

Abstract
The prominent role for the drug efflux pump ABCB1 (P-glycoprotein) in mediating resistance to chemotherapy was first suggested in 1976 and sparked an incredible drive to restore the efficacy of anticancer drugs. Achieving this goal seemed inevitable in 1982 when a series of calcium channel blockers were demonstrated to restore the efficacy of chemotherapy agents. A large number of other compounds have since been demonstrated to restore chemotherapeutic sensitivity in cancer cells or tissues. Where do we stand almost three decades since the first reports of ABCB1 inhibition? Unfortunately, in the aftermath of extensive fundamental and clinical research efforts the situation remains gloomy. Only a small handful of compounds have reached late stage clinical trials and none are in routine clinical usage to circumvent chemoresistance. Why has the translation process been so ineffective? One factor is the multifactorial nature of drug resistance inherent to cancer tissues; ABCB1 is not the sole factor. However, expression of ABCB1 remains a significant negative prognostic indicator and is closely associated with poor response to chemotherapy in many cancer types. The main difficulties with restoration of sensitivity to chemotherapy reside with poor properties of the ABCB1 inhibitors: (1) low selectivity to ABCB1, (2) poor potency to inhibit ABCB1, (3) inherent toxicity and/or (4) adverse pharmacokinetic interactions with anticancer drugs. Despite these difficulties, there is a clear requirement for effective inhibitors and to date the strategies for generating such compounds have involved serendipity or simple chemical syntheses. This chapter outlines more sophisticated approaches making use of bioinformatics, combinatorial chemistry and structure informed drug design. Generating a new arsenal of potent and selective ABCB1 inhibitors offers the promise of restoring the efficacy of a key weapon in cancer treatment – chemotherapy.
Key words: Multidrug resistance,; ABC drug efflux pump,; Combinatorial chemistry,; Drug design,; Pharmacophore modeling,; Homology modeling; High resolution structure; Pharmacokinetic interactions
Introduction
Despite its widespread use and applicability in treating all stages of cancer, i.e. from front-line therapies to palliation, the efficacy of chemotherapy remains suboptimal. One of the main reasons for the underwhelming success of chemotherapy is the resistant phenotype, which may be inherent to cancerous tissue or arise following drug administration (1–6). Resistance arises by virtue of the adaptability of cancer cells to a variable local environment (7–11). One of the types of adaptation is a network of often synergistic pathways that negate the efficacy of anticancer drugs (12, 13). These resistance pathways can range from individual factors through to the 3D tissue organization and impact on drug efficacy by alterations to drug distribution within solid tumors, affecting cellular uptake, increasing intracellular metabolism/excretion, specific mutations within target molecules, evasion of repair mechanisms, and dampening of pathways aimed at initiating cell death. Resistance mechanisms can be initiated by host factors such as high cell density, hypoxia, or stress response pathways.
One of the most widespread mechanisms of resistance is the expression of efflux pumps such as ABCB1 (P-glycoprotein), ABCC1 (MRP1) and ABCG2 (BCRP) (14–16). Expression of these proteins at the plasma membrane reduces intracellular drug concentration and is therefore a first line of cellular defense. The mechanism of resistance is a generic one owing to the ability of efflux pumps to transport an extraordinary number and range of chemicals (17–19). For example, the multidrug efflux pump ABCB1 is known to interact with over 200 compounds. The broad multispecificity of these transporters is a hallmark of their origin as environmental xenobiotic protection pathways. ABCB1 is normally expressed in a number of healthy tissues, particularly those involved in secretory roles (e.g. liver and GI tract) or in a barrier capacity (e.g. blood–brain and blood–testes) (20–23). Expression of ABCB1 in these tissues is regulated by endogenous transcription factors such as the nuclear orphan receptor family (24). However, cancers arising from these tissues frequently display inherent resistance, which is present prior to chemotherapy exposure. Consequently, overexpression of ABCB1 in cancer cells following exposure to chemotherapy agents is thought to be achieved by virtue of stress response pathways rather than a classical induction process (24).
Overexpression of ABCB1 has been demonstrated to generate a resistant phenotype in cultured cancer cell lines and various tumor models (13, 17). In addition, expression of ABCB1 has been cataloged in a large number of human cancer types including several leukemia types and solid tumors from the breast, colon, and adrenal tissues (25–28). A link between expression and a resistant phenotype is well established in acute myelogenous (AML), myelodysplastic syndrome (MDS), and acute lymphoblastic (ALL) leukemias (29, 30). However, the role of ABCB1, and other multidrug efflux pumps, in conferring resistance in many solid tumors continues to be vigorously debated (4, 15, 31, 32). The inability to unequivocally quantify the role of ABCB1 in clinical drug resistance has arisen for a number of reasons including: (1) variability in the methods used to detect ABCB1 expression, (2) the presence of other, often synergistic resistance pathways, (3) the poor inhibition of its activity in vivo, (4) poor study design, and (5) variability of ABCB1 expression patterns in tumors. A comprehensive understanding of resistance pathways in the clinical setting is therefore required to confidently assign the relative contributions of specific resistance factors.
Despite the controversy surrounding the contribution of ABCB1 to resistance in cancer, there is an apparent need to modulate its behavior. Inhibition of the efflux protein leads to increased drug accumulation in cultured cancer cells and improved intratumor distribution in animal models and patients (33–37). Consequently, the development of potent inhibitors of ABCB1 could prove highly beneficial in chemotherapeutic management of cancer. In addition, the ability of ABCB1 to influence drug pharmacokinetics in a nononcology setting renders it a target for specific modulation to regulate drug absorption, distribution, and elimination.
Inhibition of ABCB1 was first reported in 1982 using the calcium channel blocker verapamil and this strategy was rapidly progressed to clinical trials (38–40). Unfortunately, the use of verapamil was doomed owing to its poor potency to inhibit ABCB1, whereas its effects on calcium channels (particularly in cardiac tissue) occurred at low plasma concentrations. Similar effects were reported with a number of other ABCB1 “inhibitors” that were already in clinical usage for various unrelated settings (41–44). These compounds inhibited ABCB1 primarily by acting as substrates that could compete for transport by the protein. Unfortunately, drugs belonging to this class of ABCB1 inhibitor were united in displaying poor potency of action, which directly resulted in unacceptable levels of systemic toxicity (45–47). Subsequent generations of ABCB1 inhibitors (Fig. 18.1) have been explored using chemical modification of the first generation agents, combinatorial chemistry to identify new chemical moieties, and, more recently, the use of natural products to uncover novel lead compounds (48–56).
Fig. 18.1.
Inhibitors of ABCB1. A flow-chart outlining the four generations of inhibitors against the multidrug efflux pump ABCB1 that have been developed over the last 30 years.
Despite these significant efforts, only a small selection of compounds have progressed through to late stage clinical trials; of particular note are Tariquidar (XR9576) (36, 57–59) and the nonimmunosuppresive cyclosporin A derivative, Valspodar (PSC833) (60–62). The various generations of ABCB1 inhibitors have failed to deliver a method of clinical intervention to restore sensitivity of chemotherapy for a number of reasons:
Poor selectivity leading to unwanted actions.
Low affinity for ABCB1 requiring high plasma concentrations, thereby producing toxic side-effects.
Interaction of drugs with other ABC transporters – for example, the perturbation of bile formation or toxicity to stem cells.
Interaction with cytochrome P450, thereby producing elevated systemic concentrations of anticancer drug, which necessitates dose reduction.
Inability to modulate ABCB1 function in vivo.
Decreased elimination of anticancer drugs because of ABC transporter inhibitions in “physiological sites”.
The failure of clinical trials thus far has engendered a degree of pessimism regarding the ability to inhibit ABCB1 effectively in vivo, and some skepticism regarding its role in drug resistance. This does, however, seem rather premature given the small number of compounds that have been subjected to exhaustive characterization. In addition, the power of combinatorial chemistry has not been fully exploited and the newest generation of compounds (from natural sources) not yet fully characterized, although there have been suggestions that inhibitor development is redundant because of the recent emergence of novel nongenotoxic anticancer agents that are directed against specific cellular targets rather than the more generic proliferation process. However, the majority of these compounds are cytostatic and many of these novel cytostatic anticancer drugs are themselves subjected to efflux by transporters such as ABCB1 (25, 63, 64). Consequently, the role of genotoxic drugs in cancer treatment in the near future should not be dismissed. Thus the problem of ABCB1 mediated transport appears to be a phenomenon that must be dealt with and the desirability of a clinically useable and efficacious inhibitor remains high in oncological circles.
Providing a greater knowledge of the nature of drug–ABCB1 interaction remains an important goal for future anticancer drug development. Understanding pharmacophoric elements of ABCB1 substrates and elucidating the molecular interactions with protein structural elements in the drug binding site will prove useful to ensure new compounds can evade the influence of this protein. The focus of this review is twofold. First we shall compile the data available on defining the pharmacophoric elements of ABCB1 substrates and strategies to improve the number of compounds available for testing. The second half of the review will focus on compiling data about the molecular properties of the drug binding sites of ABCB1 and exploring the possibilities for using structural data to inform inhibitor design.
Drug Design for Inhibition of ABCB1
Understanding the factors that determine substrate specificity is crucial for successful drug targeting and in the rationale for the design of novel inhibitors. High resolution structural data coupled with the large volume of functional biochemical data on ABCB1 would serve as the ideal template for understanding drug–protein interaction with a view to create a design of novel inhibitors. However, the refractory nature of membrane proteins to atomic structure resolution studies has meant that high resolution data for ABCB1 has not yet been obtained. In its absence, two distinct lines of investigation have been employed to explore ABCB1–drug interactions. The first has extrapolated a molecular model of ABCB1 enabling docking studies to characterize the drug–protein interactions. This approach has been facilitated by high resolution structural templates, such as the bacterial ABC transporter Sav1866 (65), which have identical topology and a high level of sequence or structural homology. The homology modeling approach of protein-structure based drug design will be addressed in Subheading 18.3 of this chapter. The second line of investigation is a protein-structure independent method that instead exploits knowledge of the different substrates, their physicochemical parameters or affinities for the protein to generate a model of drug interaction.
What Defines Substrate Recognition?
The first explanation for substrate recognition was proposed by Emil Fischer in 1894 who provided a structural rationale for the interaction between an enzyme and its substrate (66). The elegantly simple Lock-and-Key model postulated that the enzyme contained a rigid binding pocket that interacted with a specific ligand and allowed subsequent release of the enzymatic product. Although this model has been refined over the intervening decades, classic Lock-and-Key type ligand interactions cannot account for the broad multispecificity exhibited by tranporters such as ABCB1. Currently, there are three general models that address multispecific ligand interactions. The first, proposes that the binding pocket, although still an essentially rigid region, contains different interaction sites that allow a range of structurally distinct ligands to bind (67). The second model, based on Koshland’s Induced-Fit model (68), proposes that conformational flexibility within the protein allows the binding pocket to reconfigure and accommodate structurally diverse ligands. The third and most recent model is based on the the observation that a single ligand may bind to a protein in multiple and different orientations (69). The Differential Ligand Positioning model proposes that a single ligand might be able to interact with a number of spatially distinct regions of the binding site thus allowing multiple ligand molecules to bind simulatenously.
Although the precise mechanism(s) employed by ABCB1 to interact with ligands has not been elucidated the last three decades of biochemistry have provided significant insight. The multipilicity of ABCB1 drug ligand binding sites was first shown by Tamai and Safa (70) and, to date, there are at least four known distinct drug binding sites (71–77). Biochemical studies have established that the drug binding sites show a range of different behaviors with noncompetitive inhibition for certain substrates, indicative of overlapping substrate specificities; competitive inhibition for other drug ligands, such as vinblastine and doxorubicin; and cooperative allostery between certain substrates, e.g. ATP, vinblastine and verapamil (73, 78–81). In addition there appear to be multiple binding sites on individual transmembrane segments that have the ability to simultaneously bind distinct drugs or multiple molecules of the same drug (71, 82–84). As a consequence, it is likely that the TM segments that contribute to the drug-binding pocket have a high degree of conformational mobility to allow drug molecules to form the required binding sites and to allow for different orientations of drug molecules within the binding pocket. Thus, it is likely that ABCB1 employs a combination of the general multispecific interaction models described above, thereby maximizing the range of ligands with which it can interact. However, this mobility and flexibility in substrate binding creates unique challenges for inhibitor design.
What Governs ABCB1–Drug Ligand Interactions?
Since ABCB1 was first discovered by Juliano and Ling (85), many studies have sought to clarify the basic functional and structural features of ligands that govern interaction. Identification of the basic chemical feature responsible for mediating ligand–protein interaction is key to developing a framework for interpretative and prognostic evaluations of new compounds. Structure–activity relationships (SAR) have exploited three decades of pharmacological studies on ABCB1 in an attempt to correlate substrate activity with specific molecular descriptors. However, interpreting the large volume of data collected on ABCB1 is highly complex due to a number of intrinsic issues. These stem from largely technical issues including the use of different assays, parameters reported (e.g. IC50, KD, KM & KI), drug solubilities, and variable drug partition coefficients. Despite this, some general features of ABCB1 substrates that were first noted remain relevant in that they tend to be lipophilic and amphiphilic, have a large molecular volume, contain electronegative and hydrogen bonding groups, and occasionally a weakly cationic group. More detailed molecular descriptors have since been revealed by a number of different approaches (for detailed reviews see (86–88)).
Prior to the introduction of automated and semiautomated computational pharmacophoric and 3D quantitative structure activity relationships (3D-QSAR), modeling techniques SARs were determined by correlation of substrate activities with molecular descriptors. Zamora and coworkers provided one of the first SAR studies and described the requirement of a basic nitrogen atom and two planar aromatic domains based on investigations using verapamil, indole alkaloids, lysosomotrophic agents and amines (89). This feature set was further probed by Pearce and and coworkers in 1989 using a series of reserpine and yohimbine analogs that demonstrated that these domains also adopted well-defined conformations (90). However, the requirement of the basic nitrogen atom was called into question by a number of studies that used a broader array of ligands and showed that compounds, such as steroid hormones, could also interact with ABCB1 (91–93).
In 1997 Bain and coworkers examined 44 compounds, mostly pesticides, and proposed that substrates and inhibitors could be differentiated on the basis of the number of rings, molecular weight, and hydrogen bonding potential (94). They suggested that transported substrates displayed higher molecular weight and hydrogen bonding potential than nontransported substrates. In addition, the transported substrates acted primarily as hydrogen-donors rather than acceptors. A study by Seeling examined the structure of a hundred chemically diverse compounds and sought to more clearly define the number of electron donor groups and their fixed spatial distance (95). Seelig’s analysis proposed a general pattern for ABCB1 substrate recognition comprising two or three electron-donor (or hydrogen-bonding acceptor) groups with a fixed spatial separation of 2.5 ± 0.3 Å (as a type-I pattern) or 4.6 ± 0.6 Å (as a type-II pattern), respectively. Ecker and coworkers (96) subsequently followed Seelig’s work and suggested a correlation between the total electron donating strength of a ligand and its potency as an inhibitor.
Ultimately, although SAR data has provided valuable insight into the molecular descriptors of known substrates and inhibitors, it has not provided a platform for the a priori development of novel ligands. SAR studies are constrained by the chemical data upon which they are constructed and, as a consequence, have a limited application for directing ligand screening beyond existing ABCB1 SAR chemical space. This is an issue of critical importance for a multispecific transporter such as ABCB1 and has driven the development of computational tools for applying substrate structure to new inhibitor design.
From Substrates to Templates – How Can We Design New Inhibitors?
Substrate based inhibitor design exploits the “learnt” rules for ligand–protein interactions and applies them in inhibitor selection and design. But what are the rules for ABCB1, which has defied a simple classification for ligand recognition elements and demonstrated a breadth of acceptable substrate types? It contains several distinct binding sites and may interact with a broad range of compounds without strict structural constraints. Various clinically used compounds were investigated for their ability to inhibit ABCB1 in vivo and a number of potential modulators were identified. Early attempts with these compounds to block ABCB1 in cultured cell lines and in vitro assays were highly successful and led to the first phase I clinical trials in 1985 (38). However, this and many subsequent trials with first generation ABCB1 inhibitors were plagued by failure in restoring anticancer drug efficacy. The clinical failure of these inhibitors led to the first SAR studies and provided the first insight into the molecular features crucial for interaction with ABCB1. Zamora and coworkers (89) provided the first SAR derived descriptors, however, these were not sufficiently stringent to be applied to drug development. Although they had failed clinically, the first generation ABCB1 inhibitors were effective in vitro, and thus they were used as the templates for the second generation of inhibitors designed through quantitative structure relationships (QSAR) studies.
QSAR in Inhibitor Design
QSAR is based on the pharmacological principle that drug structure does not necessarily correlate with biological activity (97). QSAR studies examine a range of related compounds for their quantitative effects on a specific target (i.e. degree of agonism or antagonism). 3D-QSAR modeling determines a mathematical model that describes drug potency as a function of the three dimensional interactions with protein based on an aligned training set of compounds. The relationship between the change in the 3D spatial interaction fields and experimentally determined variations in the target feature is calculated by statistical analysis. A number of 3D-QSAR approaches are available and include comparative molecular field analysis (CoMFA) (98), comparative molecular similarity index analysis (CoMSIA) (99), and GOLPE (100) (for detailed discussion and reviews of these techniques see (101, 102)). Quantitative models such as 3D-QSAR can be applied to de novo computational screening to lead the synthesis of higher potency lead compounds. In combination with in vitro testing and analysis, for refinement of the quantitative model, this technique has been used in the design of improved inhibitors and higher affinity ligands (103). Activity predictions by 3D-QSAR models require that the ligands be accurately aligned and, consequently, this limits their application in automated chemical compound database screening. Although 2D-QSAR models can be used for database screening, they lack highly useful 3D information crucial for subsequent drug design.
QSAR studies led to modified versions of several first generation lead compounds including indoles such as reserpine (104) and 1,4 dihydropyridines (53), phenothiazine derivatives such as transflupentixal (49), a nonimmunosuppresive cyclosporin A derivative PSC833 (56), and the verapmil derived, triazine-based S9788 (105). Detailed in vitro assays provided information on the affinity of interaction with the drug and ABCB1 compared to first generation compounds and was used to further refine and optimize the design of second generation ABCB1 inhibitors.
Phase I and II clinical trials were undertaken with the most promising second generation ABCB1 inhibitors. However, unfavorable pharmacokinetic interactions led to elevated drug plasma levels and reduced systemic clearance of anticancer drugs, producing significant toxicity in patients (61, 106–108). This necessitated a reduction in the administered dose of chemotherapeutic drugs, which in turn reduced the overall efficacy of anticancer drug treatment. Concomitant inhibition of ABCB1 and cytochrome P450-3A isoform (CYP3A), which is responsible for the metabolism of almost 50% of all clinically employed drugs by the second generation inhibitors, resulted in higher and prolonged plasma levels of anticancer drugs because of impaired metabolism and elimination. It was subsequently determined that ABCB1 and CYP3A have a considerable overlap in substrate specificities and this highlighted one of the limitations of drug design methodologies, namely, the inability to predict undesirable pharmacokinetic interactions (109, 110). Consequently, although 3D-QSAR strategies led to an improvement of first generation compounds, the second generation compounds ultimately failed to provide an ideal route to ABCB1 inhibition without compromising anticancer drug efficacy. Thus, it was necessary to approach inhibitor development from a broader chemical space approach such as pharmacophore modeling.
What Is Pharmacophore Modeling?
The pharmacophore concept was first introduced by Paul Erhlich in the early 1900s (111). The pharmacophore is a description of the molecular framework which contains the essential features responsible for a drug’s biological activity. With the benefit of nearly a century’s additional knowledge, the underlying concept has been expanded to include our understanding of three dimensional substrate structures and the arrangement of their essential molecular features. In current terminology, the pharmacophore is a representation of the spatial arrangement of structural features required for biological activity. The determination of a pharmacophore requires knowledge of (1) the three dimensional structure and bioactive conformation of molecules, (2) key atomic features, and (3) the determination of the relationship between those features and biological activity. Once developed, pharmacophoric models can be highly valuable tools to provide insight into drug molecule interactions and aid in the design of higher potency inhibitors.
Seelig’s SAR data (95) was suggestive of a fairly simple pharmacophoric distinction between substrates and inhibitors of ABCB1, whereas the spatial requirements would be indicative of discrete binding sites. SAR data can be analyzed and interpreted for small numbers of compounds (<500), but as datasets grow in size and complexity computational approaches are better suited to generating pharmacophore models. There are a range of programs that are widely used for pharmacophore generation including ALADDIN, COMPASS, SCAMPI, PARM, and DANTE (112); however, the most commonly used are DISCO (113), GASP (114), and Catalyst/HIPHOP (115). These software packages utilize different algorithms to determine a common set of molecular features on the basis of comparisons of interacting compounds (substrates or inhibitors). A consequence of this is that most of the pharmacophore models generated are based on the alignment of a small number (i.e. the training set) of energy minimized conformations of known substrates. However, this means that the dynamic nature of biologically active substrates cannot be fully predicted. One program, Catalyst/HypoGen, employs a combination of QSAR and pharmacophore methods (116). This requires a wide range of interacting and noninteracting compounds combined with experimentally determined activity data, which provides a more robust pharmacophore. The Catalyst/HypoGen pharmacophore is capable of predicting the potential capacity for a query compound to interact, as in a traditional pharmacophore model. However, it can also estimate its potential activity based on a regression of the training dataset, as with the 3D-QSAR model. Many pharmacophore models reported in current literature claim to be reasonably accurate at predicting ABCB1 substrates. Ekins and coworkers generated pharmacophore-QSAR models to rank ABCB1 inhibitors on the basis of modulating substrate transport (117). A single substrate pharmacophore was produced by overlaying verapamil and digoxin based structures, followed by fitting vinblastine, and the generated pharmacophore revealed multiple hydrophobic and hydrogen-bond acceptor features as important characteristics of ABCB1 substrates. An ensemble model of 100 pharmacophores was generated by Penzotti and coworkers and consisted of a set of 2, 3, and 4-point pharmacophores for discrimination between interacting and noninteracting ABCB1 compounds with potential ABCB1 ligands required to match at least 20% of the pharmacophores in the ensemble (118). Screening of ligands, also referred to as virtual screening, is a data mining approach that applies the pharmacophoric model to screen commercial chemical compound databases to identify molecules that can potentially interact with the protein. Potential compounds can then be purchased and directly tested using in vitro assays. Consequently, this approach has become a frequently used strategy for the identification of novel lead ligands. Several ABCB1 pharmacophores have been used in screening databases. Rebizter and coworkers used a propafenone based pharmacophore model to screen the Derwent World Drug Index (119). This identified 19 new potential ABCB1 substrates but the study did not report subsequent experimental verification (120). More recently, a pharmacophore model generated from 131 propafenone ABCB1 inhibitors was used to screen the SPECS database (134,000 compounds) and successfully identified two lead compounds with submicromolar range affinities (121). Despite these promising leads, none of these compounds have yet made the transition from the laboratory to the clinic.
Limitations of In Silico Approaches to Inhibitor Design
Regardless of the type of model used for drug/inhibitor design, the predictive and interpretative qualities are ultimately constrained by the dataset upon which they were constructed. Acquiring robust datasets is especially important in these studies, where a variety of expression systems and experimental models are available. The majority of QSAR and pharmacophore studies on ABCB1 have focused on datasets gathered from a single species or cell type, and frequently from a single laboratory (122–125). The promiscuity of transport exhibited by ABCB1 means that there are relatively few noninteracting compounds included in the development of pharmacophoric datasets. As a consequence, this has meant that the models generated have facilitated understanding of ABCB1–ligand interactions; they have not been highly effective in prospective ligand discoveries.
Despite this, the information gained from these in silico studies have improved the consensus picture for ABCB1 inhibitor design, although it still remains somewhat broad. Strong inhibitors are characterized by high lipophilicity (and/or molar refractivity) and possess at least two H-bond acceptors. Other features, such as H-bond donors and π–π-stacking, are also proposed to serve as additional interaction features. Pharmacophoric models indicate that there are also steric constraints for interaction (120, 126–130).
Combinatorial Chemistry
Combinatorial chemistry was responsible for the a priori development of the third generation of ABCB1 inhibitors generated with the objective of improving potency without unwanted pharmacokinetic interactions. Four promising lead compounds (Elacridar (50), Zosuquidar (131), Tariquidar (54), and Ontogen (52)) were developed by high throughput screening approaches using SAR analyzes. Their nanomolar potency and efficacy in experimental systems (in vitro and in vivo) led to a rapid progression to clinical trials. Tariquidar garnered the greatest interest because of its high potency (100–1,000-fold greater than the previous generation inhibitors), its discrimination between ABCB1 and ABCC1, and its long effective duration (35, 132, 133). However, despite its success in phase I and II clinical trials, phase III trials were suspended due to unfavorable toxicity reports in the treatments of lung carcinoma and the future of this inhibitor is currently unclear. Clinical trials for other third generation ABCB1 inhibitors are proceeding and although the initial reports are promising, with minimal adverse pharmacokinetic interactions reported, these trials are still at relatively early stages with small patient sample sizes and no unambiguous reports on improvements in anticancer drug efficacy (134–137). Although the pharmaceutical armory is small, there remains a paucity of extensive inhibitor characterization in the clinical setting. More attention should be devoted to trials with large patient populations and a broad range of cancer types, with detailed information on the class of resistance and greater use of surrogate assays.
Nucleotide Binding Domain Targeted Inhibition
Drug binding sites within the transmembrane domain of ABCB1 have been the main target of substrate based inhibitor design. However the inherent plasticity of these sites has rendered it difficult to identify compounds that can modulate drug efflux by this route. However, targeting the drug binding site need not be the only strategy to attain pharmacological inhibition of ABCB1.
The high conservation of the nucleotide binding domains (NBDs) amongst ABC transporters and their fundamental requirement to provide the mechanistic driving force for efflux indicates that they are ideal targets for the inhibition of ABCB1. This is underpinned by a wealth of biochemical and structural data which has meant that our understanding of the catalytic cycle of the NBDs is well understood (for review see (138–140)). For example, the distinct and specific motifs contained within the NBDs are amenable to in silico ligand design techniques. In addition, the presence of two NBDs per transporter also increases the number of potential sites for inhibitor binding, whereas inactivation of only a single NBD would be required to impair ABCB1 mediated drug transport.
Several classes of drug, such as the flavonoids, have been observed to interact with the NBDs (141–143). Flavonoids are a large class of naturally occurring compounds widely present in the green plant world with more than 6,500 different compounds described (143, 144). Some naturally occurring flavonoids and their hydrophobic derivatives (e.g. aurones) have been observed to inhibit the transport function of ABCB1 by interaction with NBD 2 and the cytosolic regions of the protein. It has been shown that although some flavonoids can inhibit the labeling of ABCB1 with their photoactive substrates (145–147), indicating that they may bind directly to the substrate binding site, others bind directly to the purified recombinant C-terminal nucleotide-binding domain from mouse ABCB1 (NBD2). Moreover, it appears that the binding domain may overlap the ATP binding site and vicinal steroid binding site (142). However, flavonoids are also potent inhibitors of drug metabolizing enzymes (148, 149) and pharmacokinetic interactions with anticancer drugs are likely to prevent a clinical application in their current form.
ABCB1 Inhibitors – Where to Now?
Systematic chemical modification and combinatorial chemistry produced the first three generations of potent ABCB1 inhibitors. Unfortunately, the majority of these inhibitors have also been reported to have caused undesirable pharmacokinetic anticancer drug interactions thus limiting their clinical application. Rapid technological advancements have made automated and semiautomated in silico approaches, such as pharmacophore and QSAR modelling, feasible for screening vast compound databases and developing higher potency inhibitors. However, despite the identification of a number of potential lead compounds these approaches have not yet led to the production of compounds for clinical trials. Because of these obstacles to ABCB1 inhibition some recent studies have returned to screening herbal and fruit extracts for lead compounds. These approaches are reminiscent of the first generation inhibitor screening methods of broadly sampling the existing chemical space in an attempt to identify lead compounds. A more rational approach may be the utilization of protein structure modelling exploiting high resolution homologous template structures. By developing models of protein interaction with potential ligands, in combination with complementary physicochemical (QSAR) and pharmacophoric descriptors this may offer a different platform for the design of potential lead inhibitors.
Structure Informed Drug Design
Has Structure Informed Drug Design Been Employed Successfully?
High resolution crystal structures have been used for rational drug design as they can provide detailed molecular information on interactions between the substrate and protein at the active site. This method was used to develop the well known antiinfluenza drugs Relenza and Tamiflu as well as the anticancer drug Imatinib. All three compounds function by targeting the active site of a critical protein involved in illness progression and are the success stories of structure-informed drug design.
The antiinfluenza drugs Relenza and Tamiflu target the enzyme neuraminidase, which is responsible for viral release from sites of infection such as the lungs (150). The enzyme cleaves sialic acid residues on a surface receptor involved in anchoring newly formed influenza viral particles, thereby facilitating virus release from infected cells. Several high resolution crystal structures of neuraminidase have been solved, but of particular significance were those structures containing bound sialic acid substrate (151–153). This provided critical insight into specific residues that interact with sialic acid at the enzyme active site. A combination of computational chemistry and examination of the crystal structures of neuraminidase revealed that the C-4 hydroxyl group of sialic acid provided a significant contact point between the substrate and the protein (154). Substitution of the C-4 hydroxyl group with a larger, basic guanidinyl group gave rise to 4-deoxy-4-guanidino-Neu5Ac2en (commonly referred to as Zanamivir or Relenza), which displayed antiviral activity (155). Moreover, structural (156) and molecular modelling (157) data of the complex demonstrated that Zanamivir bound directly at the active site of neuraminidase. Subsequent clinical trials demonstrated that Zanamivir is clinically effective for the treatment and prevention of influenza (158, 159).
The development and success of Zanamivir provided a platform from which to produce other neuraminidase-targeted antiinfluenza drugs. Further structure–activity relationship studies with Neu5Ac-based derivatives led to a number of improvements to the compound (160, 161). These included mimicking the sialic acid transition state, optimizing the hydrophobic nature of the drug thereby increasing membrane association, and, finally, the development of a prodrug form, converted to an active form in vivo. The final outcome was the antiinfluenza drug Oseltamivir or Tamiflu.
Structure-based drug design has therefore been successfully applied to therapy against influenza by virtue of the high resolution crystal structures of neuraminidase solved in complex with the substrate and the inhibitor. Furthermore, identification of neuraminidase as a target for structure-informed drug design was possible owing to a thorough understanding of both the viral lifecycle and the mechanism of substrate recognition by the protein.
Can the Approach Be Used for ABCB1?
The success of structure-based drug design relies upon having (1) high resolution protein structures (2) detailed information on the substrate binding site(s), and (3) an understanding of the substrate–protein interactions. These three requirements are essential for the successful development of inhibitors and our progress towards these goals for ABCB1 will be discussed.
Structural Information on ABCB1
A myriad of challenges face structural biologists when attempting to crystallize membrane proteins. These include protein expression, efficient protein extraction from the lipid bilayer, high purity, and sample homogeneity. Such issues are responsible for the lack of crystal structures for any full-length eukaryotic ABC tranporters. In the interim however, electron microscopy (EM) and homology modelling have been used to provide low to medium resolution structural information of ABCB1.
Electron microscopy of ABCB1 in the presence and absence of nucleotides revealed that the protein undergoes a significant reorganization in the transmembrane domains (162–167). This was interpreted as corresponding to the opening of a central pore (167), thought to allow hydrophobic drug access to the extracellular environment during the drug translocation process of ABCB1. Currently the highest resolution structure of ABCB1, determined using 2D crystals and cryo-electron microscopy, is approximately 8 Å, which is too low for structure informed drug design (164–167). However, the use of EM to monitor multiple conformational states has been of great benefit in understanding the dynamic aspects of the drug translocation process and this structural information still plays a crucial role in the validation of homology models.
Homology Models of ABCB1
To date EM has provided the only direct structural information on ABCB1. In constrast several high resolution crystal structures of prokaryotic ABC transporters have been solved in recent years (65, 168–172). These structures in conjuction with the parameters obtained from EM studies have been avidly used to produce homology models of ABCB1 (173–177). Homology models provide an invaluable starting point for the interpretation of biochemical data in the absence of high resolution structures (130, 173). The quality and accuracy of a model is entirely dependent on the resolution of the template crystal structure and the template-sample sequence alignment. As a consequence, protein homology models are viewed as approximations of protein structures, comparable to medium resolution images and not suitable for use in structure-informed drug design. Furthermore, the reliability of ABCB1 homology models was recently confounded by the withdrawal of three MsbA structures. In addition, the only available structure for an ABC multidrug efflux pump, namely Sav1866, revealed an unexpected domain organization (65). This called for the refinement and reinterpretation of all previous ABCB1 models (65, 177–179). Despite this, the quality of subsequent models has been improved owing to the higher resolution of the Sav1866 structure (∼3 Å) and better sequence alignment with ABCB1 (56% and 52.8% for transmembrane domains 1 and 2 of Sav1866 and ABCB1, respectively) (175). Furthermore, cross-linking data suggest that the unusual “domain swapping” architecture of Sav1866 is also adopted by ABCB1 (180). The crystal structure of Sav1866, therefore, appears to provide a more reliable template for the computational modelling of ABCB1.
Two groups have currently produced homology models of ABCB1 using the Sav1866 crystal structure as a template (173, 175). Both homology models were qualitatively compared to the ABCB1 EM model and were shown to be in reasonable correspondence (165). O’Mara and coworkers suggested that in the ATP bound state, the translocation pore is lined with polar residues. In contrast, the nucleotide free configuration has hydrophilic residues shifted to the interhelical regions with the hydrophobic residues being exposed to the translocation pore (175). This considerable molecular rearrangement is in agreement with substantial biochemical data demonstrating dramatic conformational changes in the TMDs caused by ATP binding (for review (18)).
A large hydrophobic cavity within the transmembrane region of the protein was observed in the homology model created by Globisch and coworkers (173). Their study incorporated data from numerous investigations on cross-linking within ABCB1 TM segments to validate the model. According to the cross-linking data the transition from the “open to inside” to “closed to inside” conformation is thought to occur during the early stages of the ATP hydrolytic cycle. In the latter state the main protein cavity, as determined by SiteID and SiteFinder programmes, has mainly hydrophobic to neutral surface properties.
It is tempting to postulate that based on both models ABCB1 assumes a conformation corresponding to an intermediate stage in the translocation process (driven by events at the NBDs) wherein the central pore develops a low/intermediate affinity for hydrophobic drugs. Such conformational changes in ABCB1 could generate the changes in substrate affinity during drug translocation (80). However, the specific conformational transitions have yet to be shown experimentally. Therefore, whereas homology models have thus far been unsuitable for structure-based inhibitor design, they have provided a means for interpreting conformational changes within the translocation pore of the protein. Such information can help in the quest to understand how ABCB1 can recognize and transport its vast array of substrates.
Substrate and Modulator Binding Sites: Biochemical Approaches
Although the homology models reveal topology and conformational shifts during translocation, they do not reveal molecular detail or location of the drug binding sites. Understanding the interaction between the protein and its substrate is crucial for structure-informed inhibitor design. The drug binding sites lie within the TM region of ABCB1 and multiple residues within the TM helices appear to contribute to the drug binding site(s) (181–183). This section outlines our current understanding of the underlying mechanism for drug recognition by ABCB1.
Location and Number of Drug Binding Sites
Locating the drug binding site(s) in ABCB1 is complicated by its promiscuity and the complexity of the drug–protein interactions. For example, (1) how many sites are there, (2) are they located at distinct regions or within a single large domain, and (3) can drugs bind to more than one site? One technique that has proven invaluable in the quest to uncover the drug binding sites is cysteine-scanning mutagenesis. This technique requires a cysteine-less protein template, and fortuitously, ABCB1 is fully functional in the absence of cysteine (184). The substitution of residues at specific positions in ABCB1 with cysteine was used to determine the residues that are critical for protein function and drug binding. Using an array of ABCB1 single-cysteine mutants and thiol-reactive substrates, (e.g. MTS-verapamil and dibromobimane), Loo and Clarke outlined a potential drug binding domain within ABCB1 (83, 185–190). The investigations suggested that TM helices 4–6 of TMD1 and 9–12 of TMD2 contributed residues to a drug binding pocket in ABCB1 (Fig. 18.2). This was in agreement with earlier findings, using photoaffinity labeling, which indicated that the N- and C-terminal ends of the TMDs are involved in drug binding (82, 181, 183). In addition, certain residues (e.g. residue Ser222) are within the binding site of more than one unrelated substrate, suggesting a degree of redundancy between regions of interaction. These combined studies provided the field with valuable information about the drug binding regions and the substrate-induced conformational changes. However, the highly reactive nature of the thiol-labeling compounds used, the complexities of the substrate–ABCB1 interaction, and the conformational flexibility of the protein make it difficult to distinguish between binding to the “true” sites from binding at intermediate stages of the translocation pathway.
Fig. 18.2.
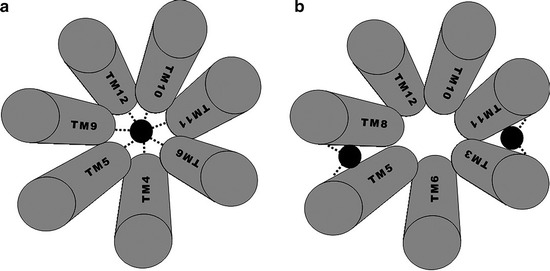
Potential drug binding sites in ABCB1. A simplified view of the transmembrane helices that are suggested to contribute to the drug binding site(s) of ABCB1. The black circle represents the drug substrate. (a) Represents the helices demonstrated by Loo et al. to form the drug binding site. (b) Illustrates the helices demonstrated by Ecker et al. to be involved in drug binding. The dashed lines demonstrate the interface at which the drug is believed to interact.
An alternative method used to identify the drug binding sites of ABCB1 involved propafenone-analogs and matrix-assisted laser desorption-ionization-time-of-flight (MALDI-TOF) mass spectrometry. This technique identified residues in transmembrane helices 3, 5, 6, 8, 10, 11 and 12 as contributing to a putative drug-binding domain for propafenone analogs although the primary sites appear to be formed at the interfaces between TM5-8 and TM3-11 (Fig. 18.2) (84, 191). The binding regions, although distinct for these compounds, also encompass residues proposed to be involved in the binding of other ligands such as vinblastine, cyclosporin, verapamil, and colchicine (192).
Predictive methods have also been used to locate the substrate binding regions in ABCB1. Globisch and coworkers used 3D structural information along with SiteID and Site Finder programs in an attempt to pinpoint binding regions and pockets (173). The programs located three binding regions and a central binding cavity. A number of residues in the vicinity of the predicted binding regions have previously been identified to contribute to the binding domain of propafenone and its chemical derivatives. Unfortunately, the binding sites identified are large, dispersed regions rather than distinct “sites” within ABCB1, thereby precluding any realistic attempts at drug-docking.
The substrate-induced fit model, in which substrate binding induces unique conformational changes in the flexible TMDs resulting in the formation of a unique drug binding site for that substrate, has been used to rationalize the large number of residues involved (189). Furthermore, an alternative substrate would induce a different conformational change creating a binding pocket specific for that substrate but within the same binding region. The model was supported by the fact that the TMs are quite flexible at 37 °C (from cross-linking data) and it was suggested that substrate binding reduces the flexibility of the TMs, therefore ensuring localisation of specific residues to the binding pocket(s) (193, 194). A challenge for the research field will be to verify, or refute, this intriguing potential mechanism.
Modulator Sites – Are They Distinct from the Drug Binding Site(s)?
The previous experiments suggested that there is one generic site in ABCB1 that accommodates many different compounds. However, a series of experiments, carried out by Dey and coworkers, demonstrated that the ABCB1 inhibitor cis-(Z)-flupentixol binds to a site distinct from the substrate binding domain, thereby preventing translocation and promoting substrate dissociation (195). Furthermore, conformational changes generated by flupentixol binding to ABCB1, as demonstrated by altered susceptibility to proteolytic digestion and UIC2 antibody binding, are distinct from that induced by ABCB1 substrates or competitive modulators (195–197). cis-(Z)-flupentixol does not interact with ABCB1 substrates in a classical competitive manner. Given that it binds at a distinct site, the interaction with the substrates is defined as an allosteric one. Earlier radioligand binding studies also identified a site that bound nontransported modulators of ABCB1 (Nicardipine and GF120918), which was distinct from the [3H]-vinblastine interaction site (73). Similarly, studies on Hoechst33342 transport also identified a potential modulator specific site that recognizes prazosin and progesterone (198). The discovery of a potentially less promiscuous, allosteric modulator site(s) in ABCB1 may provide an alternative and possibly less complicated avenue along which structure-based inhibitor design may proceed.
Conclusion
Currently, there remains a certain degree of pessimism as to whether the activity of ABCB1 can be modulated pharmacologically to restore the efficacy of chemotherapy. The pessimism has intensified following the failure of the much vaunted Tariquidar in clinical trials. However, this was one of the very few molecules that have reached advanced clinical trials. The prominent role of this transporter in healthy tissue (pharmacokinetic regulator) and in disease (e.g. resistance in cancer and epilepsy) surely warrants greater efforts to produce novel inhibitors.
The power of chemistry and bioinformatics appear to have met their match with the promiscuity exhibited by ABCB1 with respect to substrate binding. Identifying the pharmacophoric elements of substrates may prove untenable if the drug binding domain has a seemingly limitless plasticity or malleability. Generating detailed mechanistic information and protein elements of the binding domain are a priority for future rational inhibitor design.
Clearly, we cannot yet pinpoint the site of the drug or modulator binding to ABCB1 with any surety. Similarly, the precise molecular mechanism by which the protein can recognize such a wide array of compounds remains elusive. Biochemical data has brought us to the cusp of this “holy grail” of information. Provision of a high resolution structure containing a bound substrate/modulator would provide an enormous stimulus to reveal the hidden secrets of drug binding to ABCB1 and facilitate structure-informed inhibitor design.
Contributor Information
Jun Zhou, Phone: 86222350-4946, FAX: 96222350-8800, Email: junzhou@nankai.edu.cn.
Richard Callaghan, Email: richard.callaghan@ndcls.ox.ac.uk.
References
- 1.Boyer J, Allen WL, McLean EG, et al. Pharmacogenomic identification of novel determinants of response to chemotherapy in colon cancer. Cancer Res. 2006;66:2765–2777. doi: 10.1158/0008-5472.CAN-05-2693. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham L, Aplenc R. Pharmacogenetics of acute lymphoblastic leukemia treatment response. Expert Opin Pharmacother. 2007;8:2519–2531. doi: 10.1517/14656566.8.15.2519. [DOI] [PubMed] [Google Scholar]
- 3.Ferraldeschi R, Baka S, Jyoti B, et al. Modern management of small-cell lung cancer. Drugs. 2007;67:2135–2152. doi: 10.2165/00003495-200767150-00003. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 5.Longley DB, Allen WL, Johnston PG. Drug resistance, predictive markers and pharmacogenomics in colorectal cancer. Biochim Biophys Acta. 2006;1766:184–196. doi: 10.1016/j.bbcan.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Surowiak P. Prediction of the response to chemotherapy in ovarian cancers. Folia Morphol (Warsz) 2006;65:285–294. [PubMed] [Google Scholar]
- 7.Calzada MJ, del Peso L. Hypoxia-inducible factors and cancer. Clin Transl Oncol. 2007;9:278–289. doi: 10.1007/s12094-007-0055-y. [DOI] [PubMed] [Google Scholar]
- 8.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 9.De Luca A, Carotenuto A, Rachiglio A, et al. The role of the EGFR signaling in tumor microenvironment. J Cell Physiol. 2008;214:559–567. doi: 10.1002/jcp.21260. [DOI] [PubMed] [Google Scholar]
- 10.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 12.Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14(Suppl 1):35–48. doi: 10.1159/000086183. [DOI] [PubMed] [Google Scholar]
- 13.Mellor HR, Callaghan R. Resistance to chemotherapy in cancer: a complex and integrated cellular response. Pharmacology. 2008;81:275–300. doi: 10.1159/000115967. [DOI] [PubMed] [Google Scholar]
- 14.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 15.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 16.Modok S, Mellor HR, Callaghan R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr Opin Pharmacol. 2006;6:350–354. doi: 10.1016/j.coph.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Ambudkar SV, Dey S, Hrycyna CA, et al. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 18.Callaghan R, Ford RC, Kerr ID. The translocation mechanism of P-glycoprotein. FEBS Lett. 2006;580:1056–1063. doi: 10.1016/j.febslet.2005.11.083. [DOI] [PubMed] [Google Scholar]
- 19.Pauwels EK, Erba P, Mariani G, Gomes CM. Multidrug resistance in cancer: its mechanism and its modulation. Drug News Perspect. 2007;20:371–377. doi: 10.1358/dnp.2007.20.6.1141496. [DOI] [PubMed] [Google Scholar]
- 20.Cordon-Cardo C, O'Brien JP, Boccia J, et al. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38:1277–1287. doi: 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- 21.Cordon-Cardo C, O'Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci USA. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fromm MF. Importance of P-glycoprotein for drug disposition in humans. Eur J Clin Invest. 2003;33(Suppl 2):6–9. doi: 10.1046/j.1365-2362.33.s2.4.x. [DOI] [PubMed] [Google Scholar]
- 23.van der Valk P, van Kalken CK, Ketelaars H, et al. Distribution of multi-drug resistance-associated P-glycoprotein in normal and neoplastic human tissues. Analysis with 3 monoclonal antibodies recognizing different epitopes of the P-glycoprotein molecule. Ann Oncol. 1990;1:56–64. [PubMed] [Google Scholar]
- 24.Callaghan R, Crowley E, Potter S, Kerr ID. P-glycoprotein: so many ways to turn it on. J Clin Pharmacol. 2008;48:365–378. doi: 10.1177/0091270007311568. [DOI] [PubMed] [Google Scholar]
- 25.Burger H, van Tol H, Boersma AW, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 26.Chan HS, Grogan TM, Haddad G, DeBoer G, Ling V. P-glycoprotein expression: critical determinant in the response to osteosarcoma chemotherapy. J Natl Cancer Inst. 1997;89:1706–1715. doi: 10.1093/jnci/89.22.1706. [DOI] [PubMed] [Google Scholar]
- 27.Chan HS, Haddad G, Thorner PS, et al. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N Engl J Med. 1991;325:1608–1614. doi: 10.1056/NEJM199112053252304. [DOI] [PubMed] [Google Scholar]
- 28.van den Heuvel-Eibrink MM, Sonneveld P, Pieters R. The prognostic significance of membrane transport-associated multidrug resistance (MDR) proteins in leukemia. Int J Clin Pharmacol Ther. 2000;38:94–110. doi: 10.5414/cpp38094. [DOI] [PubMed] [Google Scholar]
- 29.Pallis M, Russell N. Strategies for overcoming p-glycoprotein-mediated drug resistance in acute myeloblastic leukaemia. Leukemia. 2004;18:1927–1930. doi: 10.1038/sj.leu.2403511. [DOI] [PubMed] [Google Scholar]
- 30.van der Holt B, Lowenberg B, Burnett AK, et al. The value of the MDR1 reversal agent PSC-833 in addition to daunorubicin and cytarabine in the treatment of elderly patients with previously untreated acute myeloid leukemia (AML), in relation to MDR1 status at diagnosis. Blood. 2005;106:2646–2654. doi: 10.1182/blood-2005-04-1395. [DOI] [PubMed] [Google Scholar]
- 31.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 32.Merino V, Jimenez-Torres NV, Merino-Sanjuan M. Relevance of multidrug resistance proteins on the clinical efficacy of cancer therapy. Curr Drug Deliv. 2004;1:203–212. doi: 10.2174/1567201043334650. [DOI] [PubMed] [Google Scholar]
- 33.Ak I, Aslan V, Vardareli E, Gulbas Z. Assessment of the P-glycoprotein expression by 99mTc-MIBI bone marrow imaging in patients with untreated leukaemia. Nucl Med Commun. 2003;24:397–402. doi: 10.1097/00006231-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Jekerle V, Wang JH, Scollard DA, et al. 99mTc-Sestamibi, a sensitive probe for in vivo imaging of P-glycoprotein inhibition by modulators and mdr1 antisense oligodeoxynucleotides. Mol Imaging Biol. 2006;8:333–339. doi: 10.1007/s11307-006-0057-0. [DOI] [PubMed] [Google Scholar]
- 35.Martin C, Berridge G, Mistry P, et al. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br J Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pusztai L, Wagner P, Ibrahim N, et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer. 2005;104:682–691. doi: 10.1002/cncr.21227. [DOI] [PubMed] [Google Scholar]
- 37.Walker J, Martin C, Callaghan R. Inhibition of P-glycoprotein function by XR9576 in a solid tumour model can restore anticancer drug efficacy. Eur J Cancer. 2004;40:594–605. doi: 10.1016/j.ejca.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 38.Benson AB, III, Trump DL, Koeller JM, et al. Phase I study of vinblastine and verapamil given by concurrent iv infusion. Cancer Treat Rep. 1985;69:795–799. [PubMed] [Google Scholar]
- 39.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]
- 40.Tsuruo T, Iida H, Yamashiro M, Tsukagoshi S, Sakurai Y. Enhancement of vincristine- and adriamycin-induced cytotoxicity by verapamil in P388 leukemia and its sublines resistant to vincristine and adriamycin. Biochem Pharmacol. 1982;31:3138–3140. doi: 10.1016/0006-2952(82)90097-1. [DOI] [PubMed] [Google Scholar]
- 41.Ganapathi R, Grabowski D. Enhancement of sensitivity to adriamycin in resistant P388 leukemia by the calmodulin inhibitor trifluoperazine. Cancer Res. 1983;43:3696–3699. [PubMed] [Google Scholar]
- 42.Goldberg H, Ling V, Wong PY, Skorecki K. Reduced cyclosporin accumulation in multidrug-resistant cells. Biochem Biophys Res Commun. 1988;152:552–558. doi: 10.1016/S0006-291X(88)80073-1. [DOI] [PubMed] [Google Scholar]
- 43.Ramu A, Fuks Z, Gatt S, Glaubiger D. Reversal of acquired resistance to doxorubicin in P388 murine leukemia cells by perhexiline maleate. Cancer Res. 1984;44:144–148. [PubMed] [Google Scholar]
- 44.Tsuruo T, Iida H, Kitatani Y, et al. Effects of quinidine and related compounds on cytotoxicity and cellular accumulation of vincristine and adriamycin in drug-resistant tumor cells. Cancer Res. 1984;44:4303–4307. [PubMed] [Google Scholar]
- 45.Bartlett NL, Lum BL, Fisher GA, et al. Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. J Clin Oncol. 1994;12:835–842. doi: 10.1200/JCO.1994.12.4.835. [DOI] [PubMed] [Google Scholar]
- 46.Berman E, McBride M, Lin S, Menedez-Botet C, Tong W. Phase I trial of high-dose tamoxifen as a modulator of drug resistance in combination with daunorubicin in patients with relapsed or refractory acute leukemia. Leukemia. 1995;9:1631–1637. [PubMed] [Google Scholar]
- 47.Verweij J, Herweijer H, Oosterom R, et al. A phase II study of epidoxorubicin in colorectal cancer and the use of cyclosporin-A in an attempt to reverse multidrug resistance. Br J Cancer. 1991;64:361–364. doi: 10.1038/bjc.1991.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deferme S, Van Gelder J, Augustijns P. Inhibitory effect of fruit extracts on P-glycoprotein-related efflux carriers: an in-vitro screening. J Pharm Pharmacol. 2002;54:1213–1219. doi: 10.1211/002235702320402053. [DOI] [PubMed] [Google Scholar]
- 49.Ford JM, Prozialeck WC, Hait WN. Structural features determining activity of phenothiazines and related drugs for inhibition of cell growth and reversal of multidrug resistance. Mol Pharmacol. 1989;35:105–115. [PubMed] [Google Scholar]
- 50.Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–4602. [PubMed] [Google Scholar]
- 51.Kane GC, Lipsky JJ. Drug-grapefruit juice interactions. Mayo Clin Proc. 2000;75:933–942. doi: 10.4065/75.9.933. [DOI] [PubMed] [Google Scholar]
- 52.Newman MJ, Rodarte JC, Benbatoul KD, et al. Discovery and characterization of OC144–093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res. 2000;60:2964–2972. [PubMed] [Google Scholar]
- 53.Nogae I, Kohno K, Kikuchi J, et al. Analysis of structural features of dihydropyridine analogs needed to reverse multidrug resistance and to inhibit photoaffinity labeling of P-glycoprotein. Biochem Pharmacol. 1989;38:519–527. doi: 10.1016/0006-2952(89)90393-6. [DOI] [PubMed] [Google Scholar]
- 54.Roe M, Folkes A, Ashworth P, et al. Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg Med Chem Lett. 1999;9:595–600. doi: 10.1016/S0960-894X(99)00030-X. [DOI] [PubMed] [Google Scholar]
- 55.Sadzuka Y, Sugiyama T, Sonobe T. Efficacies of tea components on doxorubicin induced antitumor activity and reversal of multidrug resistance. Toxicol Lett. 2000;114:155–162. doi: 10.1016/S0378-4274(99)00290-8. [DOI] [PubMed] [Google Scholar]
- 56.Twentyman PR, Bleehen NM. Resistance modification by PSC-833, a novel non-immunosuppressive cyclosporin [corrected] Eur J Cancer. 1991;27:1639–1642. doi: 10.1016/0277-5379(91)90435-G. [DOI] [PubMed] [Google Scholar]
- 57.Boniface GR, Ferry D, Atsmon J, et al. XR9576 (tariquidar), a potent and specific P-glycoprotein inhibitor, has minimal effects on the pharmacokinetics of paclitaxel, doxorubicin, and vinorelbine and can be administered with full-dose chemotherapy in patients with cancer. Proc Am Soc Clin Oncol. 2002;39:2173. [Google Scholar]
- 58.Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- 59.Stewart A, Steiner J, Mellows G, et al. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res. 2000;6:4186–4191. [PubMed] [Google Scholar]
- 60.Baer MR, George SL, Dodge RK, et al. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood. 2002;100:1224–1232. [PubMed] [Google Scholar]
- 61.Giaccone G, Linn SC, Welink J, et al. A dose-finding and pharmacokinetic study of reversal of multidrug resistance with SDZ PSC 833 in combination with doxorubicin in patients with solid tumors. Clin Cancer Res. 1997;3:2005–2015. [PubMed] [Google Scholar]
- 62.Gruber A, Bjorkholm M, Brinch L, et al. A phase I/II study of the MDR modulator Valspodar (PSC 833) combined with daunorubicin and cytarabine in patients with relapsed and primary refractory acute myeloid leukemia. Leuk Res. 2003;27:323–328. doi: 10.1016/S0145-2126(02)00181-9. [DOI] [PubMed] [Google Scholar]
- 63.Kamath AV, Chong S, Chang M, Marathe PH. P-glycoprotein plays a role in the oral absorption of BMS-387032, a potent cyclin-dependent kinase 2 inhibitor, in rats. Cancer Chemother Pharmacol. 2005;55:110–116. doi: 10.1007/s00280-004-0873-3. [DOI] [PubMed] [Google Scholar]
- 64.Kitazaki T, Oka M, Nakamura Y, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49:337–343. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 65.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 66.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber Dtsch Chem Ges. 1894;27:2985–2993. doi: 10.1002/cber.18940270364. [DOI] [Google Scholar]
- 67.McFarland BJ, Strong RK. Thermodynamic analysis of degenerate recognition by the NKG2D immunoreceptor: not induced fit but rigid adaptation. Immunity. 2003;19:803–812. doi: 10.1016/S1074-7613(03)00320-0. [DOI] [PubMed] [Google Scholar]
- 68.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sethi DK, Agarwal A, Manivel V, Rao KV, Salunke DM. Differential epitope positioning within the germline antibody paratope enhances promiscuity in the primary immune response. Immunity. 2006;24:429–438. doi: 10.1016/j.immuni.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 70.Tamai I, Safa AR. Azidopine noncompetitively interacts with vinblastine and cyclosporin A binding to P-glycoprotein in multidrug resistant cells. J Biol Chem. 1991;266:16796–16800. [PubMed] [Google Scholar]
- 71.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci USA. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferry DR, Russell MA, Cullen MH. P-glycoprotein possesses a 1, 4-dihydropyridine selective drug acceptor site which is allosterically coupled to a vinca alkaloid selective binding site. Biochem Biophys Res Commun. 1992;188:440–445. doi: 10.1016/0006-291X(92)92404-L. [DOI] [PubMed] [Google Scholar]
- 73.Martin C, Berridge G, Higgins CF, et al. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 74.Orlowski S, Mir LM, Belehradek J, Garrigos M. Effects of steroids and verapamil on P-glycoprotein ATPase activity: progesterone, desoxycorticosterone and verapamil are mutually non-exclusive modulators. Biochem J. 1996;317:515–522. doi: 10.1042/bj3170515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J. 1998;333:351–358. doi: 10.1042/bj3330351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scala S, Akhmed N, Rao US, et al. P-glycoprotein substrates and antagonists cluster into two distinct groups. Mol Pharmacol. 1997;51:1024–1033. doi: 10.1124/mol.51.6.1024. [DOI] [PubMed] [Google Scholar]
- 77.Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 78.Lu L, Leonessa F, Clarke R, Wainer IW. Competitive and allosteric interactions in ligand binding to P-glycoprotein as observed on an immobilized P-glycoprotein liquid chromatographic stationary phase. Mol Pharmacol. 2001;59:62–68. doi: 10.1124/mol.59.1.62. [DOI] [PubMed] [Google Scholar]
- 79.Martin C, Berridge G, Mistry P, et al. Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry. 2000;39:11901–11906. doi: 10.1021/bi000559b. [DOI] [PubMed] [Google Scholar]
- 80.Martin C, Higgins CF, Callaghan R. The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry. 2001;40:15733–15742. doi: 10.1021/bi011211z. [DOI] [PubMed] [Google Scholar]
- 81.Wang EJ, Casciano CN, Clement RP, Johnson WW. Two transport binding sites of P-glycoprotein are unequal yet contingent: initial rate kinetic analysis by ATP hydrolysis demonstrates intersite dependence. Biochim Biophys Acta. 2000;1481:63–74. doi: 10.1016/s0167-4838(00)00125-4. [DOI] [PubMed] [Google Scholar]
- 82.Bruggemann EP, Germann UA, Gottesman MM, Pastan I. Two different regions of P-glycoprotein [corrected] are photoaffinity-labeled by azidopine. J Biol Chem. 1989;264:15483–15488. [PubMed] [Google Scholar]
- 83.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 84.Pleban K, Kopp S, Csaszar E, et al. P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: a combined photoaffinity labeling-protein homology modeling approach. Mol Pharmacol. 2005;67:365–374. doi: 10.1124/mol.104.006973. [DOI] [PubMed] [Google Scholar]
- 85.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 86.Stouch TR, Gudmundsson O. Progress in understanding the structure-activity relationships of P-glycoprotein. Adv Drug Deliv Rev. 2002;54:315–328. doi: 10.1016/S0169-409X(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 87.Raub TJ. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol Pharmacol. 2006;3:3–25. doi: 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- 88.Cramer J, Kopp S, Bates SE, Chiba P, Ecker GF. Multispecificity of drug transporters: probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem. 2007;2:1783–1788. doi: 10.1002/cmdc.200700160. [DOI] [PubMed] [Google Scholar]
- 89.Zamora JM, Pearce HL, Beck WT. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells. Mol Pharmacol. 1988;33:454–462. [PubMed] [Google Scholar]
- 90.Pearce HL, Safa AR, Bach NJ, et al. Essential features of the P-glycoprotein pharmacophore as defined by a series of reserpine analogs that modulate multidrug resistance. Proc Natl Acad Sci USA. 1989;86:5128–5132. doi: 10.1073/pnas.86.13.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ueda K, Okamura N, Hirai M, et al. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem. 1992;267:24248–24252. [PubMed] [Google Scholar]
- 92.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 94.Bain LJ, McLachlan JB, LeBlanc GA. Structure-activity relationships for xenobiotic transport substrates and inhibitory ligands of P-glycoprotein. Environ Health Perspect. 1997;105:812–818. doi: 10.2307/3433698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem. 1998;251:252–261. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- 96.Ecker G, Huber M, Schmid D, Chiba P. The importance of a nitrogen atom in modulators of multidrug resistance. Mol Pharmacol. 1999;56:791–796. [PubMed] [Google Scholar]
- 97.Martin YC, Kofron JL, Traphagen LM. Do structurally similar molecules have similar biological activity? J Med Chem. 2002;45:4350–4358. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 98.Cramer RD, III, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 99.Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 100.Baroni M, Costantino G, Cruciani G, et al. Generating optimal linear PLS estimations (GOLPE): an advanced chemometric tool for handling 3D-QSAR problems. Quant Struct Act Relat. 1993;12:9–20. doi: 10.1002/qsar.19930120103. [DOI] [Google Scholar]
- 101.Van Drie JH. Pharmacophore discovery: a critical review. In: Bultinck P, editor. Computational medicinal chemistry for drug discovery. New York: Marcel Dekker; 2004. pp. 437–460. [Google Scholar]
- 102.Chang C, Swaan PW. Computational approaches to modeling drug transporters. Eur J Pharm Sci. 2006;27:411–424. doi: 10.1016/j.ejps.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 103.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152:21–37. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bhat UG, Winter MA, Pearce HL, Beck WT. A structure-function relationship among reserpine and yohimbine analogues in their ability to increase expression of mdr1 and P-glycoprotein in a human colon carcinoma cell line. Mol Pharmacol. 1995;48:682–689. [PubMed] [Google Scholar]
- 105.Dhainaut A, Regnier G, Atassi G, et al. New triazine derivatives as potent modulators of multidrug resistance. J Med Chem. 1992;35:2481–2496. doi: 10.1021/jm00091a017. [DOI] [PubMed] [Google Scholar]
- 106.Boote DJ, Dennis IF, Twentyman PR, et al. Phase I study of etoposide with SDZ PSC 833 as a modulator of multidrug resistance in patients with cancer. J Clin Oncol. 1996;14:610–618. doi: 10.1200/JCO.1996.14.2.610. [DOI] [PubMed] [Google Scholar]
- 107.Rowinsky EK, Smith L, Wang YM, et al. Phase I and pharmacokinetic study of paclitaxel in combination with biricodar, a novel agent that reverses multidrug resistance conferred by overexpression of both MDR1 and MRP. J Clin Oncol. 1998;16:2964–2976. doi: 10.1200/JCO.1998.16.9.2964. [DOI] [PubMed] [Google Scholar]
- 108.Stupp R, Bauer J, Pagani O, et al. Ventricular arrhythmia and torsade de pointe: dose limiting toxicities of the MDR-modulator S9788 in a phase I trial. Ann Oncol. 1998;9:1233–1242. doi: 10.1023/A:1008495919071. [DOI] [PubMed] [Google Scholar]
- 109.Kim RB, Wandel C, Leake B, et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16:408–414. doi: 10.1023/A:1018877803319. [DOI] [PubMed] [Google Scholar]
- 110.Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995;13:129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- 111.Erhlich P (1909) Current status of Chemotherapy Dtsch Chem Ges 42:17
- 112.van Drie JH. Pharmacophore discovery – lessons learned. Curr Pharm Des. 2003;9:1649–1664. doi: 10.2174/1381612033454568. [DOI] [PubMed] [Google Scholar]
- 113.Martin YC, Bures MG, Danaher EA, et al. A fast new approach to pharmacophore mapping and its application to dopaminergic and benzodiazepine agonists. J Comput Aided Mol Des. 1993;7:83–102. doi: 10.1007/BF00141577. [DOI] [PubMed] [Google Scholar]
- 114.Jones G, Willett P, Glen RC. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput Aided Mol Des. 1995;9:532–549. doi: 10.1007/BF00124324. [DOI] [PubMed] [Google Scholar]
- 115.Clement OO, Mehl AT. HipHop: pharmacophore based on multiple common-feature alignments. In: Guner OF, editor. Pharmacophore Perception, Development, and Use in Drug Design. San Diego: IUL; 2000. pp. 69–84. [Google Scholar]
- 116.Li H, Sutter J, Hoffman R. HypoGen: an automated system for generating 3D predictive pharmacophore models. In: Guner OF, editor. Pharmacophore perception, development, and use in drug design. San Diego: IUL; 2000. pp. 173–189. [Google Scholar]
- 117.Ekins S, Kim RB, Leake BF, et al. Application of three-dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Mol Pharmacol. 2002;61:974–981. doi: 10.1124/mol.61.5.974. [DOI] [PubMed] [Google Scholar]
- 118.Penzotti JE, Lamb ML, Evensen E, Grootenhuis PD. A computational ensemble pharmacophore model for identifying substrates of P-glycoprotein. J Med Chem. 2002;45:1737–1740. doi: 10.1021/jm0255062. [DOI] [PubMed] [Google Scholar]
- 119.Rebitzer S, Annibali D, Kopp S, et al. In silico screening with benzofurane- and benzopyrane-type MDR-modulators. Farmaco. 2003;58:185–191. doi: 10.1016/S0014-827X(03)00021-1. [DOI] [PubMed] [Google Scholar]
- 120.Langer T, Eder M, Hoffmann RD, Chiba P, Ecker GF. Lead identification for modulators of multidrug resistance based on in silico screening with a pharmacophoric feature model. Arch Pharm (Weinheim) 2004;337:317–327. doi: 10.1002/ardp.200300817. [DOI] [PubMed] [Google Scholar]
- 121.Pleban K, Kaiser D, Kopp S, et al. Targeting drug-efflux pumps – a pharmacoinformatic approach. Acta Biochim Pol. 2005;52:737–740. [PubMed] [Google Scholar]
- 122.Bednarczyk D, Ekins S, Wikel JH, Wright SH. Influence of molecular structure on substrate binding to the human organic cation transporter, hOCT1. Mol Pharmacol. 2003;63:489–498. doi: 10.1124/mol.63.3.489. [DOI] [PubMed] [Google Scholar]
- 123.Chang C, Swaan PW, Ngo LY, et al. Molecular requirements of the human nucleoside transporters hCNT1, hCNT2, and hENT1. Mol Pharmacol. 2004;65:558–570. doi: 10.1124/mol.65.3.558. [DOI] [PubMed] [Google Scholar]
- 124.Geldenhuys WJ, Malan SF, Murugesan T, Van der Schyf CJ, Bloomquist JR. Synthesis and biological evaluation of pentacyclo[5.4.0.0(2, 6).0(3, 10).0(5, 9)]undecane derivatives as potential therapeutic agents in Parkinson's disease. Bioorg Med Chem. 2004;12:1799–1806. doi: 10.1016/j.bmc.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 125.Yates CR, Chang C, Kearbey JD, et al. Structural determinants of P-glycoprotein-mediated transport of glucocorticoids. Pharm Res. 2003;20:1794–1803. doi: 10.1023/B:PHAM.0000003377.39548.f6. [DOI] [PubMed] [Google Scholar]
- 126.Pajeva IK, Wiese M. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: explanation of structural variety (hypothesis) J Med Chem. 2002;45:5671–5686. doi: 10.1021/jm020941h. [DOI] [PubMed] [Google Scholar]
- 127.Garrigues A, Loiseau N, Delaforge M, et al. Characterization of two pharmacophores on the multidrug transporter P-glycoprotein. Mol Pharmacol. 2002;62:1288–1298. doi: 10.1124/mol.62.6.1288. [DOI] [PubMed] [Google Scholar]
- 128.Ekins S, Crumb WJ, Sarazan RD, Wikel JH, Wrighton SA. Three-dimensional quantitative structure-activity relationship for inhibition of human ether-a-go-go-related gene potassium channel. J Pharmacol Exp Ther. 2002;301:427–434. doi: 10.1124/jpet.301.2.427. [DOI] [PubMed] [Google Scholar]
- 129.Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos. 2006;34:1976–1984. doi: 10.1124/dmd.106.012351. [DOI] [PubMed] [Google Scholar]
- 130.Ecker GF, Stockner T, Chiba P. Computational models for prediction of interactions with ABC-transporters. Drug Discov Today. 2008;13:311–317. doi: 10.1016/j.drudis.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 131.Slate DL, Bruno NA, Casey SM, et al. RS-33295-198: a novel, potent modulator of P-glycoprotein-mediated multidrug resistance. Anticancer Res. 1995;15:811–814. [PubMed] [Google Scholar]
- 132.Kohler S, Stein WD. Optimizing chemotherapy by measuring reversal of P-glycoprotein activity in plasma membrane vesicles. Biotechnol Bioeng. 2003;81:507–517. doi: 10.1002/bit.10488. [DOI] [PubMed] [Google Scholar]
- 133.Mistry P, Stewart AJ, Dangerfield W, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61:749–758. [PubMed] [Google Scholar]
- 134.van Zuylen L, Sparreboom A, van der Gaast A, et al. The orally administered P-glycoprotein inhibitor R101933 does not alter the plasma pharmacokinetics of docetaxel. Clin Cancer Res. 2000;6:1365–1371. [PubMed] [Google Scholar]
- 135.Fracasso PM, Goldstein LJ, de Alwis DP, et al. Phase I study of docetaxel in combination with the P-glycoprotein inhibitor, zosuquidar, in resistant malignancies. Clin Cancer Res. 2004;10:7220–7228. doi: 10.1158/1078-0432.CCR-04-0452. [DOI] [PubMed] [Google Scholar]
- 136.Le LH, Moore MJ, Siu LL, et al. Phase I study of the multidrug resistance inhibitor zosuquidar administered in combination with vinorelbine in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2005;56:154–160. doi: 10.1007/s00280-004-0942-7. [DOI] [PubMed] [Google Scholar]
- 137.Chi KN, Chia SK, Dixon R, et al. A phase I pharmacokinetic study of the P-glycoprotein inhibitor, ONT-093, in combination with paclitaxel in patients with advanced cancer. Invest New Drugs. 2005;23:311–315. doi: 10.1007/s10637-005-1439-x. [DOI] [PubMed] [Google Scholar]
- 138.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 139.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6:13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 140.Senior AE, Gadsby DC. ATP hydrolysis cycles and mechanism in P-glycoprotein and CFTR. Semin Cancer Biol. 1997;8:143–150. doi: 10.1006/scbi.1997.0065. [DOI] [PubMed] [Google Scholar]
- 141.Boumendjel A, Di Pietro A, Dumontet C, Barron D. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med Res Rev. 2002;22:512–529. doi: 10.1002/med.10015. [DOI] [PubMed] [Google Scholar]
- 142.Conseil G, Baubichon-Cortay H, Dayan G, et al. Flavonoids: a class of modulators with bifunctional interactions at vicinal ATP- and steroid-binding sites on mouse P-glycoprotein. Proc Natl Acad Sci USA. 1998;95:9831–9836. doi: 10.1073/pnas.95.17.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Morris ME, Zhang S. Flavonoid–drug interactions: effects of flavonoids on ABC transporters. Life Sci. 2006;78:2116–2130. doi: 10.1016/j.lfs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 144.Harborne JB, Williams CA. Advances in flavonoid research since 1992. Phytochemistry. 2000;55:481–504. doi: 10.1016/S0031-9422(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 145.Castro AF, Altenberg GA. Inhibition of drug transport by genistein in multidrug-resistant cells expressing P-glycoprotein. Biochem Pharmacol. 1997;53:89–93. doi: 10.1016/S0006-2952(96)00657-0. [DOI] [PubMed] [Google Scholar]
- 146.Jodoin J, Demeule M, Beliveau R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim Biophys Acta. 2002;1542:149–159. doi: 10.1016/S0167-4889(01)00175-6. [DOI] [PubMed] [Google Scholar]
- 147.Zhang S, Morris ME. Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. J Pharmacol Exp Ther. 2003;304:1258–1267. doi: 10.1124/jpet.102.044412. [DOI] [PubMed] [Google Scholar]
- 148.Cermak R. Effect of dietary flavonoids on pathways involved in drug metabolism. Expert Opin Drug Metab Toxicol. 2008;4:17–35. doi: 10.1517/17425255.4.1.17. [DOI] [PubMed] [Google Scholar]
- 149.Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006;20:187–210. doi: 10.1016/j.tiv.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 150.von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 151.Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303:41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- 152.Varghese JN, Laver WG, Colman PM. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 A resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- 153.Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA, Colman PM. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins. 1992;14:327–332. doi: 10.1002/prot.340140302. [DOI] [PubMed] [Google Scholar]
- 154.Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 155.Gubareva LV, Webster RG, Hayden FG. Comparison of the activities of zanamivir, oseltamivir, and RWJ-270201 against clinical isolates of influenza virus and neuraminidase inhibitor-resistant variants. Antimicrob Agents Chemother. 2001;45:3403–3408. doi: 10.1128/AAC.45.12.3403-3408.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Smith BJ, McKimm-Breshkin JL, McDonald M, et al. Structural studies of the resistance of influenza virus neuramindase to inhibitors. J Med Chem. 2002;45:2207–2212. doi: 10.1021/jm010528u. [DOI] [PubMed] [Google Scholar]
- 157.Malaisree M, Rungrotmongkol T, Decha P, et al. Understanding of known drug–target interactions in the catalytic pocket of neuraminidase subtype N1. Proteins. 2008;71:1908–1918. doi: 10.1002/prot.21897. [DOI] [PubMed] [Google Scholar]
- 158.Cooper NJ, Sutton AJ, Abrams KR, et al. Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta-analyses of randomised controlled trials. BMJ. 2003;326:1235. doi: 10.1136/bmj.326.7401.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Eiland LS, Eiland EH. Zanamivir for the prevention of influenza in adults and children age 5 years and older. Ther Clin Risk Manag. 2007;3:461–465. [PMC free article] [PubMed] [Google Scholar]
- 160.McKimm-Breschkin JL, Sahasrabudhe A, Blick TJ, et al. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2en-derived inhibitors. J Virol. 1998;72:2456–2462. doi: 10.1128/jvi.72.3.2456-2462.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mendel DB, Tai CY, Escarpe PA, et al. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob Agents Chemother. 1998;42:640–646. doi: 10.1128/aac.42.3.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee J-Y, Urbatsch IL, Senior AE, Wilkens S. Projection structure of P-glycoprotein by electron microscopy. Evidence for a closed conformation of the nucleotide binding domains. J Biol Chem. 2002;277:40125–40131. doi: 10.1074/jbc.M206871200. [DOI] [PubMed] [Google Scholar]
- 163.Lee J-Y, Urbatsch IL, Senior AE, Wilkens S. Nucleotide-induced structural changes in p-glycoprotein observed by electron microscopy. J Biol Chem. 2008;283:5769–5779. doi: 10.1074/jbc.M707028200. [DOI] [PubMed] [Google Scholar]
- 164.Rosenberg MF, Callaghan R, Ford RC, Higgins CF. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J Biol Chem. 1997;272:10685–10694. doi: 10.1074/jbc.272.3.1665. [DOI] [PubMed] [Google Scholar]
- 165.Rosenberg MF, Callaghan R, Modok S, Higgins CF, Ford RC. Three-dimensional structure of P-glycoprotein: the transmembrane regions adopt an asymmetric configuration in the nucleotide-bound state. J Biol Chem. 2005;280:2857–2862. doi: 10.1074/jbc.M410296200. [DOI] [PubMed] [Google Scholar]
- 166.Rosenberg MF, Kamis AB, Callaghan R, Higgins CF, Ford RC. Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J Biol Chem. 2003;278:8294–8299. doi: 10.1074/jbc.M211758200. [DOI] [PubMed] [Google Scholar]
- 167.Rosenberg MF, Velarde G, Ford RC, et al. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001;20:5615–5625. doi: 10.1093/emboj/20.20.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 169.Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 170.Hvorup RN, Goetz BA, Niederer M, et al. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 171.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 172.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 173.Globisch C, Pajeva IK, Wiese M. Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem. 2008;3:280–295. doi: 10.1002/cmdc.200700249. [DOI] [PubMed] [Google Scholar]
- 174.Jones PM, George AM. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc Natl Acad Sci USA. 2002;99:12639–12644. doi: 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O'Mara ML, Tieleman DP. P-glycoprotein models of the apo and ATP-bound states based on homology with Sav1866 and MalK. FEBS Lett. 2007;581:4217–4222. doi: 10.1016/j.febslet.2007.07.069. [DOI] [PubMed] [Google Scholar]
- 176.Ravna AW, Sylte I, Sager G. Molecular model of the outward facing state of the human P-glycoprotein (ABCB1), and comparison to a model of the human MRP5 (ABCC5) Theor Biol Med Model. 2007;4:33. doi: 10.1186/1742-4682-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stenham DR, Campbell JD, Sansom MS, et al. An atomic detail model for the human ATP binding cassette transporter P-glycoprotein derived from disulfide cross-linking and homology modeling. FASEB J. 2003;17:2287–2289. doi: 10.1096/fj.03-0107fje. [DOI] [PubMed] [Google Scholar]
- 178.Dawson RJ, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 179.Vandevuer S, Van Bambeke F, Tulkens PM, Prevost M. Predicting the three-dimensional structure of human P-glycoprotein in absence of ATP by computational techniques embodying crosslinking data: insight into the mechanism of ligand migration and binding sites. Proteins. 2006;63:466–478. doi: 10.1002/prot.20892. [DOI] [PubMed] [Google Scholar]
- 180.Zolnerciks JK, Wooding C, Linton KJ. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 2007;21:3937–3948. doi: 10.1096/fj.07-8610com. [DOI] [PubMed] [Google Scholar]
- 181.Greenberger LM. Major photoaffinity drug labeling sites for iodoaryl azidoprazosin in P-glycoprotein are within, or immediately C-terminal to, transmembrane domains 6 and 12. J Biol Chem. 1993;268:11417–11425. [PubMed] [Google Scholar]
- 182.Greenberger LM. Identification of drug interaction sites in P-glycoprotein. Methods Enzymol. 1998;292:307–317. doi: 10.1016/S0076-6879(98)92024-9. [DOI] [PubMed] [Google Scholar]
- 183.Greenberger LM, Lisanti CJ, Silva JT, Horwitz SB. Domain mapping of the photoaffinity drug-binding sites in P-glycoprotein encoded by mouse mdr1b. J Biol Chem. 1991;266:20744–20751. [PubMed] [Google Scholar]
- 184.Taylor AM, Storm J, Soceneantu L, et al. Detailed characterization of cysteine-less P-glycoprotein reveals subtle pharmacological differences in function from wild-type protein. Br J Pharmacol. 2001;134:1609–1618. doi: 10.1038/sj.bjp.0704400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem J. 2006;396:537–545. doi: 10.1042/BJ20060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Loo TW, Clarke DM. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J Biol Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 188.Loo TW, Clarke DM. Identification of residues in the drug-binding domain of human P-glycoprotein. Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J Biol Chem. 1999;274:35388–35392. doi: 10.1074/jbc.274.50.35388. [DOI] [PubMed] [Google Scholar]
- 189.Loo TW, Clarke DM. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J Biol Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 190.Loo TW, Clarke DM. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J Biol Chem. 2001;276:14972–14979. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- 191.Ecker GF, Csaszar E, Kopp S, et al. Identification of ligand-binding regions of P-glycoprotein by activated-pharmacophore photoaffinity labeling and matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Mol Pharmacol. 2002;61:637–648. doi: 10.1124/mol.61.3.637. [DOI] [PubMed] [Google Scholar]
- 192.Loo TW, Clarke DM. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J Membr Biol. 2005;206:173–185. doi: 10.1007/s00232-005-0792-1. [DOI] [PubMed] [Google Scholar]
- 193.Loo TW, Clarke DM. Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J Biol Chem. 1997;272:20986–20989. doi: 10.1074/jbc.272.34.20986. [DOI] [PubMed] [Google Scholar]
- 194.Loo TW, Clarke DM. The packing of the transmembrane segments of human multidrug resistance P-glycoprotein is revealed by disulfide cross-linking analysis. J Biol Chem. 2000;275:5253–5256. doi: 10.1074/jbc.275.8.5253. [DOI] [PubMed] [Google Scholar]
- 195.Maki N, Hafkemeyer P, Dey S. Allosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J Biol Chem. 2003;278:18132–18139. doi: 10.1074/jbc.M210413200. [DOI] [PubMed] [Google Scholar]
- 196.Maki N, Dey S. Biochemical and pharmacological properties of an allosteric modulator site of the human P-glycoprotein (ABCB1) Biochem Pharmacol. 2006;72:145–155. doi: 10.1016/j.bcp.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 197.Maki N, Moitra K, Ghosh P, Dey S. Allosteric modulation bypasses the requirement for ATP hydrolysis in regenerating low affinity transition state conformation of human P-glycoprotein. J Biol Chem. 2006;281:10769–10777. doi: 10.1074/jbc.M512579200. [DOI] [PubMed] [Google Scholar]
- 198.Shapiro AB, Fox K, Lam P, Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]