

Abstract
By now, it is well established that the error rate of the RNA-dependent RNA polymerase (RdRp) that replicates RNA virus genomes is a primary driver of the mutation frequencies observed in RNA virus populations—the basis for the RNA quasispecies. Over the last 10 years, a considerable amount of work has uncovered the molecular determinants of replication fidelity in this enzyme. The isolation of high- and low-fidelity variants for several RNA viruses, in an expanding number of viral families, provides evidence that nature has optimized the fidelity to facilitate genetic diversity and adaptation, while maintaining genetic integrity and infectivity. This chapter will provide an overview of what fidelity variants tell us about RNA virus biology and how they may be used in antiviral approaches.
Keywords: West Nile Virus, Mutation Frequency, Coxsackie Virus, Chikungunya Virus, Phosphoryl Transfer
RNA Virus Mutation Rates and Mutators/Antimutators in the Microbial World
RNA viruses possess the highest mutation rates in nature. As calculated by Drake et al. (1998), Drake and Holland (1999), even the comparatively modest retroviruses, generating 0.1 mutations per genome per replication cycle, lead to enormous variation in viral progeny. RNA virus mutation rates are therefore magnitudes higher than DNA organisms, such as Escherichia coli (0.0025) or Saccharomyces cerevisiae (0.0027). This is in large part due to the lack of proofreading mechanisms in their RNA-dependent RNA polymerases (RdRps).
Are mutation rates fixed for a given organism? High- and low-fidelity variants in E. coli and yeast were already described as early as the 1970s (Flury et al. 1976; Gillin and Nossal 1976). Three principal mechanisms contribute to fidelity in E. coli: intrinsic polymerase fidelity (how many correct versus incorrect nucleotides are incorporated), exonuclease proofreading activity, and DNA mismatch repair (Schaaper 1993). These mechanisms can be altered to obtain variants with altered replication fidelity. Interestingly, work in E. coli also suggested that mutation rates in a given organism are not fixed, but are dynamic. For instance, at least one percent of natural isolates are mutator variants (Jyssum 1960; Gross and Siegel 1981; LeClerc et al. 1996). Remarkably, by passaging E. coli for 10,000 generations in a limited glucose environment, Sniegowski et al. (1997) showed that some bacterial populations develop mutation rates one- or twofold higher than wild type, suggesting that in fluctuating environments mutator alleles are potentially beneficial (Taddei et al. 1997).
Following work in bacteria, studies in T4 bacteriophage and herpes simplex virus demonstrated that these observations could be extended to DNA viruses (Muzyczka et al. 1972; Hall et al. 1984). In some cases, these strains were missing essential proofreading functions, and in other cases, point mutations in the polymerase led to altered correct nucleotide incorporation rates (Muzyczka et al. 1972; Reha-Krantz et al. 1991). For retroviruses, some base analog-resistant HIV strains were reported with reverse transcriptase mutations (e.g., M184V) that altered the fidelity of this RNA-dependent DNA polymerase. However, the measurement of the fidelities of these strains was controversial and not clearly established until recently (Wainberg et al. 1996; Bakhanashvili et al. 1996; Dapp et al. 2013; Keulen et al. 1999; Mansky et al. 2000). The most recent discoveries that intrinsic RdRp fidelity can be altered allowed researchers to examine how restricting (high-fidelity/antimutator) or expanding (low-fidelity/mutator) viral population diversity impacts viral pathogenesis, adaptability, and evolution.
RNA Virus High-Fidelity Variants/Antimutators
The first bona fide high-fidelity variant of an RNA virus was isolated independently by two laboratories (Pfeiffer and Kirkegaard 2003; Vignuzzi et al. 2006), by serially passaging poliovirus in the presence of ribavirin to select for resistant variants. The incorporation of ribavirin directly into nascent genomes during poliovirus replication results in transition mutations, principally A → G, G → A or C → U, with detrimental effects to the virus. Sequencing of the polymerase gene of the ribavirin-resistant population revealed one amino acid change, G64S. It was hypothesized that resistance was due to (a) a more active polymerase, simply generating more progeny genomes permitting better survival; (b) a polymerase that no longer recognized ribavirin as a base analog; or (c) an overall increase in polymerase fidelity and selection of the correct nucleotide. Several observations supported this last mechanism: G64S exhibited the same replication as wild-type virus in single-cycle infections; G64S generated fewer escape mutants to antiviral compounds; and G64S was also resistant to base analogs of different structure (Pfeiffer and Kirkegaard 2003; Vignuzzi et al. 2006). In parallel, a biochemical study confirmed that G64S was a high-fidelity enzyme by directly demonstrating that the rate of incorrect nucleotide incorporation was fourfold lower than wild type (Arnold et al. 2005), and direct sequencing of genomes revealed that the mutation frequency was sixfold lower in the G64S population (Vignuzzi et al. 2006). Additionally, introducing the amino acids A, T, V, or L at position 64 also generated high-fidelity variants (Vignuzzi et al. 2008).
Following these studies in poliovirus, performing passages in mutagens to generate resistant variants became a general strategy for isolating high-fidelity/antimutator variants (Beaucourt et al. 2011). Coxsackie virus B3, also from the Picornaviridae family, was treated with moderate concentrations of ribavirin or 5-azacytidine (5-AZC, another RNA mutagen). Within 10–20 passages, a new mutation, A372V, arose in the RdRp. This mutation alone conferred resistance to the effects of three RNA mutagens at high concentrations, and its increased fidelity was confirmed by sequencing and biochemical assays (Levi et al. 2010). Guided by the high-fidelity poliovirus studies, Sadeghipour et al. (2013) identified G64R and G64T as ribavirin-resistant variants of human enterovirus 71 (HEV71). Although altered growth kinetics make results more difficult to interpret, at least one variant, G64R, was a bona fide antimutator. In addition, serial passaging of HEV71 in ribavirin led to another mutation in the RdRp, S264L, that conferred resistance and increased fidelity (Sadeghipour et al. 2013). Recently, another group found a mutation in the HEV71 RdRp, L123F, that increased fidelity (Meng and Kwang 2014). Finally, a high-fidelity polymerase variant was found for foot-and-mouth disease virus (FMDV, also in the Picornaviridae family), selected by serial passage in the RNA mutagen, 5-fluorouracil (5-FU). The resistance phenotype correlated with the appearance of mutation R84H in the RdRp. This mutation also conferred cross-resistance to ribavirin and 5-AZC, and mutation frequencies were significantly lower than wild-type virus (Zeng et al. 2013). The same group later isolated a quadruple polymerase mutant (D5N:A38V:M194I:M296V or DAMM) that exhibited a twofold decrease in replication errors, yet was not cross-resistant to other mutagens (Zeng et al. 2014), which raises questions as to whether DAMM is truly high-fidelity.
More recently, a number of high-fidelity RdRp variants have been described outside of the picornavirus family. A high-fidelity chikungunya virus C483Y was obtained during serial passage in ribavirin and 5-FU (Coffey et al. 2011). As with picornavirus high-fidelity variants, this variant was resistant to multiple RNA mutagens and generated populations with more restricted genetic diversity than wild-type virus. A mutagen-resistant influenza A virus variant, PB1-V43I, was also shown to increase fidelity (Cheung et al. 2014). Two mutations in the West Nile virus NS5 RdRp were also shown to confer mutagen resistance and fidelity increases (Van Slyke et al. 2015). Thus, mutagen resistance has proven to be a useful strategy for isolating variants with high-fidelity polymerases.
RNA Virus Low-Fidelity Variants/Mutators
The first RdRp variant that was confirmed to have lower fidelity was the FMDV M296I mutant, which, unexpectedly, was isolated in a screen for resistance to ribavirin. Interestingly, the mutant spectrum generated by this variant did not contain the bias toward transition mutations expected after exposure to ribavirin, and the complexity of the mutant spectrum was similar to the wild-type virus in the absence of ribavirin (Sierra et al. 2007). Further passage of M296I in ribavirin resulted in selection of a triple mutant, M296I:P44S:P169S, that was resistant to ribavirin without acquiring resistance to other mutagens such as 5-FU (Agudo et al. 2010). Collectively, these data support the hypothesis that these variants specifically resist ribavirin incorporation during RNA synthesis. Indeed, biochemical studies showed that while the M296I polymerase incorporated twofold less RTP than wild-type enzyme, it generated twofold more A → G transitions. This explains both the lack of mutagenesis in the presence of ribavirin as well as the lack of lower mutant frequencies.
Although informative, this FMDV M296I variant appears to be an exception to the rule. Generally, one would expect mutagen sensitivity, rather than resistance, to be a hallmark of low fidelity. However, selection for mutagen-sensitive variants is more challenging, so how to generate low-fidelity variants? In one case, influenza viruses with mutator phenotypes were isolated by selecting for escape variants by single plaque transfers during sequential treatment with monoclonal antibodies (Suárez et al. 1992). This work preceded other studies of RdRp fidelity by over a decade and did not go further to confirm whether the mutator status mapped to the polymerase genes. For the most part, low-fidelity RdRp variants have been identified by performing site-directed mutagenesis on targeted residues near the catalytic site. This approach was proven successful with both HIV and poliovirus polymerases (Martin-Hernandez et al. 1996; Korneeva and Cameron 2007). Additionally, poliovirus fidelity was lowered by introducing an amino acid change (T362I) near the catalytic site of the poliovirus RdRp (Liu et al. 2013). An initial study on mutagens of Coxsackie virus B3 uncovered a low-fidelity RdRp variant, S299T (Levi et al. 2010). A more recent study targeted residues in Coxsackie virus B3 based on their implication in the closely related poliovirus fidelity network and on predicted polymerase structure during catalysis (Arnold et al. 2005; Gong and Peersen 2010). Nine lower fidelity variants were isolated that presented elevated mutation frequencies (Gnädig et al. 2012) (Fig. 1a). For FMDV, 5 low-fidelity variants were recently shown to increase mutation frequencies and render the variants more susceptible to mutagenesis (Xie et al. 2014). In order to obtain low-fidelity variants for chikungunya virus, all possible amino acids were swapped into polymerase position 483, which was previously shown to result in a high-fidelity enzyme (C483Y) (Rozen-Gagnon et al. 2014) (Fig. 1b). Three variants proved to be low-fidelity: C483A, C483G, and C483W. Since the cysteine in position 483 is highly conserved among the alphaviruses, similar mutations in Sindbis virus at the analogous position 482 also generated two mutators, C482A and C482G. For West Nile virus, a T248I mutation in the methyl transferase domain of the NS5 protein was shown to decrease replication fidelity, likely through an interaction with the NS5 RdRp (Van Slyke et al. 2015). Finally, Eckerle and colleagues mutagenized the active sites needed for coronavirus 3-5′ exonuclease activity. Ablating coronavirus (CoV) proofreading activity resulted in 15-fold (murine hepatitis virus; MHV-CoV) (Eckerle et al. 2007) or a 21-fold (severe acute respiratory syndrome; SARS-CoV) (Eckerle et al. 2010) increases in mutation frequencies.
Fig. 1.
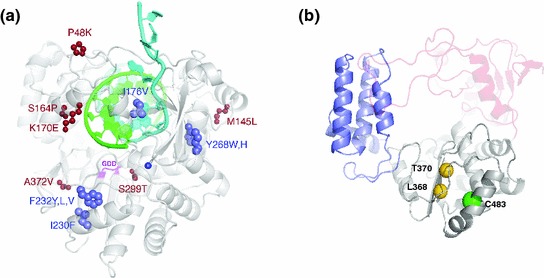
Viral polymerase structures. a Coxsackie virus B3 structure depicting the positions of all viable low-fidelity mutants (blue) likely to favor or alter different conformational states of the polymerase active site. The locations of compensatory mutations are shown in red. Adapted from (Liu et al. 2013). b Structural homology model of the CHIK nsp4 core polymerase showing the predicted locations of C483 (green sphere) and two nearby residues (L368 and T370, shown as gold spheres) that are the structural equivalents of known fidelity-altering sites in Coxsackie virus polymerase (positions I230 and F232, respectively, panel a) (Liu et al. 2013). Adapted from (Gnädig et al. 2012). Three domains are depicted in this figure: (1) the polymerase palm domain (gray), where the fidelity-altering mutations are located, is modeled with fairly high confidence because of the large number of conserved polymerase sequence motifs (motifs A–D); (2) the thumb domain (purple); (3) the fingers (red). Domains where the modeling is weak are shown as semitransparent
Is There a Natural Range in RdRp Fidelity?
The majority of viral fidelity variants identified have been significantly attenuated in vivo; as a consequence, fidelity variants are rarely observed in natural isolates. However, the phenotypic and genotypic assays used to discriminate between these fidelity changes may not be sensitive enough to detect subtle differences among natural isolates. Furthermore, given the quasispecies nature of RNA virus populations, it is not known whether fidelity variants exist at low frequencies within the mutant spectrum and whether they would modulate overall population fidelity. For instance, Coxsackie virus B3 mutator strains that presented mutation frequencies nearer to wild-type values did not display attenuated phenotypes (Gnädig et al. 2012). This raises the possibility that slightly elevated mutation rates could be advantageous in conditions that require more rapid adaptation. Interestingly, the A372V and S299T Coxsackie virus variants, respectively, presenting moderately higher and lower fidelities, exist as natural isolates. Threonine at position 299 is found in 5 % of the isolates, while valine at position 372 is found in 86 % (Harrison et al. 2008). It is thus possible that at any given time, a RNA virus quasispecies contains a subpopulation of mutators and antimutators, whose frequencies may fluctuate when environmental pressures require more rapid evolution or better maintenance of genetic integrity. Importantly, the study of fidelity variants in tissue culture has been nearly exclusively performed in highly permissible, immortalized cell lines during only a few replication cycles. Recent studies show that the mutation rates and frequencies of viruses can dramatically change depending on cell type (Rozen-Gagnon et al. 2014; Combe and Sanjuan 2014). Therefore, under more stringent selective pressures, the fidelity of RNA viruses may be adjusted. Indeed, evolution of mutation rates has been observed in bacteria during colonization in vivo or in conditions of starvation (Sniegowski et al. 1997). A similar phenomenon may occur for RNA viruses, but this remains to be demonstrated.
RdRp Structure
The error-prone viral RdRp is a primary source of the diversity we observe in RNA virus populations. Indeed, most RNA virus fidelity variants isolated have contained mutations in the RdRp (the only exceptions thus far have been the mutator coronaviruses lacking exonuclease activity) (Eckerle et al. 2007, 2010). The RdRp has a structure conserved across virus families. RdRp structures have been solved for members of the Flaviviridae, including hepatitis C virus, bovine viral diarrhea virus, Japanese encephalitis virus, West Nile virus, and dengue virus (Lu and Gong 2013; Yap et al. 2007; Malet et al. 2007; Choi et al. 2004; Bressanelli et al. 1999; Lesburg et al. 1999); the Calciviridaie, including rabbit hemorrhagic disease virus, Norwalk virus, sapovirus, reovirus λ3, and bacteriophage ϕ6 (Ng et al. 2002, 2004; Fullerton et al. 2007; Butcher et al. 2001; Salgado et al. 2004; Tao et al. 2002); the Picornaviridae, including Coxsackie virus B3, poliovirus, FMDV, HEV71, human rhinovirus, and encephalomyocarditis virus (Chen et al. 2013; Gruez et al. 2008; Love et al. 2004; Ferrer-Orta 2004; Appleby et al. 2005; Thompson and Peersen 2004; Hansen et al. 1997; Vives-Adrian et al. 2014); the Birnaviridae, including infectious bursal disease virus (Pan et al. 2007); and the Orthomyxoviridae, including influenza A virus (He et al. 2008). All these polymerases have a “right-handed” architecture, which is comprised of finger, palm, and thumb domains. The RdRps exist in a closed hand formation, accomplished by connections between the finger and thumb domains [for review see (Ferrer-Orta et al. 2006; Ng et al. 2008)]. Loops extending from the fingers (fingertips) surround the active site and create the entrance to the template channel, where template recognition occurs (Butcher et al. 2001; Ferrer-Orta 2004; O’Farrell et al. 2003). The template channel itself connects the fingers to the active site, located in the palm. The palm is made up of a three-stranded β-sheet and 3 α-helices and is a highly conserved feature. This catalytic domain contains 7 conserved motifs (A–G) present in all RdRps thus far, which catalyze the nucleotidyl transfer reaction.
The enzymatic function relies on a two-metal-ion mechanism proposed for all polymerases. This mechanism was first shown for RdRps using the poliovirus RdRp (Ng et al. 2008; Steitz 1998; Arnold et al. 1999). The incoming nucleotide enters the active site with metal ion B, which orients the NTP in the active site using the β- and γ-phosphates of the NTP and a conserved aspartic acid (Asp) in motif A. Following this, metal ion A binds the α-phosphate group of the NTP, the Asp of motif A, and another conserved Asp in motif C. In addition, metal ion A binds the 3′-OH of the nascent RNA strand, lowering the 3′-OH affinity for the H to allow nucleophilic attach of the NTP α-phosphate (Ng et al. 2008; Steitz 1998). Therefore, two protons are transferred during this reaction (Castro et al. 2007): one from the nascent RNA 3′-OH to an unknown acceptor and one to the pyrophosphate (PPi) leaving group from a general acid, usually a lysine, in Motif D (Castro et al. 2009; Cameron et al. 2009). The presence of this general acid greatly enhances the efficiency of catalysis (Castro et al. 2009) (Fig. 1a, b).
Structural and Kinetic Basis of Fidelity
The structures of wild-type and RdRp fidelity variants are essentially identical, having only one or few amino acid changes, indicating that structural dynamics of nucleotide addition are more likely to alter fidelity than large-scale structural changes (Cameron et al. 2009; Marcotte et al. 2007). The isolation of the high-fidelity variant G64S and the design of novel substrates for biochemical studies uncovered the mechanisms of nucleotide addition. This novel symmetrical substrate, called sym/sub, was a self-complementary 10-nucleotide heteropolymeric RNA primer. Regardless of the orientation of enzyme binding, both 3′-OHs permit extension of the substrate (Arnold and Cameron 2000). Using sym/sub, five kinetic steps are observed during the incorporation of a single nucleotide to a nascent RNA, called the single-nucleotide addition cycle. This cycle, which is most likely conserved for all RdRps, begins with (1) NTP binding to the enzyme-template complex (ERnNTP), followed by a conformational change (2), which allows the complex becomes catalytically active (*ERnNTP). (3) Phosphodiester bonds are then formed between the incoming nucleotide and the nascent strand (*ERn+1PPi), leading to a second conformational change (4) (ERn+1PPi), followed by (5) pyrophosphate release (ERn+1)(Arnold and Cameron 2004; Arnold et al. 2004)(Fig. 2).
Fig. 2.

The five kinetic steps in the single-nucleotide addition cycle. ER, enzyme-template complex. *ER, active enzyme-template complex. PP, phosphodiester bond. Adapted from (Arnold and Cameron 2000, 2004)
It has been shown that steps 2 (first conformational change) and 3 (phosphoryl transfer) are partially rate-limiting in the single-nucleotide addition cycle and therefore likely to influence fidelity. Step 2 involves reorientation of the triphosphate of the NTP to be available for phosphoryl transfer, and interactions with residues in the ribose-binding pocket. These interactions should be altered for an incorrect bound nucleotide, reducing the stability of complex with the bound nucleotide and the rate of the following phosphoryl transfer (Arnold and Cameron 2004; Arnold et al. 2004; Gohara et al. 2000). Therefore, residues in the binding pocket should be crucial for determining if the correct nucleotide is present (Gohara et al. 2004). Indeed, several conserved residues play major roles in orienting and stabilizing the incoming NTP (Gohara et al. 2000, 2004). Once the triphosphate is properly oriented, it is stabilized by a hydrogen bond network within residues of the binding pocket, mostly located in motif A. Additionally, in motif A, Asp-238 interacts with the 3′OH, and in motif B, Asn-297 interacts with the 2’OH to tightly hold the incoming triphosphate in the proper orientation. Asn-238 was also shown to be crucial for NTP orientation in the FMDV RdRp (Ferrer-Orta et al. 2007). Furthermore, Asp-238 and motif A are conserved in all animal virus RdRps (Koonin 1991). Based on this work, it was proposed that the binding pocket can be divided into a universal portion (motif A) and an adapted portion (motif B). These motifs intersect in the nucleotide-binding pocket, where motif A carries out the universal enzymatic functions and motif B is involved with nucleotide selection. Motif A residue Asp-238 presumably links the rate of phosphoryl transfer to the selection of the nucleotide, reducing the transfer efficiency when the incorrect nucleotide is bound (Gohara et al. 2004). Both the hydrogen bond network and interactions between the triphosphate and these specific binding pocket residues determine the stability of the active complex and the rate of the following phosphoryl transfer.
Recent work also links motif D to the efficiency and fidelity of newly incorporated nucleotides. A conserved lysine (L359 in poliovirus) was shown to act as a general acid to protonate the PPi (Castro et al. 2009). New results indicate that this motif D lysine might interact with the β-phosphate of the NTP to achieve an active (closed) enzyme conformation. Furthermore, binding of the incorrect NTP shifts the RdRp conformation away from the active state, and the motif D lysine must be protonated in order to achieve the RdRp active state (Yang et al. 2012). This may explain why poliovirus mutants in motif D have altered fidelity phenotypes (Liu et al. 2013; Castro et al. 2009; Yang et al. 2012). The dynamic changes of motif D (shown by nuclear magnetic resonance) (Cameron et al. 2009) were previously masked in crystal structures of RdRps in elongation complexes (Gong and Peersen 2010; Ferrer-Orta et al. 2007).
Although residue 64 is remote from the active site (in the fingers domain), G64S polymerase was shown to have a lower equilibrium constant for the conformational change that orients and stabilizes the incoming NTP (step 2). In fact, this residue is indirectly connected to motif A via hydrogen bonds; substitution of an S in this position ablates hydrogen bonding, explaining the reduced equilibrium constant for step 2 (Arnold et al. 2005). Although it is unclear how G64S could be connected to motif D, a conformational change was found to occur in motif D for G64S when the incorrect nucleotide is bound that does not occur for the wild-type polymerase. This decreases G64S′ ability to switch to the catalytically active form when the incorrect nucleotide is bound, leading to a higher fidelity polymerase (Arnold et al. 2005; Yang et al. 2010, 2012). Similar mechanisms were shown to be involved for the equivalent mutation in FMDV (G62S) (Ferrer-Orta et al. 2010).
In Vitro Trends of Fidelity Variants
It is somewhat difficult to make generalizations from biochemical experiments examining fidelity, which are only available for poliovirus, FMDV, and Coxsackie virus B3. Although there are some exceptions (Arias et al. 2008), it seems that higher fidelity polymerases tend to be kinetically slower, and low-fidelity polymerases tend to be faster. Notably, this was shown to be the case for high-fidelity poliovirus G64S and FMDV G62S (Arnold et al. 2005; Yang et al. 2010; Ferrer-Orta et al. 2010), and low-fidelity poliovirus and CVB3 strains (Liu et al. 2013; Gnädig et al. 2012). The kinetic proofreading hypothesis predicts that there is most likely a trade-off between fast or faithful replication (Coffey et al. 2011; Hopfield 1974). Without proofreading mechanisms (which is the case for the majority of RNA viruses), increased accuracy may only be achieved by reducing the rate of polymerization. This allows decreased stability of incorrect nucleotides and higher dissociation constants, resulting in a preference for the correct nucleotide. This phenomenon is well supported from structural and biochemical studies with poliovirus (Arnold et al. 2005; Castro et al. 2009; Yang et al. 2010, 2012). It has also been shown that biochemical fidelity assays correlate well with in vitro tissue culture measurements of mutation frequencies (Gnädig et al. 2012).
However, there are discrepancies between relative rates of nucleotide incorporation in biochemical assays using RdRp enzymes versus overall virus growth in tissue culture. For example, even though biochemical data suggested that G46S poliovirus had a 2.5-fold reduced yield from assembled elongation complexes, it grew identically to wild type in one-step growth curves. This is not necessarily surprising; given that a single-nucleotide incorporation cycle is cell-free solution is not equivalent to a complete viral life cycle within infected cells (including binding, entry, replication, packaging, and egress). Interestingly, growing G64S in competition with wild type did reveal a reduced relative fitness (Arnold et al. 2005). This in vitro growth pattern has since developed as a recurring trend. Most RdRp fidelity variants (high and low) grow similarly to wild type in isolation, but in competition with wild type suffer replicative fitness costs (Zeng et al. 2014; Coffey et al. 2011; Gnädig et al. 2012; Furió et al. 2005; Levi et al. 2010). Some studies with reverse transcriptase anti/mutators show that the further the mutation frequency from wild type, the lower the relative fitness in competition assays (Dapp et al. 2013; Furió et al. 2007). Important exceptions to this general rule are the MHV and SARS coronavirus mutators lacking 5-3′ exonuclease activity. These variants exhibited clear and stable defects (up to one log) in replication compared to the wild type (Eckerle et al. 2007, 2010; Graham et al. 2012).
In Vivo Trends of Fidelity Variants
Fidelity variants generate more or fewer mutations than wild-type viruses without presenting altered consensus sequences with respect to their parental strains. Therefore, they have proven to be invaluable tools to address the role of the mutant spectrum in virus fitness and the behavior of quasispecies in vivo. Although major growth defects are generally not observed in vitro, in vivo attenuation seems to be the general rule for fidelity variants (Fig. 3). The precedent for in vivo attenuation was set with the G64S poliovirus high-fidelity variant. Pfeiffer and Kirkegaard (2005) demonstrated that G64S was less pathogenic than wild-type virus in mice, despite not finding significant replication differences in vitro. Vignuzzi et al. (2006) also demonstrated an attenuated phenotype for G64S, where the 50 % lethal dose (LD50) in mice was 300-fold higher than in wild type. Furthermore, while both virus populations could colonize and infect spleens, kidneys, muscles, and intestines when a systemic inoculation was performed, G64S was unable to disseminate more distally to infect the CNS or to be shed in feces. Sequencing of individual genomes from the high-fidelity population in vivo confirmed its restricted genetic diversity. In this study, the restricted quasispecies of G64S was artificially expanded by mutagen treatment to present the same number of mutations as wild type. Importantly, the expanded G64S population (G64SeQS) recovered the ability to infect and colonize the spinal cord and brain and had a similar lethal dose compared to wild type. Furthermore, coinfections of a restricted G64S population with either wild type or G64SeQS allowed the genetically restricted G64S population to invade the spinal cord and brain. This seminal work provided strong evidence that not only consensus changes impact virulence and pathogenesis, but that mutant spectrum complexity can also be critical to pathogenesis and/or virus survival in vivo (although this is not always the case). This study also provided indirect evidence that cooperative interactions within the quasispecies may play a role in virus infection and disease progression. Since this first report, altering mutation frequency has been linked to attenuation in animal models for several variants. A recent study with FMDV also demonstrated that the higher their fidelity, the greater the attenuation in a newborn mouse model (Zeng et al. 2014). For HEV71, the higher fidelity G64R and S264L variants alone or in combination were attenuated in mice (Sadeghipour and McMinn 2013). More recently, another HEV71 antimutator, L123F, also exhibited reduced virulence and delayed symptoms in the AG129 mouse model (Meng and Kwang 2014). Finally, the chikungunya virus, high-fidelity variant, C483Y, was more quickly cleared in the newborn mouse model and was moderately attenuated in mosquitoes (Coffey et al. 2011). Significant attenuation was also observed in colonized and field mosquitoes infected with West Nile virus high-fidelity variants (Van Slyke et al. 2015). While the high-fidelity influenza A virus replicated to wild-type-like titers in the lungs of infected mice, lethality and neurotropism were reduced ten-fold (Cheung et al. 2014).
Fig. 3.
Schematic depicting the effects of viral diversity, due to viral polymerase fidelity changes, on mutagenic drug resistance and attenuation in vivo. The numbers in the viral diversity schematic are arbitrary illustrations meant to represent genetically inter-connected progeny genomes, comprising populations with low, normal, and high degrees of diversity
Similar trends have been observed with low-fidelity/mutator variants. Mice inoculated with the SARS-CoV strain lacking exonuclease activity (ExoN) exhibited reduced symptoms and more rapid clearance of virus from the lungs. Furthermore, the ExoN mutant retained elevated mutation frequencies in vivo (Graham et al. 2012). Coxsackie virus B3 mutator strains exhibited reduced titers in several target organs in the mouse model and were unable to establish persistent infection. Generally, the degree of attenuation correlated with the extremity of the mutator phenotype (Gnädig et al. 2012). A recent study demonstrated higher survival in mice inoculated with a low-fidelity poliovirus (Liu et al. 2013). For the alphaviruses, the low-fidelity variants of chikungunya virus showed reduced titers in all tissues tested in mice, including the primary target tissue (muscle). Similarly, a Sindbis virus mutator was attenuated in mice, presenting fewer neurological symptoms (such as limb paralysis) and was also attenuated in the fruit fly model, Drosophila melanogaster (Rozen-Gagnon et al. 2014) (Fig. 3). The low-fidelity NS5 methyl transferase variant of West Nile virus was also shown to be compromised in the mosquito host (Van Slyke et al. 2015).
Some exceptions to this rule do exist: In FMDV, the high-fidelity R84H presented a 1.4-fold increase in mutation frequency that did not result attenuation in mice (Zeng et al. 2013). A possible explanation for this lack of mutation is that these variants manifest only moderate differences in mutation frequencies (under twofold), compared to the attenuated variants described for FMDV. Therefore, there may be a threshold for mutation frequency, below which no attenuation is observed. In support of this threshold, a recent study demonstrated that for a range of low-fidelity FMDV RdRp variants, the more extreme mutators were attenuated in vivo, while those presenting twofold increases in mutation frequency retained virulence (Xie et al. 2014). Similar data were found in Coxsackie virus B3 (Gnädig et al. 2012), and only mutators with the highest mutation frequencies were attenuated. Consequently, this threshold should be different depending on the virus and its ability to cope with increased mutational load (Graci et al. 2012).
Fidelity Variants as Vaccine Candidates
Based on the nearly universal attenuation of fidelity variants in vivo, such strains are being explored as live attenuated vaccines (LAVs). Very few groups have used fidelity variants to elicit protection, but these studies have been promising. A first study examined immunogenicity and protection of the original high-fidelity G64S poliovirus, as well as high-fidelity strains G64A/V/T/L, in mice. The authors observed reduced viral shedding and high neutralizing antibody titers (indeed, higher than those induced by inoculation with the Sabin vaccine strain). Importantly, immunization conferred long-term protection; 6 months after a single immunization with fidelity variants, the majority of mice survived lethal challenge with wild-type poliovirus (Vignuzzi et al. 2008). A second study evaluated the SARS-CoV ExoN mutator as a vaccine candidate and showed that the mutator remains attenuated even in immunocompromised or aged mice (groups of concern during routine immunizations). In addition, immunization with the mutator strain elicited antibody responses and conferred resistance against lethal challenge; no virus was detectable in the lungs of immunized mice (Graham et al. 2012). LAVs have proven more efficacious than subunit vaccine counterparts, but pose safety concerns [for review see (Lauring et al. 2010)]. One principal concern in LAVs is reversion to virulence, which has been observed for a number of LAVs, including the Sabin strains of poliovirus (Cann et al. 1984). While mutator strains raise the question of safety and stability as LAVs because they generate more mutations than wild-type viruses, the SARS-CoV ExoN mutator was shown to remain stable in mouse models. Likewise, when the ExoN mutator was allowed to establish a persistent infection in SCID mice for up to 30 days, no reversion was detected at inactivated exonuclease sites. Interestingly, one of the attenuating mutations present in the RdRp of the Sabin strain of poliovirus was shown to decrease fidelity. This raises the possibility that in currently used vaccines, attenuation is already achieved in part by altering the natural fidelity of an RNA virus (although whether the strain including all attenuating mutations retains this lower fidelity has not been shown) (Liu et al. 2013). In addition, the high-fidelity G64S/A/V/T/L polioviruses passaged in mice did not revert at position 64 and remained attenuated. For LAV development high-fidelity variants may be of particular interest, since lowered mutation frequencies make mutations conferring virulence less likely to occur. Indeed, under selective pressure to revert in vivo, high-fidelity variants were less able to do so, whereas the wild-type virus quickly reverted at a significantly higher number of sites (Vignuzzi et al. 2008). Since fidelity-altering mutations map to single or few residues, combining these with other conventional attenuating mutations may be a good strategy to further reduce reversion to virulence.
Fidelity Variants and Lethal Mutagenesis
Viruses appear to have fine-tuned their mutation rate in order to maximize adaptation, while at the same time maintaining genomic integrity. Deviation from these optimized mutation rates have been shown to be attenuating. The proposed explanation for the observed attenuation is mutational meltdown, losing the ability to maintain genetic information due to the extrinsic increase in mutation rates (Eigen 1971; Domingo et al. 2005). Importantly, a consequence of the study of lethal mutagenesis as an antiviral approach was the discovery of the high-fidelity variants, which generally were more resistant to mutation-inducing drugs. This discovery further boosted interest in determining how viral mutation rates could be modulated intrinsically, leading to the isolation of more fidelity variants. Large panels of high- and low-fidelity variants are now available to feedback into the research on lethal mutagenesis as an antiviral approach: Due to their altered sensitivity to such compounds, they are excellent tools to investigate the mutagenic activities of known compounds or identify new mutagenic compounds. Indeed, the first high- and low-fidelity variants of Coxsackie virus B3 were key in identifying a previously unknown mutagenic activity for amiloride compounds (Levi et al. 2010). Similar screens using compound libraries may thus identify other classes of antivirals with mutagenic effects (Table 1).
Table 1.
List of published high- and low-fidelity polymerase variants
| Virus | Position RdRp | Phenotype | Reference |
|---|---|---|---|
| Poliovirus | G64S | High-fidelity | Pfeiffer and Kirkegaard (2003), Vignuzzi et al. (2006) |
| CVB3 | A372V | High-fidelity | Levi et al, plos pathogens |
| I176V | Low-fidelity | Gnädig et al. (2012) | |
| L241I | Low-fidelity | Gnädig et al. (2012) | |
| S164P | Low-fidelity | Gnädig et al. (2012) | |
| P48K | Low-fidelity | Gnädig et al. (2012) | |
| A239G | Low-fidelity | Gnädig et al. (2012) | |
| Y268W | Low-fidelity | Gnädig et al. (2012) | |
| Y268H | Low-fidelity | Gnädig et al. (2012) | |
| F232Y | Low-fidelity | Gnädig et al. (2012) | |
| I230F | Low-fidelity | Gnädig et al. (2012) | |
| HEV71 | G64R | High-fidelity | Sadeghipour et al. (2013) |
| S264L | High-fidelity | Sadeghipour et al. (2013) | |
| L123F | High-fidelity | Meng and Kwang (2014) | |
| FMDV | R84H | High-fidelity | Zeng et al. (2013) |
| D5N/A38V/M194I/M296V | High-fidelity | Zeng et al. (2014) | |
| M296I | Low-fidelity | Arias et al. (2008) | |
| M296I/P44S/P169S | Low-fidelity | Arias et al. (2008) | |
| D165E | Low-fidelity | Xie et al. (2014) | |
| K172R | Low-fidelity | Xie et al. (2014) | |
| K177R | Low-fidelity | Xie et al. (2014) | |
| G361S | Low-fidelity | Xie et al. (2014) | |
| Y241F | Low-fidelity | Xie et al. (2014) | |
| FLU | unknown | Low-fidelity | Suárez et al. (1992) |
| PB1-V43I | High-fidelity | Cheung et al. (2014) | |
| CHIKV | C483Y | High-fidelity | Coffey et al. (2011) |
| C483A | Low-fidelity | Rozen-Gagnon et al. (2014) | |
| C483G | Low-fidelity | Rozen-Gagnon et al. (2014) | |
| C483W | Low-fidelity | Rozen-Gagnon et al. (2014) | |
| SINV | C482A | Low-fidelity | Rozen-Gagnon et al. (2014) |
| C482G | Low-fidelity | Rozen-Gagnon et al. (2014) | |
| WNV | T248I | Low-fidelity | Van Slyke et al. (2015) |
| V793I | High-fidelity | Van Slyke et al. (2015) | |
| G806R | High-fidelity | Van Slyke et al. (2015) | |
| MHV-CoV | 3-5′ exonuclease (ExoN) | Low-fidelity | Eckerle et al. (2007) |
| SARS-CoV | 3-5′ exonuclease (ExoN) | Low-fidelity | Eckerle et al. (2010) |
Footnotes
Antonio Borderia and Kathryn Rozen-Gagnon have contributed equally to this work.
Contributor Information
Esteban Domingo, Phone: +3434911964540, Email: edomingo@cbm.uam.es.
Peter Schuster, Phone: +43430043-1-4277 52 736, Email: pks@tbi.univie.ac.at.
Marco Vignuzzi, Email: marco.vignuzzi@pasteur.fr.
References
- Agudo R, Ferrer-Orta C, Arias A, la Higuera de I, Perales C, Pérez-Luque R, Verdaguer N, Domingo E (2010) A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. Plos Pathogens 6:e1001072 [DOI] [PMC free article] [PubMed]
- Appleby TC, Luecke H, Shim JH, Wu JZ, Cheney IW, Zhong W, Vogeley L, Hong Z, Yao N. Crystal structure of complete rhinovirus RNA polymerase suggests front loading of protein primer. J Virol. 2005;79:277–288. doi: 10.1128/JVI.79.1.277-288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias A, Arnold JJ, Sierra M, Smidansky ED, Domingo E, Cameron CE. Determinants of RNA-dependent RNA polymerase (in) fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J Virol. 2008;82:12346–12355. doi: 10.1128/JVI.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub) J Biol Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D pol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Ghosh SKB, Cameron CE. Poliovirus RNA-dependent RNA Polymerase (3Dpol) divalent cation modulation of primer, template, and nucleotide selection. J Biol Chem. 1999;274:37060–37069. doi: 10.1074/jbc.274.52.37060. [DOI] [PubMed] [Google Scholar]
- Arnold JJ, Gohara DW, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D pol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+ Biochemistry. 2004;43:5138–5148. doi: 10.1021/bi035213q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J Biol Chem. 2005;280:25706–25716. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhanashvili M, Avidan O, Hizi A (1996) Mutational studies of human immunodeficiency virus type 1 reverse transcriptase: the involvement of residues 183 and 184 in the fidelity of DNA synthesis. FEBS Lett 391:257–262 [DOI] [PubMed]
- Beaucourt S2P, Border 237 AAV, Coffey LL, Gn 228 Dig NF, Sanz-Ramos M, Beeharry Y, Vignuzzi M (2011) Isolation of fidelity variants of RNA viruses and characterization of virus mutation frequency. J Vis Exp 52 [DOI] [PMC free article] [PubMed]
- Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. A mechanism for initiating RNA-dependent RNA polymerization. Nature. 2001;410:235–240. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- Cameron CE, Moustafa IM, Arnold JJ (2009) Dynamics: the missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr Opin Struct Biol 19:768–774 [DOI] [PMC free article] [PubMed]
- Cann AJ, Stanway G, Hughes PJ, Minor PD, Evans DMA, Schild GT, Almond JW (1984) Reversion to neurovirulence of the live-attenuated Sabin type 3 oral poliovirus vaccine. Nucleic Acids Res 12:7787–7792 [DOI] [PMC free article] [PubMed]
- Castro C, Smidansky E, Maksimchuk KR, Arnold JJ, Korneeva VS, Götte M, Konigsberg W, Cameron CE. Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc Natl Acad Sci. 2007;104:4267–4272. doi: 10.1073/pnas.0608952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Götte M, Konigsberg W, Cameron CE. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat Struct Mol Biol. 2009;16:212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Wang Y, Shan C, Sun Y, Xu P, Zhou H, Yang C, Shi P-Y, Rao Z, Zhang B et al (2013) Crystal structure of enterovirus 71 RNA-dependent RNA polymerase complexed with its protein primer VPg: implication for a trans mechanism of VPg uridylylation. J Virol 87:5755–5768 [DOI] [PMC free article] [PubMed]
- Cheung PPH, Watson SJ, Choy K-T, Fun Sia S, Wong DDY, Poon LLM, Kellam P, Guan Y, Malik Peiris JS, Yen H-L. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat Commun. 2014;5:4794. doi: 10.1038/ncomms5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Groarke JM, Young DC, Kuhn RJ, Smith JL, Pevear DC, Rossmann MG (2004) The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Nat Acad Sci USA 101:4425–4430 [DOI] [PMC free article] [PubMed]
- Coffey LL, Beeharry Y, Bordería AV, Blanc H, Vignuzzi M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc Natl Acad Sci USA. 2011;108:16038–16043. doi: 10.1073/pnas.1111650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combe M, Sanjuan R (2014) Variation in RNA virus mutation rates across host cells. PLoS Pathog 10:e1003855 [DOI] [PMC free article] [PubMed]
- Dapp MJ, Heineman RH, Mansky LM (2013) Interrelationship between HIV-1 fitness and mutation rate. J Mol Biol 425:41–53 [DOI] [PMC free article] [PubMed]
- Domingo E, Escarmís C, Lázaro E, Manrubia SC. Quasispecies dynamics and RNA virus extinction. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle LD, Lu X, Sperry SM, Choi L, Denison MR. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J Virol. 2007;81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell T et al (2010) Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog 6:e1000896 [DOI] [PMC free article] [PubMed]
- Eigen M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971;58:465–523. doi: 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- Ferrer-Orta C. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem. 2004;279:47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N (2006) A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol 16:27–34 [DOI] [PubMed]
- Ferrer-Orta C, Arias A, Pérez-Luque R, Escarmís C, Domingo E, Verdaguer N (2007) Sequential structures provide insights into the fidelity of RNA replication. Proc Nat Acad Sci 104:9463–9468 [DOI] [PMC free article] [PubMed]
- Ferrer-Orta C, Sierra M, Agudo R, la Higuera de I, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N (2010) Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J Virol 84:6188–6199 [DOI] [PMC free article] [PubMed]
- Flury F, Von Borstel RC, Williamson DH (1976) Mutator activity of petite strains of Saccharomyces cerevisiae. Genetics 83:645–653 [DOI] [PMC free article] [PubMed]
- Fullerton SWB, Blaschke M, Coutard B, Gebhardt J, Gorbalenya A, Canard B, Tucker PA, Rohayem J (2007) Structural and functional characterization of sapovirus RNA-dependent RNA polymerase. J Virol 81:1858–1871 [DOI] [PMC free article] [PubMed]
- Furió V, Moya A, Sanjuán R. The cost of replication fidelity in an RNA virus. Proc Natl Acad Sci. 2005;102:10233–10237. doi: 10.1073/pnas.0501062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furió V, Moya A, Sanjuán R. The cost of replication fidelity in human immunodeficiency virus type 1. Proc Biol Sci. 2007;274:225–230. doi: 10.1098/rspb.2006.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillin FD, Nossal NG (1976) Control of mutation frequency by bacteriophage T4 DNA polymerase. I. The CB120 antimutator DNA polymerase is defective in strand displacement. J Biol Chem 251:5219–5224 [PubMed]
- Gnädig NF, Beaucourt S, Campagnola G, Bordería AV, Sanz-Ramos M, Gong P, Blanc H, Peersen OB, Vignuzzi M (2012) Coxsackievirus B3 mutator strains are attenuated in vivo. Proc Natl Acad Sci USA 109:E2294–E2303 [DOI] [PMC free article] [PubMed]
- Gohara DW, Crotty S, Arnold JJ, Yoder JD, Andino R, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): structural, biochemical, and biological analysis of conserved structural motifs A and B. J Biol Chem. 2000;275:25523–25532. doi: 10.1074/jbc.M002671200. [DOI] [PubMed] [Google Scholar]
- Gohara DW, Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D pol): kinetic, thermodynamic, and structural analysis of ribonucleotide selection. Biochemistry. 2004;43:5149–5158. doi: 10.1021/bi035429s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci. 2010;107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graci JD, Gnädig NF, Galarraga JE, Castro C, Vignuzzi M, Cameron CE. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J Virol. 2012;86:2869–2873. doi: 10.1128/JVI.05712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RL, Becker MM, Eckerle LD, Bolles M, Denison MR, Baric RS. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat Med. 2012;18:1820–1826. doi: 10.1038/nm.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MD, Siegel EC (1981) Incidence of mutator strains in Escherichia coli and coliforms in nature. Mutat Res Lett 91:107–110 [DOI] [PubMed]
- Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of coxsackievirus B3 RNA-dependent rna polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on picornaviridae polymerases. J Virol. 2008;82:9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JD, Coen DM, Fisher BL, Weisslitz M, Randall S, Almy RE, Gelep PT, Schaffer PA (1984) Generation of genetic diversity in herpes simplex virus: an antimutator phenotype maps to the DNA polymerase locus. Virology 132:26–37 [DOI] [PubMed]
- Hansen JL, Long AM, Schultz SC. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure. 1997;5:1109–1122. doi: 10.1016/S0969-2126(97)00261-X. [DOI] [PubMed] [Google Scholar]
- Harrison DN, Gazina EV, Purcell DF, Anderson DA, Petrou S. Amiloride derivatives inhibit coxsackievirus B3 RNA replication. J Virol. 2008;82:1465–1473. doi: 10.1128/JVI.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z et al (2008) Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature 454:1123–1126 [DOI] [PubMed]
- Hopfield JJ (1974) Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Nat Acad Sci 71:4135–4139 [DOI] [PMC free article] [PubMed]
- Jyssum K (1960) Observations on two types of genetic instability in Escherichia coli. Acta Pathologica Microbiol Scand 48:113–120 [DOI] [PubMed]
- Keulen W, van Wijk A, Schuurman R, Berkhout B, Boucher Charles AB (1999) Increased polymerase fidelity of lamivudine-resistant HIV-1 variants does not limit their evolutionary potential. Aids 13:1343–1349 [DOI] [PubMed]
- Koonin EV. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J Gen Virol. 1991;72(Pt 9):2197–2206. doi: 10.1099/0022-1317-72-9-2197. [DOI] [PubMed] [Google Scholar]
- Korneeva VS, Cameron CE. Structure-function relationships of the viral RNA-dependent RNA polymerase: fidelity, replication speed, and initiation mechanism determined by a residue in the ribose-binding pocket. J Biol Chem. 2007;282:16135–16145. doi: 10.1074/jbc.M610090200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Jones JO, Andino R. Rationalizing the development of live attenuated virus vaccines. Nat Biotechnol. 2010;28:573–579. doi: 10.1038/nbt.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeClerc JE, Li B, Payne WL, Cebula TA (1996) High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211 [DOI] [PubMed]
- Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- Levi LI, Gnädig NF, Beaucourt S, McPherson MJ, Baron B, Arnold JJ, Vignuzzi M (2010) Fidelity Variants of RNA Dependent RNA Polymerases Uncover an Indirect, Mutagenic Activity of Amiloride Compounds 6:e1001163 [DOI] [PMC free article] [PubMed]
- Levi LI, Gnädig NF, Beaucourt S, McPherson MJ, Baron B, Arnold JJ, Vignuzzi M. Fidelity variants of RNA dependent RNA polymerases uncover an indirect, mutagenic activity of amiloride compounds. PLoS Pathog. 2010;6:e1001163. doi: 10.1371/journal.ppat.1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yang X, Lee CA, Moustafa IM, Smidansky ED, Lum D, Arnold JJ, Cameron CE, Boehr DD. Vaccine-derived mutation in motif D of poliovirus RNA-dependent RNA polymerase lowers nucleotide incorporation fidelity. J Biol Chem. 2013;288:32753–32765. doi: 10.1074/jbc.M113.484428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus. Structure. 2004;12:1533–1544. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Lu G, Gong P (2013) Crystal structure of the full-length Japanese encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog 9:e1003549 [DOI] [PMC free article] [PubMed]
- Malet H, Egloff MP, Selisko B, Butcher RE, Wright PJ, Roberts M, Gruez A, Sulzenbacher G, Vonrhein C, Bricogne G et al (2007) Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J Biol Chem 282:10678–10689 [DOI] [PubMed]
- Mansky LM, Bernard LC (2000) 3′-Azido-3′-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J Virol 74:9532–9539 [DOI] [PMC free article] [PubMed]
- Marcotte LL, Wass AB, Gohara DW, Pathak HB, Arnold JJ, Filman DJ, Cameron CE, Hogle JM. Crystal structure of poliovirus 3CD protein: virally encoded protease and precursor to the RNA-dependent RNA polymerase. J Virol. 2007;81:3583–3596. doi: 10.1128/JVI.02306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Hernandez AM, Domingo E, Menendez-Arias L (1996) Human immunodeficiency virus type 1 reverse transcriptase: role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J 15:4434–4442 [PMC free article] [PubMed]
- Meng T, Kwang J. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. J Virol. 2014;88:5803–5815. doi: 10.1128/JVI.00289-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzyczka N, Poland RL, Bessman MJ (1972) Studies on the biochemical basis of spontaneous mutation. I. A comparison of the deoxyribonucleic acid polymerases of mutator, antimutator, and wild type strains of bacteriophage T4. J Biol Chem 247:7116–7122 [PubMed]
- Ng K, Cherney MM, Vázquez AL (2002) Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J Biol Chem 277:1381–1387 [DOI] [PubMed]
- Ng K, Pendás-Franco N, Rojo J, Boga JA (2004) Crystal structure of Norwalk virus polymerase reveals the carboxyl terminus in the active site cleft. J Biol Chem 279:16638–16645 [DOI] [PubMed]
- Ng KKS, Arnold JJ, Cameron CE. Structure-function relationships among RNA-dependent RNA polymerases. Curr Top Microbiol Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell D, Trowbridge R, Rowlands D, Jäger J (2003) Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation. J Mol Biol 326:1025–1035 [DOI] [PubMed]
- Pan J, Vakharia VN, Tao YJ (2007) The structure of a birnavirus polymerase reveals a distinct active site topology. Proc Nat Acad Sci 104:7385–7390 [DOI] [PMC free article] [PubMed]
- Pfeiffer JK, Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci USA. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005;1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz LJ, Stocki S, Nonay RL, Dimayuga E, Goodrich LD, Konigsberg WH, Spicer EK. DNA polymerization in the absence of exonucleolytic proofreading: in vivo and in vitro studies. Proc Natl Acad Sci. 1991;88:2417–2421. doi: 10.1073/pnas.88.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen-Gagnon K, Stapleford KA, Mongelli V, Blanc H, Failloux A-B, Saleh M-C, Vignuzzi M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014;10:e1003877. doi: 10.1371/journal.ppat.1003877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghipour S, McMinn PC. A study of the virulence in mice of high copying fidelity variants of human enterovirus 71. Virus Res. 2013;176:265–272. doi: 10.1016/j.virusres.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghipour S, Bek EJ, McMinn PC. Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J Virol. 2013;87:1759–1769. doi: 10.1128/JVI.02139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado PS, Makeyev EV, Butcher SJ, Bamford DH, Stuart DI, Grimes JM. The structural basis for RNA specificity and Ca2+ inhibition of an RNA-dependent RNA polymerase. Structure. 2004;12:307–316. doi: 10.1016/j.str.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Schaaper RM (1993) Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem 268:23762–23765 [PubMed]
- Sierra M, Airaksinen A, Gonzalez-Lopez C, Agudo R, Arias A, Domingo E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J Virol. 2007;81:2012–2024. doi: 10.1128/JVI.01606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- Steitz TA (1998) Structural biology: a mechanism for all polymerases. Nature 391:231–232 [DOI] [PubMed]
- Suárez P, Valcárcel J, Ortín J. Heterogeneity of the mutation rates of influenza A viruses: isolation of mutator mutants. J Virol. 1992;66:2491–2494. doi: 10.1128/jvi.66.4.2491-2494.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B (1997) Role of mutator alleles in adaptive evolution. Nature 387:700–702 [DOI] [PubMed]
- Tao Y, Farsetta DL, Nibert ML, Harrison SC (2002) RNA synthesis in a cage–structural studies of reovirus polymerase lambda3. Cell 111:733–745 [DOI] [PubMed]
- Thompson AA, Peersen OB. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Slyke GA, Arnold JJ, Lugo AJ, Griesemer SB, Moustafa IM, Kramer LD, Cameron CE, Ciota AT. Sequence-specific fidelity alterations associated with west nile virus attenuation in mosquitoes. PLoS Pathog. 2015;11:e1005009. doi: 10.1371/journal.ppat.1005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Wendt E, Andino R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat Med. 2008;14:154–161. doi: 10.1038/nm1726. [DOI] [PubMed] [Google Scholar]
- Vives-Adrian L, Lujan C, Oliva B, van der Linden L, Selisko B, Coutard B, Canard B, van Kuppeveld FJM, Ferrer-Orta C, Verdaguer N (2014) The crystal structure of a cardiovirus RNA-dependent RNA polymerase reveals an unusual conformation of the polymerase active site. J Virol 88:5595–5607 [DOI] [PMC free article] [PubMed]
- Wainberg MA, Drosopoulos WC, Salomon H, Hsu M, Borkow G, Parniak MA, Gu Z, Song Q, Manne J, Islam S et al (1996) Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science 271:1282–1285 [DOI] [PubMed]
- Xie X, Wang H, Zeng J, Li C, Zhou G, Yang D, Yu L (2014) Foot-and-mouth disease virus low-fidelity polymerase mutants are attenuated. Arch Virol 159:2641–2650 [DOI] [PubMed]
- Yang X, Welch JL, Arnold JJ, Boehr DD. Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry. 2010;49:9361–9371. doi: 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Smidansky ED, Maksimchuk KR, Lum D, Welch JL, Arnold JJ, Cameron CE, Boehr DD. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure. 2012;20:1519–1527. doi: 10.1016/j.str.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TL, Xu T, Chen Y-L, Malet H, Egloff M-P, Canard B, Vasudevan SG, Lescar J (2007) Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol 81:4753–4765 [DOI] [PMC free article] [PubMed]
- Zeng J, Wang H, Xie X, Yang D, Zhou G, Yu L. An increased replication fidelity mutant of foot-and-mouth disease virus retains fitness in vitro and virulence in vivo. Antiviral Res. 2013;100:1–7. doi: 10.1016/j.antiviral.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Zeng J, Wang H, Xie X, Li C, Zhou G, Yang D, Yu L (2014) Ribavirin-resistant variants of foot-and-mouth disease virus: the effect of restricted quasispecies diversity on viral virulence. J Virol 88:4008–4020 [DOI] [PMC free article] [PubMed]