

Abstract
Over the past 20 years, the growing awareness that purinergic signaling events literally shape the immune and inflammatory responses to infection and allergic reactions warranted the development of animal models to assess their importance in vivo in acute lung injury and chronic airway diseases. The pioneer work conducted with the adenosine deaminase (ADA)-deficient mouse provided irrefutable evidence that excess adenosine (ADO) accumulating in the lungs of asthmatic patients, constitutes a powerful mediator of disease severity. These original studies launched the development of murine strains for the two major ectonucleotidases responsible for the generation of airway ADO from ATP release: CD39 and CD73. The dramatic acute lung injury and chronic lung complications, manifested by these knockout mice in response to allergens and endotoxin, demonstrated the critical importance of regulating the availability of ATP and ADO for their receptors. Therapeutic targets are currently evaluated using knockout mice and agonists/antagonists for each ADO receptor (A1R, A2AR, A2BR, and A3R) and the predominant ATP receptors (P2Y2R and P2X7R). This chapter provides an in-depth description of each in vivo study, and a critical view of the therapeutic potentials for the treatment of airway diseases.
Keywords: Adenosine deaminase, CD73, CD39, Fibrosis, Pulmonary edema
Introduction: Pulmonary Edema
Pulmonary edema is a life-threatening complication of acute lung injury, acute respiratory distress syndrome and pneumonia, which affects both barriers separating the bloodstream from the airspace: hyper-permeability of the pulmonary endothelial and epithelial barriers to macromolecules and inflammatory cells, and reduction of alveolar fluid clearance capacity mediated by ion channels (review: [1]). This complication is routinely documented in murine models targeting the enzymes and receptors regulating purinergic signaling. Yet, considerable discrepancies are reported between the beneficial or detrimental effects of receptor agonists/antagonists. The following section summarizes the current knowledge on the purinergic regulation of pulmonary edema taking place at the endothelial and epithelial barriers, which reconciles all apparent disagreements. This information will allow the reader to appreciate the impact of excess airway ATP and ADO in respiratory complications in vivo, the usefulness of certain animal models for drug development, and the appropriate administration route for therapeutic compounds.
The Permeability of the Endothelial Barrier
Until recently, our understanding of endothelial barrier regulation was limited to ADO, which plays an essential role in resealing the gate after the transmigration of inflammatory cells (review: [2]). Then again, we have learned through the first seven chapters of this book that purinergic signaling generally initiates an ATP-mediated pro-inflammatory response, which is followed by an ADO-mediated anti-inflammatory response. Therefore, several groups tested the impact of P2 receptor agonists on the regulation of endothelial barrier permeability. While the use of stable analogues and P2 receptor antagonists clearly demonstrated the sensitivity of the barrier to ATP, these studies provided contradictory conclusions (review: [1]). In this section, we provide a thorough analysis of all data which reconciles the literature under a unifying concept: opposing regulations of barrier permeability by P2Y1Rs (ADP) and P2Y2Rs (ATP), depending on local ATP/ADP concentrations. While these studies were conducted on various endothelia, these receptors are co-localized on pulmonary endothelial cells [3].
In human pulmonary artery and lung microvascular endothelial cultures, the low micromolar ATP concentrations induced a dose-dependent increase in transendothelial electrical resistance (TER) [4]. Likewise, 10 μM ATP reduced the thrombin-mediated barrier permeability of cultured human umbilical vein endothelial cells [5]. Similar data were obtained with non-hydrolysable ATP analogs known to interact with P2Y receptors, ruling out the possibility that these effects were mediated by ADO after ATP metabolism by the CD39-CD73 tandem [4]. This ATP concentration also inhibited the passage of macromolecules across endothelial monolayers, thus labeling this nucleotide as barrier protective [6]. Pharmacological analysis ascribed this response to P2Y2Rs which initiates a signaling cascade involving the activation of phospholipase C (PLC), but not PKC or Ca2+ mobilization. Incidentally, Kolosova et al. demonstrated that the enhancement of barrier integrity by low micromolar ATP involves reorganization of the intercellular tight junctions, which occurs independently of Ca2+ mobilization or ERK1/2 [4] (Fig. 8.1). The signaling cascade is reminiscent of P2Y2R-mediated induction of cell migration during epithelial repair (see 10.1007/978-94-007-1217-1_6 for details). The P2Y2Rs located near tight junctions are coupled to G proteins Gq or Gi2 (not G12 or G13), which induce protein kinase A (PKA) activation, leading to the dephosphorylation of myosin light chain, and phosphorylation of vasodilator-stimulated phosphoprotein (VASP). When phosphorylated, VASP inhibits actin polymerization and the formation of stress fibers that would disrupt the barrier.
Fig. 8.1.
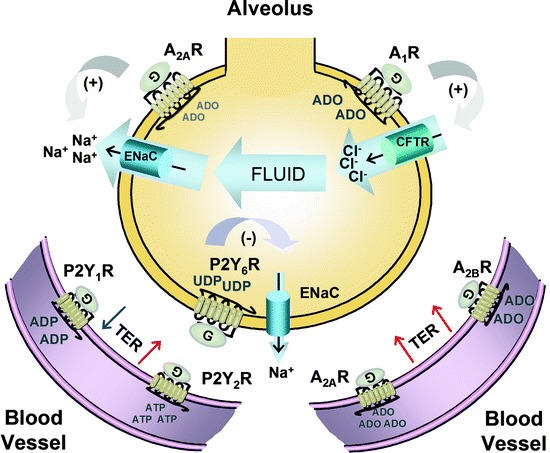
Purinergic regulation of fluid fluxes and barrier permeability in the lung. In the circulation, nucleotides regulate endothelial permeability to fluid and leukocytes by the balancing activities of ATP, ADP and ADO receptors. Whereas P2Y1R activation reduces the transmembrane electrical resistance (TER), P2Y2Rs, A2ARs and A2BRs all protect the lungs against vascular leakage. A second barrier protects the airways against excess fluid accumulation: the epithelial barrier. Alveolar cells express A1Rs and A2ARs which regulate fluid fluxes in and out of the airspace along ion gradients generated by the CFTR chloride (Cl–) channel and the ENaC sodium (Na+) channel. During viral infection, pulmonary edema is caused by the production, release and conversion of UTP into UDP, which activates P2Y6Rs to inhibit Na+ and fluid absorption via ENaC
The barrier resistance of human umbilical vein endothelial cells is reduced by selective agonists of P2Y1Rs [7]. Contrary to P2Y2Rs, the signaling mechanism is Ca2+-dependent [7] and requires myosin light chain phosphorylation [8]. Incidentally, the use of ionomycin to trigger Ca2+ entry into the cells facilitated the passage of albumin across aortic endothelial monolayers [6]. On the other hand, this receptor is only activated by ADP, released at high micromolar concentrations by platelets and red blood cells. While CD39 expressed on the endothelial surface generates ADP from ATP, the enzyme does not allow this product to accumulate significantly before further dephosphorylation into AMP. This is an important point, since the affinity of P2Y1Rs for ADP (EC50 = 8 μM) is considerably lower than P2Y2Rs for ATP (EC50 = 0.2 μM) (review: [9]). Under normal conditions, low micromolar ATP would prevent pulmonary edema primarily via P2Y2Rs. On the other hand, massive local ADP release from platelets or red blood cells squeezing into the pulmonary vascular bed would transiently raise barrier permeability via P2Y1Rs, before the nucleotide is dephosphorylated by CD39. In vascular beds, P2Y1Rs have been shown to facilitate the passage of circulating cells through the narrow vessels by inducing nitric oxide-mediated vasodilatation [10]. However, conditions reducing CD39 activity, such as hypoxia/oxidative stress (see 10.1007/978-94-007-1217-1_4), may overwhelm the protective effects of P2Y2Rs and cause pulmonary edema.
The highly efficient surface conversion of ATP and ADP into ADO generates a secondary wave of purinergic signaling for the regulation of endothelial barrier integrity. An in vivo study comparing the extent of hypoxia-induced vascular leakage among the knockout mice for each ADO receptor revealed that only the A2BR−/− mice respond to hypoxia by significantly worse lung edema than wild-type mice, which was prevented by the A2BR agonist, BAY60-6583 [11]. The capacity of A2BR activation to raise endothelial TER was confirmed on human microvascular endothelial cultures using an antagonist (PSB 1115) and by RNA silencing technique [11]. In fact, Rounds et al. recently showed that A2ARs, A2BRs and ADO transporters work in concert to prevent pulmonary edema through endothelial barrier enhancement [12]. The adenosine deaminase inhibitor, pentostatin, raised surface ADO levels by tenfold, which enhanced the formation of endothelial adherens junction and focal adhesion through Rac1 GTPase activation. The fact that A1Rs and A3Rs were ineffective at regulating endothelial barrier permeability supports the well-established notion that barrier resistance is cyclic AMP (cAMP)-dependent (review: [2]). Under baseline conditions, ADO-mediated TER would be maintained by the high-affinity A2ARs (EC50 = 0.56−0.95 μM) (review: [9]), whereas crisis situations raising circulating ADO levels (i.e. hypoxia or infection) would recruit the low-affinity A2BRs (EC50 = 16.2−64.1 μM) (review: [9]) for additional protection against vascular leakage.
The therapeutic potential of A2BR agonists for lung edema was demonstrated in a study conducted with endotoxemic pigs [13]. Endotoxin-induced acute lung injury and increase in lung extravascular water were, both, reduced by intravenously infused ADO. A similar response was obtained with the ADO receptor agonist, 2-chloroadenosine, in endotoxemic guinea-pigs [14]. Interestingly, the microvascular endothelial cells express predominantly ADO receptors causing an increase in cyclic AMP (cAMP) production (A2ARs and A2BRs), which leads to PKA-dependent VASP phosphorylation, like for the abovementioned P2Y2Rs [15]. These data suggest that circulating nucleotides normally maintain TER by successive waves of ATP- and ADO-mediated signals, which by the same token, would regulate leukocyte migration induced by inflammatory mediators.
Fluid Clearance on Alveolar Epithelia
The respiratory tract is frequently subjected to mechanical stress and infections, which stimulate the release of nucleotides into the airways. Decades of research clearly established that extracellular nucleotides are the major regulators of airway hydration (review: [16]). More importantly, they showed that the epithelial barrier regulates fluid fluxes by purinergic regulation of ion channels. The vectorial movements of Na+ and Cl– ions across the barrier create an osmotic gradient for water to follow. The absorption of Na+ by the epithelial sodium channel (ENaC) mediates fluid clearance, and Cl− secretion by the Ca2+-dependent Cl– channels (CaCC) and cystic fibrosis transmembrane regulator (CFTR) hydrates airway surfaces. This subject is covered in details in 10.1007/978-94-007-1217-1_5 for the large airways, where nucleotides are identified as stimulators of fluid accumulation into the airspace. This concept seems to apply to the regulation of alveolar fluid clearance (AFC), as intra-tracheal instillation of ATP caused lung edema in mice [17]. This initial observation launched various initiatives to clarify the involvement of purinergic signaling in the impaired epithelial fluid clearance observed during pulmonary edema.
In mice, the respiratory syncytial virus (RSV) causes a transient reduction in AFC, likely to promote clearance by the mucociliary escalator [18]. Using in vivo and in vitro studies, Matalon et al. showed that RSV impairs Na+-driven AFC through ENaC in distal lung and upper airway epithelia [19–21]. In the H441 Clara cells, RSV-mediated ENaC inhibition was partially prevented by blockers of nitric oxide production (1,400 W), or de novo UTP synthesis (A77-1726). During RSV infection, the accumulation of both purines (ATP, ADP) and pyrimidines (UTP, UDP) in the alveolar space [20], and the prevention of fluid accumulation by P2Y receptor antagonists [19], support a role for UTP synthesis and release in the purinergic regulation of AFC.
The predominant P2Y receptor capable of mediating pyrimidinergic signals in the respiratory tract is the P2Y2R, which is activated by ATP and UTP, not by ADP or UDP (10.1007/978-94-007-1217-1_7, Table 7.1). On the other hand, only soluble enzymes degrading pyrimidines (UDP-glucose pyrophosphorylase or apyrase) prevented RSV-induced edema [19]. In addition, intratracheal UTP and UDP, both, reproduced RSV-mediated AFC reduction [18]. And, UTP is readily dephosphorylated into UDP by ectonucleotidases on airway surfaces [22, 23]. Contrary to the endothelial surface, the ectonucleotidase population of the epithelial cells allows for significant accumulation of UDP before it is dephosphorylated further into UMP and uridine (see 10.1007/978-94-007-1217-1_2 for details). These results raised the possibility that UTP and UDP, both, mediated their effects through P2Y6R activation. This receptor is functionally expressed on the apical surface of airway and alveolar epithelial cells (review: [24]). In A549 human alveolar Type 2 cells, P2Y6Rs activation accounts for the majority of Ca2+-dependent ATP secretion [25]. The identity of this receptor was later confirmed using mice infected with the influenza A virus [26]. Like RSV, this virus induced hypoxemia and inhibited AFC, and was associated with a rapid increase in ATP and UTP concentrations in the BAL fluid. The use of various ion channel blockers and P2Y receptor antagonists confirmed that viral infections induce de novo UTP synthesis and secretion, followed by UTP dephosphorylation into UDP and P2Y6R activation. This signaling cascade inhibits Na+ absorption by ENaC, allowing fluid to accumulate into the alveolar space.
This purinergic receptor does not support the ATP-mediated pulmonary edema observed in mice during mechanical ventilation [17]. On the other hand, most purinergic networks accommodate a dual regulation of cellular functions mediated by the balancing effects of nucleotide (P2) receptors and ADO (P1) receptors. Davis et al. demonstrated that CFTR inhibitors, or agents preventing the formation of ADO from ATP metabolism, improved AFC during viral infection [26]. The use of selective P1 receptor antagonists identified the A1R as the culprit responsible for the CFTR-mediated reduction of AFC in infected mice. These findings were quite surprising considering the well established role of A2BRs in the stimulation of fluid secretion by CFTR in human airways (see 10.1007/978-94-007-1217-1_5). As will be described below, the purinergic network of the alveolar wall differs from the airways in terms of ectonucleotidases (see 10.1007/978-94-007-1217-1_2), as well as purinoceptors.
We are grateful for Factor et al., who meticulously determined the identity and polarity of the ADO receptors on alveolar epithelial cells [27]. Based on laser capture microdissection and quantitative PCR, they showed that all four receptors are detected on the murine epithelial barrier. Contrary to the upper airways, alveolar cells predominantly express the A2AR, with an mRNA level at least fivefold higher than for the other receptors. Membrane fractionation indicated that A1Rs and A2ARs are concentrated on the apical surface, whereas A2BRs and A3Rs were undetected by Western blot. However, one must keep in mind that in vivo measurements of AFC will include the A2BR-mediated CFTR activation and fluid secretion taking place in the airways. Incidentally, the lung edema induced, in mice, by mechanical ventilation was significantly reduced by intratracheal instillation of an A2BR antagonist (PSB1115) [28]. To specifically study the purinergic regulation of alveolar ion channels, Factor et al. used two approaches: the alveolar cell monolayers and the isolated perfused lungs [27]. Both models revealed that low ADO concentrations (0.01−10 nM) and A2AR agonists (CGS 21680) stimulate AFC via ENaC activation. In contrast, low micromolar ADO (≥1 μM) and A1R agonists (CCPA) reduce AFC via CFTR activation [27], as reported during viral infection [26]. Since both ADO receptors enhance opposing fluid fluxes across the epithelial barrier, the net directionality will depend on the local fluctuations in alveolar ADO concentrations.
Collectively, these studies support a complex purinergic regulation of AFC which involves P2Y6Rs (UDP), A2ARs (ADO) and A1Rs (ADO) (Fig. 8.1). In healthy lungs, the mechanical stress induced by normal rhythmic breathing induces the release of purines and pyrimidines into the airspace. Under these conditions, only the ADO concentration maintained by ATP release/metabolism reaches the activation thresholds of purinergic receptors, which mediates a balanced ENaC/CFTR activation to optimize AFC. During mechanical ventilation, the excessive ATP release raises ADO levels, tilting the balance toward the CFTR-mediated fluid accumulation into the lung. In addition, mechanical ventilation activates lung phosphoinositides 3-kinase gamma, which degrades cAMP [29], thereby weakening the protective effects of the ADO-A2AR-cAMP pathway against lung edema. Consequently, patients with chronic illnesses associated with high airway ADO levels would be particularly at risk of developing pulmonary edema during mechanical ventilation, combining a dominant A1R-mediated flooding and a weak A2AR-mediated AFC. Likewise, bacterial infection causes ATP release from alveolar epithelial cells [30, 31], and thus initiates the same A1R/A2AR imbalance toward flooding. In the case of viral infection, the de novo synthesis and secretion of UTP provides additional UDP for P2Y6R activation, which causes alveolar flooding via ENaC inhibition.
In terms of therapeutic application, simultaneous inhibition of CFTR (CFTRinh−172), and prevention of ENaC inhibition using a blocker of UTP de novo synthesis (A77-1726), restored normal AFC in infected mice [26]. While this approach seem attractive for the treatment of pulmonary edema, clinical studies reported considerable side-effects during treatments involving A77-1726, or its prodrug leflunomide (review: [32]). Inhibition of UTP synthesis within the cells is expected to impair critical functions, including DNA synthesis. Instead, the development of drugs targeting A1Rs, A2ARs or P2Y6Rs would allow more flexibility in dose optimization with minimal side-effects. Since CFTR and ENaC are primarily regulated by P2Y2Rs and A2BRs in the large airways, targeting these receptors would not compromise the clearance of pathogens by the mucociliary escalator.
This section emphasizes the distinct purinergic regulations of barrier permeability on the endothelial and epithelial barriers separating the bloodstream from the alveolar space. Armed with this information, we will begin our description of the murine models which were developed to appreciate the contribution of impaired ATP and ADO regulation in the development of acute and chronic disorders, with a critical eye on the therapeutic strategies proposed to restore lung homeostasis.
Murine Models of Aberrant Purine Regulation
Murine models were developed to determine the contribution of aberrant ATP and/or ADO concentrations to the development and progression of complex disorders, including respiratory diseases. Most mammalian cells regulate surface ATP and ADO by the sequential activities of CD39, CD73 and ADA. First, CD39 dephosphorylates ATP and ADP into AMP, and then CD73 converts AMP into ADO. Finally, cell surface ADA converts a fraction of the extracellular ADO into inosine, whereas the remaining portion is transported back into the cells through concentrative transporters (see 10.1007/978-94-007-1217-1_2 for details). Consequently, animal models were designed to target each of these ectonucleotidases. In the following section, we compare the phenotype of these mice to chronic respiratory diseases bearing the same metabolic disorders, to gain an appreciation of their role in the lung complications, and determine the potential of purinergic-based therapeutic approaches.
Too Much Adenosine in ADA-Deficient Mice
Humans with mutations in the ada gene resulting in a lack of enzyme activity suffer from severe combined immunodeficiency (ADA-SCID) [33]. Because ADA-deficient patients are extremely rare, considerable effort was expended in the generation of ada gene-targeted mice to better understand this genetic disease. Unfortunately, mice in which the ada gene was deleted in all tissues suffered from perinatal lethality [34, 35]. Surprisingly, the cause of death was not immunodeficiency, as they survive quite well in a clean animal facility, but rather hepatotoxicity. In an attempt to create viable ada −/− mice, Blackburn et al. engineered an ada −/− mouse strain that expressed an ada mini-gene in the placenta using trophoblast-specific controlling elements in the ada promoter [36]. These mice were viable and had normal liver function. However, they died at about 3 weeks of age from respiratory distress, characterized by severe inflammation, rapid and labored breathing. Their lifespan was extended by treatment with polyethylene glycol-conjugated bovine ADA (PEG-ADA) [37], the same material used for enzyme replacement therapy in ADA-deficient patients [38]. These mice received PEG-ADA intramuscularly every 4 days after birth, which partially suppressed the ADO-mediated lung inflammation. Contrary to intranasal instillation, intramuscular injection of PEG-ADA suppresses early events in the development of lung inflammation, like leukocyte and dendritic cell migration (see 10.1007/978-94-007-1217-1_7 for details). For the purpose of this review, these mice will be referred to as “ADA-deficient mice”. A second strain with partial ADA activity was generated by ectopic expression of a minigene in the gastrointestinal tract of ADA-deficient mice [39]. They developed a milder pulmonary disease and had a lifespan of about 5 months. They will be referred to as “partially ADA-deficient” mice.
The ADA-deficient mice appeared normal at birth, but their lungs exhibited larger alveolar spaces by day 5 [36, 37]. There was no inflammatory cell in the lungs at that time, so the defect in alveogenesis preceded the onset of inflammation, which occurred around day 10. By day 18, abnormal alveogenesis was obvious, and was characterized by thickened vascular smooth muscles and marked enlargement of the alveolar spaces. There was also hypertrophy of bronchial epithelia with increased mucus production, and cell debris accumulated in the airways. Most inflammatory cells in bronchoalveolar (BAL) fluid were macrophages, in the form of enlarged and foamy multinucleated giant cells, clustered around bronchioles and pulmonary vessels. Few eosinophils accumulated in the interstitium and the luminal space. It was suspected that these structural changes and inflammatory responses were caused by the excess ADO measured in the total lung tissue of ADA-deficient mice. Whereas lung ADO levels were barely detectable in wild-type mice, they rose about 20-fold in ADA-deficient mice, reaching a concentration of 4 nmol·mg−1 protein. The concentration of the other ADA substrate, deoxyADO was also elevated, but to a lesser extent (<0.05 nmol⋅mg−1 protein), suggesting that the toxicity of this molecule for DNA synthesis played a minor role in lung pathology. The critical role of ADO was clearly demonstrated by the fact that ADA-deficient mice were essentially “cured” by PEG-ADA treatments. The improvements in lung physiology and histology correlated with the decrease in lung ADO concentrations.
Extensive gene profiling was conducted to gain insight into the lung pathology of the ADA-deficient mice. In the initial report [40], data were generated with Atlas mouse cDNA expression arrays containing 1,176 known genes and RNA isolated at day 18 of life, when the mice were near death. The two genes, whose expression levels were most altered in ADA-deficient mice, were vascular endothelial growth factor (VEGF) which was down-regulated by about 50%, and monocyte chemoattractant protein-3 (MCP-3) which was up-regulated about tenfold. These data are consistent with the inhibitory effect of ADO on the secretion of VEGF from neutrophils, a potent inducer of endothelial barrier permeability [41]. Therefore, these mice are not expected to develop vascular leakage or excessive leukocyte infiltration, as indicated by the vast majority of resident macrophages in the BAL fluid [36, 37]. Other genes related to eosinophil trafficking, cell adhesion, inflammation and fibrosis were also significantly up-regulated in the lungs of the ADA-deficient mice. Importantly, the expression of all these genes was largely normalized by treating the mice with PEG-ADA under dosing conditions that prevented lung pathology. The second study was more extensive and used arrays containing 7,056 oligonucleotides (70-mers) that were produced at the UCSF Sandler Center of Basic Research in Asthma [42]. The aim of this study was to identify the pathways causing abnormal alveogenesis in ADA-deficient mice. Therefore, RNA samples were collected at earlier time points: at birth and at 5 and 10 days of life. This study showed that ADA deficiency up-regulates genes involved in apoptosis, but down-regulates genes coding growth factors and their receptors, as well as surfactant proteins and angiogenic factors. As for the first study, these changes in gene expression were largely reversed by treatment with PEG-ADA. These data suggest that excess ADO, reported in the airways of patients with asthma and COPD [43, 44] impacts the developing lung via multiple pathways including apoptosis, proliferation and migration, which would promote the development of airway remodeling and sub-epithelial fibrosis. Yet, the short lifespan of the ADA-deficient mice did not allow for adequate documentation of these manifestations.
The long-term consequences of high lung ADO concentrations were investigated using two different approaches: untreated partially-deficient ADA mice [39] and ADA-deficient mice treated with low doses of PEG-ADA over the course of 16 weeks [45]. Under these conditions, both cohorts eventually maintained comparable lung ADO levels (≤1.0 nmol⋅mg−1 protein), which were at least fourfold lower than untreated ADA-deficient mice. As a consequence, both cohorts developed severe lung pathology and died from respiratory distress at about 5 months of age. These models allowed the authors to analyze the long-term impact of high ADO concentrations on the respiratory system to better understand the implications for chronic airway diseases. After completion of the 16 weeks of treatment, the ADA-deficient mice exhibited lung complications comparable to untreated partially ADA-deficient mice [45]. While the inflammatory response was still dominated by multinucleated macrophages, BAL fluid also accumulated lymphocytes, neutrophils and eosinophils. The histology revealed extensive remodeling, characterized by epithelial cell hyperplasia and hypertrophy, pulmonary fibrosis, matrix deposition and smooth muscle cell thickening. These observations were confirmed by real-time PCR analysis of the lung tissue, which showed that chronically-elevated ADO up-regulates several pro-fibrotic factors (TGFβ1, osteopontin, plasminogen activator inhibitor-1 and matrix metalloproteinase-2) and pro-inflammatory Th2 cytokines (IL-1β and IL-13) [45]. Functional experiments conducted on dendritic cells showed that chronic exposure to excess ADO reduces the capacity to initiate and amplify a Th1 immune response [46]. These complications were largely reversed by 5 weeks of high-dose PEG-ADA in ADA-deficient mice initially treated 13 weeks with low-dose PEG-ADA [45].
The partially ADA-deficient mice also adopted Th2-skewed lung inflammatory responses typical of pulmonary fibrosis and asthma [47–49]. The transcript of IL-13 was undetected in the lung tissue of wild-type animals, but easily measured in partially ADA-deficient mice [50]. Other Th2 cytokines were up-regulated (IL-4 and IL-5), whereas the Th1 cytokine, IFNγ, was down-regulated, compared to wild-type mice [50]. Interestingly, IL-13 transgenic mice share many lung complications with partially ADA-deficient mice [51, 52]. They develop inflammatory infiltrates enriched in macrophages and eosinophils, airway epithelial cell hypertrophy and fibrosis, mucus cell metaplasia and hypersecretion. Furthermore, IL-13 transgenic mice exhibit high lung ADO levels and a reduced ADA expression. The fact that most lung complications of IL-13 transgenic mice and partially-deficient ADA mice were resolved by PEG-ADA treatment supports the existence of a positive amplification pathway for IL-13 and ADO accumulation in the lungs. Whereas high ADO levels up-regulate IL-13, the overexpression of this cytokine promotes ADO accumulation by a reduction in ADA activity. These findings are consistent with an in vitro study conducted on cultures of human bronchial epithelia cells, whereby chronic exposure to excess ADO raised IL-13 mRNA levels by sixfold [53]. This Th2 cytokine, which promotes eosinophilia, remodeling and fibrosis (review: [54]), likely plays a major role in the development of lung complications associated with excess airway ADO.
Neovascularization is a major complication of chronic inflammatory disorders, including asthma (review: [55]). Inflammation stimulates the growth of new blood vessels, which enhances inflammatory cell recruitment, airway obstruction and hyper-responsiveness. The ADA-deficient mice exhibited significantly enhanced vascularity in the trachea than wild-type mice as early as 18 days after birth, which was quantified by immunolocalization of the endothelial cell marker CD31 [56]. This complication was prevented by PEG-ADA treatment, and was attributed to the up-regulation of the pro-angiogenic chemokine, CXCL1, and its receptor CXCR2. These findings are consistent with the corneal micropocket assay of angiogenesis using lung extracts from wild-type and ADA-deficient mice. The angiogenic activity of lungs extract from ADA-deficient mice was inhibited by pre-treatment with PEG-ADA, or neutralizing antibodies to either CXCL1 or CXCR2. The source of CXCL1 appears to be macrophages, which accumulate in high numbers in the lung of ADA-deficient mice. While the gene expression surveys support an increased resistance against leukocyte transmigration into the airspace of the ADA-deficient mice, the longer lifespan of partially-deficient ADA mice and PEG-ADA treated ADA-deficient mice allows sufficient time to develop intense neovascularization as zones of vulnerability for leukocyte infiltration. Interestingly, genetic inactivation of A2ARs attenuated the pathological, but not developmental, angiogenesis in mouse retina [57]. Understanding the mechanisms by which excess ADO enhances neovascularization may lead to preventive therapies for airway remodeling and chronic inflammation.
Too Little Adenosine in CD73−/− Mice
Most chronic respiratory diseases are associated with aberrances in extracellular nucleotide metabolism which maintain excess ADO in airway secretions (see 10.1007/978-94-007-1217-1_4 for details). Given the devastating consequences predicted from the ADA-deficient mice, therapeutic strategies are being formulated to restore normal airway ADO levels in these patients. The importance of careful dosing the pharmacological agents is emphasized by an animal model which restricts the production of this receptor agonist. Studies were conducted with Cd73−/− mice to determine the impact of too little ADO on the overall health of the subject, as well as airway responses to acute insults and chronic conditions.
Pulmonary hypoxia/ischemia initiates the translocation of inflammatory cells from the bloodstream into the interstitium as a pathological healing response (review: [58]). Hypoxic tissue releases ATP, which constitutes a powerful neutrophil chemoattractant to eliminate apoptotic cells as part of the repair process (see 10.1007/978-94-007-1217-1_7 for details). Without the existence of an efficient feedback mechanism, the excess recruitment of inflammatory cells would cause extensive tissue injury. Purinergic signaling provides a critical defense mechanism by terminating neutrophil transmigration across the endothelial barrier. The sequential activities of CD39 and CD73 on endothelial cells convert circulating ATP into ADO, which raises barrier resistance via A2ARs/A2BRs (Fig. 8.1). Although ADO induces the production of VEGF, a potent inducer of endothelial barrier permeability, in many circumstances, it can also block its secretion from neutrophils [41]. Incidentally, adherent neutrophils were shown to facilitate their transmigration by inhibiting CD73 activity on endothelial cells [59], which may explain the lack of CD73 on these inflammatory cells [60]. For all these reasons, ADO formation by the CD39-CD73 tandem is essential for the prevention of excessive inflammation and acute lung injury during hypoxia.
Hypoxia triggers the up-regulation of CD39, CD73 and A2BRs [11, 61, 62]. While CD39 up-regulation is mediated by the Sp1 transcription factor [61], CD73 and A2BR up-regulation require binding of the hypoxia inducible factor-1α (HIF-1α) to their promoter site [11, 62]. The higher CD39-CD73 efficiency is expected to raise the circulating ADO concentration above the activation threshold of the A2BRs to enhance barrier resistance against vascular leakage and leukocyte transmigration (review: [2]). The importance of this purinergic regulation is demonstrated in mice denied CD73. The Cd73−/− mice exhibit significant vascular leakage in multiple organs, including the lung [63]. Acute hypoxia (8% O2; 4 h) causes more severe vascular leakage in Cd73−/− mice than in wild-type mice, characterized by perivascular edema and inflammatory infiltrates. These complications are partially reversed by the intraperitoneal injection of P1 receptor agonists, or soluble 5′-nucleotidase to replace CD73 [63, 64]. These results are consistent with the relative affinities of A2ARs and A2BRs, both involved in barrier protection (Fig. 8.1). While high-affinity A2ARs maintain baseline barrier resistance, the dramatic rise in circulating ADO level caused by hypoxia recruits the activities of low-affinity A2BRs. This study suggests that therapies aiming to suppress ADO production in chronic respiratory diseases could leave the patients vulnerable to hypoxia-induced pulmonary edema, and even aggravate lung inflammation, if the suppression of CD73 activity is not carefully optimized and restricted to the airways.
Animal models are currently available to assess the potential of new therapies for acute lung injury (review: [65]). The intratracheal instillation of bleomycin ranks among the five most popular models, as it recapitulates most features: neutrophilic alveolitis, damage to alveolar epithelia and endothelia, hyaline membrane formation, and thrombus accumulation in the microvasculature. This antibiotic is isolated from Streptomyces verticillus and complexes with oxygen and metals, leading to the production of oxygen radicals, DNA breaks and ultimately cell death. Within 24 h, mice develop inflammation, depicted by the accumulation of macrophages, lymphocytes and neutrophils in the BAL fluid, which lasts about 11 days [66]. Lung tissue contains higher mRNA levels of pro-inflammatory cytokines (IL-1β and TNFα) and many pro-fibrotic factors (osteopontin, plasminogen activator inhibitor-1, TGFβ1 and tissue inhibitor of metalloproteinase-1), as well as excess collagen. Fibrosis appears around day 11, and is reversed within 10 days.
Acute lung injury reproduces the high airway ADO concentrations reported in asthmatic and COPD patients [43, 44]. The bleomycin-challenged mice maintain threefold higher total lung ADO levels, and a proportional increase in CD73 activity, during 14 days [66]. To determine whether this metabolic imbalance promotes or suppresses the pulmonary complications, Cd73−/− mice were subjected to the bleomycin challenge [66]. These mice developed more severe complications to bleomycin, and died within 18 days. However, their lung ADO levels remained normal, identifying AMP dephosphorylation as the source of the agonist. These data suggest that a transient increase in airway ADO production is a defense mechanism to suppress acute lung injury. Incidentally, enzyme replacement therapy by intranasal instillation of 5′-nucleotidase restored the ability of Cd73−/− mice to accumulate lung ADO, which reduced all symptoms of inflammation and fibrosis. The fact that 5′-nucleotidase, given intranasally, was able to reduce the number of inflammatory cells in BAL fluid suggests that airway ADO can reach the interstitium to suppress leukocyte migration, or regulates epithelial permeability. Our current knowledge does not support a role for ADO in the regulation of neutrophil epithelial transmigration (Fig. 8.1). However, Hirsh et al. demonstrated that polarized bronchial epithelia support the vectorial transport of airway ADO into the interstitial tissue via apical concentrative, and basolateral equilibrative, nucleoside transporters [53]. Hence, the benefits of aerosolized drugs, given to acutely raise ADO levels during acute lung injury, may extend beyond the airways.
The chronic model of allergic asthma supports the detrimental effects of chronic excess ADO in the airways. Mice are generally sensitized with ovalbumin (OVA) by intraperitoneal injections taking place over 14 days, followed by four challenges with nebulized OVA over an additional week [67]. Within 24 h after the last challenge, their lungs develop features of allergic asthma, including eosinophilic inflammation, goblet cell metaplasia and hyperresponsiveness to methacholine [68]. Furthermore, the BAL fluid of OVA-challenged mice contains eightfold higher ATP concentrations than in saline-challenged control mice, as reported for asthmatic patients following an allergen challenge [68]. Schreiber et al. tested the impact of preventing ADO production, from this excess nucleotide, in this model of allergic asthma [69]. The Cd73−/− mice exhibited no baseline allergen-induced inflammation, and failed to develop hyperresponsiveness to methacholine following OVA treatment. These data demonstrate that the airway ADO produced by CD73, and not excess ATP, is responsible for the hyperresponsiveness of OVA-challenged mice. Aerosolized PEG-ADA may constitute a potential therapeutic intervention for airway hyperresponsiveness in asthmatic patients.
How Informative Are CD39−/− Mice?
The impact of chronically-high airway ATP concentrations has been investigated by genetic deletion of the ATP/ADP-hydrolyzing ectonucleotidase, CD39. This choice was originally based on the predominant role of endothelial CD39 in the prevention of ADP-mediated thrombus formation (review: [70]). Consequently, it was quite surprising to find that Cd39 −/− mice exhibit no symptom of pulmonary complication, unless they are subjected to an insult. For instance, mechanical ventilation has been used as a model of acute lung injury in over 500 publications (review: [65]). Patients kept on a ventilator for less than 3 days usually recover from an acute inflammatory response, but prolonged intubation initiates tissue fibrosis and loss of lung function. For this reason, mechanical ventilation remains the focus of most animal studies on acute lung injury.
Eckle et al. demonstrated that mice subjected 90 min to high-pressure mechanical ventilation develop typical manifestations of acute lung injury, characterized by vascular leakage, pulmonary edema, neutrophil infiltration and hemorrhage [71]. Interestingly, as in the case of hypoxia, this insult up-regulated both CD39 and CD73 by threefold within 2 h, resulting in a fourfold increase in total lung ADO concentration [71]. To test whether these metabolic disruptions constitute a defense mechanism, or promote the lung damage, Cd39 −/− and Cd73 −/− mice were subjected to the same insult. These knockout mice developed the same symptoms, but they were dramatically more severe than in the wild-type mice [71]. Surprisingly, similar BAL albumin level and neutrophil myeloperoxidase activity, as well as lung water content, were detected in the ventilated Cd39 −/− and Cd73 −/− mice. During acute insults, it is widely accepted that ATP mediates predominantly pro-inflammatory effects, whereas ADO generally provides a feedback anti-inflammatory response (review: [72]). Since Cd39 −/− mice allow ATP to accumulate and prevent ADO formation, we would have expected a more severe lung inflammation than in the Cd73 −/− mice, which simply deny ADO formation. This study highlights the critical protective role of excess ADO production during an acute insult, which outweighs any short-term detrimental effect of ATP accumulation. The significance of this purinergic protection was confirmed by the beneficial effects of enzyme replacement therapy on the ventilated wild-type mice. The intraperitoneal administration of apyrase (commercial form of CD39) or 5′-nucleotidase (commercial form of CD73), significantly reduced the accumulation of BAL albumin during mechanical ventilation. The excess circulating ADO, likely, provided additional endothelial barrier protection against vascular leakage via the low-affinity A2BRs (Fig. 8.1). Incidentally, this study also showed that intraperitoneal injection of an antagonist of A2BRs, not A2ARs, before the onset of mechanical ventilation reproduced the severity of the lung edema observed in the knockout mice. Consequently, this study confirms the protective role of excessive ADO against pulmonary edema during acute lung injury. Endothelial A2BRs should be targeted at the first manifestations of acute lung injury.
Most animal models of acute lung injury are based on clinical disorders, including mechanical ventilation, gastric aspiration and sepsis (review: [65]). The intratracheal administration of bacterial lipopolysaccharide (LPS) is particularly suited to investigate the impact of purinergic signaling on neutrophil migration into the airways. Aside from the usual BAL fluid cytokines, vascular leakage and disruption of the lung architecture, the mice accumulate neutrophils in all compartments of the respiratory system. This LPS challenge also induced a coordinated up-regulation of CD39 and CD73 [73], as described above for hypoxia [74] and mechanical ventilation [71]. The exposure of Cd39 −/− mice and Cd73 −/− mice to intratracheal LPS generated more severe accumulation of neutrophils in the pulmonary tissue than in LPS-treated wild-type mice. To determine which step of neutrophil migration was affected in the knockout mice, Reutershan et al. labeled their neutrophils by intravenous injection of Alexa 633-labelled Gr-1 [73]. They showed that the absence of CD39 or CD73 raised neutrophil numbers in the interstitium and the airspace, but not in the bloodstream. Since both knockout mice respond similarly to the LPS challenge, this study suggests that the prevention of ADO formation in the Cd39 −/− mice and Cd73 −/− mice was responsible for the exaggerated lung neutrophilia. The fact that ADO deficiency allowed neutrophils to accumulate to greater extent, both, in the interstitium and BAL fluid, suggest that this P1 receptor agonist normally restrict their transmigration across the endothelial and alveolar epithelial barriers. As in the other models of acute lung injury described above, the low circulating ADO levels of Cd39 −/− mice and Cd73 −/− mice were associated with a loss of endothelial barrier resistance, as determined by extravasation of Evans blue dye. On the other hand, this study provides the first evidence of a purinergic regulation of leukocyte migration across airway epithelia.
A common pattern emerges from studies conducted with mouse models of aberrant purine regulation. The fact that Cd39 −/− mice and Cd73 −/− mice respond similarly to acute insults indicates that circulating/airway ADO, not ATP, significantly influences the short term and reversible inflammatory responses. In these situations, a robust and transient increase in circulating ADO appears essential to raise endothelial barrier integrity, which restricts pulmonary edema and the infiltration of inflammatory cells. In contrast, models of chronic airway diseases clearly show that the sustained elevation of airway ADO is associated with a wide array of detrimental effects, ranging from severe inflammation to airway remodeling and hyperresponsiveness. Therefore, any therapeutic strategy that will be envisioned to reduce purine-mediated lung complications must be carefully tailored with the most appropriate murine model.
Murine Models Targeting Adenosine Receptors
Can A1Rs Go Either Way?
Ischemia-reperfusion injury causes significant morbidity and mortality, which remains a major obstacle after lung transplant. Neely et al. provided evidence that A1R antagonists attenuate inflammation and injury in a feline model of ischemic lung injury [75]. In these animals, ischemia-reperfusion induced lung edema and the infiltration of inflammatory cells (neutrophils, macrophages and red blood cells) in the alveolar space. Intralobar arterial infusion of an A1R antagonist, xanthine amine congener (XAC) or 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), before the onset of ischemia fully prevented alveolar injury. This study suggests that a proactive treatment with A1R antagonists may reduce the ischemia-reperfusion injury developing after lung transplant surgery.
Bacterial LPS initiates a number of pro-inflammatory responses, such as priming of neutrophils and macrophages, and the EGFR-mediated signaling cascades of cytokine and growth factor secretion from airway epithelial cells (see 10.1007/978-94-007-1217-1_6 and 10.1007/978-94-007-1217-1_7 for details). Yet, the specific mechanism by which LPS initiates these cascades of pathophysiological events in the lung has not been described. Neely et al. used an intralobar arterial injection of LPS as a model of acute lung injury in spontaneously breathing cats [76]. These animals developed a dose-dependent lung injury characterized by the presence of macrophages, neutrophils and red blood cells in the alveoli, alveolar edema and necrosis. Pre-treatment by intravenous bolus injection of DPCPX prevented all aspects of the acute lung injury. This study suggests that A1R antagonists may decrease the symptoms of adult respiratory distress syndrome induced by sepsis. Based on the human model of fluid flux regulation, this approach may prevent edema by suppressing A1R-mediated CFTR activity and fluid secretion in the alveolar space (Fig. 8.1). On the other hand, since few studies documented purinergic signaling in cats, it would be reassuring to repeat these experiments in mice, especially in the light of the following study.
In mice, the administration of aerosolized LPS triggers classic manifestations of acute lung injury, summarized by microvascular leakage and neutrophil accumulation in the vascular, interstitial and alveolar compartments. Interestingly, a study showed that LPS also causes a gradual increase in the total lung mRNA and protein level of A1Rs. This response was considerably weaker in mice depleted in neutrophils by injection of anti-granulocyte antibodies [77]. Therefore, the authors tested whether this receptor regulates neutrophil recruitment by exposing knockout mice to aerosolized LPS. These A1R−/− mice developed exaggerated neutrophil migration and microvascular permeability to LPS, supporting an anti-inflammatory role for this ADO receptor. Incidentally, the pre-treatment of wild-type mice by intraperitoneal injection of the new A1R agonist, named 2'Me-2-chloro-N6-cyclopentyladenosine (2′Me-CCPA), significantly reduced vascular leakage and neutrophil counts in the interstitium and alveolar space. This agonist also reduced the accumulation of pro-inflammatory cytokines (TNF-α, IL-6 and CXCL2/3) in the BAL fluid. A combination of in vivo and in vitro experiments revealed that A1Rs protect against the loss of endothelial and epithelial barrier permeability induced by LPS through inhibition of cytoskeletal rearrangement. Also, pre-treatment of neutrophils with 2′Me-CCPA reduced their transmigration across endothelial cells, supporting a role for A1Rs on the inflammatory cells. Since the receptor specificity of 2′Me-CCPA has been fully assessed [78], this study supports the therapeutic potential of the agonist for the treatment of acute lung injury regarding airway bacterial infection.
Chronic airway diseases are associated with higher mRNA levels of A1Rs in the animal models [39, 79, 80], as well as bronchial epithelial and airway smooth muscle cells from asthmatic patients [81]. An up-regulation of this receptor in asthma supports a role in the pathophysiology of this disease, and the potential of selective antagonists for the treatment of asthmatics. In the dust mite-conditioned allergic model of asthma, rabbits receiving aerosolized antisense oligodeoxynucleotides targeting A1Rs were desensitized to subsequent challenges with ADO or dust mite [82]. The intratracheal ingestion of the water soluble A1R antagonist, L-97-1, also attenuated bronchoconstriction in this model [83]. In addition, allergic animals pre-treated with L-97-1 accumulated significantly less BAL eosinophils and neutrophils in response to a dust mite challenge than the untreated allergic animals [83]. Whether ADO-induced bronchoconstriction is mediated via A1Rs expressed on airway smooth muscle, inflammatory cells or nerve endings is not clear. A recent study conducted with A1R−/− mice supports a neuronal component [84]. But the fact that aerosolized and gastric deliveries of the A1R antagonist reduce hyperresponsiveness supports a multi-cellular mechanism (see 10.1007/978-94-007-1217-1_9 for details). Detrimental roles for the A1Rs in chronic diseases also include the capacity to raise the expression of MUC2 and MUC5AC [85], which contributes to mucus overproduction, a major feature of asthma.
Murine models of chronically-high ADO concentrations in the lung are associated with enhanced or induced A1Rs expression, depending on the cell type [86]. In the ADA-deficient mice, the transcript levels of A1Rs were threefold higher in lung tissue, and 50-fold higher in BAL cell pellet, compared to wild-type mice. Immunolocalization showed enhanced protein expression on macrophages, and the receptor appeared on endothelial and epithelial cells. For these reasons, ADA/A1R double knockout mice were developed to determine the impact of A1R up-regulation on airway defenses. The genetic removal of A1Rs from the ADA-deficient mice resulted in an additional twofold increase in the total lung ADO level. Compared to the ADA-deficient mice, they had more severe pulmonary inflammation, mucus metaplasia and alveolar destruction, exaggerated expression of Th2 cytokines (IL-4 and IL-13) and matrix metalloproteinases. This study suggests that, in the presence of chronically-high ADO, A1Rs are anti-inflammatory in the respiratory system. On the other hand, it is important to note that these double knockout mice also exhibit a twofold decrease in A2AR transcript, which mediates potent anti-inflammatory responses in the lung, as described in the section below. A weaker protection by A2AR may account, at least partially, for the severity of lung complications in ADA/A1R knockout, compared to ADA-deficient mice.
These animal models provide important information on the contribution of A1Rs to acute and chronic lung disorders. However, work is needed to determine the role of this receptor, which is expected to appear on alveolar and airway epithelial cells of patients with high lung ADO content, as in asthma and COPD. The murine study, which used the new selective A1R agonist, 2′Me-CCPA [77], supports the benefits of this compound in chronic diseases. And yet, the delivery route is a key determinant in treatment design, as vascular administration reduces lung inflammation, whereas aerosolized agonists may induce bronchoconstriction. The A1R provides an excellent example for the importance of understanding the physiological background to develop a safe and efficient treatment.
The All Mighty A2ARs
The potent anti-inflammatory activities mediated by A2ARs on various cell types motivated the development of animal models to evaluate the potential of agonists for the treatment of inflammation and injury in pulmonary disorders (review: [87, 88]). Sharma et al. conducted a series of studies to investigate the impact of A2ARs on the acute lung injury caused by ischemia-reperfusion [89]. Perfused isolated lungs developed significant dysfunction (higher airway resistance, pulmonary artery pressure and lower compliance), tissue injury (vascular leakage and edema), and the accumulation of cytokines (TNFα, KC, MIP-2 and RANTES) in the BAL fluid. The addition of the selective A2AR agonist, ATL313, to the perfusate after ischemia reduced all reperfusion-related complications by at least 50%. This study shows that A2ARs expressed by resident cells, such as alveolar macrophages, endothelial and epithelial cells, play a major role in the anti-inflammatory properties of these receptors during acute lung injury.
The number of circulating neutrophils is regulated by IL-17A, a pro-inflammatory cytokine released by CD4+ T lymphocytes (review: [90]). Sharma et al. used an in vivo murine model of ischemia-reperfusion to test whether the anti-inflammatory roles of the A2AR include the suppression of CD4+ T cell activities [91]. As in the case of isolated lungs [89], perfusion of the agonist, ATL313, significantly prevented lung injury and the loss of compliance in mice [91]. The significant reduction of neutrophil infiltration was demonstrated by immunohistochemistry and BAL fluid myeloperoxidase activity. The mice subjected to ischemia-reperfusion after depletion of the CD4+ T cells or neutrophils also had significantly reduced lung injury and BAL fluid levels of inflammatory mediators, including IL-17A. However, ATL313 further reduced the symptoms only in neutrophil-depleted mice. This study shows that A2AR activation attenuates ischemia-reperfusion injury by inhibiting CD4+ T cell activation, and the subsequent neutrophil infiltration in the lung. Systemic administration of A2AR agonists, before a transplant procedure, could offer two waves of protection against excessive inflammation and injury.
The airway defenses supported by the A2AR are also sensitive to acute lung injury initiated within the airspace. Aerosolized LPS was associated with an up-regulation of the ADO receptor [92]. Therefore, the consequences of this deregulation were determined by exposing A2AR−/− mice to aerosolized LPS. Consistent with the anti-inflammatory role of this receptor, knockout mice accumulated more neutrophils into the lungs in response to LPS than the wild-type mice. Accordingly, intranasal instillation of the selective agonist, ATL 202, only reduced neutrophil transmigration in wild-type mice [92]. Together, bone marrow transplant between wild-type and A2AR−/− mice, and conditional knockout mice lacking A2ARs on macrophages, demonstrated that A2AR overexpression on macrophages plays a major role in the anti-inflammatory properties of airway ADO [92]. Since LPS was added to the airways, it is not expected to affect endothelial barrier permeability. Instead, LPS stimulates the secretion of pro-inflammatory cytokines (i.e. IL-1β, TNFα) from alveolar macrophages, which bind specific receptors on the epithelia to stimulate the release of neutrophil chemoattractants. On macrophages, A2AR activation suppresses the production and secretion of these cytokines [93–95]. During an acute airborne insult, the overexpression of A2ARs on resident macrophages would prevent the development of excessive inflammatory responses.
The intense preclinical research on A2AR agonists currently conducted in murine models of chronic airway diseases is not surprising, given the multiple roles of this P1 receptor in the prevention of excessive and damaging airway inflammation. Interestingly, contrary to the up-regulating effect of acute insults, chronic exposures to ragweed or Th2 cytokines (IL-4 or IL-13) are all associated with a down-regulation of A2ARs [79, 80, 96]. Similar findings were observed in the ADA-deficient model of chronically-elevated lung ADO [39]. The ragweed sensitization/challenge caused more severe airway inflammation and hyperresponsiveness in the A2AR−/− mice than in the wild-type mice [96]. Altogether, these studies suggest that the down-regulation of A2ARs contributes to disease severity in Th2 driven chronic lung disorders. Therefore, preclinical studies support a substantial benefit for A2AR-based aerosolized therapies in chronic diseases. In mice subjected to the OVA sensitization/challenge model of allergic asthma, the intratracheal instillation of CGS 21680 before the last challenge significantly reduced lung inflammation, quantified in BAL fluid in terms of leukocyte numbers, neutrophil myeloperoxidase and eosinophil peroxidase activities [97]. A similar protective effect of intratracheal CGS 21680 was reported in Norway rats subjected to this OVA exposure protocol [97]. On the other hand, airway hyperresponsiveness and mucus hypersecretion are not treated by A2AR agonists, in accordance with the lack of regulation of these functions by this receptor [84, 98]. Aerosolized A2AR agonists are expected to target specifically the activities of immune and inflammatory cells, and their recruitment (see 10.1007/978-94-007-1217-1_7 for details).
Lung transplant is currently the preferred option for various end-stage pulmonary diseases. While remarkable progress in the outcome has been made, chronic allograft rejection still leads to the development of bronchiolitis obliterans, a chronic inflammatory disorder. Given the well established anti-inflammatory profile of A2AR signaling, a study examined the therapeutic potential of A2AR agonists in a murine model of transplant rejection [99]. Non-revascularized tracheal transplants exhibited precocious signs of rejection in A2AR knockout mice, compared to wild-type mice, manifested by enhanced inflammation, epithelial loss and fibrosis. Pre-treatment of the wild-type mice with an A2AR agonist (ATL 313) before receiving the transplant significantly protected against the infiltration of inflammatory cells (lymphocytes, macrophages and neutrophils) and luminal occlusion by collagen deposition, measured over 21 days. This approach shows potential for the prevention of bronchiolitis obliterans in lung transplant.
Collectively, these studies support an anti-inflammatory role for A2ARs in acute and chronic lung diseases. The expression of this receptor on key regulatory and effector cells, such as lymphocytes, neutrophils and macrophages, suppresses their migration and the secretion of cytokines and chemokines to dampen immune responses. Hence, there is substantial preclinical evidence that A2AR agonists may be useful to treat inflammatory features in acute lung injuries and chronic lung diseases.
A2BRs: Balancing Clearance and Inflammation
Over the past 5 years, models of acute lung injury applied to A2BR−/− mice have assigned a protective role to the receptor (review: [100]). The first model considered was the hypoxia-induced vascular leakage and recruitment of inflammatory mediators to the lungs. Initial profiling of each ADO receptor was obtained by silencing RNA techniques in human microvascular endothelial cells. The results showed that only A2BR elimination resulted in a significant increase in endothelial leakage in response to hypoxia [11]. The same selective response was reported in knockout mice exposed to normobaric hypoxia [11]. Only the A2BR−/− mice exhibited enhanced pulmonary edema and vascular leakage, compared to the hypoxic wild-type mice. Interestingly, hypoxia selectively up-regulated A2BRs, likely as a defense mechanism against vascular leakage [11]. Therefore, agonists and antagonists were evaluated in wild-type mice for their capacity to prevent acute lung injury. Pretreatment of the mice by intraperitoneal injection of an agonist (BAY60-6583) almost completely prevented hypoxia-induced injury, whereas an antagonist (PSB1115) aggravated these lung complications. To determine the relative contribution of A2BRs expressed on pulmonary or hematopoietic cells to the vascular leakage during hypoxia, the authors generated bone marrow chimeric mice. Mice expressing A2BRs on the tissues, but not on the hematopoietic cells, showed reduced Evan's blue extravasation in response to BAY 60-6583, whereas the opposite chimera offered no protection. Taken together, these studies provide pharmacological and genetic evidence for vascular A2BR signaling as a central control point of hypoxia-associated vascular leak. These data are consistent with the purinergic regulation of vascular permeability in human lungs (Fig. 8.1), thereby supporting a role for A2BR agonists in the treatment of hypoxia-related acute lung injury.
Bacterial infection triggers an acute increase in A2BR expression throughout the respiratory system, by a mechanism involving cytokine release and autocrine responses. In cultured human microvascular endothelial cells and A549 alveolar epithelial cells, IL-1β, IL-4, IL-6 and prostaglandin E2 induce a progressive increase in A2BR mRNA levels, reaching more than sixfold higher values after 12 h, then returning to baseline within 24 h [101]. These findings were reproduced in vivo, as mice responded to LPS inhalation by a nearly fivefold increase in total lung A2BR transcript and protein levels. When the mice were pre-treated by inhalation of an A2BR antagonist (MRS 1754), they reacted to LPS by dramatically more severe lung edema, higher BAL fluid cytokine concentrations (IL-1β and TNFα) and neutrophil myeloperoxidase activity. Similar findings were obtained with A2BR−/− mice. As in the case of hypoxia [11], the bone marrow chimeric mice support a predominant role for A2BRs expressed on pulmonary cells, not hematopoietic cells, in the protection against acute lung injury. Accordingly, the A2BR agonist BAY 60-6583 clearly prevented LPS-mediated lung inflammation and edema. Collectively, these studies show that hypoxia and bacterial infection acutely up-regulate A2BRs as a defense mechanism, which offers a platform for the effective treatment of acute lung injury.
Contrary to acute lung injury, abundant literature supports pro-fibrotic and tissue destructive roles for A2BRs in chronic lung diseases, such as asthma, COPD and fibrotic pulmonary diseases [102, 103]. Numerous studies demonstrated the ability of A2BR activation to induce the expression and secretion of pro-inflammatory mediators from various cell types, including IL-4, IL-8 and IL-13 from mast cells [104–106], IL-6 and IL-19 from airway epithelial cells [107, 108] and monocyte chemoattractant protein-1 from bronchial smooth muscle cells [109]. Consequently, it is not surprising that A2BR−/− mice, subjected to the OVA sensitization/challenge model of allergic asthma, develop milder features of pulmonary inflammation and airway remodeling than wild-type mice [110]. Major inflammatory findings included a reduced production of IL-4 and lower airway eosinophil numbers in A2BR−/− mice chronically exposed to the allergen. With respect to airway remodeling, these mice presented lower levels of TGFβ, mucus cell metaplasia and thickening of the smooth muscle layer surrounding bronchial airways. Similar data were reported for mice subjected to the ragweed sensitization/NECA challenge model of allergic asthma [111]. In the sensitized mice, pretreatment by intraperitoneal injection of an A2BR antagonist (CVT-6883) significantly attenuated the airway inflammation and hyperresponsiveness induced by nebulized NECA. These studies provide evidence for the potential of A2BR antagonists in the treatment of chronic diseases, like asthma.
The contribution of A2BRs to chronic lung disease is further supported by studies conducted in the ADA-deficient model of ADO-mediated lung injury [103]. These mice develop progressive increases in total lung ADO level, in association with inflammation and tissue remodeling [37, 45]. This model is characterized by the accumulation of activated alveolar macrophages producing various inflammatory mediators, alveolar airspace destruction, mucus cell metaplasia and fibrosis. The expression of the A2BR is elevated in the lungs of ADA-deficient mice. Once lung complications are established, the selective A2BR antagonist CVT-6883 significantly reduced the production of pro-inflammatory and pro-fibrotic mediators, alveolar airspace enlargement and fibrosis [112]. Similar findings were observed in the bleomycin model of pulmonary fibrosis [112]. Interestingly, A2BRs are also up-regulated in the lungs of IL-4 [80] and IL-13 [79] overexpressing mice which develop features of asthma, COPD and pulmonary fibrosis. Moreover, high A2BR expression was recently described as a feature of patients with accelerated pulmonary fibrosis [113]. Thus, the exaggerated activation of A2BR signaling pathways appears to constitute a widespread feature of chronic airway diseases with fibrosis. In support of this, A2BR activation has been shown to promote the differentiation of human pulmonary fibroblast into collagen producing myofibroblasts [114], and to stimulate the production of pro-fibrotic fibronectin in alveolar epithelial cells [115]. These findings suggest that the sustained up-regulation and engagement of A2BRs might collectively serve to enhance chronic remodeling features of chronic lung diseases.
As in chronic lung diseases, the post-transplant bronchiolitis obliterans syndrome presents many histological characteristics of allograft injury and inflammation consistent with the pro-inflammatory and pro-fibrotic effects of A2BR activation (review: [116]). Therefore, a murine model of tracheal transplant rejection was used to determine the role of A2BRs in the development of bronchiolitis obliterans [117]. When the tracheas were transplanted into A2BR−/− mice, they exhibited less luminal obliteration and inflammatory cell infiltration (macrophages, neutrophils, CD3+ T cells) after 21 days, than in wild-type recipients. In contrast, the allografts of the A2BR−/− mice contained higher numbers of the immunosuppressive CD4/CD25/Foxp3 T regulatory (Treg) cells [117]. Therefore, apart from the well-known pro-inflammatory and pro-fibrotic effects of A2BR activation, this receptor may also stimulate the development of bronchiolitis obliterans by inhibiting the recruitment of the immunosuppressive Treg cells into the allograft.
The A3R Models the Infiltrates
As with the other ADO receptors, A3Rs play complex roles in inflammation, with both pro- and anti-inflammatory functions being described in different models [118]. Pharmacological studies have demonstrated that treatment with A3R agonists is beneficial in the feline model of reperfusion injury [119], as documented above for A1Rs [75]. In spontaneously breathing cats subjected to ischemia-reperfusion in the left lower lobe, the perfusion of IB-MECA or MRS 3558 before ischemia reduced acute lung injury, which was quantified by the number of apoptotic cells, lung edema, and BAL fluid neutrophil myeloperoxidase assay. Western blots conducted on tissue samples collected over 10 h indicated that A3R engagement activates the secondary messengers extracellular signal-regulated kinases 1 and 2 (ERK1/2), but not c-Jun amino-terminal protein kinase (JNK) and p38, which essentially shifts the signaling balance from cell death to cell survival. Therefore, A3R agonists offer a therapeutic benefit for the treatment of acute lung injury related to ischemia-reperfusion.
The role of A3Rs in acute lung injury was also recently documented in a murine model of airway infection [120]. In mice, LPS inhalation triggered general characteristics of acute lung injury, including neutrophil accumulation in all lung compartments, as well as an up-regulation of the A3R. Therefore, pharmacological experiments were initiated to determine the directionality of A3R-mediated responses, with respect to the neutrophilic inflammation. Mice pretreated by intralobar arterial infusion of the specific A3R agonist, Cl-IB-MECA, responded to aerosolized LPS by significantly lower neutrophil counts into the interstitium and alveolar airspace, but not the vasculature. Incidentally, measurement of Evans blue extravasation showed that Cl-IB-MECA inhibits the LPS-induced increases in endothelial barrier permeability. The mechanism was addressed in vitro using cultures of pulmonary microvascular endothelial cells. The A3R agonist interfered with the LPS-induced cytoskeletal rearrangement and cell retraction known to initiate vascular leakage. On the other hand, experiments conducted with bone marrow chimeric mice revealed that Cl-IB-MECA requires A3Rs expressed, both, on hematopoietic cells and pulmonary cells to suppress the transmigration of neutrophils in vivo. Possible mechanisms include cell adhesion or the secretion of mediators (see 10.1007/978-94-007-1217-1_7 for details). Regardless, this study proposes a new role for A3Rs in the recruitment of neutrophils to the airways. Whereas A3Rs expressed on neutrophils were shown to accelerate chemotaxis (review: [121]), the present study demonstrates that A3Rs also restricts neutrophil infiltration into the lung tissue and airspace. The administration of Cl-IB-MECA into the bloodstream may be useful to suppress the damaging neutrophilic inflammation associated with sepsis.
Intratracheal instillation of bleomycin ranks among the most popular models of acute lung injury. The mice transiently develop damage to the alveolar epithelial and endothelial barriers, extensive fibrosis, thrombus formation in the microvasculature and the accumulation of inflammatory cells in the interstitial space (review: [65]). Genetic deletion of the A3R yielded no significant phenotype, but the mice reacted more strongly to the bleomycin challenge [122]. After 14 days, the lungs of A3R−/− mice accumulated three times more inflammatory cells than the wild-type mice, including eosinophils. Interestingly, whereas bleomycin caused an increase in eosinophil peroxidase activity in the BAL fluid of wild-type and A3R−/− mice, the secreted enzyme activity was only higher in the wild-type mice. These data show that eosinophils require the A3R for degranulation. On the other hand, the loss of this receptor did not influence the formation of pulmonary fibrosis in response to bleomycin, as shown by measurement of collagen and α1procollagen in lung tissue. Altogether, these data suggest that A3Rs generally serve anti-inflammatory functions in the bleomycin model of acute lung injury, without affecting pulmonary fibrosis. On the other hand, lung complications associated with a predominant eosinophilic inflammation would ascribe a pro-inflammatory role to this receptor, such as asthma. This study highlights the disease-specificity of purinergic regulation which arises from the cell type specificity of receptor expression.
Chronic lung diseases are generally associated with an up-regulation of the A3R, reported in the lung biopsies of asthmatic and COPD patients [123]. The importance of this disturbance in purinergic regulation is highlighted by three murine models of Th2-mediated airway inflammation and remodeling, which are also associated with enhanced A3R expression [67, 79, 124]. This receptor affects several aspects of the lung disease. First, human and murine eosinophils abundantly express A3Rs, to inhibit chemokine-induced migration [123–125], whereas it accelerates neutrophil chemotaxis [121]. The airway epithelial cells of wild-type mice do not normally express A3Rs. However, the receptor appears selectively on the hyperplastic mucin-secreting cells of the OVA sensitized/challenge mice, IL-13 transgenic mice and ADA-deficient mice [67, 79, 124]. Young et al. determined that the receptor does not regulate the mRNA expression or packaging of mucins into granules, as wild-type and A3R−/− mice exhibit similar storage sizes following OVA sensitization/challenge [67]. In contrast, the selective A3R agonist, IB-MECA, stimulated mucin secretion in the OVA challenged, but not the untreated, animals. This study supports a role for A3R induction in the mucin hypersecretion and airway obstruction reported in many chronic airway diseases, including asthma, COPD and CF (review: [126]). On the other hand, the induction of A3R expression during mucus cell metaplasia and hyperplasia remains to be confirmed in human airways, where mucin secretion is normally mediated by ATP (P2Y2) receptors (reviews: [98, 127]) (see 10.1007/978-94-007-1217-1_5 for details).
General Patterns in Acute and Chronic Disorders
This area of purinergic research is evolving so rapidly that the notions formulated when this book was initially designed had to be revised along the way. Thankfully, we can finally discern general patterns of behavior for each ADO receptor in murine models of acute and chronic lung complications. First, the literature shows that acute and chronic disorders up-regulate all ADO receptors, except for a down-regulation of the A2AR in chronic lung diseases. Such general mobilization of the purinergic network is a testimony to the critical role of ADO in airway defenses against insults and intruders. Second, knockout mice do not present any intrinsic phenotype, but respond dramatically to insults or exposures, supporting the primary role of this network in alarm situations. This inductive behavior would prioritize the receptors responding to abnormally high ADO concentrations generated in circulation and airway secretions by ATP release and metabolism: A2ARs and A2BRs (see 10.1007/978-94-007-1217-1_7, Table 7.1).
A summary of the effects of agonists/antagonists in the murine studies identify aspects of acute and chronic lung complications regulated by each ADO receptor (Fig. 8.2). Regardless of the exposure protocol, the engagement of all receptors initiated anti-inflammatory and protective responses against acute lung injury. This came as a surprise, considering their widespread biphasic effects on immune and inflammatory cells (see 10.1007/978-94-007-1217-1_7 for details). Then again, the initial inflammatory response to an insult is the recruitment of hematopoietic cells to the lung tissue. Since they must all cross the endothelial barrier, this site constitutes a bottle neck for airway defenses to efficiently regulate the extent of airway inflammatory responses. The fact that all ADO receptors participate in the maintenance of barrier integrity identifies the endothelium as primary site for therapeutic applications regarding hypoxia, acute lung injury and sepsis. The A2AR agonists represent the safest choice because the receptor only supports protective or anti-inflammatory responses.
Fig. 8.2.
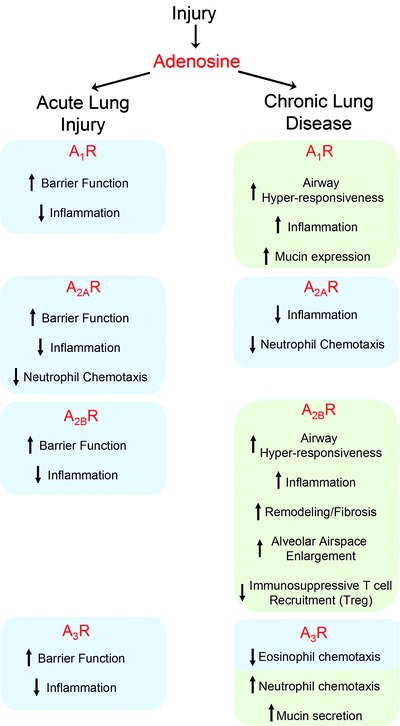
Summary of the adenosine-mediated responses in murine models of acute lung injury and chronic lung disease. The blue boxes indicate anti-inflammatory and protective effects, the green boxes indicate pro-inflammatory and damaging responses
The ADA-deficient mouse model conceptualizes the consequences of maintaining lung ADO concentrations elevated in chronic respiratory diseases, including asthma, CF and COPD. The severity of the purine-related complications results from the combined effects of A2AR down-regulation and A2BR up-regulation, which dramatically shifts the balance in favor of pro-inflammatory and remodeling responses to ADO concentrations capable of significantly activating low-affinity receptors. The murine models of chronic lung diseases revealed that ADO receptors inhibiting cAMP production (A1Rs and A3Rs) promote mucin hypersecretion and bronchoconstriction. On the other hand, A2BR activation stimulates most aspects of the inflammatory responses, remodeling and fibrosis. The ideal aerosolized therapy would combine A1R and A2BR antagonists to address all aspects of inflammatory diseases associated with airway hyperresponsiveness. On the other hand, careful dosing will be required for A2BR antagonists to minimize side-effects regarding the mucociliary clearance of allergen and infectious particles, as this receptor plays critical roles in fluid secretion and cilia beating (see 10.1007/978-94-007-1217-1_5 for details). The importance of findings in pre-clinical and cellular models will have to be validated in carefully designed clinical trials that take into account the potential impact of ADO receptors signaling in acute and chronic lung disorders.
Murine Models Targeting ATP Receptors
The field of purinergic inflammation formulated the unifying concept that ATP is released as a “danger signal” to induce inflammatory responses upon binding cell surface purinergic receptors (see 10.1007/978-94-007-1217-1_7 for details). Our current understanding of purinergic signaling in the respiratory system suggests that this concept can be extended to airway defenses against infection and obstruction (see 10.1007/978-94-007-1217-1_5 for details). The mouse models currently available for P2X and P2Y receptors, and for ectonucleotidases, highlight the airway functions particularly targeted by ATP and altered under pathological conditions. In general, these mice present no apparent phenotype or lung complication, unless they are subjected to an insult. As such, this section exposes the critical role of extracellular ATP in acute lung injury and chronic respiratory diseases.
The P2Y2R in Acute and Chronic Disorders
The most convincing evidence that the P2Y2R is engaged primarily during alarm situations is derived from studies conducted on the knockout mice [128, 129]. They are fertile, undistinguishable from their wild-type littermates, and show no abnormality in their organs, including the heart, lung, pancreas, intestines, kidneys and trachea. The fact that their airways are not obstructed is consistent with the distinct roles of P2Y2R-CaCC and A2BR-CFTR signaling pathways in mucociliary clearance (MCC), established using cultures of human airway epithelial cells (review: [24]). Under the steady-state (baseline) conditions, the surface ADO concentrations partially activate A2BRs, which maintains sufficient CFTR-mediated fluid secretion for MCC in a sterile environment. In contrast, the surface ATP concentrations periodically reach the activation threshold of P2Y2Rs during normal tidal breathing, as a result of mechanical stress-induced nucleotide release. This transient and weak activation stimulates fluid and mucin secretion to maintain a thin shield above the ciliated epithelium. Together, these ADO- and ATP-mediated signaling events continuously produce and evacuate this protective film along the ciliary escalator. On the other hand, an alarm situation, sensed by the interaction of an irritant or pathogen with the epithelium, initiates a robust and transient MCC response through ATP release and P2Y2R activation. The most powerful trigger for this flushing mechanism is a cough. The rapid metabolism of ATP into ADO by ectonucleotidases triggers a second wave of fluid secretion through enhanced A2BR-CFTR activity and ciliary beating, both gradually returning to baseline with ADO concentration. Hence, P2Y2R-mediated MCC only predominates during alarm situations.
The possibility that different P2 receptors may predominate in humans and rodents was addressed by comparing the capacity of airway epithelia from wild-type and P2Y2R−/− mice to regulate ion and fluid secretion. Measurements of ion channel activity in Ussing chambers indicated that the loss of P2Y2R decreases ATP-induced Cl– secretion by 75% in the nose and 90% in the trachea [129]. Also, mucin secretion induced by the selective P2Y2R agonist, ATPγS, was reduced by >80% in P2Y2R−/− mice, compared to their wild-type littermates [130]. These studies demonstrate that the “danger signal”, provided by stress-induced ATP release, triggers airway clearance primarily by P2Y2R activation in murine airway epithelia.
The beneficial effects of P2Y2R signaling is extended to the mobilization of cells engaged in the destruction of inhaled pathogens, including monocytes/macrophages and leukocytes. In most instances, these actions are essential to resolve airway infection. On the other hand, chronic or exaggerated activation of these functions lead to irreversible damage to the respiratory system. Whether P2Y2Rs are beneficial or detrimental in these two scenarios was examined using models of acute and chronic lung complications.
The overall impact of P2Y receptors on the susceptibility to airway infection was investigated using P2Y1R/P2Y2R double knockout mice [131]. When subjected to intra-tracheal instillation of the common airway pathogen, Pseudomonas aeruginosa, all the double knockout mice succumbed within 30 h, whereas 85% of the wild-type mice lived a normal lifespan. After 24 h, the BAL fluid of P2Y1R/P2Y2R−/− mice contained more proteins as a measure of edema, but less pro-inflammatory mediators (IL-1β, IL-6, GM-GSF and MIP-2). In contrast, the two mouse groups presented similar total cell counts and cell composition, which were primarily neutrophils (>80%). These data suggest that P2Y receptors exert a protective role during lung infection by suppressing pulmonary edema, while promoting the inflammatory responses required to eradicate the infection. The severe pulmonary edema developed by the P2Y1R/P2Y2R−/− mice is consistent with our model of fluid regulation (Fig. 8.1), whereby the loss of endothelial P2Y2Rs would prevent ATP from protecting barrier functions. On the other hand, it is intriguing that these mice accumulate BAL neutrophils to the same extent as the wild-type mice, given the critical role of P2Y2Rs for their chemotaxis (see 10.1007/978-94-007-1217-1_7 for details). Unfortunately, double knockout mice are of limited value to discriminate the roles of each receptor.
An acute model of airway infection was used to determine the impact of P2Y2Rs localized in the vasculature, on circulating cells or the endothelial surface [132]. Wild-type mice received intravenous saline, or a selective P2Y2R agonist (ATPγS), before intranasal instillation of LPS. The mice which received the P2Y2R agonist exhibited about 50% less inflammatory cells, cytokines and proteins in the BAL fluid, than the saline-treated LPS-exposed mice. The protective effect of ATPγS against vascular leakage was confirmed by Evans blue albumin extravasation into the lung tissue, pointing toward a primary function at the endothelial barrier. This was confirmed in vitro in human lung microvascular endothelial cultures, where ATPγS significantly inhibited the LPS-mediated reduction of transmembrane electrical resistance. These data suggest that intravenous P2Y2R agonists may reduce the airway inflammatory responses and lung injury caused by airborne pathogens. Using this administration route, ATPγS would not promote neutrophil chemotaxis through tissue along a chemotactic gradient [121], which is a major role postulated for this P2Y receptor.
On inflammatory cells, P2Y2R activation initiates the pro-inflammatory responses required to eradicate microbial pathogens through a series of highly coordinated events culminating in phagocytosis and destruction. In pathological circumstances, like sepsis, they release antimicrobial compounds which cause injury and death of bacteria and host cells (see 10.1007/978-94-007-1217-1_7 for details). The lung damage associated with excessive neutrophilia contributes to a wide array of respiratory complications, including ARDS, COPD and CF (review: [133]). Incidentally, genetic deletion of the P2Y2R improves the survival of mice subjected to sepsis by cecal ligation and puncture [134]. These mice exhibited fewer lung neutrophils and less pulmonary damage than wild-type mice. Since P2Y2R activation provides directionality during neutrophil chemotaxis [121], the leukocytes of P2Y2R−/− mice lack the ability to follow an ATP gradient toward the lung. This study suggests that intraperitoneal P2Y2R antagonists may improve the survival of sepsis patients.
Chronic respiratory diseases are commonly associated with elevated airway ATP concentrations, as reported in CF, IPF and COPD patients [135–138], and they are raised by allergens in asthmatic patients [68]. The expression of P2Y2Rs is also expected to be elevated in these diseases, as chronic exposure to pathogens or allergens up-regulate the receptor in animal models. For instance, house dust mites caused an increase in P2Y2R expression in eosinophils and dendritic cells from asthmatic patients, which significantly enhanced chemotaxis [139]. It is important to point out that cells from healthy subjects did not up-regulate P2Y2Rs in response to house dust mites, indicating that the purinergic regulation of inflammatory responses is not normally modified by allergens in the general population. On the other hand, rat alveolar Type 2 cells exposed overnight to LPS reacted by raising P2Y2R transcripts by threefold, which resulted in a significant increase in the ATP-mediated surfactant secretion [30]. Whereas P2Y2R overexpression may improve MCC, the pro-inflammatory reactions of immune and inflammatory cells may aggravate airway inflammation. Therefore, it is particularly important to understand the costs and benefits of P2Y2Rs in chronic diseases before we can envision therapeutic applications.
Asthmatic patients maintain normal airway ATP levels, but these concentrations rise tenfold in response to an allergen [68]. The consequences of this excess ATP were examined in the OVA sensitization/challenge model of allergic asthma [68]. The mice are sensitized by intra-peritoneal injection, and then challenged by nebulization 10 days later. After 24 h, their BAL fluid had accumulated eightfold higher ATP concentrations than in the saline-challenged control animals, as reported in the allergen-challenged asthmatic patients. They also developed all major features of allergic asthma, including eosinophil-dominant lung infiltrates, goblet cell metaplasia, hyperresponsiveness to methacholine and Th2 inflammation in the lymph nodes (IL-4, IL-5 and IL-13). These complications were significantly reduced when the OVA challenges were conducted in the presence of an ATP-metabolizing enzyme (apyrase) or P2Y receptor antagonist (PPADS or suramin). Through a series of elegant in vivo and in vitro protocols, Idzko et al. demonstrated that the excess ATP generated after an allergen challenge triggers airway inflammation by the activation of resident dendritic cells, and their mobilization to the lymph nodes to trigger a Th2 type of inflammation. It is interesting that excess ATP in the BAL fluid was not detected 10 min, but rather 24 h, after the challenge in asthmatic patients and in the mice. The mechanisms behind the delayed accumulation of ATP into the airways have not been identified. However, oxidative stress building gradually with airway inflammation has been shown to inhibit CD39 [140], and ATP-hydrolyzing ectonucleotidases expressed on airway epithelial [141], immune and inflammatory [142] cells. Consequently, this study supports the therapeutic potential of P2Y receptor antagonists for the treatment of airway inflammation in asthmatic patients.
More recently, P2Y2R−/− mice were subjected to this model of allergic asthma to identify the mechanism by which P2Y2Rs promote eosinophilia [139, 143]. The P2Y2R−/− mice exhibited a weaker capacity to recruit eosinophils in response to the OVA exposure protocol than the wild-type mice [143]. In vitro assays showed that circulating dendritic cells and bone marrow eosinophils collected from P2Y2R−/− mice fail to migrate within an ATP gradient [139]. Three findings support a role for the P2Y2R-mediated up-regulation of VCAM-1 in the purinergic stimulation of lung eosinophilia. This molecule is a potent chemoattractant and adhesion molecule for eosinophils (review: [144]). First, the OVA challenge caused the accumulation of soluble VCAM-1 in the BAL fluid, but to a lower level in P2Y2R−/− mice [143]. Second, OVA also induced an up-regulation of membrane-bound VCAM-1 at the surface of lung endothelial cells, but to a lower extent in P2Y2R−/− mice. Finally, adhesion assays confirmed that ATP promotes eosinophil adhesion to the endothelial surface through a P2Y2R-mediated increase in VCAM-1 surface expression. Collectively, these studies suggest that an amplification of the ATP-P2Y2R signaling in asthmatic patients during an allergic reaction, contributes to the development of lung eosinophilia. Accordingly, asthmatic patients may benefit from the intravenous injection of P2Y2R antagonists to suppress eosinophil recruitment to the airways.
Chronic obstructive pulmonary disease is another interesting example of P2Y2R-dominant lung pathology. The discovery that the murine model of smoke-induced lung inflammation and emphysema reproduces the high airway ATP concentrations of COPD patients provided the necessary tools to investigate the role of purinergic deregulation in the pathogenesis of this disease [145]. First, the role of ATP in the early events of airway inflammation was investigated using an acute cigarette smoke exposure protocol, which consisted of a daily 20 min session for 4 days. This acute insult caused an eightfold increase in BAL fluid ATP level, the typical macrophage and neutrophil infiltrates, and the accumulation of their cytokines (IL-1β, IL-6, IFNγ, MIP-2 and KC). Intratracheal instillation of the ATP-metabolizing apyrase, or a P2Y receptor antagonist (PPADS or suramin), before each smoke inhalation reduced all inflammatory parameters by more than 50%. This acute protocol shows that the excess ATP, accumulating after the first cigarettes, is critical to the early development of smoke-induced airway inflammation.
The contribution of ATP-induced inflammatory responses to the development of emphysema was studied using a chronic smoke exposure protocol lasting 4 months [145]. These mice developed foci of emphysema throughout the parenchyma, which was essentially prevented by oral administration of a P2Y receptor antagonists, suramin or PPADS. Real-time PCR analysis of P2 receptor expression in the inflammatory cells and lung parenchyma revealed a dramatic (eightfold) and selective up-regulation of P2Y2Rs in the neutrophils of smoke-exposed, compared to air-exposed, animals. The importance of the amplified ATP-P2Y2R signaling axis in the development of smoke-induced airway inflammation and emphysema was clearly demonstrated using P2Y2R−/− mice and chimera [137]. While the source of BAL fluid ATP has not been addressed, in vitro experiments showed that human neutrophils release ATP in response to cigarette smoke in a dose-dependent manner, and as efficiently as fMLP [137]. Also, cigarette smoke induced an ATP-dependent secretion of neutrophil elastase, which is an important mediator of lung destruction in emphysema (review: [146]). These studies suggest that the initial increase in BAL fluid ATP concentration detected after the first cigarette exposures initiates the development of airway inflammation, which is amplified over time by an up-regulation of P2Y2Rs on immune and inflammatory cells (see 10.1007/978-94-007-1217-1_7 for details). Therefore, the oral prescription of apyrase, or a P2 receptor antagonist, could prevent and reduce the irreversible lung damage and loss of lung function in COPD patients.
The P2X7R in Chronic Lung Diseases
For years, the importance of P2X7Rs for ATP-mediated inflammatory responses has been largely underestimated on the premise that the EC50 of this receptor is orders of magnitude above that of P2Y2Rs (review: [9]). However, the elegant work of Di Virgilio et al. clearly demonstrated that sites of inflammation or tissue injury accumulate ATP in the high micromolar range in vivo, above the activation threshold of P2X7Rs (10.1007/978-94-007-1217-1_7, Figs. 7.1 and 7.2). The recent finding that major pro-inflammatory cytokines involved in the initiation of airway inflammatory responses (i.e. IL-1β) depend on the P2X7R for their production, maturation and secretion (see 10.1007/978-94-007-1217-1_7 for details) is only one example of the many critical roles emerging for this receptor. Therefore, it is not surprising that the inhibition or deletion of P2X7Rs has such profound implications for airway defenses.
The mouse model of COPD, described above by Idzko et al., revealed a remarkable up-regulating effect (10- to 20-fold) of cigarette smoke on the P2X7Rs of neutrophils and macrophages within 4 days [145]. The use of selective antagonists demonstrated that P2X7R activation on these cells promotes all aspects of airway inflammation and fibrosis leading to the development of COPD and emphysema. In wild-type mice subjected to the acute smoke protocol, intratracheal injection of KN62 or A438079, before each exposure, markedly reduced the accumulation of macrophages, neutrophils and their cytokines (IL-1β, IL-6, IFNγ, MIP-2 and KC) in the BAL fluid [147]. In mice subjected to the chronic exposure protocol, these agents prevented the development of fibrosis and emphysema. The widespread effects of P2X7Rs on airway inflammation are explained by their pivotal role in the secretion of IL-1β from the resident macrophages, which initiate the events leading to leukocyte recruitment. Furthermore, P2X7R-overexpressing neutrophils and macrophages would cause considerable lung destruction by massive discharge of potent cytotoxic molecules which do not discriminate between the bacteria and host cells [146] (see 10.1007/978-94-007-1217-1_7 for details). Since smoke triggers such dramatic increases in BAL fluid ATP level [145] and P2X7R expression, an early treatment with aerosolized antagonists may reduce disease progression and prevent emphysema.
Idiopathic pulmonary fibrosis (IPF) is a fatal respiratory disorder characterized by inflammation, tissue fibrosis and destruction of the alveolar architecture. Since the IPF patients maintain high airway ATP concentrations [138], the bleomycin-induced murine model of lung fibrosis was used to test the contribution of purinergic deregulation to the pathogenesis of this disease [138]. In mice, the intratracheal administration of bleomycin initiates neutrophil recruitment within 24 h, which normalizes around day 11. First signs of fibrosis appear around day 11, and are reversed within 10 days (review: [65]). Time-course analysis indicated that bleomycin induces a transient increase in BAL fluid ATP level over the first 6 h, which is followed by the initiation of neutrophil recruitment. This time line supports a role for an ATP gradient in neutrophil recruitment to the lungs. Disruption of this ATP gradient by apyrase treatment reduced by >60% the BAL content in neutrophils, myeloperoxidase and metalloproteinases. Similar findings were obtained when comparing wild-type and P2X7R−/− mice after bleomycin exposure [138]. This study suggests that the higher availability of ATP for P2X7R activation contributes to the inflammatory processes launching tissue fibrosis and lung destruction.
In asthmatic patients, airway ATP concentrations only rise following an allergen challenge [68]. On the other hand, the purinergic network is permanently altered in these patients, as P2X7R expression is up-regulated in BAL fluid macrophages and circulating eosinophils, compared to healthy subjects [148]. A murine model of allergic asthma was used to determine the consequences for airway defenses. Mice sensitized and challenged with OVA summarize the inflammatory responses of asthmatic patients during an allergic reaction [148]. The lung infiltrates contain mainly eosinophils, followed by mast cells, neutrophils and lymphocytes, whereas BAL fluid exhibits the typical Th2 inflammation (IL-4, IL-5 and IL-13). As reported in asthmatic patients, the P2X7R was up-regulated 15-fold in the inflammatory cells and lung tissue of OVA-treated mice. The use of a selective P2X7R antagonist (KN62) on OVA-treated wild-type mice, or OVA-treated P2X7R−/− mice, was associated with milder lung inflammation compared to OVA-treated wild-type mice. The mechanism behind the pro-inflammatory role of P2X7Rs in allergic asthma was investigated using bone marrow-derived dendritic cells from wild-type and P2X7R−/− mice. The loss of P2X7Rs reduced the capacity of OVA-treated dendritic cells to induce Th2 immunity in vivo. This study highlights another critical role of P2X7Rs in the early events initiating an inflammatory response to an airborne insult, and the potential of selective antagonists for the suppression of Th2 inflammation in asthma.
Conclusion
All animal models of acute lung injury and chronic lung diseases are associated with an accumulation of extracellular purines, and an up-regulation of their receptors, except for the anti-inflammatory A2AR. Such massive amplification of purinergic signals in response to hypoxia, ischemia-reperfusion, mechanical ventilation, bacterial products and allergens supports critical and widespread functions in airway defenses. During an acute lung injury, all ADO receptors provide protection against excessive lung inflammation, damage and pulmonary edema. The intravenous administration of agonists yielded the most effective resolution of all lung complications, which identified the microvascular endothelial barrier as primary target. Treatments should focus on agonists of the high-capacity A2ARs and A2BRs to minimize receptor desensitization (10.1007/978-94-007-1217-1_7, Table 7.1). Whereas P2Y2R agonists may also be considered, possible side-effects may arise from their role in neutrophil and eosinophil recruitment. All models of chronic lung diseases are associated with a down-regulation of the anti-inflammatory A2ARs in inflammatory cells and lung tissue, which tilts the balance toward pro-inflammatory responses to ADO. Whereas A2AR agonists were shown to suppress airway inflammation, they would not address airway hyper-responsiveness, remodeling and fibrosis. Alternatively, the models of asthma, COPD and emphysema suggest that aerosolized antagonists of A2BRs, P2Y2Rs and P2X7Rs resolve most lung complications. However, the A2BR or P2Y2R antagonists must be carefully dosed to avoid any significant impairment of the mucociliary clearance mechanisms. Of particular interest are the P2X7R antagonists, which show tremendous benefits in reducing the development of COPD and emphysema in smokers through a reduction of macrophage activities responsible for the initiation of leukocyte recruitment to the airways. In asthma, aerosolized P2X7R antagonists may prevent resident dendritic cells from initiating the Th2 immunity. One important point to remember is that delivery route will dictate the cost/benefits of a purine drug depending on the local effects of the target receptor, and whether a treatment is corrective or preventive.
Contributor Information
Maryse Picher, Email: picherm@gmail.com.
Richard C. Boucher, Email: rboucher@med.unc.edu
Linda F. Thompson, Email: linda-thompson@omrf.org
Maryse Picher, Email: picherm@gmail.com.
Michael R. Blackburn, Email: michael.r.blackburn@uth.tmc.edu
References
- 1.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77:1763–1772. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5'-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balestrieri ML, Malik KU, Balestrieri C, Lee TC. Types of purinoceptors and phospholipase A2 involved in the activation of the platelet-activating factor-dependent transacetylase activity and arachidonate release by ATP in endothelial cells. Prostaglandins Other Lipid Mediat. 1998;56:363–375. doi: 10.1016/s0090-6980(98)00065-3. [DOI] [PubMed] [Google Scholar]
- 4.Kolosova IA, Mirzapoiazova T, Adyshev D, Usatyuk P, Romer LH, Jacobson JR, Natarajan V, Pearse DB, Garcia JGN, Verin AD. Signaling pathways involved in adenosine triphosphate-induced endothelial cell barrier enhancement. Circ Res. 2005;97:115–124. doi: 10.1161/01.RES.0000175561.55761.69. [DOI] [PubMed] [Google Scholar]
- 5.Gunduz D, Hirche F, Hartel FV, Rodewald CW, Schafer M, Pfitzer G, Piper HM, Noll T. ATP antagonism of thrombin-induced endothelial barrier permeability. Cardiovasc Res. 2003;59:470–478. doi: 10.1016/s0008-6363(03)00427-9. [DOI] [PubMed] [Google Scholar]
- 6.Noll T, Holschermann H, Koprek K, Gunduz D, Haberbosch W, Tillmanns H, Piper HM. ATP reduces macromolecule permeability of endothelial monolayers despite increasing [Ca2+]i. Am J Physiol. 1999;276:H1892–H1901. doi: 10.1152/ajpheart.1999.276.6.H1892. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka N, Kawasaki K, Nejime N, Kubota Y, Nakamura K, Kunitomo M, Takahashi K, Hashimoto M, Shinozuka K. P2Y receptor-mediated Ca2+ signaling increases human vascular endothelial cell permeability. J Pharmacol Sci. 2004;95:174–180. doi: 10.1254/jphs.fpj03036x. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka N, Nejime N, Kubota Y, Kagota S, Yudo K, Nakamura K, Kunitomo M, Takahashi K, Hashimoto M, Shinozuka K. Myosin light chain kinase and Rho-kinase participate in P2Y receptor-mediated acceleration of permeability through the endothelial cell layer. J Pharm Pharmacol. 2005;57:335–340. doi: 10.1211/0022357055524. [DOI] [PubMed] [Google Scholar]
- 9.Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Buvinic S, Briones R, Huidobro-Toro JP. P2Y1 and P2Y2 receptors are coupled to the NO/cGMP pathway to vasodilate the rat arterial mesenteric bed. Br J Pharmacol. 2002;136:847–856. doi: 10.1038/sj.bjp.0704789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Q, Harrington EO, Newton J, Casserly B, Radin G, Warburton R, Zhou Y, Blackburn MR, Rounds S. Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am J Physiol. 2010;298:L755–L767. doi: 10.1152/ajplung.00330.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutzsche S, Lyberg T, Bjertnaes LJ. Effects of adenosine on extravascular lung water content in endotoxemic pigs. Crit Care Med. 2001;29:2371–2376. doi: 10.1097/00003246-200112000-00021. [DOI] [PubMed] [Google Scholar]
- 14.Sakamaki F, Ishizaka A, Urano T, Sayama K, Nakamura H, Terashima T, Waki Y, Soejima K, Tasaka S, Sawafuji M, Kobayashi K, Yamaguchi K, Kanazawa M. Attenuation by intravenous 2-chloroadenosine of acute lung injury induced by live Escherichia coli or latex particles added to endotoxin in the neutropenic state. J Lab Clin Med. 2003;142:128–135. doi: 10.1016/S0022-2143(03)00105-7. [DOI] [PubMed] [Google Scholar]
- 15.Schlegel N, Waschke J. VASP is involved in cAMP-mediated Rac 1 activation in microvascular endothelial cells. Am J Physiol. 2009;296:C453–C462. doi: 10.1152/ajpcell.00360.2008. [DOI] [PubMed] [Google Scholar]
- 16.Chambers LA, Rollins BM, Tarran R. Liquid movement across the surface epithelium of large airways. Respir Physiol Neurobiol. 2007;159:256–270. doi: 10.1016/j.resp.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rich PB, Douillet CD, Mahler SA, Husain SA, Boucher RC. Adenosine triphosphate is released during injurious mechanical ventilation and contributes to lung edema. J Trauma. 2003;55:290–297. doi: 10.1097/01.TA.0000078882.11919.AF. [DOI] [PubMed] [Google Scholar]
- 18.Song W, Wei S, Matalon S. Inhibition of epithelial sodium channels by respiratory syncytial virus in vitro and in vivo. Ann NY Acad Sci. 2010;1203:79–84. doi: 10.1111/j.1749-6632.2010.05560.x. [DOI] [PubMed] [Google Scholar]
- 19.Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Nucleotide-mediated inhibition of alveolar fluid clearance in BALB/c mice after respiratory syncytial virus infection. Am J Physiol. 2004;286:L112–L120. doi: 10.1152/ajplung.00218.2003. [DOI] [PubMed] [Google Scholar]
- 20.Davis IC, Lazarowski ER, Hickman-Davis JM, Fortenberry JA, Chen F-P, Zhao X, Sorscher E, Graves LM, Sullender WM, Matalon S. Leflunomide prevents alveolar fluid clearance inhibition by respiratory syncytial virus. Am J Respir Crit Care Med. 2006;173:673–682. doi: 10.1164/rccm.200508-1200OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song W, Liu G, Bosworth CA, Walker JR, Megaw GA, Lazrak A, Abraham E, Sullender WM, Matalon S. Respiratory syncytial virus inhibits lung epithelial Na+ channels by up-regulating inducible nitric-oxide synthase. J Biol Chem. 2009;284:7294–7306. doi: 10.1074/jbc.M806816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picher M, Boucher RC. Biochemical evidence for an ecto alkaline phosphodiesterase I in human airways. Am J Respir Cell Mol Biol. 2000;23:255–261. doi: 10.1165/ajrcmb.23.2.4088. [DOI] [PubMed] [Google Scholar]
- 23.Picher M, Burch LH, Boucher RC. Metabolism of P2 receptor agonists in human airways: implications for mucociliary clearance and cystic fibrosis. J Biol Chem. 2004;279:20234–20241. doi: 10.1074/jbc.M400305200. [DOI] [PubMed] [Google Scholar]
- 24.Lazarowski ER, Boucher RC. Purinergic receptors in airway epithelia. Curr Opin Pharmacol. 2009;9:262–267. doi: 10.1016/j.coph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatur S, Groulx N, Orlov SN, Grygorczyk R. Ca2+-dependent ATP release from A549 cells involves synergistic autocrine stimulation by co-released uridine nucleotides. J Physiol. 2007;584:419–435. doi: 10.1113/jphysiol.2007.133314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolk KE, Lazarowski ER, Traylor ZP, Yu ENZ, Jewell NA, Durbin RK, Durbin JE, Davis IC. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med. 2008;178:969–976. doi: 10.1164/rccm.200803-455OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Factor P, Mutlu GM, Chen L, Mohameed J, Akhmedov AT, Meng FJ, Jilling T, Lewis ER, Johnson MD, Xu A, Kass D, Martino JM, Bellmeyer A, Albazi JS, Emala C, Lee HT, Dobbs LG, Matalon S. Adenosine regulation of alveolar fluid clearance. Proc Natl Acad Sci. 2007;104:4083–4088. doi: 10.1073/pnas.0601117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fanelli V, Puntorieri V, Assenzio B, Martin EL, Elia V, Bosco M, Delsedime L, Del Sorbo L, Ferrari A, Italiano S, Ghigo A, Slutsky AS, Hirsch E, Ranieri VM. Pulmonary-derived phosphoinositide 3-kinase gamma (PI3Kγ) contributes to ventilator-induced lung injury and edema. Intensive Care Med. 2010;36:1935–1945. doi: 10.1007/s00134-010-2018-y. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Verdugo I, Ravasio A, de Paco EG, Synguelakis M, Ivanova N, Kanellopoulos J, Haller T. Long-term exposure to LPS enhances the rate of stimulated exocytosis and surfactant secretion in alveolar type II cells and upregulates P2Y2 receptor expression. Am J Physiol. 2008;295:L708–L717. doi: 10.1152/ajplung.00536.2007. [DOI] [PubMed] [Google Scholar]
- 31.Boncoeur E, Tardif V, Tessier M-C, Morneau F, Lavoie J, Gendreau-Berthiaume E, Grygorczyk R, Dagenais A, Berthiaume Y. Modulation of epithelial sodium channel activity by lipopolysaccharide in alveolar type II cells: involvement of purinergic signaling. Am J Physiol. 2010;298:L417–L426. doi: 10.1152/ajplung.00170.2009. [DOI] [PubMed] [Google Scholar]
- 32.Chikura B, Lane S, Dawson JK. Clinical expression of leflunomide-induced pneumonitis. Rheumatology. 2009;48:1065–1068. doi: 10.1093/rheumatology/kep050. [DOI] [PubMed] [Google Scholar]
- 33.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- 34.Wakamiya M, Blackburn MR, Jurecic R, McArthur MJ, Geske RS, Cartwright J, Mitani K, Vaishnav S, Belmont JW, Kellems RE. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc Natl Acad Sci USA. 1995;92:3673–3677. doi: 10.1073/pnas.92.9.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Migchielsen AAJ, Breuer ML, van Roon MA, Riele Ht, Zurcher C, Ossendorp F, Toutain S, Hershfield MS, Berns A, Valerio D. Adenosine-deaminase-deficient mice die perinatally and exhibit liver-cell degeneration, atelectasis and small intestinal cell death. Nat Genet. 1995;10:279–287. doi: 10.1038/ng0795-279. [DOI] [PubMed] [Google Scholar]
- 36.Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- 37.Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, Kellems RE. Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J Exp Med. 2000;192:159–170. doi: 10.1084/jem.192.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hershfield MS. PEG-ADA replacement therapy for adenosine deaminase deficiency: an update after 8.5 years. Clin Immunol Immunopathol. 1995;76:S228–S232. doi: 10.1016/s0090-1229(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 39.Chunn JL, Young HWJ, Banerjee SK, Colasurdo GN, Blackburn MR. Adenosine-dependent airway inflammation and hyperresponsiveness in partially adenosine deaminase-deficient mice. J Immunol. 2001;167:4676–4685. doi: 10.4049/jimmunol.167.8.4676. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee SK, Young HWJ, Volmer JB, Blackburn MR. Gene expression profiling in inflammatory airway disease associated with elevated adenosine. Am J Physiol. 2002;282:L169–L182. doi: 10.1152/ajplung.00243.2001. [DOI] [PubMed] [Google Scholar]
- 41.Wakai A, Wang JH, Winter DC, Street JT, O’Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock. 2001;15:297–301. doi: 10.1097/00024382-200115040-00008. [DOI] [PubMed] [Google Scholar]
- 42.Banerjee SK, Young HWJ, Barczak A, Erle DJ, Blackburn MR. Abnormal alveolar development associated with elevated adenine nucleosides. Am J Respir Cell Mol Biol. 2004;30:38–50. doi: 10.1165/rcmb.2003-0102OC. [DOI] [PubMed] [Google Scholar]
- 43.Esther CR, Jr, Boysen G, Olsen BM, Collins LB, Ghio AJ, Swenberg JW, Boucher RC. Mass spectrometric analysis of biomarkers and dilution markers in exhaled breath condensate reveals elevated purines in asthma and cystic fibrosis. Am J Physiol. 2009;296:L987–L993. doi: 10.1152/ajplung.90512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esther CR, Jr, Lazaar A. Airway adenosine is elevated in COPD and correlates with disease severity. Am J Respir Crit Care Med. 2009;179:A3764. [Google Scholar]
- 45.Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol. 2005;175:1937–1946. doi: 10.4049/jimmunol.175.3.1937. [DOI] [PubMed] [Google Scholar]
- 46.Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, Girolomoni G, Norgauer J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. 2003;101:3985–3990. doi: 10.1182/blood-2002-07-2113. [DOI] [PubMed] [Google Scholar]
- 47.Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol. 1998;18:60–65. doi: 10.1165/ajrcmb.18.1.2627. [DOI] [PubMed] [Google Scholar]
- 48.Prior C, Haslam PL. In vivo levels and in vitro production of interferon-gamma in fibrotic interstitial lung diseases. Clin Exp Immunol. 1992;88:280–287. doi: 10.1111/j.1365-2249.1992.tb03074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Byrne PM. Cytokines or their antagonists for the treatment of asthma. Chest. 2006;130:244–250. doi: 10.1378/chest.130.1.244. [DOI] [PubMed] [Google Scholar]
- 50.Chunn JL, Mohsenin A, Young HWJ, Lee CG, Elias JA, Kellems RE, Blackburn MR. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am J Physiol. 2006;290:L579–L587. doi: 10.1152/ajplung.00258.2005. [DOI] [PubMed] [Google Scholar]
- 51.Elias JA, Zheng T, Lee CG, Homer RJ, Chen Q, Ma B, Blackburn M, Zhu Z. Transgenic modeling of interleukin-13 in the lung. Chest. 2003;123:339S–345S. [PubMed] [Google Scholar]
- 52.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, sub-epithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirsh AJ, Stonebraker J, van Heusden CA, Lazarowski ER, Boucher RC, Picher M. Adenosine deaminase 1 and concentrative nucleoside transporters 2 and 3 regulate adenosine on the apical surface of human airway epithelia: implications for inflammatory lung diseases. Biochemistry. 2007;46:10373–10383. doi: 10.1021/bi7009647. [DOI] [PubMed] [Google Scholar]
- 54.Kasaian MT, Miller DK. IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol. 2008;76:147–155. doi: 10.1016/j.bcp.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 55.Detoraki A, Granata F, Staibano S, Rossi FW, Marone G, Genovese A. Angiogenesis and lymphangiogenesis in bronchial asthma. Allergy. 2010;65:946–958. doi: 10.1111/j.1398-9995.2010.02372.x. [DOI] [PubMed] [Google Scholar]
- 56.Mohsenin A, Burdick MD, Molina JG, Keane MP, Blackburn MR. Enhanced CXCL1 production and angiogenesis in adenosine-mediated lung disease. FASEB J. 2007;21:1026–1036. doi: 10.1096/fj.06-7301com. [DOI] [PubMed] [Google Scholar]
- 57.Liu X, Zhou R, Pan Q, Jia XL, Gao WN, Wu J, Lin J, Chen J. Genetic inactivation of the adenosine A2A receptor attenuates pathological but not developmental angiogenesis in mouse retina. Invest Ophthalmol Vis Sci. 2010;51:6625–6632. doi: 10.1167/iovs.09-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore TM, Khimenko PL, Taylor AE. Endothelial damage caused by ischemia and reperfusion and different ventilatory strategies in the lung. Chin J Physiol. 1996;39:65–81. [PubMed] [Google Scholar]
- 59.Henttinen T, Jalkanen S, Yegutkin GG. Adherent leukocytes prevent adenosine formation and impair endothelial barrier function by ecto-5′-nucleotidase/CD73-dependent mechanism. J Biol Chem. 2003;278:24888–24895. doi: 10.1074/jbc.M300779200. [DOI] [PubMed] [Google Scholar]
- 60.Corriden R, Chen Y, Inoue Y, Beldi G, Robson SC, Insel PA, Junger WG. Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. J Biol Chem. 2008;283:28480–28486. doi: 10.1074/jbc.M800039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113:224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 65.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. 2006;176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- 67.Young HWJ, Sun C-X, Evans CM, Dickey BF, Blackburn MR. A3 Adenosine receptor signaling contributes to airway mucin secretion after allergen challenge. Am J Respir Cell Mol Biol. 2006;35:549–558. doi: 10.1165/rcmb.2006-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MAM, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC, Lambrecht BN. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 69.Schreiber R, Castrop H, Kunzelmann K. Allergen-induced airway hyperresponsiveness is absent in ecto-5′-nucleotidase (CD73)-deficient mice. Pflugers Arch. 2008;457:431–440. doi: 10.1007/s00424-008-0543-0. [DOI] [PubMed] [Google Scholar]
- 70.Atkinson B, Dwyer KM, Enjyoji K, Robson SC. Ecto-nucleotidases of the CD39/NTPDase family modulate platelet activation and thrombus formation: potential as therapeutic targets. Blood Cells Mol Dis. 2006;36:217–222. doi: 10.1016/j.bcmd.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 71.Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 72.Di Virgilio F. Purinergic signaling in the immune system. Purinergic Signal. 2007;3:1–3. doi: 10.1007/s11302-006-9048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri K-C, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009;23:473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- 74.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neely CF, Keith IM. A1 adenosine receptor antagonists block ischemia-reperfusion injury of the lung. Am J Physiol. 1995;268:L1036–L1046. doi: 10.1152/ajplung.1995.268.6.L1036. [DOI] [PubMed] [Google Scholar]
- 76.Neely CF, Jin J, Keith IM. A1-adenosine receptor antagonists block endotoxin-induced lung injury. Am J Physiol. 1997;272:L353–L361. doi: 10.1152/ajplung.1997.272.2.L353. [DOI] [PubMed] [Google Scholar]
- 77.Ngamsri K-C, Wagner R, Vollmer I, Stark S, Reutershan J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J Immunol. 2010;185:4374–4384. doi: 10.4049/jimmunol.1000433. [DOI] [PubMed] [Google Scholar]
- 78.Cappellacci L, Franchetti P, Vita P, Petrelli R, Lavecchia A, Costa B, Spinetti F, Martini C, Klotz KN, Grifantini M. 5′-Carbamoyl derivatives of 2'-C-methyl-purine nucleosides as selective A1 adenosine receptor agonists: affinity, efficacy, and selectivity for A1 receptor from different species. Bioorg Med Chem. 2008;16:336–353. doi: 10.1016/j.bmc.2007.09.035. [DOI] [PubMed] [Google Scholar]
- 79.Blackburn MR, Chun GL, Young HWJ, Chunn JL, Banerjee SK, Elias JA. Adenosine mediates IL-13-induced inflammation and remodeling in the lung: evidence for an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma B, Blackburn MR, Lee CG, Homer RJ, Liu W, Flavell RA, Boyden L, Lifton RP, Sun CX, Young HW, Elias JA. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J Clin Invest. 2006;116:1274–1283. doi: 10.1172/JCI26372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown RA, Clarke GW, Ledbetter CL, Hurle MJ, Denyer JC, Simcock DE, Coote JE, Savage TJ, Murdoch RD, Page CP, Spina D, O’Connor BJ. Elevated expression of adenosine A1 receptor in bronchial biopsy specimens from asthmatic subjects. Eur Respir J. 2008;31:311–319. doi: 10.1183/09031936.00003707. [DOI] [PubMed] [Google Scholar]
- 82.Nyce JW, Metzger WJ. DNA antisense therapy for asthma in an animal model. Nature. 1997;385:721–725. doi: 10.1038/385721a0. [DOI] [PubMed] [Google Scholar]
- 83.Nadeem A, Obiefuna PC, Wilson CN, Mustafa SJ. Adenosine A1 receptor antagonist versus montelukast on airway reactivity and inflammation. Eur J Pharmacol. 2006;551:116–124. doi: 10.1016/j.ejphar.2006.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hua X, Erikson CJ, Chason KD, Rosebrock CN, Deshpande DA, Penn RB, Tilley SL. Involvement of A1 adenosine receptors and neural pathways in adenosine induced bronchoconstriction in mice. Am J Physiol. 2007;293:L25–L32. doi: 10.1152/ajplung.00058.2007. [DOI] [PubMed] [Google Scholar]
- 85.McNamara N, Gallup M, Khong A, Sucher A, Maltseva I, Fahy J, Basbaum C. Adenosine up-regulation of the mucin gene, MUC2, in asthma. FASEB J. 2004;18:1770–1772. doi: 10.1096/fj.04-1964fje. [DOI] [PubMed] [Google Scholar]
- 86.Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest. 2005;115:35–43. doi: 10.1172/JCI22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hasko G, Pacher P. A2A receptors in inflammation and injury: lessons learned from transgenic animals. J Leukoc Biol. 2008;83:447–455. doi: 10.1189/jlb.0607359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharma AK, Linden J, Kron IL, Laubach VE. Protection from pulmonary ischemia-reperfusion injury by adenosine A2A receptor activation. Respir Res. 2009;10:58. doi: 10.1186/1465-9921-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ley K, Smith E, Stark MA. IL-17A-producing neutrophil-regulatory Tn lymphocytes. Immunol Res. 2006;34:229–242. doi: 10.1385/IR:34:3:229. [DOI] [PubMed] [Google Scholar]
- 91.Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, Kron IL, Yang Z. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2010;139:474–482. doi: 10.1016/j.jtcvs.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 93.Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabo C. Adenosine inhibits IL-12 and TNF-alpha production via adenosine A2A receptor-dependent and independent mechanisms. FASEB J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- 94.Link AA, Kino T, Worth JA, McGuire JL, Crane ML, Chrousos GP, Wilder RL, Elenkov IJ. Ligand-activation of the adenosine A2A receptors inhibits IL-12 production by human monocytes. J Immunol. 2000;164:436–442. doi: 10.4049/jimmunol.164.1.436. [DOI] [PubMed] [Google Scholar]
- 95.Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- 96.Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa J. Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am J Physiol. 2007;292:L1335–L1344. doi: 10.1152/ajplung.00416.2006. [DOI] [PubMed] [Google Scholar]
- 97.Bonneau O, Wyss D, Ferretti S, Blaydon C, Stevenson CS, Trifilieff A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am J Physiol. 2006;290:L1036–L1043. doi: 10.1152/ajplung.00422.2005. [DOI] [PubMed] [Google Scholar]
- 98.Davis CW, Dickey BF. Regulated airway goblet cell mucin secretion. Ann Rev Physiol. 2008;70:487–512. doi: 10.1146/annurev.physiol.70.113006.100638. [DOI] [PubMed] [Google Scholar]
- 99.Lau CL, Zhao Y, Kron IL, Stoler MH, Laubach VE, Ailawadi G, Linden J. The role of adenosine A2A receptor signaling in bronchiolitis obliterans. Ann Thorac Surg. 2009;88:1071–1078. doi: 10.1016/j.athoracsur.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology. 2009;24:298–306. doi: 10.1152/physiol.00022.2009. [DOI] [PubMed] [Google Scholar]
- 101.Schingnitz U, Hartmann K, Macmanus C, Eckle T, Zug S, Colgan SP, Eltzschig HK. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010;184:5271–5279. doi: 10.4049/jimmunol.0903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fozard JR, Hannon JP. Adenosine receptor ligands: potential as therapeutic agents in asthma and COPD. Pulm Pharmacol Ther. 1999;12:111–114. doi: 10.1006/pupt.1999.0191. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther. 2009;123:105–116. doi: 10.1016/j.pharmthera.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979–1986. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I. Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J Immunol. 2004;172:7726–7733. doi: 10.4049/jimmunol.172.12.7726. [DOI] [PubMed] [Google Scholar]
- 106.Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Dikov MM, Blackburn MR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J Immunol. 2008;180:7212–7220. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun Y, Wu F, Sun F, Huang P. Adenosine promotes IL-6 release in airway epithelia. J Immunol. 2008;180:4173–4181. doi: 10.4049/jimmunol.180.6.4173. [DOI] [PubMed] [Google Scholar]
- 108.Zhong H, Wu Y, Belardinelli L, Zeng D. A2B adenosine receptors induce IL-19 from bronchial epithelial cells, resulting in TNF-alpha increase. Am J Respir Cell Mol Biol. 2006;35:587–592. doi: 10.1165/rcmb.2005-0476OC. [DOI] [PubMed] [Google Scholar]
- 109.Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A2B Adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2003;30:118–125. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- 110.Zaynagetdinov R, Ryzhov S, Goldstein AE, Yin H, Novitskiy SV, Goleniewska K, Polosukhin VV, Newcomb DC, Mitchell D, Morschl E, Zhou Y, Blackburn MR, Peebles RS, Jr, Biaggioni I, Feoktistov I. Attenuation of chronic pulmonary inflammation in A2B adenosine receptor knockout mice. Am J Respir Cell Mol Biol. 2010;42:564–571. doi: 10.1165/rcmb.2008-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mustafa SJ, Nadeem A, Fan M, Zhong H, Belardinelli L, Zeng D. Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J Pharmacol Exp Ther. 2007;320:1246–1251. doi: 10.1124/jpet.106.112250. [DOI] [PubMed] [Google Scholar]
- 112.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR. Role of A2B receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:1–10. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, Gaxiola M, Perez-Padilla R, Navarro C, Richards T, Dauber J, King TE, Jr, Pardo A, Kaminski N. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS ONE. 2007;2:e482. doi: 10.1371/journal.pone.0000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol. 2005;32:2–8. doi: 10.1165/rcmb.2004-0103OC. [DOI] [PubMed] [Google Scholar]
- 115.Roman J, Rivera HN, Roser-Page S, Sitaraman SV, Ritzenthaler JD. Adenosine induces fibronectin expression in lung epithelial cells: implications for airway remodeling. Am J Physiol. 2006;290:L317–L325. doi: 10.1152/ajplung.00118.2005. [DOI] [PubMed] [Google Scholar]
- 116.Robertson AG, Griffin SM, Murphy DM, Pearson JP, Forrest IA, Dark JH, Corris PA, Ward C. Targeting allograft injury and inflammation in the management of post-lung transplant bronchiolitis obliterans syndrome. Am J Transplant. 2009;9:1272–1278. doi: 10.1111/j.1600-6143.2009.02648.x. [DOI] [PubMed] [Google Scholar]
- 117.Zhao Y, Lapar DJ, Steidle J, Emaminia A, Kron IL, Ailawadi G, Linden J, Lau CL. Adenosine signaling via the adenosine 2B receptor is involved in bronchiolitis obliterans development. J Heart Lung Transplant. 2010;29:1405–1414. doi: 10.1016/j.healun.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 119.Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wagner R, Ngamsri K-C, Stark S, Vollmer I, Reutershan J. Adenosine receptor A3 is a critical mediator in LPS-induced pulmonary inflammation. Am J Physiol. 2010;299:L502–L512. doi: 10.1152/ajplung.00083.2010. [DOI] [PubMed] [Google Scholar]
- 121.Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, Inoue Y, Woehrle T, Zhang Q, Hauser C, Junger WG. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3:ra45. doi: 10.1126/scisignal.2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, Kheradm F, Lee JJ, Blackburn MR. A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol. 2008;39:697–705. doi: 10.1165/rcmb.2007-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, Bai TR. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- 124.Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan L-NL, Chan T-S, Lee JJ, Blackburn MR. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173:1380–1389. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- 125.Knight D, Zheng X, Rocchini C, Jacobson M, Bai T, Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J Leukoc Biol. 1997;62:465–468. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- 126.Voynow JA, Rubin BK. Mucins, mucus, and sputum. Chest. 2009;135:505–512. doi: 10.1378/chest.08-0412. [DOI] [PubMed] [Google Scholar]
- 127.Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med. 2009;15:4–11. doi: 10.1097/MCP.0b013e32831da8d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Homolya L, Watt WC, Lazarowski ER, Koller BH, Boucher RC. Nucleotide-regulated calcium signaling in lung fibroblasts and epithelial cells from normal and P2Y2 receptor (-/-) mice. J Biol Chem. 1999;274:26454–26460. doi: 10.1074/jbc.274.37.26454. [DOI] [PubMed] [Google Scholar]
- 129.Cressman VL, Lazarowski E, Homolya L, Boucher RC, Koller BH, Grubb BR. Effect of loss of P2Y2 receptor gene expression on nucleotide regulation of murine epithelial Cl- transport. J Biol Chem. 1999;274:26461–26468. doi: 10.1074/jbc.274.37.26461. [DOI] [PubMed] [Google Scholar]
- 130.Ehre C, Zhu Y, Abdullah LH, Olsen J, Nakayama KI, Nakayama K, Messing RO, Davis CW. nPKCepsilon, a P2Y2-R downstream effector in regulated mucin secretion from airway goblet cells. Am J Physiol. 2007;293:C1445–C1454. doi: 10.1152/ajpcell.00051.2007. [DOI] [PubMed] [Google Scholar]
- 131.Geary C, Akinbi HT, Korfhagen T, Fabre J-E, Boucher R, Rice WR. Increased susceptibility of purinergic receptor deficient mice to lung infection with Pseudomonas aeruginosa. Am J Physiol. 2005;289:L890–L895. doi: 10.1152/ajplung.00428.2004. [DOI] [PubMed] [Google Scholar]
- 132.Kolosova IA, Mirzapoiazova T, Moreno-Vinasco L, Sammani S, Garcia JGN, Verin AD. Protective effect of purinergic agonist ATPgS against acute lung injury. Am J Physiol. 2007;294:L319–L324. doi: 10.1152/ajplung.00283.2007. [DOI] [PubMed] [Google Scholar]
- 133.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Inoue Y, Chen Y, Hirsh MI, Yip L, Junger WG. A3 and P2Y2 receptors control the recruitment of neutrophils to the lungs in a mouse model of sepsis. Shock. 2008;30:173–177. doi: 10.1097/shk.0b013e318160dad4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Esther CR, Jr, Alexis NE, Clas ML, Lazarowski ER, Donaldson SH, Ribeiro CM, Moore CG, Davis SD, Boucher RC. Extracellular purines are biomarkers of neutrophilic airway inflammation. Eur Respir J. 2008;31:949–956. doi: 10.1183/09031936.00089807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, Stoll P, Grimm M, Durk T, Zissel G, Ferrari D, Di Virgilio F, Sorichter S, Lungarella G, Virchow JC, Idzko M. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928–934. doi: 10.1164/rccm.200910-1506OC. [DOI] [PubMed] [Google Scholar]
- 137.Mortaz E, Braber S, Nazary M, Givi ME, Nijkamp FP, Folkerts G. ATP in the pathogenesis of lung emphysema. Eur J Pharmacol. 2009;619:92–96. doi: 10.1016/j.ejphar.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 138.Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VFJ, Marchand-Adam S, Crestani B, Ryffel B, Couillin I. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182:774–783. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- 139.Müller T, Robaye B, Vieira RP, Ferrari D, Grimm M, Jakob T, Martin SF, Di Virgilio F, Boeynaems JM, Virchow JC, Idzko M. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy. 2010;65:1545–1553. doi: 10.1111/j.1398-9995.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- 140.Robson SC, Kaczmarek E, Siegel JB, Candinas D, Koziak K, Millan M, Hancock WW, Bach FH. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J Exp Med. 1997;185:153–164. doi: 10.1084/jem.185.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fausther M, Pelletier J, Ribeiro CM, Sévigny J, Picher M. Cystic fibrosis remodels the regulation of purinergic signaling by NTPDase1 (CD39) and NTPDase3. Am J Physiol. 2010;298:L804–L818. doi: 10.1152/ajplung.00019.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC. CD39 and control of cellular immune responses. Purinergic Signal. 2007;3:171–180. doi: 10.1007/s11302-006-9050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vanderstocken G, Bondue B, Horckmans M, Di Pietrantonio L, Robaye B, Boeynaems J-M, Communi D. P2Y2 receptor regulates VCAM-1 membrane and soluble forms and eosinophil accumulation during lung inflammation. J Immunol. 2010;185:3702–3707. doi: 10.4049/jimmunol.0903908. [DOI] [PubMed] [Google Scholar]
- 144.Resnick MB, Weller PF. Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol. 1993;8:349–355. doi: 10.1165/ajrcmb/8.4.349. [DOI] [PubMed] [Google Scholar]
- 145.Cicko S, Lucattelli M, Müller T, Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Dürk T, Zissel G, Boeynaems JM, Sorichter S, Ferrari D, Di Virgilio F, Virchow JC, Lungarella G, Idzko M. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185:688–697. doi: 10.4049/jimmunol.0904042. [DOI] [PubMed] [Google Scholar]
- 146.Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. the role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12:361–367. [PubMed] [Google Scholar]
- 147.Lucattelli M, Cicko S, Muller T, Lommatzsch M, Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Durk T, Zissel G, Sorichter S, Ferrari D, Virgilio F, Virchow JC, Lungarella G, Idzko M. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol. 2011;44:423–429. doi: 10.1165/rcmb.2010-0038OC. [DOI] [PubMed] [Google Scholar]
- 148.Muller T, Paula Vieira R, Grimm M, Durk T, Cicko S, Zeiser R, Jakob T, Martin SF, Blumenthal B, Sorichter S, Ferrari D, di Virgilio F, Idzko M (2010) A potential role for P2X7R in allergic airway inflammation in mice and humans. Am J Respir Cell Mol Biol (in press) [DOI] [PubMed]