

Abstract
Feline infectious peritonitis (FIP) is a lethal systemic disease caused by FIP virus (FIPV). There are no effective vaccines or treatment available, and the virus virulence determinants and pathogenesis are not fully understood. Here, we describe the sequencing of RNA extracted from Crandell Rees Feline Kidney (CRFK) cells infected with FIPV using the Illumina next-generation sequencing approach. Bioinformatics analysis, based on Felis catus 2X annotated shotgun reference genome, using CLC bio Genome Workbench is used to map both control and infected cells. Kal’s Z test statistical analysis is used to analyze the differentially expressed genes from the infected CRFK cells. In addition, RT-qPCR analysis is used for further transcriptional profiling of selected genes in infected CRFK cells and Peripheral Blood Mononuclear Cells (PBMCs) from healthy and FIP-diagnosed cats.
Key words: FIPV, CRFK, PBMCs, Transcriptome, Kal’s Z test, RT-qPCR, Gene expression
Introduction
The use of a next-generation sequencing approaches in RNA sequencing has facilitated understanding and defining the expression profiles of cellular responses during pathogen infection. This method has been proven to be helpful in explaining the pathogenesis of various viruses [1, 2], including Feline Immunodeficiency Virus (FIV) [3, 4]. Furthermore, the increasing availability of complete genome sequences for a number of model organisms makes host transcriptome analysis a valuable tool for elucidating mechanisms of virus pathogenesis and host responses to virus infection. Feline infectious peritonitis virus (FIPV) is thought to be the causative agent of feline infectious peritonitis (FIP). Understanding the molecular basis of FIPV pathogenesis will provide valuable information to devise effective treatments and formulate vaccines with higher efficacy. Once established, focus can be directed at disrupting the virulent determinants or formulating new vaccine or even designing gene therapy treatment. Facilitating this, the complete 1.9X cat genome, using the Whole Genome Shotgun (WGS) approach, provides valuable information for bioinformatics analysis of feline host responses following pathogen infection. Moreover, the cat genome contigs were aligned, mapped, and annotated to NCBI annotated genome sequences of six index mammalian genomes (human, chimpanzee, mouse, rat, dog and cow) using MegaBLAST [5]. In this chapter we describe a pipeline for transcriptome analysis using FIPV infection of feline cells in culture as an example. Specifically, mRNA harvested from CRFK cells 3 h post infection with FIPV strain 79-1146 were sequenced using Illumina next-generation sequencing technology. The generated data was then analyzed using CLC bio Genomic Workbench, where the genes were compared to Felis catus 1.9X annotated shotgun reference genome. Kal’s Z-test on expression proportions [6] was used to determine significantly expressed genes. Genes expressed with a False Discovery Rate (FDR) <0.05 and >1.99- and <-1.99-fold change were considered for further analysis.
Materials
RNA Extraction and Next-Generation Sequencing
CRFK cell line.
Virus strain: FIPV 79–1146.
Maintenance medium : Minimal essential medium, 10 % fetal bovine serum, 200 μM nonessential amino acids, 200 units/ml penicillin, 200 μg/ml streptomycin, 0.5 μg/ml antimycotic.
Dulbecco’s Phosphate Buffered Saline containing no calcium and magnesium (D-PBS).
TrypLE™ Express solution or 0.25 % trypsin, 0.02 % EDTA in PBS.
RNase AWAY (Life Technologies) or similar.
RNeasy mini kit (Qiagen) or similar.
UV/Visible spectrophotometer.
Illumina Agilent RNA 6000 Nano kit.
Agilent 2100 Bio-analyzer.
Paired-End Sequencing Preparation Kit (Illumina), or similar.
Validation of Results by RT-qPCR
QiaAmp Viral RNA Mini Kit (Qiagen) or similar.
RNeasy kit (Qiagen).
RNase AWAY.
Quantitative PCR machine (e.g., CFX 96 Real-Time PCR Detection System (Bio-Rad)).
RNeasy miniplus kit (Qiagen) or similar.
NanoDrop Nanophotometer or spectrophotometer.
SensiFAST™ SYBR No-ROX One Step kit (Bioline).
Primers specific for genes of interest and reference genes.
BD Vacutainer (BD) EDTA-K2 tubes.
Ficoll-Paque Plus.
Methods
Transcriptome Analysis of FIPV Infected Cells
To take advantages of this technology, simultaneous analysis of virus–host interactions is investigated in one single experiment where both the transcription of viral genomes and host cell responses are scrutinized. Figure 1 illustrates the work flow for transcriptome analysis of this study.
Fig. 1.
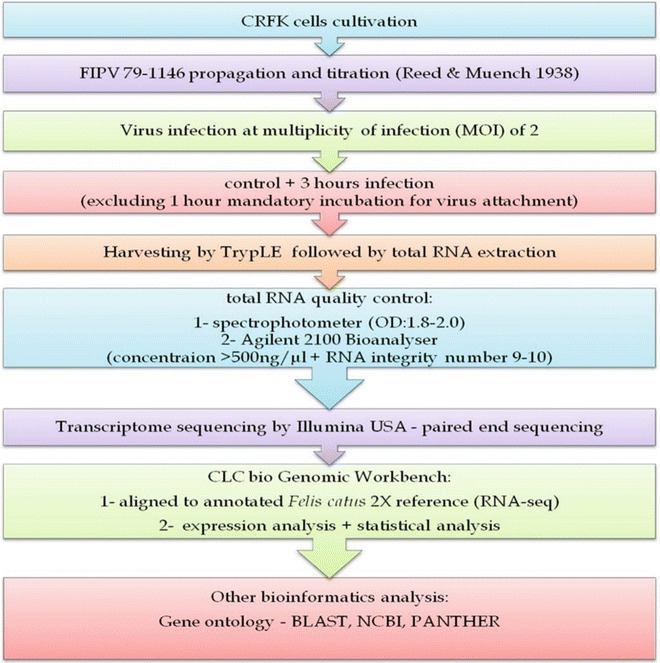
Work flow for transcriptome analysis of CRFK cells infected with FIPV 79–1146
Infection of Cells
Seed CRFK cells into 75 cm2 flasks and incubate at 37 °C with 5 % CO2 until cells reach 60–70 % confluency.
Remove media and wash the cells with D-PBS.
Infect the flasks with virus at multiplicity of infection (MOI) 2 in 2 ml, or 2 ml D-PBS as a mock control, and incubate at 37 °C with 5 % CO2 for 1 h to allow attachment. Perform inoculations in duplicates, one for RNA extraction and the other for CPE visualization.
Add 10 ml of maintenance medium with 10 % FBS and incubate the flasks for 3 h.
Following 3 h of incubation, remove inoculum, wash the cells with D-PBS.
Add 2 ml TrypLE and incubate for 1–2 min until cells detach.
Transfer cells to a centrifuge tube and pellet the cells by centrifugation at 120 × g for 5 min and discard supernatant.
Add 10 ml D-PBS and repeat centrifugation in order to remove every trace of medium and TrypLE, which could reduce RNA yield.
Discard the supernatants and store the cell pellets at −80 °C until RNA purification.
RNA Purification from Infected Cells
The RNeasy kit was used to extract and purify RNA samples in this study (see Note 1) but other RNA extraction protocols may also be suitable.
Spray all micropipettes, gloves, working area, and other things with RNase AWAY to remove any RNase and DNA contamination.
Extract RNA using RNeasy kit according to the manufacturer’s instruction.
Aliquot the eluted RNAs (500 μl) into three different tubes to avoid repeated thawing and freezing of the sample which could affect the quality of the RNA.
Use two tubes for quality control analysis with spectrophotometer and Illumina Agilent 2100 bio-analyzer and store the third one at −80 °C for sequencing.
Total RNA Quality Analysis
Determine the quality of extracted RNA by measuring the absorbance at 260 and 280 nm in UV/Visible spectrophotometer.
Consider the samples with the absorbance ratio value (A260/A280) of 1.8 to 2.0 for further analysis with Illumina Agilent 2100 Bio-analyzer to determine both the RNA quality and quantity.
In order to ensure the samples are of highest quality and quantity for transcriptome sequencing, use Agilent RNA 6000 Nano kit together with Agilent 2100 Bio-analyzer to conduct quality and quantity analysis to the extracted total RNA samples (see Note 2).
Load and prime gel–dye mixtures, then load RNA 6000 Nano marker, ladder, and samples in the specified manner.
Vortex the chip and insert in the Agilent 2100 Bio-analyzer machine. Analyze the chips based on the method recommended by the manufacturer. Verify whether the run is successful and whether the sample is properly prepared and handled by means of properly pipetted into the wells (see Note 3).
Preparing Samples for Paired-End Sequencing
Perform the following steps using the reagents provided in the paired end sample preparation kit, according to manufacturer’s instructions.
Fragment genomic DNA into fragments of less than 800 bp.
Perform end repair of DNA fragments to generate 5′-phosphorylated blunt ends.
Add an “A” base to the 3′ ends to make 3′-dA overhang.
Ligate adapters to the ends of the DNA fragments.
Purify ligation products by removing un-ligated adapters.
Enrich the Adapter-Modified DNA Fragments by PCR.
Obtain the Genomic DNA library.
CLC Bio Genomics Workbench (GWB) Software Analysis
Figure 2 represents the work flow of raw data analysis by CLC bio GWB software and the settings opted for each process. Unless stated otherwise, the settings for the raw data analysis were based from CLC bio manual (see Note 4).
Conduct RNA-seq analysis with the settings stated in the Fig. 2, using trimmed raw sequences. Quality trim limit = 0.01, ambiguity trim maximum value = 2, Adapter trim = Illumina adapter, trim both strands. Map to annotated reference—Felis catus 2X, Minimum length fraction and minimum similarity fraction = 0.9, Maximum number of hits/read = 1, Type of organism = eukaryote, Paired settings = default.
Reads Per Kilobase of transcript per Million (RPKM) are chosen as the expression value for comparison (see Note 5).
Once finished, conduct quality control to the expression analysis control versus 3 h infection (Cv3) result.
Subsequently, conduct statistical analysis to the Cv3 expression analysis based on CLC bio support recommendation.
Determine upregulated and downregulated genes from the final Cv3 expression analysis result by selecting the criteria as in Fig. 2. Upregulated = proportions fold change > 1.99 and Downregulated = proportions fold change < −1.99.
Fig. 2.

Work flow and settings for CLC bio GWB software analysis
Basic Local Alignment Search Tool (BLAST)
Once data has been obtained, import the raw data (~17.3 Gigabyte) into the CLC bio GWB. Once imported, subject the raw data to sequence reads trimming by quality trimming, ambiguity trimming and adapter trimming with the settings as in Fig. 2. The program uses the modified-Mott trimming algorithm for this purpose (see manufacturers instructions) (see Note 6).
BLAST the list of genes that were upregulated and downregulated, using the built-in BLAST program in the CLC bio GWB.
Based on the BLAST result, select homologous sequence with the lowest e-value, highest score and lowest percentage of gaps to the query sequence as the gene identity.
Briefly, opt blastn: DNA sequence and database program and references mRNA sequences (refseq_rna) or nucleotide collection (nr) database for analysis. In silico analysis which is also a part of bioinformatics analysis is able to analyze the interactions of different genes by integrating data available on bioinformatics databases (see Note 7).
Validation of Results by Real-Time RT-qPCR Analysis
Real-Time RT-qPCR Analysis of FIPV Infected CRFK Cells
In order to validate the transcriptome results, analyze the expression profiles of genes of interest (for examples A3H, PD-1, and PD-L1 genes) using real-time PCR.
Seed CRFK cells into 6-well plates and incubate at 37 °C 5 % CO2 until 60–70 % confluent.
Wash cells once with D-PBS and then infect with FIPV strain 79–1146 at MOI 2 in 1 ml, or with 1 ml D-PBS as a negative control.
Incubate cells at 37 °C 5 % CO2 for desired time.
Extract viral RNA from cells using QiaAmp Viral RNA Mini Kit, according to the manufacturer’s instructions.
Design primers for qPCR specific for the genes of interest, e.g., Table 1.
Perform the RT-qPCR reactions using for example SensiFAST™ SYBR No-ROX One Step kit on Real-Time System, with Thermal Cycler. The reaction mixture of 20 μl contains 10 μl 2× SensiFAST SYBR No-ROX One-Step mix, 0.5 μl forward and reverse primers (5 nM for GADPH, PD-L1, and A3H, 3 nM for PD-1, and 10 nM for YWHAZ), 0.2 μl RT, 0.4 μl RiboSafe RNase inhibitor, 2.4 μl H2O, and 6 μl extracted RNA.
The RT-qPCR reaction conditions are as follows; one cycle at 45 °C for 10 min, one cycle at 95 °C for 2 min, and 35 cycles at 95 °C for 5 s; then 57 °C (YWHAZ), 58 °C (PD-L1), 59 °C (GAPDH), 64 °C (A3H), and 65 °C (PD-1) for 20 s; and finally, at 72 °C for 5 s (Table 2). One cycle for the dissociation curve for all reactions is added and melting curve analysis is performed.
Analyze the data generated from the technical triplicate experiment with 2ΔΔCT method, selecting appropriate reference genes, e.g., GAPDH and /or YWHAZ, using Bio-Rad CFX Manager.
Table 1.
Sequence of primers for RT-qPCR
| Target gene | Accession number | Sequence 5′–3′ | References |
|---|---|---|---|
| GAPDH | NM 001009307 |
F: AGTATGATTCCACCCACGGCA R: GATCTCGCTCCTGGAAGATGGT |
[7] |
| YWHAZ | EF458621 |
F: ACAAAGACAGCACGCTAATAATGC R: CTTCAGCTTCATCTCCTTGGGTAT |
[9] |
| PD-1 | EU295528 |
F: GAGAACGCCACCTTCGTC R: TGGGCTCTCATAGATCTGCGT |
[10] |
| PD-L1 | EU246348 |
F: CGATCACAGTGTCCAAGGACC R: TCCGCTTATAGTCAGCACCG |
[10] |
| A3H | EF173020 |
F: ACCCACAATGAATCCACTACAG R: AGGCAGTCTTTGTGAATTAGGG |
[11] |
F forward primer, R reverse primer
Table 2.
Amplification program for one step RT-qPCR assay
| No. | Step | Temperature (°C) | Time |
|---|---|---|---|
| 1 | Reverse transcription | 48 | 45 min |
| 2 | Initial denaturation | 94 | 02 min |
| 3 | Denaturation | 94 | 15 s |
| 4 | Annealing | 62 | 15 s |
| 5 | Extension | 72 | 15 s |
| 6 | Repeat steps 3–5 for 35 cycles | ||
| 7 | Final extension | 72 | 05 min |
Real-Time RT-qPCR Analysis of Peripheral Blood Mononuclear Cells from FIP Diagnosed Cats
Besides FIPV infected cell culture, feline Peripheral Blood Mononuclear Cells (PBMCs) can be used to analyze the transcriptome results, providing valuable in vivo results for comparison.
Select five healthy cats and five infected cats. Perform the sampling according to internationally recognized guidelines and recommended by the Animal Care and Use Committee.
Draw 2–5 ml of cat blood and store at 4 °C in BD Vacutainer® EDTA-K2 tubes.
Isolate PBMCs using the Ficoll-Paque™ Plus method, according to the manufacturer’s protocol.
Isolate total RNA from PBMCs using an RNeasy mini plus kit, as described by the manufacturer.
Measure and assess the RNA quantity and purity using NanoDrop.
Store the isolated RNA samples at −80 °C for further analysis, or immediately use for real-time RT-qPCR analysis, as steps 6–9 in Subheading 3.2.1.
Notes
The wash step is performed in order to remove traces of serum, calcium, and magnesium that would inhibit the action of dissociation agent.
http://www.chem.agilent.com/Library/usermanuals/Public/G2938-90034_RNA6000Nano_KG.pdf.
The first feature of a successful total RNA run is that the electropherogram must contain three peaks where one peak represents marker peak while the others two are 18S and 28S ribosomal peaks. Absence of one or both of the ribosomal peaks indicates poor sample preparation or poor sample pipetting technique. The second feature of a successful run is a complete ladder electropherogram. A complete ladder electropherogram must feature one marker peak and six RNA peaks where all seven peaks are well resolved.
Adapters used by common high-throughput sequencing vendors such as Illumina and SOLiD were predefined and are available by the software. Removing the adapters will increase the specificity of the raw sequence reducing false match.
Perform expression analysis based on the method described by Mortazavi et al. in 2008 [7] and CLC bio manual, CLC bio tutorials and recommendations from CLC bio support services.
In short, high quality trim value allowed low quality base or base with low Phred quality score to be included in the sequence. The ambiguity trimming trims the sequence ends based on the presence of ambiguous nucleotides usually denoted as N making the sequence more specific.
-
Other Bioinformatics analysis: A gene in eukaryotic organism is commonly regulated by other genes and proteins in its system. The interactions among genes expressed and between gene expressed and other genes can be elucidated by means of computer or in silico analysis. In silico analysis, a part of bioinformatics analysis, able to do this by integrating data with available data on bioinformatics databases. Such integration will allow a researcher to make accurate predictions and designing experiments to test the hypothesis. The main objective of in silico analysis is gene ontology which is defined as the process of elucidating associated pathways, molecular function, biological process, cellular components and protein products of a gene [8].
The bioinformatics database used to analyze gene identity and gene interaction is Protein Analysis Through Evolutionary Relationships Classification System or in short known as PANTHER (http://www.pantherdb.org/). It is a unique resource that classifies genes by their functions, using published scientific experimental evidence and evolutionary relationships to predict function even in the absence of direct experimental evidence and is a part of the Gene Ontology Reference Genome Project (http://www.geneontology.org/GO.refgenome.shtml#curation).
PANTHER provides tools for gene expression analysis for data interpretation (http://www.pantherdb.org/tools/genexAnalysis.jsp). Multiple gene lists will be mapped to PANTHER molecular function, biological process, and cellular component categories as well as to biological pathways. The gene expression data interpretation is conducted by comparing genes in a given list and statistically compares the list to the reference list to look for under and over represented functional categories. The step-by-step method and the statistical test employed are described in detail at http://www.pantherdb.org/tips/tips_binomial.jsp. For the statistical analysis, a cutoff of less than 0.05 is selected as significant p-value.
Acknowledgments
The authors wish to thank laboratory personnel at Virology Lab, Faculty of Veterinary Medicine, UPM and Laboratory of Vaccines and Immunotherapeutics, Institute of Bioscience, UPM. This project was funded by Fundamental Research Project No: 01-11-08-6390FR, Ministry of Higher Education, Malaysia. The funding source has no role in this study.
Contributor Information
Helena Jane Maier, Email: helena.maier@pirbright.ac.uk.
Erica Bickerton, Email: erica.bickerton@pirbright.ac.uk.
Paul Britton, Email: paul.britton@pirbright.ac.uk.
Abdul Rahman Omar, Email: aro@upm.edu.my.
References
- 1.Nanda S, Havert MB, Calderón GM, et al. Hepatic transcriptome analysis of hepatitis C virus infection in chimpanzees defines unique gene expression patterns associated with viral clearance. PLoS One. 2008;3:e3442. doi: 10.1371/journal.pone.0003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assarsson E, Greenbaum JA, Sundström M, et al. Kinetic analysis of a complete poxvirus transcriptome an immediate early class of gene. Proc Natl Acad Sci U S A. 2008;105:2140–2145. doi: 10.1073/pnas.0711573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dowling RJ, Bienzle D. Gene-expression changes induced by Feline immunodeficiency virus infection differ in epithelial cells and lymphocytes. J Gen Virol. 2005;86:2239–2248. doi: 10.1099/vir.0.80735-0. [DOI] [PubMed] [Google Scholar]
- 4.Ertl R, Birzele F, Hildebrandt T, et al. Viral transcriptome analysis of feline immunodeficiency virus infected cells using second generation sequencing technology. Vet Immunol Immunopathol. 2011;143:314–324. doi: 10.1016/j.vetimm.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Schwartz S, Wagner L, et al. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 6.Kal AJ, Van Zonneveld AJ, Benes V, et al. Dynamics of gene expression revealed by comparison of serial analysis of gene expression transcript profiles from yeast grown on two different carbon sources. Mol Biol Cell. 1999;10:1859–1872. doi: 10.1091/mbc.10.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 8.Ashburner M, Ball CA, Blake J, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Livak K, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 10.Li MM, Emerman M. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J Virol. 2011;85:8197–8207. doi: 10.1128/JVI.00624-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harun MSR, Choong OK, Selvarajah GT, et al. Transcriptional profiling of feline infectious peritonitis virus infection in CRFK cells and in PBMCs from FIP diagnosed cats. Virol J. 2013;10:329. doi: 10.1186/1743-422X-10-329. [DOI] [PMC free article] [PubMed] [Google Scholar]