

Abstract
A large number of viruses can individually and concurrently cause various respiratory illnesses. Metagenomic sequencing using next-generation sequencing (NGS) technology is capable of identifying a variety of pathogens. Here, we describe a method using a large panel of oligo probes to enrich sequence targets of 34 respiratory DNA and RNA viruses that reduces non-viral reads in NGS data and achieves high performance of sequencing-based pathogen identification. The approach can be applied to total nucleic acids purified from respiratory swabs stored in viral transport medium. Illumina TruSeq RNA Access Library procedure is used in targeted sequencing of respiratory viruses. The samples are subjected to RNA fragmentation, random reverse transcription, random PCR amplification, and ligation with barcoded library adaptors. The libraries are pooled and subjected to two rounds of enrichments by using a large panel of oligos designed to capture whole genomes of 34 respiratory viruses. The enriched libraries are amplified and sequenced using Illumina MiSeq sequencing system and reagents. This method can achieve viral detection sensitivity comparable with molecular assay and obtain partial to complete genome sequences for each virus to allow accurate genotyping and variant analysis.
Electronic supplementary material:
The online version of this chapter (10.1007/978-1-4939-8682-8_10) contains supplementary material, which is available to authorized users.
Key words: Targeted sequencing, Next-generation sequencing, Genotyping, Respiratory disease, Respiratory virus, Viral enrichment, Pathogen discovery, Virome
Introduction
In targeted DNA or RNA sequencing, a panel of oligonucleotide probes are used to capture nucleotide contents containing interested sequences, the enriched targets are sequenced using next-generation sequencing (NGS) to achieve sensitive detection and sequence-based analysis [1]. Several methods with comparable performance had been developed and successfully applied to sequencing of exomes, transcriptomes, cancer genes, significant pathogens, etc. [2–8]. Utilizing TruSeq RNA Access protocol and a large panel of oligos for whole-genome capture of 34 human respiratory viral pathogens (TruSeq RVP) enables enriching sequences of respiratory DNA and RNA viruses out of complex clinical specimens and production of genome sequences for genotyping and genetic variant analysis. The process includes collection, transportation, and storage of respiratory specimens, extraction of DNA and RNA, preparation of TruSeq fragment library, RVP enrichment, NGS and sequence data analysis (Fig. 1). The laboratory procedures require approximately 5 days from specimens to NGS run excluding specimen acquisition, NGS instrument run time and post-run data processing and analyses.
Fig. 1.
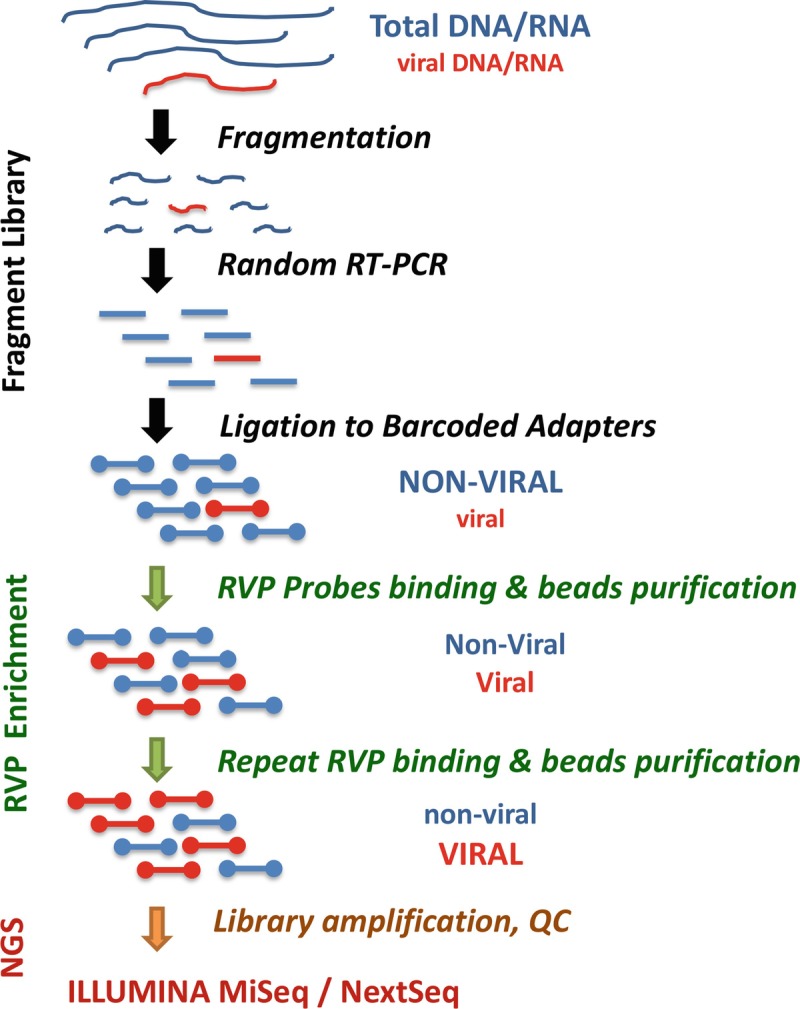
Outline of the targeted sequencing of respiratory viruses in total nucleic acids from clinical specimens. The method uses TruSeq RNA Access Library Prep protocol and custom RVP oligos for genome-wide capture of 34 human respiratory viruses for next-generation sequencing (NGS)-based detections
The method is expected to detect all commonly known respiratory viral pathogens that are diagnosed with molecular tests such as Luminex xTAG RVP assay, FilmArray Respiratory Panel (RP) tests, multiplex real-time PCR tests for respiratory viral infection with comparable sensitivity and accuracy [9–11]. The large capture panel and the scheme of genome-wide capture allow detection of most known respiratory viruses and viruses with high sequence divergences. The method is robust in handling specimens with viruses of a wide range of concentrations as well as specimens of compromised quality, e.g., degraded nucleic acids or high non-viral contents. The high capture efficiency and the superior sensitivity from this method make it a powerful tool for discovery of respiratory virome. However, there is an elevated risk of false positivity which demands more stringent contamination control procedure for the investigation of clinical specimens (see Note 1).
Materials
Perform the procedures in a Biosafety Level 2 (BSL-2) laboratory with a certified biosafety cabinet (BSC). Use clinical specimens and agents that can cause infection within the biosafety cabinet.
Nucleic Acids from Respiratory Specimens
Extract and purify DNA and RNA contents from respiratory swabs stored in a viral transport medium. Divide purified DNA/RNA into small aliquots, e.g., 10 μL per vial and store at −80 °C (see Note 2).
Library Preparation
Illumina TruSeq RNA Access Library Prep Kit (see Note 3). In this protocol, Coding Exome Oligos provided with the TruSeq RNA Access Library Prep kit are replaced with custom RVP oligos (see Supplementary file RVP capture probes for oligo sequences). The RVP oligos were designed to target whole genomes of 34 human respiratory viral pathogens (Table 1).
Illumina PhiX control V3 kit.
SuperScript III Reverse Transcriptase (200 U/μL) (see Note 4).
Agencourt AMPure XP beads. Store at 4 °C. Before use, equilibrate the beads at room temperature for 30 min, vortex vigorously for 1 min to thoroughly suspend the beads.
Nuclease-free water.
80% ethanol (EtOH): Prepare fresh 80% EtOH and use within a day.
0.2 M Tris–HCl, pH 7.0.
10 nM Tris–HCl, pH 8.5 with 0.1% Tween 20.
Microplate sealing film: Use one sheet to seal a 96-well microplate. Discard the seal after the removal from the plate. Use a new sheet to reseal the plate if needed.
Vortex mixer for small volumes in 96-well microplates and microcentrifuge tubes: Control speed and duration to obtain uniform mixing of solutions in microcentrifuge tubes or microplates.
Agilent 2100 Bioanalyzer, DNA 1000 (or 7500) chips and reagents. Store DNA chips at room temperature. Store the reagents at −20 °C. Before use equilibrate the reagents at room temperature for 30 min.
Magnetic beads separation stand for a 96-well microplate, magnetic beads separation rack for tubes.
Centrifuges: benchtop centrifuge with microplate carriers, microcentrifuge, mini centrifuge.
PCR Thermocycler for 96-well microplate.
Two water baths.
A set of pipettors with pipetting volumes from 2 μL to 1000 μL. One 20 μL 8-channel pipettor.
Table 1.
Respiratory viruses targeted by TruSeq RVP RNA access sequencing
| Respiratory virus (34) | Genome size | Type | Target genome sequences |
|---|---|---|---|
| Respiratory syncytial virus B (S2) | 15,190 | (−)ssRNA | NC_001803.1 |
| Respiratory syncytial virus A | 15,225 | (−)ssRNA | AY353550 |
| Influenza A (H9N2) | 13,500 | (−)ssRNA | NC_004910.1 NC_004911.1 NC_004912.1 NC_004908.1 NC_004905.2 NC_004909.1 NC_004907.1 NC_004906.1 |
| Influenza A (H2N2) | 13,460 | (−)ssRNA | NC_007378.1 NC_007375.1 NC_007376.1 NC_007374.1 NC_007381.1 NC_007382.1 NC_007377.1 NC_007380.1 |
| Influenza A (H3N2) | 13,630 | (−)ssRNA | NC_007373.1 NC_007372.1 NC_007371.1 NC_007366.1 NC_007369.1 NC_007368.1 NC_007367.1 NC_007370.1 |
| Influenza A (H1N1) | 13,590 | (−)ssRNA | NC_002023.1 NC_002021.1 NC_002022.1 NC_002017.1 NC_002019.1 NC_002018.1 NC_002016.1 NC_002020.1 |
| Influenza A (H5N1) | 13,590 | (−)ssRNA | NC_007357.1 NC_007358.1 NC_007359.1 NC_007362.1 NC_007360.1 NC_007361.1 NC_007363.1 NC_007364.1 |
| Influenza A (H7N9) | 13,590 | (−)ssRNA | KC885955 KC885956 KC885957 KC885958 KC885959 KC885960 KC885961 KC885962 |
| Influenza B | 14,450 | (−)ssRNA | NC_002204.1 NC_002205.1 NC_002206.1 NC_002207.1 NC_002208.1 NC_002209.1 NC_002210.1 NC_002211.1 |
| Parainfluenza 1 | 15,600 | (−)ssRNA | NC_003461.1 |
| Parainfluenza 2 | 15,650 | (−)ssRNA | NC_003443.1 |
| Parainfluenza 3 | 15,460 | (−)ssRNA | NC_001796.2 |
| Parainfluenza 4 | 17,050 | (−)ssRNA | NC_021928.1 |
| Human metapneumovirus | 13,340 | (−)ssRNA | NC_004148.2 |
| Adenovirus C | 35,937 | dsDNA | NC_001405.1 |
| Adenovirus B | 35,343 | dsDNA | NC_011203.1 |
| Adenovirus E | 35,994 | dsDNA | NC_003266.2 |
| Human coronavirus HKU1 | 29,930 | (+)ssRNA | NC_006577.2 |
| Human coronavirus NL63 | 27,550 | (+)ssRNA | NC_005831.2 |
| Human coronavirus 229E | 27,320 | (+)ssRNA | NC_002645.1 |
| Human coronavirus OC43 | 30,738 | (+)ssRNA | AY391777.1 |
| Rhinovirus A | 7150 | (+)ssRNA | NC_001617.1 |
| Rhinovirus C | 7100 | (+)ssRNA | NC_009996.1 |
| Rhinovirus B14 | 7210 | (+)ssRNA | NC_001490.1 |
| Human Bocavirus 1 | 5299 | ssDNA | NC_007455.1 |
| Human Bocavirus 2 | 5196 | ssDNA | NC_012042.1 |
| Human Bocavirus 3 | 5242 | ssDNA | NC_012564.1 |
| Human Bocavirus 4 | 5104 | ssDNA | NC_012729.2 |
| KI polyomavirus | 5040 | dsDNA | NC_009238.1 |
| WU polyomavirus | 5229 | dsDNA | NC_009539.1 |
| HPeV type 1 | 7296 | (+)ssRNA | FM242866.1 |
| HPeV type 6 | 7347 | (+)ssRNA | AB252582.1 |
| Human enterovirus C104 | 7408 | (+)ssRNA | AB686524.1 |
| Human enterovirus C109 | 7354 | (+)ssRNA | GQ865517.1 |
NGS Run Using MiSeq or NextSeq System
0.5% Tween 20.
Illumina MiSeq Reagent kit v3 or v2 (see Note 5).
Molecular biology grade water.
Illumina MiSeq or NextSeq sequencing system.
Methods
Carry out all procedures at room temperature unless otherwise specified. Brief centrifugation or quick spin is done at 280 × g for 1 min in a benchtop centrifuge with microplate carriers for 96-well microplate or by a touch-spin using a microcentrifuge or a mini-centrifuge for microcentrifuge tubes. Thaw frozen reagents at room temperature completely, vortex and centrifuge briefly, then keep them at room temperature or on ice until use. Immediately return all reagents to their original storage condition after use except for Resuspension Buffer which is kept at 4 °C after the initial use.
TruSeq RVP RNA Access Library Preparation
Fragment RNA
Label a clean 96-well microplate as DFP, add 8.5 μL of Elute/Prime/Fragment High Mix and 8.5 μL of RNA samples (see Note 6) into each well, seal and shake the DFP plate at 1,600 rpm for 20 s.
Put the DFP plate in a thermocycler for incubation, using parameters: lid temperature at 100 °C, incubation at 94 °C for 8 min and then at 4 °C holding.
Remove the DFP plate from thermocycler when it reaches 4 °C, centrifuge, then proceed to step 3.1.2 immediately.
Synthesize First Strand cDNA
Remove the seal from the DFP plate, add 7.2 μL of First Strand Synthesis Act D Mix and 0.8 μL of SuperScript III Reverse Transcriptase into each well, seal and shake the plate at 1,600 rpm for 20 s.
Put the DFP plate in the thermocycler for incubation, using parameters: lid temperature at 100 °C, incubation at 25 °C for 10 min, 42 °C for 15 min, 70 °C for 15 min, and finally at 4 °C holding.
Remove the DFP plate from the thermocycler when it reaches 4 °C, centrifuge, then proceed to step 3.1.3 immediately.
Synthesize Second Strand cDNA
Remove the seal from the DFP plate, add 5 μL of Resuspension Buffer and 20 μL of Second Strand Marking Master Mix into each well, seal and shake the plate at 1,600 rpm for 20 s.
Put the DFP plate in the thermocycler for incubation, using parameters: lid temperature at 30 °C, incubation at 16 °C for 60 min and then at 24 °C for 2 min.
-
Immediately following the incubation, remove the DFP plate from thermocycler, remove the seal, add 90 μL of well-mixed AMPure XP beads to each well, seal and shake the plate at 1,800 rpm for 2 min.
Follow steps 4–10 in Subheading 3 for AMPure XP beads binding and washing.
Incubate the plate at room temperature for 5 min.
Centrifuge the plate briefly, place the plate on a magnet stand for 5 min.
Remove the seal from the plate, discard all supernatants from each well.
With the plate on the magnet stand, add 200 μL of 80% EtOH to each well.
Incubate the plate at room temperature for 30 s, then remove all supernatants.
Repeat steps 7 and 8 once. Using a 20 μL multi-channel pipettor, carefully remove the remaining 80% EtOH from the wells.
Keep the plate stand at room temperature for 5 min to allow the EtOH to evaporate, then remove the plate from the magnet stand.
Add 17.5 μL of Resuspension Buffer into each well of the DFP plate, seal and shake the plate at 1,900 rpm for 2 min.
Incubate the DFP plate at room temperature for 2 min.
Centrifuge the DFP plate, and then place the plate on the magnet stand for 5 min.
With the DFP plate on the magnet stand, transfer 15 μL of clear supernatant from each well of the DFP plate to a new 96-well microplate labeled as ALP, without touching the magnetic beads. Seal the ALP plate. Discard the DFP plate.
Store the ALP plate at −20 °C and continue to step 3.1.4 within 7 days.
Adenylate 3’End
If the ALP plate has been stored at −20 °C, thaw it at room temperature. Set up two water baths, one at 37 °C, the other at 70 °C.
Centrifuge the ALP plate. Remove the seal from the plate, add 2.5 μL of Resuspension Buffer and 12.5 μL of A-Tailing Mix to each well, seal the plate and shake it at 1,800 rpm for 2 min.
Incubate the ALP plate at 37 °C for 30 min, and then immediately transfer the plate to 70 °C and incubate for 5 min. After incubation, immediately place the plate on ice for at least 1 min.
Ligate Adapters
Remove the seal from the ALP plate, add 2.5 μL of Resuspension Buffer, 2.5 μL of Ligation Mix, and 2.5 μL of RNA Adapter Index to each well, seal and shake the plate at 1,800 rpm for 2 min, and then centrifuge the ALP plate.
Incubate the ALP plate at 30 °C for 10 min in a water bath, then move the plate to room temperature, and keep for 5 min to allow the ALP plate temperature drop to room temperature before proceeding to the next step.
Remove the seal from the ALP plate, add 5 μL of Stop Ligation Buffer to each well, seal and shake the plate at 1,800 rpm for 2 min, then centrifuge the plate.
Remove the seal from the ALP plate, add 42 μL of well-mixed AMPure XP beads to each well of the ALP plate, seal and shake the ALP plate at 1,800 rpm for 2 min.
Follow 3.1.3. steps 4 to 10 for AMPure XP beads binding and washing.
Add 52.5 μL of Resuspension Buffer into each well of the ALP plate, seal and shake the ALP plate at 1,800 rpm for 2 min.
Incubate the ALP plate at room temperature for 2 min.
Centrifuge the ALP plate, remove the seal of the ALP plate, then place the plate on the magnet stand for 5 min.
Carefully transfer 50 μL of clear supernatant from each well of the ALP plate to a new 96-well microplate labeled as CAP. Discard the ALP plate.
Add 50 μL of well-mixed AMPure XP beads to each well of the CAP plate, seal and shake at 1,800 rpm for 2 min.
Follow 3.1.3. steps 4 to 10 for AMPure XP beads binding and washing.
Add 22.5 μL of Resuspension Buffer into each well of the CAP plate, seal and shake the plate at 1,900 rpm for 2 min.
Incubate the CAP plate at room temperature for 2 min.
Centrifuge the CAP plate, remove the seal from the CAP plate, then place the plate on the magnet stand for 5 min.
Transfer 20 μL of clear supernatant from each well of the CAP plate to a new 96-well microplate labeled as PCR.
First PCR Amplification
Add 5 μL of PCR Primer Cocktail and 25 μL of PCR Master Mix to each well of the PCR plate.
Seal the PCR plate and shake at 1,600 rpm for 20 s. Centrifuge the PCR plate.
Put the PCR plate in a thermocycler for PCR amplification, using parameters: lid temperature at 100 °C, 98 °C for 30 s, then 15 cycles of 98 °C for 10 s, 60 °C for 30 s and 72 °C for 30 s, followed by 72 °C for 5 min and finally at 4 °C holding.
Remove the PCR plate from the thermocycler, centrifuge the plate, remove the seal, add 50 μL of well-mixed AMPure XP beads to each well, seal and shake the plate at 1,800 rpm for 2 min.
Follow 3.1.3. steps 4 to 10 for AMPure XP beads binding and washing.
Add 17.5 μL of Resuspension Buffer into each well of the PCR plate, seal and shake the plate at 1,900 rpm for 2 min.
Incubate the PCR plate at room temperature for 2 min.
Centrifuge the PCR plate, remove the seal, then place the plate on the magnet stand for 5 min.
Transfer 15 μL of clear supernatant from each well of the PCR plate to a new 96-well microplate labeled as TSP1. Discard the PCR plate.
Quality Examination of the Library
Use 1 μL of each library from the TSP1 plate to measure DNA concentration and size distribution on Agilent 2100 Bioanalyzer with DNA 1000 (or 7500) kit (see Note 7). Determine DNA concentration in ng/μL for each library. The expected size distribution for the library is 200−500 bp with apparent peak at approximately 260 bp (Fig. 2).
Fig. 2.
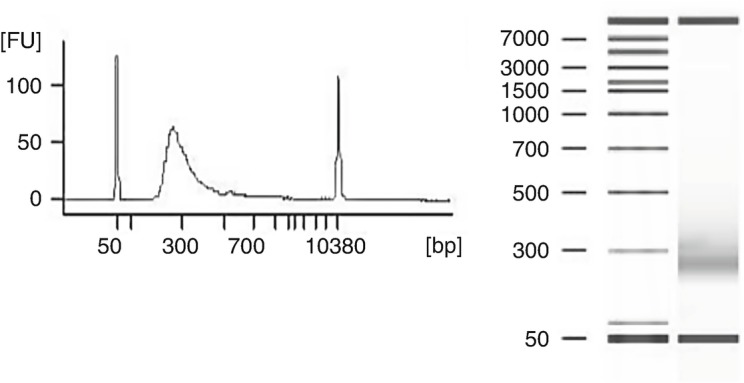
Analysis of fragment library using Agilent 2100 Bioanalyzer and DNA 7500 kit
First Hybridization
Combine up to four libraries (200 ng of DNA for each library) to one pool. If the total volume of a pool is greater than 45 μL, use a centrifugal ultrafiltration unit with a molecular weight cutoff of 30 kDa to concentrate the mixture. Bring the total volume to 45 μL with Resuspension Buffer if a pool volume is less than 45 μL. Save the library pools in a new 96-well microplate labeled as RAH1.
Add 50 μL of Capture Target Buffer 3 and 5 μL of the RVP oligos to each pool of the RAH1 plate.
Seal and shake the plate at 1,200 rpm for 1 min, then centrifuge the plate.
Put the plate in a thermocycler for incubation, using parameters: lid temperature at 100 °C, incubation at 95 °C for 10 min, 18 steps of decreasing temperatures starting at 94 °C, decreased by 2 °C at each step and kept for 1 min at each temperature, then final incubation at 58 °C for 90 min (see Note 8).
During the incubation, prepare Pre-Mixed Elution in a microcentrifuge tube (to be used in 3.1.9. step 7): mix 28.5 μL of Enrichment Elution Buffer 1 and 1.5 μL of 2 N NaOH for each pool, and keep at room temperature until use.
Once the incubation is over, immediately remove the plate from the thermocycler and proceed to step 3.1.9.
First Capture
Centrifuge the RAH1 plate, remove the seal, and transfer the first pool content to a clean 1.5 mL microcentrifuge tube labeled as S1, the second pool content to tube S2, and so on (see Note 9). Discard the RAH1 plate.
Add 250 μL of well-mixed Streptavidin magnetic beads to each S tube. Shake S tubes at 1,200 rpm for 5 min. Incubate at room temperature for 25 min. Centrifuge S tubes briefly, then put S tubes in a magnet rack for 2 min. Discard all supernatants with all S tubes on the magnet rack.
Remove all S tubes from the magnet rack, add 200 μL of Enrichment Wash Solution, shake at 1,800 rpm for 4 min.
Heat S tubes in a 50 °C water bath for 20 min. Then put S tubes on the magnet rack for 2 min. Remove supernatants with S tubes in the magnet rack.
Remove all S tubes from the magnet rack, add 200 μL of Enrichment Wash Solution, shake at 1,800 rpm for 4 min. Quick spin, then transfer the mixture from each S tube into a new 96-well microplate labeled as RAW1. Seal the RAW1 plate. Discard S tubes.
Incubate the RAW1 plate at the 50 °C water bath for 20 min. Immediately put the plate on the magnet stand for 2 min, then remove the supernatants with the plate on the magnet stand.
Add 23 μL of Pre-Mixed Elution to each well on the RAW1 plate, seal the plate and shake at 1,800 rpm for 2 min, incubate the plate at room temperature for 2 min, centrifuge and place the plate on the magnet stand for 2 min.
Transfer 21 μL of the clear supernatant from each well of the RAW1 plate to a new 96-well microplate labeled as RAH2. Discard the RAW1 plate.
Add 4 μL of Elute Target Buffer 2 to each well of the RAH2 plate to neutralize the elution.
Seal the RAH2 plate, shake at 1,800 rpm for 1 min, and then centrifuge the plate.
Second Hybridization
Remove seal from the RAH2 plate, add 20 μL of Resuspension Buffer, 50 μL of Capture Target Buffer 3 and 5 μL of RVP oligos into each well.
Follow 3.1.8. steps 3 to 6 for hybridization, then proceed immediately to step 3.1.11.
Second Capture
Centrifuge the RAH2 plate, remove the seal and transfer the contents from each well to clean 1.5 mL tubes labeled as S1, S2, and so on. Discard the RAH2 plate.
Follow 3.1.9. steps of 2 to 10 for capture, except that the new microplates are labeled as RAW2 and RAC1 respectively.
Remove the seal from the RAC1 plate, add 45 μL of well-mixed AMPure XP beads to each well, seal and shake the plate at 1,800 rpm for 1 min.
Follow 3.1.3. steps 4 to 10 for AMPure XP beads binding and washing.
Add 27.5 μL of Resuspension Buffer into each well, seal and shake the RAC1 plate at 1,900 rpm for 1 min.
Keep the RAC1 plate at room temperature for 2 min.
Centrifuge the RAC1 plate, remove the seal and place the plate on the magnet stand for 2 min.
Transfer 25 μL of the clear supernatant from each well of the RAC1 plate to a new 96-well microplate labeled as RAA. Seal the RAA plate. Discard the RAC1 plate.
Store the RAA plate at −20 °C and continue within a week.
Second PCR Amplification and Cleanup
Thaw the RAA plate, centrifuge the plate, remove the seal, add 5 μL of PCR Primer Cocktail and 20 μL of Enhanced PCR Mix. Seal the plate, shake at 1,200 rpm for 1 min, and then centrifuge the plate.
Put the RAA plate in thermocycler for PCR amplication, using parameters: lid temperature at 100 °C, 98 °C for 30 s, 17 cycles of 98 °C for 10 s, 60 °C for 30 s and 72 °C for 30 s, followed by 72 °C for 5 min and finally at 4 °C holding.
Remove the seal of the RAA plate, add 90 μL of well-mixed AMPure XP beads to each pool, seal and shake the plate at 1,800 rpm for 1 min.
Follow 3.1.3. steps 4 to 10 for AMPure XP beads binding and washing.
Add 32 μL of Resuspension Buffer into each well of the RAA plate, seal and shake the plate at 1,900 rpm for 1 min.
Leave the RAA plate at room temperature for 2 min.
Centrifuge the RAA plate, remove the seal, and then place the plate on the magnet stand for 2 min.
Transfer 30 μL of clear supernatant from each well of the RAA plate to a new 96-well microplate labeled as RAL. Discard the RAA plate.
Quality Examination of the Finial Library
Use 1 μL of the final library in the RAL plate to measure DNA concentration and size distribution on Agilent 2100 Bioanalyzer with DNA 1000 (or 7500) kit. Determine DNA concentration in nM for each pool. The expected size distribution for the library is 200−500 bp with apparent peak at approximately 300 bp (Fig. 3) (see Note 10).
Fig. 3.

Analysis of post-enrichment TruSeq RVP RNA Access library using Agilent 2100 Bioanalyzer and DNA 7500 kit
Dilution and Denaturation of the Libraries for NGS
The enriched libraries can be sequenced on Illumina Miseq or NextSeq systems. The loading concentration for libraries on MiSeq is 6−15 pM for reagents kit v2 or v3. We use loading concentration in the range of 8–14 pM for sequencing with MiSeq and reagents kit v3.
Dilute each library in the RAL plate from the concentration determined with Agilent 2100 Bioanalyzer to 1 nM with Resuspension Buffer. Prepare 22 μL of 1 nM of each library and pool all of them together into a new 1.5 mL tube labeled as 1 nM lib pool mix. Mix the tube at 1,200 rpm for 30 s and centrifuge. Keep the tube on ice until use. Store the RAL plate at −20 °C.
Prepare fresh 0.2 N NaOH by adding 200 μL of 1 N NaOH into 800 μL of nuclease-free water in a clean 1.5 mL tube labeled as 0.2 N NaOH. Mix the tube at 1,200 rpm for 30 s and centrifuge.
In a new 1.5 mL tube labeled as 20 pM lib pool mix, add 20 μL of 1 nM lib pool mix and 20 μL of 0.2 N NaOH. Mix the tube at 1,200 rpm for 30 s and centrifuge. Keep it at room temperature for 5 min. Add 20 μL of 0.2 M Tris-HCl, pH 7.0 into the tube. Mix the tube at 1,200 rpm for 30 s and centrifuge. Add 940 μL of prechilled HT1. Mix the tube at 1,200 rpm for 30 s and centrifuge. Keep the 20 pM lib pool mix tube on ice. Store the 1 nM lib pool mix tube at −20 °C.
In a new 1.5 mL tube labeled as loading conc. Pool, dilute the 20 pM denatured pool mix with prechilled HT1 using following chart as guide. Keep the loading conc. Pool tube on ice.
| Loading concentration (pM) | 6 | 8 | 10 | 12 | 14 | 20 |
|---|---|---|---|---|---|---|
| 20 pM lib pool mix (μL) | 180 | 240 | 300 | 360 | 420 | 600 |
| Pre-chilled HT1 (μL) | 420 | 360 | 300 | 240 | 180 | 0 |
-
5.
In a clean 1.5 mL tube labeled as 20 pM PhiX control (see Note 11), add 2 μL of 10 nM PhiX library, 3 μL of 10 nM Tris-Cl, pH 8.5 with 0.1% Tween 20, and 5 μL of 0.2 N NaOH. Mix the tube at 1,200 rpm for 30 s and centrifuge. Incubate at room temperature for 5 min. Add 990 μL of prechilled HT1. Mix the tube at 1,200 rpm for 30 s and centrifuge. Keep this tube on ice.
-
6.
Add 6 μL of 20 pM PhiX control into 600 μL of the loading conc. pool tube. Mix the tube at 1,200 rpm for 30 s and centrifuge. Keep the tubes on ice until NGS run. (The mixture inside of the tube will be loaded into the reagent cartridge for sequencing run).
NGS and Sequence-Based Detection of Respiratory Pathogens
Sequence the lib pool with PhiX control using Illumina MiSeq or NextSeq system. The acquired sequence data can be applied to bioinformatics data analysis pipeline, which may include data quality processing, the removal of human genome and transcriptome sequences, de novo assembly, and comparison of sequence contigs and unassembled reads with sequence databases using BLAST programs [12] (see Note 12).
Notes
The method we described in this chapter is for research use only. It is not validated for clinical diagnosis purpose. The potential contamination and cross-contamination can come from specimen collection, shipping and handling, storage, laboratory surfaces and instruments, reagents, viral cultures, nucleic acids or PCR amplification products, etc. Conduct the experiments in a clean laboratory room where high titer cultures, high concentration nucleic acids and PCR amplifications are not present. Clean frequently used instruments and devices, e.g., centrifuge, thermocycler, mixer, pipettors, on a regular basis to prevent accumulative contaminations and before each experiment to minimize carry-over of nucleic acids from the previous run. Use newly opened consumable materials and freshly prepared reagents. We highly recommend dividing original specimens, DNA/RNA extracts, and stock solutions into small aliquots or fractions to avoid accumulative contamination and for independent repeating of the test when trouble-shooting or verification of results is needed.
Respiratory swabs can be stored in transport medium such as Copan Universal Transport Medium (UTM) (Murrieta, CA, USA) or PrimeStore Molecular Transport Medium (MTM) (Longhorn Vaccines and Diagnostics LLC, Bethesda, MD, USA). We use QIAGEN QIAamp Viral RNA Mini Kit in DNA/RNA preparation. Carrier RNA is used in DNA/RNA extraction with QIAamp Viral RNA kit to increase RNA yield and protect purified RNA from degradation. Store RNA extracts in small aliquots and minimize freeze thaw cycles. Use RNase-decontamination products, for example, RNase AWAY made by Molecular Bio-Products, Inc., to clean the surface of the hood, benches, instruments, etc. When handle RNA samples, always wear gloves and change them often. During the procedure, work quickly and keep tubes closed and plates covered.
Most reagents used in this method are parts in Illumina TruSeq RNA Access Library Prep Kit. The user’s guide that we used with modification is RS-301-9001DOC Part# 15049525 Rev. B. The plate names in the protocol are DFP (Depleted RNA Fragmentation Plate), ALP (Adaptor Ligation Plate), CAP (Clean up ALP Plate), RAH1 (RNA Access Hybridization 1), RAW1 (RNA Access Wash 1), RAH2 (RNA Access Hybridization 2), RAW2 (RNA Access Wash 2), RAC1 (RNA Access Clean-up 1), RAA (RNA Access Amplification), TSP1 (Target Sample Plate 1) and RAL (RNA Access Library).
SuperScript II Reverse Transcriptase and the new SuperScript IV Reverse Transcriptase can also be used. SuperScript III Reverse Transcriptase is the most often used reverse transcriptase. It is advised that SuperScript IV can obtain good yield and length of cDNA synthesis with improvements on thermal stability, speed, and resistance to inhibitors.
The TruSeq RNA Access library has size range around 300 bp. Both Illumina MiSeq Reagent kits v2 and v3, which can read 150 bp and 300 bp from either end, can be used in sequencing the libraries.
The TruSeq RNA Access Library guide recommends using 10−100 ng of RNA as starting materials. Since total nucleic acids extracted from respiratory specimens contain both DNA and RNA and have highly variable concentrations from specimen to specimen, we use 8.5 μL of DNA/RNA for each sample in RNA fragmentation.
Agilent 2100 Bioanalyzer can analyze up to 12 samples in one run. An alternative high-throughput system, the Agilent TapeStation 4200, can analyze up to 96 samples in one run.
The total hybridization time is about 2 h. Over hybridization may cause a high degree of nonspecific binding.
Instead of using microcentrifuge tubes and a magnetic beads separation rack, a 0.8 mL 96 deep well plate and a magnetic beads separation stand for 96-well plate can be used to process a large number of samples.
After two rounds of hybridization and capture and the second PCR amplification, the post-enrichment library appears having broadened size distribution with the peak shifted slightly to a higher molecular weight as compared with the fragment library shown in Fig. 2.
We always include Illumina PhiX Control v3 library in MiSeq runs. The PhiX control can be used for quick assessment of data quality and for technical trouble shooting. Besides, the addition of PhiX library can improve base calling quality for libraries with high G/C or A/T contents or low diversity sequences. The spiking ratio can be increased or reduced based on the needs and the nature of the specimens.
Using this method, partial to near-complete viral genome sequences can be obtained to allow genotyping and recombination and variant analysis. The method can detect both RNA and DNA viruses. During interpretation of the results, take cautious consideration of probable contamination and cross-contamination. If necessary, verify the results with additional tests such as molecular assays and/or by repeating the experiment with archived specimens or DNA/RNA aliquots.
Electronic Supplementary Material
(XLS 855 kb)
Acknowledgments
We thank Darrell Dinwiddie for help in designing the RVP oligos. This work is supported by the Global Emerging Infections Surveillance and Response System (GEIS), a Division of the Armed Forces Health Surveillance Center. The views expressed here are those of the authors and do not reflect the official policy of the Department of the Army, Department of Defense or U.S. Government. We declare that no conflict of interest exists. This is the work of U.S. government employees and may not be copyrighted (17 USC 105).
Contributor Information
Andrés Moya, Email: andres.moya@uv.es.
Vicente Pérez Brocal, Email: perez_vicbro@gva.es.
Jun Hang, Email: jun.hang.civ@mail.mil.
References
- 1.Garcia-Garcia G, Baux D, Faugere V, et al. Assessment of the latest NGS enrichment capture methods in clinical context. Sci Rep. 2016;6:20948. doi: 10.1038/srep20948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terracciano I, Cantarella C, Fasano C, et al. Liquid-phase sequence capture and targeted re-sequencing revealed novel polymorphisms in tomato genes belonging to the MEP carotenoid pathway. Sci Rep. 2017;7(1):5616. doi: 10.1038/s41598-017-06120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JS, Mackie RS, Harrison T, et al. Targeted enrichment for pathogen detection and characterization in three felid species. J Clin Microbiol. 2017;55(6):1658–1670. doi: 10.1128/JCM.01463-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deviatkin AA, Lukashev AN, Markelov MM, et al. Enrichment of viral nucleic acids by solution hybrid selection with genus specific oligonucleotides. Sci Rep. 2017;7(1):9752. doi: 10.1038/s41598-017-10342-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonfiglio S, Vanni I, Rossella V, et al. Performance comparison of two commercial human whole-exome capture systems on formalin-fixed paraffin-embedded lung adenocarcinoma samples. BMC Cancer. 2016;16:692. doi: 10.1186/s12885-016-2720-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wylie TN, Wylie KM, Herter BN, et al. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015;25(12):1910–1920. doi: 10.1101/gr.191049.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briese T, Kapoor A, Mishra N, et al. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. MBio. 2015;6(5):e01491–e01415. doi: 10.1128/mBio.01491-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehler JW, Hall AT, Rolfe PA, et al. Development and evaluation of a panel of filovirus sequence capture probes for pathogen detection by next-generation sequencing. PLoS One. 2014;9(9):e107007. doi: 10.1371/journal.pone.0107007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malhotra B, Swamy MA, Reddy PV, et al. Evaluation of custom multiplex real - time RT - PCR in comparison to fast - track diagnostics respiratory 21 pathogens kit for detection of multiple respiratory viruses. Virol J. 2016;13:91. doi: 10.1186/s12985-016-0549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhary ML, Anand SP, Tikhe SA, et al. Comparison of the conventional multiplex RT-PCR, real time RT-PCR and Luminex xTAG(R) RVP fast assay for the detection of respiratory viruses. J Med Virol. 2016;88(1):51–57. doi: 10.1002/jmv.24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babady NE, Mead P, Stiles J, et al. Comparison of the Luminex xTAG RVP fast assay and the Idaho technology FilmArray RP assay for detection of respiratory viruses in pediatric patients at a cancer hospital. J Clin Microbiol. 2012;50(7):2282–2288. doi: 10.1128/JCM.06186-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilianski A, Carcel P, Yao S, et al. Pathosphere.org: pathogen detection and characterization through a web-based, open source informatics platform. BMC Bioinformatics. 2015;16:416. doi: 10.1186/s12859-015-0840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS 855 kb)