

Abstract
The etiologic agent of severe acute respiratory syndrome (SARS) has been identified as a new type of coronavirus, known as SARS-coronavirus (SARS-CoV). Although the SARS epidemic has subsided, many authorities, including the World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC), have warned of the possible re-emergence of this highly infectious disease. Although antibody-based diagnosis of SARS has been demonstrated to be a reliable proof of SARS infection, it is not sensitive enough for detection during the early phase of the disease. To date, based on the publicly released full genomic sequences of SARS-CoV, various molecular detection methods based on reverse-transcription polymerase chain reaction (RT-PCR) have been developed. Although most of the assays have initially been focused on RNA extracted from nasopharyngeal aspirates, urine, and stools, several of the more recently developed assays have been based on the analysis of RNA extracted from plasma and serum. Such assays allow the more standardized quantitative expression of viral loads and are potentially useful for early SARS diagnosis. In this chapter, two real-time quantitative RT-PCR systems for the quantification of SARS-CoV RNA in serum are discussed. The two RT-PCR systems, one aimed toward the nucleocapsid region and the other toward the polymerase region of the virus genome, have a detection rate of up to 80% during the first week of illness. These quantitative systems are potentially useful for the early diagnosis of SARS and can also provide viral load information that might assist clinicians in making a prognostic evaluation of an infected individual.
Key Words: Serum RNA, SARS-CoV RNA, viral RNA extraction, RNA quantification, real-time quantitative reverse-transcription PCR
Introduction
The identification of severe acute respiratory syndrome-coronavirus (SARS-CoV) as the etiologic agent of SARS has led scientists to develop rapid and sensitive diagnostic tests (1–6). Although the SARS epidemic has subsided, many authorities, including the World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC), have warned of the possible re-emergence of this highly infectious disease. Thus, the sensitive tests for SARS are of great public health importance. However, a number of diagnostic tests are not very sensitive during the early phase of the disease. For instance, the use of nasopharyngeal aspirates using an early generation reversetranscription polymerase chain reaction (RT-PCR) for SARS-CoV has a sensitivity of only 32% on d 3 of the disease (7). This severe limitation has restricted our ability to identify patients in a prompt manner and to institute isolation and treatment.
Based on the publicly released full genomic sequences of SARS-CoV (8–10), various molecular detection methods based on RT-PCR have been developed. These PCR-based diagnostic tests are used to detect SARS-CoV RNA in patients' specimens in which viral RNA is reverse-transcribed into DNA and then different regions of the SARS-CoV genome are specifically amplified by PCR. Several RT-PCR protocols developed by members of the WHO laboratory network are available on the WHO website (http://www.who.int/csr/sars/ primers/en/).
RT-PCR is mainly divided into qualitative (conventional) and quantitative approaches. Conventional RT-PCR approaches are normally qualitative in nature and require time-consuming and contamination-prone post-PCR analysis. Real-time quantitative RT-PCR has overcome many of these shortcomings and has been increasingly adopted by various laboratories for SARS diagnosis (2, 11–15). With suitable instrumentation, this technology allows data to be recorded and analyzed during PCR cycling. Furthermore, it runs as a closedtube system, and postamplification manipulation can be eliminated. Thus, this methodology reduces the risk of contamination and minimizes hands-on time. The entire amplification process requires only 3 h and allows such technology to be used for high-throughput application.
During the SARS outbreak, the PCR-based testing for SARS was focused mainly on the analysis of nasopharyngeal aspirates, urine, and stools (7, 11). An early study reported that SARS-CoV RNA was detected in 32% of nasopharyngeal aspirates from SARS patients studied at a mean of 3.2 d after the onset of illness, and the detection rate increased to 68% at d 14 (7). In the same study, SARS-CoV RNA was detected in 97% of stool samples collected at a mean of 14.2 d after symptom onset. Similarly, viral RNA was detected in 42% of urine samples collected from the SARS patients at a mean of 15.2 d after onset (7). Despite the high sensitivity of stool sample testing, early detection of SARS-CoV still suffers from a lack of high sensitivity. Although most of the assays have been predominantly focused on RNA extracted from nasopharyngeal aspirates, urine, and stools, the quantitative interpretation of these data can be difficult because of the inability to standardize such data as a result of numerous factors, such as sampling technique for nasopharyngeal aspirates, urine volume, variations of bowel transit time (e.g., during diarrhea), or stool consistency. On the other hand, plasma/serum-based assays may allow the precise and standardized quantitative expression of viral loads, thus enabling the assessment of disease severity and prognosis. Detection of viral nucleic acids in plasma/serum has been well established for viral load studies for numerous other viruses (16, 17). At the beginning of the SARS outbreak, a single report showed the relatively low sensitivity of detecting SARS-CoV RNA in plasma using an ultracentrifugation-based approach, with low concentrations of SARS-CoV detected in the plasma of a patient 9 d after disease onset (2). Subsequently, together with the improvement of viral RNA extraction in which plasma or serum requires no ultracentrifugation, two real-time quantitative RT-PCR assays, one aimed toward the polymerase region and the other toward the nucleocapsid region of the virus genome (Fig. 1), were developed for measuring the concentration of SARS-CoV RNA in serum/plasma samples from SARS patients (13,14). In these assays, the absolute calibration curves are constructed by serial dilutions of high-performance liquid chromatography (HPLC)-purified, single-stranded synthetic DNA oligonucleotides specifying the studied amplicons (Fig. 2). Previous studies have shown that such single-stranded oligonucleotides reliably mimic the products of the reverse-transcription step and produce calibration curves that are identical to those obtained using T7-transcribed RNA (18, 19). The use of such calibration methodology significantly simplifies the process of obtaining a calibration curve when compared with the labor-intensive preparation of calibration curve involving amplicon subcloning and in vitro transcription.
Fig. 1.
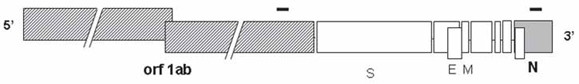
The locations of the polymerase chain reaction (PCR) amplicons within the genomic organization of severe acute respiratory syndrome (SARS)-coronavirus (CoV). The black bars represent the two PCR amplicons located within the genomic structure of SARS-CoV. The size of the open reading frames (ORFs) is drawn to scale, except for orf1ab. The size of the genome is 29.3 kb. Shaded boxes represent ORFs encoding the viral polymerase, whereas the filled box (N) represents the nucleocapsid region. S represents spike protein; E represents envelope protein; M represents membrane protein.
Fig. 2.

Detection of severe acute respiratory syndrome (S ARS)-coronavirus (CoV) RNA by real-time quantitative reverse-transcription-polymerase chain reaction (PCR) for the nucleocapsid region of the viral genome. An amplification plot of ARn, which is the fluorescence intensity over the background (y-axis) against the PCR cycle number (x-axis). Each plot corresponds to a particular input synthetic DNA oligonucleotide target quantity marked by a corresponding symbol.
The sensitivities of the amplification steps of these assays are sufficient to detect 5 copies of the targets in the reaction mixtures, corresponding to 74 copies/mL of serum (13). Using these RT-PCR assays, SARS-CoV RNA was detected in 75-78% of serum samples from SARS patients during the first week of illness (13). In the same study, data showed that median concentrations of serum SARS-CoV RNA in patients who required intensive care unit (ICU) admission during the course of hospitalization were significantly higher than in those who did not require intensive care (13) (Fig. 3). This quantitative test thus provides the viral load information allowing clinicians to make a prognostic evaluation of the infected individual.
Fig. 3.
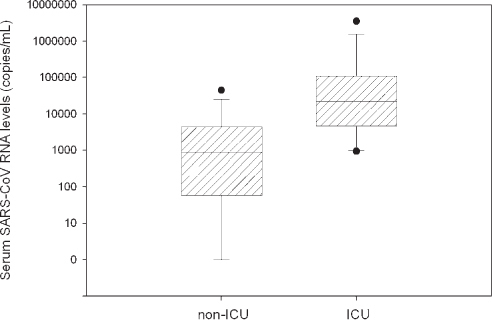
Serum severe acute respiratory syndrome (SARS)-coronavirus (CoV) RNA levels in SARS patients on the day of hospital admission. Box plot of SARS-CoV RNA levels (common logarithmic scale) in sera of SARS patients requiring and not requiring intensive care unit admission. A real-time quantitative reverse-transcription polymerase chain reaction assay towards the nucleocapsid region of the SARS-CoV genome was used for quantification. The horizontal lines denote the medians. The lines inside the boxes denote the medians. The boxes mark the interval between the 25th and 75th percentiles. The whiskers denote the interval between the 10th and 90th percentiles. The filled circles mark the data points outside the 10th and 90th percentiles.
Recent reports revealed that the clinical course of pediatric SARS patients was less severe in comparison with adult SARS patients (20, 21). On the whole, the outcomes of pediatric SARS patients were favorable. With the use of the real-time quantitative RT-PCR assay, SARS-CoV RNA has recently been shown to be detectable in the plasma samples of pediatric patients during different stages of SARS (Fig. 4) (14). No significant difference in plasma SARS-CoV viral load has been observed between pediatric and adult SARS patients taken within the first week of admission and at d 7 after fever onset (14). Overall, viremia appears to be a consistent feature in both pediatric and adult SARS patients.
Fig. 4.

Serial analysis of plasma severe acute respiratory syndrome (SARS)-coronavirus (CoV) RNA levels in pediatric SARS patients. Plots of plasma SARS-CoV RNA levels (y-axis) against time after the onset of fever (d 1 refers the day of fever onset) (x-axis). The duration of fever and the periods of steroid and ribavirin treatment are indicated for each case. The arrows in patient 2 indicate the time of intravenous methylprednisolone treatment.
The relatively high detection rate of SARS-CoV in plasma and serum during the first week of illness suggests that plasma/serum-based RT-PCR should be incorporated into the routine diagnostic workup of suspected or confirmed SARS patients both in adult and pediatric populations. This approach opens up numerous interesting research opportunities. For example, this assay can be used to monitor the effect or lack of effects of anti-viral agents. Also, it would be valuable to explore the potentially damaging effect of giving steroids at a time when the viral load is still relatively high. We are aware that many of these questions might not be answerable with retrospectively collected samples. Nonetheless, the development of animal models (5, 6) might allow the testing of some of these hypotheses in a controlled manner.
In this chapter, the two systems discussed as follows are based on the development of the quantifications of SARS-CoV RNA in serum. The SARSN RT-PCR system was designed to amplify the nucleocapsid region and SARSpol1 system was designed to amplify the polymerase region of the virus. These SARS-CoV RT-PCR systems are useful for the early diagnosis of SARS and can provide viral load information, helping clinicians to make a prognostic evaluation of the infected individual.
Materials
Sample Collection
Plain collection tubes for serum collection.
RNase Away (Invitrogen, Carlsbad, CA).
RNA Extraction
QIAamp viral RNA Mini Kit (Qiagen, Hilden, Germany).
Absolute ethanol.
Real-Time Quantitative RT-PCR
Amplification Reagents
Primers (see Note 1):
SARSpol1:
Forward: ′-GAGTGTGCGCAAGTATTAAGTGA-′.
Reverse: ′-TGATGTTCCACCTGGTTTAACA-′.
-
b.
SARSN:
-
i.
Forward: ′-TGCCCTCGCGCTATTG-′.
-
ii.
Reverse: ′-GGCCTTTACCAGAAACTTTGC-′.
Dual-labeled fluorescent probes (see Note 1:
SARSpol1: ′-(FAM)ATGGTCATGTGTGGCGGCTCACTA(TAMRA)-′.
SARSN: ′-(FAM)TGCTAGACAGATTGAACCAGCTTG(TAMRA)-′, where FAM is 6-carboxy-fluorescein; TAMRA is 6-carboxy-tetramethyl-rhodamine.
The amplicon locations of the SARSN and SARSpol1 RT-PCR systems are shown in Fig. 1.
Calibrators:
Synthetic DNA oligonucleotide corresponding to part of the polymerase gene of SARS-CoV genome, HPLC-purified (Proligos, Singapore) (see Note 2): ′-AACGAGTGTGCGCAAGTATTAAGTGAGATGGTCATGTGTGG CGGCTCACTATATGTTAAACCAGGTGGAACATCATCCGG-′.
Synthetic DNA oligonucleotide corresponding to part of the nucleocapsid gene of SARS-CoV genome, HPLC-purified (Proligos, Singapore) (see Note 2): ′-GAAACTGCCCTCGCGCTATTGCTGCTAGACAGATTGAACC AGCTTGAGAGCAAAGTTTCTGGTAAAGGCCAACAA-′.
-
4.
RNase-free water.
-
5.
EZ rTth RNA PCR reagent set (Applied Biosystems, Foster City, CA).
Instrumentation for Quantitative Analysis
ABI Prism 7700 Sequence Detector (Applied Biosystems).
Methods
Prevention of Contamination
Because of the high sensitivity of RT-PCR-based approaches, strict precautions should be applied to prevent the RT-PCR assay from contamination (22). These precautions include:
Aerosol-resistant pipet tips should be used for all liquid handling.
Separate areas should be used for the RNA extraction step, the setting up of amplification reactions, the addition of template, and the carrying out of amplification reactions.
Real-time PCR approaches obviate the need for post-PCR processing and further reduce the risk of contamination.
The assay should include a further level of anticontamination measure in the form of pre-amplification treatment using uracil N-glycosylase, which destroys any carried-over uracil containing PCR products (23).
Multiple negative water blanks should be included in every analysis so as to detect the possibility of reagent contamination.
Sample Collection
Collect blood samples in plain tubes. To ensure a sufficient amount of serum for RNA analysis, at least 3 mL of blood should be taken for each sample (see Note 3). The blood samples should be processed as soon as possible to maximize the chance of obtaining good-quality viral RNA. If the processing procedures cannot take place immediately, the blood samples should be stored with extra care (see Note 4).
Centrifuge the blood samples at 1600g for 10 min at 4°C.
Transfer serum into new tubes.
Store the serum at -80°C until RNA extraction (see Note 5.
RNA Extraction
The RNA extraction should be performed in a clean and separate area to minimize the chance of cross-contamination.
Add 1.12 mL of AVL buffer to 0.28 mL of serum, mix, and incubate at room temperature for 10 min.
Add 1.12 mL of absolute ethanol to the mixture and mix.
Apply the mixture to an RNeasy mini column and wash the column according to the manufacturer's recommendations.
To elute RNA, add 50 μL of AVE buffer onto the silica-gel membrane and incubate for 1 min at room temperature. Centrifuge the RNeasy column for 1 min at 6000g.
Store the extracted RNA at -80°C until use.
Real-Time Quantitative RT-PCR
Serum SARS-CoV RNA is quantified by using one-step real-time quantitative RT-PCR (24). In this method, the rTth (Thermus thermophilus) DNA polymerase functions both as a reverse transcriptase and a DNA polymerase (25) (see Note 7). Instead of using a two-step RT-PCR approach, one-step RT-PCR, incorporating both the reverse transcription and PCR steps in a single tube, should be used to reduce both hands-on time and the risk of contamination. In this protocol, the quantification of SARS-CoV RNA is described as follows.
Quantification of pol and N Genes of SARS-CoV Genome
Prepare calibration curves by serially diluting the synthetic DNA oligonucleotides specifying the amplicons with concentrations ranging from 1.0 × 107 copies to 5 copies (see Note 3).
Set up the RT-PCR reaction mixture for the Pol and N genes according to Table 1.
Add 11.25 μL of sample RNA, synthetic DNA oligonucleotides (for calibration curve), or RNase-free water (for negative blanks) into the reaction mixture.
Perform the real-time RT-PCR reactions in the ABI Prism 7700 Sequence Detector with cycling conditions shown in Table 2.
Table 1.
Composition of Reverse-Transcription Polymerase Chain Reaction Mix for Amplification of Pol and N Genes of Severe Acute Respiratory Syndrome-Coronavirus
| Component | Volume for one reaction (μL) | Final concentration |
|---|---|---|
| 5X TaqMan EZ buffer | 5 | 1X |
| Mn(OAc)2 (25 mM) | 3 | 3 mM |
| dATP(10mM) | 0.75 | 300 μM |
| dCTP (10 mM) | 0.75 | 300 μM |
| dGTP(10mM) | 0.75 | 300 μM |
| dUTP (20 mM) | 0.75 | 600 μM |
| Forward primer (10 μM) | 0.5 | 300 nM |
| Forward primer (10 μM) | 0.5 | 300 nM |
| Probe (5 μM) | 0.5 | 100 nM |
| rTth DNA Polymerase (2.5 U/μL) | 1 | O.1 U/μL |
| AmpErase uracil-N-glycosylase | ||
| (1 U/μL) | 0.25 | 0.01 U/μL |
| Total volume | 13.75 |
Table 2.
Cycling Profile for Amplification of Pol and N Genes of Severe Acute Respiratory Syndrome-Coronavirus
| Step | Temperature | Time |
|---|---|---|
| Uracil N-glycosylase (UNG) treatment | 50°C | 2min |
| Reverse transcription | 60°C | 30 min |
| Deactivation of UNG | 95°C | 5 min |
| Denaturation | 94°C | 20 s |
| 40 Cycles | ||
| Annealing/extension | 56°C | 1 min |
Data Analysis
Amplification data are analyzed and stored by the Sequence Detection System Software (v1.9; Applied Biosystems). The SARS-CoV RNA concentration is expressed as copies per milliliter of serum (copies/mL). The calculation is shown as follows:
 |
in which C represents the SARS-CoV RNA concentration in serum (copies/ mL), Q represents the target quantity (copies/ μJ.L) determined by a sequence detector in a PCR, VRNA represents the total volume of RNA obtained after extraction (typically 50 μL), VSerum represents the volume of serum extracted (typically 0.28 mL).
The validations of the two real-time RT-PCR systems are as described previously (13).
Notes
Primers and probes are designed with the use of the Primer Express®Software v2.0 (Applied Biosystems). Certain precautions for the design are listed as follows:
The amplicon length should be less than 100 bp, ideally no longer than 80 bp. Short amplicon length is preferable for several reasons, including: (1) the synthetic oligonucleotides specifying the amplicon used as a calibration curve are commercially available with size of up to 100 nucleotides, and (2) amplification with shorter amplicon length is more efficient than that of longer amplicon lengths (18).
To avoid false-positive results arising from co-amplification of genes with high homology, it is necessary to perform a BLASTN search with the primer and probe sequences against the National Center for Biotechnology Information (NCBI) GenBank database. The result of such a search will provide information regarding the specificity of the amplification.
-
2.
Generally, in vitro-transcribed RNA is used as a calibration curve for an absolute RNA quantification. This in vitro-transcribed RNA is usually generated by subcloning the amplicon behind a T7 RNA polymerase promoter in a plasmid vector. However, this procedure is labor-intensive and time-consuming, which is unsuitable when a large number of RT-PCR systems must be constructed in a short period of time. An alternative method can be used to construct a calibration curve, using synthetic single-stranded oligonucleotides corresponding to the amplified sequence. Previous data have shown that such single-stranded oligonucleotides reliably mimic the products of the reverse transcription step and produce calibration curves that are essentially identical to those obtained using T7-transcribed RNA (18).
-
3.
Our studies have revealed that 0.28 mL of plasma is the minimal sample volume for the robust detection of SARS-CoV RNA in serum. Thus, 3 mL of blood samples should be sufficient for testing.
-
4.
When plain blood samples are left unprocessed, their corresponding serum RNA concentrations will fluctuate over time (26). This artifactual fluctuation may be owing to several factors, such as release of RNA from necrotic and/or apoptotic blood cells and the stability of the original and the newly released RNA. To guarantee a reliable serum RNA concentration, we recommend all blood samples be stored at 4°C and be processed as soon as possible.
-
5.
According to our pre-analytical studies, no significant differences in viral RNA concentrations were observed between serum stored at -20°C and -80°C (unpublished data). Nonetheless, we routinely store serum samples at -80°C.
-
6.
The use of one-step, one-enzyme RT-PCR with rTth polymerase has several advantages over the two-enzyme RT-PCR: (1) it has been reported that the Tth polymerase is more resistant to inhibitors present in biological specimens than Taq polymerase (27); (2) the rTth polymerase is thermostable and thus allows the reverse transcription to perform at high temperature (60°C), thereby minimizing the secondary structures present in the RNA; and (3) as both reverse-transcription and PCR are carried out in a single tube, this reduces both hands-on time and the risk of contamination.
Acknowledgments
This work is supported by a Special Grant for SARS Research (CUHK 4508/ 03M) from the Research Grants Council of the Hong Kong Special Administrative Region (China).
References
- 1.Peiris J. S., Guan Y., Yuen K. Y. Severe acute respiratory syndrome. Nat. Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drosten C., Gunther S., Preiser W., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Peiris J. S., Lai S. T., Poon L. L. M., et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poutanen S. M., Low D. E., Henry B., et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 5.Fouchier R. A., Kuiken T., Schutten M., et al. Aetiology: Koch’s postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuiken T., Fouchier R. A. M., Schutten M., et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peiris J. S., Chu C. M., Cheng V. C., et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsui S. K. W., Chim S. S. C., Lo Y. M. D., The Chinese University of Hong Kong Molecular SARS Research Group Coronavirus genomic-sequence variations and the epidemiology of the severe acute respiratory syndrome. N. Engl. J. Med. 2003;349:187–188. doi: 10.1056/NEJM200307103490216. [DOI] [PubMed] [Google Scholar]
- 9.Rota P. A., Oberste M. S., Monroe S. S., et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 10.Marra M. A., Jones S. J., Astell C. R., et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 11.Poon L. L. M., Wong O. K., Luk W., Yuen K. Y., Peiris J. S., Guan Y. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome (SARS) Clin. Chem. 2003;49:953–955. doi: 10.1373/49.6.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon L. L. M., Chan K. H., Wong O. K., et al. Early diagnosis of SARS coronavirus infection by real time RT-PCR. J. Clin. Virol. 2003;28:233–238. doi: 10.1016/j.jcv.2003.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng E. K. O., Hui D. S. C., Chan K. C. A., et al. Quantitative analysis and prognostic implication of SARS coronavirus RNA in the plasma and serum of patients with severe acute respiratory syndrome. Clin. Chem. 2003;49:1976–1980. doi: 10.1373/clinchem.2003.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng E. K. O., Ng P. C., Hon K. L., et al. Serial analysis of the plasma concentration of SARS coronavirus RNA in pediatric patients with severe acute respiratory syndrome. Clin. Chem. 2003;49:2085–2088. doi: 10.1373/clinchem.2003.024588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung E. C. W., Chim S. S. C., Chan P. K., et al. Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin. Chem. 2003;49:2108–2109. doi: 10.1373/clinchem.2003.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J. R., Bai Y. J., Zhang Q. H., Wan Y., Li D., Yan X. J. Detection of hepatitis B virus DNA by real-time PCR using TaqMan-MGB probe technology. World. J. Gastroenterol. 2005;11:508–510. doi: 10.3748/wjg.v11.i4.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castelain S., Descamps V., Thibault V., et al. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J. Clin. Virol. 2004;31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Bustin S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 19.Ng E. K. O., Tsui N. B. Y., Lau T. K., et al. mRNA of placental origin is readily detectable in maternal plasma. Proc. Natl. Acad. Sci. USA. 2003;100:4748–4753. doi: 10.1073/pnas.0637450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu W. K., Cheung P. C., Ng K. I., et al. Severe acute respiratory syndrome in children: Experience in a regional hospital in Hong Kong. Pediatr. Crit. Care Med. 2003;4:279–283. doi: 10.1097/01.PCC.0000077079.42302.81. [DOI] [PubMed] [Google Scholar]
- 21.Hon K. L., Leung C. W., Cheng W. T., et al. Clinical presentations and outcome of severe acute respiratory syndrome in children. Lancet. 2003;361:1701–1703. doi: 10.1016/S0140-6736(03)13364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borst A., Box A. T. A., Fluit A. C. False-positive results and contamination in nucleic acid amplification assays: suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 2004;23:289–299. doi: 10.1007/s10096-004-1100-1. [DOI] [PubMed] [Google Scholar]
- 23.Longo M. C., Berninger M. S., Hartley J. L. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene. 1990;93:125–128. doi: 10.1016/0378-1119(90)90145-H. [DOI] [PubMed] [Google Scholar]
- 24.Gibson U. E., Heid C. A., Williams P. M. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 25.Myers T. W., Gelfand D. H. Reverse transcription and DNA amplification by a Thermus thermophilus DNA polymerase. Biochemistry. 1991;30:7661–7666. doi: 10.1021/bi00245a001. [DOI] [PubMed] [Google Scholar]
- 26.Tsui N. B. Y., Ng E. K. O., Lo Y. M. D. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 2002;48:1647–1653. [PubMed] [Google Scholar]
- 27.Poddar S. K., Sawyer M. H., Connor J. D. Effect of inhibitors in clinical specimens on Taq and Tth DNA polymerase-based PCR amplification of influenza A virus. J. Med. Microbiol. 1998;0:1131–1135. doi: 10.1099/00222615-47-12-1131. [DOI] [PubMed] [Google Scholar]