

Abstract
Cytokine storm defines a dysregulation of and an excessively exaggerated immune response most often accompanying selected viral infections and several autoimmune diseases. Newly emerging and re-emerging infections of the respiratory tract, especially influenza, SARS, and hantavirus post considerable medical problems. Their morbidities and mortalities are often a direct result of cytokine storm. This chapter visits primarily influenza virus infection and resultant cytokine storm. It provides the compelling evidence that illuminates cytokine storm in influenza pathogenesis and the clear findings that cytokine storm is chemically tractable by therapy directed toward sphingosine-1-phosphate receptor (S1PR) modulation, specifically S1P1R agonist therapy. The mechanism(s) of how S1P1R signaling works and the pathways involved are subjects of this review.
Keywords: Influenza Virus, Alveolar Macrophage, Severe Acute Respiratory Syndrome, Influenza Infection, Influenza Virus Infection
Introduction
Newly emerging and re-emerging infections of the respiratory tract pose considerable medical and public health concerns as well as economic hardships to humans and countries. The last century witnessed at least five pandemics: 1918/1919, H (viral hemagglutinin) 1 N (viral neuraminidase) 1 Spanish influenza; 1957, H2N2 Asia influenza; 1968, H3N2 Hong Kong influenza; 1977, H1N1 Russian influenza; and 1997, H5N1 bird influenza (reviewed Wright et al. 2007). In twenty-first century alone, two viral pandemics have already occurred—the first was in 2002 when the new viral pandemic, severe acute respiratory syndrome (SARS), appeared (reviewed Oldstone 2010), followed by the first influenza virus pandemic in 2009, H1N1 swine influenza (Dawood et al. 2012). Moreover, Hantaviruses have infected humans in the past and recently in an outbreak at Yellowstone National Park. These viral infections loom as important zoonotic human diseases with the threat of human to human transmission and excessively high mortality rates. For example, 1918/1919 H1N1 influenza infections caused the greatest loss of life from any infectious disease or medical condition known, visiting roughly 5 % of the world’s population and killing 2 % or 40–50 million persons (Ahmed et al. 2007; Johnson and Mueller 2002). The most recent influenza pandemic, 2009 H1N1 swine influenza, rapidly infected millions worldwide with estimates exceeding 290,000 deaths of which more than 201,000 resulted from respiratory failure and over 83,000 from cardiovascular complications (Dawood et al. 2012). All of the above diseases in humans (Arankalle et al. 2010; Cheng et al. 2010; Lee et al. 2011) and experimental animals (Baskin et al. 2009; Kobasa et al. 2007; Marcelin et al. 2011; Zhang et al. 2012) are accompanied by early exacerbation and dysregulation of innate immune responses, a combination of events called “cytokine storm.” Severe disease and death following infection correlated strongly with the cytokine storm.
Susceptibility or resistance to any viral infection is determined by the balance between the virulence of the infecting agent and the resistance of the host including the aggressiveness of the latter’s immune response against the virus infection. When the immune response is limited due to either host genetics, acquired defects like lymphoid diseases, immaturity of the immune system in fetuses, newborns, or young children, or loss of immune vigor in the aged, the advantage is firmly in the virus’s court. However, usually when the infection occurs in individuals with a developed and competent immune system, the advantage is the host’s, unless the infecting virus overwhelms the individual’s immune system or the immune response becomes hyperactive resulting in an excessive innate and adoptive immune reaction, the “cytokine storm” phenomenon. Cytokine storm leads to immune-mediated injury (immunopathology).
When available, vaccination is useful in protecting groups of previously uninfected (naive) individuals from acute viral respiratory diseases. By this means, the spread of infection is diminished. Additionally antiviral drugs, which were developed as effective therapies to diminish or in some instances prevent ongoing infections, are reasonably effective, nevertheless come with two marked limitations. First, antiviral drugs exert selective pressure on viral progeny, promoting their mutation and selection thereby creating a new generation that is more fit and resistant to the drug (Nguyen et al. 2012; Orozovic et al. 2011). Second, the injury associated with these acute viral respiratory diseases, including influenza, results from a combination of the virus’s intrinsic virulence in lysing cells it infects and the intensity of the immune response which can damage tissues and promote a cytokine storm. Antiviral drugs are effective against the virus but not against cytokine storm or immune-mediated injury.
Recently, while studying human H1N1 2009 influenza virus infection in mice (Walsh et al. 2011; Teijaro et al. 2011) and ferrets (Teijaro et al. 2013), we uncovered the first direct and definitive experimental evidence that cytokine storm, per se, was a major factor in the causation of morbidity and mortality from influenza virus and some other acute, severe respiratory infections rather than just the accompanying phenomena. Further, we documented that cytokine storm was chemically treatable using an immunomodulatory small molecule, sphingosine-1-phosphate agonist, which dramatically inhibited the production of cytokines/chemokines and the innate cellular response, thereby blunting both the innate as well as the adoptive antiviral T cell response (Marsolais et al. 2009; Walsh et al. 2011; Teijaro et al. 2011). These events successfully limited immunopathologic injury. Nevertheless, a sufficient host T cell response remained and coupled with the antiviral antibody response curtailed the acute infection while providing recall immunologic memory to any renewed insult by the virus. This review focuses primarily on our experimental work that provided these conclusions.
Influenza Virus Infection
Epidemiologic and Experimental Evidence for Cytokine Storm
An overly aggressive innate immune response, the early recruitment of inflammatory leukocytes to the lung and dysregulated immune gene expression were key contributors to morbidity from the 1918/1919 influenza virus onslaught, as suggested by experimental infection of macaques with the 1918 H1N1 virus strain (Kobasa et al. 2007; Cilloniz et al. 2009). Clinical studies of humans infected by H5N1 bird influenza virus revealed a significant association between excessive early cytokine responses and immune cell recruitment as strongly predictive of poor medical outcomes (de Jong et al. 2006). Recently, similar results for influenza virus infections were reported for experimental animal models (Baskin et al. 2009; Marcelin et al. 2011, Zhang et al. 2012) and for humans (Arankalle et al. 2010; Cheng et al. 2010; Lee et al. 2011). Among reports of H1N1 2009 pandemic influenza infections in humans, that of Arankalle et al. (2010) is illuminating. Analyzing viral events and cytokine storms in critically ill-hospitalized patients, the investigators showed that those who died had no difference in influenza viral load from those who recovered. However, the patients who recovered and left the hospital had significantly lower cytokine storm profiles than the population who succumbed from the infection. My colleague, Hugh Rosen, and I reasoned that calming the host’s aggressive and exaggerated cytokine storm response might provide the opportunity to shift the balance from severe morbidity and mortality to survival. Our laboratories started jointly about 7 years ago to test this hypothesis (Marsolais et al. 2008). We selected the molecule sphingosine 1-phosphate (S1P) and sought to determine if harmful immunologic processes accompanying H1N1 2009 influenza infection could be modulated by S1P receptors in the lung. We selected S1P agonists because of their documented history of modulating lymphoid trafficking by inducing sequestration of lymphocytes in secondary lymphoid regions. By that means, S1P agonists limit the migration of effector lymphocytes to areas where such cells mediate immunologic injury (Rosen et al. 2007, 2009, 2013; see Chaps. 1 (Rosen) and 6 (Cyster) in this volume)). S1P is a signaling lipid present at a concentration of 1–3 nM in plasma and approximately 100 nM in lymph. Physiologically, S1P levels are under tight homeostatic control, and S1P signals through specific S1P receptors of which there are five (S1P receptors 1–5). These five specific S1P receptors are coupled to different G proteins for the purpose of regulating a variety of downstream pathways specific for many cells, tissues, and organs (Rosen et al. 2007, 2009, 2013).
Tracking and Kinetics of Influenza Virus-Specific CD8 and CD4 T Cells in the Lung and their Modulation by S1P Agonist
Infiltration of lymphoid cells into pulmonary tissues accompanies influenza virus infection. To identify and quantitate CD8 and CD4 cells that specifically recognize influenza and separate these virus-specific effector T cells from the majority of CD8 and CD4 bystander T cells nonspecifically drawn into the lung by chemotoxic attractants released during virally induced damage of infected pulmonary epithelial cells, we took advantage of the wealth of reagents we and others created for lymphocytic choriomeningitis virus (LCMV). Our colleague Yoshi Kawaoka and his co-workers used reverse genetics (Marsolais et al. 2009) to place the MHC Db-restricted immunodominant LCMV CD8 T cell epitope glycoprotein (Gp) aa31-41 and the MHC IAb restricted immunodominant CD4 T cell epitope Gp aa65-77 into the neuroaminidase stalk of WSN influenza virus. This technology generated a recombinant WSN Flu/LCMV virus that replicated in vivo displaying the same pulmonary geography as wild-type (wt) WSN virus. The experimental plan utilized GFP- or RFP-labeled, cloned LCMV recognition lymphocytes obtained from T cell receptor mice in which >98 % of CD8 fluoroprobe-labeled T lymphocytes recognized LCMV Gp aa31-41, and >97 % of CD4 fluoroprobe-labeled T lymphocytes recognized Gp aa65-77. Such GFP/RFP-labeled, virus-specific lymphocytes were adoptively transferred into naïve H-2b C57Bl/6 mice where they resided in secondary lymphoid tissues as resting lymphocytes. Two days later the recombinant WSN Flu/LCMV was administered intratracheally. Virus replication in pulmonary epithelial cells (Fig. 1a) was followed by the infiltration of virus-specific CD8 T cells (red) and virus-specific CD4 T cells (green) (Fig. 1b) at day 6 (Marsolais et al. 2009). Kinetic study of infiltrating virus-specific CD8 T cells showed their arrival by day 4, peak amounts at days 6–8, and significant numerical decrease at day 10 postinfection (Fig. 1c). There are five S1P receptors, i.e., S1P1, S1P2, S1P3, S1P4, and S1P5. Administration of S1P permissive agonist AAL-R, which signals on S1P1, S1P3, S1P4, and S1P5 receptors but not the S1P2 receptor, significantly reduced the numbers of virus-specific CD8 T cells entering the lung (Fig. 1d). The result was significant protection from pulmonary tissue injury (Fig. 1e) and related mortality (Fig. 1f) when compared to the effects of vehicle alone or use of a control isomer, AAL-S, that is not able to be phosphorylated and cannot signal S1P receptors. Blunting of innate cytokine and chemokine responses following AAL-R treatment was evident and remarkable at day 2 postinfluenza infection (Fig. 1g). All these observations were initially made with murine H1N1 WSN virus and later confirmed by use of the non-murine adopted human pathogenic H1N1 influenza viruses A/Wisconsin/WSLH34934/09 and A/California/04/09 (Walsh et al. 2011). In studies with all these influenza viruses, although cytokine/chemokine expression was significantly blunted by S1P agonist AAL-R, AAL-R-treated mice terminated the virus infection, displayed robust virus-specific CTL responses 7 days after influenza infection, as measured by 51chromium release assay, and also mounted vigorous specific memory T cell responses upon rechallenge with virus 40 days after the infection. Further, the kinetics, titers of neutralizing anti-influenza antibodies in sera, or immunoglobulin subtypes of either AAL-R or AAL-S or vehicle-treated mice were equivalent. Together, these results document the validity of our premise. That is, the permissive S1P agonist AAL-R, which signals via S1P1, S1P3, S1P4, and S1P5 receptors, when given locally into the respiratory tract, down-modulated numbers of virus-specific T cells, decreased innate cytokine/chemokine expression in the lung parenchyma, and reduced the supply of innate inflammatory cells—NK, PMN, and macrophages (Marsolais et al. 2009; Walsh et al. 2011)—sufficiently to abort cytokine storm. The successful outcome was protection of the host from influenza virus infection while still providing an antiviral response that curtailed and eventually impeded the influenza infection. Our data indicated that 23 of 28 mice (82 %) receiving AAL-R were protected (P = <0.001; only five of 28 died from the infection) when compared to a dose of virus that killed approximately 80 % of vehicle- or AAL-S-treated mice (22 of 28 mice died) (Walsh et al. 2011). Interestingly, when an optimal dose of the currently used antiviral drug Tamiflu was administered by itself, protection was significantly less effective 50 % (14 lived of 28 mice treated) compared to survival after AAL-R therapy alone (80 %). These results document a prominent role for cytokine storm as the cause of death from influenza infection. Most important is the benefit of S1P agonist therapy for the victims of multiple influenza virus strains and especially those that are resistant to anti-neuraminidase therapy. Although greater benefit was obtained in blocking cytokine storms with the S1P agonist than with Tamiflu (82 % vs. 50 %) protection, administering both the antiviral drug and the S1P agonist as combined therapy was optimal, yielding a 96 % protection rate from influenza virus challenge (27 of 28 mice survived the infection) (Walsh et al. 2011).
Fig. 1.
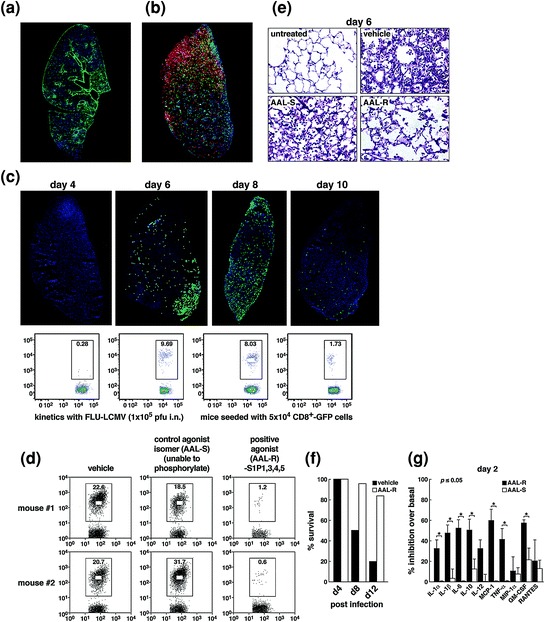
Panel a: Distribution of viral antigen (green, left); Panel b: Virus-specific CD4 T cells (green, right) and CD8 T cells (red, left) in the lung 7 days following influenza/LCMV infection; Panel c: Kinetics of virus-specific CD8 T cell infiltration into the lung analyzed by immunohistochemistry (upper panels) or FACS (lower panels); Panel d: The S1P permissive agonist AAL-R significantly blunts infiltration of virus-specific CD8 T cells into the lung following influenza/LCMV recombinant virus infection; Panel e: Significant reduction of pulmonary tissue injury and preservation of alveolar air space in influenza-infected mice treated with AAL-R; Panel f: Significant protection from mortality accompanying influenza virus infection with AAL-R; and Panel g: AAL-R significantly dampens cytokine and chemokine content at day 2 following influenza virus infection. Figure reprinted from Marsolais et al. (2009), with permission from PNAS
Pulmonary Injury and Disease Associated with Influenza and Resultant Cytokine Storm are Treatable with a Single S1P1 Receptor Agonist Molecule
All five S1P receptors couple to different G proteins require many downstream signaling pathways (Fig. 2a) (Rosen et al. 2007, 2009, 2013, Chap. 1 in this volume). The biological functions of these various S1P receptors are dependent on the cell/tissue location of the receptors, their expression, and their activation. Knowing that a broadly permissive S1P agonist AAL-R, which signals via S1P1, S1P3, S1P4, and S1P5 but not S1P2 receptors, significantly downregulated the cytokine storm and protected mice from the effects of a pathogenic human H1N1 influenza infection (Fig. 1, Panels c–g) (Walsh et al. 2011), we repeated the experiments shown in Fig. 1, Panels e–g, using two S1P1-specific agonists, CYM-5542 (Walsh et al. 2011), or RP-002 (Teijaro et al. 2011). The results are displayed in Fig. 2 and indicate that either of the two specific S1P1 receptor agonists whose signal is entirely restricted to S1P1 receptors were as effective as the broadly permissive AAL-R that signals on S1P1, S1P3, S1P4, and S1P5. The S1P1-specific agonists CYM-5442 were administered intratracheally (2 mg/kg) and RP-002 intratracheally (3 mg/kg) or orally (6 mg/kg) (Teijaro et al. 2011). Both S1P1 receptor agonist molecules provided protection against a lethal intranasal challenge with human H1N1 A/Wisconsin/WSLH34934/09 or A/California/04/09 (Fig. 2b) and blunted cytokine storm (Fig. 2c and d). The S1P1 receptor agonists significantly inhibited secretions of cytokines and chemokines associated with influenza virus infection, namely IFN-α, CCL-2, IL-6, TNF-α, CCL-3, CCL-5, CxCl-2, IL-1α, and IFN-γ. Observations from several experiments indicated that amounts of these cytokines/chemokines were inhibited to a degree similar to that from AAL-R treatment. The S1P1 selective agonists also significantly blunted the accumulation of innate infiltrating inflammatory cells (Fig. 2, Panel d). Notable were the reductions of macrophages/monocytes (marked by CD11b+, LyG6−, F480+), neutrophils (CD11b+, LyG6+−, F480−), and natural killer cells (NK1.1+, CD3−). Correspondingly, the quantity of activation marker CD69 was significantly reduced following S1P1 agonist therapy. Further, pulmonary tissues also reflected S1P1’s beneficial outcome, since histologic study of mice given this remedy manifested mostly open alveolar air spaces, diminished to negligible inflammatory cell infiltrates and neither edema nor hemorrhage in the lung tissue. Importantly, S1P1 agonist treatment did not enhance viral titers. Influenza infection was effectively terminated, and both anti-influenza neutralizing antibodies and anti-influenza virus CD8 T cells were generated. Although numbers of T cells were reduced compared to infected mice not receiving S1P1 agonist therapy, they were sufficient to terminate the infection. Further, immune memory was established following this S1P1-specific therapy.
Fig. 2.
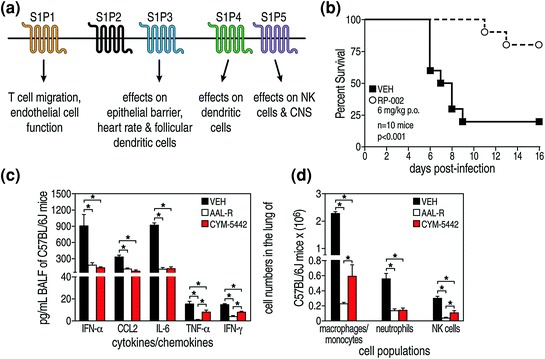
S1P1 specific agonists CYM-5442 and RP-002 are therapeutically equivalent to the permissive AAL-R agonist that utilizes S1P1, S1P3, S1P4, and S1P5 receptor signaling to blunt cytokine storm and also protect mice from a lethal challenge by human pandemic H1N1 2009 A/Wisconsin/WSLH34939/09 (shown) or A/California 04/09 (not shown) viruses. Panel a: Cartoons of the five S1P receptors and their biologic effects. AAL-R signals on receptors S1P1, S1P3, S1P4, and S1P5 but not S1P2. Panel b: S1P1 receptor-specific agonist RP-002 given orally protects mice challenged with H1N1 human 2009 influenza virus A/Wisconsin. Treatment with S1P1 receptor-specific agonist CYM-5442 after mice are challenged with 2009 influenza A/Wisconsin inhibits their cytokine/chemokine response (cytokine storm) equivalently to treatment with the permissive AAL-R (Panel c) and also impedes the recruitment of innate immune cells into their lungs (Panel d). BALF Bronchial lavage fluid, * = p < 0.01. Figure reprinted from Teijaro et al. (2011), with permission from Elsevier
Thus, severe pulmonary injury and disease associated with influenza infection and resultant cytokine storm were treatable with a preparation composed of only S1P1 receptor agonist molecules, thereby avoiding signaling through S1P2, S1P3, S1P4, and S1P5 receptors. Pharmaceutically this may be of importance if/when individually S1P2, S1P3, S1P4, or S1P5 signaling might lead to unwanted harmful biologic effects.
S1P1 Receptors are Located on Pulmonary Endothelial Cells, Which Serve as the Gatekeepers for Cytokine Storm
Having identified S1P1rec signaling as the primary pathway for the initiation of cytokine storm, we sought to identify the cell or cells in the lung that expressed the S1P1 receptor. Since epithelial cells are the primary cells infected by influenza viruses, we suspected that S1P1 receptors might be located on those cells. To determine which pulmonary cell types bear the S1P1 receptor, we took advantage of eGFP-S1P1 receptor knock-in mice made by Stuart Cahalan in the Rosen laboratory (Cahalan et al. 2011, see Cahalan Chap. 4, this volume). In this strain of mice, the native S1P1 receptor was homologously replaced with a functional fused eGFP-tagged S1P1 receptor (Cahalan et al. 2011). Utilizing this mouse model, we could directly detect eGFP-S1P1 receptor protein expression on pulmonary cells by using antibodies to specific pulmonary cell markers and flow cytometry (Fig. 3, Panel a). Additional substantiation came from biochemical analysis of these purified pulmonary cells (Fig. 3, Panel b). S1P1-eGFP receptor expression was plentiful on lung lymphatic (CD45−, CD31+, GP38+) and vascular (CD45−, CD31+, GP38−) endothelial cells but, surprisingly, was absent on pulmonary epithelial cells (CD45−, CD31−, EpCAM+) (Fig. 3, Panel a) (Teijaro et al. 2011). These results were confirmed by doing Western blots on >98.5 % pure populations of pulmonary endothelial and epithelial cells (Fig. 3, Panel b). As expected and previously reported, CD4 T cells (CD4+, CD3+), CD8 T cells (CD8+, CD3+), and B cells (B200+, CD19+) also expressed the S1P1-eGFP receptor (Fig. 3, Panel a). In contrast, pulmonary leukocytes, including macrophages/monocytes (CD11c+, CD11b−, F480+), dendritic cells (CD11c+, IA-IE+, CD205+, F480−), neutrophils, NK cells (NK1.1+, CD3−) (Fig. 3, Panel a), and immature lymphoid cells (LIN−, SCA-1+) failed to express significant levels of eGFP-S1P1 receptor protein. S1P1-eGFP receptor expression was similar whether cells were harvested from mice that were uninfected or infected with influenza virus. Other experiments in infected mice indicated that S1P1-eGFP receptor expression was not altered during influenza virus infection. Importantly, S1P1 agonist treatment of infected eGFP-S1P1 receptor knock-in mice did not lessen expression of the S1P1-eGFP receptors indicating that administration of specific S1P1 agonist does not degrade the endothelial S1P1 receptor. These results signify that the functional agonism of S1P1, not its antagonist effect of receptor degradation, was the mechanism by which S1P1 receptor blocking molecules CYM-5442 and RP-002 suppressed cytokine storms. In other studies, pulmonary endothelial cells were processed to a greater than 98.5 % purity during the first 48 h following influenza virus infection and treated with S1P1 agonist. Assessment of both RNA and protein levels showed that the S1P1 agonist CYM-5442 effectively decreased amounts of cytokines and chemokines made by vascular as well as lymphatic pulmonary endothelial cells (Fig. 3b).
Fig. 3.
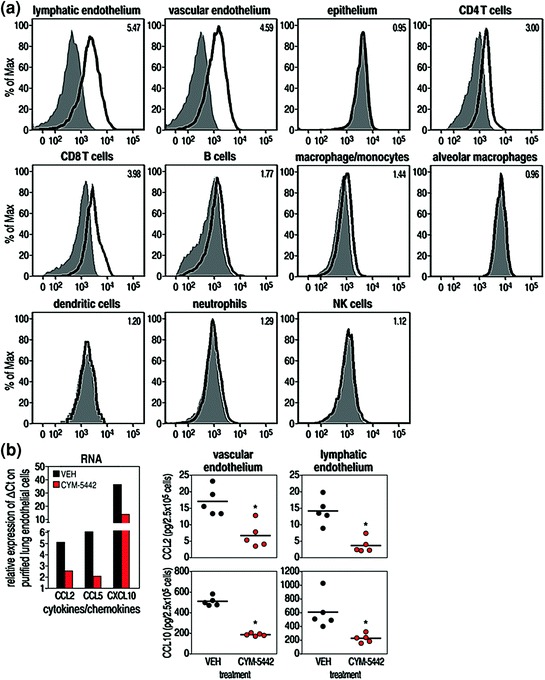
S1P1 is expressed on endothelial cells and lymphocytes isolated from eGFP-S1P1 receptor knock-in mice. Panel a: Cell populations purified using antibodies to specific cell surface markers and FACS. Purity of all cell populations exceeded 98.5 %. See Teijaro et al. (2011) for details about reagents and experiments. As seen in Panel a, only endothelial cells (lymphoid and vascular) and lymphocytes (CD4 + T cells, CD8 + T cells) expressed the GFP-S1P1 receptor marker. Pulmonary epithelial cells, the primary target for influenza virus, do not express the S1P1 receptor. Panel b:S1P1 agonism inhibits chemokine expression in endothelial cells following influenza virus infection. * = p < 0.01. See Teijaro et al. (2011) for details. Figure reprinted from Teijaro et al. (2011), with permission from Elsevier
T and B lymphocytes as well as pulmonary endothelial cells were the only cells within the lung that expressed measurable amounts of S1P1-eGFP protein (Fig. 3 Panel a). We therefore determined whether lymphocytes expressing S1P1 receptors were involved in S1P1 agonist inhibition of cytokine storm or were merely bystander cells accompanying the innate immune response to influenza virus infection. Since Rag2-/- mice are deficient in lymphocytes, we reasoned that if such mice, when infected with influenza virus, generated a cytokine storm that could be blocked by S1P1 agonist, then lymphocytes were ruled out as initiators of cytokine storms. Our experiments documented that cytokine storm occurred in Rag2-/- mice infected with influenza virus. Importantly, treatment of infected Rag2-/- mice with the S1P1 agonist CYM-5442 significantly reduced cytokines and chemokines in the bronchial lavage fluids as well as minimalizing the infiltration of innate cells (macrophages/monocytes and NK cells). Recently, John Teijaro (2013), utilizing cell sorting and a biochemical approach, found S1P1 receptor on plasmacytoid dendritic cells (pDC) whose expression was undetectable in the S1P1-γ GFP transgenic mouse model.
Type I Interferon Signaling is Essential for the Cytokine/Chemokine Response of Cytokine Storm but is not Involved in Recruitment of Innate Inflammatory Cells into the Lung
As observed in Fig. 4a and b and detailed in Teijaro et al. 2011, amounts of type I interferon and almost exclusively the interferon-α species were elevated early after acute influenza virus infection. The release and action of type I interferon was crucial for the production of pro-inflammatory cytokines/chemokines, since blockage of the type I interferon response by using monoclonal antibody to interferon-α-β receptor (IFNAR1) significantly reduced the quantity of pulmonary cytokines/chemokines associated with acute influenza infection (Fig. 4b). Further, treatment with S1P1 receptor agonist inhibited the production of interferon-α in the pulmonary bronchial lavage fluid early after initiating influenza virus infection. Proof that this blunting of interferon-α production was a mechanism by which S1P1 receptor agonist inhibited cytokine storm derived from use of IFNAR1 receptor knock-out mice infected with H1N1 virus and treated with S1P1 receptor agonist CYM-5442. Such studies showed a significant reduction of cytokines/chemokines (IFN-α, CCL-2, IL-6 (shown Fig. 4c), IFN-γ, CCL-5, CxCl-0, not shown) in the bronchial lavage fluid when compared to results from similar experimentation in mice with an intact interferon-α-β receptor signaling ability (Fig. 4c). Of interest, blockage of interferon-α-β receptor signaling did not retard pulmonary infiltration by the inflammatory cells—macrophages, monocytes, neutrophils, or NK cells—following S1P1 receptor agonist therapy (Fig. 4d). Thus, the infiltration of innate inflammatory cells was blunted only in interferon-α-β sufficient mice but not in interferon-α-β receptor knock-out mice. This outcome indicates that regulation of such cell recruitment into the lung was primarily mediated by endothelial cells and was independent of type I interferon signaling (Teijaro et al. 2011). Cytokine/chemokine production in the lung was also mediated by pulmonary endothelial cells, and S1P1 receptor agonism of such cells inhibited interferon-α production leading to the significantly diminished inflammatory cytokine/chemokine responses we observed.
Fig. 4.
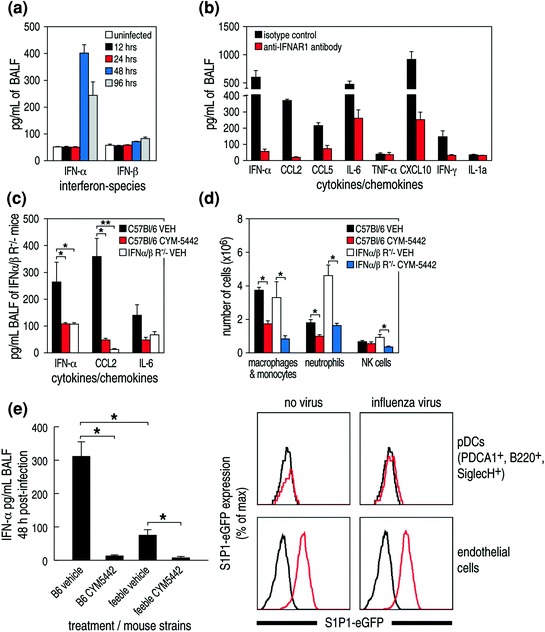
Interferon-α is the predominant type 1 interferon produced early following virus infection (Panel a) and is associated with the dysregulation of cytokines and chemokines that causes a cytokine storm. Antibody to interferon type 1-α-β receptor significantly blocks release of cytokines and chemokines (Panel b). Panel c: S1P1 agonist suppression of cytokines is dependent on interferon 1. Panel d: Innate inflammatory cell recruitment is independent of interferon-α-β receptor signaling. Panel e, left: The majority (75–85 %) of interferon-α released following influenza viral infection is from plasmacytoid dendritic cells (pDCs, use of feeble mice—see text). Panel e, right: Data from S1P1-eGFP knock-in mice indicating that S1P1 receptors are not present on surfaces of pulmonary pDCs but are found, as expected, on surfaces of pulmonary endothelial cells. However, utilizing more sensitive techniques S1P1 receptors are found on pDCs (see text). Figure reprinted from Teijaro et al. (2011), with permission from Elsevier
Influenza virus infection induces a robust interferon type I response, despite the early induction of the viral NS1 protein that suppresses the cellular induction of and response to interferon I (Fernandez-Sesma 2007). The predominant type I interferon produced early following influenza virus infection is alpha, not beta (Fig. 4a). However, the cellular sources of interferon type I-α produced and amounts made by various cell populations have not been clear. The two major pulmonary cell populations known to make type I interferon in vivo following respiratory viral infections are pDCs and alveolar macrophages (Kumagai et al. 2007). To judge the contribution of pDCs to interferon-α production in the lung, we utilized a novel mouse model recently developed at Scripps by Bruce Beutler and termed “feeble.” Feeble mice have a specific genetic defect that prevents their pDCs from producing type I interferon and pro-inflammatory cytokines upon activation of TLR7 and TLR9 ligands by influenza virus stimulation (Blasius et al. 2010). Importantly, there is no disruption of the numbers or vitality of pDCs, and the feeble mouse defect is specifically restricted to pDCs. As displayed in Fig. 4e, when wild type or feeble mice received H1N1 human 2009 swine influenza with or without S1P1 agonist CYM5442 treatment, 75–85 % of total interferon-α was produced by pDCs. Further, interferon-α release was significantly inhibited by the S1P1 agonist CYM-5442. These results were confirmed using a pDC depletion antibody (anti-PDCS-1 clone 120.68), which again resulted in significant depletion of pDCs in the lung and corresponding reductions in interferon-α, CCL2, CCL5, and IL-6. Thus, pDCs are the essential and major producers of interferon-α (75–85 %) and involved in amplification of cytokine/chemokine volumes following influenza infection. Other depletion studies indicate that most of the remaining interferon-α production (~15–25 %) was by alveolar macrophages.
Since S1P1 agonist therapy diminished interferon-α production, and the majority of interferon-α produced was by pDCs, it was important to learn whether or not pDCs expressed S1P1 receptors on their surfaces. We know that alveolar macrophages, the other main albeit minority producers of interferon-α do not express S1P1 receptors (Fig. 2a). Plasmacytoid dendritic cells were recently found to express S1P1 receptors (Teijaro 2013). However, using the S1P1 eGFP receptor knock-in mice and pDCs of more than 98.5 % purity failed to show that these cells expressed S1P1 receptors (Fig. 4e). Thus, the S1P1-specific receptor is found primarily on pulmonary endothelial cells with lesser amounts on pulmonary pDCs. S1P1 receptor agonist acts directly on pulmonary endothelial and pDCs and likely indirectly on alveolar macrophages. We have, as yet, been unable to detect S1P1 receptor on alveolar macrophages.
Working Model for the Initial Production of Cytokine Storm and its Chemical Tractability by Single S1P1 Molecules
A working model based on the accumulated data for the earliest events of influenza infection is provided in Fig. 5. Although there are several possible scenarios, I selected the simplest one in which S1P1 agonist signals a factor(s) that blocks [negative signal(s)] the release of interferon-α from pulmonary pDCs and the migration of innate inflammatory cells from blood vessels into the lung. This model is based on presence of S1P1 receptors on pulmonary endothelial and pDCs but their absence on lung epithelial cells and the findings that alveolar macrophages which produce type I interferon do not display S1P1 receptors on their surfaces. However, pulmonary endothelial cells and pulmonary pDCs do express S1P1 receptors on their surfaces. Cytokines/chemokines elicit the initial cytokine storm reflective of factors (viral or nonviral) produced by the influenza virus-infected lung epithelial cells per se. These factors likely activate pulmonary pDCs and alveolar macrophages to release interferon-α. As chemotoxic factors are then liberated into the site of action, infiltrating cells of the innate immune system (macrophages/monocytes, NK cells, leukocytes) are drawn into the inflammatory milieu where they release additional cytokines/chemokines.
Fig. 5.
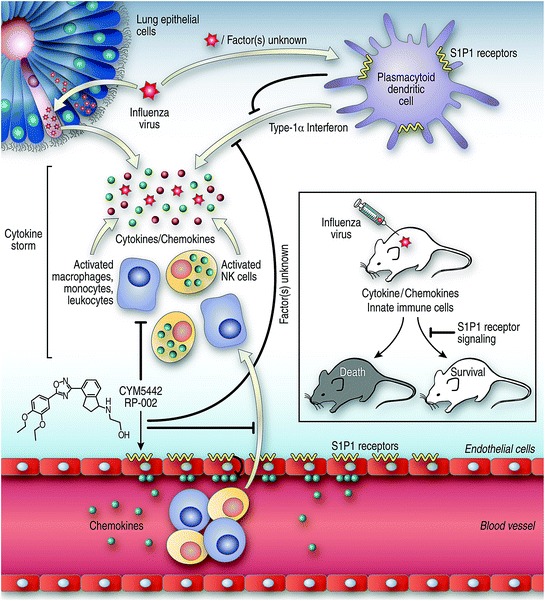
Schematic of data presented in the text: Proposed pathways and cell–cell crosstalk in the lung following influenza virus infection and S1P1 receptor agonist signaling. Initial events: Viruses infect lung epithelial cells that release one or more (currently unknown) factors that signal plasmacytoid cells (primary cell-type involved) and alveolar macrophages to release type 1 interferon-α, which dysregulates cytokines/chemokines to elicit cytokine storm. Released chemokines attract innate immune inflammatory cells that become activated and release additional chemokines/cytokines to amplify cytokine storm. Therapeutic control of cytokine storm: Pulmonary endothelial cells and pDCs contain S1P1 receptors on their surfaces, but S1P1 receptors are absent from lung epithelial cells and alveolar macrophages. The S1P1 receptor agonist signals pulmonary endothelial cells and pDCs likely to release factors that negatively regulate the cytokine storm in terms of both its cytokine/chemokine release and infiltration of innate inflammatory cells
After that initial response, a second stage occurs by day 4–8 (see 2.2) via a mechanism described in our publication (Marsolais et al. 2009). Here influenza virus-specific T cells are activated and expand numerically in the mediastinal lymph nodes and pulmonary tissues. These T cells of the adoptive immune system produce additional inflammatory molecules and lyse influenza virus-infected epithelial cells thereby augmenting cytokine storm and immune-mediated injury. This second phase of tissue injury is primarily influenza virus–specific T cell-mediated and signals through S1P1 but also likely progresses via S1P3 and 4 receptors, as by our preliminary results. However, additional data are required to ensure these observations. What is clear is that treatment with S1P permissive-AAL-R agonists signaling via S1P1,3,4,5 receptors affect adoptive immune T cell-mediated immunopathology by downregulating MHC and co-stimulatory molecules of DCs located in the mediastinal lymph nodes and the lung parenchyma thereby blunting the arming, expansion, and migration of virus-specific CD4 and CD8 T cells into the lung (Marsolais et al. 2009).
The cytokine pathways blunted by S1P1 agonist signaling are displayed in Fig. 5 by the symbol ⊢.
Conclusions and Future Studies
Conclusions
Cytokine storm plays an essential role in the pathogenesis and clinical outcome of influenza virus infection. Blockade of cytokine storm provides greater protection than does antiviral therapy, like that with a neuraminidase inhibitor, and does so without compromising the control and clearance of viruses. Moreover, optimal therapy is achieved by combining S1P agonists with anti-neuraminidase treatment. For the foregoing observations, human pathogenic H1N1 09 influenza virus and mouse adapted H1N1 influenza virus were used.
Sphingosine-1-phosphate (S1P) receptor agonists blunt cytokine storm. Importantly, cytokine storm is chemically disarmed by administering one of the five S1P receptors: S1P1.
The molecular mechanism of this event involves S1P1 receptor signaling on pulmonary endothelial cells and pulmonary pDCs but not virally infected epithelial cells or alveolar macrophages. Pulmonary endothelial cells are the major gateway combined with pulmonary pDCs to precipitate a cytokine storm. S1P1 agonism suppresses the recruitment of both cytokines, innate and adoptive immune cells. Blunting of innate immune cell function and virus-specific T cell activity lessens morbidity and prevents mortality associated with experimental models of influenza virus infection in mice and ferrets. In both species, there is a sufficient antiviral response remaining to terminate the virus infection and provide immune memory upon rechallenge.
Immune cell infiltration and cytokine production are distinct events, but both are orchestrated by endothelial and pDC cells. Pro-inflammatory cytokine responses depend on type I interferon signaling; IFN-α is the predominant interferon made. The predominant pulmonary cell making type I interferon is the pDC (over 75–85 %); alveolar macrophages make most of the rest.
Future Studies
Investigate interferon I as to the cellular source and signaling pathway(s) in the influenza system.
Dissect crosstalk and signaling between pulmonary endothelial cells, infected epithelial cells, and interferon-producing pDCs. Identify the molecules involved. See if these molecules provide potential therapeutic targets.
Determine generalities for other acute respiratory infections, e.g., Hantavirus, respiratory syncytial virus, SARS, pneumococcal pneumonia, in which cytokine storm plays a prominent role.
Study an animal model (subhuman primates) more reflective of influenza in humans. Results from our studies in ferrets (Teijaro et al. 2013) mirror the protection supplied by S1P1 agonist therapy in defending against influenza virus infection in the murine model.
Define the S1P pathway and design a genetic screen to identify humans who are the most susceptible to cytokine storm.
Develop specific S1P receptor agonists and antagonists for human therapeutics, focusing initially on S1P1 molecules.
Acknowledgments
The experimental work described in this chapter was initiated with and carried out as a collaboration between my laboratory and that of Hugh Rosen. Graduate student (Stuart Cahalan) and former postdoctoral fellows (David Marsolais, Kevin Walsh, and John Teijaro) were instrumental in the findings presented. John Teijaro continues his independent work in this area as a faculty member at Scripps. Yoshi Kawaoka and colleagues in his laboratories have been valued collaborators. This work was funded by NIH grants AI009484 (MBAO), AI074564 (MBAO, HR), MH084512 (HR), and was also supported by the NIH/NIAID under Award Number U54 AI057160 to the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Diseases Research (MBAO).
Contributor Information
Michael B. A. Oldstone, Phone: +1858-784-8054, Email: mbaobo@scripps.edu
Hugh Rosen, Phone: +18587842396, Email: hrosen@scripps.edu.
Michael B. A. Oldstone, Phone: +1-858-7848054, FAX: +1-858-7849981, Email: mbaobo@scripps.edu
References
- Ahmed R, Oldstone MBA, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol. 2007;8:229–288. doi: 10.1038/ni1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arankalle VA, Lole KS, Arya RP, Tripathy AS, Ramdasi AY, Chadha MS, Sangle SA, Kadam DB. Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PLoS ONE. 2010;5:e13099. doi: 10.1371/journal.pone.0013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, García-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, Aicher LD, Rosenzweig ER, Murali-Krishna K, Clark EA, Kotur MS, Fornek JL, Proll S, Palermo RE, Sabourin CL, Katze MG. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci USA. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, Ross C, Li X, Smart NG, Beutler B. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci USA. 2010;107:19973–19978. doi: 10.1073/pnas.1014051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, Yeager A, Clemons B, Scott F, Rosen H. Actions of a picomolar short-acting S1P1 agonist in S1P1-eGFP knock-in mice. Nat Chem Biol. 2011;7:254–256. doi: 10.1038/nchembio.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XW, Lu J, Wu CL, Yi LN, Xie X, Shi XD, Fang SS, Zan H, Kung HF, He ML. Three fatal cases of pandemic 2009 influenza A virus infection in Shenzhen are associated with cytokine storm. Respir Physiol Neurobiol. 2010;175:185–187. doi: 10.1016/j.resp.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Cillóniz C, Shinya K, Peng X, Korth MJ, Proll SC, Aicher LD, Carter VS, Chang JH, Kobasa D, Feldmann F, Strong JE, Feldmann H, Kawaoka Y, Katze MG. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009;5:e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Mølbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, Widdowson MA. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sesma A. The influenza virus NS1 protein: inhibitor of innate and adaptive immunity. Infect Disord Drug Targets. 2007;7:336–343. doi: 10.2174/187152607783018754. [DOI] [PubMed] [Google Scholar]
- Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:10–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, Ngai KL, Lui GC, Wong BC, Lee SK, Choi KW, Hui DS. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS ONE. 2011;6:e26050. doi: 10.1371/journal.pone.0026050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G, Aldridge JR, Duan S, Ghoneim HE, Rehg J, Marjuki H, Boon AC, McCullers JA, Webby RJ. Fatal outcome of pandemic H1N1 2009 influenza virus infection is associated with immunopathology and impaired lung repair, not enhanced viral burden, in pregnant mice. J Virol. 2011;85:11208–11219. doi: 10.1128/JVI.00654-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H. Local not systemic modulation of dendritic cell S1P receptors in lung blunts virus-specific immune responses to influenza. Mol Pharmacol. 2008;74:896–903. doi: 10.1124/mol.108.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA. 2009;106:1560–1565. doi: 10.1073/pnas.0812689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HT, Fry AM, Gubareva LV. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther. 2012;17:159–173. doi: 10.3851/IMP2067. [DOI] [PubMed] [Google Scholar]
- Oldstone MBA (2010) Severe acute respiratory syndrome (SARS). Viruses, plague and history, Oxford Press, pp 251–283
- Orozovic G, Orozovic K, Lennerstrand J, Olsen B. Detection of resistance mutations to antivirals oseltamivir and zanamivir in avian influenza A viruses isolated from wild birds. PLoS ONE. 2011;6:e16028. doi: 10.1371/journal.pone.0016028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–662. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro JR, Walsh KB, Long JP, Tordoff KP, Stark GV, Kawaoka Y, Rosen H, Oldstone MB (2013) H1N1 2009 influenza virus infection of ferrets is successfully treated with sphingosine-1-phosphate 1 receptor agonist RP-002. Virology Submitted [DOI] [PMC free article] [PubMed]
- Teijaro JR (2013) Unpublished data
- Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, Kawaoka Y, Rosen H, Oldstone MB. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011;108:12018–12023. doi: 10.1073/pnas.1107024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PF, Neumann G, Kawaoka Y (2007) Orthomyxoviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, (eds) Fields virology, Chap 48. Wolters Kluwer, Lippincott Williams & Wilkins, Philadelphia, pp 1691–1740
- Zhang Y, Sun H, Fan L, Ma Y, Sun Y, Pu J, Yang J, Qiao J, Ma G, Liu J. Acute respiratory distress syndrome induced by a swine 2009 H1N1 variant in mice. PLoS ONE. 2012;7:e29347. doi: 10.1371/journal.pone.0029347. [DOI] [PMC free article] [PubMed] [Google Scholar]