

Abstract
The renin-angiotensin system (RAS) constitutes a key hormonal system in the physiological regulation of blood pressure via peripheral and central mechanisms. Dysregulation of the RAS is considered a major factor in the development of cardiovascular pathologies and pharmacologic blockade of this system by the inhibition of angiotensin-converting enzyme (ACE) or antagonism of the angiotensin type 1 receptor (AT1R) is an effective therapeutic regimen. The RAS is now defined as a more complex system composed of different angiotensin peptides with diverse biological actions mediated by distinct receptor subtypes. The classic RAS comprises the ACE-Ang II-AT1R axis that promotes vasoconstriction, water intake, sodium retention and increased oxidative stress, fibrosis, cellular growth, and inflammation. The non-classical or alternative RAS is composed primarily of the ACE2-Ang-(1-7)-AT7R pathway that opposes many actions of the Ang II-AT1R axis. In lieu of the complexity of this system, the chapter discusses the current evidence on the enzymatic cascade of the Ang-(1-7) axis of the RAS regarding the peptidases that contribute to the formation and degradation of the peptide in the circulation and various tissues.
Keywords: Angiotensin-(1-7), Neprilysin, Dipeptidyl peptidase 3, ACE, ACE2, Renin, Thimet oligopeptidase, Prolyl carboxypeptidase, Prolyl oligopeptidase
Introduction
The renin-angiotensin system (RAS) was originally characterized as a circulating endocrine system initiated by the protease renin to hydrolyze angiotensinogen to the inactive peptide Angiotensin I (Ang I) and the subsequent conversion to Ang II by angiotensin-converting enzyme (ACE) (Fig. 1). Ang II recognizes the angiotensin type 1 receptor (AT1R) to invoke both peripheral and central mechanisms in the regulation of blood pressure. Chronic activation of the ACE-Ang II-AT1R pathway may also be associated with various pathological responses including fibrosis, inflammation, metabolic dysregulation, heart failure, cancer, aging, and diabetic injury [1–6]. Although the blockade of the Ang II axis through ACE inhibitors or AT1R receptor antagonists are effective therapies for the treatment of cardiovascular disease, there is now abundant evidence for alternative pathways within the RAS that may contribute to the beneficial actions of conventional RAS blockade. Indeed, our original identification and quantification of the endogenous expression of Ang-(1-7) in the brain, circulation, and peripheral tissues provided a compelling case for a functional non-classical RAS pathway [7]. Subsequent studies in both experimental models and humans that ACE inhibition augments the circulating levels of Ang-(1-7) further supported the concept that Ang-(1-7) may oppose the actions of the Ang II-AT1R pathway [8]. Blockade of the AT1R also increases the formation of Ang-(1-7) through ACE2-dependent conversion of Ang II, as well as shunts Ang II to the AT2R pathway that shares similar properties to the Ang-(1-7) system [9–11]. As the functional actions of the RAS now reflect a far more complex array of peptide ligands and distinct receptors than previously envisaged, we present a comprehensive review of the enzymatic pathways involved in the formation and metabolism of the Ang-(1-7) axis in the circulation, central and peripheral tissues as well as cerebrospinal fluid (CSF) and urine. The review includes the roles of renin and ACE in regard to the generation of Ang I and Ang II as substrates for the subsequent processing to Ang-(1-7), as well as the influence of ACE and dipeptidyl peptidase I (DPP3) to metabolize Ang-(1-7). The pathways for the generation of the novel analog [Ala1]-Ang-(1-7) (alamandine) whereby the aspartic acid is decarboxylated to alanine and the functional consequences of this substitution are discussed in Chap. 10.1007/978-3-030-22696-1_2.
Fig. 1.
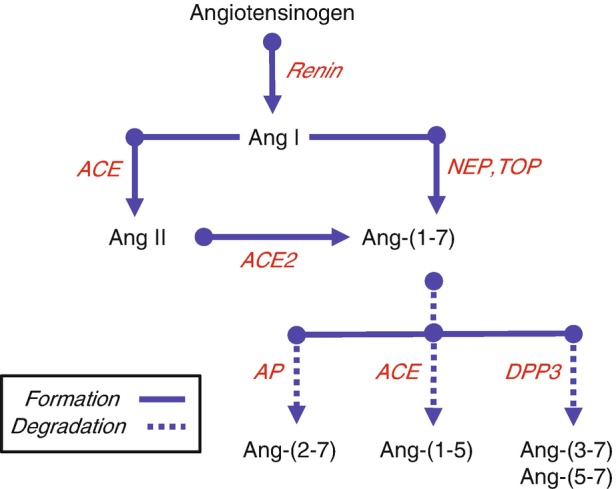
Processing cascade for Angiotensin-(1-7). Renin cleaves angiotensinogen to angiotensin I (Ang I), which is further processed to the biologically active peptide Ang II by angiotensin-converting enzyme (ACE). Ang II undergoes further processing by the carboxypeptidase ACE2 to form Ang-(1-7). Ang-(1-7) is also formed through non-Ang II pathways by the direct processing of Ang I by the endopeptidases neprilysin (NEP) and thimet oligopeptidase (TOP). Ang-(1-7) is subsequently metabolized by ACE to Ang-(1-5), dipeptidyl peptidase 3 (DPP3) to Ang-(3-7) and Ang-(5-7), or by aminopeptidase A (APA) to Ang-(2-7). (Adapted from Chappell [89])
Angiotensin-(1-7) Forming Pathways
Renin
Renin [EC 3.4.23.15, 35 kDa] belongs to the class of aspartyl-type acid proteases but exhibits a more neutral pH optima [6.5–8.0] than other proteases in this group. The only known substrate for renin is angiotensinogen and the enzyme hydrolyzes the Leu10-Leu11/Val11 bond of the precursor protein to form the inactive peptide Ang I. Renin-dependent formation of Ang I is considered the enzymatic initiator of the RAS cascade to ultimately generate Ang II (Fig. 1). The enzyme is synthesized predominantly in the juxtaglomerular (JG) cells of the kidney and is stored in both inactive (pro-renin) and active forms for the regulated release of the protease into the circulation. The collecting duct (CD) cells are another source of renin within the kidney that primarily secretes the active form of renin into the tubular fluid [12–14]. The tubular secretion of renin may contribute to the processing of angiotensinogen and subsequent Ang II and Ang-(1-7) formation to influence distal nephron function. The regulation of JG renin appears to be quite distinct from CD renin; Ang II negatively regulates JG renin release through inhibition of cAMP levels that constitutes the short negative feedback loop while CD renin release is stimulated by Ang II that may reflect increased protein kinase C (PKC) activity [14].
In regard to the generation of Ang-(1-7), renin is required for the formation of the peptide via the processing of Ang I or Ang II. Since renin is typically secreted from the kidney and other tissues, the formation of Ang-(1-7) is likely to occur in the extracellular compartments including the blood, interstitial fluid, renal tubular fluid, and CSF. However, there is evidence for alternative renin isoforms in the kidney, brain, heart , and adrenal gland [15–17]. The renin isoform lacks the pre-pro domain of the protein that includes the secretory signal, thus the isoform should reside within the cell. Peters and colleagues originally reported that truncated renin localized to the mitochondria and that the renin isoform was internalized by the mitochondria [18–21]. We recently reported the presence of active renin in a purified preparation of mitochondria from the sheep renal cortex, as well as evidence for mitochondrial Ang II and Ang-(1-7) [50–60 fmol/mg protein] [22]. Moreover, the endopeptidases neprilysin and thimet oligopeptidase processed Ang I directly to Ang-(1-7) in mitochondria suggesting an Ang I-dependent pathway for the intracellular formation of Ang-(1-7) (see Neprilysin and Thimet Oligopeptidase sections). In renal NRK-52 epithelial cells, immunocytochemical staining for renin with the Inagami antibody was evident in the nucleus of these cells and active renin was confirmed by aliskerin-sensitive conversion of angiotensinogen to Ang I. Isolated nuclei also contained Ang I, Ang II, and Ang-(1-7) [5–20 fmol/mg protein]; however, the processing pathways within the nuclear compartment remain to be defined [23]. Ishigami et al. reported a truncated renin transcript expressed in the proximal tubules of the mouse kidney [17]. Targeted expression of this renin isoform within the proximal tubules was associated with a sustained elevation in blood pressure, but no change in the circulating levels of renin [17]. Although the intracellular localization of the renin isoform or tubular content of angiotensins were not determined, overexpression of renin in tubules augmented blood pressure that suggests the primary intracellular generation of Ang II rather than Ang-(1-7). Indeed, Zhou and colleagues find that the intracellular expression of Ang II in the proximal tubules also resulted in a sustained increase in blood pressure [24] that is consistent with the demonstration of the intracellular AT1R and a local RAS within the kidney [25–28].
Non-renin pathways for the processing of angiotensinogen to Ang I or Ang II include tonin, cathepsin G, and cathepsin A; however, the participation of these enzymes in the endogenous generation of angiotensins has yet to be firmly established [29]. Moreover, it is not known whether there is a direct pathway for the formation of Ang-(1-7) from angiotensinogen and whether this contributes to the intracellular content of the peptide. Non-renin pathways for the formation of Ang-(1-7) (or Ang II) may also potentially occur through the processing of Ang-(1-12), a novel precursor peptide originally identified in rat urine by Nagata and colleagues [30]. In this regard, neprilysin converts Ang-(1-12) to Ang-(1-7) in renal cortical membranes and by recombinant forms of both human and mouse neprilysin by a two-step process that involves Ang I as an intermediate product; however, the role of Ang-(1-12) in the generation of endogenous Ang-(1-7) in the circulation and tissues is presently unknown [31].
Endopeptidases
Neprilysin
Neprilysin (EC 3.4.24.11; 95 kDa) is a metalloendopeptidase of the type II membrane-anchored family of enzymes [32]. The peptidase was initially characterized in brain homogenates or membrane preparations to hydrolyze the opiate pentapeptide enkephalin which explained its original characterization as an “enkephalinase.” Inhibitors against neprilysin were originally developed to prolong the analgesic actions of the opiate peptides, although neprilysin was subsequently found to be expressed in a number of peripheral tissues [32]. Cardiovascular interest in neprilysin initially reflected its ability to metabolize the family of natriuretic peptides including ANP, BNP, and uroguanylin. Moreover, McKinnie et al. report that neprilysin inactivates apelin, suggesting that beneficial cardiovascular effects of neprilysin inhibition may also reflect the protection of endogenous apelin [33]. Neprilysin inhibitors were considered a potential therapeutic approach to prolong the natriuretic and vasorelaxant properties of these peptides; however, Ang II is also a substrate for neprilysin hydrolysis of the Tyr4-Ile5 bond to form Ang-(1-4) and combined inhibitors to neprilysin and ACE termed vasopeptidase inhibitors were developed to prevent the potential increase in circulating and renal Ang II by neprilysin inhibitors alone [34–36]. Although the vasopeptidase inhibitors were potent agents to lower blood pressure and reduce cardiac and renal damage, the first clinical agent omapatrilat was withdrawn due to a greater incidence of angioedema in patients. Subsequently, a new generation of agents have been developed that combine a neprilysin inhibitor and an AT1R antagonist to obviate the inhibitory effects on ACE, yet maintain blockade of the Ang II-AT1R axis. Indeed, the combined neprilysin/AT1R blockade may be a promising therapeutic approach for the treatment of heart failure [37].
Neprilysin is located on the vascular surface of blood vessels and is responsible for the direct conversion of Ang I to Ang-(1-7) in the circulation , particularly under conditions of chronic ACE inhibition (Fig. 1) [32]. Neprilysin hydrolyzes the Pro7-Phe8 bond of Ang I to generate Ang-(1-7), as well as the Tyr4-Ile5 bond to form Ang-(1-4) that is consistent with the enzyme’s preference for aromatic and hydrophobic residues [38]. The generation of circulating Ang-(1-7) from infused Ang I in ACE-blocked Wistar Kyoto (WKY) and spontaneously hypertensive rats (SHR) was abolished by the potent and selective neprilysin inhibitor SCH39370 [39]. We further demonstrated that administration of another selective neprilysin inhibitor CGS24592 partially attenuated the blood pressure-lowering effects of the ACE inhibitor lisinopril in the SHR [40]. The neprilysin inhibitor SCH39370 also reduced Ang-(1-7) levels in the rat hindlimb preparation perfused with Krebs buffer containing the ACE inhibitor lisinopril [41]. Finally, Campbell and colleagues found that a dual ACE/neprilysin inhibitor lowered circulating levels of Ang-(1-7) in the SHR using a combined HPLC-RIA approach to quantify an array of angiotensins using an N-terminally directed antibody [42].
Neprilysin is also highly expressed on the apical surface of proximal tubules within the kidney [32]. We demonstrated a neprilysin-dependent pathway in isolated proximal tubules from sheep kidney and rat cortical membranes for Ang I processing to Ang-(1-7) (Fig. 2) [44, 45]. In isolated tubules, we note that neprilysin blockade did not abolish the formation of Ang-(1-7) as ACE-dependent generation of Ang II contributed to Ang-(1-7) via the ACE2 pathway (Fig. 2). Velez and colleagues applied HPLC-MS analysis of Ang I processing in rat glomeruli , isolated mouse podocytes, and human glomerular endothelial cells to reveal the predominant conversion of Ang I to Ang-(1-7) by neprilysin [46–48]. Poglitsch and colleagues recently show that the renal levels of Ang-(1-7) were significantly reduced in mice chronically treated with the neprilysin inhibitor LBQ657, but increased in ACE2-deficient mice utilizing UHPLC-MS to quantify angiotensins in the kidney [43]. Moreover , the neprilysin inhibitor markedly reduced Ang I to Ang-(1-7) conversion in both mouse and human kidney homogenates [43]. Indeed, these studies support our earlier findings that despite almost complete depletion of renal Ang II in ACE knockout mice, kidney levels of Ang-(1-7) were not diminished supporting an ACE (Ang II)-independent pathway for the generation of Ang-(1-7) in this tissue [49]. Finally, neprilysin activity in isolated mitochondria from the sheep kidney was partially responsible for conversion of Ang I to Ang-(1-7), and was the sole Ang-(1-7) forming activity in a 100,000 xg membrane fraction consistent with the membrane-anchored form of the peptidase [22].
Fig. 2.
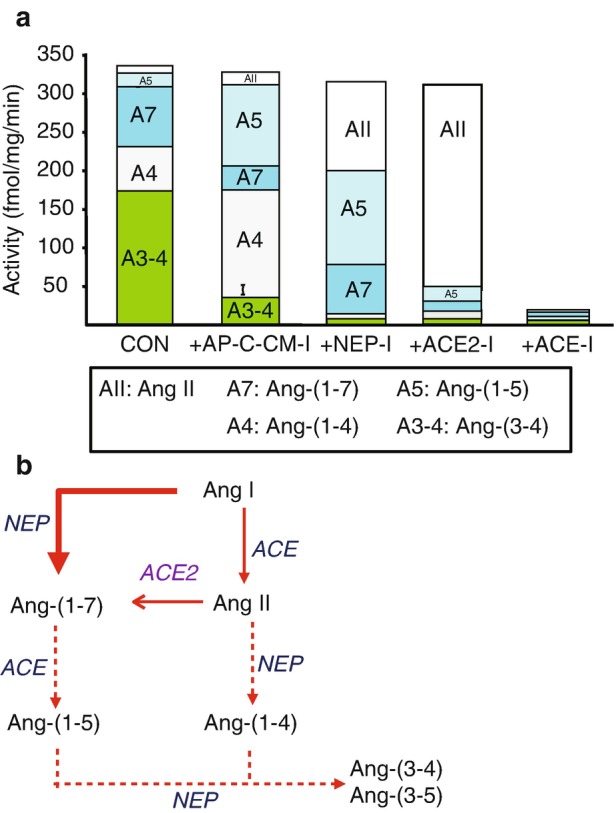
Ang I processing to Ang-(1-7) and Ang II in sheep proximal tubules. 125I-Ang I (AI) was incubated with 50 μg of proximal tubules membranes for 30 minutes at 37 °C and the metabolites identified by HPLC. Panel a: Quantification of the peptidase activities for 125I-AII metabolism from the sheep proximal tubule membranes expressed as the rate of metabolism products formed (fmol/mg/min). Conditions: Control (no inhibitors); +AP,CYS,CHM-I (inhibitors for aminopeptidase, carboxypeptidase, chymase, cysteine proteases); +NEP-I (addition of neprilysin inhibitor); +ACE-I (addition of ACE inhibitor); +ACE2-I (addition of ACE2 inhibitor). Panel b: Metabolism pathway for Ang I to Ang-(1-7), Ang II and degradation products in sheep proximal tubules. Data shown are mean values; n = 5. (Adapted from Shaltout et al. [43])
Thimet Oligopeptidase
Thimet oligopeptidase (EC 3.4.24.15, 80 kDa) is a soluble metalloendopeptidase that resides within intracellular compartments of the cell. Similar to neprilysin, thimet oligopeptidase is an endopeptidase that prefers aromatic and hydrophobic residues and cleaves multiple peptide substrates [50]. Thimet oligopeptidase hydrolyzes Ang I exclusively at the Pro7-Phe8 bond to form Ang-(1-7) (Fig. 1). In contrast to neprilysin, thimet oligopeptidase does not process Ang I to Ang-(1-4), although the peptidase does cleave Ang II to Ang-(1-4) (Fig. 1). The current data suggest that thimet oligopeptidase may be responsible for the intracellular processing of Ang I to Ang-(1-7). Pereira et al. find that thimet oligopeptidase was the primary activity in tissue homogenates of brain medulla that processed Ang I to Ang-(1-7) [51]. Both neprilysin and thimet oligopeptidase contributed to the processing of Ang I to Ang-(1-7) within isolated mitochondria, and thimet oligopeptidase was the sole Ang-(1-7) forming activity in the 100,000 xg soluble fraction of renal mitochondria [22]. In human proximal tubule HK-2 cells, cytosolic thimet oligopeptidase was the primary activity responsible for the generation of Ang-(1-7) from exogenous Ang I [52]. Chronic treatment of the HK-2 cells with the cell-permeable metallopeptidase inhibitor JMV-390 reduced the intracellular levels of Ang-(1-7) which may reflect the inhibition of thimet oligopeptidase, although the JMV inhibitor may target other metallopeptidases or cell mechanisms that influence the intracellular expression of the peptide [53]. Moreover, thimet oligopeptidase activity in isolated nuclei of NRK-52 renal epithelial cells processed Ang I exclusively to Ang-(1-7) and this peptidase may contribute to the nuclear levels of Ang-(1-7) within the cell [23]. Suski et al. reported that Ang I was primarily converted to Ang-(1-7) in vascular smooth muscle cells (VSMC) as characterized by HPLC-MS analysis [54]; these data confirm our earlier study that thimet oligopeptidase directly processed Ang I to Ang-(1-7) in rat VSMC by HPLC characterization [55]. Although the RAS was originally characterized as a classic endocrine or circulating system, there is compelling evidence for the intracellular expression of angiotensins and angiotensin receptors that include Ang-(1-7) and the MasR [9, 56–61]. The mechanisms for intracellular expression of Ang-(1-7) are currently unknown; however, thimet oligopeptidase should be considered as one candidate processing enzyme in the cellular Ang-(1-7) axis.
Prolyl Oligopeptidase
Prolyl oligopeptidase (EC 3.4.24.16, 75 kDa), also known as prolyl endopeptidase (PEP), is a soluble intracellular serine peptidase that cleaves the Pro7-Phe8 bond of both Ang I and Ang II to form Ang-(1-7). Thus, prolyl oligopeptidase may function as both an endopeptidase and a monocarboxypeptidase in the processing of angiotensins, as well as the hydrolysis of other peptides that contain a Pro-XX motif including TRH, substance P, oxytocin, bradykinin and vasopressin [62]. Although prolyl oligopeptidase is considered a cytosolic peptidase, membrane and nuclear forms of the enzyme have been described in both central and peripheral cells suggesting a role for the enzyme in the intracellular expression of peptides [62]. Santos et al. report that prolyl oligopeptidase activity was involved in the extracellular conversion of Ang I to Ang-(1-7) in human aortic endothelial cells, although it remains unclear as to whether the peptidase was secreted into the media or that the peptidase resides on the plasma membrane of endothelial cells [63]. Domenig et al. found that although neprilysin was the predominant Ang-(1-7) forming activity in the kidney, prolyl oligopeptidase activity contributed approximately 20% and 10% to Ang-(1-7) in the mouse and human kidney, respectively, based on the inhibition by Z-prolyl prolinal (ZPP) [43]. Prolyl oligopeptidase would appear to be an ideal enzymatic candidate for the processing of both Ang I and Ang II to Ang-(1-7); however, treatment approaches with more selective inhibitors against the peptidase have generally revealed beneficial effects that contrast with the expected actions of Ang-(1-7), particularly regarding inflammation [62, 64, 65].
Carboxypeptidases
Angiotensin-Converting Enzyme 2
ACE2 is a membrane-bound monocarboxypeptidase (EC 3.4.17.23; 90-120 kDa) that converts Ang II directly to Ang-(1-7) (Fig. 1). ACE2 was initially characterized as an ACE homolog (~40% homology) that cleaved Ang I to the nonapeptide Ang-(1-9), but not directly to Ang II [66]. Subsequent studies found that Ang II exhibits far better kinetic values as a substrate for human ACE2 that would favor processing of Ang II over that of Ang I [67]. Among a number of peptide substrates (> 100) that were screened for human ACE2, Vickers et al. reported that only apelin 13 exhibited comparable kinetic values to that of Ang II [68]. In the murine heart, Ang II was primarily converted to Ang-(1-7) by ACE2 and that in the presence of the ACE2 inhibitor MLN-4760 or in ACE2 null mice there was little metabolism of Ang II [68]. In contrast, under identical kinetic conditions, Ang I was primarily converted to Ang-(1-9) by carboxypeptidase A and not ACE2 in both the wild-type and ACE2 knockout mice [68].
In comparison to ACE, the circulating levels of ACE2 are typically quite low and the extent this reflects a reduced degree of shedding or simply the lower vascular expression of the peptidase is not clear. Rice et al. reported that the molar concentration of ACE in human serum at 7 nM while ACE2 content was >200-fold lower at 33 pM and detectable in <10% of their patient population [69]. In comparison, circulating neprilysin content (290 pM) was also lower than ACE and evident in <30% of these patients [69]. Serum and urinary ACE2 activities are elevated in diabetes, heart failure, and hypertension [70–72]. Circulating ACE2 activity increased approximately three-fold in the diabetic hypertensive mRen2.Lewis rat; however, serum ACE activity also increased in the diabetic rats and may mitigate against the elevated levels of ACE2 (Fig. 3) [70]. As measured under identical kinetic conditions, serum ACE activity for Ang I was far higher than ACE2 for Ang II suggesting that the capacity to generate Ang II [or metabolize Ang-(1-7)] remains greater than the capability to form Ang-(1-7) from Ang II (Fig. 3) [70]. Moreover, the serum ACE:ACE2 ratio in both male and female normotensive Lewis and female mRen2.Leiws essentially reflected the ratio found in human plasma [69, 70]. Serum ACE2 activity in the male hypertensive mRen2.Lewis was significantly higher than that in the females, despite higher blood pressure and greater circulating levels of Ang II in the males [70]. Whether the higher serum levels of ACE2 in the male mRen2.Lewis reflect a compensatory effect to reduce hypertension and vascular damage or that shedding away from the vascular wall (and attenuated local metabolism of Ang II) contributes to the increase in blood pressure is currently not known [70].
Fig. 3.
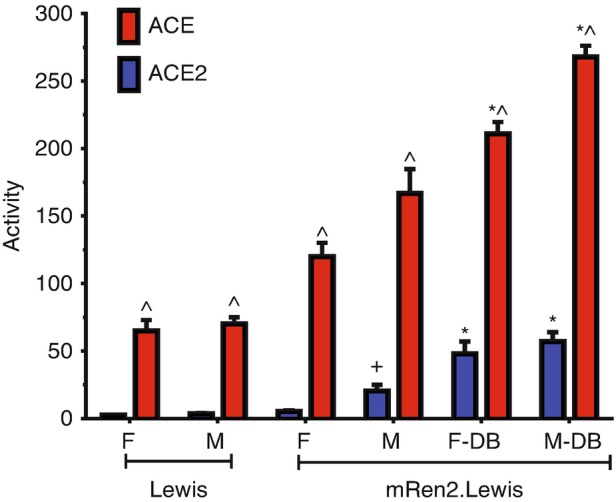
Diabetes increases both circulating ACE and ACE2 activities in the hypertensive mRen2.Lewis rat. Comparison of circulating ACE2 and ACE activities in female and male normotensive Lewis, hypertensive mRen2.Lewis, and diabetic (DB) mRen2.Lewis rats. Data are mean ± SEM; 5-6 per group ∗P < 0.05 versus non-diabetic mRen2.Lewis; ^P < 0.05 versus ACE2; +P < 0.05 versus female mRen2.Lewis. Activities are expressed as fmol/minute/ml serum. (Adapted from Yamaleyeva et al. [71])
ACE2 constitutes a key enzymatic component of the RAS as a single catalytic step efficiently metabolizes Ang II to attenuate the Ang II-AT1R pathway, and generates Ang-(1-7) that would activate the Ang-(1-7)-AT7/MasR axis (Fig. 1). In isolated proximal tubules, Ang II is processed to Ang-(1-7), Ang-(1-4), Ang-(1-5), and Ang-(3-4) (Fig. 4) [44]. The selective neprilysin inhibitor SCH39370 essentially abolished Ang-(1-4) confirming a role for neprilysin to metabolize Ang II. Subsequent addition of an ACE inhibitor attenuates Ang-(1-5) and reveals higher levels of Ang-(1-7) that emphasize the importance of the ACE pathway to metabolize Ang-(1-7) in the kidney (Fig. 4a) [44]. Finally, Ang-(1-7) formation is essentially abolished by the ACE2 inhibitor MLN4760 (Fig. 4a). We also show that the MLN4760 (MLN) alone significantly attenuates Ang II metabolism by the isolated proximal tubules and increases the peptide’s half-life by >2.5-fold, although it clearly does not abolish Ang II metabolism by other peptidases (Fig. 4b) [44]. Moreover, ACE2 protein expression and activity (Ang II to Ang-(1-7) conversion) is markedly reduced in the renal tubules, circulation, and brain medulla in a sheep model of fetal programming; the downregulation of this key peptidase may contribute to an altered ratio of Ang II to Ang-(1-7) in these important cardiovascular tissues that reflect fetal programming events [73]. Indeed, our findings are consistent with studies that demonstrate an exaggerated response to exogenous Ang II or in conditions of an activated RAS in ACE2-deficient animals [72–77]. Overexpression of ACE2 or administration of the soluble form of the peptidase which retains full enzymatic activity attenuates the Ang II-dependent increase in blood pressure and indices of target organ injury [78–86]. The premise of ACE2 supplementation is that sufficiently high levels of ACE2 are administered to reduce the Ang II: Ang-(1-7) ratio thereby attenuating the actions of Ang II-AT1R axis while amplifying those of the Ang-(1-7)-AT7/MasR effects (Fig. 5). Oudit and colleagues find that chronic administration of soluble ACE2 attenuated various indices of cardiac and renal injury, inflammation , and fibrosis in both type 1 and type 2 diabetic mice [79, 80]. Surprisingly, the administration of ACE2 reduced tissue levels of Ang II in the heart and kidney and increased the tissue contents of Ang-(1-7) [79]. Scholey and colleagues also report that ACE 2 given subcutaneously by an osmotic pump attenuated several indices of renal damage in the transgenic Col4A3-/- mouse, a model of Alport syndrome, as well as tended to lower blood pressure [87]. The renal protective effects of soluble ACE2 treatment were associated with a marked reduction in the ratio of Ang II: Ang-(1-7) in the kidney [87]. However, Wysocki et al. observed that neither the administration of ACE2 nor the chronic overexpression of the soluble peptidase by minicircle DNA conveyed any protective effects against diabetic nephropathy in diabetic mice [88]. In this study, plasma angiotensin peptides were quantified by UHPLC-MS with sufficient sensitivity to detect peptides in the low pM range and resolve related N-terminal metabolites of Ang I, Ang II, and Ang-(1-7) that include Ang-(2-10), Ang-(2-8), Ang-(3-8), Ang-(2-7), and Ang-(3-7), respectively; these metabolites are not distinguished by direct RIAs or ELISAs unless coupled to HPLC/UHPLC separation prior to immunoassay. This analysis revealed that chronic ACE2 treatment reduced the total plasma ratio of Ang II: Ang-(1-7) approximately four-fold in the diabetic mice; however , the effect of ACE2 on renal peptide content was not addressed [88]. Moreover, it should be noted that the N-terminal metabolites Ang-(2-10), Ang-(2-8) or Ang III and Ang-(2-7) were the major components in plasma in this study that contrasts with the accepted profile of angiotensins in both the circulation and tissues [89].
Fig. 4.


Ang II processing to Ang-(1-7) and other metabolites in sheep proximal tubules. 125I-Ang II (AII) was incubated with 50 μg of proximal tubules membranes for 30 minutes at 37 °C and the metabolites identified by HPLC. Panel a: Quantification of the peptidase activities for 125I-AII metabolism from the sheep proximal tubule membranes expressed as the rate of metabolism products formed (fmol/mg/min). Conditions: Control (no inhibitors); +AP,CYS,CHM-I (inhibitors for aminopeptidase, carboxypeptidase, chymase, cysteine proteases); +NEP-I (addition of neprilysin inhibitor); +ACE-I (addition of ACE inhibitor); +ACE2-I (addition of ACE2 inhibitor). Panel b: Influence of ACE2 inhibition on half-life (t1/2) of 125I-Ang II (AII) in proximal tubules. Conditions: Control (no inhibitors); +MLN (only the ACE2 inhibitor). Data are mean values; n = 5; ∗P < 0.05 versus Control. Panel c: Metabolism pathway for Ang II to Ang-(1-7) and degradation products in sheep proximal tubules. (Adapted from Shaltout et al. [43])
Fig. 5.

Scheme depicting the influence of exogenous ACE2 on the ratio of Ang II to Ang-(1-7). Upper panel: Predominance of ACE activity results in a higher ratio of Ang II to Ang-(1-7) that may contribute to increased blood pressure (+BP), oxidative stress (ROS), fibrosis, and inflammation, but reduced levels of nitric oxide (-NO). Lower panel: Treatment with a soluble form of recombinant ACE2 (rACE2) or overexpression of the peptidase reduces the Ang II to Ang-(1-7) ratio that may lower BP, ROS, fibrosis and inflammation, but increase levels of NO
In regard to the benefits of an activated ACE2 pathway, several compounds have been identified that may act as allosteric activators of ACE2 including xanthenone (XNT) and diminazene aceturate (DIZE) to promote a higher ratio of Ang-(1-7) to Ang II (89) [1]. Chronic treatment with DIZE ameliorated the extent of pulmonary hypertension and fibrosis, renal tissue injury, and myocardial infarction consistent with enhanced levels of Ang-(1-7) and a reduction in Ang II [90–93]. Interestingly, DIZE treatment was also associated with increased mRNA levels of ACE2 suggesting that DIZE may exhibit actions apart from the direct activation of the peptidase. However, the stimulatory effects of DIZE on either ACE2 activity or expression have not been confirmed by others. Haber et al. found no effect of DIZE on soluble ACE2 activity or an influence on Ang II-dependent hypertension using similar doses of DIZE as previously reported [94]. Velkosa et al. also show no direct effect of various concentrations of DIZE on renal ACE2 activity ex vivo, as well as no in vivo effect on cardiac ACE2 activity or Ang-(1-7) content in the 5/6 nephrectomized rat following a 2-week administration of DIZE [95]. Indeed, this study reported that DIZE normalized the marked increase in cardiac ACE activity and Ang II suggesting that ACE may be a more relevant target than ACE2 to impact the cardiac RAS [95]. DIZE also failed to increase ACE2 activity or enhance the local vascular actions of Ang-(1-7) in a preparation of isolated pig coronary arteries [96].
Conceptually, the use of ACE2 as a therapeutic agent to chronically alter the balance of Ang II and Ang-(1-7) is challenging. ACE activity in the circulation and the vasculature surface is significant with a very high capacity to generate Ang II. This reflects not simply the abundance of ACE but the marked capability of angiotensinogen and renin to generate the ACE substrate Ang I. Moreover, reduced Ang II levels by exogenous ACE2 should stimulate the generation of Ang II that reflects inactivation of negative feedback mechanisms on renin. Therefore, it is difficult to conceive that sufficiently high levels of ACE2 can be achieved to chronically reduce Ang II and increase Ang-(1-7) except with the possible addition of an ACE inhibitor. In this case, supplementation of ACE2 may degrade residual levels of Ang II and the circulating levels of Ang-(1-7) may be augmented, particularly as the Ang-(1-7)-degrading pathway in the circulation is attenuated by the ACE inhibitor. In this regard, it is worth noting that Jin and colleagues have recently developed a Fc fusion protein to ACE2 that markedly prolongs the activity of the enzyme by reducing its clearance; however, the ACE2-fusion protein was assessed in an Ang II-infusion model that has suppressed endogenous Ang II, and other models of hypertension need to be examined [97].
It is presently unclear how increased circulating ACE2 augments Ang-(1-7) tissue levels as the intracellular mechanisms for Ang-(1-7) generation are not known. One possibility is that administered ACE2 increases circulating levels of Ang-(1-7) and the peptide is subsequently internalized by the MasR into a stable or protected intracellular compartment. Indeed, Gironacci and colleagues describe internalization of the Mas receptor following stimulation by Ang-(1-7); however, the assessment of intracellular Ang-(1-7) was not determined [98]. Alternatively, ACE2 treatment may alter that intracellular signaling milieu to attenuate oxidative stress or inflammation that impacts the local generation of Ang II and Ang-(1-7). Similar to neprilysin and ACE, ACE2 is primarily expressed as an ectocellular membrane-anchored peptidase that should readily access circulating or interstitial levels of Ang II. We observed that isolated nuclei from sheep renal cortex expressed ACE2 activity that converted Ang II to Ang-(1-7) and that the ACE2 inhibitor MLN4760 increased the oxidative stress response to Ang II suggesting that intracellular ACE2 may regulate the cellular balance of Ang II and Ang-(1-7) [99]. Tikellis et al. show that the renal content of Ang-(1-7) was reduced in ACE2-/- mice to a similar extent as that following chronic treatment with the potent ACE inhibitor perindopril [72]. Lavrentyev and colleagues also found that siRNA treatment against ACE2 reduced cellular levels of Ang-(1-7) in rat aortic smooth muscle cells [100]. Finally, Mompeon et al. recently demonstrate that estradiol increased the cellular levels of Ang-(1-7) that was associated with higher levels of both ACE and ACE2 by activation of the ERα subtype in human umbilical vein endothelial cells [101]. Overall, these studies suggest that intracellular expression of Ang-(1-7) is dependent in part on the processing of Ang II by ACE2 either intracellularly or extracellular conversion with the subsequent uptake of Ang-(1-7) by the Mas receptor.
Prolyl Carboxypeptidase
Prolyl carboxypeptidase is a monocarboxypeptidase [EC 3.4.16.2, Angiotensinase C, 58 kDa] with specificity for the C-terminal hydrolysis of the Pro-X bond where X is a hydrophobic residue [102]. The enzyme is capable of converting Ang II to Ang-(1-7), as well as degrading α-MSH, but activating the pre-kallikrein protease. In contrast to prolyl oligopeptidase and ACE2, the pH optima for prolyl carboxypeptidase is in the more acidic range of pH 4-5. Indeed, Grobe et al. find that prolyl carboxypeptidase was responsible for Ang II to Ang-(1-7) conversion in mouse renal cortex and urine at pH < 6.0 while ACE2 was predominant at pH >7.0 [103]. Velez et al. report that the mixed prolyl oligopeptidase/prolyl carboxypeptidase inhibitor ZPP partially blocked Ang II to Ang-(1-7) conversion, but had no effect on Ang I metabolism and concluded that the Ang-(1-7) forming enzymes from Ang II in human glomerular endothelial cells were prolyl carboxypeptidase and ACE2 [46]. Xu et al. find increased plasma levels of prolyl carboxypeptidase by ELISA in a cohort of obese diabetic patients; however, the circulating levels of Ang II or Ang-(1-7) were not evaluated in this study [104]. Interestingly, prolyl carboxypeptidase knockout mice exhibit higher blood pressure, increased oxidative stress, reduced vascular eNOS expression, renal damage, and cardiac dysfunction; however, neither circulating nor cardiac levels of Ang II and Ang-(1-7) were altered in this transgenic mouse as compared to wild-type [105, 106]. Finally, Jeong et al. report the prolyl carboxypeptidase knockout mice were protected against the metabolic effects of diet-induced obesity which runs counter to the expected effects of a higher Ang II: Ang-(1-7) ratio and suggest non-RAS targets of prolyl carboxypeptidase [107].
Angiotensin-(1-7) Degrading Pathways
Angiotensin-Converting Enzyme
The predominant pathway of the classical RAS for the conversion of Ang I to the bioactive peptide Ang II is catalyzed by the metallopeptidase ACE [EC 3.4.15.1], a dipeptidyl carboxypeptidase that cleaves two residues from the carboxy terminus of Ang I (Fig. 1) [108]. The peptidase is a membrane-bound, glycosylated protein (120–180 kDa) that is expressed in multiple tissues [108]. Soluble forms of the enzyme are present in the circulation, CSF, lymph fluid, and urine that retain peptidase activity [108]. The soluble form of ACE arises from the hydrolysis of the membrane-anchoring or stalk region of the protein that may reflect the processing by A Disintegrin and Metalloproteinase (ADAM) family of metallo-enzymes, although the precise role of ACE shedding in cardiovascular disease is presently unclear. Somatic ACE is characterized by two active sites termed N and C terminal domains that likely arose from the gene duplication of germinal or testicular ACE that contains only the single C terminal active site. In addition to forming Ang II, ACE degrades a number of other peptides that exhibit cardiovascular actions including bradykinin, substance P, and acetyl-SDKP [108]. Indeed, the cardioprotective effects of ACE inhibitors may reflect the protection of these peptides from metabolism, as well as the inhibitory effects on Ang II generation. In lieu of the increased circulating levels of Ang-(1-7) to ACE inhibitors, we postulated that Ang-(1-7) may be an endogenous substrate for ACE and demonstrated that ACE hydrolyzes Ang-(1-7) at the Ile5-His6 bond to yield the pentapeptide Ang-(1-5) and the dipeptide His-Pro (Fig. 1) [109]. Treatment with the potent ACE inhibitor lisinopril markedly reduced the clearance of the peptide and addition of the ACE inhibitor was required to demonstrate the accumulation of Ang-(1-7) derived from either Ang II or Ang I in isolated proximal tubules and following infusion of Ang I in SHR and WKY [44, 110]. Thus, the reduced metabolism of Ang-(1-7) likely contributes to the elevation in circulating levels of Ang-(1-7) following the chronic treatment with ACE inhibitors in experimental animals and in humans. These data suggest a pivotal role for ACE to regulate the balance of Ang II and Ang-(1-7) tone as the two peptides exhibit strikingly different actions from one another.
Dipeptidyl Peptidase 3
ACE clearly plays a role in the metabolism of Ang-(1-7), but there are other potential pathways that may regulate endogenous levels of the peptide [8]. Marshall and colleagues reported that ACE and a second peptidase activity in the sheep cerebrospinal fluid (CSF) degraded Ang-(1-7) [111–113]. Interestingly, the non-ACE degrading activity (subsequently identified as dipeptidyl peptidase 3, DPP3) accounted for a greater contribution of the metabolism of Ang-(1-7) than ACE [113]. Moreover, this activity was inversely correlated to CSF levels of Ang-(1-7) in control and betamethasone-exposed sheep, a model of fetal programming that exhibits elevated blood pressure and an attenuated baroreflex (Fig. 6). The Ang-(1-7)-degrading activity was also evident in sheep brain and kidney cortex, as well as in the human proximal tubule HK-2 cell line [52, 113]. The enzyme activity exhibited unusual characteristics as Ang I and other peptides equal to or greater than 10 residues were not substrates for the peptidase [52, 113]. Moreover, the inhibitor JMV-390, originally developed to block the metallo-endopeptidases neprilysin, thimet oligopeptidase and neurolysin, potently inhibited the Ang-(1-7)-degrading activity in the brain and kidney [IC50 < 1 nM]. Conversely , specific inhibitors against the three endopeptidases did not attenuate the Ang-(1-7) degrading activity [113]. Interestingly, the peptidase activity accounted for the sole degradative pathway in the cytosolic fraction and the media of the HK-2 cells while thimet oligopeptidase was responsible for intracellular generation of Ang-(1-7) from Ang I [52]. Utilizing the HK-2 cells as the source of the Ang-(1-7) degrading activity, we purified the peptidase from the cell cytosol by ion exchange and hydrophobic interaction chromatography and identified the enzyme as dipeptidyl peptidase 3 (EC 3.4.14.4, 85 kDa) [53].
Fig. 6.

Betamethasone-exposed (BMS) offspring exhibit higher mean arterial pressure (MAP) and greater CSF dipeptidyl aminopeptidase (DPP 3) activity than non-exposed sheep. MAP was higher in BMS animals at 6 months of age (a). CSF Ang-(1-7) peptide levels were lower in BMS animals (b). CSF DPP 3 was two-fold higher in BMS animals as compared to controls (c). Ang-(1-7) peptide levels negatively correlate with DPP 3 activity in the CSF (r = −0.81, P = 0.01) (d). Data are mean ± SEM; 4-5 per group; ∗P < 0.05 or ∗∗∗P < 0.001 vs. controls. (Adapted from Marshall et al. [113])
DPP 3 belongs to a family of metallo-aminopeptidases that sequentially cleave 2 residues from the N-terminus of peptides ≤8 residues in length that explains our previous results that Ang I, apelin-13 and neurotensin were not substrates for the Ang-(1-7)-degrading activity in the CSF and brain [113]. We obtained a human recombinant form of DPP 3 that hydrolyzed Ang-(1-7) in two steps [55]. DPP 3 initially cleaved Ang-(1-7) at the Arg2-Val3 bond to form Ang-(3-7) and the dipeptide Arg1-Asp2. Ang-(3-7) is then very rapidly cut at Tyr4-Ile5 to form Ang-(5-7) and Val3-Tyr4. The kinetic analysis of DPP 3 hydrolysis revealed a higher efficiency constant [kcat/Km] for Ang-(3-7) than Ang-(1-7) [53]. The preferred hydrolysis of Ang-(3-7) by DPP 3 explains the inability to demonstrate the accumulation of Ang-(3-7) following the initial metabolism of Ang-(1-7). In regard to an in vivo role for DPP 3 to modulate Ang-(1-7), human HK-2 cells were treated with varying doses of the JMV-390 inhibitor and we assessed both the endogenous content of Ang-(1-7) and the intracellular DPP-3/Ang-(1-7) degrading activity in the cells. Treatment with 20 nM and 200 nM JMV-390 reduced DPP3 activity by >30% and > 80%, respectively, as compared to control suggesting that the inhibitor effectively penetrates the cells [53]. The lower dose of JMV increased the cellular content Ang-(1-7) approximately two-fold, although this did not reach statistical significance. The higher dose of JMV, however, significantly reduced the intracellular levels of the peptide [53]. The higher JMV dose may block other peptidases including thimet oligopeptidase that may be involved in the intracellular generation of Ang-(1-7) in the renal cells [53]. Thus, the blockade of Ang-(1-7)-forming enzymes by the high-dose JMV may override any protective effects of DPP 3 inhibition.
Aminopeptidase A
Both Ang II and Ang-(1-7) share the same N-terminal sequence and are likely substrates for N-terminal directed metabolism. Aminopeptidase A [EC 3.4.11.7, 50 kDa] was characterized as a classic angiotensinase that hydrolyzed the Asp1-Arg2 bond to Ang II to form Ang-(2-8) or Ang III [114]. Grobe and colleagues applied “in situ” MALDI to characterize both renal and cardiac metabolism of exogenous Ang II and Ang-(1-7) [115, 116]. Ang-(1-7) was the primary product from Ang II in the renal cortex while Ang III was the major metabolite in the medulla [115]. In the heart, Ang III and Ang-(1-7) were products of Ang II metabolism catalyzed by Aminopeptidase A and ACE2, respectively [116]. These data confirm earlier HPLC-based studies on the contribution of ACE2 to Ang-(1-7) formation in the mouse and human heart [68, 117]. In mouse podocytes, Aminopeptidase A contributed to the metabolism of Ang-(1-7) to Ang-(2-7) and the subsequent conversion to Ang-(3-7) by arginine aminopeptidase (EC 3.4.11.6) – an identical pathway for the metabolism of Ang II to Ang III and Ang-(3-8) [46, 114]. Aminopeptidase A is widely distributed in tissues predominantly in a membrane-bound form, although soluble forms are present in the circulation, urine, and CSF [114]. Aminopeptidase A knockout mice show an increase in blood pressure, an enhanced pressor response to Ang II and greater susceptibility to glomeruli injury that would be consistent with a role of the peptidase in the metabolism of Ang II; however, it is not known the extent that circulating endogenous levels of Ang-(1-7) are altered to potentially mitigate against the cardiovascular effects of higher Ang II [118, 119].
AGE-Induced Peptidase
The progression of epithelial to mesenchymal transition (EMT) in renal injury is an important process that leads to increased fibrosis and loss of epithelial function [120–122]. We assessed the role of Ang-(1-7) in EMT of renal epithelial NRK-52e cells provoked by either advanced glycation end products (AGEs) or TGF-β [123]. Treatment with Ang-(1-7) abolished EMT in the NRK-52e cells by the inhibition of the non-canonical ERK 1/2 signaling pathway stimulated by AGE [123]. In addition to stimulating EMT, AGE exposure reduced the intracellular levels of Ang-(1-7), but not Ang II (Fig. 7a, b, respectively). The intracellular processing of Ang I to Ang-(1-7) by thimet oligopeptidase tended to be reduced by AGE; however, Ang-(1-7) metabolism was significantly increased by AGE exposure (Fig. 7c, d, respectively). AGE-induced EMT may reflect lower Ang-(1-7) expression in the renal epithelial cells that may be permissive for the progression of EMT and increased fibrosis [120]. Although our data suggest that DPP 3 is not responsible for the AGE-induced metabolism of Ang-(1-7) in the NRK-52 cells, a distinct endopeptidase activity may participate in the cellular metabolism of the peptide [123].
Fig. 7.

Advanced glycation end products (AGEs) reduce cellular expression of Ang-(1-7) in NRK-52e renal epithelial cells. The AGE methyl glyoxal albumin (MGA) reduces Ang-(1-7) content (a) but not Ang II levels (b) in NRK-52e cells. MGA tended to reduce processing of Ang I to Ang-(1-7) (c), but significantly increased the metabolism of Ang-(1-7) (d) in the 100,000 xg cytosolic fraction of NRK-52e cells. Data are means ± SEM; n = 5. ∗P < 0.05 versus Control. Activities are expressed as fmol/minute/mg protein. (Adapted from Alzayadneh and Chappell [124])
Conclusion
Accumulating evidence from multiple laboratories clearly indicates that activation of the Ang-(1-7)-AT7/MasR axis appears to be a potentially important therapeutic target in the treatment of cardiovascular disease and other pathologies [125]. In lieu of the described peptidases that degrade Ang-(1-7) and the apparent reliance on Ang II as an endogenous substrate for the generation of the peptide in several tissues, the development of selective non-peptide analogs of Ang-(1-7) that target both the Mas receptor and the newly described Mas-related receptor D (MRG-D) [125] may be of particular benefit in terms of their oral availability, greater resistance to peptidase metabolism, improved selectivity, their potential ability to accumulate within the cell and to cross the blood-brain barrier.
A functional intracellular AT7/MasR was first identified on isolated nuclei from the ovine kidney that was linked to NO generation following exposure to low pM doses of Ang-(1-7) [124]. These findings were recently corroborated in the brain that revealed evidence of the MasR on nuclei and mitochondria, as well as Ang-(1-7)-dependent stimulation of NO [126]. Moreover, AT7/MasR binding on renal nuclei and the Ang-(1-7)-evoked NO response were attenuated in aged sheep and in adult sheep exposed to betamethasone in utero as compared to the younger non-exposed animals [9, 99]. Conversely, intracellular levels of the AT1R that were associated with stimulation of oxidative stress on isolated nuclei were increased in the older animals and in the betamethasone-exposed sheep [9, 98]. Abadir et al. originally reported an increased ratio of AT1R: AT2R receptors in isolated renal mitochondrial of aging mice [127]. Valenzuela et al. also find reduced an increased AT1R:AT2R ratio in brain mitochondria of aging mice that may lead to higher cellular levels of oxidative stress [128, 129]. Overall, these novel findings of nuclear and mitochondrial angiotensin receptors support extensive evidence for an intracellular RAS within various tissues although the exact role of an activated intracellular RAS to influence cardiovascular disease and other pathologies is not resolved and requires further study. Nevertheless, peptidase-resistant and cell-permeable agonists of the Ang-(1-7) axis may provide additional cardioprotective effects to conventional approaches to block the RAS and may expand the targeted therapies required to combat cardiovascular disease.
Acknowledgments
I am most fortunate to have shared a moment in time with a number of very talented graduate students and postdoctoral fellows including Karl Pendergrass, Liliya Yamaleyeva, Hossam Shaltout, TanYa Gwathmey, Sarah Lindsey, Jonathan Cohen, Shea Gilliam, Allyson Marshall, Ebaa Al-Zayadneh, Bryan Wilson, and Nildris Cruz-Diaz, as well as Brian Westwood and Nancy Pirro whose research efforts to the studies in this review I gratefully acknowledge. Portions of this work were supported by grants from the National Institute of Health grants (HL-56973, HL-51952, HD-084227, HD-047584, HL-091797, HL-146818, GM102773 and HD-017644) and the American Heart Association (AHA-151521, AHA-355741 and AHA-18TPA34170522). An unrestricted grant from the Farley-Hudson Foundation (Jacksonville, NC), Groskert Heart Fund, and the Wake Forest Venture Fund is also acknowledged.
Contributor Information
Robson Augusto Souza Santos, Email: robsonsant@gmail.com.
Mark C. Chappell, Email: mchappel@wakehealth.edu
Literature Cited
- 1.Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RA, et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension (Dallas, TX: 1979) 2008;51(5):1312–1317. doi: 10.1161/hypertensionaha.107.108944. [DOI] [PubMed] [Google Scholar]
- 2.Aroor AR, Demarco VG, Jia G, Sun Z, Nistala R, Meininger GA, et al. The role of tissue renin-angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front Endocrinol. 2013;4:161. doi: 10.3389/fendo.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98(1):121–128. doi: 10.1016/j.amjcard.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 4.Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116(6):960–975. doi: 10.1161/circresaha.116.303587. [DOI] [PubMed] [Google Scholar]
- 5.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 6.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11(2):180–186. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chappell MC, Brosnihan KB, Diz DI, Ferrario CM. Identification of angiotensin-(1-7) in rat brain. Evidence for differential processing of angiotensin peptides. J Biol Chem. 1989;264(28):16518–16523. [PubMed] [Google Scholar]
- 8.Santos RA. Angiotensin-(1-7) Hypertension (Dallas, TX: 1979) 2014;63(6):1138–1147. doi: 10.1161/hypertensionaha.113.01274. [DOI] [PubMed] [Google Scholar]
- 9.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the angiotensin converting enzyme 2-angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol. 2014;4:201. doi: 10.3389/fendo.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carey RM, Padia SH. Role of angiotensin AT(2) receptors in natriuresis: Intrarenal mechanisms and therapeutic potential. Clin Exp Pharmacol Physiol. 2013;40(8):527–534. doi: 10.1111/1440-1681.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sumners C, de Kloet AD, Krause EG, Unger T, Steckelings UM. Angiotensin type 2 receptors: blood pressure regulation and end organ damage. Curr Opin Pharmacol. 2015;21:115–121. doi: 10.1016/j.coph.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension (Dallas, TX: 1979) 2011;57(4):859–864. doi: 10.1161/hypertensionaha.110.167957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension (Dallas, TX: 1979) 2011;57(3):355–362. doi: 10.1161/hypertensionaha.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension (Dallas, TX: 1979) 2011;57(3):594–599. doi: 10.1161/hypertensionaha.110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters J, Clausmeyer S. Intracellular sorting of renin: cell type specific differences and their consequences. J Mol Cell Cardiol. 2002;34(12):1561–1568. doi: 10.1006/jmcc.2002.2079. [DOI] [PubMed] [Google Scholar]
- 16.Lavoie JL, Liu X, Bianco RA, Beltz TG, Johnson AK, Sigmund CD. Evidence supporting a functional role for intracellular renin in the brain. Hypertension (Dallas, TX: 1979) 2006;47(3):461–466. doi: 10.1161/01.HYP.0000203308.52919.dc. [DOI] [PubMed] [Google Scholar]
- 17.Ishigami T, Kino T, Chen L, Minegishi S, Araki N, Umemura M, et al. Identification of bona fide alternative renin transcripts expressed along cortical tubules and potential roles in promoting insulin resistance in vivo without significant plasma renin activity elevation. Hypertension (Dallas, TX: 1979) 2014;64(1):125–133. doi: 10.1161/hypertensionaha.114.03394. [DOI] [PubMed] [Google Scholar]
- 18.Peters J, Wanka H, Peters B, Hoffmann S. A renin transcript lacking exon 1 encodes for a non-secretory intracellular renin that increases aldosterone production in transgenic rats. J Cell Mol Med. 2008;12(4):1229–1237. doi: 10.1111/j.1582-4934.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clausmeyer S, Sturzebecher R, Peters J. An alternative transcript of the rat renin gene can result in a truncated prorenin that is transported into adrenal mitochondria. Circ Res. 1999;84(3):337–344. doi: 10.1161/01.RES.84.3.337. [DOI] [PubMed] [Google Scholar]
- 20.Wanka H, Kessler N, Ellmer J, Endlich N, Peters BS, Clausmeyer S, et al. Cytosolic renin is targeted to mitochondria and induces apoptosis in H9c2 rat cardiomyoblasts. J Cell Mol Med. 2009;13(9a):2926–2937. doi: 10.1111/j.1582-4934.2008.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wanka H, Staar D, Lutze P, Peters B, Hildebrandt J, Beck T, et al. Anti-necrotic and cardioprotective effects of a cytosolic renin isoform under ischemia-related conditions. J Mol Med (Berlin, Germany) 2016;94(1):61–69. doi: 10.1007/s00109-015-1321-z. [DOI] [PubMed] [Google Scholar]
- 22.Wilson BA, Nautiyal M, Gwathmey TM, Rose JC, Chappell MC. Evidence for a mitochondrial angiotensin-(1-7) system in the kidney. Am J Physiol Renal Physiol. 2016;310(7):F637–Ff45. doi: 10.1152/ajprenal.00479.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alzayadneh EM, Chappell MC. Nuclear expression of renin-angiotensin system components in NRK-52E renal epithelial cells. J Renin Angiotensin Aldosterone Syst: JRAAS. 2015;16(4):1135–1148. doi: 10.1177/1470320313515039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li XC, Cook JL, Rubera I, Tauc M, Zhang F, Zhuo JL. Intrarenal transfer of an intracellular fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am J Physiol Renal Physiol. 2011;300(5):F1076–F1088. doi: 10.1152/ajprenal.00329.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103(47):17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XC, Hopfer U, Zhuo JL. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2012;303(12):F1617–F1628. doi: 10.1152/ajprenal.00219.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT(1a) receptors induces blood pressure responses to intracellular angiotensin II in AT(1a) receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2013;304(8):R588–R598. doi: 10.1152/ajpregu.00338.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davisson RL, Ding Y, Stec DE, Catterall JF, Sigmund CD. Novel mechanism of hypertension revealed by cell-specific targeting of human angiotensinogen in transgenic mice. Physiol Genomics. 1999;1(1):3–9. doi: 10.1152/physiolgenomics.1999.1.1.3. [DOI] [PubMed] [Google Scholar]
- 29.Campbell DJ. Angiotensin II generation in vivo: does it involve enzymes other than renin and angiotensin-converting enzyme? J Renin Angiotensin Aldosterone Syst: JRAAS. 2012;13(2):314–316. doi: 10.1177/1470320312447162. [DOI] [PubMed] [Google Scholar]
- 30.Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun. 2006;350(4):1026–1031. doi: 10.1016/j.bbrc.2006.09.146. [DOI] [PubMed] [Google Scholar]
- 31.Westwood BM, Chappell MC. Divergent pathways for the angiotensin-(1-12) metabolism in the rat circulation and kidney. Peptides. 2012;35(2):190–195. doi: 10.1016/j.peptides.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner AJ. Neprilysin. In: Barrett A, Woessner J, Rawlings N, editors. Handbook of proteolytic enzymes. 2. London: Elsevier Academic Press; 2004. pp. 419–426. [Google Scholar]
- 33.McKinnie SM, Fischer C, Tran KM, Wang W, Mosquera F, Oudit GY, et al. The metalloprotease neprilysin degrades and inactivates apelin peptides. Chembiochem: Eur J Chem Biol. 2016;17(16):1495–1498. doi: 10.1002/cbic.201600244. [DOI] [PubMed] [Google Scholar]
- 34.Cataliotti A, Boerrigter G, Chen HH, Jougasaki M, Costello LC, Tsuruda T, et al. Differential actions of vasopeptidase inhibition versus angiotensin-converting enzyme inhibition on diuretic therapy in experimental congestive heart failure. Circulation. 2002;105(5):639–644. doi: 10.1161/hc0502.102962. [DOI] [PubMed] [Google Scholar]
- 35.Tikkanen I, Tikkanen T, Cao Z, Allen TJ, Davis BJ, Lassila M, et al. Combined inhibition of neutral endopeptidase with angiotensin converting enzyme or endothelin converting enzyme in experimental diabetes. J Hypertens. 2002;20(4):707–714. doi: 10.1097/00004872-200204000-00029. [DOI] [PubMed] [Google Scholar]
- 36.Kubota E, Dean RG, Hubner RA, Casley DJ, Johnston CI, Burrell LM. Differential tissue and enzyme inhibitory effects of the vasopeptidase inhibitor omapatrilat in the rat. Clin Sci (London, England: 1979) 2003;105(3):339–345. doi: 10.1042/cs20030097. [DOI] [PubMed] [Google Scholar]
- 37.Hubers SA, Brown NJ. Combined angiotensin receptor antagonism and neprilysin inhibition. Circulation. 2016;133(11):1115–1124. doi: 10.1161/circulationaha.115.018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt 1):45–51. doi: 10.1042/bj20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertension (Dallas, TX: 1979) 1992;19(6 Pt 2):692–696. doi: 10.1161/01.HYP.19.6.692. [DOI] [PubMed] [Google Scholar]
- 40.Iyer SN, Chappell MC, Averill DB, Diz DI, Ferrario CM. Vasodepressor actions of angiotensin-(1-7) unmasked during combined treatment with lisinopril and losartan. Hypertension (Dallas, TX: 1979) 1998;31(2):699–705. doi: 10.1161/01.HYP.31.2.699. [DOI] [PubMed] [Google Scholar]
- 41.Chappell MC, Gomez MN, Pirro NT, Ferrario CM. Release of angiotensin-(1-7) from the rat hindlimb: influence of angiotensin-converting enzyme inhibition. Hypertension (Dallas, TX: 1979) 2000;35(1 Pt 2):348–352. doi: 10.1161/01.HYP.35.1.348. [DOI] [PubMed] [Google Scholar]
- 42.Campbell DJ, Anastasopoulos F, Duncan AM, James GM, Kladis A, Briscoe TA. Effects of neutral endopeptidase inhibition and combined angiotensin converting enzyme and neutral endopeptidase inhibition on angiotensin and bradykinin peptides in rats. J Pharmacol Exp Ther. 1998;287(2):567–577. [PubMed] [Google Scholar]
- 43.Domenig O, Manzel A, Grobe N, Konigshausen E, Kaltenecker CC, Kovarik JJ, et al. Neprilysin is a mediator of alternative renin-angiotensin-system activation in the murine and human kidney. Sci Rep. 2016;6:33678. doi: 10.1038/srep33678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaltout HA, Westwood BM, Averill DB, Ferrario CM, Figueroa JP, Diz DI, et al. Angiotensin metabolism in renal proximal tubules, urine, and serum of sheep: evidence for ACE2-dependent processing of angiotensin II. Am J Physiol Renal Physiol. 2007;292(1):F82–F91. doi: 10.1152/ajprenal.00139.2006. [DOI] [PubMed] [Google Scholar]
- 45.Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1---7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol. 2000;279(5):F841–F850. doi: 10.1152/ajprenal.2000.279.5.F841. [DOI] [PubMed] [Google Scholar]
- 46.Velez JC, Ierardi JL, Bland AM, Morinelli TA, Arthur JM, Raymond JR, et al. Enzymatic processing of angiotensin peptides by human glomerular endothelial cells. Am J Physiol Renal Physiol. 2012;302(12):F1583–F1594. doi: 10.1152/ajprenal.00087.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwacke JH, Spainhour JC, Ierardi JL, Chaves JM, Arthur JM, Janech MG, et al. Network modeling reveals steps in angiotensin peptide processing. Hypertension (Dallas, TX: 1979) 2013;61(3):690–700. doi: 10.1161/hypertensionaha.111.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velez JC, Ryan KJ, Harbeson CE, Bland AM, Budisavljevic MN, Arthur JM, et al. Angiotensin I is largely converted to angiotensin (1-7) and angiotensin (2-10) by isolated rat glomeruli. Hypertension (Dallas, TX: 1979) 2009;53(5):790–797. doi: 10.1161/hypertensionaha.109.128819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Modrall JG, Sadjadi J, Brosnihan KB, Gallagher PE, Yu CH, Kramer GL, et al. Depletion of tissue angiotensin-converting enzyme differentially influences the intrarenal and urinary expression of angiotensin peptides. Hypertension (Dallas, TX: 1979) 2004;43(4):849–853. doi: 10.1161/01.HYP.0000121462.27393.f6. [DOI] [PubMed] [Google Scholar]
- 50.Barrett AJ, Chen JM. Thimet oligopeptidase. In: Barrett A, Woessner J, Rawlings N, editors. Handbook of Proteolytic enzymes. 2. London: Elsevier Academic Press; 2004. pp. 352–356. [Google Scholar]
- 51.Pereira MG, Souza LL, Becari C, Duarte DA, Camacho FR, Oliveira JA, et al. Angiotensin II-independent angiotensin-(1-7) formation in rat hippocampus: involvement of thimet oligopeptidase. Hypertension (Dallas, TX: 1979) 2013;62(5):879–885. doi: 10.1161/hypertensionaha.113.01613. [DOI] [PubMed] [Google Scholar]
- 52.Wilson BA, Cruz-Diaz N, Marshall AC, Pirro NT, Su Y, Gwathmey TM, et al. An angiotensin-(1-7) peptidase in the kidney cortex, proximal tubules, and human HK-2 epithelial cells that is distinct from insulin-degrading enzyme. Am J Physiol Renal Physiol. 2015;308(6):F594–F601. doi: 10.1152/ajprenal.00609.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cruz-Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, Chappell MC. Identification of dipeptidyl peptidase 3 as the angiotensin-(1-7) degrading peptidase in human HK-2 renal epithelial cells. Peptides. 2016;83:29–37. doi: 10.1016/j.peptides.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suski M, Gebska A, Olszanecki R, Stachowicz A, Uracz D, Madej J, et al. Influence of atorvastatin on angiotensin I metabolism in resting and TNF-alpha-activated rat vascular smooth muscle cells. J Renin Angiotensin Aldosterone Syst: JRAAS. 2014;15(4):378–383. doi: 10.1177/1470320313475907. [DOI] [PubMed] [Google Scholar]
- 55.Chappell MC, Tallant EA, Brosnihan KB, Ferrario CM. Conversion of angiotensin I to angiotensin-(1-7) by thimet oligopeptidase (EC 3.4. 24.15) in vascular smooth muscle cells. J Vasc Med Biol. 1994;5:129–137. [Google Scholar]
- 56.Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension (Dallas, TX: 1979) 2007;50(4):596–599. doi: 10.1161/hypertensionaha.106.076216. [DOI] [PubMed] [Google Scholar]
- 57.Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol. 2012;302(5):R518–R530. doi: 10.1152/ajpregu.00525.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol. 2012;302(5):R494–R509. doi: 10.1152/ajpregu.00487.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (London, England: 1979) 2012;123(5):273–284. doi: 10.1042/cs20120089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol. 2012;302(5):R482–R493. doi: 10.1152/ajpregu.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides. 2012;38(2):437–445. doi: 10.1016/j.peptides.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Myohanen TT, Garcia-Horsman JA, Tenorio-Laranga J, Mannisto PT. Issues about the physiological functions of prolyl oligopeptidase based on its discordant spatial association with substrates and inconsistencies among mRNA, protein levels, and enzymatic activity. J Histochem Cytochem: Off J Histochem Soc. 2009;57(9):831–848. doi: 10.1369/jhc.2009.953711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santos RA, Brosnihan KB, Jacobsen DW, DiCorleto PE, Ferrario CM. Production of angiotensin-(1-7) by human vascular endothelium. Hypertension (Dallas, TX: 1979) 1992;19(2 Suppl):Ii56–Ii61. doi: 10.1161/01.hyp.19.2_suppl.ii56. [DOI] [PubMed] [Google Scholar]
- 64.Penttinen A, Tenorio-Laranga J, Siikanen A, Morawski M, Rossner S, Garcia-Horsman JA. Prolyl oligopeptidase: a rising star on the stage of neuroinflammation research. CNS Neurol Disord Drug Targets. 2011;10(3):340–348. doi: 10.2174/187152711794653742. [DOI] [PubMed] [Google Scholar]
- 65.Lawandi J, Gerber-Lemaire S, Juillerat-Jeanneret L, Moitessier N. Inhibitors of prolyl oligopeptidases for the therapy of human diseases: defining diseases and inhibitors. J Med Chem. 2010;53(9):3423–3438. doi: 10.1021/jm901104g. [DOI] [PubMed] [Google Scholar]
- 66.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87(5):E1–E9. doi: 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 67.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 68.Garabelli PJ, Modrall JG, Penninger JM, Ferrario CM, Chappell MC. Distinct roles for angiotensin-converting enzyme 2 and carboxypeptidase A in the processing of angiotensins within the murine heart. Exp Physiol. 2008;93(5):613–621. doi: 10.1113/expphysiol.2007.040246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension (Dallas, TX: 1979) 2006;48(5):914–920. doi: 10.1161/01.hyp.0000244543.91937.79. [DOI] [PubMed] [Google Scholar]
- 70.Yamaleyeva LM, Gilliam-Davis S, Almeida I, Brosnihan KB, Lindsey SH, Chappell MC. Differential regulation of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis rats with early-onset diabetes. Am J Physiol Renal Physiol. 2012;302(11):F1374–F1384. doi: 10.1152/ajprenal.00656.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, Klein AL, et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. 2009;15(7):565–571. doi: 10.1016/j.cardfail.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57(4):1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 73.South AM, Shaltout HA, Washburn LK, Hendricks AS, Diz DI, Chappell MC. Fetal programming and the angiotensin-(1-7) axis: a review of the experimental and clinical data. Clin Sci (Lond) 2019;113(1):55–74. doi: 10.1042/CS20171550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Soler MJ, Wysocki J, Ye M, Lloveras J, Kanwar Y, Batlle D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72(5):614–623. doi: 10.1038/sj.ki.5002373. [DOI] [PubMed] [Google Scholar]
- 75.Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168(6):1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 77.Oudit GY, Kassiri Z, Patel MP, Chappell M, Butany J, Backx PH, et al. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc Res. 2007;75(1):29–39. doi: 10.1016/j.cardiores.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 78.Chhabra KH, Xia H, Pedersen KB, Speth RC, Lazartigues E. Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. Am J Physiol Endocrinol Metab. 2013;304(8):E874–E884. doi: 10.1152/ajpendo.00490.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59(2):529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhong JiuChang, Basu Ratnadeep, Guo Danny, Chow Fung L., Byrns Simon, Schuster Manfred, Loibner Hans, Wang Xiu-hua, Penninger Josef M., Kassiri Zamaneh, Oudit Gavin Y. Angiotensin-Converting Enzyme 2 Suppresses Pathological Hypertrophy, Myocardial Fibrosis, and Cardiac Dysfunction. Circulation. 2010;122(7):717–728. doi: 10.1161/CIRCULATIONAHA.110.955369. [DOI] [PubMed] [Google Scholar]
- 81.Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension (Dallas, TX: 1979) 2010;55(1):90–98. doi: 10.1161/hypertensionaha.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye M, Wysocki J, Gonzalez-Pacheco FR, Salem M, Evora K, Garcia-Halpin L, et al. Murine recombinant angiotensin-converting enzyme 2: effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension (Dallas, TX: 1979) 2012;60(3):730–740. doi: 10.1161/hypertensionaha.112.198622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, et al. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol. 2005;90(5):783–790. doi: 10.1113/expphysiol.2005.031096. [DOI] [PubMed] [Google Scholar]
- 84.Diez-Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27(1):12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- 85.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182(8):1065–1072. doi: 10.1164/rccm.200912-1840OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension (Dallas, TX: 1979) 2007;49(4):926–931. doi: 10.1161/01.hyp.0000259942.38108.20. [DOI] [PubMed] [Google Scholar]
- 87.Bae EH, Fang F, Williams VR, Konvalinka A, Zhou X, Patel VB, et al. Murine recombinant angiotensin-converting enzyme 2 attenuates kidney injury in experimental Alport syndrome. Kidney Int. 2017;91(6):1347–1361. doi: 10.1016/j.kint.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 88.Wysocki J, Ye M, Khattab AM, Fogo A, Martin A, David NV, et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017;91(6):1336–1346. doi: 10.1016/j.kint.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310(2):H137–H152. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qi Y, Zhang J, Cole-Jeffrey CT, Shenoy V, Espejo A, Hanna M, et al. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension (Dallas, TX: 1979) 2013;62(4):746–752. doi: 10.1161/hypertensionaha.113.01337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Maria ML, Araujo LD, Fraga-Silva RA, Pereira LA, Ribeiro HJ, Menezes GB, et al. Anti-hypertensive effects of diminazene aceturate: an angiotensin- converting enzyme 2 activator in rats. Protein Pept Lett. 2016;23(1):9–16. doi: 10.2174/0929866522666151013130550. [DOI] [PubMed] [Google Scholar]
- 92.Macedo LM, Souza AP, De Maria ML, Borges CL, Soares CM, Pedrino GR, et al. Cardioprotective effects of diminazene aceturate in pressure-overloaded rat hearts. Life Sci. 2016;155:63–69. doi: 10.1016/j.lfs.2016.04.036. [DOI] [PubMed] [Google Scholar]
- 93.Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187(6):648–657. doi: 10.1164/rccm.201205-0880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: studies in vivo, ex vivo, and in vitro. Hypertension (Dallas, TX: 1979) 2014;63(4):774–782. doi: 10.1161/hypertensionaha.113.02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Velkoska E, Patel SK, Griggs K, Burrell LM. Diminazene aceturate improves cardiac fibrosis and diastolic dysfunction in rats with kidney disease. PLoS One. 2016;11(8):e161760. doi: 10.1371/journal.pone.0161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raffai G, Khang G, Vanhoutte PM. Angiotensin-(1-7) augments endothelium-dependent relaxations of porcine coronary arteries to bradykinin by inhibiting angiotensin-converting enzyme 1. J Cardiovasc Pharmacol. 2014;63(5):453–460. doi: 10.1097/fjc.0000000000000069. [DOI] [PubMed] [Google Scholar]
- 97.Liu Pan, Wysocki Jan, Souma Tomokazu, Ye Minghao, Ramirez Veronica, Zhou Bisheng, Wilsbacher Lisa D., Quaggin Susan E., Batlle Daniel, Jin Jing. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney International. 2018;94(1):114–125. doi: 10.1016/j.kint.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 98.Cerniello FM, Carretero OA, Longo Carbajosa NA, Cerrato BD, Santos RA, Grecco HE, et al. MAS1 receptor trafficking involves ERK1/2 activation through a beta-arrestin2-dependent pathway. Hypertension (Dallas, TX: 1979) 2017;70(5):982–989. doi: 10.1161/hypertensionaha.117.09789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gwathmey TM, Shaltout HA, Rose JC, Diz DI, Chappell MC. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension (Dallas, TX: 1979) 2011;57(3):620–626. doi: 10.1161/hypertensionaha.110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lavrentyev EN, Estes AM, Malik KU. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ Res. 2007;101(5):455–464. doi: 10.1161/circresaha.107.151852. [DOI] [PubMed] [Google Scholar]
- 101.Mompeon A, Lazaro-Franco M, Bueno-Beti C, Perez-Cremades D, Vidal-Gomez X, Monsalve E, et al. Estradiol, acting through ER alpha, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1-7 production. Mol Cell Endocrinol. 2016;422:1–8. doi: 10.1016/j.mce.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Tan F, Erdos EG. Lysosomal Pro-X carboxypeptidase. In: Barrett A, Woessner J, Rawlings N, editors. Handbook of proteolytic enzymes. 2. London: Elsevier Academic Press; 2004. pp. 1936–1938. [Google Scholar]
- 103.Grobe N, Weir NM, Leiva O, Ong FS, Bernstein KE, Schmaier AH, et al. Identification of prolyl carboxypeptidase as an alternative enzyme for processing of renal angiotensin II using mass spectrometry. Am J Physiol Cell Physiol. 2013;304(10):C945–C953. doi: 10.1152/ajpcell.00346.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu S, Lind L, Zhao L, Lindahl B, Venge P. Plasma prolylcarboxypeptidase (angiotensinase C) is increased in obesity and diabetes mellitus and related to cardiovascular dysfunction. Clin Chem. 2012;58(7):1110–1115. doi: 10.1373/clinchem.2011.179291. [DOI] [PubMed] [Google Scholar]
- 105.Adams GN, LaRusch GA, Stavrou E, Zhou Y, Nieman MT, Jacobs GH, et al. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood. 2011;117(14):3929–3937. doi: 10.1182/blood-2010-11-318527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maier C, Schadock I, Haber PK, Wysocki J, Ye M, Kanwar Y, et al. Prolylcarboxypeptidase deficiency is associated with increased blood pressure, glomerular lesions, and cardiac dysfunction independent of altered circulating and cardiac angiotensin II. J Mol Med (Berlin, Germany) 2017;95(5):473–486. doi: 10.1007/s00109-017-1513-9. [DOI] [PubMed] [Google Scholar]
- 107.Jeong JK, Szabo G, Raso GM, Meli R, Diano S. Deletion of prolyl carboxypeptidase attenuates the metabolic effects of diet-induced obesity. Am J Physiol Endocrinol Metab. 2012;302(12):E1502–E1510. doi: 10.1152/ajpendo.00544.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, et al. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013;65(1):1–46. doi: 10.1124/pr.112.006809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension (Dallas, TX: 1979) 1998;31(1 Pt 2):362–367. doi: 10.1161/01.HYP.31.1.362. [DOI] [PubMed] [Google Scholar]
- 110.Yamada K, Iyer SN, Chappell MC, Ganten D, Ferrario CM. Converting enzyme determines plasma clearance of angiotensin-(1-7) Hypertension (Dallas, TX: 1979) 1998;32(3):496–502. doi: 10.1161/01.HYP.32.3.496. [DOI] [PubMed] [Google Scholar]
- 111.Marshall AC, Shaltout HA, Pirro NT, Rose JC, Diz DI, Chappell MC. Antenatal betamethasone exposure is associated with lower ANG-(1-7) and increased ACE in the CSF of adult sheep. Am J Physiol Regul Integr Comp Physiol. 2013;305(7):R679–R688. doi: 10.1152/ajpregu.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marshall AC, Shaltout HA, Pirro NT, Rose JC, Diz DI, Chappell MC. Enhanced activity of an angiotensin-(1-7) neuropeptidase in glucocorticoid-induced fetal programming. Peptides. 2014;52:74–81. doi: 10.1016/j.peptides.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marshall AC, Pirro NT, Rose JC, Diz DI, Chappell MC. Evidence for an angiotensin-(1-7) neuropeptidase expressed in the brain medulla and CSF of sheep. J Neurochem. 2014;130(2):313–323. doi: 10.1111/jnc.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang JO, Cooper MD. Aminopeptidase A. In: Barrett A, Woessner J, Rawlings N, editors. Handbook of proteolytic enzymes. 2. London: Elsevier Academic Press; 2004. pp. 299–301. [Google Scholar]
- 115.Grobe N, Elased KM, Cool DR, Morris M. Mass spectrometry for the molecular imaging of angiotensin metabolism in kidney. Am J Physiol Endocrinol Metab. 2012;302(8):E1016–E1024. doi: 10.1152/ajpendo.00515.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alghamri MS, Morris M, Meszaros JG, Elased KM, Grobe N. Novel role of aminopeptidase-A in angiotensin-(1-7) metabolism post myocardial infarction. Am J Physiol Heart Circ Physiol. 2014;306(7):H1032–H1040. doi: 10.1152/ajpheart.00911.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation. 2003;108(14):1707–1712. doi: 10.1161/01.cir.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 118.Velez JCQ, Arif E, Rodgers J, Hicks MP, Arthur JM, Nihalani D, et al. Deficiency of the angiotensinase aminopeptidase A increases susceptibility to glomerular injury. J Am Soc Nephrol: JASN. 2017;28(7):2119–2132. doi: 10.1681/asn.2016111166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitsui T, Nomura S, Okada M, Ohno Y, Kobayashi H, Nakashima Y, et al. Hypertension and angiotensin II hypersensitivity in aminopeptidase A-deficient mice. Mol Med (Cambridge, Mass) 2003;9(1-2):57–62. doi: 10.1007/BF03402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chappell MC, Al Zayadneh EM. Angiotensin-(1-7) and the regulation of anti-fibrotic signaling pathways. J Cell Signal. 2017;2(1):1–3. [DOI] [PMC free article] [PubMed]
- 121.Inoue T, Umezawa A, Takenaka T, Suzuki H, Okada H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015;87(1):233–238. doi: 10.1038/ki.2014.235. [DOI] [PubMed] [Google Scholar]
- 122.Tennakoon Anusha, Izawa Takeshi, Kuwamura Mitsuru, Yamate Jyoji. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis. Journal of Clinical Medicine. 2015;5(1):4. doi: 10.3390/jcm5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alzayadneh EM, Chappell MC. Angiotensin-(1-7) abolishes AGE-induced cellular hypertrophy and myofibroblast transformation via inhibition of ERK1/2. Cell Signal. 2014;26(12):3027–3035. doi: 10.1016/j.cellsig.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, et al. Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol. 2010;299(5):F983–F990. doi: 10.1152/ajprenal.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1-7)/MAS Axis of the renin-angiotensin system: focus on angiotensin-(1-7) Physiol Rev. 2018;98(1):505–553. doi: 10.1152/physrev.00023.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Costa-Besada Maria A., Valenzuela Rita, Garrido-Gil Pablo, Villar-Cheda Begoña, Parga Juan A., Lanciego Jose L., Labandeira-Garcia Jose L. Paracrine and Intracrine Angiotensin 1-7/Mas Receptor Axis in the Substantia Nigra of Rodents, Monkeys, and Humans. Molecular Neurobiology. 2017;55(7):5847–5867. doi: 10.1007/s12035-017-0805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011;108(36):14849–14854. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Valenzuela R, Costa-Besada MA, Iglesias-Gonzalez J, Perez-Costas E, Villar-Cheda B, Garrido-Gil P, et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016;7(10):e2427. doi: 10.1038/cddis.2016.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Villar-Cheda B, Costa-Besada MA, Valenzuela R, Perez-Costas E, Melendez-Ferro M, Labandeira-Garcia JL. The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 2017;8(9):e3044. doi: 10.1038/cddis.2017.439. [DOI] [PMC free article] [PubMed] [Google Scholar]