

Abstract
With a few exceptions, all secretory and plasma membrane proteins studied to date are synthesized in the endoplasmic reticulum (ER) membrane. Then, they are transported by successive vesicle budding and fusion from the ER through the Golgi stacks to the plasma membrane following the general intracellular transport route referred to as secretory pathway (originally reviewed in 1). Gap junction connexins have been shown to follow this pathway.
Keywords: Sodium Dodecyl Sulfate, Signal Recognition Particle, Wheat Germ Extract, Translation Reaction, Immunoprecipitation Buffer
Introduction
With a few exceptions, all secretory and plasma membrane proteins studied to date are synthesized in the endoplasmic reticulum (ER) membrane. Then, they are transported by successive vesicle budding and fusion from the ER through the Golgi stacks to the plasma membrane following the general intracellular transport route referred to as secretory pathway (originally reviewed in 1). Gap junction connexins have been shown to follow this pathway.
A large portion of the current knowledge on the synthesis and translocation of secretory and transmembrane proteins (reviewed in 2–4) has been obtained by synthesizing these proteins in a cell-free translation system. In principle, the system consists of a translation competent cell-lysate supplemented with ER-derived membrane vesicles referred to as microsomes (5), and a synthetic protein-encoding RNA. The cell-free translation system has been found to accurately reproduce the steps involved in translation, translocation, as well as co- and posttranslational protein modifications that occur in vivo and therefore has become a standard assay system to study those processes. Because secretory as well as transmembrane proteins were found to use the same translocation machinery in the ER membrane (3,6,7), the lysate system proved suitable to study both types of proteins.
Expressing a protein in a cell-free system appears attractive, as protein biosynthesis can be studied independently from the complex mechanisms occurring in a living cell. In addition, the system is readily accessible to scientific manipulations. Other advantages of this method include speed, a relative ease to interpret results because only one or a small number of known RNA species are present and will be translated, simple detection of the synthesized radiolabeled proteins on sodium dodecyl sulfate (SDS)-polyacrylamide gels, no detectable endogenous protease activity, and a wide range of co- and post-translational protein modifications, as the system is derived from eukaryotic cells. The system, however, is not suitable for the production of large quantities of protein. Yields of protein obtained with this method will be only in the picomole range. In addition, the standard cell-free translation system will not allow the study of processes occurring in intracellular compartments downstream of the ER, although a modification of the method has been described in which the microsomal vesicles are replaced by detergent-permeabilized whole cells (8). Transport through the Golgi apparatus, as well as insertion into the plasma membrane, was observed in this modified system.
In this chapter, the synthesis and membrane integration of proteins into microsomes is described first, with special emphasis on connexins. Second, posttranslational protein assays are described that allow differentiation between cytoplasmic, transmembrane, and secretory proteins, and allow detection of posttranslational modifications such as signal peptide processing and core glycosylation. Third, methods are described to analyze potential oligomerization of the newly synthesized proteins, and finally protein detection methods are discussed.
Another description of these methods, with special emphasis on the analysis of homo- as well as hetero-oligomerization, and the characterization of connexin-specific assembly signals, has been published recently (9).
Materials
Reagents
Reagents required for molecular biology, including restriction endonucleases, modifying enzymes, plasmids, connexin cDNAs, and in vitro transcription vectors.
In Vitro Transcription
Transcription kit containing 5× transcription buffer, 0.1 M DL-dithiothreitol (DTT), ribonucleotide trisphosphates (rNTPs), acetylated bovine serum albumin (BSA), and control DNA (Promega).
Sterile, high-quality deionized water.
RNase inhibitor such as RNasin (Promega).
SP6, T3, and/or T7 RNA polymerase (Promega).
DNA grade agarose for agarose gel electrophoresis.
Ethidium bromide.
In Vitro Translation/Membrane Integration
Reticulocyte lysate or wheat germ extract, including amino acid mixture minus methionine, or cysteine, and control RNA (Promega).
Canine pancreatic rough microsomes (Promega).
Isotopes: This example uses high-quality [35S]methionine (SJ1515, Amersham).
Reagents for Posttranslational Assays
Buffer 1 for microsomal membrane pelleting: 150 mM K-acetate (KOAc), 50 mM triethanolamine (TEA)-acetic acid, pH 7.0, and 2.5 mM Mg-acetate (MgOAc).
1 M NaOH for alkali extraction.
Sucrose for gradients.
1 M CaCl2 to stabilize microsomes.
Protease K or trypsin (Boehringer Mannheim).
Appropriate protease inhibitors such as diisopropyl fluorophosphate (DFP) or phenylmethylsulfonyl fluoride (PMSF) (both from Sigma). Caution: DFP is highly toxic and should be used only under a chemical hood. However, it is approx 10 times more efficient than PMSF and therefore preferable.
Detergents such asTritonX-100 (TX-100), NonidetP-40 (NP-40), deoxycholate (DOC), and SDS.
Buffer 2 for membrane lysis: 150mMNaCl, 1% NP-40,0.5% DOC, 50mMTris, pH7.5.
Endoglycosidase F or H (Boehringer Mannheim).
1 × PBS.
Linear 5–20% sucrose gradients containing 150 mM NaCl, 50 mM Tris, pH 7.6; and the respective detergent used for solubilization.
Scintillation fluid for aqueous solutions.
Standard proteins with known sedimentation coefficient (S values) such as myoglobin, 2S; ovalbumin, 3.5S; BSA, 4.3S; catalase, 11.5S.
Connexin-specific antibodies (Zymed Laboratories).
Immunoprecipitation buffer containing 150 mM NaCl, 50 mM Tris, pH 7.6, and either no detergent, nonionic detergent such as TX-100, or ionic detergent (SDS), respectively.
Protein A-sepharose beads (Pharmacia).
10% and 12.5% standard SDS Laemmli acrylamide gels. Caution: Acrylamide is a cumulative neurotoxin. It is recommended that gloves be worn whenever it is handled.
Protein sample buffer containing 3% SDS, 0.5% β-mercaptoethanol.
Mixture of prestained SDS gel marker proteins (Bio-Rad).
1 M sodium salicylate (Sigma).
Equipment
Adjustable heating block or bath.
Agarose gel apparatus.
Micropipets and 0.5- and 1.5-mL microcentrifuge tubes. Tips and tubes must be autoclaved.
Airfuge or tabletop ultracentrifuge (both from Beckman Instruments).
Ultracentrifuge and SW55Ti rotor, or equivalent.
Refractometer.
Liquid scintillation counter.
Laboratory rocker or shaker.
SDS-PAGE apparatus such as Bio-Rad Mini Protean II.
Equipment to dry SDS-polyacrylamide gel electrophoresis (PAGE) gels.
X-ray film, X-ray cassettes including intensifying screens.
Densitometer.
Methods
Connexin Protein Synthesis and Membrane Integration
Cell-free translation systems consist of a translation competent cell lysate, containing ribosomes, precursor tRNAs, an energy generating system, factors involved in the translation process, a mixture of unlabeled amino acids except in general methionine, and radioactively labeled methionine for protein detection. On addition of a synthetic RNA that encodes a protein, the RNA will be translated into protein. If the RNA encodes a secretory or transmembrane protein and the lysate is supplemented with microsomes (see Subheading 3.1.3.) the protein will cotranslationally translocate into the microsomes. The lysate can then be electrophoresed on an SDS polyacrylamide gel and the translated proteins can be visualized by autoradiography using X-ray film or a phosphor imager system (see Fig. 1).
Fig. 1.
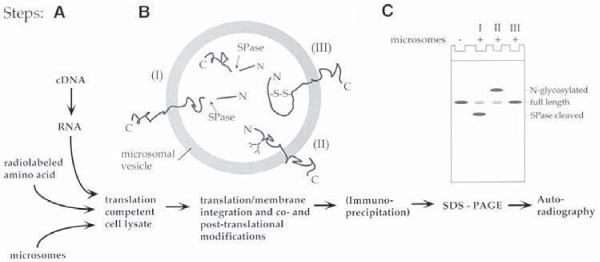
Schematic representation of the components of a cell-free translation system and of the steps involved in a cell-free translation reaction. Secretory and transmembrane proteins can be expressed and cotranslationally translocated across and into membranes when the lysate system is supplemented with endoplasmic reticulum derived membrane vesicles (microsomes) (A). Secretory and transmembrane proteins will be co- and posttranslationally modified inside the microsomes (B). Modifications include signal peptidase (SPase)-mediated cleavage of N-terminal signal peptides (I), core glycosylation (II), and disulfide bridge formation (III). Polypeptide modifications I and II, leading to an increased (I), or decreased (II) mobility on SDS protein gels, can readily be detected by SDS-PAGE and autoradiography (C).
In Vitro Transcription
Before a desired protein can be translated a synthetic RNA transcript has to be synthesized. Several vectors are commercially available that have an SP6, T7, or T3 bacteriophage promoter cloned upstream of a multiple cloning site suitable for inserting the cDNA encoding the desired protein. These bacteriophage promoters are highly specific for their RNA polymerases. Therefore, even cDNAs encoding proteins that are toxic for E. coli can be cloned and amplified in these vectors. All three bacteriophage RNA polymerases are commercially available in very good qualities (also as complete transcription kits) allowing efficient synthesis of transcripts of up to 5–10 kb in length (10–12) (see Note 1).
Synthetic RNAs can be synthesized as capped, as well as uncapped RNAs. While capping is required for transfection of mammalian cells (13), uncapped RNAs will be translated efficiently in the lysate system. A somewhat higher translation efficiency of capped RNAs is in general compensated by the more efficient synthesis of uncapped RNAs. Poly(A) tails are also not required for efficient translation in lysate systems.
Although the vectors contain a transcription termination signal downstream of the cloning site, linearization of the plasmid downstream of the inserted cDNA is desirable and substantially increases the yield of RNA copies. Standard procedures for synthetic RNA synthesis are supplied in Promega Biotech’s Technical Manual “Transcription in vitro Systems” (part no. TM016).
A typical transcription reaction used for the synthesis of connexin cRNAs (14–16) is as follows (see Note 2): 10 μL of 5× transcription buffer 5 μLof 0.1M DTT 10 μLof2.5mM rNTPs 1 μL of 1 mg/mL of acetylated BSA 1 U/μL of transcription volume RNasin 0.3 U/μL of transcription volume SP6-polymerase 0.1–1 μg of linearized plasmid DNA Water to 50 μL
Mix at room temperature and incubate at least 1 h at 40°C.
If the synthetic RNA is to be synthesized with a 5′ CAP structure, only the concentration of rGTP is decreased fivefold, and 1 μLof1 mM CAP analog (m7G[5′]ppp[5′]G) (Pharmacia) is added to the transcription protocol given previously.
Synthesized RNAs can be used without further purification in following translation reactions. However, if additional background bands appear on the gels, phenolchloroform purification, followed by ethanol precipitation (17) is recommended (see Note 3).
Efficiency of RNA synthesis, and quality of the RNA transcripts can be checked by electrophoresing a small aliquot (1–2 μL) on a freshly prepared standard agarose gel prepared with TAE or Tris/Borate/EDTA (TBE) buffer. To recognize the newly synthesized RNA on the gel and to avoid confusion with the linearized DNA that was used as a template and is also present in the transcription reaction, a lane containing linearized plasmid alone should be electrophoresed as a control in parallel as well.
RNA can be visualized by standard ethidium bromide staining (17) (see Note 4).
In Vitro Translation
Two types of translation-competent cell lysates are commercially available. Both lysates are prepared from cells highly active in protein biosynthesis. Reticulocyte lysate is prepared from the red blood cells of rabbits that have been injected with phenylhydrazine to destroy their mature red blood cells, and wheat germ extract is prepared from sprouting wheat seeds. Both extracts are depleted of endogenous RNAs and produce only minimal amounts of endogenous proteins. However, it is always advisable to run a control translation reaction without adding any synthetic RNA with each new batch of lysate. Translation in wheat germ lysates is sensitive to the concentration of potassium and magnesiumions and their concentration has to be optimized for each RNA. For most RNAs optimal potassium ion concentrations range from 120–160 mM, and optimal magnesium ion concentrations range from 1.5 to 4 mM. Although both lysates can be prepared in the laboratory (18,19), best connexin translation results have been obtained with Promega Biotech’ nuclease-treated Rabbit Reticulocyte Lysate System. Protocols for standard translation reactions are provided in the Promega Biotech Technical Manuals “Rabbit Reticulocyte Lysate System” (part no. TM232), and “Wheat Germ Extract” (part no. TM230). A standard protocol for connexin translation is given in Subsection 3.1.3.
Membrane Protein Translocation
Secretory as well as transmembrane proteins in general are cotranslationally translocated into the membrane of the ER. Several protocols have been developed to prepare ER membrane-derived vesicles (microsomes) from cells highly active in protein secretion. Most common, and the source of commercially available microsomes (e.g., Promega Biotech), are microsomes prepared from the acinar cells of canine pancreas (see Note 5). This organ is low in RNase concentration, soft in texture, and the secretory acinar cells are very rich in rough ER membranes. The microsomes are depleted of endogenous RNAs and give only few background bands. However, it is always advisable to run a control translocation reaction without adding any synthetic RNA with each new batch of microsomes (see Note 6).
The microsomes are patches of rough ER membranes that vesiculate upon their isolation (see Fig. 2). The microsomes have been described to contain all the components required for cotranslational protein translocation, including signal recognition particle (SRP), ribosomes, energy supplying molecules, and other factors (see Note 7). Furthermore, the microsomes have signal peptidase (20), and core glycosylation activity (21). N-terminal signal peptides will be cleaved from secretory and certain transmembrane proteins, and certain aspar-agine residues located in the lumen of the microsomes can be glycosylated. In Fig. 3, the expected translation products of a secretory (A), a transmembrane protein with an N-terminal cleavable signal sequence (B), a transmembrane protein without cleavable signal sequence (C), an N-glycosylated membrane protein without cleavable signal sequence (D), and a cytoplasmic protein (E) are schematically shown. A translation reaction in the presence of microsomes (shown in Fig. 3, lanes 2, 6, 10, 14, and 18) should always be accompanied by a parallel translation reaction in the absence of microsomes (shown in Fig. 3, lanes 1, 5, 9, 13, and 17) to indicate the electrophoretic mobility of unmodified, full-length polypeptides.
Standard connexin translation reactions are 10–25 μL in volume and mixed as follows: 25 μL of reticulocyte lysate 2.5 μL of amino acid mixture minus methionine 2.5 μL of [35S]methionine (15 μCi/μL) (see Note 8) 0.5–2 μg of synthetic RNA water to 47.5 μL
Divide into two aliquots of 25 and 22.5 μL volume.
Add 2.5 μL (corresponding to 1 Eq/10 μL reaction volume) of microsomes to the smaller sample.
Incubate at 30–37°C for 30–60 min (see Note 9).
Fig. 2.
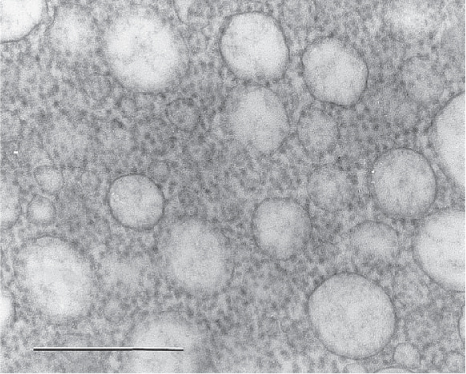
Electron micrograph of negatively stained microsomal vesicles prepared from the endoplasmic reticulum of canine pancreatic acinar cells. Such microsomes are competent to cotranslationally translocate secretory and membrane proteins. Note that each microsomal vesicle has many ribosomes bound to its surface, indicating that each vesicle has multiple protein insertion sites. This is a prerequisite for the assembly of an oligomeric connexon within microsomes. Scale bar = 500 nm.
Fig. 3.
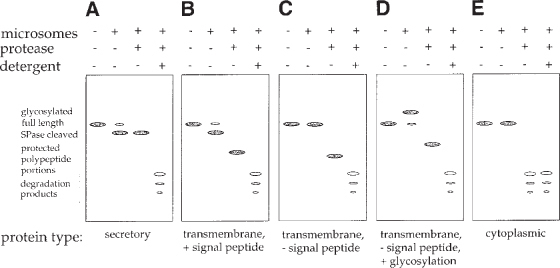
Schematic representation of the protein band patterns expected for different types of proteins after translating these proteins in translation competent cell lysates supplemented with microsomes, SDS-PAGE, and autoradiography. The expected translation products of a secretory protein (A), a transmembrane protein with an N-terminal cleavable signal peptide (B), a transmembrane protein without cleavable signal peptide (C), a core glycosylated membrane protein without cleavable signal peptide (D), and a cytoplasmic protein (E) are shown. For each protein type the translation products generated in the absence (−) (lanes 1) and in the presence of (+) microsomes (lanes 2) are shown on each gel. Furthermore, polypeptide domains located inside the microsomes and protected from proteolytic degradation are shown after addition of protease to the lysate when translation is completed (lanes 3), and addition of protease in combination with detergent (lanes 4). Full-length, unmodified polypeptides are generated in the absence of microsomes (lanes 1). Secretory and transmembrane proteins encoding a signal sequence can be recognized by their increased electrophoretic mobility on SDS protein gels when translated in the presence of microsomes (lanes 2, gels A and B). Proteins not encoding a cleavable signal sequence migrate unaltered (gel C). Core glycosylated proteins can be recognized by their decreased mobility (gel D). Note that in a core glycosylated protein encoding a signal peptide, increase and decrease in electrophoretic mobility will interfere. In addition, deglycosylation is necessary to correctly recognize this protein type (see Fig. 7). Membranetranslocated proteins (gels A–D) can be distinguished from non-translocated proteins (gel E) by the generation of protease-protected polypeptide portions (lanes 3, gels A–D). Protected portions are degraded when the microsomal membrane seal is resolved by the addition of detergent (lanes 4, gels A–D).
Increasing Translation Efficiency
A problem sometimes encountered with soluble as well as membrane proteins is that the synthetic RNA encoding the desired protein is not translated efficiently in translation-competent cell lysates. This was also found for connexin polypeptides, and may be related to the length of the 5′ untranslated region, the structure of the translation initiation sequence adjacent to the translation Start-AUG (22), or the absence of required factors. Optimization of the translation initiation sequence by site directed mutagenesis as well as adding subcellular fractions can improve translation efficiency. However, connexin translation efficiency was not substantially increased by these approaches (M. Falk, unpublished data).
Another approach is based on cloning the 5′ untranslated region of an efficiently translated protein, such as globin, upstream of the cloned cDNA. Cloning the connexin cDNAs into the transcription vector pSP64T (23) that encodes the 5′ noncoding region of Xenopus β-globin (24) immediately downstream of an SP6 promoter was found to dramatically increase the translation efficiency of the connexins as well as other cDNAs (14–16,25) (Fig. 4). An advanced version of pSP64T, pSPUTK is commercially available from Stratagene. Reducing the length of the 5′ untranslated portion of the desired cDNA may further increase translation efficiency to prevent scanning ribosomes from unspecifically falling off the template cDNA.
Fig. 4.
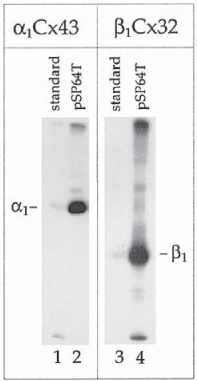
Increasing translation efficiency of connexins in cell-free translation systems. α1Cx43 and β1Cx32 cDNAs were cloned into standard transcription vectors (lanes 1, 3), and into the transcription vector pSP64T (lanes 2, 4) that contains the 5′ noncoding region of Xenopus β-globin as a translation enhancer upstream of the cDNA. cDNAs were transcribed and translated in rabbit reticulocyte lysates in the absence of microsomes. In each lane 0.5 μL of translation reaction was analyzed on SDS-polyacrylamide gels and translation products were visualized by autoradiogra- phy. Note the dramatically increased translation efficiency of transcripts containing the translation enhancer sequence (lanes 2, 4 vs 1, 3). Additional bands detectable on the gels represent aggregates of translation products, and translation products initiated at AUG codons located downstream from the connexin authentic initiation codons, and were generated mostly due to nonoptimized conditions. Gels were purposely overexposed to visualize these products. (Compare Fig. 5 for connexin-expression profiles obtained after optimizing translation conditions.) Substantially increasing the translation efficiency of connexins allowed the subsequent synthesis and assembly of oligomeric connexons in cell-free translation systems, and to analyze the assembly behavior of different connexin isotypes.
Another approach to enhance translation efficiency is based on using the CAP independent translation initiation sequences (IRES = Internal Ribosomal Entry Site elements; or CITE = CAP-Independent Translation Enhancer) from picornaviruses (see ref. 26 for review). Several vectors using these sequences, such as Novagen’ pCITE-1, are commercially available. In general it is advisable to ensure that no AUG codon in any of the three possible reading frames is encoded in the 5′ untranslated region upstream of the Start-AUG to prevent ribosomes scanning along the cDNA from initiating at wrong, upstream AUG codons.
Synthesis of Full-Length, Membrane Integrated Connexins in Cell-Free Translation Systems
We found that the translation of connexin polypeptides in standard cell-free translation systems supplemented with ER-derived microsomes resulted in a complete but inappropriate proteolytic processing that affected all connexin polypeptides upon their membrane integration (reported in 14,16) (see Fig. 5). A careful analysis of the cleavage reaction revealed that the relatively weak hydrophobic character of the first transmembrane spanning domain, which acts as internal signal anchor sequence, is responsible for this inappropriate cleavage. Our results indicate that the connexin signal anchor sequence is aberrantly recognized and positioned as a cleavable signal peptide within the endoplasmic reticulum translocon, and that this mispositioning enabled signal peptidase to access the cleavage sites (16). These observations provide direct evidence for the involvement of yet uncharacterized cellular factors in the membrane integration process of connexins that are absent or inactive in the standard cell-free translation system (14,16).
Fig. 5.
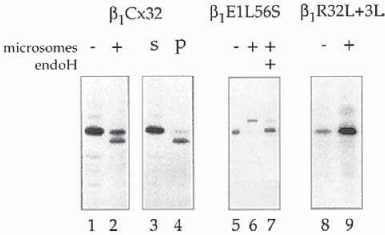
Synthesis of cleaved and full-length membrane-integrated connexins. Translation of connexins in cell-free translation systems supplemented with pancreatic microsomes results in a complete, aberrant processing by the ER resident protease signal peptidase that removes an N-terminal portion including the N-terminal domain, and the first transmembrane spanning domain of connexins (lane 2). In the absence of microsomes no cleavage occurs (lane 1). The cleaved connexins pellet (p) together with the microsomes (lane 4), while the full-length connexins stay in the soluble lysate fraction (s) (lane 3). This result indicates that all membrane-integrated connexin polypeptides are cleaved; and that the full-length connexins also synthesized in the presence of microsomes (lane 2) are synthesized on nonmembrane bound ribosomes, and have failed to insert cotranslationally into the microsomal membranes. Full-length, glycosylated, and membrane integrated connexins were synthesized when an N-glycosylation site was introduced into the first extracellular loop (L56S amino acid exchange) (lane 6). Endoglycosidase H (endoH) removes the carbohydrate sidechain. The electrophoretic mobility of the deglycosylated polypeptides corresponds to the mobility of full-length connexins (lane 7). Full-length, membrane-integrated connexins are also synthesized when the length and hydrophobicity of the first transmembrane spanning domain is increased (R32L + 3L amino acid exchange) (lane 9).
Two different methods have been found that prevent the cleavage and allow the efficient synthesis of full-length, correctly integrated connexin polypeptides into microsomes. Single amino acid exchanges, introduced by sitedirected mutagenesis into the first extracellular loops of α1Cx43, β1Cx32, as well as β2Cx26 (Glu57 to Ser in α1Cx43, Leu56 to Ser in β2Cx32, and Leu54 to Ser in β2Cx26), completely inhibited the cleavage reaction, most likely because of a steric hindrance between oligosaccharyl transferase (OST), the enzyme that recognizes the core glycosylation sequence and transfers core glycosyl groups from dolichol onto asparagine (see Subheading 3.2.3.), and signal pep- tidase (15,16, M. Falk, unpublished results). The point mutations result in the creation of core glycosylation sites within the connexin sequences and the binding of OST to the nascent connexin polypeptides (see Subheading 3.2.3.). However, core glycosyl groups were added only to Cx32, and Cx26 polypeptides during translation, and not to the Cx43 sequence (15,16, M. Falk, unpublished results).
The other method is based on increasing the hydrophobic character of the signal anchor sequence (the first transmembrane spanning domain) of connexins. Increasing the length of the hydrophobic core of the first transmembrane spanning domain in Cx32 from 18 amino acids (Val23 to Ala40) in the wild-type protein to 21 amino acids and exchanging the central Arg32 with an uncharged amino acid (leucine) completely abolished the cleavage as well (Fig. 7 in ref. 16).
Fig. 7.
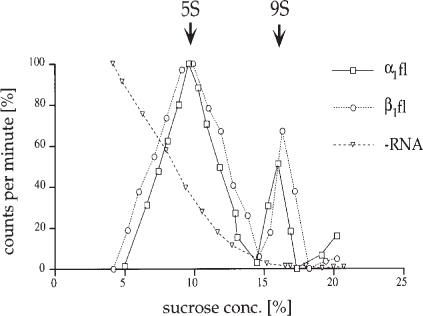
Assembly of homo-oligomeric connexons in a cell-free translation system. Reticulocyte lysates were supplemented with microsomes and programmed with αCx43 RNA, α1Cx32 RNA, and without (−) RNA in control. After translation was full-length (fl) completed microsomes were harvested, lysed in nonionic detergent, and assembly of connexins was analyzed by hydrodynamic analysis on linear sucrose gradients. Gradients were fractionated from the bottom, and sucrose concentration, radioactivity, and connexin protein content was determined in each fraction. Radioactivity recovered from each fraction was plotted vs the sucrose concentration after subtracting the lowest counts from each fraction. 9S particles represent assembled connexons, while 5S particles represent unassembled connexin polypeptides. More than 30% of the connexin polypeptides were recovered as assembled hexameric gap junction connexons. (Reproduced with kind permission fromref. 15).
Combined Transcription/Translation Systems
A few years ago combined transcription/translation systems were introduced that synthesize protein from an added cDNA in one step. These systems (e.g., TNT lysate from Promega) were primarily designed to test if a cloned cDNA or open reading frame produced a protein with the expected molecular weight. Such systems are probably better not used for the integration of connexins into microsomes because the combination of different reactions into one vial may complicate the interpretation of results, and may generate less clean translation reactions.
Assaying for Membrane Integration and Transmembrane Orientation
After translation of a membrane protein is completed its membrane integration can be analyzed. Great care should be taken in the interpretation of results that indicate potential membrane integration. This is especially true for membrane proteins that are normally not modified during or after their membrane integration by signal peptide cleavage, or core glycosylation. Electrophoretic mobility of such proteins will remain unaltered (see Fig. 3). Three methods (see Subheadings 3.2.1.–3.2.3.) are commonly used to determine membrane translocation that produce more or less convincing results. For polytopic membrane proteins with extended hydrophobic regions such as the connexins, a combination of different methods is generally required to obtain convincing results. We have used all three methods with connexins (14,16).
Alkali Extraction
This method is based on the perforation of membranes at alkaline pH. When microsomes are exposed to alkaline pH after a completed translation reaction, a secretory protein, translocated into the lumen of the microsomes, would be extracted from the microsomes, while a transmembrane protein would still remain with the microsomal membranes. In control experiments, microsomes treated at neutral pH are assayed. Furthermore, it is important to assay well-characterized transmembrane, secretory, and cytoplasmic control proteins in parallel for a valid interpretation of results (see ref. 14). This is especially important for proteins with extended hydrophobic regions, such as the connexins, as they tend to adhere strongly to hydrophobic surfaces. Therefore, it can be difficult to determine if a protein is indeed translocated into the membranes, or only adhered to the microsomal membrane surface (see Note 10).
We have used a modification of the original method described by Gilmore and Blobel (27) with connexins. Alkali-treated and control microsomes were pelleted after treatment with an Airfuge ultracentrifuge (Beckman Instruments, Palo Alto, CA). This centrifuge is very well suited for the small volume translation reactions. The standard A-100/18 rotor holds six tubes with a maximum capacity of 175 μL each. Alternatively, a tabletop microcentrifuge for Eppendorf tubes or a Beckman TL-100 tabletop ultracentrifuge can be used, although centrifugation times have to be extended substantially.
Following translation, 15 μL translocation reaction aliquots are adjusted to 50 μL volume with buffer 1. For alkali extractions, aliquots are adjusted to pH 11.5 with 1 M NaOH, and adjusted to 50 μL volume.
Samples are incubated on ice for 10 min, overlaid onto a 100 μL cushion of 0.5 M sucrose in buffer 1 or a 100 μL cushion of 0.2 M sucrose in 30 mM HEPES adjusted to pH 11.5, 150 mM KOAc, 2.5 mM MgOAc, and fractionated into a supernatant and pellet fraction.
Proteins in the supernatant fractions are then precipitated with trichloroacetic acid (TCA) and neutralized with saturated Tris base, before processing for SDS-PAGE.
At neutral as well as alkaline pH, membrane-integrated connexin polypeptides remain predominantly associated with the microsomal membranes and are detected in the pellet fraction. A similar result is obtained with the transmembrane control protein acetylcholine receptor subunit α7, while the secretory control proteins prolactin and yeast α-factor were largely extracted from the microsomal membranes at alkaline pH and were detected in the supernatant fractions (see Fig. 4 in ref. 14, and Fig. 6).
Fig. 6.
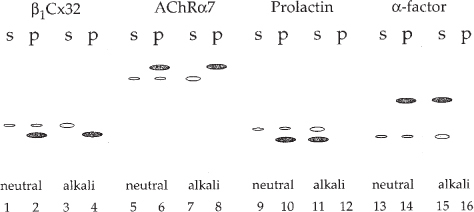
Assaying for protein membrane integration. Schematic representation of the protein band patterns expected for transmembrane, and secretory proteins after incubating the microsomes at neutral and alkaline pH. At neutral as well as alkaline pH, membrane-integrated connexin polypeptides remained associated with the microsomal membranes and were detected in the pellet fraction (p) (lanes 2 and 4). Full-length, not membrane integrated connexins were detected in the soluble supernatant fraction (s) (lanes 1 and 3). A similar result was obtained with the glycosylated, transmembrane control protein acetylcholine receptor subunit α7 (AchRα7) (⩼lanes 5–8), while the secretory control proteins prolactin and yeast α-factor were extracted from the microsomal membranes at alkaline pH and were detected in the supernatant fractions (lanes 10, 14, and 12, 16).
Protease Protection Assays
Translocation of many transmembrane proteins can be assayed better using a protease protection assay. Microsomes are incubated with externally added protease after translation is completed. Domains of a membrane integrated polypeptide protruding from the outside surface of the microsomes will be digested, while transmembrane domains, and domains located in the lumen of the microsomes are protected from proteolytic degradation (see Fig. 3, lanes 3, 7, 11, and 15). In control, detergent is added to an aliquot of the translocation reaction prior to addition of protease that resolves the vesicle barrier (see Fig. 3, lanes 4, 8, 12, and 16). After the digest is completed protected polypeptide domains can be characterized by immunoprecipitation analysis using domain- specific antipeptide antibodies (ref. 14, and Subheading 3.3.2.).
The following method has been used to determine membrane integration and transmembrane orientation of connexin polypeptides integrated into micorosomal vesicles:
10 μL Aliquots of the connexin translocation reactions are diluted to 50 μL in buffer 1 (see Subheading 3.2.1.).
To increase the seal of the vesicles, microsomes are stabilized by the addition of CaCl2 to a final concentration of 10 mM and incubated for 10 min on ice.
Either water or proteinase K (predigested for 30 min at 30°C) is added to final concentrations of 0.1 mg/mL and 0.5 mg/mL, or trypsin (predigested in the same way) is added to final concentrations of 0.5 mg/mL and 1 mg/mL from 10s× stock solutions in water, respectively. In addition, where indicated, NP-40 is added to a final concentration of 1% from a 10% (w/v) stock in water. All digests are incubated for 1 h at 37°C (see Note 11).
Protease activity is blocked by the addition of 5 mM DFP, 1 % SDS (final concentrations) and boiling for 5 min, following the method described by Chavez and Hall (28) (see Note 12).
Samples are then diluted 10× with buffer, chilled on ice, and processed for immunoprecipitation analysis, as described in Subheading 3.3.2.
Immunoprecipitated polypeptide fragments (see Note 13) are analyzed on special 20% SDS-gels allowing the resolution of small polypeptide fragments (29) (see Fig. 3).
N-Glycosylation Tagging
Probably the most convincing method to determine the integration of a protein into the membrane bilayer, which in addition allows determination of the overall transmembrane topology of a membrane protein, is to introduce core glycosylation sites into the polypeptide sequence. This approach is based on the activity of oligosaccharyl transferase (OST) that is present in the lumen of the ER and within microsomes. The enzyme transfers mannose-rich carbohydrate moieties onto asparagine residues that are part of N-glycosylation consensus sequences (Asp/X/Ser or Thr) (30). Since glycosylation can occur only at sites located at the lumenal face of the ER (or the microsomes), glycosylation at any given site can be taken as proof of the ER lumenal localization (which coincides with the extracellular face) of that polypeptide domain. Each core glycosylation group added to the polypeptide chain increases its molecular mass by ir~2.5 kDa. The accompanying reduction in electrophoretic mobility on SDS-polyacrylamide gels is generally large enough to be readily visible with proteins up to ir70 kDa in size.
We have used this method to demonstrate a cotranslational membrane integration of α1Cx43, β1Cx32, and β2Cx26 into microsomes, and that the transmembrane topology of connexin polypeptides integrated into microsomes is identical to their topology in the plasma membrane (16, M. Falk, unpublished results).
N-Glycosylation consensus sequences were introduced into the extracellular loops E1 and E2, and into the intracellular loop of α1Cx43, β1Cx32, and β2Cx26 by substituting specific amino acid residues using site directed mutagenesis (15,16, M. Falk, unpublished results). Whereas the mutants with the consensus sites in the extracellular loop E1 or E2 were efficiently glycosylated after cell-free translation in the presence of microsomes resulting in a reduced mobility on SDS-polyacrylamide gels, the mutants with the consensus sites in the intracellular loop were not glycosylated. In control aliquots, endogly-cosidase H (endoH) digestion was used to remove the carbohydrate side chains. Electrophoretic mobility was shifted back to the electrophoretic mobility of wild-type connexin proteins (see Fig. 1 in ref. 15, Fig. 2B in ref. 16, and Fig. 5, lanes 5–7).
Synthesis of Oligomeric Connexons in Microsomes
It was commonly thought that the oligomerization of membrane proteins does not occur in cell-free translation/membrane integration systems owing to the low probability for multiple polypeptide insertions into a microsomal vesicle (31). However, micrographs of thin-sectioned microsomes showed that each microsomal vesicle has many ribosomes bound to its membrane surface (Fig. 2), indicating that each vesicle has multiple protein insertion sites, and as such would potentially allow the integration of several polypeptides into the same vesicle. Later, functional expression and assembly of a Shaker type K+-channel (Shaker H4, an oligomeric structure consisting of four identical copies of a protein traversing the membrane bilayer six times) (32), assembly of a human HLA-DR histocompatibility molecule (an α/β/Γ-heterotrimer) (33,34), and assembly of the asialoglycoprotein receptor (an α/β hetero-oligomer) (35), although probably not into a functional receptor molecule (36), were reported to occur during cell-free expression in microsomes. These observations, combined with the expression of gap junction connexons reported in Falk et al. (15), provide compelling evidence that the assembly of functional membrane structures consisting of several subunit proteins can take place in microsomes, and that the cell-free translation system can be used to study protein oligomerization processes.
Substantially increasing the translation efficiency of connexins in lysates (see Subsection 3.1.4.) was a crucial prerequisite to achieve multiple connexin polypeptide insertions into individual microsomes and the successful assembly into connexons within microsomes. Furthermore, the increased translation efficiency allowed subsequent analysis of the assembly behavior of connexin isotypes, and determination of assembly signals within the connexin polypeptide sequences (15,37,38, M. Falk, unpublished results).
Translation and oligomeric assembly analysis conditions are as follows (see also ref. 15):
Translation reactions are generally 50–100 μL in volume for subsequent hydrodynamic oligomerization analysis. Volumes of 10–25 μL were used for the assembly analysis of connexins by immunoprecipitation.
To maximize translation efficiency, rabbit reticulocyte lysates are programmed with large amounts of the appropriate cRNA (typically 2–4 μg of RNA, as estimated from an ethidium bromide stained agarose gel, per 100 μL reaction volume).
To maximize connexin polypeptide membrane integration, low amounts of microsomes are used in the translation reactions. Typical concentrations are 5 Eq/100 μL reaction volume.
Translation reactions are incubated at 30°C for 1–3 h at 30°C to allow complete posttranslational folding and association of the newly synthesized polypeptides before subsequent oligomerization analysis (see Note 14).
Assembly Assayed by Hydrodynamic Analysis
Connexon assembly was assayed by hydrodynamic analysis using linear 5–20% sucrose gradients. This technique is generally used for assaying assembly of protein subunits into oligomeric structures and was used for connexins by others before (39–41).
Nonmembrane integrated connexin polypeptides that are synthesized to a certain extent in the cell-free translation reactions as a byproduct on nonmembrane bound, free ribosomes (see Subheadings 3.1.3. and 3.2.2., and Fig. 5) are separated from membrane integrated connexins prior to gradient analysis by pelleting the microsomes through a 0.5 M sucrose cushion made in 1× PBS, using an Airfuge ultracentrifuge (Beckman Instruments, Palo Alto, CA) as described in Subheading 3.2.1.
To remove unincorporated radioactive label, microsomes are washed twice in 0.25 M sucrose, 1× PBS and pelleted as before.
Microsomes are then solubilized in 1% TX-100 (or other detergents) for 30 min at 4°C.
Detergent-insoluble material is precipitated by a high-speed centrifugation (15 min, 30 psi) using the Airfuge ultracentrifuge.
Supernatants are loaded on top of 5-mL linear 5–20% (w/v) sucrose gradients containing 150 mM NaCl; 50 mM Tris, pH 7.6; and the respective detergent used for solubilization.
After centrifugation for 16 h at 4°C in a SW55Ti rotor (Beckman Instruments Inc.), at 43,000 rpm (gav = 160,000; gmax = 210,000), gradients are fractionated by puncturing the bottom of the tube with a 26-gauge needle, and approx 20 0.25-mL fractions are collected.
Aliquots of 25 μL of the fractions are analyzed by liquid scintillation counting and SDS-PAGE. In general, between 2000 and 15,000 counts per minute (cpm) are obtained per 5S peak fraction.
The cpm recorded in each fraction are corrected by the background activity (fraction with the lowest cpm), and plotted in percent activity per fraction (Fig. 7).
Aliquots of all fractions are also analyzed by immunoprecipitation (see Subheading 3.3.2.) using connexin-specific antibodies.
To verify assembly of connexins into connexons, control aliquots of the translocation reactions are solubilized in 0.1% SDS to resolve the oligomeric assemblies and analyzed in parallel as described previously, except that the gradients are prepared with 0.1 % SDS, and run at 20°C to prevent precipitation of the SDS in the gradients.
The refractive index of each fraction is measured using a refractometer and converted into the corresponding sucrose concentrations using a standard conversion table.
Standard proteins with known sedimentation coefficients (myoglobin, 2S; ovalbumin, 3.5S; BSA, 4.3S; catalase, 11.5S) and gap junction connexons consisting of α1Cx43, β1Cx32, or β2Cx26 expressed and purified from baculovirus infected insect cells (42) are analyzed on separate gradients to compare the connexin specific S-values with corresponding sucrose concentrations.
Dependent on the detergent used for the solubilization of microsomes, up to 32% of the membrane-integrated connexin polypeptides were recovered as hexameric connexons (ref. 15, and Fig. 7).
Assembly Assayed by Coimmunoprecipitation
Connexin specific monoclonal and antipeptide antibodies directed against different regions of α1Cx43, β1Cx32, and β2Cx26 that display no detectable cross-reactivity with other connexin isotypes are used for the immunoprecipita- tion of connexin polypeptides from in vitro translation reactions (see Note 15). Oligomerization of connexin polypeptides into homo- and hetero-oligomeric complexes can be analyzed by immunoprecipitation following general methods described by Harlow and Lane (43). Connexins are translated in different combinations together with other connexin isotypes, connexin mutants, or nonconnexin transmembrane proteins. Connexin polypeptides are then immunoprecipitated either from complete translocation reactions or from microsomes that had been pelleted through sucrose cushions as described in Subheading 3.2.1., followed by their resuspension in 1× PBS, 0.25 M sucrose (see Fig. 8).
Microsomes are solubilized for 30 min on ice in immunoprecipitation buffer containing 1 50 mMNaCl, 50 mMTris, pH 7.6; 1% TX-100, which lysed the microsomal vesicles without disrupting the oligomeric connexin complexes.
Insoluble material is precipitated by high-speed centrifugation using the Airfuge ultracentrifuge.
Aliquots of the supernatant corresponding to 10–25 μL of translocation reaction, a connexin-specific antibody against α1Cx43, β1Cx32, or β2Cx26 (see above), and preswollen Protein A-Sepharose (where required) are incubated together in 1 mL of immunoprecipitation buffer for 2 h at room temperature, or at 4°C overnight with shaking (see Note 16).
Beads are sedimented by centrifugation and washed 2× with immunoprecipitation buffer prior to the addition of SDS protein sample buffer.
Precipitated antigens and associated polypeptides are detected by SDS-PAGE and autoradiography (see Note 17).
As controls, aliquots of the translocation reactions or resuspended microsomes are solubilized in immunoprecipitation buffer containing 0.1% SDS, or no detergent (see Fig. 8 for a schematic representation).
Fig. 8.
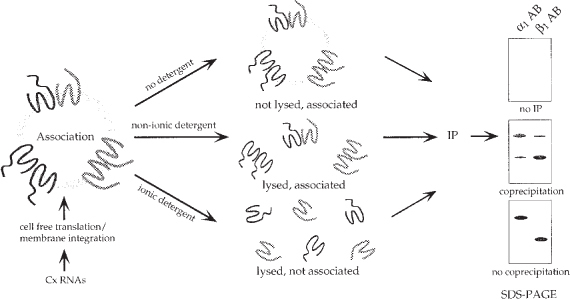
Schematic representation of a connexin assembly assay. Oligomerization of connexins was analyzed by co-immunoprecipitation. Different connexin isotypes were translated simultaneously in a cell-free translation reaction in the presence of microsomes. Connexin assembly into homo-and hetero-oligomeric complexes was allowed when several connexin polypeptides inserted into the same microsomal vesicle. Microsomes were lysed in nonionic detergent that kept the oligomeric complexes intact. Co-immunoprecipitation using connexin specific antibodies indicated co-assembly of the different connexin isotypes. In control aliquots microsomes were not lysed in detergent. No immunoprecipitation occurred under theses conditions due to the relatively large size of the microsomes. Or microsomes were lysed in SDS that also disrupted the oligomeric connexin complexes. No co-immunoprecipitation occurred under these conditions.
Connexin Protein Detection Methods
Connexin translation products were analyzed qualitatively on 10% and 12.5% SDS Laemmli Bio-Rad minigels (acrylamide/bis-acrylamide ratio 29:1). Samples are solubilized in SDS sample buffer containing 3% SDS and 5% β-mercaptoethanol and analyzed without heating to prevent unspecific aggregation of connexin polypeptides, a phenomenon often observed with polytopic membrane proteins.
Following electrophoresis, gels are soaked for 10 min in 1 M sodium salicylate (Sigma) to enhance 35S and 3H autoradiography, dried, and exposed to Kodak X-AR film at −70°C using an intensifying screen.
To quantitatively analyze connexin expression and compare amounts of different translation products with each other, autoradiographs are scanned densitome- trically using an AlphaImager 2000 Digital Imaging and Analysis System (Alpha Innotech Corporation, San Leandro, CA) as described in Falk and Gilula (16).
Because the specific radioactivity of a radiolabeled amino acid incorporated during cell-free translation is provided by the manufacturer, and the frequency of this amino acid in the connexin polypeptide sequence is known, the amount of connexin polypeptides synthesized in the cell-free system can be determined quantitatively using a liquid scintillation counter. We have used this method to quantify the number of polypeptides adsorped to antibody coated Protein A-Sepharose beads. Technical details are given in Kahn et al. (25).
Notes
Bacteriophage RNA polymerases are active for several hours and yield of RNA transcripts increases with longer incubation times.
If several different transcripts will be synthesized, it is desirable to mix all components except the individual cDNAs to reduce pipetting steps, then aliquot the mix, and finally add cDNAs.
Shorter RNA transcripts (1–2 kb in length) can be frozen and thawed several times without significant degradation. Longer transcripts (5–10 kb) are best used fresh to avoid degradation by the freeze-thaw cycle.
Great care has to be taken to avoid any possible contamination with RNases. Only autoclaved materials should be used, and tubes and pipet tips should not be touched with bare hands. All electrophoresis equipment needs to be cleaned thoroughly immediately before use, and buffers should be prepared fresh to reduce cRNA degradation.
When purchased, microsomes are relatively costly. I have prepared microsomes from fresh canine pancreas following the protocol of Walter and Blobel (21) with great success. The prepared microsomes had at least a similar translocation efficiency, and low backgrounds comparable to those of purchased microsomes, and were very efficient in connexin protein integration. Inside-out microsomes were not detected in the preparations (see Fig.2).
Microsomes are quite stable when stored at −70°C (2–3 yr). However, they should not be thawed and refrozen more than once or twice. Therefore, when first used, aliquoting them out into smaller volumes is highly recommended.
To investigate if the membrane translocation of a protein is dependent on signal recognition particle (SRP) wheat germ extract has to be used as a lysate, in combination with SRP-depleted microsomes and purified SRP. SRPs present in the wheat germ lysate do not interact with mammalian microsomes. Although we have used this methodology to prove that the membrane integration of connexins into microsomes is SRP-mediated (14), details of this specific application are not described in this more general chapter. However, protocols for the high-salt extraction of microsomes and the preparation of SRP are described in several publications by Peter Walter and co-workers (University of California, San Francisco).
To reduce background, a highly purified methionine, such as SJ1515 (Amersham Biotech) should be used in the translation reactions. Translabel (ICN) was found to produce additional bands, and therefore should be avoided for this application.
To avoid nonspecific aggregation of connexin polypeptides that failed to integrate into microsomes during translation and are detected as a protein band on top of the separating gel, shorter incubation times of only 30–45 min are recommended.
The posttranslational membrane integration of α1Cx43 and β2Cx26 recently reported by two laboratories (27,28) is solely based on the results obtained by pelleting microsomes through sucrose cushions, and a protease protection assay using an inadequate protease (Staphylococcus aureus V8 protease; see below). Therefore, the observation may reflect a strong adhesion of these connexin isotypes to the outer microsomal membrane surface owing to their strong overall hydrophobicity, rather than real posttranslational membrane translocation. Further experiments using adequate technology (see Subheadings 3.2.1.-3.2.3.) and control proteins are required to convincingly demonstrate a posttranslational ER membrane integration of connexin polypeptides.
Proteases with a very low specificity, such as proteinase K or trypsin, are recommended for this assay because exposed polypeptide domains should be degraded completely. More specific proteases, such as Staphylococcus aureus V8 protease, which cleaves only peptide bonds C-terminally at glutamic acid and with a 3000-fold lower rate at aspartic acid (44) that have been used in connexin protease protection assays previously (44) are not suitable. The large polypeptide fragments generally produced by V8 protease do not make it possible to determine if a detected polypeptide fragment was indeed protected from proteolytic degradation and located inside the microsomal vesicle, or simply did not contain a protease V8 specific cleavage site (see ref. 47 for further reading).
Protease activity has to be blocked completely before the microsomes are resolved in protein sample buffer; otherwise the protected polypeptide fragments will be degraded. This is especially important when the protected fragments will be characterized subsequently by immunoprecipitation. Proteinase K is extremely active, exhibiting residual activity even in protein sample buffer. A combination of either DFP or PMSF, together with SDS, and heating to at least 80°C as described above was found to efficiently block proteinase K, as well as trypsin activity.
The main disadvantage of protease protection assays is that sometimes the protease concentration cannot be increased high enough to obtain a complete digest of exposed polypeptide domains, before encountering leak of protease activity into the lumen of the microsomal vesicles. As a result, exposed but tightly membrane-associated domains may remain undigested. This was observed, for example, with a large portion of the C-terminal domain of the coronavirus glycoprotein E1 (13). Although potentially accessible to proteolytic degradation the intracellular loop region of connexins was found to be relatively resistant to protease digestion (48,49), which resulted in the precipitation of connexin polypep- tide fragments that included the intracellular loop domain in such protease protection assays (see Fig. 7 in ref. 14). This result intially misled us to believe that the transmembrane topology of connexins integrated into microsomes may be partially inverted (50). The assumption was revised (14), and the correct trans- membrane topology of connexins integrated into microsomes was proven (16).
Previously, 3–5 mM (final conc.) oxidized gluthatione (GSSG; Sigma, St. Louis, MO) added to the translation reactions was described to promote proper folding of membrane proteins by maintaining oxidized conditions in the translation reactions (34,51). However, addition of GSSG was not found to be necessary for connexon assembly.
Several researchers have used chemical crosslinking to detect connexin-connexin interactions (39,41,45). However, because this technique requires a chemical alteration of the polypeptides that may produce misleading results, especially if the concentration of the connexin polypeptides is low compared to the amount of total protein present in the microsomes, this technique was not further developed for the assembly analysis of cell-free expressed connexins.
Antibodies are used in combination with Protein A-Sepharose beads (Sigma). However, the cleanest immunoprecipitations are obtained with connexin-specific antibodies covalently bound to protein G-Sepharose beads (Pharmacia, Piscataway, NJ). To link the antibodies to the beads, 1 mL of protein G bead slurry is washed and incubated in 10 mM sodium phosphate buffer, pH 7.3, with 15 mg of monoclonal antibodies for 1 h at room temperature with continuous rocking. Antibody-bead complexes are washed twice in 0.1 M sodium borate, pH 9.0, and bound antibodies are covalently crosslinked to the Protein A with 20 mM dimethyl pimelimidate · 2HCl (DMP, Pierce, IL), in 0.1 M sodium borate, pH 9.0, for 30 min at room temperature with continuous rocking. The reaction was quenched by washing twice with 0.2 M ethanolamine, pH 8.0, for 1 h. Antibody-bead complexes were resuspended in 1× PBS, 0.01% Thimerosal and stored at 4°C until used.
Cell-free expressed connexons were found to be functional by means of channel activity. Single-channel activities were characterized by electrophysiological analysis of channels obtained after fusion of microsomes containing cell-free expressed connexins with planar lipid membranes. This method requires special experimental setup and is not described in this chapter. However, the experimental conditions are described in detail in Buehler et al. (52) and Falk et al. (15).
References
- 1.Pfeffer S. R., Rothman J. E. Biosynthetic protein transport and sorting by the endoplasmic reticulum and Golgi. Annu. Rev. Biochem. 1987;56:829–852. doi: 10.1146/annurev.bi.56.070187.004145. [DOI] [PubMed] [Google Scholar]
- 2.Rapoport T. A., Jungnickel B., Kutay U. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu. Rev. Biochem. 1996;65:271–303. doi: 10.1146/annurev.bi.65.070196.001415. [DOI] [PubMed] [Google Scholar]
- 3.High S., Laird V. Membrane protein biosynthesis—all sewn up? Trends Cell Biol. 1997;7:206–210. doi: 10.1016/S0962-8924(97)01035-0. [DOI] [PubMed] [Google Scholar]
- 4.Johnson A. E. Protein translocation at the ER membrane: a complex process becomes more so. Trends Cell Biol. 1997;7:90–95. doi: 10.1016/S0962-8924(97)01029-5. [DOI] [PubMed] [Google Scholar]
- 5.Blobel G., Dobberstein B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975;67:852–862. doi: 10.1083/jcb.67.3.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borel A. C., Simon S. M. Biogenesis of polytopic membrane proteins: Membrane segments of p-glycoprotein sequentially translocate to span the ER membrane. Biochemistry. 1996;35:10587–10594. doi: 10.1021/bi960950q. [DOI] [PubMed] [Google Scholar]
- 7.Mothes W., Heinrich S. U., Graf R., Nilsson I. M., von Heijne G., Brunner J., Rapoport T. A. Molecular mechanism of membrane protein integration into the endoplasmic reticulum. Cell. 1997;89:523–533. doi: 10.1016/S0092-8674(00)80234-2. [DOI] [PubMed] [Google Scholar]
- 8.Jadot M., Hofmann M. W., Graf R., Quader H., Martoglio B. Protein insertion into the endoplasmic reticulum of permeabilized cells. FEBS Lett. 1995;371:145–148. doi: 10.1016/0014-5793(95)00881-9. [DOI] [PubMed] [Google Scholar]
- 9.Falk M. M. Cell-free synthesis for analyzing the membrane integration, oligomerization, and assembly characteristics of gap junction connexins. Methods. 2000;20:165–179. doi: 10.1006/meth.1999.0934. [DOI] [PubMed] [Google Scholar]
- 10.Falk M. M., Grigera P. R., Bergmann I. E., Zibert A., Multhaup G., Beck E. Foot-and-mouth disease virus protease 3C induces Specific proteolytic cleavage of host cell histone H3. J. Virol. 1990;64:748–756. doi: 10.1128/jvi.64.2.748-756.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falk M. M., Sobrino F., Beck E. VPg gene amplification correlates with infective particle formation in foot-and-mouth disease virus. J. Virol. 1992;66:2251–2260. doi: 10.1128/jvi.66.4.2251-2260.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zibert A., Maass G., Strebel K., Falk M. M., Beck E. Infectious foot-and-mouth disease virus derived from a cloned full-length cDNA. J. Virol. 1990;64:2467–2473. doi: 10.1128/jvi.64.6.2467-2473.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer T., Tamura T., Falk M. M., Niemann H. Membrane integration and cellular transport of the coronavirus glycoprotein E1, a class III membrane glycoprotein. J. Biol. Chem. 1988;263:14956–14963. doi: 10.1016/S0021-9258(18)68131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falk M. M., Kumar N. M., Gilula N. B. Membrane insertion of gap junction connexins: polytopic channel forming membrane proteins. J. Cell Biol. 1994;127:343–355. doi: 10.1083/jcb.127.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falk M. M., Buehler L. K., Kumar N. M., Gilula N. B. Cell-free synthesis and assembly of connexins into functional gap junction membrane channels. EMBO J. 1997;10:2703–2716. doi: 10.1093/emboj/16.10.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falk M. M., Gilula N. B. Connexin membrane protein biosynthesis is influenced by polypeptide positioning within the translocon and signal peptidase access. J. Biol. Chem. 1998;273:7856–7864. doi: 10.1074/jbc.273.14.7856. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J., Fritsch E. F., Maniatis T. eds., editors. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 18.Jackson R. J., Hunt T. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. Methods Enzymol. 1983;96:50–74. doi: 10.1016/S0076-6879(83)96008-1. [DOI] [PubMed] [Google Scholar]
- 19.Erickson A., Blobel G. Cell-free translation of messenger RNA in a wheat germ system. Methods Enzymol. 1983;96:38–50. doi: 10.1016/S0076-6879(83)96007-X. [DOI] [PubMed] [Google Scholar]
- 20.Evans E. A., Gilmore R., Blobel G. Purification of microsomal signal peptidase as a complex. Proc. Natl. Acad. Sci. USA. 1986;83:581–585. doi: 10.1073/pnas.83.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter P., Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 1983;96:84–93. doi: 10.1016/S0076-6879(83)96010-X. [DOI] [PubMed] [Google Scholar]
- 22.Kozak M. The scanning model for translation: an update. J. Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieg P. A., Melton D. A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams J. G., Kay M., Patient R. K. The nucleotide sequence of the major β-globin mRNA from Xenopus laevis. Nucleic Acids Res. 1980;8:4247–4258. doi: 10.1093/nar/8.18.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahn T. W., Beachy R. N., Falk M. M. Cell-free expression of a GFP-fusion protein allows protein quantitation in vitro and in vivo. Curr. Biol. 1997;7:R207–R208. doi: 10.1016/S0960-9822(06)00100-X. [DOI] [PubMed] [Google Scholar]
- 26.Jackson R. J., Howell M. T., Kaminski A. The novel mechanism of initiation of picornavirus RNA translation. Trends Biochem. Sci. 1990;15:477–483. doi: 10.1016/0968-0004(90)90302-R. [DOI] [PubMed] [Google Scholar]
- 27.Gilmore R., Blobel G. Translocation of secretory proteins across the microsomal membrane occurs through an environment accessible to aqueous perturbants. Cell. 1985;42:497–505. doi: 10.1016/0092-8674(85)90107-2. [DOI] [PubMed] [Google Scholar]
- 28.Chavez R. A., Hall Z. W. Expression of fusion proteins of the nico-tinic acetylcholine receptor from mammalian muscle identifies the membrane-spanning regions in the a and Γ subunits. J. Cell Biol. 1992;116:385–393. doi: 10.1083/jcb.116.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas J. O., Kornberg D. An octamer of histones in chromatin and free in solution. Proc. Natl. Acad. Sci. USA. 1975;72:2626–2630. doi: 10.1073/pnas.72.7.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hart G. W., Brew K., Grant G. A., Bradshaw R. A., Lennarz W. J. Primary structural requirements for the enzymatic formation of the N-glycosidic bond in glycoproteins. J. Biol. Chem. 1978;254:9747–9753. [PubMed] [Google Scholar]
- 31.Anderson D. J., Blobel G. In vitro synthesis, glycosylation, and membrane insertion of the four subunits of Torpedo acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 1981;78:5598–5602. doi: 10.1073/pnas.78.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg R. L., East J. E. Cell-free expression of functional Shaker potassium channels. Nature. 1992;360:166–169. doi: 10.1038/360166a0. [DOI] [PubMed] [Google Scholar]
- 33.Kvist S., Wiman K., Claesson L., Peterson P. A., Dobberstein B. Membrane insertion and oligomeric assembly of HLA-DR histocompatibility an-tigens. Cell. 1982;29:61–69. doi: 10.1016/0092-8674(82)90090-3. [DOI] [PubMed] [Google Scholar]
- 34.Qu D., Green M. Folding and assembly of a human MHC class II molecule in a cell-free system. DNA Cell Biol. 1995;14:741–751. doi: 10.1089/dna.1995.14.741. [DOI] [PubMed] [Google Scholar]
- 35.Sayer J. T., Doyle D. Assembly of a heterooligomeric asialoglyco-protein receptor complex during cell-free translation. Proc. Natl. Acad. Sci. USA. 1990;87:4854–4858. doi: 10.1073/pnas.87.12.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yilla M., Doyle D., Sawyer J. T. Early disulfide bond formation prevents heterotypic aggregation of membrane proteins in a cell-free translation system. J. Cell Biol. 1992;188:245–252. doi: 10.1083/jcb.118.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falk M. M., Buehler L. K., Kumar N. M., Gilula N. B. Cell-free expression of functional gap junction channnels. In: Werner R., editor. Gap Junctions. Amsterdam, Berlin, Oxford, Tokyo, Washington DC: IOS Press; 1998. pp. 135–139. [Google Scholar]
- 38.Yeager M., Unger V., Falk M. M. Synthesis, assembly, and structure of gap junction intercellular channels. Curr. Opin. Struct. Biol. 1998;8:517–524. doi: 10.1016/S0959-440X(98)80131-0. [DOI] [PubMed] [Google Scholar]
- 39.Musil L. S., Goodenough D. A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 40.Kistler J., Goldie K., Donaldson P., Engel A. Reconstitution of native-type noncrystalline lens fiber gap junctions from isolated hemichannels. J. Cell. Biol. 1994;126:1047–1058. doi: 10.1083/jcb.126.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cascio M., Kumar N. M., Safarik R., Gilula N. B. Physical characterization of gap junction membrane connexons (hemi-channels) isolated from rat liver. J. Biol. Chem. 1995;270:18643–18648. doi: 10.1074/jbc.270.31.18643. [DOI] [PubMed] [Google Scholar]
- 42.Stauffer K. A., Kumar N. M., Gilula N. B., Unwin N. Isolation and purification of gap junction channels. J. CellBiol. 1991;115:141–150. doi: 10.1083/jcb.115.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harlow E., Lane D. ed., editors. Antibodies. A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 44.Zhang J.-T., Chen M., Foote C. I., Nicholson B. J. Membrane integration of in vitro-translated gap junctional proteins: co-and posttranslational mechanisms. Mol. Biol. Cell. 1996;7:471–482. doi: 10.1091/mbc.7.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad S., Diez J. A., George H., Evans W. H. Synthesis and assembly of connexins in vitro into homomeric and heteromeric functional gap junction hemichannels. Biochem. J. 1999;339:247–253. doi: 10.1042/0264-6021:3390247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorensen S. B., Sorensen T. L., Breddam K. Fragmentation of proteins by S. aureus strain V8 protease. FEBS Lett. 1991;294:195–197. doi: 10.1016/0014-5793(91)80667-R. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto T., Aprin M., Gaetani S. Use of proteases for the study of membrane insertion. Methods Enzymol. 1983;96:121–150. doi: 10.1016/S0076-6879(83)96013-5. [DOI] [PubMed] [Google Scholar]
- 48.Zimmer D. B., Green C. R., Evans W. H., Gilula N. B. Topological analysis of the major protein in isolated intact rat liver gap junctions and gap junction-derived single membrane structures. J. Biol. Chem. 1987;262:7751–7763. [PubMed] [Google Scholar]
- 49.Milks L. C., Kumar N. M., Houghten R., Unwin N., Gilula N. B. Topology of the 32-kd liver gap junction protein determined by site-directed antibody localizations. EMBO J. 1988;7:2967–2975. doi: 10.1002/j.1460-2075.1988.tb03159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falk M. M., Kumar N. M., Gilula N. B. Biosynthetic membrane integration of connexin proteins. Prog. Cell Res. 1995;4:319–322. [Google Scholar]
- 51.Marquardt T., Hebert D. N., Helenius A. Posttranslational folding of influenza hemagglutinin in isolated endoplasmic reticulum-derived microsomes. J. Biol. Chem. 1993;268:19618–19625. [PubMed] [Google Scholar]
- 52.Buehler L. K., Stauffer K. A., Gilula N. B., Kumar N. M. Single channel behavior of recombinant β2 gap junction connexons reconstituted into planar lipid bilayers. Biophys. J. 1995;68:1767–1775. doi: 10.1016/S0006-3495(95)80353-X. [DOI] [PMC free article] [PubMed] [Google Scholar]