

Abstract
Finding new therapeutic targets of glomerulosclerosis treatment is an ongoing quest. Due to a living environment of various stresses and pathological stimuli, podocytes are prone to injuries; moreover, as a cell without proliferative potential, loss of podocytes is vital in the pathogenesis of glomerulosclerosis. Thus, sufficient understanding of factors and underlying mechanisms of podocyte injury facilitates the advancement of treating and prevention of glomerulosclerosis. The clinical symptom of podocyte injury is proteinuria, sometimes with loss of kidney functions progressing to glomerulosclerosis. Injury-induced changes in podocyte physiology and function are actually not a simple passive process, but a complex interaction of proteins that comprise the anatomical structure of podocytes at molecular levels. This chapter lists several aspects of podocyte injuries along with potential mechanisms, including glucose and lipid metabolism disorder, hypertension, RAS activation, micro-inflammation, immune disorder, and other factors. These aspects are not technically separated items, but intertwined with each other in the pathogenesis of podocyte injuries.
Keywords: Podocyte injury, Glomerular sclerosis
Introduction
The Structure and Physiology of Podocytes
The glomerular filtration membrane constituted by three components, porous endothelial cells, glomerular basement membrane (GBM), and epithelial cells in the GBM, which also called podocytes. Podocytes are highly differentiated epithelial cells consist of three distinct parts: cell body, major processes, and foot processes (FPs). Podocytes have a voluminous cell body, which is at the central position of the cell and lies in the urinary space. The cell body contains a nucleus, abundant endoplasmic reticulum, a well-developed Golgi system, lysosomes, and mitochondria. The densely distributed organelles in the cell body suggest a high level of anabolic and catabolic activity. Microtubules and intermediate filaments, such as vimentin and desmin, are the dominated cytoskeleton components in cell body to accounts for the unique shape of podocytes (Pavenstadt et al. 2003). From the cell body, podocytes give rise to primary processes that reach to glomerular capillary, forming an affixation by FPs.
Podocytes are polarized epithelial cells which contain a apical/luminal and a basal cell membrane. The apical surface domain forms a few fingerlike protrusions which bulge into Bowman’s space. The apical domain is negatively charged, which limits the passage of albumin (also negatively charged) and maintain the separation of adjacent podocytes by anion charge. The basal cell membrane mediates the affixation to the GBM by α3β1 integrin and α- and β-dystroglycans, which play the function by connecting to certain matrix proteins within the GBM (Kreidberg et al. 1996; Raats et al. 2000). Both of apical and basal membranes contain numerously distributed cholesterol-rich domains, and it was found that specific membrane proteins of podocytes are obviously arranged in rafts (Schwarz et al. 2001; Simons et al. 2001).
FPs functionally consist of a luminal or apical membrane domain and a basal cell membrane domain. FPs are characterized by a podosome-like cortical network of short, branched actin filaments and by the presence of highly ordered, parallel contractile actin filament bundles, which are thought to modulate the permeability of the filtration barrier through changes in FP morphology (Greka and Mundel 2012). The foot processes of neighboring podocytes are bridged by slit diaphragm (SD), which is the site of convective fluid flow through the visceral epithelium and the final barrier to urinary protein loss. Similar to the apical membrane domain of podocytes, the SD is also covered by a thick surface coat mainly constituted by sialoglycoproteins, including podoendin, podocalyxin, and others, which are responsible for the high negative surface charge of the podocytes. In addition, several molecules, including ZO-1 (zonula occludens protein), nephrin, CD2AP (CD2-associated protein), FAT, and P-cadherin, have all been shown to be expressed within the SD, and some of those molecules play a major role for its integrity (Pavenstadt et al. 2003).
The unique shape of podocyte and the maintenance of its processes are owing to a well-developed cytoskeleton, which serves as the podocyte’s “backbone.” And also, the actin-rich cytoskeleton makes podocytes to be able to alter shape continually and dynamically. The cytoskeleton is comprised by microfilaments (7–9 nm diameter), intermediate filaments (10 nm diameter), and microtubules (24 nm diameter), which are mainly defined by their diameter. Microtubules and intermediate filaments are predominant cytoskeletal constituents in the cell body and the primary processes. In the FPs, microfilaments are the main cytoskeletal component, which consist of a network with densely accumulated F-actin and myosin. FP actin cytoskeleton is extensively distributed in all three domains of FPs, resulting to an important role of actin for the function and dysfunction of podocytes. FP effacement requires the activation of actin filaments reorganization, a process which is regulated by multiple signaling events involving integrin activation, G protein-coupled receptor (GPCR) and growth factor receptor, and calcium (Ca2+) influx pathways as upstream modulators of the actin cytoskeleton (Takeda et al. 2001).
The complex architecture of podocytes, in particular on the maintenance of highly ordered, parallel, contractile actin filament bundles in FPs, is required for the highly specialized functions of podocytes, which include (i) a size barrier to protein; (ii) charge barrier to protein; (iii) maintenance of the capillary loop shape; (iv) counteracting the intraglomerular pressure; (v) synthesis and maintenance of the GBM; (vi) production and secretion of vascular endothelial growth factor (VEGF) required for GEN integrity (Shankland 2006).
Podocyte is the most differentiated cell type in the glomerulus, which plays a crucial role in the glomerular filtration barrier. Podocyte foot processes with the interposed SD represent the last filtration barrier of GBM. The SD is a subtle signal transduction unit characterized by a modified adherens junction that bridges the 30–50-nm-wide filtration slits (Reiser et al. 2000). Transmembrane proteins such as nephrin and FAT constitute the rod-like units of SD which are connected by numerous linear bar, forming a network with pores the same size as or smaller than albumin (Mundel and Kriz 1995). Meanwhile, negatively charged apical domain of SD works as a charge barrier to prevent the albumin loss. Thus, the podocyte is the important size and charge barrier of GBM, and podocytes’ damage leads to the disruption of GBM integrity and proteinuria.
Podocytes stabilize glomerular architecture owing to FPs counteract distensions of the glomerular basement membrane, which is regulated by vasoactive hormones. In this regard, they are responsible for 40% of the hydraulic resistance of the filtration barrier (Pavenstadt 2000). ANG II regulates the contractile state of their foot processes by activating a Cl− conductance and increasing [Ca2+]i, cAMP in podocytes, thereby modulating the ultrafiltration coefficient Kf. Other agonists such as AVP, oxytocin, norepinephrine, and parathormone have also been reported to modulate [Ca2+]i in podocytes. Vasoactive hormones may also alter charge properties of the podocyte and thereby enhance urinary protein excretion (Pavenstadt 2000).
VEGF family consists of five secreted homodimeric glycoproteins: VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor. In human and murine kidneys, VEGF-A isoform is constitutively expressed in podocytes, while playing its role mainly by contact with VEGFR-1 and VEGFR-2 predominately localized on the glomerular endothelial cells. It was assumed that VEGF-A is critical for the regulation of endothelial cell survival, proliferation, differentiation, and migration as well as endothelium-dependent vasodilatation and vascular permeability (Advani 2014). The complex paracrine signaling pathway between podocytes and glomerular endothelial cells plays a central role in maintaining the structure and integrity of the kidney filtration barrier.
The Role of Podocyte Injury in the Progresses of Glomerulosclerosis
Podocyte injury is the common pathological process in many glomerular diseases such as minimal change disease, membranous glomerulopathy, focal segmental glomerulosclerosis (FSGS), diabetic nephropathy (DN), and lupus nephritis. Physiological stresses or pathological stimuli like mechanical stress, oxidative stress, and immunologic stress disrupt the homeostasis of glomerular filtration barrier. Transcapillary pressure increment is induced by glomerular hypertension/hyperfiltration, and podocyte processes’ elongation is induced by capillary expansion contribute to cytoskeletal dysregulation and intrinsic stress (Neal et al. 2007). Pathological factors, such as ischemia–reperfusion, chemical/toxic substances from the primary urine, usually cause reactive oxygen species (ROS) production in podocytes (Chen et al. 2013). It was also reported that aldosterone and angiotensin II promoted receptor-mediated ROS generation in podocytes (Liu et al.2013). Immunologic stress is induced by cytokine/complement, such as CC chemokine receptor 2, tumor necrosis factor, and sublytic C5b-9-mediated intracellular stress in podocytes (Nagata 2016).
Typical electron microscopy manifestations of podocyte injury include microcystic, pseudocystic changes, vacuolization, the presence of cytoplasmic inclusion bodies, and detachment from the GBM. Besides those changes, foot process effacement is the most characteristic change in podocyte injury. The damage of SD proteins contributes to cytoskeleton disorganization, leading to podocyte effacement and proteinuria (Shankland 2006). SD between adjacent podocytes is constituted predominantly by SD proteins including nephrin, podocin, CD2AP, Neph1, and FAT1. Mutations/abnormalities of those proteins result proteinuria and kidney disease. Studies have shown that SD proteins regulate cytoskeleton organization and podocyte shape by interacting with proteins associated with actin cytoskeleton. FAT-1 is an organizer of actin polymerization. CD2AP connects the nephrin complex with the actin-modifying proteins WASP, CAPZ, cortactin, and the Arp2/3 complex.
Reduced podocyte number causes proteinuria and glomerulosclerosis. Podocyte detachment, podocyte apoptosis, and the lack of adequate podocyte proliferation are three main reasons leading to the decrease in podocyte number also called “podocytopenia.” The lack of charge- and size-selective barriers induced by podocyte loss leads to proteinuria. Studies have demonstrated the correlation of podocytes reduction with the onset and progression of glomerulosclerosis. Because podocytes counteract the outward forces of glomerular pressures and maintain capillary loop shape, podocyte loss results to local bulging of the GBM when glomerular pressures increase in many renal diseases. The denuded GBM tends to form a synechia attachment by contacting with the parietal epithelial cells and Bowman’s capsule, which is thought to be the first “committed step” of focal segmental glomerular sclerosis (FSGS) (Kriz et al. 1994, 1998a, b).
Podocytes maintain a healthy intraglomerular environment by cross talk with glomerular endothelial cells. Endothelial cell swelling and attenuation of fenestrae are observed in podocyte injury models by ultrastructural study (Kriz et al. 2013). It was illustrated that podocyte injury disrupts intracapillary homeostasis, causing thrombotic micro-angiopathy and mesangial abnormalities by reducing VEGF signaling (Eremina et al. 2008; Kobayashi et al. 2015).
Glomerulosclerosis is a terminal consequence of podocyte injury. The classic type of glomerulosclerosis, as defined by segmental obliteration of glomerular capillaries by the extracellular matrix, has been believed to progress to complete sclerosis without regression (Nagata 2016). In early stage of FSGS, cellular lesions including transformed podocytes were accompanied by segmental sclerosis, supporting the fact that podocyte damage might be an early event of glomerulosclerosis. In a recent elegant review by Kim JS and his colleagues, the essential steps of glomerulosclerosis were suggested as follows: (1) increased glomerular capillary pressure and filtration flow through podocyte slits, (2) foot process effacement as an adaptive response, (3) podocyte hypertrophy and glomerulomegaly, (4) mismatch between glomerular tuft growth and podocyte hypertrophy, (5) stretching and attenuation of podocyte cell body, (6) pseudocysts formation by hindered flow of filtrates beneath the podocyte that is partially detached on bare areas of GBM, (7) complete podocyte detachment by enlarged pseudocysts and adhesion to Bowman’s capsule, (8) glomerular tuft’s adhesion to Bowman’s capsule, (9) spreading of filtrates to interstitium out of nephron through adhesion structure, and (10) interstitial proliferation and nephron degeneration (Kim et al. 2016).
The Role of Glucose Metabolism Disorder in Podocyte Injury
Podocytes’ injury and depletion was a crucial step in the development of albuminuria in DN. In DN, the number of podocyte-specific markers and podocytes number is decreased, which leads to the occurrence of albuminuria and further develops into glomerulosclerosis. Hyperglycemia is the main pathological change of diabetes and plays an important role in promoting the occurrence and development of DN. Increased intracellular glucose could induce multiple cell and molecular events in podocyte: (1) generation of reactive oxygen species (ROS) and advanced glycation end products (AGEs), (2) increased flux of polyols and hexosamines, (3) activation of protein kinase C (PKC), (4) increased cytokines and growth factors, (5) aberrant Notch signaling, and (6) activate the renal RAS. These abnormal molecular pathological changes mediate the functional and morphological changes of podocytes in a direct or indirect way, including podocyte hypertrophy, epithelial mesenchymal transition (EMT), podocyte detachment, and podocyte apoptosis.
Podocyte injury is a key factor in the development of DN. Recent studies in both type 1 and type 2 diabetes have proposed that a reduction in the number of podocytes may lead to the development of proteinuria. It is reported that the structure and function of podocytes are abnormal under high glucose conditions, such as podocyte fusion, septal injury, and podocyte loss. There has been evidence that podocytes possess a completely functional system for glucose uptake (Lewko et al. 2005). Coward et al. have revealed that the cultured human podocytes express glucose transporter (GLUTs) in two forms: GLUT1 and GLUT4, which participate in insulin-dependent glucose transport to the cell (Coward et al. 2005, 2007). In addition, the podocyte split protein, such as Nefin, is also involved in glucose transport. Schiffer et al. have demonstrated that podocytes also express another insulin-sensitive glucose transporter, GLUT8 (Schiffer et al. 2005). GLUT1 is the primary glucose transporter in most cells as well as in podocytes (Coward et al. 2005, 2007). In diabetes, hyperglycemia and alteration of glucose transporter cause increased intracellular glucose concentration in podocyte and lead to severe impairment of the glomerular filtration barrier. Conversely, Zhang et al. found that enhancement of GLUT1 expression in diabetic podocyte significantly reduced the mesangial expansion and fibronectin accumulation by inhibiting the expression of vascular endothelial growth factor (VEGF) (Zhang et al. 2010). Similarly to other cells, under high glucose condition, podocyte can undergo many pathological changes induced by aberrations in various cellular and molecular events. High glucose induces generation of advanced glycation end products (AGEs) and reactive oxygen species (ROS), increased flux of polyols and hexosamines, increased activity of protein kinase C (PKC), upregulated expression of cytokines and growth factors including vascular endothelial growth factor (VEGF), and transforming growth factor-beta (TGF-β), induces aberrant Notch signaling, and activates the renal RAS (Anil Kumar et al. 2014).
Pathomechanism of podocyte injury in DN mainly includes podocyte hypertrophy, EMT, podocyte detachment, and podocyte apoptosis. Podocyte hypertrophy is the initial stage of podocyte injury in early DN. Hyperglycemia upregulated the expression of cyclin-dependent kinase p27kip1, which leads to further cell cycle arrest and hypertrophy. It was found that p27kip1-/- mice had significantly improved renal damage in DN (Wolf et al. 2005). Several studies suggested high glucose-induced podocyte hypertrophy by activating mTORC1 pathway (Fantus et al. 2016). In addition, hyperglycemia increased expression of nuclear STAT3 via the activation of the upstream signal transduction element Gp130, which eventually leads to podocyte hypertrophy. Excessive hypertrophy could result in degenerative changes in podocyte structure and functions, leading to its detachment from glomerular basement membrane (GBM). Previous studies have shown that phenotype conversion of podocyte was involved in the early stage of podocyte deletion in DN by inducing podocyte detachment or podocyte apoptosis. Podocyte EMT is a manifestation of podocyte phenotype conversion and one of the initiating factors leading to a variety of glomerular diseases. When EMT occurs, the cells lose their original characteristics, resulting in disappearance of intercellular contact, impaired cell polarity, and expression of mesenchymal markers such as alpha smooth muscle actin (alpha-SMA) and fibroblast-specific protein 1 (FSP1). EMT is also an explanation for podocyte depletion in DN (Yamaguchi et al. 2009). Emerging evidence suggested that podocytes could undergo EMT in DN, characterized by loss of epithelial features such as nephrin and P-cadherin, while expressing mesenchymal markers such as FSP-1, type I collagen, and fibronectin (Reidy and Susztak 2009). Xing et al. (2015) demonstrated that stimulation with high glucose for 48 h could activate the PI3 K/AKT pathway in podocyte, and thereby induce the protein expressions of α-SMA and desmin. Dai et al. (2012) suggested that connective tissue growth factor (CTGF) and integrin-linked kinase (ILK) were involved in high glucose-induced phenotypic alterations of podocytes. Lv et al. (2013) findings elaborated that Rac1/PAK1 signaling contributed to high glucose-induced podocyte EMT via promoting β-catenin and Snail transcriptional activities, which could be a potential mechanism involved in podocytes injury in response to stimuli under diabetic conditions. Guo et al. indicated high glucose can also activate β-catenin and Snail expressions by upregulating GSK-3β. In addition, hyperglycemia-induced podocyte detachment by decreasing the expression of key proteins involved in the foot process actin cytoskeleton, split diaphragm (SD) integrity, and podocyte–GBM interactions. A3b1 integrins are the important transmembrane protein involved in anchoring foot processes in the GBM. High glucose regulates the expression of integrin subunits and inhibits the synthesis of agrin. Therefore, high glucose affects not only the structure of podocytes, but also their ability to adhere to GBM (Chen et al. 2000; Han et al. 2006; Yard et al. 2001). It is found that high glucose can alter podocyte adhesion by decreasing expression of integrin α3β1v which was an important receptor that could tightly connect podocyte with the GBM and participated in the adhesion function of podocyte. In addition, α-Actinin, an actin filament for protein crosslink, is also an important factor required for podocyte adhesions (Dandapani et al. 2007). High glucose and AGE treatment resulted in α-actinin-4 expression changes and induces cytoplasmatic translocation in podocyte (Ha 2006). There are some evidences that podocyte apoptosis played a role in reduction in density and number of glomerular in DN. High glucose led to podocyte apoptosis by increased production of ROS, activation of poly(ADP-ribose) polymerase, NF-kB, and p38 MAP kinase (Susztak et al. 2006; Szabo et al. 2006). In diabetes, the surface receptors of the AGEs are upregulated in the podocytes (Tanji et al. 2000). Binding AGEs to receptors activates activated transcription factor FOXO4, which also induced podocyte apoptosis via p38 protein kinase signaling pathways (Cohen et al. 2005). In addition, high glucose increased the protein expression of Nestin, which is a VI intermediate filament protein-related cell cytoskeleton, thereby increased podocyte apoptosis rate (Liu et al. 2012). High glucose increased the expression of TGF-β1 in podocyte. TGF-β1 could induce podocyte apoptosis by directly activate Smad7, p38 MAP kinase, and Notch pathway (Li et al. 2004).
In addition to its direct effects, elevated glucose may act indirectly, via the proinflammatory response, Ang II-dependent pathways, and lipid accumulation. Under high glucose conditions, secretion of the MCP-1 protein by cultured podocytes was increased rapidly (Han et al. 2004), and similar effect was observed in podocyte stimulated with AGEs (Gu et al. 2006). Podocyte can also express TNF-α, a cytokine produced by various immune cells, in response to high glucose stimulation and in diabetic conditions (Ikezumi et al. 2008; Ruster et al. 2009). High glucose could stimulate activity and expression of the local RAS components in podocyte, including Ang II and its AT1 receptors (Yoo et al. 2007). Following that, it was recently demonstrated that local RAS activation would lead to podocyte injury through a variety of pathways. Ang II could induce podocyte apoptosis through activation of NADPH oxidase and production of ROS, and upregulate the expression of GLUT transporters (Gill and Wilcox 2006; Nose et al. 2003). In addition, Ma et al. found that lipid accumulation in podocytes was increased under the high glucose stimulation, which is mediated through the disruption of low-density lipoprotein receptor (LDLr) pathway (Zhang et al. 2015a). Interestingly, reducing lipid accumulation in podocytes decreased the protein expression of SMA and increased the expression of nephrin in podocyte. These studies reveal that high glucose-induced lipid accumulation is involved in the podocyte injury in DN. Therefore, the above shows that high glucose could induce various other metabolic disorders and indirectly lead to podocyte injury.
Lipid Metabolism Disorder in Podocyte Injury
Lipid metabolism disorder is commonly observed in patients with chronic kidney disease (CKD), accompanied by increased fasting triglyceride levels and decreased high-density lipoprotein cholesterol (HDL-C) (Bianchi et al. 2016). It is increasingly recognized that dysregulation of lipid metabolism is involved in the development and progression of CKD, such as obesity-related renal disease and DN (de Vries et al. 2014). Podocytes, as specialized cells of glomerulus, play an important role in the pathologist of CKD when they are injured (Fiorina et al. 2014). And excessive lipid accumulation in podocytes can lead to cellular dysfunction and death, which is called lipotoxicity.
Cholesterol
Between neighboring podocytes, there is a unique interdigitating structure bridged by SD, maintaining the proper glomerular filtration (Ruotsalainen et al. 1999). Researches have revealed that SD is a lipid raft structure containing multiple podocyte-specific proteins, such as podocin and nephrin (Schermer and Benzing 2009). In particular, podocin can recruit and bind to cholesterol to form SD, and this binding can influence the composition of lipid membrane, allowing cholesterol to contact with the ion-channel transient receptor potential canonical 6 (TRPC6) (Huber et al. 2006). This suggests that cholesterol homeostasis is essential for glomerular functions. However, excessive cholesterol can also negatively disrupt the mutual binding of podocyte SD proteins, or interfere with the binding between podocyte SD proteins and caveolin-1, a lipid raft-associated protein, binding nephrin, and Cluster of Differentiation 2 (CD20)-associated protein (Sorensson et al. 2002)
The content and distribution of cellular cholesterol regulated by cholesterol synthesis and intracellular trafficking.
It is regulated by some functional proteins such as ATP-binding cassette transporter A1 (ABCA1) involving cholesterol efflux, 3-hydroxy-3-methyl-glutaryl CoA reductase (HMG-CoA reductase, HMGCR) regulating cholesterol synthesis and low-density lipoprotein receptor (LDLR) mediating cholesterol influx. The expression of HMGCR and LDLR is regulated by some transcription factors, such as the sterol regulatory element-binding protein (SREBP), under negative feedback loops. When cells are rich in cholesterol or its derivatives, the transcription of LDLR gene or other genes necessary to lipid synthesis are suppressed. As a result, the cells are not able to generate and uptake cholesterol, and then establish cholesterol homeostasis. In contrast, when intracellular sterols are exhausted, the transcriptions of SREBP target genes will be activated, increasing intracellular cholesterol (Zhang et al. 2016). This enables cellular cholesterol homeostasis despite physiological fluctuations in cholesterol requirements and exogenous supply.
However, it is demonstrated that the cellular cholesterol imbalance of podocytes can induce proteinuric glomerular diseases (Merscher et al. 2014). It is demonstrated that human glomerular podocytes express ABCA1, HMGCR, and LDLR (Merscher-Gomez et al. 2013). Ma et al. found that some pathogenic factors such as inflammation can disrupt LDLR pathway (Zhang et al. 2015b). Thus, excessive lipid accumulates in podocytes, resulting in effacement of the foot processes and epithelial mesenchymal transition of podocytes (Zhang et al. 2015b). It is recently demonstrated that human podocytes treated with the sera from diabetic kidney disease (DKD) patients had increased cholesterol accumulation compared with human podocytes exposed to the sera of patients with diabetes, but no DKD. This was associated with a reduction of ABCA1 and an impairment of cholesterol efflux (Merscher-Gomez et al. 2013). Besides, it is showed that c-x-c motif ligand 16 (CXCL16) is the main scavenger receptor for oxidized LDL (oxLDL) in human podocyte (Gutwein et al. 2009). The expression of glomerular CXCL16 was increased in patients with membranous nephropathy, accompanied with higher levels of oxLDL (Gutwein et al. 2009). And in diabetic db/db mice, CXCL16 pathway was activated, in parallel with increased cholesterol accumulation in kidney (Hu et al. 2018). In vitro, oxLDL can induce loss of nephrin expression from cultured podocytes (Bussolati et al. 2005).
In summary, cholesterol metabolism disorder can destroy the structure and function of podocytes, leading to the progression of CKD.
Fatty Acids and Triglycerides
In addition to hypercholesterolemia, free fatty acids (FFAs) can also affect podocyte function in kidney disease. The essential role of fatty acids is to form the phospholipid bilayers of the cell membranes and act as phospholipid messengers, transmitting vital intracellular signals (Lee 2011). Normal cellular fatty acid homeostasis reflects a balance between generation or delivery and utilization. SREBP-1c is involved in fatty acid and TG synthesis, targeting lipogenic enzymes including acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) (Horton et al. 2002). FFAs can be transported into cells by the scavenger receptor platelet glycoprotein 4 (also called as CD36) or via the assistance of vascular endothelial growth factor B (VEGF-B) (Hagberg et al. 2010; Masuda et al. 2009). Cellular FFAs are esterified or transported into the mitochondria for oxidation and subsequent energy production (Lee 2011).
Palmitic and stearic acids, belonging to saturated FFAs (SFAs), and oleic acid, belonging to monounsaturated FFAs (MUFAs), account for 70–80% of plasma FFAs (Raclot et al. 1997). SFAs can induce insulin resistance and cell death, involving the pathogenesis of diabetes mellitus type 2 (T2DM) (Lennon et al. 2009; Sieber et al. 2010). In contrast, MUFAs can prevent SFA-induced lipotoxicity (Sieber et al. 2010). In human podocytes, insulin resistance can be induced by palmitic acid (Lennon et al. 2009). It is observed that insulin sensitivity in glomeruli of obese and diabetic rats is reduced (Mima et al. 2011). Podocyte-specific insulin receptor knockout mice develop albuminuria and glomerulosclerosis, indicating that normal insulin signaling is critical for podocyte function and survival (Welsh et al. 2010). These findings imply that FFAs play potential roles in insulin resistance, promoting the development and progression of obesity-related renal disease and DN.
In the tubulointerstitial and glomerular segment of renal biopsies obtained from patients with DN, endoplasmic reticulum (ER) stress is observed (Sieber et al. 2010). Importantly, in a T1D mouse model, the progression of DN can be attenuated by ameliorating ER stress (Qi et al. 2011). ER dyshomeostasis can decrease the ER folding capacity, thereby leading to accumulation of unfolded and misfolded proteins. This in turn initiates the unfolded protein response (UPR), adaptively maintaining proper ER function (Ma and Hendershot 2001). But if ER stress persists, apoptosis will be induced by the proapoptotic transcription factor C/EBP homologous protein (CHOP) (Rasheva and Domingos 2009). In podocytes, ER stress induced by palmitic acid results in the upregulation of several UPR markers/effectors, such as the ER chaperone heavy chain-binding protein (BiP), and CHOP, while monounsaturated palmitoleic and oleic acids only upregulated BiP but not CHOP (Sieber et al. 2010). As BiP can attenuate palmitic acid-induced apoptosis (Laybutt et al. 2007), the beneficial effect of MUFAs may own to the upregulation of BiP. In addition to the unfolded proteins, alterations in ER membrane lipid composition can also sensitively affect the expression of the ER stress sensor inositol requiring enzyme 1 (IRE-1) (Promlek et al. 2011). It is shown that small molecule compound 4m8C, specific IRE-1 inhibition, can attenuate palmitic acid-induced podocyte death (Sieber and Jehle 2014).
Enhanced FFA uptake by podocytes is induced by increased expression of CD36 and a decrease in fatty acid β-oxidation, leading to excessive intracellular lipid accumulation (Soetikno et al. 2013). In animal model of type 1 diabetes (T1D), increased expression of SREBP-1 in renal results in upregulation of enzymes responsible for FFA synthesis and as a consequence of a high level of triglyceride (TG) in renal (Hashizume and Mihara 2012). Accumulated lipids in podocytes limited mitochondrial fatty acids β-oxidation. It induced mitochondrial damage and inhibition of AMP kinase (AMPK) activity, leading to endoplasmic reticulum (ER) stress, autophagy, and apoptosis in podocytes. As a result, mitochondrial dysfunction caused decreased podocyte density and increased in foot process width, together with inflammation (Szeto et al. 2016). Renal accumulation of TG is associated with reduced expression of the ultrasensitive energy sensor AMPK strongly. This suggests that the imbalance between energy-generating and energy-consuming pathways might be related to podocyte dysfunction in DKD and other disorders in CKD, due to lipid accumulation (Wahl et al. 2016). And hypertriglyceridemia can also increase podocytic de novo expression of desmin, which represents podocyte injury (Joles et al. 2000).
Gangliosides and Sphingolipids
Since the first description that glycosphingolipid accumulation in the renal results in glomerular hypertrophy in streptozotocin (STZ)-induced diabetic mice, several studies have highlighted the role of sphingolipids and gangliosides in podocyte biology (Merscher-Gomez et al. 2013).
Analysis of kidney biopsy compartments from 14 patients with Fabry disease using unbiased quantitative stereology indicated age-dependent accumulation of globotriaosylceramide (Gb3) in podocytes (Najafian et al. 2011). In vitro, globotriaosylsphingosine (known as lysoglobotriaosylceramide) acts as a profibrotic metabolite in cultured human podocytes (Sanchez-Nino et al. 2011). Ganglioside GM3 (GM3) is a receptor for soluble Flt1, locating in lipid raft domains in the SD of podocytes. Binding of soluble Flt1 to GM3 plays essential roles in autocrine preservation of the podocyte actin cytoskeleton and in prevention of proteinuria (Jin et al. 2012). O-acetylated disialosyllactosylceramide (GD3), a sialic-acid-containing lipid, was identified as a podocyte-specific ganglioside in rat (Reivinen et al. 1992). Treating mice with an antibody against GD3 caused nephrin phosphorylation and dislocation from the podocyte SD (Simons et al. 2001).
It is an emerging concept that sphingolipids act as modulators of podocyte function in FSGS and other glomerular diseases. Patients with FSGS are more likely to have recurrence of proteinuria after kidney transplantation. And the number of acid sphingomyelinase-like phosphodiesterase 3b (SMPDL3b) positive podocytes is decreased in patients with recurrent proteinuria (Fornoni et al. 2011).
To sum up, lipid metabolism disorder is involved in the pathogenesis of podocyte injury. Cholesterol helps form SD between podocytes, maintaining the proper glomerular filtration. LDL-cholesterol uptake is mediated via the LDLR or CXCL16 and may cause ER stress. Cholesterol metabolism is regulated by several nuclear receptors and transcription factors, including SREBP. Excessive cholesterol accumulation in podocytes may contribute to kidney disease. Free fatty acids are primarily transported via CD36, causing oxidative and ER stress based on the degree of saturation. Sphingolipids and gangliosides also play a role in podocyte biology. Binding of soluble Flt1 to GM3 plays essential roles in autocrine preservation of the podocyte actin cytoskeleton and in prevention of proteinuria (Fig. 10.1).
Fig. 10.1.

Lipid metabolism disorder is involved in the pathogenesis of podocyte injury
Role of Hypertension in the Damage of Podocytes
Hypertension has become the second leading cause of end-stage renal disease (ESRD) after diabetes mellitus (Udani et al. 2011). High blood pressure can affect renal vessels, glomeruli, and tubulointerstitium. Recently, more and more studies have indicated that podocyte damage play an important role in hypertensive nephrosclerosis. Decreased intrarenal podocyte and increased urinary podocyte were observed in hypertensive nephrosclerosis (Wang et al. 2009). As terminally differentiated cells, podocyte loss leads to denudation of the glomerular basement membrane (GBM) and focal adhesion of the tufts to Bowman’s capsule, which finally results in glomerulosclerosis and reduced filtration (Cellesi et al. 2015).
Podocyte loss in hypertension includes detachment of viable cells and apoptosis (Kriz et al. 2013). The major factor for podocyte loss in hypertension is the capillary hypertension, which cause glomerular hypertrophy and hyperfiltration (Kriz and Lemley 2015). Glomerular hypertrophy results in relatively decreased podocyte density. Puelles et al. (2016) examined the effect of hypertension on podocyte depletion using kidneys obtained from autopsy, and they did not observe a difference in total podocyte number solely driven by hypertension, while the relative podocyte depletion is associated with glomerular hypertrophy which resulted in the reductions in podocyte density. Hyperfiltration gives rise to increased shear stress by elevating driving force and augmenting GBM area. Podocytes cultured in vitro are sensitive to shear stress, which induces reorganization of cytoskeleton (Friedrich et al. 2006), and this helps them to cover an expanding GBM which further leads to foot process effacement. In desoxycorticosterone-trimethylacetate (DOCA) hypertensive mice, chloride intracellular channel 5A, which is highly enriched in podocytes foot process, protects against hypertension-induced podocyte injury through weakening the tensile strength of the actin cytoskeleton in Rac1-dependent manner (Tavasoli et al. 2016). This has been considered to be the protective response for podocyte to escape detachment. However, this strategy is not always successful and finally results in podocyte detachment from GBM, as seen in progressive stage of fawn-hooded hypertensive (FHH) (Kriz et al. 1998c) and DOCA hypertensive rat model (Kretzler et al. 1994). Apoptosis is another cause for podocyte loss under shear stress in hypertension, which is before or in conjunction with cell detachment (Kriz et al. 2013; Ying et al. 2000).
Besides mechanical stress, renin–angiotensin–aldosterone system (RAAS) plays central role in the pathogenesis of hypertensive nephrosclerosis, mainly through its actions on the subtype 1 receptor. Mechanical strain increased angiotensin II production and upregulation of angiotensin receptor 1 (AT1) in cultured podocytes, while the increased apoptosis induced by mechanical strain was also in an angiotensin II-dependent manner (Durvasula et al. 2004). Increased angiotensin II results in decreased expression of podocin and integrin β1, which are both vital in viable podocytes adhesion to the GBM and interaction of podocytes with other GBM components. This might elucidate that the elevated intraglomerular pressure is translated into a maladaptive response in podocyte probably due to the activation of local tissue angiotensin system. Furthermore, angiotensin II is also considered to be associated with the rearrangement of the actin cytoskeleton (Macconi et al. 2000). Aldosterone, an important mediator of the effect of angiotensin, has become a hot spot concern in hypertensive nephropathy. Using only the inhibition of aldosterone by eplerenone dramatically alleviated podocyte injury in Dahl salt-hypertensive rats, an animal model inclined to hypertensive glomerulosclerosis (Nagase et al. 2006). In a double-blind, randomized, placebo-controlled trial, additional use of low-dose eplerenone to renin–angiotensin system inhibitors has renoprotective effects in hypertensive patients with non-diabetic chronic kidney disease (Ando et al. 2014). These findings suggested that aldosterone plays an important role in hypertension-induced podocyte injury. The underlying mechanism is primarily due to aldosterone-induced mitochondrial dysfunction, which increased oxidative stress. In uninephrectomized rats infused with aldosterone and fed with high-salt diet, podocyte-associated proteins nephrin and podocin were dramatically decreased, along with reduced nicotinamide-adenine dinucleotide phosphate oxidase activation, increased oxidative stress, and enhanced aldosterone effector kinase Sgk1. Thus, podocyte is the prominent target for aldosterone by inducing oxidative stress and Sgk1 (Shibata et al. 2007). Selective mineralocorticoid receptor (MR) antagonist eplerenone also ameliorated the salt-induced proteinuria and podocyte injury in hypertensive rat model (Nagase et al. 2007).
After detachment from GBM, podocyte moves through meshes of Bowman’s capsule to the urine and might keep alive. Unfortunately, the detection of viable podocytes in the urine is a complex procedure, which is still unavailable in all laboratories. However, elevated mRNA levels of podocin and nephrin can be examined in urine of hypertensive patients (Kelder et al. 2012). Recent studies suggested that increased podocyte-derived extracellular vesicles may predict podocyte stress and subsequent podocyte loss in hypertensive patients, which might provide a novel non-invasive detective method (Kwon et al. 2017). Podocytes, as the gatekeepers of protein in glomerular filtration barrier, are major targets of high blood pressure. In all, hypertension could cause mechanical stress and the activation of RAAS (mentioned below). Mechanical stress further induces capillary hypertension, promoting glomerular hypertrophy and hyperfiltration. These changes would lead to reduced podocyte density and the reorganization of cytoskeleton in podocytes, resulting in detachment of viable podocyte and podocyte apoptosis, progressing to final glomerulosclerosis. More studies are needed to prove that podocytes can be the detective marker for hypertensive nephrosclerosis and find the more specific method for early diagnosis and treatment.
Activation of RAS in Podocyte Injury
Hemodynamic changes and RAS of the glomeruli are key factors of CKD patients’ persistent proteinuria and disease progression. Many investigations suggest that local intrarenal RAS activation contributes to kidney tissue injury (Gurley et al. 2011), and RAS activation accelerates renal injury by various mechanisms.
Angiotensinogen (AGT), the original of RAS, transforms into Ang II through the conversion of Ang I as a result of the enzymatic cleavage process by renin and ACE. As the most active peptide of RAS, Ang II was demonstrated to induce TGF-β expression and provoke oxidative stress and inflammation, which are main factors in the initiation, development, and progression of CKD (Ruggenenti et al. 2012).
Under a condition of continuous glomerular hypertension in CKD, podocytes may undergo actin cytoskeletal reorganization, compensatory hypertrophy, weakened local adhesion ability due to downregulation of adhesion molecules of basement membrane cells, and apoptosis of podocytes induced by local Ang II activation. The continuous increase of Ang II caused by mechanical stress further affects the capillary intraglomerular pressure, resulting in a vicious circle and contributing to the pathogenesis of glomerulosclerosis (Ruster and Wolf 2011). In addition to causing podocyte lesions by altering glomerular hemodynamics, Ang II also has a direct effect on the structure and functions of podocytes, which is mentioned later in this section.
Podocytes, in possession of a complete RAS (Marquez et al. 2015), can produce functional RAS elements themselves and participate in local RAS systems as well, playing an important role in not only its own physiological process but pathological status (Fig. 10.2). It has been reported that mechanical stress and high glucose could increase the production of local Ang II and AT1 receptor (AT1R) in podocytes (Durvasula et al. 2004; Durvasula and Shankland 2008), with inducing the expression of other RAS elements (Sakoda et al. 2011).
Fig. 10.2.
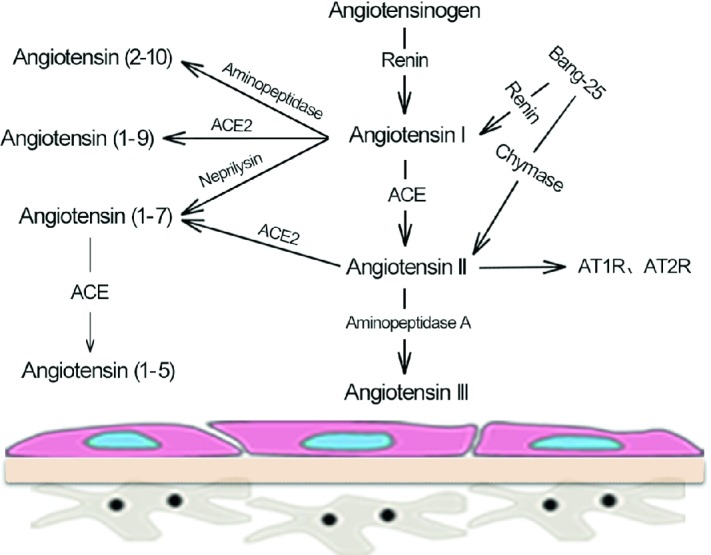
Renin–angiotensin system (RAS) in podocytes
There are also important elements of the RAS system expressing in human differentiated podocytes, including angiotensin, renin, ACE, AT1R, and AT2R subtype mRNA, but the related proteins were not detected (Liebau et al. 2006). Therefore, podocytes could not only be a target of the damage caused by Ang II, but a source of localized Ang II as well. However, it has been found that Ang II secreted by podocytes was not blocked by renin inhibitors, ACEI, and chymase inhibitors (Liebau et al. 2006), suggesting that there might be an unknown pathway for Ang II formation in podocytes.
Velez et al. (2007) used Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS) to quantify the presence of RAS-related peptide chains in rat podocytes, in order to further explore the role of podocytes in the metabolism of RAS elements. As a result, after co-incubated with Ang I, mesangial cells mainly produced Ang II while the main product of podocytes was Ang (1–7) with almost no Ang II. Furthermore, it was confirmed that podocyte-producing Ang (1–7) is mainly through the neprilysin pathway, as ACE-mediated Ang II production did not result in an increase of Ang II concentration in podocytes, which might be related to podocytes’ degradation of Ang II through ACE2 and aminopeptidase A pathways.
As a new member of RAS, ACE2 might have a negatively regulatory effect on ACE-produced Ang II of traditional RAS, mainly by accelerating the degradation of Ang II to attenuate its effect, and through the generation of Ang (1–7), which has the most expression in podocyte RAS. There has been no evidence that podocytes express the receptor of Ang (1–7), i.e., Mas, yet Ang (1–7) and its receptor seem to be involved in the renal protection for DN, such as regulating inflammation, oxidative stress, and retaining the progression of renal fibrosis.
Therefore, podocytes probably play an essential role in maintaining the balance of local RAS system in the kidney, similar to that between systemic Ang II and intrarenal RAS system, by degrading the systemic Ang II filtered from the glomeruli, and/or promoting the conversion of glomerular-filtrated Ang I and AGT to Ang (1–7), thereby regulating the damage caused by the whole systemic Ang II to the kidney.
The pathways of Ang II signaling mediating podocyte injury can be generally divided into the following aspect:
To damage the function of the pore membrane and structure of the cytoskeleton
The SD is an essential structure of the glomerular filtration barrier, which is connected to the foot processes adjacent to podocytes. Nephrin and zonula occludens (ZO)-1 are main proteins of SD, preventing macromolecules from entering the urine. It has been found that SD is susceptible to damage, leading to decreased expression of nephrin and ZO-1, and cytoskeletal reorganization of podocytes.
It has been found that the expression of nephrin in renal biopsy specimens from patients with T2DM-induced DKD was significantly reduced compared with healthy volunteers, and the patient’s urinary concentration of nephrin was significantly positively correlated with their urinary protein level (Jim et al. 2012). Ren et al. (2012) have found in vitro that Ang II could directly cause the downregulation and dephosphorylation of nephrin, which mediates podocyte injury. Besides, application of ACEI and ARB has been reported to inhibit the rearrangement of cytoplasmic ZO-1 and reduced the degree of proteinuria (Macconi et al. 2000).
-
(2)
To induce podocyte apoptosis
One of the main causes of podocyte loss in CKD patients is podocyte apoptosis, and the occurrence of urinary podocyte plays an important role in glomerular sclerosis.
Ang II reportedly could induce the apoptosis of rat podocytes cultured in vitro in a dose- and time-dependent manner, and this process required cells to be exposed to TGF-β and TGF-β antibody could inhibit apoptosis of podocytes (Ding et al. 2002). After activation of TGF-β in diabetic glomeruli, the nuclear factor κB might be inhibited via the gene Smad7, resulting in podocyte apoptosis.
The advanced glycation end products (AGEs) were also found to activate the RAS system of podocytes, upregulate Ang II levels, and induce podocyte apoptosis via a AGEs receptor-PIK3/protein kinase B (Akt)-dependent signaling pathway; ARB could attenuate Ang II-induced podocyte apoptosis.
-
(3)
To cause cell phenotypic transformation and hypertrophy
p27Kip1 encodes a protein which belongs to cyclin-dependent kinase (Cdk) inhibitor proteins, which could control the cell cycle progression at G1 phrase, thereby inhibiting cell proliferation. It was found that Ang II could directly increase the levels of p27Kip1 mRNA and protein in podocytes cultured in vitro and in vivo in DN, which was inhibited by ARB. Ang II-induced upregulation of p27Kip1 expression might lead to podocyte hypertrophy (Xu et al. 2005). It was also observed that Ang II can upregulate the expression of p27Kip1 protein, causing pathological podocyte hypertrophy similar to that in a DN text (Romero et al. 2010).
As an essential factor promoting the progression to renal fibrosis, EMT in podocytes will result in loss of epithelial markers with de novo expression of EMT markers; in more severe cases, it may lead to podocyte detachment from the glomerular basement membrane, thereby aggravating proteinuria and glomerulosclerosis (Li et al. 2015; Loeffler and Wolf 2015). A recent study reported that a high concentration of glucose and Ang II promoted EMT in podocytes, which could be reversed by silencing TCF8 (Bai et al. 2017).
-
(4)
To induce podocyte membrane depolarization and damage the charge barrier
Studies by using patch clamp recording technique in isolated glomeruli in vitro have demonstrated that Ang II could cause sustained and irreversible depolarization of podocyte membranes. Stimulation of Ang II resulted in an immediate calcium influx of cultured podocytes (Greka and Mundel 2011). Studies have confirmed that TRPC6 colocalized with podocyte nephrin and podocin, and its functional mutation could disrupt the integrity of the pore membrane, leading to proteinuria and FSGS (Reiser et al. 2005; Winn et al. 2005). Numerous studies have found that abnormal calcium signaling may be the main cause of related podocyte diseases. For example, calcium increases evoked by Ang II are primarily mediated via TRPC6 channels and this pathway could be pharmacologically targeted to abate the development of DKD (Nijenhuis et al. 2011; Sonneveld et al. 2014).
-
(5)
To induce podocyte autophagy
As terminally differentiated cells, podocytes mainly reduce intracellular accumulation of damaged DNA and macromolecular substances through autophagy rather than cell division (Pan et al. 2008). In vitro experiments, animal experiments, and human kidney biopsy indicate that podocytes have a high-level basis of autophagy, which plays an important role in maintaining the stability of podocytes. Recent research using a CKD animal model has demonstrated that autophagy is an essential intracellular process to encourage the survival of renal cells (Huber et al. 2012), while excessive and dysfunctional autophagy might result in podocyte injury (De Rechter et al. 2016). It has been found that Ang II could enhance the ROS production and increase oxidative stress in the renal system by enhancing the activity of systematic NADPH, leading to detrimental podocyte autophagy (De Rechter et al. 2016; Yadav et al. 2010), the underlying pathways of which is dependent or independent on mTOR (Mao et al. 2016). A recent study has found that autophagy could enhance the cell viability of Ang II-treated podocytes, suggesting improving autophagy may become a new targeted therapy to relieve Ang II-induced podocyte injury (Gao et al. 2017).
Traditionally concerned solely as an inactive precursor of renin, prorenin actually participates in the functional regulation of body through the hydrolysis of AGT to produce ANG I and can also bind to prorenin/renin receptor (PRR) (non-proteolytic pathway) to activate, like mitogen-activated protein kinases (MAPKs), initiating intracellular signal transductions. The plasma prorenin/renin ratio in diabetic patients was significantly higher, and the prorenin levels began to increase before the appearance of micro-albuminuria without changes in renin levels (Sakoda et al. 2011), suggesting that prorenin itself exerts somewhat important effects on DN.
Immunofluorescence double-labeling studies have showed that prorenin activated by non-proteolytic pathway coexisted with the podocyte marker nephrin, and electron microscopy also displayed that PRR was distributed on podocyte foot processes (Ichihara et al. 2006). Handle region peptide (HRP) is a polypeptide blocker of prorenin receptor. Ichihara et al. (2006) have found that gene deletion of AT1R or using ACEI inhibitor could to some extent delay the occurrence of proteinuria and glomerular sclerosis in streptomycin-induced DN rats, while continuous instillation of HRP could almost completely block the progression of DN. It is noteworthy that the MAPK signaling pathway was activated in AT1R-deficient mice, and HRP could significantly inhibit MAPK, indicative of an equally important role of prorenin coupling with PRR-induced angiotensin-independent pathway in diabetic kidney injuries. Besides, Sakoda et al. (2010) have confirmed that adding prorenin to human podocytes cultured in vitro could increase the intracellular level of Ang II and activate the MAPK intracellular signal transduction pathway, resulting in podocyte damage.
Roles of Micro-inflammation in Podocyte Injury
Definition of Micro-inflammation State
In mammalians, the acute-phase reaction is beneficial for eliminating acute insults for protection against microorganisms, limiting tissue damage, and maintaining homeostasis. This reaction would become disadvantageous under a chronic condition called micro-inflammation.
Micro-inflammation is a state with low-intensity, chronic persistent and dominant inflammation caused by the infection of non-pathogenic microorganisms, which is characterized by mild persistent elevation of inflammatory cytokines in the systemic circulation (Kaysen 2001; Schomig et al. 2000). Micro-inflammation is a continuous and relatively secretive action, the essence of which is immune inflammation.
Diagnosis and Detection of Micro-inflammation State
Micro-inflammation state has no obvious clinical symptoms, there is no specific diagnostic criteria, and the diagnosis of micro-inflammation relies mainly on the examination of circulating inflammatory biomarkers such as C-reactive protein (CRP) and serum amyloid A (SAA), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6). The acute-phase reactants including the above proteins are mainly synthesized by hepatocytes, such as complement components, coagulation proteins, and metal-binding proteins. It is important to note that when we are in the diagnosis of micro-inflammatory state, other causes and diseases of increased inflammatory markers must first be ruled out, such as connective tissue disease and recent microbial infection.
During acute-phase reaction, the concentration of CRP may increase over 1000-fold compared with normal levels (Kaysen 2001). In addition, CRP follows the course of a disease with little delay due to its short half-life. CRP is supposed to bind multiple other binding specificities such as opsonin of bacteria, immune complexes, and chromatin. CRP reflects not only the activity of inflammation, is also a sign of cytokine activation, its levels was positively associated with the degree of infection. The diagnosis of state of micro-inflammation based on CRP is the level of CRP > 8 mg/L but not more than 10–15 mg/L. SAA is a sensitive acute-phase reactant in micro-inflammatory state. The level of SAA obviously rises before other acute-phase reaction proteins.
The Mediators of Micro-inflammation State
A variety of inflammatory cytokines have emerged as being closely involved in the micro-inflammation state. Immune cells and intrinsic renal cells such as podocytes secrete proinflammatory cytokines including interleukin-1 (IL-1), IL-6, TNF-α, and monocyte chemoattractant protein-1 (MCP-1), which may contribute to the inflammatory process and aggravate diseases progression. For DN as an example, a strong induction of MCP-1 and keratinocyte chemoattractant (KC) by fetuin-A (FetA) or lipopolysaccharide (LPS) is associated with exacerbated palmitic acid-induced podocyte death. Moreover, the prevention of MCP-1 and KC secretion and inhibition of IL-1 attenuates the inflammatory and ultimate cell death response elicited by FetA alone or combined with palmitic acid. The study offers evidence that inflammation aggravates palmitic acid-induced podocyte death and the IL-1β signaling might be novel potential therapeutic targets for prevention and treatment of DN (Orellana et al. 2017).
Infiltrating macrophages/monocytes are associated with chronic, low-grade inflammation. The macrophages can interact with resident renal cells to generate a proinflammatory micro-environment that amplifies tissue injury and promotes scarring.
Macrophage-derived TNF-α had a direct role in the progression of DN. Conditional deletion of TNF-α from macrophages markedly reduced albuminuria, lessening the increase of plasma creatinine and histopathologic lesions (Awad et al. 2015). Likewise, tonicity-responsive enhancer-binding protein (TonEBP) in macrophages promotes hyperglycemia-mediated proinflammatory activation and chronic renal inflammation leading to DN and CKD (Choi et al. 2018).
Lipid-Related Inflammatory Signals
Lipids such as triglycerides and cholesterol may accumulate ectopically in the kidney, which contributes to a lipotoxicity process. Palmitic acid-treated podocytes had intracellular lipid accumulation and abnormal lipid metabolism, accompanied by the process of inflammation, insulin resistance, and rearrangements of the SD and actin cytoskeleton of podocyte. Thus, lipotoxicity accelerated podocyte damage through lipid accumulation related inflammation (Martinez-Garcia et al. 2015).
Lipoproteins including LDL, VLDL, and IDL might act as proinflammatory mediators, which promote the production of inflammatory cytokines, such as TGF-β, platelet-derived growth factor (PDGF), and IL-6 secreted from human mesangial cells. Lipoprotein-mediated cytokine production may cause recruitment of monocytes, lipid-mediated cell proliferation and apoptosis, and extracellular matrix production, thus contributing to podocyte injuries and glomerulosclerosis.
Micro-inflammation Promotes Podocyte Injuries
Micro-inflammation and Insulin Resistance of Podocytes
Chronic inflammation can reduce podocyte insulin sensitivity. Nucleotide-binding oligomerization domain-containing 2 (NOD2) is a subtype of intracellular pattern recognition receptor (PRR), playing functions in innate immunity. Of particular interest, increased levels of NOD2 were observed in DN patients and high fat diet (HFD)/STZ-induced mice models. Furthermore, HFD/STZ-induced diabetes mice models with NOD2 knock-out showed reduced podocyte injury and proteinuria compared with wild-type diabetic mice (Du et al. 2013). In vitro, NOD2 which was activated by bacterial component muramyl dipeptide in podocytes reduced insulin-induced glucose uptake and inhibited serine phosphorylation of IRS-1. Another study has explored the role of other PRR toll-like receptors (TLRs) in the db/db mice model of DN. Administration of a selective TLR2/4/6 inhibitor GIT27 improved insulin sensitivity, reduced albuminuria and urinary nephrin levels, indicative of reduced podocyte damage. TLR4 expression in podocytes was found to be highest expressed (Cha et al. 2013). Given the links between some specific PRRs activation and insulin stimulation in podocytes, how podocyte insulin responses are altered following PRRs activation and inhibition may need specifically investigated.
IKB/NF-κB is another important pathway of insulin resistance in podocyte, and NF-κB expression was increased in kidney tissues of patients with type 2 diabetes. NF-κB can increase the level of IRS serine phosphorylation and the expression of inflammatory MCP-1, IL-6, and TNF-α. Moreover, the increased expressed inflammatory factors can further activate the NF-κB. The inflammatory cytokines and the activation of NF-κB pathway form positive feedback to induce insulin resistance.
Micro-inflammation and Dyslipidemia Act Synergistically in Podocyte Injury
Xu et al. reported that chronic systemic inflammation exacerbates lipid accumulation in the kidney of ApoE knockout mice by diverting lipid from the plasma to the kidney via the SCAP-SREBP2-LDLr pathway and causing renal injury (Xu et al. 2011). Consisted with this, IL-1β stimulation in vitro increased the lipid accumulation in the podocytes by increasing the expression of lipid metabolism related proteins, for instance, LDLr, sterol regulatory element-binding protein-2 (SREBP-2) and SREBP cleavage-activating protein (SCAP), and through promoting translocation of the SCAP/SREBP-2 complex from the endoplasmic reticulum to the Golgi in the podocytes (Zhang et al. 2015b). Compared with db/db mice, podocyte injury was more severe in db/db mice with subcutaneous casein injections, which are supposed to induce inflammatory stress in vivo. Altogether, inflammation may be associated with high risk for chronic renal fibrosis.
Intrinsic Proinflammatory Signaling in Podocytes
Activation of intrinsic proinflammatory signaling in podocytes such as NF-κB signal pathway aggravates podocyte injury and proteinuria. In STZ-induced diabetic mice models with Ccr2 knock-out, transgenic CCR2 overexpression in the podocytes resulted in significantly increased albuminuria and podocyte loss, without concurrent increase in kidney macrophage infiltration or inflammatory cytokine production. These findings support that activation of CCR2 signaling cascade in podocytes mediates diabetic renal injury, which is independent of macrophage recruitment (You et al. 2017).
IL-20, a proinflammatory cytokine which is upregulated by high glucose and TGF-β1, can increase MCP-1 and TGF-β1 expression in podocytes and induce apoptosis in podocytes through activating caspase-8. In STZ-induced early DN mice models, anti-IL-20 monoclonal antibody (7E) treatment or IL-20R1-deficiency led to lower blood glucose and improved renal functions, and IL-20 is proved to be expressed in podocytes. Collectively, intrinsic proinflammatory signaling in podocytes contributes to podocyte damage (Fig. 10.3).
Fig. 10.3.
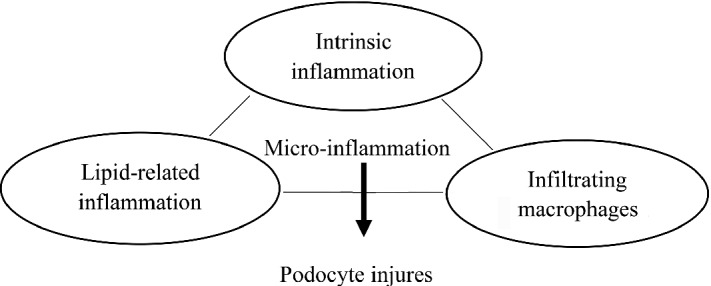
Scheme pattern of micro-inflammation-mediated podocyte injury
Immune Disorder in Podocyte Injury
Immune injuries are common causes of podocyte damage. Processes interfering with podocyte’s structural or functional integrity lead to disruption of the glomerular filtration barrier.
Immunoactive Molecules Expressed at Podocytes
Complement and Complement Regulatory Protein
Primary-cultured human podocytes synthesize and secrete complement C3 physiologically, and the stimulation of inflammatory factor INF-γ could increase the production of C3. Under physiological conditions, C3 produced by glomerular podocytes can resist the invasion of foreign pathogens and protect local tissues. C3 activation can lead to decreased immune complex formation and increased disintegration. On the other hand, C3 activation leads to increased production of vasoactive molecules and chemokines, which in turn recruits more inflammatory mediators into the glomerulus. The activation of complement would produce proinflammatory components of complement, i.e., C5a. In immune complex diseases and ischemia-reperfusion injury, C5a is an important mediator that triggers an inflammatory cascade (Heller et al. 1999).
The kidney is one of the organs that are most susceptible to abnormally activated complement, which can be seen in various glomerulonephritis. The main pathogenesis of idiopathic membranous nephropathy (IMN) is caused by the binding of IgG to the intrinsic antigen on the basement membrane side of glomerular podocytes, which combine to form an antigen–antibody complex, thereby activating the complement-forming membrane attack complex (Takano et al. 2013). In IMN, the concentrations of complement cleavage products such as C3a, C5a and C5b-9 are significantly increased. C5b-9 is the final product of complement activation in three pathways of complement activation, causing podocyte injury not through conventional lysis, but probably via the mechanism related to the activation of corresponding intracellular signaling pathways in a subdissolved form. Ronco and Debiec have confirmed that the podocyte surface antigen megalin binded to the corresponding antibody underwent an immune complex reaction, activated the complement system, and promoted the formation of the membrane attack complex C5b-9 (Ronco and Debiec 2007). As a stimulant of podocytes, c5b-9 could destroy podocyte cytoskeletal proteins, inserting in the membrane to increase cell permeability, and activating a series of transduction pathways, resulting in the diffuse thickening of GBM and defects in glomerular filtration barrier, clinically leading to significant proteinuria.
In addition, podocytes begin to express complement receptor 1 (CR1, or C3bR, or CD35) during the capillary synthesis stage of renal development and are evenly distributed on the cell membrane and the membrane of foot processes. CR1 is expressed as a cofactor of the complement factor I and expressed in most circulating cells. CR1 is the only physiological blocker of complement synthesis in podocytes and inactivates the lysate of complement to promote the clearance of immune complexes, protecting podocytes from complement-mediated damage (Alexander et al. 2007). It has been reported that the production of CR1 was reduced in several glomerular diseases, making podocytes vulnerable to complement attacks.
Complement regulatory proteins include Crry, CD59, and decay acceleration factors (DAF or CD55), which are vital to limiting the activation of podocyte complement (Cheng et al. 2018). Podocyte expression of Crry and CD59 could inhibit C3 invertase and the synthesis of C5b-9, thus to protect podocytes from injuries induced by antibody-complement activation. In addition, podocytes both in vitro and in vivo could be detected of DAF. In a mouse model of nephritis, deficiency of DAF resulted in serious podocyte foot fusion, indicating that DAF might protect podocytes from complement-mediated injury (Bao et al. 2009).
Cytokines and Chemokines
In both physiological and pathological texts, podocytes of humans, rats, and mice all express the receptors of cytokines interleukin 4 (IL-4), IL-10, and IL-13. After stimulating podocytes cultured in vitro with IL-4 and IL-13, the skeletal structure and intercellular-link protein of podocytes were damaged and the permeability increased (Ha et al. 2017; Kim et al. 2017), suggesting that IL-4 and IL-13 could damage podocytes by binding to its receptors.
In early minimal change disease (MCD), FSGS, and MN, podocytes increasingly express inflammatory mediators IL-1 α/β along with IL-1 type 1 receptor (IL-1 RI), and IL-1 RI is decreasingly expressed at late stage of the disease when glomerular cell hyperplasia and sclerosis appear (Brahler et al. 2012), indicating that these molecules participate in podocyte damage and repair, glomerular local inflammation.
In addition, both podocytes cultured in vitro and renal tissue express receptors of functional CC chemokine receptor (CCR) and CXC chemokine receptors (CXCR), which could couple with corresponding chemokines to promote the production of cytoplasm Ca2+ and ROS and be involved in podocyte injuries (Huber et al. 2002). Moreover, it has been found that podocytes themselves could produce IL-8 (ligand of CXCR1/CXCR2), thus podocytes could be activated via autocrine.
CXCL16 might play an important role in the inflammatory response of kidney diseases. Podocytes overexpress CXCL16 under the stimulation of proinflammatory factors. Soluble CXCL16 plays a chemotactic role in inflammation and immune response, while transmembrane CXCL16 removes oxLDL (Gutwein et al. 2009), which is harmful to the kidney. Therefore, abnormal expression of CXCL16 in podocytes might cause renal damage due to excessive immune-inflammatory reaction or an accumulation of oxLDL. It has been found that the expression of CXCL16 and oxLDL in the glomeruli of MN patients increased not only significantly but consistently as well (Gutwein et al. 2009). The inflammatory factor IFN-γ is the strongest stimulator of CXCL16, which upregulates several forms and overall cell expression levels of CXCL16, consequently promoting podocyte damage (Wang et al. 2014).
Toll-like Receptors (TLRs)
Under physiological conditions, podocytes of humans and mice could express TLR4. Stimulating cultured murine podocytes in vitro with the ligand of TLR4-like LPS, lipid A, and fibrins (endogenous ligand), resulted in an increasing expression of CCL and CXCL. In the mouse model of cryoglobulinemia membrane proliferative glomerulonephritis, podocytes expressed more TLR4, promoting the synthesis and secretion of chemokines and further leukocyte recruitment and glomerular injury (Banas et al. 2008). It has been shown that under the stimulation of endogenous TLR4 ligand, podocytes upregulate TLR4, promote the production of proinflammatory chemokines, and actively participate in the recruitment of inflammatory cells, all leading to glomerular injuries (Banas et al. 2008).
Apart from TLR4, other members of the TLR family have also been proved to participate in podocyte injury. A recent study has pointed out that the overexpression of TLR-8 correlates with the progression of podocyte injury in glomerulonephritis, suggesting that altered levels of urinary Tlr8 mRNA might reflect the degree of podocyte injury in murine autoimmune GN (Kimura et al. 2014).
TLR-7 and TLR-9 expressed by B cells and dendritic cells have been considered as important molecules involved in the pathogenesis of systemic lupus. Recent study demonstrated that active LN onset in childhood expressed more TLR-9, accompanied by weakened expression of podocyte SD protein nephrin, podocin, and synaptopodin; in the meantime, patients showed proteinuria and high ds-DNA antibody and low complement (Machida et al. 2010). Therefore, under pathological conditions, TLRs link the innate immune system with podocyte and glomerular injuries.
Costimulatory Factors
B7-1(CD80) belongs to the immunoglobulin superfamily, mainly expressed in antigen-presenting cells, and provides a costimulatory signal by coupling with corresponding molecular receptors expressed on T cells, i.e., CD28 and CTLA 4, regulating the immune responses induced by activated T cells. It has been found that B7-1 was expressed on podocytes of lupus nephritis (LN) (Reiser et al. 2004), and the expression of podocyte B7-1 in LN patients and LN mouse models is positively correlated with the degree of proteinuria. However, new evidence has stricken up a discordant tune (Baye et al. 2016), leading to further mandatory studies of the application of B7-1 blockers in treating proteinuric patients (Novelli et al. 2016a).
Studies have shown that under the induction of hypoxia, high glucose, or bacteriocin lipopolysaccharide (LPS), the expression of B7-1 would be induced in podocytes which does not occur under physiological conditions, and participate in podocyte cytoskeletal reorganization and the pathogenesis of proteinuria (Chang et al. 2013; Fiorina et al. 2014; Shimada et al. 2012).
In the glomerulus of nephritis, podocyte-expressed B7-1 may also recruit T cells to where GBM is damaged and promote further inflammation. Podocytes from necrotic crescentic nephritis rat model and cultured rat podocytes in vitro could express both MHC I/II molecules and intercellular adhesion molecule 1 (ICAM-1) after stimulation of IFN-γ, suggesting that cytokines could present the antigen to infiltrating T cells (Goldwich et al. 2013). Recently, it has been pointed that compared to normal people, MCD patients but not FSGS patients excreted more urinary B7-1, while podocytes of relapsed MCD patients and FSGS patients did not express B7-1, thus B7-1 might be used to identify MCD and FSGS (Novelli et al. 2016b).
Immune Disorder and Podocyte Injuries
The glomerulus is a well-recognized target of miscellaneous immune-mediated injuries, and the pathogenesis of immune-mediated glomerular disease is multifactorial (Fig. 10.4).
Fig. 10.4.
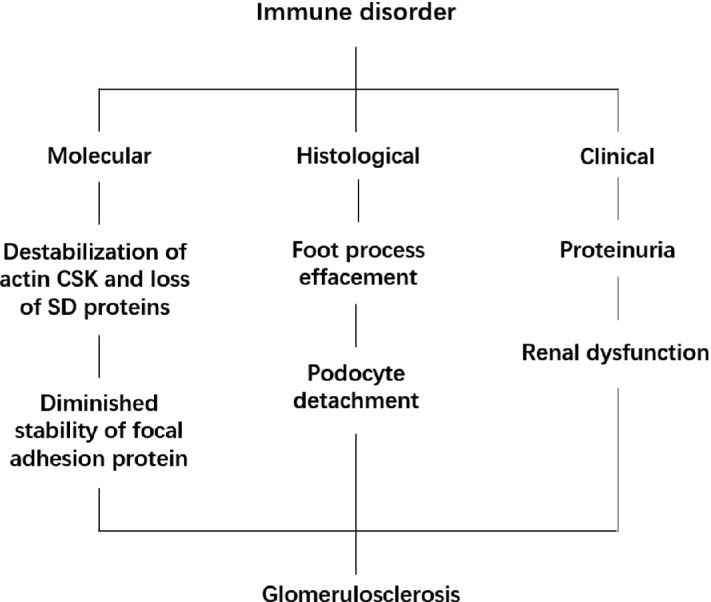
Sequences of immune-mediated podocyte injury
Anti-podocyte Antibody
In MN, the surface molecules of glomerular podocyte act as antigens and trigger systematic immune responses, resulting in the formation of in situ immune complexes. The classic animal model of MN, Heymann nephritis, reproduces typical mesangial lesions by eliciting auto-antibodies against the podocyte membrane protein megalin in rats (Ronco and Debiec 2005).
It has been reported in vivo that the occurrence of human newborns MN was due to the production of auto-antibodies against glomerular podocyte membrane proteins. Neutral endopeptidase (NEP) is a membrane protein expressed on the surface of human podocytes. Studies have shown that neonatal MN occurs due to the presence of anti-NEP auto-antibodies in children (Herrmann et al. 2012). Its origin is due to the mother’s carrying the relevant mutation gene and lacking NEP. If the mother bred a normal healthy fetus, the mother will produce an anti-NEP antibody against the fetus during pregnancy and the antibody enters the fetus through the placenta. Anti-NEP antibodies react with NEP antigens on fetal podocytes, forming an immune complex on the epithelial side, leading to neonatal MN. Although the incidence of this type of patients is very low, its pathogenesis confirms the role of anti-podocyte antigen antibodies in the development of human MN (Pozdzik et al. 2015).
T Cell Dysfunction
T cell dysfunction and the release of cytokines (circulatory factors) causing podocyte injury are associated with the formation of proteinuria in MCD patients. It is currently believed that the cytokines produced by Th1 and Th2 cells in T cells are involved in the occurrence of MCD, but the cytokines produced by Th2 cells (IL-4, IL-8, IL-13) might be more important (Mack 2009). Animal experiments have found that the injection of IL-8 to rats can reduce the content of heparin sulfate on the surface of podocytes, weaken the membrane filtration barrier of charge, and trigger proteinuria. There are also receptors for IL-4 and IL-13 on the podocyte, and the increase of circulating or local IL-4 and IL-13 can directly damage the podocyte through the receptors on the podocyte and increasing the permeability of the filtration membrane.
Shimada et al. (2011) have proposed that MCD is the result of a “two-hit” attack from podocyte immune dysfunction: The first hit is the effects of bacterial products, viruses, and various cytokines on podocytes, resulting in an abnormal expression of CD80 in podocytes, and further cytoskeleton reorganization and morphological changes of podocytes, increasing the permeability of GBM which might bring about proteinuria. However, due to the self-regulation of the body, podocytes can downregulate the expression of CD80. If the auto-regulatory function of podocytes and the body is defective, the sustained expression of CD80 would lead to proteinuria and even MCD. Moreover, Ishimoto et al. (2011) also observed increasing expression of CD80 in the urine of MCD patients; in view of the fact that the expression of CD80 in podocytes can be induced by IL-13 and bacterial products through the TLR pathway and regulated by CTLA4, suggesting that defective immune functions of podocytes is an essential cause of MCD.
Antigen–Antibody Immune Complex
Certain exogenous antigens (small molecular weight, positively charged) are implanted on the epithelial side and can also lead to the formation of in situ immune complexes. Hepatitis B virus (HBV)-associated nephropathy is often manifested as mesangial lesions (especially in children), and HBeAg plays an important role in its occurrence. The HBeAg molecule is of small mass and negatively charged and can be implanted across the glomerular basement membrane (GBM) on the epithelial side, triggering the formation of in situ immune complexes (Gupta and Quigg 2015).
Under inflammatory conditions, podocytes would inhibit the expression of MHC class II molecules, promoting the remove of immune complexes from the GBM. In some cases, podocytes might act as antigen-presenting cells themselves, taking up and processing antigens to initiate specific T cell responses. There has been evidence that transgenic mice with a loss of MHC class II exclusively in podocytes developed only a very moderate degree of nephrosclerosis and glomerular crescent formation compared to the control animals, indicative of their defective capacity to activate CD8+ T cells (Goldwich et al. 2013).
The Role of Other Factors in Podocyte Injury
Viral infection, such as human immunodeficiency virus (HIV)-1, parvovirus B19, cytomegalovirus (CMV), hepatitis B virus (HBV), and hepatitis C virus (HCV), is associated with podocyte injury. HIV-associated nephropathy (HIVAN) mostly manifests collapsing glomerulopathy or classic FSGS (Chandra and Kopp 2013). Podocyte infection is associated with podocyte injury and dedifferentiation and rapid loss of renal function. Studies have reported that HIV virus can be internalized by podocytes in vitro, which might be associated with receptors, such as viral coat protein gp120, and subsequent endocytosis, phagocytosis, or pinocytosis (Bruggeman 2017). Although the transmission of virus in vitro has been well documented, further studies are needed to demonstrate the definite mechanism by which the virus enters podocyte in vivo. Structural viral proteins, gag and pol, and non-structural proteins, vpr, nef, and tat, have been considered to be associated with HIVAN (Conaldi et al. 2002; Reid et al. 2001; Zuo et al. 2006). HBV is a major cause for membranous nephropathy and FSGS scarcely, which can be diagnosed by evidence of HBV antigen or antibodies on kidney biopsy. The possible mechanisms of HBV-induced podocyte injury might be as follows: detective infection of the cells by HBV, deposition of circulating immune complex in renal cells, effects of HBV-induced immunological mediators (Bhimma and Coovadia 2004; Sakai et al. 2011).
Podocytes are also targets of some toxicity drugs, which may further progress to glomerulosclerosis. For example, gold, bucillamine, and d-penicillamine, which are used for the treatment of rheumatoid arthritis, are confirmed to cause MN. The possible mechanism might be closely related to stress, energy metabolism, and inflammation (Fujiwara et al. 2011; Seguin et al. 2005). Other drugs, like non-steroid anti-inflammatory drugs and interferon, also can be inductor of podocyte injury. Organic solvents, like gasoline, dimethylbenzene, and formaldehyde, can induce podocyte injury including foot process fusion and decreased expression of nephrin and podocin (Qin et al. 2012).
Hypoxia can be induced by various pathogenic conditions including hypertension and diabetes. Chronic hypoxia can trigger endoplasmic reticulum (ER) stress, which result in increased ROS. Nephrin and alpha-actin-4, the structural components of SD, are subject to mutations, which cause defective protein folding in the ER of podocytes. The underling mechanism might include transient receptor protein 6 and complement complex and increased expression of MCP-1 (Chen et al. 2011; Cybulsky 2013; Maekawa and Inagi 2017). Targeting hypoxia and ER stress and the possible signal networks might be the novel target for intervention of podocyte injury in CKD.
Summary
Living in an environment of a variety of pathological stresses and stimuli, podocytes adapt to maintain the integrity and stability of the glomerular basement membrane, depending on their highly differentiated characteristics which also reflect the vulnerability of this barrier.
The different responses of podocytes to injury are associated with the pathology and prognosis of glomerular diseases. As a vital type of renal intrinsic cells, podocyte damage is an important cause of nephrotic proteinuria and glomerular sclerosis. However, as a highly differentiated terminal cell, podocyte has no proliferative potential, and loss of podocyte is associated with poor renal outcomes such as increased proteinuria, glomerulosclerosis, and renal disease progression.
Podocytes have different responses to injuries, including endoplasmic reticulum stress and autophagy reactions caused by abnormal energy metabolism. This chapter lists several aspects of podocyte injuries along with potential underlying mechanisms, including glucose and lipid metabolism disorder, hypertension, RAS activation, micro-inflammation, immune disorder, and other factors. These aspects are not technically separated items, but intertwined with each other in the pathogenesis of podocyte injuries.
Injured podocytes would undergo a series of morphological changes: FP disappearance, cellular shrink, pseudocysts form, cell hypertrophy, cytoplasmic lysosomal enrichment, etc. These changes eventually cause podocytes to detach from the GBM. Moreover, due to the lack of proliferative capacity, the number of glomerular podocytes would become less and less, until reduced by more than 20% when glomerulosclerosis occurs.
Glomerulosclerosis is not a specific disease but a state representing podocyte injury which is mediated by diverse causes. Podocytes interact with GBM and capillary loops tightly, dysfunction of which is an early event leading to glomerulosclerosis. Glomerulosclerosis seems like a station to stay in just before arriving to destination. Unanswered questions in the pathogenesis of podocyte injury and glomerulosclerosis are still ill-defined, and the causing list will continue to grow. Uncovering the selective targeting to pathogenesis and underlying mechanism of podocyte injury and glomerulosclerosis is bound to provide clues to answer for treatment and prevention of the disease in the future.
Acknowledgements
This study was supported by grants from the National Key Research and Development Program of China (2018YFC1314004).
Contributor Information
Bi-Cheng Liu, Phone: +86862583262422, Email: liubc64@163.com.
Hui-Yao Lan, Email: hylan@cuhk.edu.hk.
Lin-Li Lv, Phone: +868613770603746, Email: lvlinli@seu.edu.cn.
Kun-Ling Ma, Email: klma05@163.com.
References
- Advani A. Vascular endothelial growth factor and the kidney: something of the marvellous. Curr Opin Nephrol Hypertens. 2014;23:87–92. doi: 10.1097/01.mnh.0000437329.41546.a9. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Wang Y, Chang A, Jacob A, Minto AW, Karmegam M, et al. Mouse podocyte complement factor H: the functional analog to human complement receptor 1. J Am Soc Nephrol. 2007;18:1157–1166. doi: 10.1681/ASN.2006101125. [DOI] [PubMed] [Google Scholar]
- Ando K, Ohtsu H, Uchida S, Kaname S, Arakawa Y, Fujita T, et al. Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:944–953. doi: 10.1016/S2213-8587(14)70194-9. [DOI] [PubMed] [Google Scholar]
- Anil Kumar P, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol (Lausanne) 2014;5:151. doi: 10.3389/fendo.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015;88:722–733. doi: 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Liang S, Wang Y, Jiao B. Knocking down TCF8 inhibits high glucose- and angiotensin II-induced epithelial to mesenchymal transition in podocytes. Biosci Trends. 2017;11:77–84. doi: 10.5582/bst.2016.01224. [DOI] [PubMed] [Google Scholar]
- Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, et al. TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol. 2008;19:704–713. doi: 10.1681/ASN.2007040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Haas M, Pippin J, Wang Y, Miwa T, Chang A, et al. Focal and segmental glomerulosclerosis induced in mice lacking decay-accelerating factor in T cells. J Clin Invest. 2009;119:1264–1274. doi: 10.1172/JCI36000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baye E, Gallazzini M, Delville M, Legendre C, Terzi F, Canaud G. The costimulatory receptor B7-1 is not induced in injured podocytes. Kidney Int. 2016;90:1037–1044. doi: 10.1016/j.kint.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhimma R, Coovadia HM. Hepatitis B virus-associated nephropathy. Am J Nephrol. 2004;24:198–211. doi: 10.1159/000077065. [DOI] [PubMed] [Google Scholar]
- Bianchi S, Baronti A, Cominotto R, Bigazzi R (2016) Lipid metabolism abnormalities in Chronic Kidney Disease. G Ital Nefrol 33: S68 [PubMed]
- Brahler S, Ising C, Hagmann H, Rasmus M, Hoehne M, Kurschat C, et al. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am J Physiol Renal Physiol. 2012;303:F1473–F1485. doi: 10.1152/ajprenal.00031.2012. [DOI] [PubMed] [Google Scholar]
- Bruggeman LA. HIV-1 infection of renal cells in HIV-associated nephropathy. J Am Soc Nephrol. 2017;28:719–721. doi: 10.1681/ASN.2016111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussolati B, Deregibus MC, Fonsato V, Doublier S, Spatola T, Procida S, et al. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J Am Soc Nephrol. 2005;16:1936–1947. doi: 10.1681/ASN.2004080629. [DOI] [PubMed] [Google Scholar]
- Cellesi F, Li M, Rastaldi MP. Podocyte injury and repair mechanisms. Curr Opin Nephrol Hypertens. 2015;24:239–244. doi: 10.1097/MNH.0000000000000124. [DOI] [PubMed] [Google Scholar]
- Cha JJ, Hyun YY, Lee MH, Kim JE, Nam DH, Song HK, et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology. 2013;154:2144–2155. doi: 10.1210/en.2012-2080. [DOI] [PubMed] [Google Scholar]
- Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. 2013;6:1–5. doi: 10.1093/ckj/sft002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JM, Hwang DY, Chen SC, Kuo MC, Hung CC, Hwang SJ, et al. B7-1 expression regulates the hypoxia-driven cytoskeleton rearrangement in glomerular podocytes. Am J Physiol Renal Physiol. 2013;304:F127–F136. doi: 10.1152/ajprenal.00108.2012. [DOI] [PubMed] [Google Scholar]
- Chen HC, Chen CA, Guh JY, Chang JM, Shin SJ, Lai YH. Altering expression of alpha3beta1 integrin on podocytes of human and rats with diabetes. Life Sci. 2000;67:2345–2353. doi: 10.1016/S0024-3205(00)00815-8. [DOI] [PubMed] [Google Scholar]
- Chen S, He FF, Wang H, Fang Z, Shao N, Tian XJ, et al. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium. 2011;50:523–529. doi: 10.1016/j.ceca.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Chen S, Meng XF, Zhang C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed Res Int. 2013;2013:839761. doi: 10.1155/2013/839761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Gou SJ, Qiu HY, Ma L, Fu P. Complement regulatory proteins in kidneys of patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Clin Exp Immunol. 2018;191:116–124. doi: 10.1111/cei.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Lim SW, Salimi S, Yoo EJ, Lee-Kwon W, Lee HH, et al. Tonicity-responsive enhancer-binding protein mediates hyperglycemia-induced inflammation and vascular and renal injury. J Am Soc Nephrol. 2018;29:492–504. doi: 10.1681/ASN.2017070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MP, Chen S, Ziyadeh FN, Shea E, Hud EA, Lautenslager GT, et al. Evidence linking glycated albumin to altered glomerular nephrin and VEGF expression, proteinuria, and diabetic nephropathy. Kidney Int. 2005;68:1554–1561. doi: 10.1111/j.1523-1755.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- Conaldi PG, Bottelli A, Baj A, Serra C, Fiore L, Federico G, et al. Human immunodeficiency virus-1 tat induces hyperproliferation and dysregulation of renal glomerular epithelial cells. Am J Pathol. 2002;161:53–61. doi: 10.1016/S0002-9440(10)64156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095–3102. doi: 10.2337/diabetes.54.11.3095. [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Koziell A, Hussain S, Lennon R, Ni L, et al. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes. 2007;56:1127–1135. doi: 10.2337/db06-0693. [DOI] [PubMed] [Google Scholar]
- Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin-proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84:25–33. doi: 10.1038/ki.2012.390. [DOI] [PubMed] [Google Scholar]
- Dai HY, Zheng M, Lv LL, Tang RN, Ma KL, Liu D, et al. The roles of connective tissue growth factor and integrin-linked kinase in high glucose-induced phenotypic alterations of podocytes. J Cell Biochem. 2012;113:293–301. doi: 10.1002/jcb.23355. [DOI] [PubMed] [Google Scholar]
- Dandapani SV, Sugimoto H, Matthews BD, Kolb RJ, Sinha S, Gerszten RE, et al. Alpha-actinin-4 is required for normal podocyte adhesion. J Biol Chem. 2007;282:467–477. doi: 10.1074/jbc.M605024200. [DOI] [PubMed] [Google Scholar]
- De Rechter S, Decuypere JP, Ivanova E, van den Heuvel LP, De Smedt H, Levtchenko E, et al. Autophagy in renal diseases. Pediatr Nephrol. 2016;31:737–752. doi: 10.1007/s00467-015-3134-2. [DOI] [PubMed] [Google Scholar]
- de Vries AP, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM, et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014;2:417–426. doi: 10.1016/S2213-8587(14)70065-8. [DOI] [PubMed] [Google Scholar]
- Ding G, Reddy K, Kapasi AA, Franki N, Gibbons N, Kasinath BS, et al. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol. 2002;283:F173–F180. doi: 10.1152/ajprenal.00240.2001. [DOI] [PubMed] [Google Scholar]
- Du P, Fan B, Han H, Zhen J, Shang J, Wang X, et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013;84:265–276. doi: 10.1038/ki.2013.113. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294:F830–F839. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantus D, Rogers NM, Grahammer F, Huber TB, Thomson AW. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat Rev Nephrol. 2016;12:587–609. doi: 10.1038/nrneph.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorina P, Vergani A, Bassi R, Niewczas MA, Altintas MM, Pezzolesi MG, et al. Role of podocyte B7-1 in diabetic nephropathy. J Am Soc Nephrol. 2014;25:1415–1429. doi: 10.1681/ASN.2013050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011;3:85ra46. doi: 10.1126/scitranslmed.3002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich C, Endlich N, Kriz W, Endlich K. Podocytes are sensitive to fluid shear stress in vitro. Am J Physiol Renal Physiol. 2006;291:F856–F865. doi: 10.1152/ajprenal.00196.2005. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Tsuchiya H, Sakai N, Shibata K, Fujimura A, Koshimizu TA. Proximal tubules and podocytes are toxicity targets of bucillamine in a mouse model of drug-induced kidney injury. Eur J Pharmacol. 2011;670:208–215. doi: 10.1016/j.ejphar.2011.08.051. [DOI] [PubMed] [Google Scholar]
- Gao N, Wang H, Yin H, Yang Z. Angiotensin II induces calcium-mediated autophagy in podocytes through enhancing reactive oxygen species levels. Chem Biol Interact. 2017;277:110–118. doi: 10.1016/j.cbi.2017.09.010. [DOI] [PubMed] [Google Scholar]
- Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, et al. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol. 2013;24:906–916. doi: 10.1681/ASN.2012020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greka A, Mundel P. Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol. 2011;22:1969–1980. doi: 10.1681/ASN.2011040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Hagiwara S, Fan Q, Tanimoto M, Kobata M, Yamashita M, et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol Dial Transplant. 2006;21:299–313. doi: 10.1093/ndt/gfi210. [DOI] [PubMed] [Google Scholar]
- Gupta A, Quigg RJ. Glomerular diseases associated with hepatitis B and C. Adv Chronic Kidney Dis. 2015;22:343–351. doi: 10.1053/j.ackd.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutwein P, Abdel-Bakky MS, Schramme A, Doberstein K, Kampfer-Kolb N, Amann K, et al. CXCL16 is expressed in podocytes and acts as a scavenger receptor for oxidized low-density lipoprotein. Am J Pathol. 2009;174:2061–2072. doi: 10.2353/ajpath.2009.080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TS. High glucose and advanced glycosylated end-products affect the expression of alpha-actinin-4 in glomerular epithelial cells. Nephrology (Carlton) 2006;11:435–441. doi: 10.1111/j.1440-1797.2006.00668.x. [DOI] [PubMed] [Google Scholar]
- Ha TS, Nam JA, Seong SB, Saleem MA, Park SJ, Shin JI. Montelukast improves the changes of cytoskeletal and adaptor proteins of human podocytes by interleukin-13. Inflamm Res. 2017;66:793–802. doi: 10.1007/s00011-017-1058-y. [DOI] [PubMed] [Google Scholar]
- Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- Han SY, So GA, Jee YH, Han KH, Kang YS, Kim HK, et al. Effect of retinoic acid in experimental diabetic nephropathy. Immunol Cell Biol. 2004;82:568–576. doi: 10.1111/j.1440-1711.2004.01287.x. [DOI] [PubMed] [Google Scholar]
- Han SY, Kang YS, Jee YH, Han KH, Cha DR, Kang SW, et al. High glucose and angiotensin II increase beta1 integrin and integrin-linked kinase synthesis in cultured mouse podocytes. Cell Tissue Res. 2006;323:321–332. doi: 10.1007/s00441-005-0065-4. [DOI] [PubMed] [Google Scholar]
- Hashizume M, Mihara M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine. 2012;58:424–430. doi: 10.1016/j.cyto.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Heller T, Hennecke M, Baumann U, Gessner JE, zu Vilsendorf AM, Baensch M, et al. Selection of a C5a receptor antagonist from phage libraries attenuating the inflammatory response in immune complex disease and ischemia/reperfusion injury. J Immunol. 1999;163:985–994. [PubMed] [Google Scholar]
- Herrmann SM, Sethi S, Fervenza FC. Membranous nephropathy: the start of a paradigm shift. Curr Opin Nephrol Hypertens. 2012;21:203–210. doi: 10.1097/MNH.0b013e32835026ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI0215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZB, Ma KL, Zhang Y, Wang GH, Liu L, Lu J, et al. Inflammation-activated CXCL16 pathway contributes to tubulointerstitial injury in mouse diabetic nephropathy. Acta Pharmacol Sin. 2018;39:1022–1033. doi: 10.1038/aps.2017.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Reinhardt HC, Exner M, Burger JA, Kerjaschki D, Saleem MA, et al. Expression of functional CCR and CXCR chemokine receptors in podocytes. J Immunol. 2002;168:6244–6252. doi: 10.4049/jimmunol.168.12.6244. [DOI] [PubMed] [Google Scholar]
- Huber TB, Schermer B, Muller RU, Hohne M, Bartram M, Calixto A, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci U S A. 2006;103:17079–17086. doi: 10.1073/pnas.0607465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara A, Suzuki F, Nakagawa T, Kaneshiro Y, Takemitsu T, Sakoda M, et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2006;17:1950–1961. doi: 10.1681/ASN.2006010029. [DOI] [PubMed] [Google Scholar]
- Ikezumi Y, Suzuki T, Karasawa T, Kawachi H, Nikolic-Paterson DJ, Uchiyama M. Activated macrophages down-regulate podocyte nephrin and podocin expression via stress-activated protein kinases. Biochem Biophys Res Commun. 2008;376:706–711. doi: 10.1016/j.bbrc.2008.09.049. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Shimada M, Araya CE, Huskey J, Garin EH, Johnson RJ. Minimal change disease: a CD80 podocytopathy? Semin Nephrol. 2011;31:320–325. doi: 10.1016/j.semnephrol.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Jim B, Ghanta M, Qipo A, Fan Y, Chuang PY, Cohen HW, et al. Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: a cross sectional study. PLoS ONE. 2012;7:e36041. doi: 10.1371/journal.pone.0036041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Sison K, Li C, Tian R, Wnuk M, Sung HK, et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell. 2012;151:384–399. doi: 10.1016/j.cell.2012.08.037. [DOI] [PubMed] [Google Scholar]
- Joles JA, Kunter U, Janssen U, Kriz W, Rabelink TJ, Koomans HA, et al. Early mechanisms of renal injury in hypercholesterolemic or hypertriglyceridemic rats. J Am Soc Nephrol. 2000;11:669–683. doi: 10.1681/ASN.V114669. [DOI] [PubMed] [Google Scholar]
- Kaysen GA. The microinflammatory state in uremia: causes and potential consequences. J Am Soc Nephrol. 2001;12:1549–1557. doi: 10.1681/ASN.V1271549. [DOI] [PubMed] [Google Scholar]
- Kelder TP, Penning ME, Uh HW, Cohen D, Bloemenkamp KW, Bruijn JA, et al. Quantitative polymerase chain reaction-based analysis of podocyturia is a feasible diagnostic tool in preeclampsia. Hypertension. 2012;60:1538–1544. doi: 10.1161/HYPERTENSIONAHA.112.201681. [DOI] [PubMed] [Google Scholar]
- Kim JS, Han BG, Choi SO, Cha SK. Secondary focal segmental glomerulosclerosis: from podocyte injury to glomerulosclerosis. Biomed Res Int. 2016;2016:1630365. doi: 10.1155/2016/1630365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Chung JJ, Akilesh S, Koziell A, Jain S, Hodgin JB, et al. B cell-derived IL-4 acts on podocytes to induce proteinuria and foot process effacement. JCI Insight. 2017;2:pii: 81836. doi: 10.1172/jci.insight.81836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Ichii O, Miyazono K, Nakamura T, Horino T, Otsuka-Kanazawa S, et al. Overexpression of toll-like receptor 8 correlates with the progression of podocyte injury in murine autoimmune glomerulonephritis. Sci Rep. 2014;4:7290. doi: 10.1038/srep07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Ueno T, Ohashi K, Yamashita H, Takahashi Y, Sakamoto K, et al. Podocyte injury-driven intracapillary plasminogen activator inhibitor type 1 accelerates podocyte loss via uPAR-mediated beta1-integrin endocytosis. Am J Physiol Renal Physiol. 2015;308:F614–F626. doi: 10.1152/ajprenal.00616.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, et al. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- Kretzler M, Koeppen-Hagemann I, Kriz W. Podocyte damage is a critical step in the development of glomerulosclerosis in the uninephrectomised-desoxycorticosterone hypertensive rat. Virchows Arch. 1994;425:181–193. doi: 10.1007/BF00230355. [DOI] [PubMed] [Google Scholar]
- Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol. 2015;26:258–269. doi: 10.1681/ASN.2014030278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Elger M, Nagata M, Kretzler M, Uiker S, Koeppen-Hageman I, et al. The role of podocytes in the development of glomerular sclerosis. Kidney Int Suppl. 1994;45:S64–S72. doi: 10.1038/ki.1994.47. [DOI] [PubMed] [Google Scholar]
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- Kriz W, Hosser H, Hahnel B, Gretz N, Provoost AP. From segmental glomerulosclerosis to total nephron degeneration and interstitial fibrosis: a histopathological study in rat models and human glomerulopathies. Nephrol Dial Transplant. 1998;13:2781–2798. doi: 10.1093/ndt/13.11.2781. [DOI] [PubMed] [Google Scholar]
- Kriz W, Hosser H, Hahnel B, Simons JL, Provoost AP. Development of vascular pole-associated glomerulosclerosis in the Fawn-hooded rat. J Am Soc Nephrol. 1998;9:381–396. doi: 10.1681/ASN.V93381. [DOI] [PubMed] [Google Scholar]
- Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol. 2013;304:F333–F347. doi: 10.1152/ajprenal.00478.2012. [DOI] [PubMed] [Google Scholar]
- Kwon SH, Woollard JR, Saad A, Garovic VD, Zand L, Jordan KL, et al. Elevated urinary podocyte-derived extracellular microvesicles in renovascular hypertensive patients. Nephrol Dial Transplant. 2017;32:800–807. doi: 10.1093/ndt/gfw077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lee HS. Mechanisms and consequences of hypertriglyceridemia and cellular lipid accumulation in chronic kidney disease and metabolic syndrome. Histol Histopathol. 2011;26:1599–1610. doi: 10.14670/HH-26.1599. [DOI] [PubMed] [Google Scholar]
- Lennon R, Pons D, Sabin MA, Wei C, Shield JP, Coward RJ, et al. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant. 2009;24:3288–3296. doi: 10.1093/ndt/gfp302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewko B, Bryl E, Witkowski JM, Latawiec E, Golos M, Endlich N, et al. Characterization of glucose uptake by cultured rat podocytes. Kidney Blood Press Res. 2005;28:1–7. doi: 10.1159/000080889. [DOI] [PubMed] [Google Scholar]
- Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, et al. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176–178. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- Li G, Li CX, Xia M, Ritter JK, Gehr TW, Boini K, et al. Enhanced epithelial-to-mesenchymal transition associated with lysosome dysfunction in podocytes: role of p62/Sequestosome 1 as a signaling hub. Cell Physiol Biochem. 2015;35:1773–1786. doi: 10.1159/000373989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau MC, Lang D, Bohm J, Endlich N, Bek MJ, Witherden I, et al. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol Renal Physiol. 2006;290:F710–F719. doi: 10.1152/ajprenal.00475.2004. [DOI] [PubMed] [Google Scholar]
- Liu W, Zhang Y, Hao J, Liu S, Liu Q, Zhao S, et al. Nestin protects mouse podocytes against high glucose-induced apoptosis by a Cdk5-dependent mechanism. J Cell Biochem. 2012;113:3186–3196. doi: 10.1002/jcb.24195. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hitomi H, Diah S, Deguchi K, Mori H, Masaki T, et al. Roles of Na(+)/H(+) exchanger type 1 and intracellular pH in angiotensin II-induced reactive oxygen species generation and podocyte apoptosis. J Pharmacol Sci. 2013;122:176–183. doi: 10.1254/jphs.12291FP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler I, Wolf G. Epithelial-to-mesenchymal transition in diabetic nephropathy: fact or fiction? Cells. 2015;4:631–652. doi: 10.3390/cells4040631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z, Hu M, Zhen J, Lin J, Wang Q, Wang R. Rac1/PAK1 signaling promotes epithelial-mesenchymal transition of podocytes in vitro via triggering beta-catenin transcriptional activity under high glucose conditions. Int J Biochem Cell Biol. 2013;45:255–264. doi: 10.1016/j.biocel.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/S0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- Macconi D, Ghilardi M, Bonassi ME, Mohamed EI, Abbate M, Colombi F, et al. Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J Am Soc Nephrol. 2000;11:477–489. doi: 10.1681/ASN.V113477. [DOI] [PubMed] [Google Scholar]
- Machida H, Ito S, Hirose T, Takeshita F, Oshiro H, Nakamura T, et al. Expression of toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol Dial Transplant. 2010;25:2530–2537. doi: 10.1093/ndt/gfq058. [DOI] [PubMed] [Google Scholar]
- Mack M. Podocyte antigens, dendritic cells and T cells contribute to renal injury in newly developed mouse models of glomerulonephritis. Nephrol Dial Transplant. 2009;24:2984–2986. doi: 10.1093/ndt/gfp380. [DOI] [PubMed] [Google Scholar]
- Maekawa H, Inagi R. Stress signal network between hypoxia and ER stress in chronic kidney disease. Front Physiol. 2017;8:74. doi: 10.3389/fphys.2017.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao N, Tan RZ, Wang SQ, Wei C, Shi XL, Fan JM, et al. Ginsenoside Rg1 inhibits angiotensin II-induced podocyte autophagy via AMPK/mTOR/PI3 K pathway. Cell Biol Int. 2016;40:917–925. doi: 10.1002/cbin.10634. [DOI] [PubMed] [Google Scholar]
- Marquez E, Riera M, Pascual J, Soler MJ. Renin-angiotensin system within the diabetic podocyte. Am J Physiol Renal Physiol. 2015;308:F1–F10. doi: 10.1152/ajprenal.00531.2013. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia C, Izquierdo-Lahuerta A, Vivas Y, Velasco I, Yeo TK, Chen S, et al. Renal lipotoxicity-associated inflammation and insulin resistance affects actin cytoskeleton organization in podocytes. PLoS ONE. 2015;10:e0142291. doi: 10.1371/journal.pone.0142291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda D, Hirano K, Oku H, Sandoval JC, Kawase R, Yuasa-Kawase M, et al. Chylomicron remnants are increased in the postprandial state in CD36 deficiency. J Lipid Res. 2009;50:999–1011. doi: 10.1194/jlr.P700032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Pedigo CE, Mendez AJ. Metabolism, energetics, and lipid biology in the podocyte-cellular cholesterol-mediated glomerular injury. Front Endocrinol (Lausanne) 2014;5:169. doi: 10.3389/fendo.2014.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes. 2013;62:3817–3827. doi: 10.2337/db13-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima A, Ohshiro Y, Kitada M, Matsumoto M, Geraldes P, Li C, et al. Glomerular-specific protein kinase C-beta-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–896. doi: 10.1038/ki.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl) 1995;192:385–397. doi: 10.1007/BF00240371. [DOI] [PubMed] [Google Scholar]
- Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50:877–883. doi: 10.1161/HYPERTENSIONAHA.107.091058. [DOI] [PubMed] [Google Scholar]
- Nagata M. Podocyte injury and its consequences. Kidney Int. 2016;89:1221–1230. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Najafian B, Svarstad E, Bostad L, Gubler MC, Tondel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79:663–670. doi: 10.1038/ki.2010.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal CR, Muston PR, Njegovan D, Verrill R, Harper SJ, Deen WM, et al. Glomerular filtration into the subpodocyte space is highly restricted under physiological perfusion conditions. Am J Physiol Renal Physiol. 2007;293:F1787–F1798. doi: 10.1152/ajprenal.00157.2007. [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A, Mori Y, Uchiyama-Tanaka Y, Kishimoto N, Maruyama K, Matsubara H, et al. Regulation of glucose transporter (GLUT1) gene expression by angiotensin II in mesangial cells: involvement of HB-EGF and EGF receptor transactivation. Hypertens Res. 2003;26:67–73. doi: 10.1291/hypres.26.67. [DOI] [PubMed] [Google Scholar]
- Novelli R, Gagliardini E, Ruggiero B, Benigni A, Remuzzi G. Another piece of the puzzle of podocyte B7-1 expression: lupus nephritis. Nephron. 2016;133:129–138. doi: 10.1159/000446324. [DOI] [PubMed] [Google Scholar]
- Novelli R, Gagliardini E, Ruggiero B, Benigni A, Remuzzi G. Any value of podocyte B7-1 as a biomarker in human MCD and FSGS? Am J Physiol Renal Physiol. 2016;310:F335–F341. doi: 10.1152/ajprenal.00510.2015. [DOI] [PubMed] [Google Scholar]
- Orellana JM, Kampe K, Schulze F, Sieber J, Jehle AW. Fetuin-A aggravates lipotoxicity in podocytes via interleukin-1 signaling. Physiol Rep. 2017;5:pii: e13287. doi: 10.14814/phy2.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St Germain C, et al. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci U S A. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavenstadt H. Roles of the podocyte in glomerular function. Am J Physiol Renal Physiol. 2000;278:F173–F179. doi: 10.1152/ajprenal.2000.278.2.F173. [DOI] [PubMed] [Google Scholar]
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Pozdzik AA, Debiec H, Brocheriou I, Husson C, Rorive S, Broeders N, et al. Anti-NEP and anti-PLA2R antibodies in membranous nephropathy: an update. Rev Med Brux. 2015;36:166–171. [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. 2011;22:3520–3532. doi: 10.1091/mbc.e11-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles VG, Cullen-McEwen LA, Taylor GE, Li J, Hughson MD, Kerr PG, et al. Human podocyte depletion in association with older age and hypertension. Am J Physiol Renal Physiol. 2016;310:F656–F668. doi: 10.1152/ajprenal.00497.2015. [DOI] [PubMed] [Google Scholar]
- Qi W, Mu J, Luo ZF, Zeng W, Guo YH, Pang Q, et al. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism. 2011;60:594–603. doi: 10.1016/j.metabol.2010.07.021. [DOI] [PubMed] [Google Scholar]
- Qin W, Xu Z, Lu Y, Zeng C, Zheng C, Wang S, et al. Mixed organic solvents induce renal injury in rats. PLoS ONE. 2012;7:e45873. doi: 10.1371/journal.pone.0045873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raats CJ, van den Born J, Bakker MA, Oppers-Walgreen B, Pisa BJ, Dijkman HB, et al. Expression of agrin, dystroglycan, and utrophin in normal renal tissue and in experimental glomerulopathies. Am J Pathol. 2000;156:1749–1765. doi: 10.1016/S0002-9440(10)65046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raclot T, Langin D, Lafontan M, Groscolas R. Selective release of human adipocyte fatty acids according to molecular structure. Biochem J. 1997;324(Pt3):911–915. doi: 10.1042/bj3240911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci U S A. 2001;98:9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy K, Susztak K. Epithelial-mesenchymal transition and podocyte loss in diabetic kidney disease. Am J Kidney Dis. 2009;54:590–593. doi: 10.1053/j.ajkd.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reivinen J, Holthofer H, Miettinen A. A cell-type specific ganglioside of glomerular podocytes in rat kidney: an O-acetylated GD3. Kidney Int. 1992;42:624–631. doi: 10.1038/ki.1992.327. [DOI] [PubMed] [Google Scholar]
- Ren Z, Liang W, Chen C, Yang H, Singhal PC, Ding G. Angiotensin II induces nephrin dephosphorylation and podocyte injury: role of caveolin-1. Cell Signal. 2012;24:443–450. doi: 10.1016/j.cellsig.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M, Ortega A, Izquierdo A, Lopez-Luna P, Bosch RJ. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): implications for diabetic nephropathy. Nephrol Dial Transplant. 2010;25:2447–2457. doi: 10.1093/ndt/gfq104. [DOI] [PubMed] [Google Scholar]
- Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol. 2005;16:1205–1213. doi: 10.1681/ASN.2004121080. [DOI] [PubMed] [Google Scholar]
- Ronco P, Debiec H. Target antigens and nephritogenic antibodies in membranous nephropathy: of rats and men. Semin Immunopathol. 2007;29:445–458. doi: 10.1007/s00281-007-0091-2. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P, Cravedi P, Remuzzi G. Mechanisms and treatment of CKD. J Am Soc Nephrol. 2012;23:1917–1928. doi: 10.1681/ASN.2012040390. [DOI] [PubMed] [Google Scholar]
- Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestila M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A. 1999;96:7962–7967. doi: 10.1073/pnas.96.14.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol. 2011;22:1189–1199. doi: 10.1681/ASN.2010040384. [DOI] [PubMed] [Google Scholar]
- Ruster C, Bondeva T, Franke S, Tanaka N, Yamamoto H, Wolf G. Angiotensin II upregulates RAGE expression on podocytes: role of AT2 receptors. Am J Nephrol. 2009;29:538–550. doi: 10.1159/000191467. [DOI] [PubMed] [Google Scholar]
- Sakai K, Morito N, Usui J, Hagiwara M, Hiwatashi A, Fukuda K, et al. Focal segmental glomerulosclerosis as a complication of hepatitis B virus infection. Nephrol Dial Transplant. 2011;26:371–373. doi: 10.1093/ndt/gfq600. [DOI] [PubMed] [Google Scholar]
- Sakoda M, Ichihara A, Kurauchi-Mito A, Narita T, Kinouchi K, Murohashi-Bokuda K, et al. Aliskiren inhibits intracellular angiotensin II levels without affecting (pro)renin receptor signals in human podocytes. Am J Hypertens. 2010;23:575–580. doi: 10.1038/ajh.2009.273. [DOI] [PubMed] [Google Scholar]
- Sakoda M, Itoh H, Ichihara A. Podocytes as a target of prorenin in diabetes. Curr Diabetes Rev. 2011;7:17–21. doi: 10.2174/157339911794273955. [DOI] [PubMed] [Google Scholar]
- Sanchez-Nino MD, Sanz AB, Carrasco S, Saleem MA, Mathieson PW, Valdivielso JM, et al. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant. 2011;26:1797–1802. doi: 10.1093/ndt/gfq306. [DOI] [PubMed] [Google Scholar]
- Schermer B, Benzing T. Lipid-protein interactions along the slit diaphragm of podocytes. J Am Soc Nephrol. 2009;20:473–478. doi: 10.1681/ASN.2008070694. [DOI] [PubMed] [Google Scholar]
- Schiffer M, Susztak K, Ranalletta M, Raff AC, Bottinger EP, Charron MJ. Localization of the GLUT8 glucose transporter in murine kidney and regulation in vivo in nondiabetic and diabetic conditions. Am J Physiol Renal Physiol. 2005;289:F186–F193. doi: 10.1152/ajprenal.00234.2004. [DOI] [PubMed] [Google Scholar]
- Schomig M, Eisenhardt A, Ritz E. The microinflammatory state of uremia. Blood Purif. 2000;18:327–332. doi: 10.1159/000014457. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI200112849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin B, Boutros PC, Li X, Okey AB, Uetrecht JP. Gene expression profiling in a model of D-penicillamine-induced autoimmunity in the Brown Norway rat: predictive value of early signs of danger. Chem Res Toxicol. 2005;18:1193–1202. doi: 10.1021/tx050040m. [DOI] [PubMed] [Google Scholar]
- Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–364. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- Shimada M, Araya C, Rivard C, Ishimoto T, Johnson RJ, Garin EH. Minimal change disease: a “two-hit” podocyte immune disorder? Pediatr Nephrol. 2011;26:645–649. doi: 10.1007/s00467-010-1676-x. [DOI] [PubMed] [Google Scholar]
- Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappaB-dependent pathway. Nephrol Dial Transplant. 2012;27:81–89. doi: 10.1093/ndt/gfr271. [DOI] [PubMed] [Google Scholar]
- Sieber J, Jehle AW. Free Fatty acids and their metabolism affect function and survival of podocytes. Front Endocrinol (Lausanne) 2014;5:186. doi: 10.3389/fendo.2014.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber J, Lindenmeyer MT, Kampe K, Campbell KN, Cohen CD, Hopfer H, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010;299:F821–F829. doi: 10.1152/ajprenal.00196.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Schwarz K, Kriz W, Miettinen A, Reiser J, Mundel P, et al. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol. 2001;159:1069–1077. doi: 10.1016/S0002-9440(10)61782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetikno V, Sari FR, Sukumaran V, Lakshmanan AP, Harima M, Suzuki K, et al. Curcumin decreases renal triglyceride accumulation through AMPK-SREBP signaling pathway in streptozotocin-induced type 1 diabetic rats. J Nutr Biochem. 2013;24:796–802. doi: 10.1016/j.jnutbio.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol. 2014;184:1715–1726. doi: 10.1016/j.ajpath.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Sorensson J, Fierlbeck W, Heider T, Schwarz K, Park DS, Mundel P, et al. Glomerular endothelial fenestrae in vivo are not formed from caveolae. J Am Soc Nephrol. 2002;13:2639–2647. doi: 10.1097/01.ASN.0000033277.32822.23. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. doi: 10.2337/diabetes.55.01.06.db05-0894. [DOI] [PubMed] [Google Scholar]
- Szabo C, Biser A, Benko R, Bottinger E, Susztak K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55:3004–3012. doi: 10.2337/db06-0147. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997–1011. doi: 10.1016/j.kint.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Takano T, Elimam H, Cybulsky AV. Complement-mediated cellular injury. Semin Nephrol. 2013;33:586–601. doi: 10.1016/j.semnephrol.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Takeda T, McQuistan T, Orlando RA, Farquhar MG. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J Clin Invest. 2001;108:289–301. doi: 10.1172/JCI12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- Tavasoli M, Li L, Al-Momany A, Zhu LF, Adam BA, Wang Z, et al. The chloride intracellular channel 5A stimulates podocyte Rac1, protecting against hypertension-induced glomerular injury. Kidney Int. 2016;89:833–847. doi: 10.1016/j.kint.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Udani S, Lazich I, Bakris GL. Epidemiology of hypertensive kidney disease. Nat Rev Nephrol. 2011;7:11–21. doi: 10.1038/nrneph.2010.154. [DOI] [PubMed] [Google Scholar]
- Velez JC, Bland AM, Arthur JM, Raymond JR, Janech MG. Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am J Physiol Renal Physiol. 2007;293:F398–F407. doi: 10.1152/ajprenal.00050.2007. [DOI] [PubMed] [Google Scholar]
- Wahl P, Ducasa GM, Fornoni A. Systemic and renal lipids in kidney disease development and progression. Am J Physiol Renal Physiol. 2016;310:F433–F445. doi: 10.1152/ajprenal.00375.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Lai FM, Kwan BC, Lai KB, Chow KM, Li PK, et al. Podocyte loss in human hypertensive nephrosclerosis. Am J Hypertens. 2009;22:300–306. doi: 10.1038/ajh.2008.360. [DOI] [PubMed] [Google Scholar]
- Wang L, Sun S, Zhou A, Yao X, Wang Y. oxLDL-induced lipid accumulation in glomerular podocytes: role of IFN-gamma, CXCL16, and ADAM10. Cell Biochem Biophys. 2014;70:529–538. doi: 10.1007/s12013-014-9952-1. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- Wolf G, Schanze A, Stahl RA, Shankland SJ, Amann K. p27(Kip1) Knockout mice are protected from diabetic nephropathy: evidence for p27(Kip1) haplotype insufficiency. Kidney Int. 2005;68:1583–1589. doi: 10.1111/j.1523-1755.2005.00570.x. [DOI] [PubMed] [Google Scholar]
- Xing L, Liu Q, Fu S, Li S, Yang L, Liu S, et al. PTEN inhibits high glucose-induced phenotypic transition in podocytes. J Cell Biochem. 2015;116:1776–1784. doi: 10.1002/jcb.25136. [DOI] [PubMed] [Google Scholar]
- Xu ZG, Yoo TH, Ryu DR, Cheon Park H, Ha SK, Han DS, et al. Angiotensin II receptor blocker inhibits p27Kip1 expression in glucose-stimulated podocytes and in diabetic glomeruli. Kidney Int. 2005;67:944–952. doi: 10.1111/j.1523-1755.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- Xu ZE, Chen Y, Huang A, Varghese Z, Moorhead JF, Yan F, et al. Inflammatory stress exacerbates lipid-mediated renal injury in ApoE/CD36/SRA triple knockout mice. Am J Physiol Renal Physiol. 2011;301:F713–F722. doi: 10.1152/ajprenal.00341.2010. [DOI] [PubMed] [Google Scholar]
- Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, et al. ANG II promotes autophagy in podocytes. Am J Physiol Cell Physiol. 2010;299:C488–C496. doi: 10.1152/ajpcell.00424.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Iwano M, Suzuki D, Nakatani K, Kimura K, Harada K, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54:653–664. doi: 10.1053/j.ajkd.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Yard BA, Kahlert S, Engelleiter R, Resch S, Waldherr R, Groffen AJ, et al. Decreased glomerular expression of agrin in diabetic nephropathy and podocytes, cultured in high glucose medium. Exp Nephrol. 2001;9:214–222. doi: 10.1159/000052614. [DOI] [PubMed] [Google Scholar]
- Ying WZ, Wang PX, Sanders PW. Induction of apoptosis during development of hypertensive nephrosclerosis. Kidney Int. 2000;58:2007–2017. doi: 10.1111/j.1523-1755.2000.00373.x. [DOI] [PubMed] [Google Scholar]
- Yoo TH, Li JJ, Kim JJ, Jung DS, Kwak SJ, Ryu DR, et al. Activation of the renin-angiotensin system within podocytes in diabetes. Kidney Int. 2007;71:1019–1027. doi: 10.1038/sj.ki.5002195. [DOI] [PubMed] [Google Scholar]
- You H, Gao T, Raup-Konsavage WM, Cooper TK, Bronson SK, Reeves WB, et al. Podocyte-specific chemokine (C-C motif) receptor 2 overexpression mediates diabetic renal injury in mice. Kidney Int. 2017;91:671–682. doi: 10.1016/j.kint.2016.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Schin M, Saha J, Burke K, Holzman LB, Filipiak W, et al. Podocyte-specific overexpression of GLUT1 surprisingly reduces mesangial matrix expansion in diabetic nephropathy in mice. Am J Physiol Renal Physiol. 2010;299:F91–F98. doi: 10.1152/ajprenal.00021.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L, et al. Dysregulation of low-density lipoprotein receptor contributes to podocyte injuries in diabetic nephropathy. Am J Physiol Endocrinol Metab. 2015;308:E1140–E1148. doi: 10.1152/ajpendo.00591.2014. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L, et al. Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta Diabetol. 2015;52:1045–1056. doi: 10.1007/s00592-015-0753-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma KL, Ruan XZ, Liu BC. Dysregulation of the low-density lipoprotein receptor pathway is involved in lipid disorder-mediated organ injury. Int J Biol Sci. 2016;12:569–579. doi: 10.7150/ijbs.14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Matsusaka T, Zhong J, Ma J, Ma LJ, Hanna Z, et al. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. 2006;17:2832–2843. doi: 10.1681/ASN.2005080878. [DOI] [PubMed] [Google Scholar]