraction kits (for example Qiagen gel extraction kit). It is recommended to elute the extracted DNA in the minimal volume defined by the manufacturer. Determine the concentration of the extracted DNA spectrophotometrically (e.g., Thermo Scientific NanoDrop 2000). Store in frozen aliquots.
Inserting Polyprotein Expression Cassettes into BstEII/RsrII Digested Transfer Vectors
Digest several micrograms of the DNA (generated by DNA synthesis, SLIC, PCR assembly, or other methods of choice) encoding for the desired polyprotein with BstEII and RsrII enzymes according to the manufacturers’ recommendation. Sequential digestion is recommended as BstEII cuts optimally at 60 °C, while RsrII prefers 37 °C.
Purify digested insert DNA by using a commercial gel extraction kit. It is recommended to elute the extracted DNA in the minimal volume defined by the manufacturer. Determine the concentration of the extracted DNA spectrophotometrically.
Set up ligation reactions by mixing purified insert and vector (see Subheading 3.2) in 10–20 μL reaction volume with T4 DNA ligase and specific buffer according to the recommendations from the supplier. Perform ligation reactions at 25 °C overnight. It is recommended to analyze the ligation reaction by agarose gel electrophoresis to evaluate the ligation efficiency.
Transform regular E. coli competent cells (see Note 2) with ligation reaction by following standard transformation procedures. Incubate the transformation reaction in a 37 °C shaker for 1–2 h and plate on agar plates in a dilution series to ensure optimal colony separation.
Pick colonies, grow minicultures, and purify plasmids.
Indentify positive clones by restriction digestion and DNA sequencing of the insert.
Integrating Polyprotein Expression Constructs into the MultiBac Baculoviral Genome via Tn7 Transposition
Transform corresponding E. coli competent cells (DH10MultiBac or DH10EMBacY) with transfer plasmid by following standard transformation procedures. Incubate the transformation reaction in a 37 °C shaker overnight (see Note 5).
Plate the transformation reaction on agar plates containing antibiotics as described [9], IPTG (1 mM) and Bluo-Gal (or X-Gal) in a dilution series to ensure optimal colony separation. Incubate at 37 °C until blue and white colonies are well distinguishable.
Restreak four to eight white colonies to unambiguously confirm that they are positive (white). It is recommended to restreak also a blue colony as negative control.
Inoculate four confirmed white colonies in 2 mL aliquots of LB medium supplemented with corresponding antibiotics. After overnight incubation, use two to four of the cell cultures for bacmid purification, transfection, viral amplification, and multiprotein complex overexpression [8].
Integrating Polyprotein Expression Cassettes into MultiBac Baculoviral Genome via In Vivo Cre-LoxP Reaction
Place polyprotein expression cassette into Donor plasmid of choice by SLIC, restriction–ligation, PCR assembly, or other methods of choice. Validate resulting constructs by restriction mapping.
Transform DH10MultiBacCre electro-competent cells (these contain Cre recombinase expressed from a separate plasmid [9]) with this polyprotein expressing Donor plasmid by following standard electroporation procedures. Incubate the transformation reaction in a 37 °C shaker overnight.
Plate the transformation reaction on agar plates containing corresponding antibiotics, IPTG (1 mM) and Bluo-Gal (or X-Gal) in a dilution series to ensure optimal colony separation. Incubate at 37 °C until blue color of the colonies is clearly observed.
Restreak four to eight blue colonies on the same type of agar plates to confirm they are positive.
Inoculate four confirmed blue colonies in 2 mL aliquots of LB medium supplemented with corresponding antibiotics. After overnight incubation, use all four cell cultures (see Note 6) for bacmid purification, transfection, viral amplification, and multiprotein complex overexpression following published protocols [8].
Notes
The BstEII enzyme has the asymmetric restriction site G^GTNAC_C, the RsrII restriction enzyme has the asymmetric restriction site CG^GWC_CG. In both cases the central base can have different contexts. When constructing the ORF encoding for the polyprotein, the sites have to be chosen such as to be compatible with the transfer plasmids.
Donors and their derivatives can only be propagated in cells that express the pir gene (such as BW23473, BW23474, or PIR1 and PIR2, Invitrogen) due to the conditional origin present on these plasmids [13]. In contrast, Acceptors and their derivatives contain regular ColE1 origin of replication and can be propagated in regular E. coli strains (TOP10, HB101, or comparable) [3, 12].
The generation of DH10MultiBacCre cells by expressing Cre recombinase is detailed in ref. 9.
Plasmids pPBac and pKL-PBac rely on Tn7 transposition and a baculovirus genome in form of a bacterial artificial chromosome (bacmid) for composite baculovirus generation (i.e., Bac-to-Bac system from Invitrogen, MultiBac). Plasmid pOmni-PBac, in contrast, is a universal transfer plasmid that can access baculoviruses by both Tn7 transposition and homologous recombination [7].
Besides polyprotein expression constructs, the Tn7 attachment site (mini-attTn7) and the LoxP site can also be used for integrating single protein and multigene expression constructs (Fig. 2b).
It is recommended to check at least four blue colonies since the integration efficiency of in vivo Cre-LoxP reaction is generally lower than Tn7 transposition.
Acknowledgements
We thank all the members of the Berger laboratory for helpful discussions. YN was a fellow of the Marie-Curie training network Chromatin Plasticity and the Boehringer Ingelheim Foundation (BIF, Germany). IBE is a Hoffmann-La Roche postdoctoral fellow. ST was supported by a European Commission (EC) Marie Curie IEF postdoctoral fellowship. IB acknowledges support from the Swiss National Science Foundation (SNSF), the Agence Nationale de la Recherche (ANR), the Centre National de la Recherche Scientifique (CNRS), the EMBL and the European Commission (EC) Framework Programme (FP7) projects INSTRUCT, PCUBE, BioStruct-X, and ComplexINC.
Competing Financial Interest Statement
The authors declare competing financial interests. IB is the author on patents and patent applications containing parts of the methods and reagents here described.
References
- 1.Nie Y, Viola C, Bieniossek C. Getting a grip on complexes. Curr Genomics. 2009;10:558–572. doi: 10.2174/138920209789503923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger I, Fitzgerald DJ, Richmond TJ. Baculovirus expression system for heterologous multiprotein complexes. Nat Biotechnol. 2004;22:1583–1587. doi: 10.1038/nbt1036. [DOI] [PubMed] [Google Scholar]
- 3.Bieniossek C, Imasaki T, Takagi Y. MultiBac: expanding the research toolbox for multiprotein complexes. Trends Biochem Sci. 2012;37:49–57. doi: 10.1016/j.tibs.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieniossek C, Papai G, Schaffitzel C. The architecture of human general transcription factor TFIID core complex. Nature. 2013;493:699–702. doi: 10.1038/nature11791. [DOI] [PubMed] [Google Scholar]
- 5.Vijayachandran LS, Viola C, Garzoni F, et al. Robots, pipelines, polyproteins: enabling multiprotein expression in prokaryotic and eukaryotic cells. J Struct Biol. 2011;175:198–208. doi: 10.1016/j.jsb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorbalenya AE, Enjuanes L, Ziebuhr J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vijayachandran LS, Thimiri Govinda Raj DB, Edelweiss E, Gupta K, Maier J, Gordeliy V, Fitzgerald DJ, Berger I. Gene gymnastics: synthetic biology for baculovirus expression vector system engineering. Bioengineered. 2013;4:1–9. doi: 10.4161/bioe.22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bieniossek C, Richmond TJ, Berger I (2008) MultiBac: multigene baculovirus-based eukaryotic protein complex production. In: Coligan JE, Dunn BM, Speicher DW, Wingfield PT (eds) Current protocols in protein science. Wiley, Hoboken, NJ, USA, Unit 5.20 [DOI] [PubMed]
- 9.Fitzgerald DJ, Berger P, Schaffitzel C. Protein complex expression by using multigene baculoviral vectors. Nat Methods. 2006;3:1021–1032. doi: 10.1038/nmeth983. [DOI] [PubMed] [Google Scholar]
- 10.Bieniossek C, Nie Y, Frey D. Automated unrestricted multigene recombineering for multiprotein complex production. Nat Methods. 2009;6:447–450. doi: 10.1038/nmeth.1326. [DOI] [PubMed] [Google Scholar]
- 11.Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 12.Trowitzsch S, Bieniossek C, Nie Y. New baculovirus expression tools for recombinant protein complex production. J Struct Biol. 2010;172:45–54. doi: 10.1016/j.jsb.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Metcalf WW, Jiang W, Wanner BL. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6Kλ origin plasmids at different copy numbers. Gene. 1994;138:1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]