

Abstract
We have developed a reverse genetics system for the avian coronavirus infectious bronchitis virus (IBV) in which a full-length cDNA corresponding to the IBV genome is inserted into the vaccinia virus genome under the control of a T7 promoter sequence. Vaccinia virus as a vector for the full-length IBV cDNA has the advantage that modifications can be introduced into the IBV cDNA using homologous recombination, a method frequently used to insert and delete sequences from the vaccinia virus genome. Here, we describe the use of transient dominant selection as a method for introducing modifications into the IBV cDNA that has been successfully used for the substitution of specific nucleotides, deletion of genomic regions, and exchange of complete genes. Infectious recombinant IBVs are generated in situ following the transfection of vaccinia virus DNA, containing the modified IBV cDNA, into cells infected with a recombinant fowlpox virus expressing T7 DNA-dependant RNA polymerase.
Key words: Transient dominant selection (TDS), Vaccinia virus, Infectious bronchitis virus (IBV), Coronavirus, Avian, Reverse genetics, Nidovirus, Fowlpox virus, T7 RNA polymerase
Introduction
The Coronaviridae form part of the order , which comprises two subfamilies, the Coronavirinae and Torovirinae. There are four genera of coronaviruses, alpha-, beta-, gamma-, and deltacoronaviruses [1], which were so named for their visual resemblance to the corona of the sun in negatively stained preparations [2]. Coronaviruses are enveloped viruses with a single-stranded positive-sense RNA genome of 26–32 kb, the largest genomes of all RNA viruses currently known, that replicate in the cytoplasm of infected cells. The genome associates with the nucleoprotein (N) forming a helical nucleocapsid within the virus particles. Although common among negative-sense RNA viruses, coronaviruses are the only positive-sense RNA viruses to possess helical nucleocapsids, which are enclosed within lipid envelopes containing the spike (S) glycoprotein, membrane (M) protein, and envelope (E) protein.
Avian infectious bronchitis virus (IBV) is a gammacoronavirus that is the etiological agent of infectious bronchitis (IB), an acute and highly contagious disease of poultry characterized by nasal discharge, snicking, tracheal ciliostasis, and rales [3]. IBV replicates primarily in the respiratory tract but also in many other epithelial surfaces including oviducts, enteric surfaces, and kidneys [4–7]. Following infection with IBV, egg production and quality may be impaired in layers and weight gain in broilers is reduced [8]. Infected birds are predisposed to secondary bacterial infections such as colibacillosis and mortality in young chicks is not uncommon. Fecal excretion of the virus is a consequence of replication in the intestinal tract; however, this does not normally result in clinical disease.
Infectious bronchitis was first described in the USA in the 1930s [9–11] and is prevalent in poultry farming across the world due to the intensive nature of poultry production, estimated to involve the global production of 55 billion chickens (50 billion broilers and five billion layers) on an annual basis. According to a new market research report published by MarketsandMarkets, the global poultry meat market was worth $179,551.2 million in 2012 and is expected to reach $250,949.5 million by 2018. In a report, commissioned by Defra in 2005 [12], IBV was indicated as a major cause of ill health among chickens and was implicated as being responsible for more economic loss in the UK poultry industry than any other disease [13, 14]; IBV was estimated to cost the UK economy nearly £19 million per year, mainly due to loss of egg production, with serious implications for animal welfare. The cost of control through vaccination is approximately £5 million per year in the UK.
Coronaviruses are enveloped viruses that replicate in the cell cytoplasm. Coronavirus genomes consist of single-stranded positive-sense RNA, and are the largest of all the RNA viruses ranging from approximately 27–32 kb; the genome of IBV is 27.6 kb. Molecular analysis of the role of individual genes in the pathogenesis of RNA viruses has been advanced by the availability of full-length cDNAs, for the generation of infectious RNA transcripts that can replicate and result in infectious viruses. The assembly of full-length coronavirus cDNAs was hampered due to regions from the replicase gene being unstable in bacteria. We therefore devised a reverse genetics strategy for IBV involving the insertion of a full-length cDNA copy of the IBV genome, under the control of a T7 RNA promoter, into the vaccinia virus genome in place of the thymidine kinase (TK) gene. A hepatitis δ ribozyme (HδR) is located downstream of the coronavirus poly(A) tail followed by a T7 termination sequence. IBV infectious RNA is generated from the T7 promoter immediately adjacent to the 5′ end of the IBV cDNA using T7 RNA polymerase and terminates at the T7 termination sequence downstream of the HδR sequence, which autocleaves itself and the T7 termination sequence at the end of the poly(A) sequence, resulting in an authentic IBV genomic RNA copy. Infectious IBV is recovered in situ in cells both transfected with vaccinia virus DNA and infected with a recombinant fowlpox virus expressing T7 RNA polymerase [15].
One of the main advantages of using vaccinia virus as a vector for IBV cDNA is its ability to accept large quantities of foreign DNA without loss of integrity and stability [16]. A second and equally important advantage is the ability to modify the IBV cDNA within the vaccinia virus vector through transient dominant selection (TDS), a method taking advantage of recombinant events between homologous sequences [17, 18]. The TDS method relies on a three-step procedure. In the first step, the modified IBV cDNA is inserted into a plasmid containing a selective marker under the control of a vaccinia virus promoter. In our case we use a plasmid, pGPTNEB193 (Fig. 1; [19]), which contains a dominant selective marker gene, Escherichia coli guanine phosphoribosyltransferase (Ecogpt ; [20]), under the control of the vaccinia virus P7.5K early/late promoter.
Fig. 1.
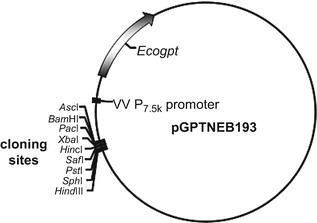
Schematic diagram of the recombination vector for insertion of genes into a vaccinia virus genome using TDS . Plasmid pGPTNEB193 contains the Ecogpt selection gene under the control of the vaccinia virus early/late P7.5K promoter, a multiple cloning region for the insertion of the sequence to be incorporated into the vaccinia virus genome, and the bla gene (not shown) for ampicillin selection of the plasmid in E. coli. For modification of the IBV genome, a sequence corresponding to the region being modified, plus flanking regions of 500–800 nucleotides for recombination purposes, is inserted into the multiple cloning sites using an appropriate restriction endonuclease . The plasmid is purified from E. coli and transfected into Vero cells previously infected with a recombinant vaccinia virus containing a full-length cDNA copy of the IBV genome
In the second step, this complete plasmid sequence is integrated into the IBV sequence within the vaccinia virus genome (Fig. 2). This occurs as a result of a single crossover event involving homologous recombination between the IBV cDNA in the plasmid and the IBV cDNA sequence in the vaccinia virus genome. The resulting recombinant vaccinia viruses (rVV) are highly unstable due to the presence of duplicate sequences and are only maintained by the selective pressure of the Ecogpt gene, which confers resistance to mycophenolic acid (MPA) in the presence of xanthine and hypoxanthine [17]. In the third step, the MPA -resistant rVVs are grown in the absence of MPA selection, resulting in the loss of the Ecogpt gene due to a second single homologous recombination event between the duplicated sequences (Fig. 3). During this third step two recombination events can occur; one event will result in the generation of the original (unmodified) IBV sequence and the other in the generation of an IBV cDNA containing the desired modification (i.e., the modification within the plasmid sequence). In theory these two events will occur at equal frequency; however in practice this is not necessarily the case.
Fig. 2.
Schematic diagram demonstrating the TDS method for integrating a modified IBV sequence into the full-length IBV cDNA within the genome of a recombinant vaccinia virus (vNotI-IBVFL). The diagram shows a potential first single-step recombination event between the modified IBV sequence within pGPTNEB193 and the IBV cDNA within vNotI-IBVFL. In order to guarantee a single-step recombination event any potential recombinant vaccinia viruses are selected in the presence of MPA ; only vaccinia viruses expressing the Ecogpt gene are selected. The main IBV genes are indicated, the replicase, spike (S), membrane (M), and nucleocapsid (N) genes. The IBV gene 3 and 5 clusters that express three and two gene products, respectively, are also indicated. In the example shown a modified region of the S gene is being introduced into the IBV genome
Fig. 3.
Schematic diagram demonstrating the second step of the TDS method. Integration of the complete pGPTNEB193 plasmid into the vaccinia virus genome results in an unstable intermediate because of the presence of tandem repeat sequences, in this example the 3′ end of the replicase gene, the S gene, and the 5′ end of gene 3. The second single-step recombination event is induced in the absence of MPA ; loss of selection allows the unstable intermediate to lose one of the tandem repeat sequences including the Ecogpt gene. The second step recombination event can result in either (I) the original sequence of the input vaccinia virus IBV cDNA sequence, in this case shown as a recombination event between the two copies of the 3′ end of the replicase gene which results in loss of the modified S gene sequence along with Ecogpt gene, or (II) retention of the modified S gene sequence and loss of the original S gene sequence and Ecogpt gene as a result of a potential recombination event between the two copies of the 5′ end of the S gene sequence. This event results in a modified S gene sequence within the IBV cDNA in a recombinant vaccinia virus
To recover infectious rIBVs from the rVV vector, rVV DNA is transfected into primary chick kidney (CK ) cells previously infected with a recombinant fowlpox virus expressing T7 RNA polymerase ( rFPV-T7; [21]). In addition, a plasmid, pCi-Nuc [15, 22], expressing the IBV nucleoprotein (N), under the control of both the cytomegalovirus (CMV) RNA polymerase II promoter and the T7 RNA promoter, is co-transfected into the CK cells. Expression of T7 RNA polymerase in the presence of the IBV N protein and the rVV DNA, containing the full-length IBV cDNA under the control of a T7 promoter, results in the generation of infectious IBV RNA, which in turn results in the production of infectious rIBVs (Fig. 4). Primary CK cells are refractory for growth of most IBV isolates; therefore rIBVs expressing S glycoproteins from such isolates cannot be recovered using CK cells. In order to recover such rIBVs, the supernatants from the transfected CK cells are used to infect 10-day-old embryonated hen’s eggs. Allantoic fluid is collected and any potential virus passed a further three times in 10-day-old embryos. RNA is extracted from the allantoic fluid of infected eggs and RT-PCR followed by sequencing is used to confirm the identity of the rIBV.
Fig. 4.
A schematic representation of the recovery process for obtaining rIBV from DNA isolated from a recombinant vaccinia virus containing a full-length IBV cDNA under the control of a T7 promoter. (a) In addition to the vaccinia virus DNA containing the full-length IBV cDNA under the control of a T7 promoter a plasmid, pCI-Nuc, expressing the IBV nucleoprotein, required for successful rescue of IBV, is transfected into CK cells previously infected with a recombinant fowlpox virus, FPV-T7, expressing T7 RNA polymerase. The T7 RNA polymerase results in the synthesis of an infectious RNA from the vaccinia virus DNA that consequently leads to the generation of infectious IBV being released from the cell. (b) Any recovered rIBV present in the media of P0 CK cells is used to infect P1 CK cells. The media is filtered through a 0.22 μm filter to remove any FPV-T7 virus. IBV-induced CPE is normally observed in the P1 CK cells following a successful recovery experiment. Any rIBV is passaged a further two times, P2 and P3, in CK cells. Total RNA is extracted from the P1–P3 CK cells and the IBV-derived RNA analyzed by RT-PCR for the presence of the required modification
The overall procedure is a multistep process which can be divided into two parts: the generation of an rVV containing the modified IBV cDNA (Fig. 5) and the recovery of infectious rIBV from the rVV vector (Fig. 4). The generation of the Ecogpt plasmids, based on pGPTNEB193, containing the modified IBV cDNA, is by standard E. coli cloning methods [23, 24] and is not described here. General methods for growing vaccinia virus have been published by Mackett et al. [25] and for using the TDS method for modifying the vaccinia virus genome by Smith [26].
Fig. 5.
Schematic detailing the multistep process of constructing a recombinant vaccinia virus. Vero cells are infected with rVV containing IBV cDNA and then transfected with a plasmid containing the IBV sequence to be inserted and the selective marker gene Ecogpt . Homologous recombination occurs and the complete plasmid sequence is inserted into the rVV . The Ecogpt gene allows positive selection of these rVV as it confers resistance to MPA in the presence of xanthine and hypoxanthine. The viruses are plaque purified three times in the presence of selection agents ensuring that no wild-type VV is present. The removal of the selection agents results in a second recombination event with the loss of the Ecogpt gene. Plaque purification in the absence of selection agents ensures not only the loss of the GPT gene but also the maintenance of a single viral population. Small stocks of rVV are grown from individual plaques which are screened through PCR for the desired modification; this is found in theoretically 50% of rVVs
Materials
Homologous Recombination and Transient Dominant Selection in Vero Cells
Vero cells.
PBSa: 172 mM NaCl , 3 mM KCl , 10 mM Na2HPO4 , and 2 mM KH2PO4 , adjusted to pH 7.2 with HCl .
1× Eagle’s Minimum Essential Medium (E-MEM) with Earle’s salts, 2 mM l- glutamine, and 2.2 g/L sodium bicarbonate .
BES medium: 1 × E-MEM , 0.3% tryptose phosphate broth (TPB), 0.2% bovine serum albumin (BSA), 20 mM N, N-Bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) , 0.21% sodium bicarbonate , 2 mM l- glutamine, 250 U/mL nystatin, 100 U/mL penicillin , and 100 U/mL streptomycin.
OPTIMEM 1 with GLUTAMAX-1 (Life Technologies).
Lipofectin (Life Technologies).
Mycophenolic acid (MPA ): 10 mg/mL in 0.1 M NaOH (30 mM); 400× concentrated.
Xanthine: 10 mg/mL in 0.1 M NaOH (66 mM); 40× concentrated. Heat at 37 °C to dissolve.
Hypoxanthine: 10 mg/mL in 0.1 M NaOH (73 mM); 667× concentrated.
Screw-top 1.5 mL microfuge tubes with gasket.
Cup-form sonicator.
2× E-MEM : 2× E-MEM , 10% fetal calf serum, 0.35% sodium bicarbonate , 4 mM l- glutamine, 1000 U/mL nystatin, 200 U/mL penicillin, and 200 U/mL streptomycin.
2% Agar.
Ecogpt selection medium: 1× E-MEM , 75 μM MPA , 1.65 mM xanthine, 109 μM hypoxanthine, 1% agar (see Note 7).
Overlay medium: 1× E-MEM , 1% agar.
1% Neutral red solution (H2O).
Extraction of DNA from Recombinant Vaccinia Virus
20 mg/mL Proteinase K.
2× Proteinase K buffer: 200 mM Tris–HCl pH 7.5, 10 mM EDTA , 0.4% SDS , 400 mM NaCl .
Phenol/ chloroform/ isoamyl alcohol (25:24:1).
Chloroform.
Absolute ethanol .
70% Ethanol.
QlAamp DNA mini kit (QIAGEN).
3 M Sodium acetate .
Production of Large Stocks of Vaccinia Virus
BHK-21 maintenance medium: Glasgow-Modified Eagle’s Medium (G-MEM), 2 mM l- glutamine, 0.275% sodium bicarbonate , 1% fetal calf serum, 0.3% TPB , 500 U/mL nystatin, 100 U/mL penicillin , and 100 U/mL streptomycin.
TE buffer: 10 mM Tris –HCl pH 9, 1 mM EDTA .
BHK-21 cells.
50 mL Falcon tubes.
Vaccinia Virus Partial Purification
30% Sucrose (w/v) in 1 mM Tris –HCl pH 9, filtered through 0.22 μm.
Superspin 630 rotor and Sorvall OTD65B ultracentrifuge or equivalent.
Analysis of Vaccinia Virus DNA by Pulsed Field Agarose Gel Electrophoresis
10× TBE buffer: 1 M Tris–HCl, 0.9 M boric acid pH 8, and 10 mM EDTA .
Pulsed field-certified ultrapure DNA-grade agarose.
DNA markers (e.g., 8–48 kb markers, Biorad).
0.5 mg/mL Ethidium bromide .
CHEF-DR® II pulsed field gel electrophoresis (PFGE) apparatus (Biorad) or equivalent.
6× Sample loading buffer: 62.5% Glycerol, 62.5 mM Tris –HCl pH 8, 125 mM EDTA , and 0.06% bromophenol blue.
Preparation of rFPV-T7 Stock Virus
Chicken embryo fibroblast (CEF) cells.
CEF maintenance medium: 1× 199 Medium with Earle’s Salts, 0.3% TPB , 2% newborn calf serum (NBCS), 0.225% sodium bicarbonate , 2 mM l- glutamine , 100 U/mL penicillin, 100 U/mL streptomycin , and 500 U/mL nystatin .
Recovery of rIBV and Serial Passage on CK Cells
Chick kidney (CK ) cells.
Stock of rFPV-T7 virus.
The rVV DNA prepared from large partially purified stocks of rVV .
Plasmid pCi-Nuc which contains IBV nucleoprotein under the control of the CMV and T7 promoters.
0.22 μm Syringe-driven filters.
5 mL Syringes.
Methods
Infection/Transfection of Vero Cells with Vaccinia Virus
Freeze-thaw the vaccinia virus containing the full-length IBV cDNA genome to be modified three times (37 °C/dry ice) and sonicate for 2 min using a cup-form sonicator, continuous pulse at 70% duty cycle, 7 output control (see Notes 1–4).
Infect 6-well plates of 40% confluent monolayers of Vero cells with the rVV at a multiplicity of infection (MOI) of 0.2. Use two independent wells per recombination (see Notes 1–4).
Incubate at 37 °C 5% CO2 for 2 h to allow the virus to infect the cells.
-
After 1 h of incubation, prepare the following solutions for transfection:
Solution A: For each transfection: Dilute 5 μg of modified pGPTNEB193 (containing the modified IBV cDNA) in 1.5 mL of OPTIMEM medium.
Solution B: Dilute 12 μL of lipofectin in 1.5 mL of OPTIMEM for each transfection.
Incubate solutions A and B separately for 30 min at room temperature, then mix the two solutions together, and incubate the mixture at room temperature for 15 min.
During the 15-min incubation, remove the inoculum from the vaccinia virus-infected cells and wash the cells twice with OPTIMEM.
Add 3 mL of the transfection mixture (prepared in step 5) to each well.
Incubate for 60–90 min at 37 °C 5% CO2 (see Note 5).
Remove the transfection mixture from each well and replace it with 5 mL of BES medium .
Incubate the transfected cells overnight at 37 °C, 5% CO2.
The following morning add the MXH selection components, MPA 12.5 μL, xanthine 125 μL, and hypoxanthine 7.4 μL, directly to each well (see Note 6).
Incubate the cells at 37 °C 5% CO2 until they display extensive vaccinia virus-induced cytopathic effect (CPE) (normally 2 days).
Harvest the infected/transfected cells into the cell medium of the wells and centrifuge for 3–4 min at 300 × g. Discard supernatant, resuspend the pellet in 400 μL 1× E-MEM , and store at −20 °C.
Plaque Purification in the Presence of GPT Selection Agents: Selection of MPA -Resistant Recombinant Vaccinia Viruses (GPT+ Phenotype)
Freeze-thaw the vaccinia virus produced from Subheading 3.1 three times and sonicate as described in the previous subheading (Subheading 3.1, step 1).
Remove the medium from confluent Vero cells in 6-well plates and wash the cells once with PBSa.
Prepare 10−1 to 10−3 serial dilutions of the recombinant vaccinia virus in 1× E-MEM .
Remove the PBSa from the Vero cells and add 500 μL of the diluted virus per well.
Incubate for 1–2 h at 37 °C 5% CO2.
Remove the inoculum and add 3 mL of the Ecogpt selection medium (see Note 7).
Incubate for 3–4 days at 37 °C 5% CO2 and stain the cells by adding 2 mL of 1× E-MEM containing 1% agar and 0.01% neutral red.
Incubate the cells at 37 °C 5% CO2 for 6–24 h and pick 2–3 well-isolated plaques for each recombinant, by taking a plug of agarose directly above the plaque. Place the plug of agar in 400 μL of 1× E-MEM .
Perform two further rounds of plaque purification for each selected recombinant vaccinia virus in the presence of Ecogpt selection medium, as described in steps 1–8 (see Note 8).
Plaque Purification in the Absence of GPT Selection Agents: Selection of MPA -Sensitive Recombinant Vaccinia Viruses (Loss of GPT+ Phenotype)
Take the MPA -resistant plaque-purified rVVs which have been plaque purified a total of three times as described in Subheading 3.2 and freeze-thaw and sonicate as described in Subheading 3.1, step 1.
Remove the medium from confluent Vero cells in 6-well plates and wash the cells with PBSa.
Prepare 10−1 to 10−3 serial dilutions of the recombinant vaccinia virus in 1× E-MEM .
Remove the PBSa from the Vero cells and add 500 μL of the diluted virus per well.
Incubate for 1–2 h at 37 °C 5% CO2.
Remove the inoculum and add 3 mL of the overlay medium (see Note 9).
Incubate for 3–4 days at 37 °C 5% CO2 and stain the cells by adding 2 mL 1× E-MEM containing 1% agar and 0.01% neutral red.
Incubate the cells at 37 °C 5% CO2 for 6–24 h and pick 3–6 well-isolated plaques for each recombinant, by taking a plug of agar directly above the plaque. Place the plug of agar in 400 μL of 1× E-MEM (see Note 8).
Perform two further rounds of plaque purification for each selected recombinant vaccinia virus in the presence of selection medium, as described in steps 1–8.
Production of Small Stocks of Recombinant Vaccinia Viruses
Take the MPA -sensitive plaque-purified rVVs which have been plaque purified a total of three times as described in Subheading 3.3 and freeze-thaw and sonicate as described in Subheading 3.1, step 1.
Remove the medium from confluent Vero cells in six-well plates and wash the cells with PBSa.
Dilute 150 μL of the sonicated rVVs in 350 μL of BES medium .
Remove the PBSa from the Vero cells and add 500 μL of the diluted rVVs per well.
Incubate at 37 °C and 5% CO2 for 1–2 h.
Add 2.5 mL per well of BES medium .
Incubate the infected Vero cells at 37 °C and 5% CO2 until the cells show signs of extensive vaccinia virus-induced CPE (approx. 4 days).
Scrape the Vero cells into the medium, and harvest into 1.5 mL screw-cap tubes with gaskets.
Centrifuge for 3 min at 16,000 × g in a benchtop centrifuge.
Discard the supernatants, resuspend the cells in a total of 400 μL of BES cell culture medium, and store at −20 °C.
DNA Extraction from Small Stocks of Recombinant Vaccinia Virus for Screening by PCR
There are two methods for DNA extraction:
- DNA extraction using phenol/ chloroform/ isoamyl alcohol.
- To 100 μL of rVV stock produced in Subheading 3.4, add 100 μL 2× proteinase K buffer and 2 μL of the proteinase K stock. Gently mix and incubate at 50 °C for 2 h.
- Add 200 μL of phenol/ chloroform/ isoamyl alcohol to the proteinase K-treated samples, mix by inverting the tube 5–10 times, and centrifuge at 16,000 × g for 5 min (see Note 10).
- Take the upper aqueous phase and repeat step 2 twice more.
- Add 200 μL of chloroform to the upper phase and mix and centrifuge as in step 2.
- Take the upper phase and precipitate the vaccinia virus DNA by adding 2.5 volumes of absolute ethanol ; the precipitated DNA should be visible. Centrifuge the precipitated DNA at 16,000 × g for 20 min. Discard the supernatant.
- Wash the pelleted DNA with 400 μL 70% ethanol and centrifuge at 16,000 × g for 10 min. Discard the supernatant, carefully, and remove the last drops of 70% ethanol using a capillary tip.
- Resuspend the DNA in 30 μL water and store at 4 °C (see Note 11).
- Extraction of rVV DNA using the Qiagen QlAamp DNA mini kit.
- Follow the blood/bodily fluid spin protocol and start with 200 μL of rVV stock produced in Subheading 3.4.
- Elute the rVV DNA in 200 μL buffer AE (provided in the kit) and store at 4 °C.
At this stage the extracted rVV DNA is analyzed by PCR and/or sequence analysis for the presence/absence of the Ecogpt gene and for the modifications within the IBV cDNA sequence. Once an rVV is identified that both has lost the Ecogpt gene and contains the desired IBV modification, large stocks are produced. Typically two rVVs will be taken forward at this stage, which ideally have been generated from different wells of the infection/transfection of Vero cells stage previously described in Subheading 3.1. Once the large stocks of the chosen rVVs have been produced, rVV DNA will be extracted and prepared for the recovery of rIBV.
Production of Large Stocks of Vaccinia Virus
Freeze-thaw and sonicate the chosen rVV stocks from Subheading 3.4 as described in Subheading 3.1, step 1.
Dilute the sonicated virus in BHK-21 maintenance medium and infect 11× T150 flasks of confluent monolayers of BHK-21 cells using 2 mL of the diluted vaccinia virus per flask at an MOI of 0.1–1.
Incubate the infected cells for 1 h at 37 °C and 5% CO2.
Add 18 mL of pre-warmed (37 °C) BHK-21 maintenance medium and incubate the infected cells at 37 °C and 5% CO2 until the cells show an advanced CPE (normally about 2–3 days postinfection). At this stage the cells should easily detach from the plastic.
Either continue to step 6 or freeze the flasks in plastic boxes lined with absorbent material and labeled with biohazard tape at −20 °C until further use.
If prepared from frozen, the flasks need to be defrosted by leaving them at room temperature for 15 min and then at 37 °C until the medium over the cells has thawed.
Tap the flasks to detach the cells from the plastic; if necessary use a cell scraper.
Transfer the medium containing the cells to 50 mL Falcon tubes and centrifuge at 750 × g for 15 min at 4 °C to pellet the cells.
Discard the supernatant (99% of vaccinia virus is cell associated) and resuspend the cells in 1 mL of TE buffer per flask.
Pool the resuspended cells, then aliquot into screw-top microfuge tubes with gasket, and store at −70 °C.
Use one 1 mL aliquot of the resuspended cells as a virus stock. Use the resuspended cells from the remaining 10 flasks for partial purification.
Vaccinia Virus Partial Purification
Freeze-thaw and sonicate the resuspended cells generated from Subheading 3.6 as described in Subheading 3.1, step 1.
Centrifuge at 750 × g for 10 min at 4 °C to remove the cell nuclei.
Keep the supernatant and add TE buffer to give a final volume of 13 mL.
Add 16 mL of the 30% sucrose solution into a Beckman ultra-clear (25 × 89 mm) ultracentrifuge tube and carefully layer 13 mL of the cell lysate from step 3 on to the sucrose cushion.
Centrifuge the samples using an ultracentrifuge at 36,000 × g, 4 °C for 60 min.
The partially purified vaccinia virus particles form a pellet under the sucrose cushion. After centrifugation, carefully remove the top layer (usually pink) and the sucrose layer with a pipette. Wipe the sides of the tube carefully with a tissue to remove any sucrose solution.
Resuspend each pellet in 5 mL TE buffer and store at −70 °C.
Extraction of Vaccinia Virus DNA from Large Partially Purified rVV Stocks
Defrost the partially purified vaccinia virus from Subheading 3.7 at 37 °C.
Add 5 mL of pre-warmed 2× proteinase K buffer and 100 μL of 20 mg/mL proteinase K to the partially purified vaccinia virus in a 50 mL Falcon tube. Incubate at 50 °C for 2.5 h (see Notes 1–4).
Transfer into a clean 50 mL Falcon tube.
Add 5 mL of phenol/ chloroform/ isoamyl alcohol, mix by inverting the tube 5–10 times, and centrifuge at 1100 × g in a benchtop centrifuge for 15 min at 4 °C. Transfer the upper phase to a clean 50 mL Falcon tube using wide-bore pipette tips (see Note 10–11).
Repeat step 3.
Add 5 mL chloroform, mix by inverting the tube 5–10 times, and centrifuge at 1100 × g for 15 min at 4 °C. Transfer the upper phase into a clean 50 mL Falcon tube.
Precipitate the vaccinia virus DNA by adding 2.5 volumes of −20 °C absolute ethanol and 0.1 volumes of 3 M sodium acetate . Centrifuge at 1200 × g, 4 °C for 60–90 min. A glassy pellet should be visible.
Discard the supernatant and wash the DNA using 10 mL −20 °C 70% ethanol . Leave on ice for 5 min and centrifuge at 1200 × g, 4 °C for 30–45 min. Discard the supernatant and remove the last drops of ethanol using a capillary tip. Dry the inside of the tube using a tissue to remove any ethanol .
Air-dry the pellet for 5–10 min.
Resuspend the vaccinia DNA in 100 μL of water. Do not pipette to resuspend as shearing of the DNA will occur.
Leave the tubes at 4 °C overnight. If the pellet has not dissolved totally, add more water.
Measure the concentration of the extracted DNA using a nanodrop or equivalent.
Store the vaccinia virus DNA at 4 °C. DO NOT FREEZE (see Note 6).
Analysis of Vaccinia Virus DNA by Pulsed Field Agarose Gel Electrophoresis (PFGE)
Prepare 2 L of 0.5× TBE buffer for preparation of the agarose gel and as electrophoresis running buffer; 100 mL is required for a 12.7 × 14 cm agarose gel and the remainder is required as running buffer.
Calculate the concentration of agarose that is needed to analyze the range of DNA fragments to be analyzed. Increasing the agarose concentration decreases the DNA mobility within the gel, requiring a longer run time or a higher voltage. However, a higher voltage can increase DNA degradation and reduce resolution. A 0.8% agarose gel is suitable for separating DNA ranging between 50 and 95 kb. A 1% agarose gel is suitable for separating DNA ranging between 20 and 300 kb.
Place the required amount of agarose in 100 mL 0.5× TBE buffer and microwave until the agarose is dissolved. Cool to approximately 50–60 °C.
Clean the gel frame and comb with MQ water followed by 70% ethanol . Place the gel frame on a level surface, assemble the comb, and pour the cooled agarose into the gel frame. Remove any bubbles using a pipette tip, allow the agarose to set (approx. 30–40 min), and store in the fridge until required.
Place the remaining 0.5× TBE buffer into the CHEF-DR® II PFGE electrophoresis tank and switch the cooling unit on. Leave the buffer circulating to cool.
Digest 1 μg of the DNA with a suitable restriction enzyme such as Sal I in a 20 μL reaction.
Add the sample-loading dye to the digested vaccinia virus DNA samples and incubate at 65 °C for 10 min.
Place the agarose gel in the electrophoresis chamber; load the samples using wide-bore tips and appropriate DNA markers (see Note 11).
The DNA samples are analyzed by PFGE at 14 °C in gels run with a 0.1–1.0-s switch time for 16 h at 6 V/cm at an angle of 120° or with a 3.0–30.0-s switch time for 16 h at 6 V/cm depending on the concentration of agarose used.
Following PFGE, place the agarose gel in a sealable container containing 400 mL 0.1 μg/mL ethidium bromide and gently shake for 30 min at room temperature.
Wash the ethidium bromide -stained agarose gel in 400 mL MQ water by gently shaking for 30 min.
Visualize DNA bands using a suitable UV system for analyzing agarose gels. An example of recombinant vaccinia virus DNA digested with the restriction enzyme Sal I and analyzed by PFGE is shown in Fig. 6.
Fig. 6.
Analysis of Sal I-digested vaccinia virus DNA by PFGE. Lane 1 shows DNA markers and Lane 2 the digested vaccinia virus DNA. The IBV cDNA used does not contain a Sal I restriction site; therefore the largest DNA fragment (~31 kb) generated from the recombinant vaccinia virus DNA represents the IBV cDNA with some vaccinia virus-derived DNA at both ends
Preparation of rFPV-T7 Stock
Infectious recombinant IBVs are generated in situ by co-transfection of vaccinia virus DNA, containing the modified IBV cDNA, and pCi-Nuc (a plasmid containing the IBV N gene) into CK cells previously infected with a recombinant fowlpox virus expressing the bacteriophage T7 DNA-dependant RNA polymerase under the direction of the vaccinia virus P7.5 early/late promoter 8 ( rFPV-T7). This protocol covers the procedure for preparing a stock of rFPV/T7 by infecting primary avian chicken embryo fibroblasts (CEFs).
Preparation of a 200 mL stock of rFPV-T7 uses ten T150 flasks containing confluent monolayers of CEFs .
Remove the culture growth medium from the cells and infect with 2 mL rFPV/T7 at an MOI of 0.1, previously diluted in CEF maintenance medium.
Incubate the infected cells for 1 h at 37 °C 5% CO2 and then without removing the inoculum add 20 mL of CEF maintenance medium.
After 4 days of infection check for CPE (90% of the cells should show CPE ). Tap the flasks to detach the cells from the plastic and disperse the cells into the medium by pipetting them up and down.
Harvest into 50 mL Falcon tubes and freeze-thaw the cells three times as described in Subheading 3.1, step 1.
Centrifuge at 750 × g, 4 °C for 5 min to remove the cell debris. Take the supernatant containing the virus stock and store at −70 °C until required.
Determine the titer of the virus stock using CEF cells. The titer should be in the order of 106–107 PFU/mL.
Infection and Transfection of CK Cells for the Recovery of rIBV
Wash 40% confluent CK cells in 6-well plates once with PBSa.
Infect the cells with rFPV-T7 at an MOI of 10 in 1 mL of CK cell culture medium. Typically we carry out ten replicates per recovery experiment.
Incubate for 1 h at 37 °C 5% CO2.
During this infection period prepare the transfection reaction solutions.
Solution A: 1.5 mL OPTIMEM, 10 μg rVV DNA, and 5 μg pCi-Nuc per replicate.
Solution B: 1.5 mL OPTIMEM and 30 μL lipofectin per replicate.
Incubate solutions A and B at room temperature for 30 min.
Mix solutions A and B together producing solution AB, and incubate for a further 15 min at room temperature.
Remove the rFPV-T7 from each well, wash the CK cells twice with OPTIMEM, and carefully add 3 mL of solution AB per well.
Incubate the transfected cells at 37 °C 5% CO2 for 16–24 h.
Remove the transfection medium from each well, replace with 5 mL of BES medium , and incubate at 37 °C 5% CO2.
Two days after changing the transfection media, when FPV/IBV-induced CPE is extensive, harvest the cell supernatant from each well and using a 5 mL syringe, filter through 0.22 μm to remove any rFPV-T7 virus present.
Store the filtered supernatant, referred to as passage 0 (P0 CKC) supernatant at −70 °C.
Serial Passage of rIBVs in CK Cells
To check for the presence of any recovered rIBVs the P0 CKC supernatant is passaged three times, P1–P3, in CK cells (Fig. 4b). At each passage the cells are checked for any IBV-associated CPE and for further confirmation RNA is extracted from P3 CKC supernatant and is analyzed by RT-PCR (see Note 12).
For passage 1 (P1):
Wash the confluent CK cells in 6-well plates once with PBSa.
Add 1 mL of the P0 CKC supernatant per well and incubate at 37 °C 5% CO2 for 1 h.
Without removing the inoculum add 2 mL of BES medium per well.
Check cells for IBV-associated CPE over the next 2–3 days using a bright-field microscope.
Harvest the supernatant from each well and store at −70 °C.
Repeat steps 1–6 for passages P2 and P3 in CK cells.
At P3 any recovered virus is used to prepare a large stock for analysis of the virus genotype and phenotype.
Notes
Vaccinia virus is classified as a category 2 human pathogen and its use is therefore subject to local regulations and rules that have to be followed.
Always discard any medium of solution containing vaccinia virus into a 1% solution of Virkon; leave at least 12 h before discarding.
Flasks of cells infected with vaccinia virus should be kept in large plastic boxes, which should be labeled with the word vaccinia and biohazard tape. A paper towel should be put on the bottom of the boxes to absorb any possible spillages.
During centrifugation of vaccinia virus-infected cells use sealed buckets for the centrifugation to avoid possible spillages.
After 2 h of incubation with the transfection mixture, the cells begin to die. It is best therefore not to exceed 90-min incubation.
It is important that after the addition of each selection agent, the medium is mixed to ensure that the selection agents are evenly distributed. This can be achieved by gently rocking/swirling the plate.
Add an equal volume of 2% agar to the 2× EMEM containing MPA , xanthine, and hypoxanthine and mix well before adding it to vaccinia virus-infected cells. There is skill to making the overlay medium and adding it to the cells before the agar sets. There are a number of methods including adding hot agar to cold medium, or pre-warming the medium to 37 °C and adding agar that has been incubated at 50 °C. Despite the method chosen it is important that all components of the overlay medium are mixed well, and the medium is not too hot when it is added to the cells. If there are problems, 1% agar can be substituted with 1% low-melting agarose.
The first recombinant event in the TDS system will not necessarily occur in the same place in every rVV . It is therefore important to pick a number of plaques from the first round of plaque purification in the presence of GPT selection agents and take a variety of them forward. The following two rounds of plaque purification in the presence of GPT selection agents ensure a single virus population and also that no carry through of the input receiver/wild-type vaccinia virus has occurred.
Previous chapters and protocols have instructed during plaque purification in the absence of GPT selection agents to plate 10−1 rVV dilution in the presence of GPT selection medium and rVV dilutions 10−2 and 10−3 in the absence. When there are no plaques in the 10−1 dilution, it means that the rVV has lost the GPT gene and the plaques are ready to amplify and check for the presence of mutations.
There are risks associated with working with phenol/ chloroform/ isoamyl alcohol and chloroform. It is important to check the local COSHH guidelines and code of practices.
Vaccinia virus DNA is a very large molecule that is very easy to shear; therefore when working with the DNA be gentle and use wide-bore tips or cut the ends off ordinary pipette tips. In addition always store vaccinia virus DNA at 4 °C; do not freeze as this leads to degradation. However, there is an exception to this if the vaccinia virus DNA has been extracted using the Qiagen QlAamp DNA mini kit, as this DNA will have already been sheared (the kit only purifies intact DNA fragments up to 50 bp). This DNA can be stored at −20 °C but it is only suitable for analysis of the rVV genome by PCR and is not suitable for the infection and transfection of CK cells for the recovery of rIBV.
There is always the possibility that the recovered rIBV is not cytopathic. In this case, check for the presence of viral RNA by RT-PCR at passage 3 (P3). It is quite common even with a cytopathic rIBV not to see easily definable IBV-induced CPE at P1 and P2. The recovery process is a low-probability event and the serial passage of rIBVs in CK cells acts as an amplification step.
Contributor Information
Daniel R. Perez, Phone: +11301-314-6811, Email: dperez1@umd.edu
Erica Bickerton, Email: erica.bickerton@pirbright.ac.uk.
Paul Britton, Email: paul.britton@pirbright.ac.uk.
References
- 1.Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol. 2009;155:133–146. doi: 10.1007/s00705-009-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tyrrell DAJ, Almeida JD, Berry DM, et al. Coronaviruses. Nature. 1968;220:650. doi: 10.1038/220819a0. [DOI] [Google Scholar]
- 3.Britton P, Cavanagh D. Avian coronavirus diseases and infectious bronchitis vaccine development. In: Thiel V, editor. Coronaviruses: molecular and cellular biology. Norfolk, UK: Caister Academic Press; 2007. [Google Scholar]
- 4.Ambali AG, Jones RC. Early pathogenesis in chicks of infection with an enterotropic strain of infectious bronchitis virus. Avian Dis. 1990;34:809–817. doi: 10.2307/1591367. [DOI] [PubMed] [Google Scholar]
- 5.Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34:439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- 6.Cavanagh D, Gelb J., Jr . Infectious bronchitis. In: Saif YM, editor. Diseases of poultry. 12th. Iowa: Blackwell Publishing; 2008. [Google Scholar]
- 7.Jones RC. Viral respiratory diseases (ILT, aMPV infections, IB): are they ever under control? Br Poult Sci. 2010;51:1–11. doi: 10.1080/00071660903541378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cook JKA, Mockett APA. Epidemiology of infectious bronchitis virus. In: Siddell SG, editor. The coronaviridae. New York and London: Plenum Press; 1995. [Google Scholar]
- 9.Schalk AF, Hawn MC. An apparently new respiratory disease of baby chicks. J Am Vet Ass. 1931;78:413–422. [Google Scholar]
- 10.Beach JR, Schalm OW. A filtrable virus distinct from that of laryngotracheitis: the cause of respiratory disease of chicks. Poult Sci. 1936;15:199–206. doi: 10.3382/ps.0150199. [DOI] [Google Scholar]
- 11.Beaudette FR, Hudson CB. Cultivation of the virus of infectious bronchitis. J Am Vet Med Assoc. 1937;90:51–60. [Google Scholar]
- 12.ZZ0102, Defra report on the Economic assessment of livestock diseases in the United Kingdom (UK) http://randd.defra.gov.uk/Default.aspx?Menu=Menu&Module=More&Location=None&ProjectID=9781&FromSearch=Y&Publisher=1&SearchText=ZZ0102&SortString=ProjectCode&SortOrder=Asc&Paging=10#Description
- 13.Bennett R. The direct costs of livestock disease: the development of a system of models for the analysis of 30 endemic livestock diseases in Great Britain. J Agric Econ. 2003;54:55–71. doi: 10.1111/j.1477-9552.2003.tb00048.x. [DOI] [Google Scholar]
- 14.Bennett R, Jpelaar JI. Updated estimates of the costs associated with thirty four endemic livestock diseases in Great Britain. J Agric Econ. 2005;56:135–144. doi: 10.1111/j.1477-9552.2005.tb00126.x. [DOI] [Google Scholar]
- 15.Casais R, Thiel V, Siddell SG, et al. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J Virol. 2001;75:12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiel V, Siddell SG. Reverse genetics of coronaviruses using vaccinia virus vectors. Curr Top Microbiol Immunol. 2005;287:199–227. doi: 10.1007/3-540-26765-4_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falkner FG, Moss B. Transient dominant selection of recombinant vaccinia viruses. J Virol. 1990;64:3108–3111. doi: 10.1128/jvi.64.6.3108-3111.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Britton P, Evans S, Dove B, et al. Generation of a recombinant avian coronavirus infectious bronchitis virus using transient dominant selection. J Virol Method. 2005;123:203–211. doi: 10.1016/j.jviromet.2004.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boulanger D, Green P, Smith T, et al. The 131-amino-acid repeat region of the essential 39-kilodalton core protein of fowlpox virus FP9, equivalent to vaccinia virus A4L protein, is nonessential and highly immunogenic. J Virol. 1998;72:170–179. doi: 10.1128/jvi.72.1.170-179.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulligan R, Berg P. Selection for animal cells that express the E.coli gene coding for xanthine-guanine phosphoribosyl transferase. Proc Natl Acad Sci U S A. 1981;78:2072–2076. doi: 10.1073/pnas.78.4.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Britton P, Green P, Kottier S, et al. Expression of bacteriophage T7 RNA polymerase in avian and mammalian cells by a recombinant fowlpox virus. J Gen Virol. 1996;77:963–967. doi: 10.1099/0022-1317-77-5-963. [DOI] [PubMed] [Google Scholar]
- 22.Hiscox JA, Wurm T, Wilson L, et al. The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J Virol. 2001;75:506–512. doi: 10.1128/JVI.75.1.506-512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ausubel FM, Brent R, Kingston RE, et al. Current protocols in molecular biology. New York: John Wiley and Sons, Inc.; 1987. [Google Scholar]
- 24.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual/second edition. 2. New York: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 25.Mackett M, Smith GL, Moss B. The construction and characterisation of vaccinia virus recombinants expressing foreign genes. In: Glover DM, editor. DNA cloning, a practical approach. Oxford: IRL Press; 1985. pp. 191–211. [Google Scholar]
- 26.Smith GL. Expression of genes by vaccinia virus vectors. In: Davison MJ, Elliot RM, editors. Molecular virology, a practical approach. Oxford: IRL Press; 1993. pp. 257–283. [Google Scholar]