

Abstract
In the last decade, molecular beacons have emerged to become a widely used tool in the multiplex typing of single nucleotide polymorphisms (SNPs). Improvements in detection technologies in instrumentation and chemistries to label these probes have made it possible to use up to six spectrally distinguishable probes per reaction well. With the remarkable advances made in the characterization of human genome diversity, it has been possible to describe empirical patterns of SNPs and haplotype variation in the genome of diverse human populations. These patterns have revealed that the human genome is structured in blocks of strong linkage disequilibrium (LD). Because SNPs tend to be in LD with each other, common haplotypes share common SNPs and thus the majority of the diversity in a region can be characterized by typing a very small number of SNPs; so-called tag SNPs. Herein lies the advantage of the multiplexing ability of molecular beacons, since it becomes possible to use as few as 30 probes to interrogate several haplotypes in a high-throughput approach. Thus, through the combined use of tag SNPs and molecular beacons it becomes possible to type individuals for clinically relevant haplotypes in a high-throughput manner at a cost that is orders of magnitude less than that for high throughput sequencing methods.
Key words: Linkage disequilibrium, single nucleotide polymorphism, tagging single nucleotide polymorphisms, DC-SIGN, Mycobacterium tuberculosis, molecular beacons, real-time PCR
Introduction
Sanjay Tyagi the inventor of molecular beacons (1) once wrote:
Imagine that you have a magic reagent to which you add a droplet of a body fluid from a patient; you wait for a moment and a glow appears in the tube holding the mixture; the glow not only tells you which pathogen is responsible for the patient’s illness, but also indicates which drugs to use to treat the disease. Also imagine that you can perform this diagnosis before any symptoms of the disease appear, improving the chances of success with the treatment, and you can perform this test on a large population with ease. The creation and development of such reagents are the promise of nucleic acid-based detection and are the aspiration of a diverse community of researchers (2).
The promise of the technologies evoked by Sanjay Tyagi is borne out in the above quotation. The sequencing of the human genome (3) furnished an unprecedented understanding of its structure and organization, but could not in itself account for human biological variation. To address the latter, a number of international consortiums or private corporations, such as the International SNP Map Working Group, SeattleSNPs PGA, and the Perlegen consortium, have multiplied efforts to resequence genes or genomic regions to characterize single nucleotide polymorphism (SNP) variations in the human genome (4 – 6). To date, more than 11 million SNPs have been recorded in dbSNP, the public repository for DNA variation data (http://www.ncbi.nlm.nih.gov/SNP/index.html) (see Chapter 3 for details). Decorating the human genome at a frequency of one in every 500–1,000 bp, they are the most common form of human variation and can serve as high-resolution genetic markers. This variation, which represents a legacy of our evolutionary past and in the future may be a treasure trove of information paving the way to personalized medicine, may at least partially explain the wide range of phenotypic differences observed among individuals and populations (7 – 9). These catalogues of sequence variation therefore provide scientists and clinicians with the precious raw material to be exploited in both human evolutionary studies and medically related research. Here the major challenges have been in devising and implementing cost-effective, easily accessible, and rapid molecular diagnostic methods that can interrogate anywhere from a few dozen to hundreds of thousands of polymorphisms. The comparison of these SNPs among large numbers of individuals can be used in therapy and drug design and even in devising new, more powerful approaches in cell-based screening approaches for drug discovery. It is these diverse and complicated needs that have driven the creation of high-throughput methods of SNP typing.
Once genome sequence diversity has been catalogued, the next step is to determine how this diversity is organized within the human genome. Eleven million SNPs discovered to date appear to be not entirely random. When a new mutation arises, it is associated with neighboring variants present on the same chromosome or haploid DNA molecule, forming what is commonly known as a “haplotype.” When two alleles lying on the same chromosome are always observed together, or at least more often than expected by chance, these two variants are said to be in linkage disequilibrium (LD). The HapMap project, a natural extension of the Human Genome Project, was a pioneer in describing empirically the patterns of SNP and haplotype variation in the human genome and in obtaining a general LD map in populations of different ethnic origins (10). HapMap data clearly demonstrate that the human genome is organized in a LD block-like structure and that these LD blocks are often disrupted by recombination hotspots (11, 12). When SNPs are in LD with each other, redundant information is contained within the haplotype (i.e., by knowing the marker at one locus, we can predict the marker that will occur at the linked loci nearby). Thus, when one infers haplotypes within a region of reasonable LD, the diversity of haplotypes is accounted for by a few common haplotypes and lots of rare ones. The common haplotypes will share a number of SNPs in common with each other, whereas the rarer haplotypes will be characterized by carrying the rarer alleles at certain loci. Thus, one can capture the majority of the diversity within a region by typing those SNPs which allow one to cover the most diversity; so-called tag SNPs.
Currently, HapMap phase II provides the most complete available resource for selecting tag SNPs genomewide (12). Importantly, tag SNPs defined on the basis of the HapMap populations have been shown to adequately capture patterns of variation in other human groups; tag SNPs are therefore highly “portable” (13 – 15). In the practical sense, the HapMap data have already proven to be useful, as attested by the increasing number of successful genomewide association studies on diseases as diverse as type 1 (16, 17) and type 2 (16, 18, 19) diabetes, coronary artery disease (20), obesity-related traits (21, 22), rheumatoid arthritis (16, 23), and human immunodeficiency virus (HIV) disease progression (24). The portability and utility of tag SNPs opens up the possibility of their usage in “lower” high-throughput methods that are cheaper to implement and broadly accessible. Indeed, with a wide range of relatively cheap and robust instruments (see Table 17.1) and multiplexing probes such as molecular beacons, cost-effective high-throughput SNP typing becomes a reality (see Fig. 17.1).
Table 17.1.
Specifications of spectrofluorometric thermal cyclers
| Company | Model | Excitation source | Fluorophore choicea | Multiplex capability | Sample capacity | Hybridization probe compatibility |
|---|---|---|---|---|---|---|
| Applied Biosystems | 7300 real-time PCR system | THL | FAM, TET, TMR, and Texas red | 4 targets | 96 wells | All types, less suited for adjacent probes |
| Applied Biosystems | 7500 real-time PCR system | THL | FAM, TET, TMR, Texas red, and Cy5 | 5 targets | 96 wells | All types, less suited for adjacent probes |
| Applied Biosystems | PRISM 7700 and 7900HT | ABLL | FAM, TET, HEX, TMR, ROX, and Texas red | 6 targets | 96 wells and 384 wells | All types |
| Applied Biosystems | StepOne real-time PCR system | LED | FAM, HEX, and ROX | 3 targets | 48 wellsC | All types |
| Bio-Rad | MiniOpticon | LED | FAM and HEX | 2 targets | 48 wells | All types, less suited for adjacent probes |
| Bio-Rad | Chromo 4 | LED | FAM, TMR, Texas red, and Cy5 | 4 targets | 96 wells | All types, less suited for adjacent probes |
| Bio-Rad | ICycler IQ5 | THL | FAM, HEX, TMR, Texas red, and Cy5 | 5 targets | 96 wells | All types |
| Cepheid | SmartCycler II | LED | FAM, Cy3, Texas red, and Cy5 | 4 targets | 16 unitsb,c | All types, less suited for adjacent probes |
| Corbett Research | Rotor-Gene 6000 | LED | CPM, FAM, TET, Texas red, Cy5, and LightCycler Red 705 | 6 targets | 72 wellsc | All types |
| Eppendorf | Mastercycler realplex 2 | LED | FAM and HEX | 2 targets | 96 wells | All types, less suited for adjacent probes |
| Eppendorf | Mastercycler realplex 4 | LED | FAM, TET, TMR, and Texas red | 4 targets | 96 wells | All types, less suited for adjacent probes |
| Idaho Technologies | R.A.P.I.D. | LED | FAM, LightCycler Red 640, and LightCycler Red 705 | 3 targets | 32 wellsc | All types |
| Roche Applied Science | LightCycler 1.5 | LED | FAM, LightCycler Red 640, and LightCycler Red 705 | 3 targets | 32 wellsc | Best suited for adjacent probes and WS-MB probes |
| Roche Applied Science | LightCycler 2.0 | LED | FAM, HEX, LightCycler Red 610, LightCycler Red 640, LightCycler Red 670, and LightCycler red 705 | 6 targets | 32 wellsc | Best suited for adjacent probes and WS-MB probes |
| Roche Applied Science | LightCycler 480 | XL | CPM, FAM, HEX, LightCycler Red 610, LightCycler Red 640, and Cy5 | 6 targets | 96 wells and 384 wellsc | All types |
| Stratagene | Mx3000P | THL | FAM, TMR, Texas red, and Cy5d | 4 targets | 96 wells | All types |
| Stratagene | Mx3005P | THL | FAM, HEX, TMR, Texas red, and Cy5d | 5 targets | 96 wells | All types |
Modified from (39)
THL tungsten-halogen lamp, ABLL argon blue-light laser, LED light-emitting diode, XL xenon lamp, WS-MB wavelength-shifting molecular beacon.
aRefer to Table 17.2 for alternative fluorophores.
bEach unit is independently programmable.
cRapid cycle capabilities
dAlternative preinstalled excitation and emission filter sets are available.
Fig. 17.1.
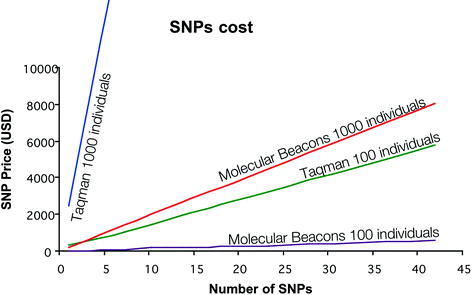
Comparative cost between TaqMan assays and molecular beacons. Regardless of the number of individuals or the number of single nucleotide polymorphisms (SNPs) to be genotyped, the cost of molecular beacons is significantly reduced with respect to TaqMan assays owing to the multiplexing power of molecular beacons in a single tube. The cost for TaqMan assays is based on the prices provided by Applied Biosystem when using 96-well plates and 25-μL PCRs. The cost of TaqMan assays can be reduced by approximately 5–10% by performing the assays in 384-well plates and 5-μL reactions.
Two principal obstacles must be overcome in the detection and analysis of SNPs. The first is the small amounts of nucleic acid present in clinical specimens. This can be overcome by use of differing nucleic acid amplification strategies, most notably polymerase chain reaction (PCR). This and other methods such as nucleic acid sequence based amplification allow the selective amplification and enrichment of a locus of interest by several-thousand-fold over other nucleic acid sequences present (25). The second obstacle is unambiguous detection of the SNP. Herein lies an intrinsic property of nucleic acid chemistry that can be exploited. A unique property of nucleic acid hybridization is its extremely high fidelity. Such molecular interactions are the most specific and stable known in nature. It becomes possible to monitor and detect hybridization of nucleic acids if it is accompanied by an assayable change in conformation. Two principal methods have emerged in detecting such assayable changes in conformation. The first, TaqMan (26), depends upon the monitoring of enzymatic nucleic acid probe cleavage, resulting in fluorescence (see Chapters 18 and 19 for details). The second, molecular beacons (1), detects a conformational change in the probe, which fluoresces upon hybridization. We will focus principally on the use of molecular beacons.
Molecular beacons are single-stranded oligonucleotide probes with a stem-and-loop structure (see Fig. 17.2). The loop is complementary to a known sequence in a target nucleic acid sequence, whereas the stem forms by the hybridization of the arm sequences on either side of the loop sequence. A fluorescent moiety is covalently linked to the extremity of one arm sequence and a quencher is covalently linked to the extremity of another arm. Thus, the fluorophore and quencher are directly juxtaposed when the stem is formed and are in extremely close proximity to each other. This association prevents fluorescence from being emitted from the fluorophore. When the loop portion of the molecule encounters a perfectly complementary target, the entire molecule undergoes a conformational change that results in the separation of the arms of the stem. This causes a restoration of fluorescence to the fluorophore as it is moved away from the quencher. Alterations to the length of the probe region strongly influence the stability and specificity of the probe–target hybrid, contributing to the extreme specificity of molecular beacons. A wide variety of differently colored fluorophores are possible with molecular beacons (27), thus enabling the simultaneous detection of multiple targets in the same solution by using molecular beacons designed to detect differing targets each labeled with a spectrally distinguishable fluorophore.
Fig. 17.2.

Principle of how molecular beacons function. (a) When the probe sequence (loop portion) encounters a target that is perfectly complementary to it, a conformational reorganization of the molecule occurs, resulting in a separation of the stem and the generation of a fluorescence signal. (b) Thermal denaturation profiles of molecular beacons when they are with wild-type or mutant targets. The wild-type target is represented by solid lines and the mutant target is represented by dashed lines. The absence of target is indicated by a dotted line. The conformational state of the molecular beacon is shown directly above the line. By careful design of molecular beacons, mismatched targets can be easily discriminated from perfectly matched targets with “windows of discrimination” as high as 10°C. The optimal temperature for the annealing step from this thermal denaturation profile is found to be 50°C and therefore is used in real-time PCR. (c) An example of how each molecular beacon, the “red”-labeled or the “green”-labeled, competes to hybridize to the same region depending on whether it is perfectly complementary to the region.
The above-mentioned properties of molecular beacons enable their use in monitoring the progress of nucleic acid amplification reactions (28 – 32), self-reporting oligonucleotide arrays, and the detection of messenger RNA in living cells (33 – 36). Molecular beacons are especially adept at the detection of SNPs since they recognize their targets with exquisite specificity unlike conventional linear probes, owing to their hairpin structure (37). Thermodynamic studies where linear and stem–loop probes were compared have revealed that this enhanced specificity is a general feature of conformationally constrained probes such as molecular beacons. Thus, specificity can be “tuned” by altering the degree to which the probes are conformationally constrained. Practically this involves altering the length of the stem structure in relation to the length of the loop. In applications such as SNP detection, molecular beacons can be designed to bind over a wide range of temperatures such that only perfectly complementary probe–target hybrids are formed. This keeps mismatched probes which vary by even as much as one base unbound and dark, whereas only perfectly complementary probe–target hybrids elicit fluorescence. Owing to these unique properties, the use of molecular beacons for SNP detection has proliferated broadly as has its expansion into a cost-effective high-throughput SNP diagnostic tool.
Materials
Reagents and Equipment
Molecular beacon probes (see Section 3.4) designed to hybridize to a target sequence carrying SNP of interest (see Note 2) (Biosearch Technologies, http://www.biosearchtech.com).
Fluorescent dyes for manual linking to molecular beacons (Glen Research or Molecular Probes/Invitrogen).
Black Hole quenchers (Biosearch Technologies, http://www.biosearchtech.com).
Buffer I: 0.1 M sodium bicarbonate, pH 8.5.
Buffer II: 10 mM tris(hydroxymethyl)aminomethane (Tris)–HCl, pH 8.0, 4 mM MgCl2, 50 mM KCl.
Buffer A: 0.1 M triethylammonium acetate, pH 6.5.
Buffer B: 0.1 M triethylamonium acetate in 75% acetonitrile, pH 6.5.
Ammonium sulfate (3 M).
Silver nitrate (0.15 M).
Dithiothreitol (0.15 M).
Sodium bicarbonate (0.2 M), pH 9.0.
1X TE buffer: 10 mM Tris–HCl, pH 7.5, 1 mM EDTA.
Sephadex G-25 column NAP-5 (GE/Amersham-Pharmacia).
Filter: 0.2-µm Centrex MF-0.4 filter (Schleicher & Schuell).
High-pressure liquid chromatography (HPLC) system Gold (Beckman Coulter)
C-18 reverse-phase column (Waters).
Molecular beacon buffer: 10 mM Tris–HCl, pH 8.0, 3.5 mM MgCl2.
Thermocycler, PRISM 7700 PCR system (Applied Biosystems).
AmpliTaq Gold DNA polymerase (Applied Biosystems). Store at –20°C.
dNTP set, 100 mM solutions (Applied Biosystems). Store at –20°C.
Spectrofluorometer, QuantaMaster (Photon Technology International).
Haploview software program (HapMap project, http://www.hapmap.org).
Zuker/mfold fold software program (http://www.bioinfo.rpi.edu/applications/mfold/).
Synthesis of Molecular Beacons
Significant advances have been made in solid-phase chemistry enabling the routine synthesis of nucleic acids coupled to fluorophore and quencher moieties (38). Almost all organic dyes that are routinely used in the visible and infrared light range are available as phosphoramidites, which can be coupled to nucleic acid oligomers during routine syntheses. This is also true for quenchers. For complex syntheses and nonstandard molecular beacons, it is also possible to use manual coupling approaches. This is done by using oligonucleotides which contain either amino or sulfahydryl functional groups at either their 5′-ends or their 3′-ends. By using succinimidyl ester, iodoacetamide derivatives, or maleimide derivatives of the fluorophores and quenchers, one can couple most commercially available dyes and quenchers to oligonucleotides possessing either amino or sulfahydryl functional groups. In Section 3.1 and 3.2 we describe a protocol for manual synthesis of modified oligonucleotides.
Matching the Fluorophore to the Instrument
With the emergence of real-time PCR as a standard instrument in most laboratories, a number of instruments with differing capabilities have become available. For high-throughput applications such as SNP typing, the principal considerations should be multiplexing abilities, throughput (number of wells), and to a certain extent cycling speed. Spectral overlap is minimized with molecular beacons since they are quenched when unbound. In addition, several instruments (Table 17.1) are able to detect up to six spectrally distinguishable dyes (Table 17.2), routinely enabling extremely powerful multiplexing capabilities.
Table 17.2.
Fluorophore labels for fluorescent hybridization probes
| Fluorophore | Alernative fluorophore | Excitation (nm) | Emission (nm) |
|---|---|---|---|
| Coumarin | Biosearch Bluea, LightCycler Cyan 500b | 430 | 475 |
| FAM | 495 | 515 | |
| TET | CAL Fluor Gold 540a | 525 | 540 |
| HEX | ATTO 532c, CAL Fluor Orange 560a, JOE, VICd | 535 | 555 |
| Cy3 | NEDd, Oyster 556f, Quasar 570a | 550 | 570 |
| TMR | Alexa 546g, CAL Fluor Red 590a | 555 | 575 |
| ROX | Alexa 568g, CAL Fluor Red 610a, LightCycler Red 610b | 575 | 605 |
| Texas red | Alexa 594g, CAL Fluor Red 610a, LightCycler Red 610b | 585 | 605 |
| LightCycler Red 640 | CAL Fluor Red 635a | 625 | 640 |
| Cy5 | ATTO 647 Nc, LC Red 670b, Oyster 645f, Quasar 670a | 650 | 670 |
| LightCycler Red 705 | Cy5.5e, Quasar 705a | 680 | 710 |
Modified from (39)
aBiosearch Blue, CAL, and Quasar fluorophores are available from Biosearch Technologies.
bLightCycler fluorophores are available from Roche Applied Science.
cATTA dyes are available from ATTO-TEC.
dVIC and NED fluorophores are available from Applied Biosystems.
eCyanine dyes are available from Amersham Biosciences.
fOyster fluorophores are available from Integrated DNA Technologies.
gAlexa fluorophores are available from Invitrogen.
To run this application one would need to have one of the instruments described in Table 17.1. The choice of the instrument depends on the task and the dyes to be used.
Methods
Coupling of Quencher
Dissolve 50–250 nmol of dry (commercially obtained or custom-made) oligonucleotide in 500 µL of buffer I. In DMSO dissolve approximately 20 mg succinimidyl ester coupled quencher and add it to a stirring solution of the oligonucleotide in 10-µL aliquots at 20-min intervals. Continue stirring for at least 12 h. Perform this reaction in the dark (see Note 1). We recommend the Black Hole family of quenchers that are available in three variants dependent on the desired wavelength for quenching (see Section 2.2).
Remove particulate material by spinning the mixture in a microcentrifuge for 1 min at 16,000 g. To remove unreacted quencher, pass the supernatant through a gel-exclusion column. Equilibrate a Sephadex G-25 column with buffer A, load the supernatant, and elute the contents of the column with 1 mL of buffer A. Filter the eluate through a 0.2-µm Centrex MF-0.4 filter.
Purify the oligonucleotides by HPLC on a C-18 reverse-phase column, utilizing a linear elution gradient of 20–70% buffer B in buffer A and run the elution for 25 min at a flow rate of 1 mL/min. Monitor the absorption of the elution stream at 260 nm and the specific quencher absorption maximum. Collect the eluate that absorbs at both wavelengths, and that therefore contains oligonucleotides with a protected sulfhydryl group at their 5′-ends and the quencher at their 3′-ends.
Precipitate the collected material with ethanol and 3 M ammonium sulfate, and spin the precipitate in a centrifuge for 10 min at 16,000g, discard the supernatant, dry the pellet, and dissolve it in 250 µL of buffer A.
Coupling of Fluorophore
To remove the trityl moiety, add 10 µL of 0.15 M silver nitrate and incubate the solution for 30 min. Add 15 µL of 0.15 M dye to this mixture and shake the mixture for 5 min. Spin the mixture for 2 min at 16,000g and transfer the supernatant to a new tube. Dissolve about 40 mg og 5-iodoactamido-reactive fluorophore in 250 µL of 0.2 M sodium bicarbonate, pH 9.0, and add it to the supernatant. Incubate the mixture for 90 min. Each of these solutions should be prepared just before use.
Remove excess uncoupled fluorophore from the reaction mixture by gel-exclusion chromatography and purify the oligonucleotides coupled to the fluorophore by HPLC, following the instructions in steps 2 and 3 in Section 3.1. Collect the fractions that absorb with a peak at 260 nm and at the specific fluorophore absorption maximum. This eluate should be fluorescent when observed with an ultraviolet lamp in a dark room.
Precipitate the collected material and dissolve the pellet in 100 µL 1X TE buffer. Determine the absorbance at 260 nm and estimate the yield (1 OD260 = 33 ng/µL). Store the purified molecular beacon for long-term storage in lyophilized form at –80°C (see Notes 1 and 2).
Characterization of Molecular Beacons
Signal-to-Background Ratio
Determine the fluorescence of 200 µL of molecular beacon buffer solution (F buffer), using 491 nm as the excitation wavelength and the emission wavelength of the fluorophore used (Fig. 17.3).
Add 10 µL of 1 µM molecular beacon to this solution and record the new level of fluorescence (F closed).
Add a twofold molar excess of a complementary oligonucleotide target and monitor the rise in fluorescence until it reaches a stable level (F open).
Calculate the signal-to-background ratio as (F open-F buffer)/(F closed-F buffer).
Fig. 17.3.

Spectrofluorometric characterization of molecular beacons. The molecular beacons are functionally characterized in the presence of perfectly complementary oligonucleotide. Here a 30-fold increase is observed.
Thermal Denaturation Profiles
Prepare two tubes containing 50 µL of 200 nM molecular beacon dissolved in molecular beacon buffer solution and add the oligonucleotide target to one of the tubes at a final concentration of 400 nM (see Fig. 17.2).
Determine the fluorescence of each solution as a function of temperature using a spectrofluorometric thermal cycler (see Table 17.1). Decrease the temperature of these tubes from 80 to 10°C in 1°C steps, with each hold lasting 1 min, while monitoring the fluorescence during each hold (see Fig. 17.2).
Design of Primers and Molecular Beacons for SNP Detection
The design of molecular beacons for SNP detection is at times challenging since the flexibility in the targeting region to be detected is virtually nil. The region where the SNP of interest occurs must be targeted and molecular beacons with as little as one base variant from this region must not bind under amplification conditions. To satisfy these constraints, the loop portion of the probe is made to be not more than 25 nucleotides in length. As a rule of thumb, the shorter the length of the loop, the more highly discriminating the probe will be. Care must be taken to ensure that the melting temperature of the probe–target hybrid is compatible with the annealing temperature of primers during PCR. With this part of the design complete, stem/arm sequences can be designed that allow the stem to dissociate at about 7–10°C above the annealing temperature of the primers during PCR. This design process is made more complex in certain examples where multiple primers are used in a single tube (as in the example given later in this chapter). The challenge when doing multiplex PCR is to optimize all the primers for all the PCRs first. This ensures that all primers make good amplicons at the same temperature. Molecular beacons can then be designed to be SNP-discriminating at the annealing temperature of the primers by alterations in loop size. It is always useful to verify the secondary structure of the designed molecular beacon to ensure that it does not contain secondary structures that restrict the loop from binding to a PCR target. The preferred program for nucleic acid secondary structure prediction is Zuker/mfold fold (http://www.bioinfo.rpi.edu/applications/mfold/). For extremely difficult situations where design for AT- or GC-rich regions makes the stability of annealing variable, this can be circumvented by a number of strategies such as sliding the loop region so the SNP is no longer at its center. A second strategy is to include the stem/arm sequences in the binding sequence so as to create an even more stable hybrid (this could be useful in AT-rich regions). Lastly, if these strategies prove unsuccessful, an additional annealing step for the purposes of detection can be programmed into the thermal cycling profile. This step can be designed to occur at a temperature where it is easier to meet SNP discrimination constraints with the molecular beacons designed. It can also potentially result in false priming so it is not a preferred approach. For detailed instructions on the general design of molecular beacons for SNP detection, see (29,32).
PCR primers were designed that consistently amplified regions no greater than 250 base pairs. Those design rules were followed to make the probes and primers shown in (see Fig. 17.4). The dedicated software package Beacon Builder (Premier Biosoft International) can be used for the design of similar molecular beacons. The window of discrimination outlined in Fig. 17.4 should be carefully studied and respected in designing molecular beacons to detect SNPs.
Fig. 17.4.

High-throughput SNP scoring of the DC-SIGN locus. (a) Eighteen molecular beacons and corresponding primers were designed to score the major and minor alleles of nine “tag” SNPs of the DC-SIGN locus. Each major and minor SNP allele had a molecular beacon labeled in a spectrally distinct color. This means that in instruments where up to six colors are spectrally distinguishable, it is possible to simultaneously detect up to six major and/or minor alleles. To score each of the alleles in a given individual, three PCR amplifications were set up with the appropriate primers (not shown) that all annealed at a similar temperature. At each annealing step, depending on the presence or absence of a particular allele, a given molecular beacon would fluoresce. By “scoring” the data for each tube, one can determine, for each individual the specific genotype for each of the nine tag SNPs. (b) The three possibilities for a given SNP locus, either a single major or a single minor allele is present, in which case a homozygous result is obtained and only a single color is observed. Alternatively, both alleles are observed, indicating that the locus is a heterozygote. (c) Haplotypes observed for the combination of these nine tag SNPs in the Cape Town population. The frequencies reported correspond to the frequencies observed for each of these haplotypes in the Cape Town population independent of their disease status. An association was observed between two DC-SIGN promoter variants (-871G and -336A) and decreased risk of developing tuberculosis. Haplotype 3 turned out to be the best predictor of an increased resistance to tuberculosis, at least in the South African population. This haplotype, which contains both -871G and -336A, was found to be more frequently observed in the control group than in people who developed tuberculosis (8.9% vs. 14.2% p = 1.6 × 10 − 3; odds ratio 1.7; 95% confidence interval 1.22–2.38.
Real-Time PCR
Prepare a 50-µL (or as little as 5-µL) reaction that contains 100 nM major allele specific molecular beacon, 100 nM minor allele specific molecular beacon, 500 nM concentration of each primer, at least 1 unit of AmpliTaq Gold DNA polymerase, and 250 µM concentration of each type of dNTP, dissolved in buffer II.
Run the PCR. The thermal cycle for most of the machines described in Table 17.1 should be 10 min at 95°C followed by 35–40 cycles at 30 s at 95°C, 45 s at 50°C (or a temperature which is compatible with the window of discrimination), and 30 s at 72°C. The fluorescence should be monitored at the appropriate channel during the 50°C annealing step (see Notes 3, 4 and 5).
Data Analysis in a Case Study Using Tag SNPs (High-Throughput SNP Scoring of the DC-SIGN Locus)
In human genetics, association studies aim to identify loci that contribute to disease susceptibility by comparing patterns of genetic variation between people with a disease (cases) and those without (controls). As mentioned earlier, several studies have revealed an interesting feature present in the structure of human genetic variation that can be utilized to dramatically reduce the cost of association studies (11, 40 – 43). Specifically, alleles at nearby loci often show strong statistical association (i.e., LD). This can be exploited to design a powerful and cost-effective way to perform association studies by using tag SNPs for a region of interest, i.e., by determining which loci within that region capture the majority of the diversity.
In this section we outline a study of the DC-SIGN gene. By using the unique multiplexing power of molecular beacons in a high-throughput assay, we are able to genotype nine tag SNPs thereby obtaining information from 54 SNPs. Thus, with three tubes per individual and with three pairs of molecular beacons per tube, we are able to score all the information of 54 SNPs.
DC-SIGN is an innate immunity gene that belongs to the C-type lectin family. C-type lectins are calcium-dependent carbohydrate-binding proteins with a wide range of biological functions, many of which are related to immunity (44). DC-SIGN as well as its homolog L-SIGN are particularly interesting, since they can act as both cell-adhesion receptors and pathogen-recognition receptors (45). DC-SIGN was originally cloned for its ability to bind and internalize the heavily glycosylated HIV gp120 protein (46). DC-SIGN strongly binds all HIV and simian immunodeficiency virus strains examined to date and plays an important role in virus adhesion to dendritic cells (47, 48). These studies have paved the way for further investigations into interactions between DC-SIGN and other pathogens and it has now become clear that this lectin recognizes a vast range of microbes, some of which are of major public health importance (48). Indeed, DC-SIGN captures bacteria such as Mycobacterium tuberculosis, Mycobacterium leprae, Helicobacter pylori, and certain Klebsiela pneumonia strains; viruses such as HIV-1, Ebola virus, cytomegalovirus, hepatitis C virus, Dengue virus, and SARS coronavirus; and parasites such as Leishmania pifanoi and Schistosoma mansoni (47, 49 – 59).
In light of the ability of DC-SIGN to interact with a large plethora of pathogens, it is plausible that variation in its gene may influence the pathogenesis of a number of infectious diseases. Indeed, multiple association studies have shown a relationship between genetic variants in the promoter region of DC-SIGN and susceptibility to several infectious diseases. Specifically, it has been shown that two promoter variants, -871G and -336A, confer protection against tuberculosis. Similarly, the -336A variant has been reported to protect against parental HIV infection and to influence the severity of dengue pathogenesis (60, 61). More recently, two other promoter variants, –139A/G and –939G/A, showed a significant association with an increased risk of developing human cytomegalovirus reactivation and disease (60).
How can one efficiently test for an association between DC-SIGN variation and susceptibility to disease? Imagine that you want to explore the relationship between DC-SIGN polymorphisms and susceptibility to tuberculosis (62). The best way to do so is to follow the strategy described below:
- Collect a cohort, from the same population (see Note 6), that includes a group of individuals that developed tuberculosis (i.e., cases) and a group of matched individuals that did not develop the disease (i.e., controls). Ideally, one would need/like to fully resequence DC-SIGN in the entire cohort to obtain the full extent of diversity present in cases and controls. Nevertheless, full resequencing approaches are unacceptably expensive and time consuming and, therefore, the most powerful and cost-effective way to perform association studies is by defining tag SNPs for a region of interest (see Section 17.1 for details). To do so, you have two alternatives:
- Begin by fully resequencing the region under study in a subset of your cohort. Typically 20–30 individuals should be enough to capture the most common haplotypes in the population. After haplotype reconstruction (see Note 7) and on the basis of the LD patterns observed, you can then identify the set of SNPs best able to characterize the diversity observed (i.e., tag SNPs) (see Note 8).
- Use publicly available datasets to identify tag SNPs. The best available resource to choose tag SNPs is the HapMap data. Go to the HapMap Web site (http://www.hapmap.org) and using the genome browser retrieve genotypic data for all the SNPs that have been typed for the region you are interested in; in this case DC-SIGN. Then, upload the data in Haploview (a free software program provided by the HapMap consortium) and run Tagger to identify tag SNPs for your region (see Note 7). The current limitation of using HapMap is that the data are restricted to three human populations – the samples came from an African population from Nigeria (Yoruba; N = 90), a mostly Utah (USA) population of European ancestry (N = 90), and a sample drawn from Japanese (N = 45) and Han Chinese (N = 45) populations. If your population is genetically distinct from these HapMap populations, you will have to follow the resequencing strategy; as the tag SNPs identified using HapMap populations might differ from those characterizing the diversity of your study-population.
Once you have identified the set of SNPs best able to characterize the full diversity observed in your population, the next step is to genotype these tag SNPs in the entire cohort. In Fig. 17.4 we present an example of a haplotyping approach scoring tag SNPs in a high-throughput assay using molecular beacons to easily test for an association between DC-SIGN variation and susceptibility to infectious diseases. This example is based on a previous study that explored the relationship between DC-SIGN polymorphisms and susceptibility to tuberculosis (63). The authors showed that nucleotide variation in the DC-SIGN promoter region is associated with susceptibility to tuberculosis. Specifically they identified a specific haplotype (Fig. 17.4) associated with decreased risk of developing tuberculosis (63 ).
Notes
Molecular beacons deteriorate as they are exposed to light. Therefore, avoid exposure to light whenever possible. Molecular beacons should be stored in aluminum-foil-wrapped test tubes at –20°C and preferably at –80°C in lyophilized form. When preparing them for use, one can resuspended them in TE buffer.
Since most oligonucleotide manufacturers worldwide can provide molecular beacons with all these functionalities, obtaining molecular beacons with diverse fluorophore and quencher combinations has become routine. These suppliers can be found at http://www.molecular-beacons.org.
At times, false amplicons may appear during PCR and may appear if the sensitivity of the PCR is reduced. Two approaches can be used to circumvent this. Firstly, DNA polymerases that are active only after activation at 95°C can be used. Secondly, paying careful attention to the design of primers that function well within the “window of discrimination” is recommended.
The real-time PCR machines and fluorescent dyes proposed in Table 17.1 and 17.2 are fairly good at discriminating between the proposed dyes. Thus, if poor discrimination is observed between major and minor alleles, tweaks to the primers and annealing temperatures can be made that permit more stringent discrimination. If these are unsuccessful, modifications to the molecular beacons themselves can be made. One modification is to increase the length of the molecular beacon stem to promote stability and increase stringency. A second modification is to use 2′-O-methyl molecular beacons, which intrinsically have a higher melting temperature than DNA-based molecular beacons. However 2′-O-methyl molecular beacons are more expensive to synthesize. Third, the stem sequence of the molecular beacon can be designed to also bind to the amplicon.
Amplicon size has a very important influence on the fluorescence signal obtained with molecular beacons. Thus, it is important to design PCRs where amplicons do not exceed 250 bp.
It is important that the groups of cases and controls are genetically matched, as population stratification between cases and controls can be a confounding factor leading to a spurious positive association. This will be particularly harmful if cases and controls are from different populations, but also in admixed populations (e.g. CAP population from South Africa). Indeed, the use of admixed populations in association-mapping studies can be very useful for identification of disease-causing genetic variants that differ in frequency across parental populations. However, when the admixture event is too recent, allelic frequencies can differ coincidentally among cases and controls, reflecting a nonuniform genetic contribution from the parental populations to each subpopulation (i.e., cases and controls), rather than a genuine association between a given genetic variant and the phenotype under study. In this case, the study cohort is said to present population stratification.
To reconstruct haplotypes we recommend the Bayesian statistical method implemented in Phase version 2.1.162 (64). Alternatively, you can use the accelerated expectation maximization algorithm implemented in Haploview version 3.163 (65). At least for regions with high levels of LD, both algorithms should give similar results.
Tag SNPsfor each population can be selected using Haploview’s Tagger in pairwise tagging mode (r2 ≥ 0.80, minor allele frequency cutoff 5%, and other settings at default value).
References
- 1.Tyagi S., Kramer F. R. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 2.Tyagi S. DNA Probes. In: Meyers R. A., editor. Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation. Chichester, UK: John Wiley & Sons Ltd; 2000. p. 4911. [Google Scholar]
- 3.Lander E. S., Linton L. M., Birren B. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.Sachidanandam R., Weissman D., Schmidt S. C. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 5.Hinds D. A., Stuve L. L., Nilsen G. B. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 6.Miller R. D., Phillips M. S., Jo I. High-density single-nucleotide polymorphism maps of the human genome. Genomics. 2005;86:117–126. doi: 10.1016/j.ygeno.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruglyak L., Nickerson D. A. Variation is the spice of life. Nat. Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 8.Miller R. D., Kwok P. Y. The birth and death of human single-nucleotide polymorphisms: new experimental evidence and implications for human history and medicine. Hum. Mol. Genet. 2001;10:2195–2198. doi: 10.1093/hmg/10.20.2195. [DOI] [PubMed] [Google Scholar]
- 9.Crawford D. C., Akey D. T., Nickerson D. A. The patterns of natural variation in human genes. Annu. Rev. Genomics Hum. Genet. 2005;6:287–312. doi: 10.1146/annurev.genom.6.080604.162309. [DOI] [PubMed] [Google Scholar]
- 10.Consortium TIH. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 11.Consortium TIH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frazer K. A., Ballinger D. G., Cox D. R. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conrad D. F., Jakobsson M., Coop G. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Neira A., Ke X., Lao O. The portability of tagSNPs across populations: a worldwide survey. Genome Res. 2006;16:323–330. doi: 10.1101/gr.4138406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eberle M. A., Ng P. C., Kuhn K. Power to detect risk alleles using genome-wide tag SNP panels. PLoS Genet. 2007;3:1827–1837. doi: 10.1371/journal.pgen.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consortium TWTCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todd J. A., Walker N. M., Cooper J. D. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxena R., Voight B. F., Lyssenko V. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 19.Zeggini E., Weedon M. N., Lindgren C. M. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samani N. J., Erdmann J., Hall A. S. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frayling T. M., Timpson N. J., Weedon M. N. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scuteri A., Sanna S., Chen W. M. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomson W., Barton A., Ke X. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fellay J., Shianna K. V., Ge D. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leone G., van Schijndel H., van Gemen B., Kramer F. R., Schoen C. D. Molecular beacon probes combined with amplification by NASBA enable homogeneous, real-time detection of RNA. Nucleic Acids Res. 1998;26:2150–2155. doi: 10.1093/nar/26.9.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak K. J. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet. Anal. 1999;14:143–149. doi: 10.1016/S1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 27.Tyagi S., Bratu D. P., Kramer F. R. Multicolor molecular beacons for allele discrimination. Nat. Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 28.El-Hajj H. H., Marras S. A., Tyagi S., Kramer F. R., Alland D. Detection of rifampin resistance in Mycobacterium tuberculosis in a single tube with molecular beacons. J. Clin. Microbiol. 2001;39:4131–4137. doi: 10.1128/JCM.39.11.4131-4137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marras S. A., Kramer F. R., Tyagi S. Genotyping SNPs with molecular beacons. Methods Mol. Biol. 2003;212:111–128. doi: 10.1385/1-59259-327-5:111. [DOI] [PubMed] [Google Scholar]
- 30.Vet J. A., Majithia A. R., Marras S. A. Multiplex detection of four pathogenic retroviruses using molecular beacons. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6394–6399. doi: 10.1073/pnas.96.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostrikis L. G., Tyagi S., Mhlanga M. M., Ho D. D., Kramer F. R. Spectral genotyping of human alleles. Science. 1998;279:1228–1229. doi: 10.1126/science.279.5354.1228. [DOI] [PubMed] [Google Scholar]
- 32.Mhlanga M. M., Malmberg L. Using molecular beacons to detect single-nucleotide polymorphisms with real-time PCR. Methods. 2001;25:463–471. doi: 10.1006/meth.2001.1269. [DOI] [PubMed] [Google Scholar]
- 33.Bratu D. P., Cha B. J., Mhlanga M. M., Kramer F.R., Tyagi S. Visualizing the distribution and transport of mRNAs in living cells. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13308–13313. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mhlanga M. M., Vargas D. Y., Fung C. W., Kramer F. R., Tyagi S. tRNA-linked molecular beacons for imaging mRNAs in the cytoplasm of living cells. Nucleic Acids Res. 2005;33:1902–1912. doi: 10.1093/nar/gki302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyagi S., Alsmadi O. Imaging native beta-actin mRNA in motile fibroblasts. Biophys. J. 2004;87:4153–4162. doi: 10.1529/biophysj.104.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vargas D. Y., Raj A., Marras S. A., Kramer F. R., Tyagi S. Mechanism of mRNA transport in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17008–17013. doi: 10.1073/pnas.0505580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonnet G., Tyagi S., Libchaber A., Kramer F. R. Thermodynamic basis of the enhanced specificity of structured DNA probes. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee L. G., Livak K. J., Mullah B., Graham R. J., Vinayak R.S., Woudenberg T. M. Seven-color, homogeneous detection of six PCR products. Biotechniques. 1999;27:342–349. doi: 10.2144/99272rr01. [DOI] [PubMed] [Google Scholar]
- 39.Marras S. A. Interactive fluorophore and quencher pairs for labeling fluorescent nucleic acid hybridization probes. Mol. Biotechnol. 2008;38:247–255. doi: 10.1007/s12033-007-9012-9. [DOI] [PubMed] [Google Scholar]
- 40.Daly M. J., Rioux J. D., Schaffner S. F., Hudson T. J., Lander E. S. High-resolution haplotype structure in the human genome. Nat. Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- 41.Dawson E., Abecasis G. R., Bumpstead S. A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002;418:544–548. doi: 10.1038/nature00864. [DOI] [PubMed] [Google Scholar]
- 42.Gabriel S. B., Schaffner S. F., Nguyen H. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 43.Reich D. E., Cargill M., Bolk S. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 44.Zelensky A. N., Gready J. E. The C-type lectin-like domain superfamily. FEBS J. 2005;272:6179–6217. doi: 10.1111/j.1742-4658.2005.05031.x. [DOI] [PubMed] [Google Scholar]
- 45.Soilleux E. J. DC-SIGN (dendritic cell-specific ICAM-grabbing non-integrin) and DC-SIGN-related (DC-SIGNR): friend or foe? Clin. Sci. (Lond) 2003;104:437–446. doi: 10.1042/CS20020092. [DOI] [PubMed] [Google Scholar]
- 46.Curtis B. M., Scharnowske S., Watson A. J. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl. Acad. Sci. U. S. A. 1992;89:8356–8360. doi: 10.1073/pnas.89.17.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geijtenbeek T. B., Kwon D. S., Torensma R. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/S0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 48.Geijtenbeek T. B., van Vliet S. J., Engering A., Hart B. A., van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu. Rev. Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 49.Alvarez C. P., Lasala F., Carrillo J., Muniz O., Corbi A. L., Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002;76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Appelmelk B. J., van Die I., van Vliet S. J., Vandenbroucke-Grauls C. M., Geijtenbeek T. B., van Kooyk Y. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 2003;170:1635–1639. doi: 10.4049/jimmunol.170.4.1635. [DOI] [PubMed] [Google Scholar]
- 51.Barreiro L. B., Quach H., Krahenbuhl J. DC-SIGN interacts with Mycobacterium leprae but sequence variation in this lectin is not associated with leprosy in the Pakistani population. Hum. Immunol. 2006;67:102–107. doi: 10.1016/j.humimm.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergman M. P., Engering A., Smits H. H. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J. Exp. Med. 2004;200:979–990. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colmenares M., Puig-Kroger A., Pello O. M., Corbi A. L., Rivas L. Dendritic cell (DC)-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC-SIGN, CD209), a C-type surface lectin in human DCs, is a receptor for Leishmania amastigotes. J. Biol. Chem. 2002;277:36766–36769. doi: 10.1074/jbc.M205270200. [DOI] [PubMed] [Google Scholar]
- 54.Geijtenbeek T. B., Van Vliet S. J., Koppel E. A. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halary F., Amara A., Lortat-Jacob H. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 2002;17:653–664. doi: 10.1016/S1074-7613(02)00447-8. [DOI] [PubMed] [Google Scholar]
- 56.Lozach P. Y., Lortat-Jacob H., de Lacroix de Lavalette A. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003;278:20358–20366. doi: 10.1074/jbc.M301284200. [DOI] [PubMed] [Google Scholar]
- 57.Marzi A., Gramberg T., Simmons G. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tailleux L., Schwartz O., Herrmann J. L. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tassaneetrithep B., Burgess T. H., Granelli-Piperno A. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin M. P., Lederman M. M., Hutcheson H. B., et al. Association of DC-SIGN promoter polymorphism with increased risk for parenteral, but not mucosal, acquisition of human immunodeficiency virus type 1 infection. J. Virol. 2004;78:14053–14056. doi: 10.1128/JVI.78.24.14053-14056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sakuntabhai A., Turbpaiboon C., Casademont I. A variant in the CD209 promoter is associated with severity of dengue disease. Nat. Genet. 2005;37:507–513. doi: 10.1038/ng1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mezger M., Steffens M., Semmler C. Investigation of promoter variations in dendritic cell-specific ICAM3-grabbing non-integrin (DC-SIGN) (CD209) and their relevance for human cytomegalovirus reactivation and disease after allogeneic stem-cell transplantation. Clin. Microbiol. Infect. 2008;14:228–234. doi: 10.1111/j.1469-0691.2007.01902.x. [DOI] [PubMed] [Google Scholar]
- 63.Barreiro L. B., Neyrolles O., Babb C. L. Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med. 2006;3:e20. doi: 10.1371/journal.pmed.0030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stephens M., Donnelly P. A. comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barrett J. C., Fry B., Maller J., Daly M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]