

Abstract
Coronavirus reverse genetic systems have become valuable tools for studying the molecular biology of coronavirus infections. They have been applied to the generation of recombinant coronaviruses, selectable replicon RNAs, and coronavirus-based vectors for heterologous gene expression. Here we provide a collection of protocols for the generation, cloning, and modification of full-length coronavirus cDNA using vaccinia virus as a cloning vector. Based on cloned coronaviral cDNA, we describe the generation of recombinant coronaviruses and stable cell lines containing coronaviral replicon RNAs. Initially, the vaccinia virus-based reverse genetic system was established for the generation of recombinant human coronavirus 229E. However, it is also applicable to the generation of other coronaviruses, such as the avian infectious bronchitis virus, mouse hepatitis virus, and SARS coronavirus.
Key Words: Coronavirus, RNA virus, reverse genetics, vaccinia virus, full-length cDNA, in vitro transcription, recombinant coronaviruses
Introduction
The extraordinarily large size of the positive-stranded coronavirus RNA genome posed a significant obstacle for the establishment of coronavirus reverse genetic systems based on cloned full-length cDNA. Conventional cloning techniques using plasmid DNA cloning vectors were not suitable to stably accommodate large coronaviral cDNAs. Moreover, in numerous cases, specific coronaviral cDNA sequences turned out to be resistant to cloning in conventional plasmid DNAs or were unstable upon propagation in prokaryotic hosts. Finally, however, several laboratories succeeded in establishing coronavirus reverse genetic systems based on full-length cDNA (1–3. Not surprisingly, those researchers had to solve the problem of “cDNA instability” and, accordingly, the solutions they provided are all based on nonconventional approaches.
The reverse genetic system described here is based on the use of a vaccinia virus as a cloning vector that replicates in eukaryotic cells. Vaccinia virus is a large DNA virus with a genome size of approximately 200 kb that is able to stably accommodate foreign DNA sequences of coronavirus genome size (27–31 bp). The basic techniques required to clone (Section 3.1), modify (Section 3.2), and rescue (Section 3.2) recombinant coronaviruses are described. One application of the system, namely, the generation of coronavirus replicon RNAs and cell lines, is described in Section 3.3.
Materials
QiaexII Gel elution kit (Qiagen).
Buffer A (10 mM Tris-Cl pH 9.0, 1 mM EDTA).
MagNA Lyser Instrument, MagNa Lyser Green Beads (Roche).
Phosphate-buffered saline (PBS).
0.25% (w/v) trypsin.
36% sucrose.
Sorvall or Beckman ultracentrifuge, AH-629 or SW-28 rotors.
RNase-free DNase.
Proteinase K, PCR grade (Roche).
Proteinase K digestion buffer (1X concentration: 100 mM Tris-Cl pH 7.5, 5 mM EDTA, 0.2% (w/v) SDS, 200 mM NaCl).
RNase-free water.
T4 DNA Ligase (high-concentrate; Fermentas).
Pulse field gel instrument and equipment.
Lipofectin, Lipofectamine2000 (Invitrogen).
Sonication water bath (Branson 3210).
RiboMax large-scale RNA production system—T7 (Promega).
m7G(5')ppp(5')G cap analog (30 mM).
LiCl solution (7.5 M LiCl, 50 mM EDTA pH 7.5).
Sodium dodecylsulfate (SDS).
Electroporation instrument (e.g., BioRad Gene Pulser, 0.4-cm electroporation cuvettes).
Cell culture medium chemicals for GPT+ selection: (a) mycophenolic acid (MPA), 10 mg/ml in 0.1 M NaOH (400X stock); (b) xanthine, 10 mg/ml in 0.1 M NaoH (40X stock) and (c) hypoxanthine, 10 mg/ml in 0.1 M NaOH (667X stock).
Cell culture medium chemicals for GPT– selection: 6-thioguanine (6-TG), 1 mg/ ml (1000X stock).
Cells: BHK-21, CV-1, D980R (4.
Viruses: Vaccinia virus vNotI/tk (5, fowlpox virus.
Methods
Cloning of Coronavirus cDNA in Vaccinia Virus
This section describes the steps involved in the cloning of a full-length coronavirus cDNA. Starting from viral RNA, a set of plasmid DNAs should be generated together covering the full-length coronavirus genomic sequence. The plasmid insert cDNAs are then assembled by in vitro ligation to obtain a full-length coronavirus cDNA fragment. This fragment will be inserted into a vaccinia virus genome, again by in vitro ligation. The cloned full-length coronavirus cDNA in vaccinia virus is then amenable to mutagenesis by vaccinia virus-mediated homologous recombination.
Generation of Plasmid DNAs Covering a Coronavirus Full-Length cDNA
Analyze the coronavirus genome for useful naturally encoded endonuclease restriction sites that can later be used to ligate cloned cDNA inserts. Preferably use restriction enzymes that produce nonpalindromic sticky ends with at least three nucleotide (nt) overhangs. Avoid the use of restriction enzymes that generate blunt ends, since ligation efficiencies of blunt end fragments are low. If there are no useful sites available at particular regions of the cDNA sequence, restriction sites may be generated that introduce silent nucleotide changes. Alternatively, introduce sites at the border of the coronavirus cDNA fragments for restriction enzymes that cleave outside of their recognition sequence and orientate the sites so that cleavage occurs in the coronavirus cDNA region (Fig. 18.1 A).
Generate a set of plasmid DNAs covering the entire coronavirus cDNA using standard plasmid DNA cloning techniques. cDNA insert fragments should have a size of approximately 5 kb. Make sure that the cDNA fragment borders are flanked by appropriate endonuclease restriction sites in order to release the cloned cDNA fragments by endonuclease digestion (see above). If particular plasmid clones appear unstable upon propagation in Escherichia coli, change the plasmid backbone, preferably to a low copy plasmid. If plasmid DNAs remain unstable upon propagation proceed with the cloning of the remaining part of the coronavirus cDNA and insert the respective unstable cDNA sequence on the vaccinia virus level by vaccinia virus-mediated recombination using RT-PCR cDNA fragments (see Section 3.2).
The cDNA fragments corresponding to the 5'- and 3'-end of the genome should contain additional sequences as follows (Fig. 18.2 ). To facilitate cloning into the vaccinia virus genome by in vitro ligation with NotI-cleaved vaccinia virus DNA (see Section 3.1.4) both end fragments should contain an EagI or Bsp120I site. (DNA cleaved with EagI or Bsp120I can be ligated with NotI cleaved DNA.) Furthermore, the 5'-end fragment should contain a promoter for the bacteriophage T7 RNA polymerase and one G nucleotide (if not yet present at the genomic 5'-end of the coronavirus genomic RNA) for the proper initiation of the in vitro transcription. The 3'-end fragment should encode a stretch of A nucleotides (approximately 20 nt) followed by a restriction site that is not present in the coronavirus cDNA sequence.
Fig. 1.
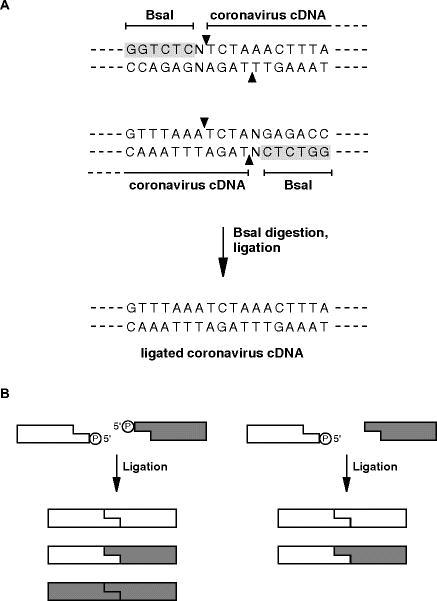
Strategies to minimize the number of possible ligation products: (A) Ligation of two coronavirus cDNA fragments using BsaI restriction endonuclease. BsaI recognition sequences can be engineered adjacent to the coronavirus cDNA to obtain BsaI-cleaved cDNA ends without heterologous sequences. The sticky ends are not palindromic and are comprised of a coronavirus-encoded sequence. The subsequent ligation reaction is directional and gives rise to only one possible reaction product. (B) The use of alkaline phosphatase to reduce the number of possible ligation products is illustrated. The left panel shows a conventional ligation using cDNA fragments with palindromic sticky ends. In this case three different ligation products are possible. The right panel shows a ligation reaction if one cDNA fragment has been dephosphorylated with alkaline phosphatase prior to the ligation reaction. In this case only two ligation products are possible.
Fig. 2.
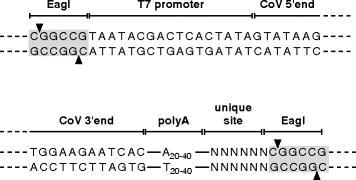
Structure of 5'- and 3'-ends of cloned full-length coronavirus cDNA. Upstream of the coronavirus 5'-end there should be an EagI or Bsp120I restriction endonuclease site to allow insertion of the cDNA into the NotI site of the vaccinia virus genomic DNA by in vitro ligation. Between the EagI or Bsp120I site and the 5'-end of the coronavirus cDNA sequence there should be a bacteriophage T7 RNA polymerase promoter and one G nucleotide (if not yet present at the 5'-end of the corona- virus genome) for the initiation of the in vitro transcription reaction. Downstream of the 3'-end of the coronavirus genome, a stretch of 20–40 A nucleotides (synthetic poly(A) tail) and a unique (i.e., not present in the coronavirus genomic sequence) restriction endonuclease site should be cloned. The unique restriction site is needed for the generation of runoff in vitro transcripts. Furthermore, an EagI or Bsp120I site is needed to insert the cDNA into the NotI site of the vaccinia virus genome by in vitro ligation. If the coronavirus sequence does not encode an EagI or Bsp120I site, the unique site is not needed since EagI or Bsp120I cleavage can be done to produce templates for the generation of runoff in vitro transcripts.
Assembly of Long cDNA Fragments by in vitro Ligation
Design a strategy for the sequential assembly of cloned cDNA inserts. Examples have been described for the construction of full-length human coronavirus 229E (HCoV-229E), avian infectious bronchitis virus (IBV), and mouse hepatitis virus, strain A59 (MHV-A59) cDNAs (3,6,7.
Liberate cloned insert cDNA fragments from plasmid DNAs by restriction endonuclease cleavage. Start with 50–100 μg plasmid DNA. Isolate the cDNA fragments by gel purification using standard agarose gels. Avoid exposure of cDNA fragments to UV light during the isolation process (see Note 1). If palindromic sticky ends are present at the cDNA fragment termini, the number of possible ligation products can be reduced by dephosporylation of one ligation partner by alkaline phosphatase treatment (Fig. 1 B). Note that the ends of cDNA fragments corresponding to the 5'- and 3'-genomic end should be cleaved with EagI or Bsp120I and dephosphorylated by alkaline phosphatase treatment.
Ligate cDNA fragments in analytical scale using high-concentrate T4 DNA ligase. Leave ligation reaction overnight at room temperature. Analyze ligation products on standard agarose gels. The samples should be heated to 65°C for 5 min before loading on the gel.
If the ligation reaction worked efficiently in the analytical scale, use the same conditions for a preparative scale ligation. Analyze an aliquot of the reaction on a standard agarose gel. If inefficient ligation is encountered repeatedly, revise the assembly strategy by using alternative restriction sites.
If the preparative ligation reaction was efficient, purify the desired ligation fragments by gel purification (avoid UV exposure; see Note 1) using the QiaexII gel elution procedure (Qiagen).
Use the ligated and purified cDNA fragments from step 5 for further ligation reactions with further cDNA fragments until a full-length cDNA fragment has been obtained. Alternatively assemble fragments to obtain a set of not more than two or three cDNA fragments together encompassing the entire coronavirus cDNA sequence (see Note 2).
Preparation of Vaccinia Virus DNA
This section describes the preparation of purified vaccinia virus DNA that can be used: (i) for the in vitro ligation with the assembled coronavirus full-length cDNA (see Section 3.1.4), and (ii) as template for in vitro transcription reactions (see Section 3.3.1). The protocol describes the vaccinia virus purification and subsequent DNA preparation in a preparative scale (virus derived from 10 to 20 150-cm2 flasks of infected BHK-21 cells). However, the protocol can also be down-scaled.
Grow 10 to 20 150-cm2 flasks of BHK-21 cells to 80% confluency and infect with vaccinia virus. Vaccinia virus infection should be done with an appropriate multiplicity of infection (MOI) to obtain complete a cytopathic effect (CPE) 3 days postinfection (p.i.).
Three days p.i. freeze cells by putting the flasks into a freezer for at least 2 h. Thaw, collect, and pellet cells (1000 rpm, 5 min, 4°C). Wash cell pellet with PBS. Resuspend concentrated infected cells in 1 ml Buffer A per 150-cm2 tissue culture flask.
Fill MagNA Lyser green bead tubes to maximum filling level of 1 ml.
Homogenize using MagNA Lyser machine (1 ×20 sec; speed 5000) (see Note 3).
Centrifuge at 1000 rpm 4°C for 2 min.
Pipette supernatant into a clean fresh tube. Treat supernatant with 0.1 volume 0.25% (w/v) trypsin and incubate at 37°C for 20 min.
Adjust the trypsin-treated cell homogenate with buffer A to a volume of 18 ml and carefully overlay an 18-ml 36% (w/v) sucrose cushion in a 36-ml ultracentrifugation tube and centrifuge (13,500 rpm at 4°C for 80 min; Sorvall or Beckman ultracentrifuge, Rotor AH 629 or SW 28).
Discard the supernatant and resuspend the pellet in 0.4 ml Buffer A.
Digest with RNase-free DNase (1–5 U) for 20 min (see Note 4) and then stop DNase treatment by adjusting the solution to 10 mM EDTA and incubate for 10 min at 65°C.
Add 1 vol of 2X Proteinase K digestion buffer (final concentration is 1X proteinase K digestion buffer) and 4 μl proteinase K; incubate at 50°C for 2 h.
Extract DNA with 1 vol phenol/chloroform/isoamylalcohol (25:24:1), mix gently (do not vortex! See Note 5), and centrifuge (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge). Take the water phase and perform a second round of DNA extraction with 1 vol chloroform/isoamylalcohol (24:1), mix gently (do not vortex!) and centrifuge (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge).
Take the water phase and add 2.5 vol 100% ethanol, mix gently (do not vortex!), and pellet the DNA by centrifugation (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge) (see Note 5).
Discard the supernatant and wash the DNA with 70% ethanol; centrifuge again (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge).
Discard the supernatant completely and resolve the DNA in 50–200 μl RNase-free water.
Ligation of Insert cDNA with Vaccinia Virus DNA
This section describes the integration of the assembled coronaviral cDNA fragments (Section 3.2) into the vaccinia virus genome by in vitro ligation. Inserted DNA fragments can be a full-length coronavirus cDNA or two or three cDNA fragments that can be ligated just prior to the ligation to the vaccinia virus genomic DNA. As parental virus we recommend the vaccinia virus vNotI/tk that encodes a unique NotI cloning site (5.
Set up a standard in vitro ligation reaction if the insert DNA consists of more than one fragment. This procedure is only recommended if the number of possible ligation products is limited (e.g., by avoiding palindromic sticky ends or by limiting the number of possible ligation products by using one dephosporylated DNA end per ligation reaction). The reaction should include a standard ligation buffer (including ATP) and can have a volume of up to 100 μl. Let the reaction go for 1–2 h at room temperature while preparing the NotI-cleaved vaccinia virus DNA (step 2). We recommend a molar ratio of insert:vaccinia virus DNA of 1:1 and a prior test of the reaction on an analytical scale.
Cleave vaccinia virus vNotI/tk DNA with NotI for 1–2 h at 37°C. The volume of the reaction can be up to 50 μl.
Mix the ligation reaction and the NotI restriction reaction and adjust buffers to 1X concentrations (1X ligation buffer and 1X NotI restriction buffer). Add fresh T4 ligase and NotI enzymes and incubate overnight at room temperature (see Note 6).
Heat the reaction to 65°C for 5 min, centrifuge (14,000 rpm, room temperature, Eppendorf centrifuge). Take the supernatant, add fresh NotI enzyme, and incubate at 37°C for 1–2 h (see Note 7).
Analyze the reaction products (or an aliquot thereof) on a pulse field gel. Prior to loading the sample(s) heat to 65°C for 5 min to achieve appropriate separation of DNA fragments in the pulse field gel.
Store ligation products at –20°C.
Rescue of Recombinant Vaccinia Viruses Containing Full-Length Coronavirus cDNA Insert
Seed CV-1 cells in a six-well dish 1 day before transfection. Cells should be 80% confluent for optimal transfection efficiency.
Infect 80% confluent CV-1 cells with fowlpox virus (MOI 1–10) for 1–2 h (see Note 8).
Transfect ligation reaction from Section 3.1.4 (without any further purification) into fowlpox virus-infected CV-1 cells using Lipofectin as described by the manufacturer (Invitrogen). Do not vortex at any time and use cut pipette tips when handling vaccinia virus DNA (see Note 5).
After 3–4 h trypsinize cells and seed them together with fresh (uninfected) CV-1 cells (4:1 excess of fresh CV-1 cells) into a 96-well plate.
At 5–10 days posttransfection collect cells and medium from wells displaying CPE (this is the first vaccinia virus stock) (see Note 9).
Transfer half of the first vaccinia virus stock to fresh CV-1 cells plated in a six-well dish. Wait until full CPE becomes apparent and collect the second vaccinia virus stock.
To analyze the obtained recombinant vaccinia viruses take half of the second vaccinia virus stock, pellet cells, and prepare DNA from the cell pellet according to Section 3.1.3, steps 10–14 (use 1X Proteinase K buffer).
To confirm the identity of recombinant vaccinia viruses perform Southern blot, PCR, and/or sequencing analyses.
Modification of Coronavirus cDNA by Vaccinia Virus-Mediated Homologous Recombination
The cloned coronavirus cDNA is amenable to mutagenesis by vaccinia virus-mediated homologous recombination. Two steps of homologous recombination are performed using appropriate plasmid DNAs (Fig. 18.3 ). The first “targeting” plasmid DNA contains a sequence of approximately 500 bp corresponding to a region encoded upstream of the region that should get targeted for mutagenesis (left flank), the E. coli guanosin-phosphoribosyltransferase (gpt) gene located downstream of a vaccinia virus promoter and a sequence of approximately 500 bp corresponding to a region encoded downstream of the region that should be targeted for mutagenesis (right flank).
Fig. 3.
Modification of the coronavirus cDNA. The modification of the cloned coronavirus cDNA by vaccinia virus-mediated homologous recombination is illustrated. The parental vaccinia virus vMHV-inf-1 is used in combination with a targeting plasmid to target the region of interest in the cloned cDNA. Upon gpt+ selection an intermediate clone is obtained that is subjected to a second round of recombination with a mutagenesis plasmid encoding the mutation of choice. The final mutant cDNA clone can be obtained after gpt– selection. Note that the intermediate clone can also be used to introduce other mutations in the targeted region by using a different mutagenesis plasmid.
The targeting plasmid is used to target the region of interest for a second round of recombination using a “mutagenesis” plasmid that contains the left and right flank of the targeting plasmid and, between the flanks, the region of interest encoding the desired mutation. Both plasmids can be constructed by standard cloning techniques. We routinely use the plasmid pGPT-1 (4 that is based on pBluescriptKS+ (Stratagene) and contains a fragment encoding the E. coli gpt gene downstream of a vaccinia virus promoter cloned into the pBluescriptKS+ multiple cloning site. The plasmid pGPT-1 is available from the authors upon request. Note that vaccinia virus-mediated homologous recombination also works when a linear DNA fragment, such as an RT-PCR product, is used instead of a plasmid DNA (7.
Vaccinia Virus-Mediated Homologous Recombination
This section describes the procedure of vaccinia virus-mediated homologous recombination. The protocol is used to generate a “transfection” stock containing recombined viruses at a ratio of approximately 1:1000 (recombinant viruses:parental viruses). The transfection stock is subsequently subjected to plaque purification under gpt+ or gpt– selection (see Sections 3.2.2. and 3.2.3) to obtain stocks of recombinant vaccinia virus clones.
Seed CV-1 cells in a six-well dish so that they are 80–95% confluent on the day of infection.
Infect with parental vaccinia virus at an MOI of 1. Incubate for 1–2 h at 37°C.
At 1–2 h postinfection transfect 5 μg of the targeting or mutagenesis plasmid using Lipofectin or Lipofectamine 2000 according to the manufacturer’s (Invitrogen) instructions.
Wash cells at 3–6 h posttransfection and culture cells for 2–3 days until full CPE becomes apparent.
Prepare a virus stock (which is the “transfection stock”) from the infected/ transfected culture by scraping the cells off the plate in 0.5 ml of the culture medium. Store at –20°C.
Targeting the Region of Interest: Selection of Recombinant gpt+ Vaccinia Viruses
Seed CV-1 cells in a six-well dish so that they are 80–95% confluent on the day of infection.
Replace culture medium with gpt+ selection medium (MEM containing 5% FCS, antibiotics and 25 μg/ml MPA, 250 μg/ml xanthine, and 15 μg/ml hypoxanthine) at least 6 h prior to infection.
Freeze-thaw the vaccinia virus transfection stock (see Section 3.2.1.) three times using dry ice on ethanol and sonicate for 1–5 min immediately prior to infection (see Note 10).
Infect CV-1 cells with different dilutions (10–2, 10––3, and 10––4) of the transfection stock (see Note 11).
Culture cells for 2–3 days in the gpt+ selection medium. Pick plaques as soon as they are easily detectable (usually on day 2 p.i.) by marking them at the bottom of the dish, followed by scraping the cells of the plaque off the dish, and aspirate in 100 μl of medium using a standard pipette (see Note 12).
Perform another two rounds of gpt+ plaque selection (steps 4 and 5) by infecting the CV-1 cells with 5–20 μl of a picked plaque. Always perform freeze-thaw cycles and sonication prior to infection.
After the third round of plaque selection infect CV-1 cells in a six-well dish with half of a picked plaque and culture cells until full CPE. Store half of this stock for further use and use the other half for DNA preparation and analysis of the recombinant vaccinia virus clone (i.e., PCR, Southern blot, sequencing).
Inserting the Mutation of Choice: gpt Negative Selection
Seed D980R cells in a six-well dish so that they are 60–80% confluent on the day of infection (see Note 13). Cells can be seeded in gpt– selection medium containing 0.5–1 μg/ml 6-TG. The cells should be cultured in gpt– selection for at least 6 h prior to infection.
Perform plaque selection as described in Section 3.2.2. steps 4–7. The only differences are the cells (D980R cells) and the gpt– selection medium.
Rescue of Recombinant Coronaviruses from Cloned Full-Length cDNA
The rescue of recombinant coronaviruses is based on two steps. First, a full-length coronavirus RNA is produced using the genomic DNA of a vaccinia virus containing the full-length coronavirus cDNA insert as a template for in vitro transcription. Second, the recombinant full-length RNA is transfected into eukaryotic cells. Within these cells the coronavirus replication cycle will be initiated by translation of replicase gene products from the transfected RNA and, finally, recombinant coronaviruses are released into the tissue culture supernatant.
Generation of Infectious Full-Length Coronavirus RNA by in vitro Transcription
Prepare vaccinia virus DNA from purified virus stocks as described in Section 3.1.3.
Cleave the vaccinia virus DNA (1–10 μg) with the restriction enzyme for which a unique recognition site downstream of the synthetic poly(A) tail has been introduced (Fig. 18.2 ).
Extract DNA with 1 vol phenol/chloroform/isoamylalcohol (25:24:1), mix gently, and centrifuge (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge). Take the water phase and perform a second round of DNA extraction with 1 vol chloroform/isoamylalcohol (24:1), mix gently, and centrifuge (14,000 rpm, 5 min, room temperature, Eppendorf centrifuge).
Take water phase and precipitate the cleaved vaccinia virus DNA by adding 1/20 vol of 5 M NaCl and 2.5 vol of 100% ethanol and centrifuge (14,000 rpm, 5 min, Eppendorf centrifuge). Do not overdry vaccinia virus DNA.
Wash DNA pellet with 70% ethanol; centrifuge again.
Completely remove the supernatant and resolve the DNA in 10–20 μl RNase-free water.
Set up the in vitro transcription reaction using the RiboMax Kit (Promega) as follows (see Note 14):
| 5X transcription buffer | 10 μl |
| m7G(5')ppp(5')G cap analog (30 mM) | 5 μl |
| GTP (100 mM) | 0.7 μl |
| ATP, CTP, UTP (100 mM), each | 3.75 μl |
| Template DNA (1–10 μg) | x μl |
| RNase-free water | y μl |
| Enzyme mix (RNasin, T7 RNA pol.) | 5 μl |
| Total | 50 μl |
-
7.
Incubate at 30°C for 2 h.
-
8.
Add 2 μl of RNase-free DNase, incubate at 37 °C for 20 min. Either store the reaction at –80 °C until transfection or (optional) precipitate the RNA (steps 9–11).
-
9.
Add half the volume of LiCl solution and freeze the sample for at least 30 min.
-
10.
Pellet RNA by centrifugation (14,000, 15 min, 4 °C, Eppendorf centrifuge)
-
11.
Wash RNA pellet (should appear yellowish) with 70% ethanol and resolve in RNase-free water. Store at –80 °C.
-
12.
Analyze the RNA on an agarose gel containing 1% SDS. Stain the RNA after gel electrophoresis with ethidium bromide.
Rescue of Recombinant Coronaviruses
One day before RNA transfection seed BHK-21 (see Note 15) cells so that there are 5 ×106 to 1 ×107 BHK-21 cells for each transfection. RNA transfection will be performed by electroporation.
Trypsinize, collect, and pellet 5 ×106 to 1 ×107 BHK-21 cells (centrifuge 1000 rpm, 5 min, 4 °C). Perform all further steps on ice.
Wash cells with 20 ml ice-cold PBS. Make sure that cells are well separated. Take one drop to count the cells and pellet the rest again (1000 rpm, 5 min, 4 °C).
Resolve 5 ×106 to 1 ×107 BHK-21 cells in 0.8 ml ice-cold PBS and fill into a 0.4-cm electroporation cuvette.
Add RNA and electroporate with two pulses (settings on BioRad Gene Pulser: Resistance = ∞, 230 V, high-capacity 1000 μF) (see Note 15).
Transfer the electroporated cells from into a 10-cm culture dish with 10 ml warm culture medium and add 1 ×106 fresh cells that are susceptible for the coronavirus that should be rescued (e.g., murine 17Cl1 cells for MHV rescue, human MRC-5 cells for the rescue of HCoV-229E).
Change the medium after 3–6 h when cells have attached to the bottom of the culture dish.
Recombinant coronaviruses should be released into the tissue culture medium between days 1 and 3 postelectroporation. Check for released virus on days 1–3 by transferring part of the supernatant onto susceptible fresh cells. Store culture supernatant for further analysis at –80 °C.
Coronavirus Replicon RNAs
Replicon RNAs are autonomously replicating RNAs encoding: (i) all replicative proteins required for the expression of a functional replication complex, and (ii) cis-acting elements required for the recognition of the replicon RNA by the replicase complex. Usually replicon RNAs are devoid of sequences leading to production of progeny particles. Coronavirus replicon RNAs differ from those of other positive-stranded RNA viruses in that they have to encode the nucleocapsid protein, which has been shown to be important for efficient coronavirus RNA replication (8,9.
It has been shown for HCoV-229E and SARS-CoV replicons that stable cell lines can be generated if the replicon RNA mediates the expression of a selection marker (4,10. Two selection markers, conferring neomycin/G418 (4 or blasticidin (10 resistance, have been used successfully for establishing stable coronavirus replicon cell lines. The HCoV-229E replicon RNA encodes the neomycin resistance gene inserted downstream of the nonstructural protein (Nsp) 1 and a sequence encoding a “2A-like” autoprocessing peptide. The 2A-like autoprocessing peptide mediates a co-translational liberation of a slightly modified Nsp1 carboxyterminus and subsequent translation of the neomycin resistance gene. In order to ensure translation of the remaining Nsps of the replicase gene (Nsps 2–16), an internal ribosomal entry site (IRES) derived from the encephalomyocarditis virus (EMCV) has been placed upstream of the Nsp2-coding sequence.
The SARS-CoV replicon RNA contains a gene encoding a fusion protein comprising the green fluorescent protein (GFP) and the blasticidin deaminase (GFP-BlaR) that has been cloned downstream of the replicase gene as a separate transcription unit under the control of the transcription regulatory sequence (TRS) of the SARS-CoV spike gene. In both cases, transfection of in vitro synthesized replicon RNA into eukaryotic cells and subsequent selection using G418 or blasticidin resulted in the establishment of stable cell lines containing actively replicating coronavirus replicon RNAs. To facilitate the detection of replicon-containing cell lines, green fluorescence resulting from replicon-mediated GFP expression has been used. For the SARS-CoV replicon RNA this has been achieved by the use of the GFP-BlaR fusion protein (10. To achieve GFP expression by the HCoV-229E replicon RNA, the GFP gene has been inserted as a separate transcription unit downstream from the replicase gene, driven by the TRS of the HCoV-229E spike gene (4.
Coronavirus replicon cell lines can be used as a noninfectious system to analyze coronavirus replication and transcription or to identify and evaluate replicase inhibitors. The following protocols describe the generation of corona- virus replicon cell lines and their use in the evaluation of coronavirus replicase inhibitors.
Generation of Coronavirus Replicon Cell Lines
Based on a full-length coronavirus cDNA cloned in vaccinia virus, a replicon RNA-encoding cDNA can be generated using vaccinia virus-mediated homologous recombination as described in Section 3.2.
Generate replicon RNA by in vitro transcription as described in Section 3.3.1.
Introduce the replicon RNA into a host cell line of choice (see Note 16) by electroporation as described in Section 3.3.2 (steps 1–5).
Plate the transfected cells in normal growth medium. Change the medium after 3–6 h when cells have attached to the bottom of the culture dish and continue to culture the cells for 1–2 days in growth medium without selection pressure. Split if necessary.
Start the selection of stable lines at antibiotic concentrations only slightly above the level at which nontransfected cells die (see Note 17).
Increase the antibiotic concentration gradually during the following 2–3 weeks until resistant colonies appear.
Pick colonies for subculture in separate wells and test them for maintenance of replicon RNA. Expression of a reporter protein, such as GFP, by the replicon RNA facilitates the screening of replicon RNA-containing resistant colonies.
When stable clones have been obtained, further culturing can be done under low selection pressure (see note 18). Replicon cells can be stored in liquid nitrogen.
Identification and Evaluation of Coronavirus Replicase Inhibitors Using Replicon Cell Lines
Seed the replicon cells in selection medium so that they are 50–70% confluent on the next day. You can use 96- , 24- , or 6-well dishes.
Prior to adding antiviral compounds, wash the cells and culture them in standard medium without selection drugs.
Add graded doses of antiviral compound(s) to the cells and culture them for 1–3 days (see Note 19). For comparison include nontreated cells and culture them under identical conditions.
In order to assay for cytotoxicity of candidate inhibitors and to determine the selectivity index, include a cytotoxicity/cell viability test. This can be done with replicon cells or the respective parental cell line.
Determine GFP expression on days 1, 2, and 3 posttreatment by fluorescence microscopy and flow cytometry (see Note 20).
Notes
We found that DNA that has been exposed to UV light is difficult and sometimes impossible to clone into vaccinia virus DNA. When purifying DNA fragments from agarose gels, cut small slices at the edges of the fragment band out of the gel and stain them with ethidium bromide. Use UV light to visualize the borders of the DNA band in the slices and mark the position. Insert the slices back into the gel and cut the piece of agarose between the two marked positions out of the gel. The DNA recovered from those agarose pieces have not been exposed to UV light and are easily clonable in vaccinia virus.
It is possible to insert more than one DNA fragment into the vaccina virus genome by in vitro ligation (37). Up to three DNA fragments can be ligated prior to adding NotI-cleaved vaccinia virus DNA (see Section 3.1.4, step 1). However, we recommend this procedure only if the fragments can be efficiently ligated (a full-length cDNA fragment should be visible in agarose gels). Furthermore, the number of possible ligation products should be minimized using strategies illustrated in Fig. 18.1.
In order to prepare vaccinia virus, DNA the virus particles have to be liberated from cells and cell debris. This can be achieved by using a tight Dounce homogenisator or, as described here, by using the MagNA Lyser protocol. To establish appropriate conditions we recommend doing a pilot experiment in which several conditions are compared. After homogenization check for virus titers and decide for the most vigorous homogenization conditions that still leave the virus particles intact.
This step results in DNase digestion of free DNA (mostly of cellular origin) and will leave the DNA in virus particles intact.
Vaccinia virus genomic DNA has a size of approximately 200 kb. Standard pipette tips are usually too narrow and pipetting will result in shearing the DNA. To avoid this, cut the pipette tips to generate an opening of about 2–3 mm and avoid vigorous pipetting. Avoid drying the DNA. If the large vaccinia virus DNA is overdried it will no longer be possible to dissolve it in water.
The ligation of insert DNA fragment(s) and the NotI-cleaved vaccinia virus arms is facilitated by adding NotI into the ligation reaction (11. Religated vaccinia virus arms are recleaved by the NotI enzyme allowing a new round of ligation. The 5'- and 3'-ends of the insert fragment are cleaved with EagI or Bsp120I and dephosphorylated. Therefore: (i) insert-insert ligation is not possible (owing to dephophorylated ends), and (ii) Eag or Bsp120I-NotI ligation products (i.e., insert-vector ligation) are not recleavable with NotI. As a result the ligation-restriction reaction will drive the overall reaction toward an accumulation of insert-vector ligation products.
The ligation-restriction reaction may still contain a small proportion of unrecleaved vector-vector ligation products. These products may lead to functional vaccinia virus genomes without the insert cDNA fragment and may cause a high background in the rescue of recombinant vaccinia viruses (Section 3.1.5). We therefore recommend an additional NotI cleavage reaction after the ligation-restriction reaction.
Since the vaccinia virus genomic DNA is not infectious, a helper virus has to be provided to rescue recombinant vaccinia viruses from DNA. We recommend fowlpox virus as a helper virus, since a fowlpox virus infection is abortive in mammalian cells, but can still serve to rescue vaccinia virus from DNA. Therefore, the recovered viruses will be vaccinia virus only (and no fowlpox virus).
On days 2–4 p.i. the cells may look heavily infected, most likely owing to fowlpox virus infection. However, recombinant vaccinia virus cannot yet be expected in the cell culture. Just continue to cultivate the cells; most cell layers will recover. Usually, the first vaccinia virus-mediated CPE can be expected on day 5 p.i. and a peak is observed around day 7 p.i.. If cells get too confluent the medium can be changed.
Freeze-thawing and sonication is needed to release and separate vaccinia virus particles from the cells and cell debris. For selection of pure recombinant vaccinia virus this procedure is critical.
A ratio of 1:1000 of recombinant vaccinia viruses:parental vaccinia viruses can be expected. Thus, under selection pressure, single plaques should appear on CV-1 cell layers that have been infected with a 10–3 dilution of the transfection stock. At 2 h p.i., an overlay of 1% low-melting agarose in selection medium can be made. This is done to reduce the risk of contamination of recombinant plaques with parental virus. Because most vaccinia virus is contained within an infected cell within the time frame (2–3 days) of the selection, agarose overlays are usually not necessary.
Six plaques is a reasonable number to pick. There is a limited risk of picking “false” plaques or plaques contaminated by parental virus that necessitates selection of a few plaques in parallel.
D980R cells grow fast and vaccinia virus plaques are not as easily recognizable as on CV-1 cells. During the selection it is necessary to keep cells in good condition to facilitate the formation of easily detectable plaques. The risk of overgrowth can be reduced by seeding cells at a lower density, and cell death from starvation can be reduced by replacing the medium with fresh selection medium on day 2 p.i..
It is possible, although not recommended by the manufacturer, to use the Promega RiboMax Kit to generate capped in vitro transcripts. One simply has to add a cap structure analog to the reaction. In the given in vitro transcription protocol the ratio of m7G(5')ppp(5')G cap analog to GTP is 2:1.
The optimal conditions for the electoporation of long RNA molecules are dependent on the cells and the electroporation device. BHK-21 cells are known to be suitable for efficient RNA electroporation and should be the first choice. We recommend doing a pilot optimization to determine optimal conditions for RNA transfection. It is now well established that the coronavirus nucleocapsid (N) protein facilitates the rescue of recombinant coronaviruses in several systems (2,6,7. We recommend co-electroporating an mRNA encoding the coronavirus N protein (5–10 μg N mRNA produced by in vitro transcription). It is even more efficient to generate and use a BHK-21-derived cell line stably expressing the N protein (7.
We observed replicon RNA replication in a wide variety of eukaryotic host cells. Although coronaviruses are usually species specific, coronavirus replicon RNAs are able to replicate in many cell lines once introduced into the host cell cytoplasm by transfection. Cell lines tested in our laboratory (using the HCoV-229E replicon) include cells of human (e.g., MRC-5, HeLa cells) and animal (e.g., BHK-21, 17clone1, L929 cells) origin.
We recommend determining the lowest concentration of the selection drug where nontransfected cells die for the cell line of choice.
Replicon cell lines based on commonly used cells such as baby hamster kidney (BHK) or Chinese hamster ovary (CHO) cells are generally easy to culture. To increase the number of cells expressing a high level of replicon-derived transcripts it is important to split the lines often enough to maintain them constantly subconfluent. GFP is a convenient marker to determine the percentage of GFP-expressing cells by flow cytometry.
Depending on the cell density and the stability of the compound it might be necessary to change the medium daily.
GFP is a valuable reporter protein to determine the percentage of green fluorescent cells as a marker for the percentage of cells with actively replicating RNA or to determine the mean fluorescence as a value that indicates GFP expression levels. Some inhibitors may lead to a reduced overall number of green fluorescent cells, whereas some inhibitors may just reduce the mean fluorescence. To generate more quantitative data on the inhibitory effect of a compound and to gain some insight into the kinetics of inhibition, other reporter proteins, such as luciferase proteins or alkaline phosphatase, may be used.
Acknowledgment
This work was supported by the Swiss National Science Foundation and the European Commission (SARS-DTV SP22-CT-2004-511064).
References
- 1.Almazan F., Gonzalez J.M., Penzes Z., et al. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Aca.d Sci. USA. 2000;97(10):5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yount B., Curtis K. M., Baric R. S. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J. Virol. 2000;74(22):10600–10611. doi: 10.1128/JVI.74.22.10600-10611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiel V., Herold J., Schelle B., Siddell S. G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001;82(Pt 6):1273–1281. doi: 10.1099/0022-1317-82-6-1273. [DOI] [PubMed] [Google Scholar]
- 4.Hertzig T., Scandella E., Schelle B., et al. Rapid identification of coronavirus replicase inhibitors using a selectable replicon RNA. J. Gen. Virol. 2004;85(Pt 6):1717–1725. doi: 10.1099/vir.0.80044-0. [DOI] [PubMed] [Google Scholar]
- 5.Merchlinsky M., Moss B. Introduction of foreign DNA into the vaccinia virus genome by in vitro ligation: recombination-independent selectable cloning vectors. Virology. 1992;190(1):522–526. doi: 10.1016/0042-6822(92)91246-Q. [DOI] [PubMed] [Google Scholar]
- 6.Casais R., Thiel V., Siddell S.G., Cavanagh D., Britton P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001;75(24):12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coley S. E., Lavi E., Sawicki S. G., et al. Recombinant mouse hepatitis virus strain A59 from cloned, full-length cDNA replicates to high titers in vitro and is fully pathogenic in vivo. J. Virol. 2005;79(5):3097–3106. doi: 10.1128/JVI.79.5.3097-3106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almazan F., Galan C., Enjuanes L. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 2004;78(22):12683–12688. doi: 10.1128/JVI.78.22.12683-12688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schelle B., Karl N., Ludewig B., Siddell S. G., Thiel V. Selective replication of coronavirus genomes that express nucleocapsid protein. J. Virol. 2005;79(11):6620–6630. doi: 10.1128/JVI.79.11.6620-6630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge F., Luo Y., Liew P. X., Hung E. Derivation of a novel SARS-coronavirus replicon cell line and its application for anti-SARS drug screening. Virology. 2007;360(1):150–158. doi: 10.1016/j.virol.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiel V., Siddell S. G. Reverse genetics of coronaviruses using vaccinia virus vectors. Curr. Top. Microbiol. Immunol. 2005;287:199–227. doi: 10.1007/3-540-26765-4_7. [DOI] [PMC free article] [PubMed] [Google Scholar]