

Abstract
Viruses are intracellular parasites that hijack the cellular machinery for their own replication. Therefore, an obligatory step in the virus life cycle is the delivery of the viral genome inside the cell. Enveloped viruses (i.e., viruses with a lipid envelope) use a two-step procedure to release their genetic material into the cell: (i) they first bind to specific surface receptors of the target cell membrane and then, (ii) they fuse the viral and cell membranes. This last step may occur at the cell surface or after internalization of the virus particle by endocytosis or by some other route (e.g., macropinocytosis). Remarkably, the virus-cell membrane fusion process goes essentially along the same intermediate steps as other membrane fusions that occur for instance in vesicular fusion at the nerve synapsis or cell-cell fusion in yeast mating. Specialized viral proteins, fusogens, promote virus-cell membrane fusion. The viral fusogens experience drastic structural rearrangements during fusion, liberating the energy required to overcome the repulsive forces that prevent spontaneous fusion of the two membranes. This chapter describes the different types of viral fusogens and their mode of action, as are currently known.
Keywords: Class I fusion protein, Class II fusion protein, Class III fusion protein, Enveloped virus, Fusion pore, Glycoprotein, Membrane, Membrane fusion intermediate, Post-entry events, Viral fusogen, Virus entry
Introduction
Enveloped viruses are characterized by having a lipid bilayer (envelope) surrounding the virus particle (virion) (see 10.1007/978-94-007-6552-8_11). One or several virus-encoded glycoproteins are inserted into the envelope. These proteins are exposed at the virion surface and are responsible therefore of the initial interactions of the virus with the target cell, leading to “virus entry”. In fact, it is only the cargo inside the envelope layer and not the virus itself what is actually discharged inside the cell. The virus glycoproteins are also the main targets of the neutralizing antibody response produced by the host in its defense against the virus.
To infect a new cell, the virus particle must first attach to the cell surface through non-covalent interactions of one or more of the viral glycoproteins anchored into the lipid bilayer with specific cell surface receptors. These interactions are described in detail in 10.1007/978-94-007-6552-8_15. Suffice to say here that virus-receptor binding is one of the factors that can influence virus tropism; i.e., which cell types are actually infected by the virus.
For enveloped viruses, fusion of the viral and cell membranes is an obligatory step that follows virus binding to cells. Virus-cell fusion is therefore the step at which the virus particle loses its individuality. Membrane fusion may proceed at the cell surface or alternatively after internalization of the virus particle, generally by endocytosis. In either case, fusion is driven by specialized viral glycoproteins (fusogens) which are activated (triggered) by specific events occurring either at the cell surface or inside the endosome. The viral fusogens are in metastable conformations in the virus particle. Once triggered, they initiate a series of conformational changes (in most cases irreversibly) that facilitate approximation of the two membranes, followed by fusion. At the end of the fusion process, the viral fusogens adopt highly stable conformations. The free energy liberated during the transition from the metastable pre-fusion to the highly stable post-fusion conformation drives the fusion process.
General Principles of Membrane Fusion
Protein-Free Membrane Fusion
Lipid mixing occurs spontaneously in monolayers but several forces prevent the spontaneous mixing of lipids between bilayer membranes [1]. Most important among them are: (i) hydrophobic effects that seek to minimize solvent-exposed apolar surfaces, (ii) elastic forces that prevent monolayer deformation and (iii) electrostatic repulsions between negatively charged phospholipids. Nevertheless, fusion between protein-free lipid bilayers (e.g., liposomes) can be induced under certain conditions. For instance, certain phospholipids (e.g., phosphatidylcholine, PC) induce positive curvature of the lipid monolayer whereas others (e.g., phosphatidylethanolamine, PE) induce negative curvature. The distribution of PC and PE between the two leaflets of the lipid bilayer can either promote or inhibit spontaneous protein-free membrane fusion. Also direct dehydration between bilayers promotes fusion by bringing the two membranes into very close contact. However, under most physiological conditions, specialized proteins are needed to overcome the repulsive forces that prevent membrane fusion.
Independently of the driving machinery, fusion of two lipid bilayers occurs in a stepwise manner that includes the formation of an hourglass-like structure, known as the lipid stalk (Fig. 16.1a) [2]. This stalk is then expanded forming what is called the hemifusion diaphragm, in which lipids of the two distal leaflets of the bilayers are now in direct contact. Finally, rupture of the hemifusion diaphragm leads to formation of the fusion pore that is then expanded to complete membrane fusion and content mixing of the two compartments. However, opening of the fusion pore may be a reversible step and does not always lead to full fusion [3]. Energy is therefore required through all steps of the fusion process, including expansion of the fusion pore. Hence, proteins when present must operate from the initial stages of membrane deformation to final merging of the two membranes.
Fig. 16.1.
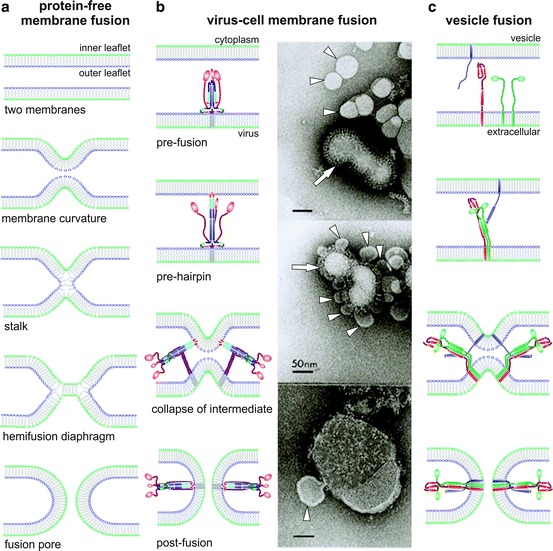
Steps of the membrane fusion process. (a) Diagram of the fusion steps between two protein-free lipid bilayers. From top to bottom: the lipids (represented by heads and tails) of the two bilayers are initially curved into nipple-like structures that approach the two membranes. This is followed by formation of the stalk in which the two proximal leaflets are fused. This stalk is then expanded forming a hemifusion diaphragm in which lipids of the distal leaflets of the bilayers are now in direct contact. Finally, rupture of the hemifusion diaphragm leads to formation of the fusion pore. (b) Diagram of the virus-cell fusion process: As an example, fusion mediated by the influenza HA is illustrated. Left panels: HA is a homotrimer that initially binds to the cell surface (not shown) by interactions of each subunit head with sialic acid. Then, the virus is internalized by endocytosis. For simplicity, only 1 HA trimer in the pre-fusion conformation is shown, anchored into the viral membrane. After endosome acidification, the HA globular head falls apart, allowing refolding of the molecule to produce three long α-helices. The fusion peptides, placed at the N-terminal end of each α-helix, insert into the target membrane. This intermediate, dubbed pre-hairpin, refolds to bring the two membranes into proximity leading to formation of the lipid stalk followed by formation of the hemifusion diaphragm (not shown). Finally, the fusion pore is formed by the concerted action of several HA molecules that adopt a very stable post-fusion conformation. Right panels: The upper panel shows a mixture of an influenza virus particle (strain X31, H3N2, white arrow) and liposomes (some of them indicated by white arrowheads) made with lipids commonly found in cell membranes, incubated at neutral pH. Note the glycoprotein spikes (mostly haemagglutinin, HA) sticking out of the viral membrane in contrast with the smooth surface of liposomes. The middle panel shows the same virus/liposome mixture after incubation for 5–10 s at pH 5.0 followed by neutralization. Note binding of liposomes to the virus surface and initiation of virus-liposome fusion. The lower panel shows the virus/liposome mixture after incubation for 5 min at pH 5.0 followed by neutralization. Note that the virus has fused with several liposomes, yielding a large vesicle with viral glycoproteins disperse throughout the surface and with a small liposome still in the process of fusion. The HA spikes also have changed morphology after exposure to low pH and fusion. (Courtesy of L.J. Calder and S.A. Wharton, Division of Virology, MRC National Institute for Medical Research, London, UK). (c) Diagram of vesicle fusion at the synaptic junction: Initially one of the SNARE proteins (synaptobrevin, blue) is inserted into the vesicle membrane, while three other SNAREs (two SNAP 25, green and one syntaxin, red) are inserted in the plasma membrane. After an initial interaction, refolding of the SNAREs leads to formation of a bundle of four parallel α-helices that drives approximation of the two membranes and formation of the stalk and hemifusion intermediates (not shown). Completion of the SNARE complex results in formation of the fusion pore. In the two lower panels of parts (b) and (c), two HAs and two SNARE complexes are shown surrounding the fusion pore, although the actual number of molecules involved in fusion pore formation is likely to be higher
Virus-Induced Membrane Fusion
Enveloped viruses contain specialized surface glycoproteins that mediate: (i) initial binding of virus to the cell surface and (ii) fusion of the virus and cell membranes, either at the cell surface or after endocytosis [4]. The initial attachment of the virus to the cell surface may involve only one type of viral glycoprotein (e.g., influenza virus) or it may require the combined action of several viral glycoproteins (e.g., herpesvirus). However, the actual process of membrane fusion is driven by a specialized type of viral glycoproteins (the viral fusogens) which may or may not have participated additionally in attachment.
Although the sequences of viral fusion proteins vary considerably, they all share certain structural characteristics and are subject to analogous structural rearrangements during the fusion process. As an example, Fig. 16.1b (left panels) illustrates the structural changes that the influenza virus haemagglutin (HA) undergo during fusion of the viral and the endosomal membrane. In this case, influenza HA also mediates the initial interaction of the virus with sialic acid of glycoproteins or glycolipids at the cell surface (see 10.1007/978-94-007-6552-8_15). After this initial binding, the virus is internalized by endocytosis. Acidification of the endosome (probably by fusion with lysosomes) triggers HA to start the conformational changes depicted in Fig. 16.1b (left panels) and described next.
Independently of the triggering event, all viral fusion proteins undergo conformational changes upon activation that lead to the formation of an extended unstable intermediate, dubbed pre-hairpin (Fig. 16.1b, “pre-hairpin”). Formation of the pre-hairpin intermediate involves very large-scale structural rearrangements in the viral fusogens with exposure of hydrophobic segments or loops (the fusion peptide, FP). Since the hydrophobic fusion peptide cannot be exposed to a hydrophilic environment it inserts into the target membrane. At this point the viral and target membranes are bridged by two separate segments of the same polypeptide; one is the fusion peptide bound to the target membrane and the other is the transmembrane (TM) region of the viral fusogen inserted into the viral membrane. The pre-hairpin intermediate may have a relatively long half-life; for the human immunodeficiency virus (HIV) gp41 protein, the half-life seems to be several minutes [5], but in other cases, it may only be a few seconds [6]. The pre-hairpin bridge then collapses bringing into close apposition the viral and target membranes, which are distorted probably into protein-free nipple-like configurations (Fig. 16.1b, “collapse of intermediate”). This is followed by the formation of a lipid stalk and a hemifusion diaphragm (in analogy with the protein-free membrane fusion path), which allows lipid mixing between the two proximal leaflets of the viral and target membranes. Finally, the hemifusion diaphragm opens to form a transient fusion pore that may flicker open and closed until it expands [7], leaving the viral fusogen in a highly stable post-fusion hairpin conformation inserted into the target membrane (Fig. 16.1b, “post-fusion”).
Influenza HA dependent membrane fusion can be reproduced in the test tube with purified virus and liposomes and observed by electron microscopy, as illustrated in Fig. 16.1b (right panels). After mixing of virions (arrows) and liposomes (arrowheads) they remain separated (upper panel), since the former lack the influenza virus receptor (sialic acid). However, a brief pulse at low pH (middle panel) exposes the fusion peptide of the influenza HA which is then inserted into the membrane of multiple liposomes. Longer pulses of low pH result in fusion of the virus with multiple liposomes leading to formation of large vesicles (lower panel; note two vesicles, one small and one large, caught in the process of fusion).
The fusion peptides probably insert only into the outer leaflet of the cell target membrane. Due to the large number of fusogen molecules present at the viral surface, multiple fusion peptides may interact with the external leaflet of the target membrane upon formation of the pre-hairpin intermediate, potentially initiating membrane deformation. This suggests that cooperativity between several viral fusogens may be required for membrane fusion. In fact, fusion mediated by the influenza HA is positively affected by protein density [8]. It is estimated that 4–6 HA molecules are required for fusion, forming a protein ring at the periphery of the fusion pore. Also, electron microscopy (10.1007/978-94-007-6552-8_3) and X-ray crystallography (10.1007/978-94-007-6552-8_4) results indicate that the E1 glycoprotein of Semliki Forest alphavirus interacts cooperatively during membrane insertion and fusion [9].
Despite the above arguments in favor of cooperativity, calculations of the energy barrier that must be overcome en route to a hemifusion diaphragm is estimated to be about 40–50 kcal.mol−1. A free energy of roughly this magnitude could be recovered from the collapse of one or two pre-hairpin intermediates, depending on the interactions driving such collapse. In fact, experiments with HIV suggest that only one or two active envelope glycoproteins are sufficient for fusion [10], although later estimates have increased this number [11]. It may be that the fusion proteins of HIV and other retroviruses have evolved to manage with a single fusion protein, as the number of envelope glycoproteins in the virus particle (estimated 15–20, in contrast to hundreds in other viruses) is rather sparse.
Formation of the pre-hairpin structure and refolding of this intermediate entails some of the most drastic protein rearrangements ever found in biology. Pre-hairpin collapse involves folding back of the membrane proximal domain of the viral fusogen onto a trimeric core whose distal end from the viral membrane is inserted into the target membrane (Fig. 16.1b). Zippering together of these two domains brings the membranes into close proximity. Dehydration of the initial contact site induces monolayer rupture resulting in lipid stalk formation and hemifusion. However, formation of the fusion pore requires further structural rearrangements, including interactions between regions adjacent to the fusion peptide and the transmembrane region [12, 13] and, probably, additional contacts between these two hydrophobic regions that are now inserted into the same membrane. For instance, membrane fusion by the influenza HA with a glycosyl phosphotidylinositol (GPI) anchor replacing the TM region halts at the hemifusion stage [14].
Finally, enlargement of the initial fusion pore is probably the most energy demanding step and requires the coordinated action of several fusogen molecules that surround the early nipple-like fusion intermediate [15].
Vesicle and Cell-Cell Fusion
This topic is brought here only to emphasize the analogies and differences between membrane fusions promoted by unrelated proteins. Vesicle fusion is required for essential biological processes, such as exocytosis and synaptic transmission. Cell-cell fusion is involved in hypodermal cell fusion in C. elegans, sperm-egg fusion, yeast mating (mating of two haploid yeast cells to produce a diploid cell), placenta formation in mammals, and muscle and bone formation.
In all cases, membrane fusion follows the same steps already described in previous sections; i.e., deformation and approximation of the two membranes, formation of the stalk and hemifusion intermediates and finally formation and enlargement of the fusion pore (Fig. 16.1c). In analogy with virus-cell fusion, vesicle and cell-cell fusion requires formation of highly stable protein assemblies that provide the energy necessary to overcome the repulsive forces of membranes in close proximity [16]. Also, vesicle and cell-cell fusion, as viral fusion, requires higher order multimerization of the fusogens that delineate the hemifusion diaphragms and the fusion pores [17].
The main difference between virus-cell fusion and vesicle or cell-cell fusion is that in the former process the protein fusogen is present only in the viral membrane. In contrast, the proteins involved in vesicle fusion and cell-cell fusion are initially inserted in the two membranes predestined to fuse (Fig. 16.1c). In synaptic vesicles, the main proteins responsible of membrane fusion are the so-called SNARE (soluble N-ethylamine sensitive factor attachment receptor protein) proteins [18] which share a conserved 60–70 amino acid motif. These proteins, when they find each other refold into a highly stable four-helix parallel coiled-coil bundle that resembles the six-helix bundle formed by the heptad repeats (structural motifs with a repeating pattern of seven amino acids) of certain viral fusogens (see below). Formation of the four-helix bundle leads to membrane apposition and hemifusion, as with the collapse of the pre-hairpin intermediate of viral fusogens. However, a unique characteristic of vesicle fusion is that the protein machinery involved in the process is disassembled, once fusion is finished to be reused in subsequent fusion events. This is accomplished by the ATPase (adenosine triphosphatase) N-ethylmaleimide sensitive factor (NSF) [19].
In contrast to vesicle fusion, cell-cell fusion entails the same set of fusion proteins in the two membranes. For instance, the exceptional process of hypodermal cell fusion in C. elegans to form a large multinucleated syncytium of all skin cells is driven by the EFF-1 protein [20]. Unlike SNAREs and viral fusogens, EFF-1 has a homotypic fusion machinery in the opposite membrane. In other words, both membranes must have EFF-1 for fusion to occur. Nevertheless, cell-cell fusion is a multistep process that goes along the same lipid intermediates as viral and vesicle fusion.
Viral Fusion Proteins
Based on biosynthetic and structural characteristics, viral fusogens have been classified into three categories (Table 16.1). Class I fusion glycoproteins are characterized by being synthesized as inactive precursors that require proteolytic processing to become fusion-competent. They are all homotrimers that upon fusion refold into hairpins containing a long central coiled-coil core structure (formed by helices that are coiled together). Class II fusion glycoproteins are derived from longer polyprotein precursors that are proteolytically processed during biosynthesis. The class II fusion proteins form icosahedral scaffolds of protein dimers at the viral surface. During fusion, these proteins undergo an oligomeric rearrangement, converting the metastable prefusion dimer into a stable hairpin homotrimer composed of β-sheet structures. Finally, class III glycoproteins are not proteolytically processed. Their post-fusion hairpin trimer displays a central α-helical coiled-coil, as class I glycoproteins, but the fusion domain exposes two fusion loops located at the tip of an elongated β-sheet, revealing a striking convergence with class II fusion proteins.
Table 16.1.
Classification of viral fusion proteins
| Class | Virus family | Representative | Viral fusogen | Involved in attachment |
|---|---|---|---|---|
| I | Orthomyxoviridae | Influenza virus | Haemaglutinin (HA) | Yes |
| Retroviridae | Human immunodeficiency virus (HIV) | Envelope glycoprotein;gp 41 subunit | Yes | |
| Filoviridae | Ebola virus | GP glycoprotein | Yes | |
| Coronaviridae | Severe acute respiratory syndrome (SARS) virus | S glycoprotein | Yes | |
| Paramyxoviridae | Sendai virus | F glycoprotein | No | |
| II | Alphaviridae | Semliki Forest Virus | E1 glycoprotein | No |
| Flaviviridae | Dengue virus | E glycoprotein | Yes | |
| III | Rhabdoviridae | Vesicular stomatitis virus | G glycoprotein | Yes |
| Baculoviridae | Baculovirus | Gp64 glycoprotein | Yes | |
| Herpesviridae | Herpes simplex virus | gB glycoprotein | No |
Class I Viral Fusion Proteins
The first atomic structure of any viral or cellular glycoprotein was determined by X-ray crystallography and reported in 1981 by the laboratories of Wiley and Skehel [21]. It was the structure of the influenza haemagglutinin (HA) trimeric ectodomain (the domain that protrudes from the plasma membrane), as released from the virus particles by bromelain treatment, which cleaves the HA polypeptides near the TM region.
Influenza HA is synthesized in the infected cell as a polypeptide precursor (HA0) of about 550 amino acids that is cleaved proteolytically to generate the HA1 (roughly the N-terminal two thirds) and HA2 (the C-terminal third) chains that remain covalently linked by a disulfide bond. At the newly created HA2 N-terminus there is a stretch of hydrophobic amino acids, called the fusion peptide, which is inserted into the target membrane during fusion. The overall structure of the influenza HA is that of an elongated spike sticking out of the membrane. The distal head, formed exclusively by HA1 sequences, bears the receptor (sialic acid) binding site, formed by a shallow pocket exposed on its outward-forming surface. The stem, made largely by HA2 amino acids, is a trimeric α-helical coiled-coil. The structural rearrangements of the influenza HA during membrane fusion are shown in Fig. 16.1b.
As influenza, other viruses also contain class I fusion glycoproteins that have both receptor and membrane fusion activities (Table 16.1). For instance, the envelope glycoprotein of HIV that is also proteolitically processed and that binds to protein receptors (CD4) and chemokine co-receptors before engaging in membrane fusion at the cell surface. Similarly, the receptor-binding proteins of filovirus and coronavirus mediate additionally viral-cell membrane fusion.
In contrast, the attachment and fusion activities reside in two different surface glycoproteins of paramyxoviruses. The attachment protein (named HN, H or G) is required for the initial interaction of the virus with the cell surface (see 10.1007/978-94-007-6552-8_15 for virus receptor usage). Once the virus is bound to the cell, the other major viral glycoprotein (called F, for fusion) is triggered to promote fusion of the viral and cell membranes. Structure determination of prototypic paramyxovirus F proteins in the pre-fusion metastable conformation [22] and in the post-fusion state [23] by X-ray crystallography, as well as identification of fusion intermediates [24], has provided the most complete picture of the membrane fusion process driven by class I fusion glycoproteins, as depicted in Fig. 16.2.
Fig. 16.2.
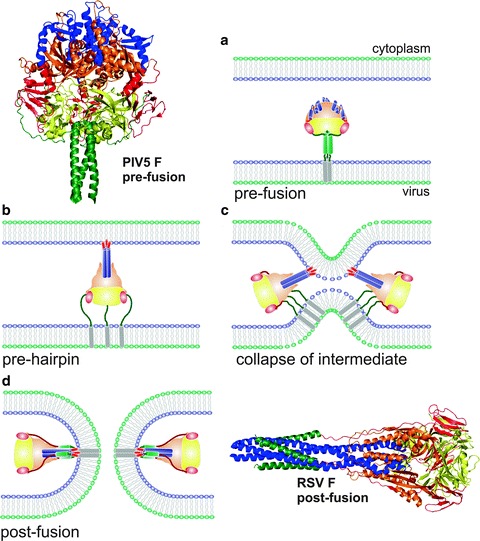
Membrane fusion mediated by a class I fusion protein (Paramyxovirus). The atomic structures of the pre-fusion form of Parainfluenza virus type 5 (PIV5) [22] (upper left) and the post-fusion form of Respiratory Syncytial Virus (RSV) [53] (lower right) F proteins are shown as ribbons. The same protein regions are highlighted with identical colors in the two conformations. (a–d) Diagram of the fusion process denoting: (a) the pre-fusion paramyxovirus F protein trimer inserted in the viral membrane before activation, (b) formation of the pre-hairpin structure which includes refolding of the long central HRA α-helices (blue) with the fusion peptide (red) inserted into the cell membrane, (c) collapse of the pre-hairpin to approach the two membranes, and (d) formation of the fusion pore and stabilization of the F trimer in the post-fusion conformation. In the last two steps, two F protein molecules are represented to indicate the cooperation needed to drive the fusion process
The paramyxovirus F protein, as other class I glycoproteins, is synthesized as an inactive precursor (F0) that is translocated co-translationally to the lumen of the endoplasmic reticulum where it assembles into a trimer. Each F protein subunit is proteolytically cleaved during transport to the cell surface, generating two chains, F2 N-terminal and F1 C-terminal that remain linked by one or more disulfide bonds. F1 (equivalent to the HA2 chain of influenza virus) has a hydrophobic fusion peptide at the N-terminus and two heptad repeat sequences (HRA and HRB) in its ectodomain. HRA is adjacent to the fusion peptide and HRB is proximal to the transmembrane (TM) region, which is placed near the F1 C-terminus.
The pre-fusion three-dimensional (3D) structure of the parainfluenza virus type 5 (PIV5) F protein contains a large globular head connected to a short trimeric coiled-coil made by the HRB region [22] (Fig. 16.2). Comparison with the post-fusion structure of the F ectodomain from other paramyxovirus (for instance respiratory syncytial virus (RSV (Fig. 16.2)) and functional studies using peptide inhibitors [24] provided the following model of membrane fusion: Upon virus binding to cells through the attachment protein, F is activated and initiates a series of conformational changes, including separation of the HRB coils and refolding of HRA sequences to form a very elongated trimeric coiled-coil. The fusion peptides -now at the N-terminus of the HRAs- insert into the target cell membrane, resulting in formation of the pre-hairpin intermediate. This step is followed by zipping of the C-terminal part of the molecule along the core coiled-coil to bring together the two membranes and the fusion and TM domains, in analogy with the process described before for influenza HA. However, in the case of the paramyxovirus F the HRB sequences wrap around the HRA coiled-coil forming an extremely stable six-helix bundle (6HB) in the post-fusion hairpin. Formation of this 6HB provides most of the energy required to overcome membrane repulsion. The 6HB structure is shared by other class I fusion glycoproteins, such as the gp41 chain of the HIV envelope glycoprotein.
While activation of influenza HA requires exposure to the endosomal low pH (probably by protonation of key amino acid residues), the event that triggers paramyxovirus F proteins is still ill-defined. Cell-cell fusion of transfected cells that express paramyxovirus F requires in most cases co-expression of the homotypic attachment protein, suggesting that an interaction of the two proteins is needed for membrane fusion. Two alternative models (“clamp” and “provocateur”) have been proposed to explain the requirement of the attachment protein for fusion:
The clamp model postulates that HN (or the equivalent attachment protein depending on the virus) is complexed with F in the virus particle, retaining the latter in the metastable configuration. Conformational changes in HN upon receptor binding release F from the complex to initiate membrane fusion.
Alternatively, the provocateur model postulates that HN and F do not interact in the virus before contacting the cell. Concomitantly to the structural changes induced in HN upon receptor binding, HN binds to F and this interaction triggers F for fusion [25].
Intriguingly, the F protein of viruses belonging to the Pneumovirinae subfamily of paramyxoviruses (e.g., RSV and human metapneumovirus, hMPV) do not require co-expression of the attachment protein (G) for cell-cell fusion [26]. Furthermore, deletion mutant viruses have been obtained in which the entire G gene is obliterated. These mutants still infect cells in vitro, although less efficiently than the wild type virus and are attenuated in animal models of infection [27]. Activation of the F protein of those deletion mutants cannot rely on interactions with the G protein and therefore alternative regulatory mechanisms should control membrane fusion. Of note, a unique characteristic of the RSV F protein is the presence of two proteolytic cleavage sites (instead of one, as in all other paramyxovirus) in the F0 protein precursor [26]. The presence of a double cleavage site in F has been found to influence membrane fusion activation by a still poorly understood G independent mechanism [28].
Class II Viral Fusion Proteins
In contrast to class I fusion proteins, the so-called class II fusion proteins (Table 16.1) are derived from a polyprotein precursor that is cleaved during biosynthesis to generate the E1 protein of alphaviruses (e.g., Semliki Forest virus (SFV)) or the E protein of flaviviruses (e.g., dengue virus and tick-borne encephalitis virus). Both proteins fold co-translationally with a companion or regulatory protein, termed p62 for alphaviruses and prM for flaviviruses [9].
In alphavirus, the p62-E1 complex is transported to the plasma membrane where they are incorporated into new budding icosahedral virus particles as dimers of p62-E1. p62 is then proteolitically processed (and then named E2) but remains bound to the virus where it covers most of the fusion protein E1 and specially its fusion loop. E2 mediates binding to the cell surface receptor.
In contrast, flavivirus particles bud into the endoplasmic reticulum as immature virions containing prM-E protein complexes. The immature viruses are then transported to the exterior through the exocytic pathway where prM is processed and separated from E [29]. The latter protein is then arranged in E-E homodimers at the virion surface with icosahedral symmetry. The flavivirus fusion E protein is additionally responsible for receptor binding.
The first structure of any class II glycoprotein, solved by X-ray crystallography, was that of the tick-borne encephalitis (TBE) flavivirus E protein ectodomain [30], solubilized from virions by limited trypsin digestion. Similar structures have now been solved for the E ectodomain of dengue virus types 2 and 3 [9]. The polypeptide chain of the E protein follows a complex path, resulting in three globular domains, essentially constituted by β-sheets (Fig. 16.3). The first domain is a β-barrel with up-and-down topology (red). Two adjacent strands in domain I are extended, forming domain II (yellow) which is a long “finger-like” structure that runs parallel to the viral membrane. At the tip of domain II is the hydrophobic fusion loop which remains buried in the virion from the hydrophilic environment by interaction with domain III (blue) of the adjacent monomer in the E-E dimer. Domain I is also connected to domain III which bridges the E ectodomain with the so-called stem region that extends to the TM region of the protein.
Fig. 16.3.
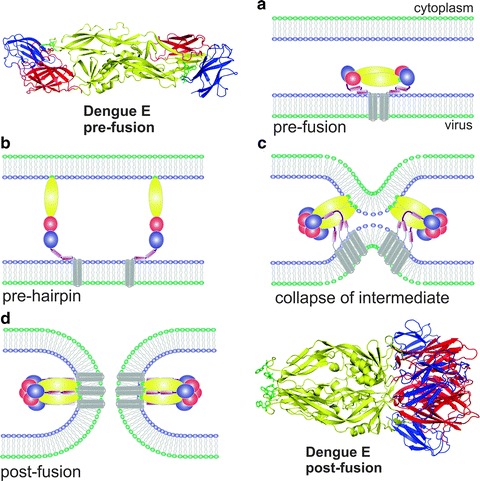
Membrane fusion mediated by a class II fusion protein (Flavivirus). Ribbon representation of the atomic structures of the dengue virus E protein dimer in the pre-fusion conformation [54] (upper left) and the E protein trimer in the post-fusion conformation [55] (lower right). Domains I, II and III of the E glycoprotein are colored red, yellow and blue, respectively. (a–d) Diagram of the fusion process denoting: (a) the structure of the flavivirus E glycoprotein dimer already in the endosome before activation, (b) dissociation of the E protein subunits, refolding of the fusion domain (yellow) and insertion of the fusion loop (green) into the endosomal membrane, (c) formation and refolding of the E protein trimer to approach the two membranes, and (d) formation of the fusion pore and stabilization of the E trimer in the post-fusion conformation. In the last two steps, two E protein molecules are represented to indicate the cooperation needed to drive the fusion process
Unlike the class I fusion proteins, which are trimeric in their pre- and post-fusion conformations, class II fusion glycoproteins undergo major oligomeric transformations during fusion. As in the case of influenza virus, the flavivirus E protein first binds to a cell surface receptor which induces endocytosis of the virion. Once in the acidic endosome, the E-E homodimer dissociates, resulting in disassembly of the icosahedral scaffold. The individual subunits swing outward by the hinge region that connects domains I and II, and the fusion loops insert into the target membrane. Lateral interactions between monomers facilitates reclustering into trimers [31]. These rearrangements lead to the formation of an extended trimeric structure, analogous to the pre-hairpin intermediate of class I fusion glycoproteins, in which two different regions of each E polypeptide are inserted into the two membranes to be fused. Collapse of the extended intermediate can proceed by rotation of domain III in each subunit about the segment that links it to domain I and zipping up of the stem alongside the clustered domains II. This refolding brings the two membranes together to initiate formation of the lipid stalk, the hemifusion diaphragm and the fusion pore.
The structure of the fusion E1 glycoprotein of alphaviruses (SFV) was found unexpectedly very similar to that of the flavivirus E protein, despite the lack of detectable sequence conservation. E1 has also three discernible domains, equivalent to those of flavivirus E. The only significant difference is the association of E1 with E2 in the virus particle. E2 interacts with the cell surface receptor to initiate the endocytic internalization of the SFV virion [32]. In the acidic endosome, E2 separates from E1 and it is probably degraded. Upon low pH exposure, E1 undergoes similar conformational changes to those of the flavivirus E protein, leading to fusion of the viral and endosomal membranes. Electron microscopy and X-ray crystallography results provide support for interactions between adjacent E1 trimers when the fusion loops are inserted in the target membrane to produce rings of five or six trimers. It has been postulated that these fivefold interactions would act at the fusion site to induce the formation of a nipple-like curvature in the viral and target membranes, favoring membrane fusion [33]. Although there is no direct evidence, it is likely that the flavivirus E protein forms similar rings of trimers during fusion.
Class III Viral Fusion Proteins
The best characterized members of the so-called class III fusion viral glycoproteins are the rhabdovirus (e.g., vesicular stomatitis virus, VSV) G glycoprotein, the herpesvirus gB glycoprotein and the baculovirus gp64 glycoprotein. The pre-fusion [34] and post-fusion [35] structures of the VSV_G glycoprotein ectodomain have been solved by X-ray crystallography while only the post-fusion conformations of gB [36] and gp64 [37] are known.
Class III fusion glycoproteins are expressed from individual mRNAs and do not require proteolytic processing of either a protein precursor (as in class I proteins) or an accompanying protein (as in class II proteins) for activity. Class III proteins are trimeric before and after fusion and share structural characteristics with both class I and class II fusion glycoproteins, as described below.
The rhabdovirus G protein possesses both receptor binding and fusion promoting activities. As in the case of influenza virus, binding of rhabdovirus G to a poorly characterized receptor at the cell surface induces endocytosis of the virus particle. Acidification of the endosome triggers G for membrane fusion. However, and in contrast with all other fusion proteins, the low pH inactivation of rhabdovirus G is reversible. Thus, virions inactivated by prolonged incubation at pH <6 can be reactivated by raising the pH to neutral [38]. This reversibility may be required to allow G to be transported through the acidic Golgi apparatus and to recover its native fusion-competent state when incorporated to new virions [39]. Given this reversibility, it is believed that the energy released during the structural transition of a single trimer from the pre-fusion to the post-fusion conformation is probably small, compared with the energetic barrier of the fusion reaction. In agreement with this hypothesis, the estimated number of rhabdovirus spikes required for fusion is higher (at least 15 trimers) than for other enveloped viruses.
A soluble ectodomain of the VSV_G glycoprotein, released from purified virions treated with thermolysin, was used to solve the structures of the pre-fusion and post-fusion conformations, after exposure to high and low pH, respectively [34, 35]. Several domains could be observed in both structures that are rearranged in their relative orientations during transit from the pre- to the post-fusion structure (Fig. 16.4). In the pre-fusion conformation, the fusion domain contains two fusion loops reminiscent of class II proteins that are oriented downward towards the viral membrane. After low pH exposure, the fusion domain moves upward by flipping relative to the central core of the trimer. Thus, an intermediate equivalent to the pre-hairpin structure of class I proteins is formed. This is followed by the reversal of the molecule around a central rigid block formed by lengthening of the central helix and refolding of the three C-terminal segments into helices that position themselves in the grooves of the central core in an anti-parallel manner. This six-helix bundle has obvious resemblance with that of the class I proteins.
Fig. 16.4.
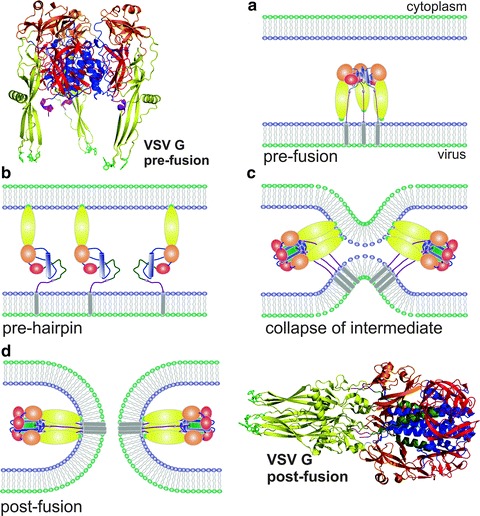
Membrane fusion mediated by a class III fusion protein (Vesicular Stomatitis Virus, VSV). Ribbon representations of the VSV glycoprotein (G), in the pre- (upper left) and post-fusion (lower right) conformations. Domains are colored similarly in all images. The fusion domain is colored in yellow and the fusion loops in green. (a–d) Diagram of the fusion process denoting: (a) the structure of the VSV G glycoprotein trimer already in the endosome before activation, (b) dissociation of the G protein subunits, refolding of the fusion domain (yellow) and insertion of the fusion loop (green) into the endosomal membrane, (c) formation and refolding of the G protein trimer to approach the two membranes, and (d) formation of the fusion pore and stabilization of the G primer in the post-fusion conformation. In the last two steps, two G protein molecules are represented to indicate the cooperation needed to drive the fusion process
It is likely that the transition of VSV_G from the pre-fusion to the post-fusion conformation involves disassembly of the trimer into monomers and reassembly into trimers upon interaction of the fusion loops with the target membrane [40]. It is also likely that cooperativity between G glycoproteins is needed to overcome the energy barrier, as mentioned above. As for class II glycoproteins, lattices of G proteins have been observed in virions, particularly in the planar base of the rhabdovirus bullet-shape particle, which may act to induce nipple-like deformations in the viral and target membranes.
Although it has not been reported, it is likely that low pH exposure also leads to reversible inactivation of the baculovirus gp64 glycoprotein. This protein, like VSV_G, is involved in both receptor binding and membrane fusion after endosome acidification, since baculoviruses also use an endocytic route of entry [41]. In contrast, membrane fusion mediated by herpesvirus gB can occur either at the plasma membrane or in endosomes, depending on the virus and the target cell type. In either case, attachment of herpesvirus to host cells follows a complex mechanism in which several viral glycoproteins interact with cell surface molecules. Some of these interactions trigger fusion, whereas others simply serve to tether the virus to the cell and are dispensable for fusion. In any case, the gB protein, shared by all viruses of the Herpesviridae family, is responsible for fusion [42].
Other Viral Fusion Proteins
Poxviruses (vaccinia virus is the best known member) represent an extreme case among enveloped viruses, regarding the number of viral glycoproteins required for entry. As for herpes virus, entry can occur by fusion at the plasma membrane or in a low pH-dependent manner from within an intracellular particle, depending on the virus strain and the cell type. Vaccinia virus internalization is believed to occur by macropinocytosis (a type of non-specific endocytosis). Four vaccinia virus proteins are involved in attachment to cell surface proteoglycans or laminin [43]. Eleven or 12 other relatively small glycoproteins, ranging in size between 35 and 377 amino acids, form the so-called entry fusion complex (EFC) that mediates membrane fusion [44]. These proteins have N- or C-terminal transmembrane domains but no sequence similarity with the fusion peptide of other viral fusion glycoproteins has been found in any of them. Therefore, the actual mechanism of vaccinia virus membrane fusion remains to be elucidated but it seems to be different from that of other enveloped viruses. By using conditional lethal mutants of each of the 11 proteins that make the EFC, it was found that eight of them were required to reach the hemifusion step and the other three were needed for completion of virus entry [44]. It is likely that hydrophobic regions of several proteins may assemble in the EFC to form a hydrophobic surface that could bind to the target membrane and drive membrane fusion by some novel mechanism.
Finally, the fusion-associated small transmembrane (FAST) proteins of reoviruses are brought here -despite not being involved in virus entry and reovirus being a non-enveloped virus- because they induce cell-cell fusion and therefore facilitates dissemination of virus to neighboring cells. The FAST proteins are small non structural proteins (98–148 amino acids, depending on the viral strain) that are expressed on the surfaces of virus-infected cells, where they induce cell-cell fusion and syncytia (multinucleate cells) formation. Purified FAST proteins, when reconstituted into liposome membranes, induce fusion indicating that they are bona fide fusogens [45]. The orientation of the FAST polypeptides in the cell membrane is also unique among viral fusogens, with a relatively short N-terminal ectodomain followed by a transmembrane region and a long C-terminal cytoplasmic tail. Although they lack a fusion peptide, a relatively hydrophobic region near the N-terminus which is additionally myristoylated seems to insert into the target membrane to drive membrane fusion, at least for certain FAST proteins [46] .
Early Post-Entry Events
Once membrane fusion has been completed, the viral genome -generally in complex with other proteins or inside a viral nucleocapsid (see Chaps. 10.1007/978-94-007-6552-8_2 and 10.1007/978-94-007-6552-8_11)- is found for a second time in a cytoplasmic environment. The first time is when the genome assembles in the cytosol of the infected cell or when it is trafficking from the nucleus to the cell exterior, depending on the virus. However, the fate of the incoming genome is now very different and characteristic for each virus.
Most RNA viruses replicate in the cell cytoplasm, although there are exceptions like influenza virus or borna virus that do so in the nucleus. If the RNA is of positive polarity, like in flavivirus, the genome may act as mRNA to be translated by the cell protein synthesis machinery. In most cases the primary translational product is a polyprotein that matures into the different viral gene products by proteolytic processing [47]. In the case of negative-stranded RNA viruses, like paramyxoviruses, the first step after entry is the transcription of the viral genome to yield the different mRNAs that are translated into the distinct viral gene products [48].
For RNA viruses that replicate in the nucleus, the nucleoprotein complex of the viral genome and associated proteins has to be transported to the cell nucleus for transcription. Most of the viral proteins required in the nucleus have their own nuclear localization signal (NLS), which is a cluster of basic amino acids. However, actual import of the viral ribonucleoprotein into the cell nucleus may require additional interactions with certain host factors. For instance, the NLS of the influenza nucleoprotein interacts with karyopherin α and this in turn with karyopherin β which mediates interactions with the proteins of the nuclear pore to promote nuclear import of the viral ribonucleoproteins by an energy-dependent process [49].
Retroviruses represent a special case in which a RNA viral genome (diploid), packed in a capsid inside the virus envelope has to be transcribed to DNA before integration into the cell host genome. In this case, fusion of the virus and cell membrane delivers the capsid into the cell cytoplasm, where it interacts with cytoskeleton and other cell components for transport to the vicinity of the nucleus where reverse transcription and uncoating takes place. Then, the resulting pre-integration complexes are transported through the nuclear pore inside the nucleus for integration into the host genome [50].
Most DNA viruses replicate in the nucleus, with exceptions like poxviruses (e.g., vaccinia virus). In the case of herpesviruses, the capsids that contain the viral genome are transported from the site of release to the nuclear pore where importin β promotes nuclear import of the viral DNA by an energy-dependent mechanism [51]. In contrast, the virus core of vaccinia virus is released into the cytoplasm. The virus core has all the machinery for transcription of the early genes that ensues further progress on DNA replication and transcription of the remaining genome [52].
Perspectives and Conclusions
As noted, viral-cell membrane fusion is the last extracellular step of enveloped viruses and therefore still amenable to inhibition by chemical or biological products without the drawback of the membrane permeability barrier. Hence, it is not surprising that viral fusogens have received recently very much attention as ideal targets for the development of effective antivirals, some of them already in clinical use (e.g., the T20 peptide for HIV or the Synagis antibody for respiratory syncytial virus) (see also 10.1007/978-94-007-6552-8_20). The development of high throughput technologies for screening of large libraries of chemical or biological molecules (e.g., antibodies) should provide in the near future a plethora of drugs to fight some of the important diseases caused by enveloped virus.
A critical step in the process of viral induced membrane fusion is the activation of the viral fusogen. While most viruses that enter the cell through low pH endocytosis rely on protonation of certain residues (mostly histidines) of the viral fusogen to trigger fusion, the activating step in the case of other viruses is still ill-defined. Better understanding of fusion triggering may therefore bring new possibilities for the manipulation of virus entry. Finally, determination of the structures of certain fusion intermediates could provide additional targets for antivirals.
Learning how virus-cell membrane fusion proceeds may also be relevant for other spheres of Biology. For instance, regulation of vesicle fusion could benefit from knowledge acquired in the field of Virology and find applications in studies of synaptic transmission and Neurobiology.
Acknowledgements
Current research activities in the Biología Viral laboratory are funded by grants GR09/0039 from Instituto de Salud Carlos III and SAF2009-11632 from Plan Nacional de I + D + i.
Contributor Information
Mauricio G. Mateu, Email: mgarcia@cbm.uam.es
José A. Melero, Email: jmelero@isciii.es
References and Further Reading
- 1.Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nat Struct Mol Biol. 2008;15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Huang HW. Observation of a membrane fusion intermediate structure. Science. 2002;297:1877–1879. doi: 10.1126/science.1074354. [DOI] [PubMed] [Google Scholar]
- 3.Chanturiya A, Chernomordik LV, Zimmerberg J. Flickering fusion pores comparable with initial exocytotic pores occur in protein-free phospholipid bilayers. Proc Natl Acad Sci U S A. 1997;94:14423–14428. doi: 10.1073/pnas.94.26.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 2008;105:15382–15387. doi: 10.1073/pnas.0807771105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmerberg J, Blumenthal R, Sarkar DP, Curran M, Morris SJ. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Kurteva S, Ren X, Lee S, Sodroski J. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J Virol. 2006;80:4388–4395. doi: 10.1128/JVI.80.9.4388-4395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magnus C, Rusert P, Bonhoeffer S, Trkola A, Regoes RR. Estimating the stoichiometry of human immunodeficiency virus entry. J Virol. 2009;83:1523–1531. doi: 10.1128/JVI.01764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci U S A. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010;6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 15.Chernomordik LV, Kozlov MM. Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem. 2003;72:175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- 16.Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J Biol Chem. 1997;272:28036–28041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- 17.Rickman C, Hu K, Carroll J, Davletov B. Self-assembly of SNARE fusion proteins into star-shaped oligomers. Biochem J. 2005;388:75–79. doi: 10.1042/BJ20041818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 20.Mohler WA, Shemer G, del Campo JJ, Valansi C, Opoku-Serebuoh E, Scranton V, Assaf N, White JG, Podbilewicz B. The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev Cell. 2002;2:355–362. doi: 10.1016/S1534-5807(02)00129-6. [DOI] [PubMed] [Google Scholar]
- 21.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 22.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connolly SA, Leser GP, Jardetzky TS, Lamb RA. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J Virol. 2009;83:10857–10868. doi: 10.1128/JVI.01191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Reyes L, Ruiz-Arguello MB, Garcia-Barreno B, Calder L, Lopez JA, Albar JP, Skehel JJ, Wiley DC, Melero JA. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc Natl Acad Sci U S A. 2001;98:9859–9864. doi: 10.1073/pnas.151098198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. Recombinant human Metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J Virol. 2004;78:12877–12887. doi: 10.1128/JVI.78.23.12877-12887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rawling J, Cano O, Garcin D, Kolakofsky D, Melero JA. Recombinant sendai viruses expressing fusion proteins with two furin cleavage sites mimic the syncytial and receptor-independent infection properties of respiratory syncytial virus. J Virol. 2011;85:2771–2780. doi: 10.1128/JVI.02065-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830–1834. doi: 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- 30.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 31.Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, Rey FA. The fusion glycoprotein shell of Semliki forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/S0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 33.Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. Conformational change and protein-protein interactions of the fusion protein of Semliki forest virus. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 34.Roche S, Rey FA, Gaudin Y, Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 35.Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 36.Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A. 2009;106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol. 2008;15:1024–1030. doi: 10.1038/nsmb.1484. [DOI] [PubMed] [Google Scholar]
- 38.Roche S, Gaudin Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology. 2002;297:128–135. doi: 10.1006/viro.2002.1429. [DOI] [PubMed] [Google Scholar]
- 39.Albertini AA, Baquero E, Ferlin A, Gaudin Y. Molecular and cellular aspects of rhabdovirus entry. Viruses. 2012;4:117–139. doi: 10.3390/v4010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albertini AA, Merigoux C, Libersou S, Madiona K, Bressanelli S, Roche S, Lepault J, Melki R, Vachette P, Gaudin Y. Characterization of monomeric intermediates during VSV glycoprotein structural transition. PLoS Pathog. 2012;8:e1002556. doi: 10.1371/journal.ppat.1002556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hefferon KL, Oomens AG, Monsma SA, Finnerty CM, Blissard GW. Host cell receptor binding by baculovirus GP64 and kinetics of virion entry. Virology. 1999;258:455–468. doi: 10.1006/viro.1999.9758. [DOI] [PubMed] [Google Scholar]
- 42.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho Y, Hsiao JC, Yang MH, Chung CS, Peng YC, Lin TH, Chang W, Tzou DL. The oligomeric structure of vaccinia viral envelope protein A27L is essential for binding to heparin and heparan sulfates on cell surfaces: a structural and functional approach using site-specific mutagenesis. J Mol Biol. 2005;349:1060–1071. doi: 10.1016/j.jmb.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 44.Laliberte JP, Weisberg AS, Moss B. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog. 2011;7:e1002446. doi: 10.1371/journal.ppat.1002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Top D, de Antueno R, Salsman J, Corcoran J, Mader J, Hoskin D, Touhami A, Jericho MH, Duncan R. Liposome reconstitution of a minimal protein-mediated membrane fusion machine. EMBO J. 2005;24:2980–2988. doi: 10.1038/sj.emboj.7600767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Top D, Read JA, Dawe SJ, Syvitski RT, Duncan R. Cell-cell membrane fusion induced by p15 fusion-associated small transmembrane (FAST) protein requires a novel fusion peptide motif containing a myristoylated polyproline type II helix. J Biol Chem. 2012;287:3403–3414. doi: 10.1074/jbc.M111.305268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenbach BD, Thiel H-J, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Philadelphia: Lippincott, Williams and Wilkins; 2012. [Google Scholar]
- 48.Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. 5. Philadelphia: Lippincott, Williams and Wilkins; 2012. [Google Scholar]
- 49.Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem. 2003;278:28193–28200. doi: 10.1074/jbc.M303571200. [DOI] [PubMed] [Google Scholar]
- 50.Arhel N. Revisiting HIV-1 uncoating. Retrovirology. 2010;7:96. doi: 10.1186/1742-4690-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields virology. 5. Philadelphia: Lippincott, Williams and Wilkins; 2012. [Google Scholar]
- 52.Broyles SS. Vaccinia virus transcription. J Gen Virol. 2003;84:2293–2303. doi: 10.1099/vir.0.18942-0. [DOI] [PubMed] [Google Scholar]
- 53.McLellan JS, Yang Y, Graham BS, Kwong PD. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol. 2011;85:7788–7796. doi: 10.1128/JVI.00555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
Further Reading
- Palfreyman MT, Jorgensen EM (2009) In vivo analysis of membrane fusion. In: Encyclopedia of Life Sciences (ELS). John Wiley & Sons, Chichester. doi:10.1002/9780470015902.a0020891
- Grove J, Marsh M. The cell biology of receptor-mediated virus entry. J Cell Biol. 2011;7:1071–1082. doi: 10.1083/jcb.201108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A. Virus entry and uncoating. In: Knipe DM, Howley PM, editors. Fields virology. 5. Philadelphia: Lippincott, Williams and Wilkins; 2007. [Google Scholar]
- Also especially recommended for further reading are references [4, 9, 15, 42, 44] listed above