

Abstract
Bioterrorism is the deliberate release of biological toxins, pathogenic viruses, bacteria, parasites, or other infectious agents into the public sphere with the objective of causing panic, illness, and/or death on a local, regional, or possibly national scale. The list of potential biological agents compiled by the Centers for Disease Control and Prevention is long and diverse. However, a trait common to virtually all the potential bioterrorism agents is the fact that they are likely to be disseminated by either aerosol or in food/water supplies with the intention of targeting the mucosal surfaces of the respiratory or gastrointestinal tracts, respectively. In some instances, inhalation or ingestion would mimic the natural route by which humans are exposed to these agents. In other instances, (e.g., the inhalation of a toxin is normally associated with food borne illness), it would represent an unnatural route of exposure. For most potential bioterrorism agents, the respiratory or gastrointestinal mucosa may simply serve as a route of entry by which they gain access to the systemic compartment where intoxication/replication occurs. For others, however, the respiratory or gastrointestinal mucosa is the primary tissue associated with pathogenesis, and therefore, the tissue for which countermeasures must be developed.
Keywords: Mucosal Immunity, Severe Acute Respiratory Syndrome, Staphylococcal Enterotoxin, Yersinia Pestis, Eastern Equine Encephalitis Virus
Introduction
“The ability of our nation to detect and counter bioterrorism depends to a large degree on the information generated by biomedical research on disease-causing microorganisms and the immune system’s response to them.” Dr. Anthony Fauci, Director, National Institutes of Allergy and Infectious Disease, National Institutes of Health, USA.
In 1999, the Centers for Disease Control (CDC) convened a special expert panel to identify known biological weapons or potential biological threat agents (“biothreats”) which, if used for nefarious purposes, posed a significant threat to civilian populations (Rotz et al. 2002). The panel established a list of potential bioterrorism agents, including toxins, viruses, bacteria, and protozoa, that were classified into three categories (A, B, and C). The classification system was based on the following four criteria: (i) the potential to cause morbidity and mortality in healthy individuals; (ii) the potential to be disseminated within the public sphere; (iii) the perceived threat and potential to elicit fear or panic; (iv) the capacity of local, state, and federal public health networks to respond and control an event. In response to the Public Health Security and Bioterrorism Preparedness and Response Act of 2002, the Department of Health and Human Services (DHHS) and the United States Department of Agriculture (USDA) expanded this original list to include emerging infectious diseases, as well as threats to livestock and economically important crops. The members of this expanded list are collectively referred to as Select Agents and Toxins, the possession and use of which is now regulated by the National Select Agent Registry under the auspices of the CDC. The National Institutes of Health (NIH), specifically the National Institute of Allergy and Infectious Diseases (NIAID), considers a subset of the select agents as “Priority Pathogens” for which there is a particular need for countermeasures, including vaccines. A complete list of these agents with their respective designations (Category A–C) is presented in Table 1.
Table 1.
Category A–C Biothreats
| Toxins |
| Botulinum neurotoxinsa,A |
| Shigatoxina |
| Tetrodotoxina |
| T-2 toxina |
| Staphylococcal enterotoxinsa,B |
| Ricina,A |
| Diacetoxyscirpenola |
| Conotoxinsa |
| Abrina |
| Saxitoxina |
| Shiga-like ribosome inactivating proteinsa |
| Clostridium perfringens epsilon toxina,B |
| Viruses |
| Eastern equine encephalitisa,B |
| Hendra virusb |
| Variola major (smallpox)a,A |
| Variola minor (alastrim)a,A |
| Monkeypoxa,A |
| Filoviruses |
| Ebola virusa,A |
| Marburg virusa,A |
| Arenaviruses |
| Junina,A |
| Machupoa,A |
| Guanaritoa,A |
| Flexala,A |
| Sabiaa,A |
| Lassaa,A |
| Japanese encephalitis virusB |
| Venezuelan equine encephalitisb,B |
| DengueA |
| LaCrosseB |
| California encephalitisB |
| Western equine encephalitisB |
| Bunyaviruses |
| HantavirusesA |
| Rift Valley Feverb,A |
| ChikungunyaC |
| Hepatitis AB |
| Yellow feverC |
| RabiesC |
| Nipah virusb |
| Tick-borne encephalitis complex (flavivirus) |
| Central European tick-borne encephalitisa |
| Kyasanur forest diseasea,B |
| Omsk hemorrhagic fevera |
| Far eastern tick-borne encephalitisa |
| Russian spring/summer encephalitis a |
| Avian influenza virus (highly pathogenic)c |
| Reconstructed 1918 influenza virusa |
| Cercopithecine herpesvirus 1 (Herpes B virus)a |
| Severe acute respiratory syndrome (SARS)C |
| CalicivirusesB |
| InfluenzaC |
| West nile virusB |
| Crimean-congo hemorrhagic fevera,C |
| Bacteria/Rickettsia |
| Bacillus anthracis b,A |
| Brucella abortus b,B |
| Brucella melitensis b,B |
| Brucella suis b,B |
| Burkholderia mallei b,B |
| Burkholderia pseudomallei b,B |
| Coxiella burnetii a,B |
| Francisella tularensis a,A |
| Yersinia pestis a,A |
| Rickettsia prowazekii a,B |
| Rickettsia rickettsii a |
| Pathogenic vibriosB |
| Shigella speciesB |
| Salmonella speciesB |
| Listeria monocytogenes B |
| Yersinia enterolitica B |
| Campylobacter jejuni B |
| Multi-drug resistant tuberculosisC |
| Other RickettsiasC |
| Chlamydia psittaci A |
| Diarrheagenic E. coli B |
| Botulinum toxin-producing species of Clostridium a |
| Protozoa |
| Cryptosporidium parvum B |
| Cyclospora cayatanensis B |
| Entamoeba histolytica B |
| ToxoplasmaB |
| Giardia lamblia B |
| Fungi |
| Coccidioides posadasii a |
| Coccidioides immitis a |
| MicrosporidiaB |
Listing of the biological agents considered to be a threat to human health as a compilation from a number of sources including the 1) select agents and toxins provided by the U.S. Department of Health and Human Services (DHHS), Centers for Disease Control and Prevention (CDC) and the U.S. Department of Agriculture (USDA) and 2) the priority pathogens from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIAID) (www3.niaid.nih.gov/topics/BiodefenseRelated/Biodefense/research/CatA.htm)
aSelect Agents and Toxins designated by DHHS/CDC
bOverlap select agents and toxins that are designated and regulated by both DHHS/CDC and the U.S. Department of Agriculture (USDA)
cSelect agents designated and regulated by the USDA
A, B, C NIH/NIAID priority pathogens group A, B or C
Category A Agents
The CDC Category A agents include four pathogens that are highly infectious as aerosols. These are Bacillus anthracis, Yersinia pestis, smallpox (variola major) and Francisella tularensis. Among these, smallpox poses the greatest threat to public safety due to its highly pathogenic nature, its capacity to spread person-to-person, and the fact that herd immunity to the virus has waned since routine vaccination was discontinued more than three decades ago (Artenstein 2008). Anthrax also poses a significant and real threat to public health because B. anthracis spores are highly infectious, though the disease itself is not communicable. Also among the list of Category A agents is botulinum neurotoxin (BoNT), one of the most lethal known biological toxins. While BoNT can be aerosolized and is highly toxic following inhalation, the CDC’s primary concern is the possible use of the toxin to contaminate food/water supplies (Sobel et al. 2002). The toxin is extremely potent via the oral route (~LD50 of 0.001 μg/kg) and has a long history as a bioweapon. Botulinum neurotoxin has been produced in large scale quantities by numerous governments as well as terrorist organizations (Sobel et al. 2002).
Category B Agents
The Category B agents are defined as being moderately easy to disseminate, capable of inflicting moderate morbidity/low mortality and requiring specific enhancements to standard diagnostic capacity, as well as enhanced disease surveillance. Whereas, the Category A agents are primarily threats by aerosol, the majority of the Category B agents target the gastrointestinal tract. These include food and water safety threats (e.g., Salmonella and Shigella species, pathogenic vibrios, enterotoxigenic E. coli), as well as toxins (e.g., ricin, staphylococcal enterotoxin B). In general, the Category B agents are not communicable from person to person but tend to be relatively easy to disperse at doses sufficient to be highly debilitating, and to necessitate prolonged medical attention. Many of the food/water safety threats are endemic in developing countries and are already a focus of ongoing biomedical research.
Category C Agents
At present, the Category C agents are not considered as high risk, but rather as emerging diseases that may pose a threat to public health in the future. The CDC restricts this category to Nipah virus and hantavirus, whereas the NIAID Category C priority pathogens include yellow fever, influenza, rabies, tick-borne encephalitis viruses, severe acute respiratory syndrome associated coronavirus (SARS-CoV), as well as certain other types of drug/antibiotic-resistant pathogens, such as tuberculosis-causing mycobacteria. In addition, the NIAID Category C agents include pathogens commonly found circulating in the general population (e.g., Hepatitis A), and which possess the potential to cause morbidity among the larger population.
Assessing the Risk Posed by Biothreat Agents
It is generally assumed that biothreat agents will be disseminated by one of the two routes: aerosol or introduction into food/water supplies. Aerosol dissemination of pathogenic organisms, from either a point source such as a contaminated letter sent through the postal service, or from a planned aerial attack remain the greatest type of biological threat to the general public. No other modality of dispersion, except perhaps the widespread contamination of drinking water, could do more harm than aerosol dissemination of an infectious or toxic agent (Henderson 1999). Indeed, when engineered with the intent of causing mass casualties, biological agents pose health risks similar in magnitude to chemical or nuclear threats. Past weapons programs in the US and in the former Union of Soviet Socialist Republics (USSR) developed industrial processes for producing biological preparations that were optimized for aerosol delivery using sophisticated drying, milling, and packaging processes (Kortepeter et al. 2001). These preparations were designed for dispersion as highly respirable particles containing extremely large concentrations of stable, viable biological agent. Although many of the known biological weapons programs have been discontinued, aerosol delivery of an infectious or highly toxic biological agent continues to be a potential threat that rivals other weapons of mass destruction in terms of potential morbidity and mortality, as well as widespread panic.
The actual efficiency of aerosol to disseminate biological agents depends on the sophistication of the sample preparation, as well as biological and physical stressors in the environment. The infectivity of many of these pathogens is highly dependent on their size, hydrophobicity, and aggregation, as well as environmental conditions such as humidity and temperature. Humidity and temperature can affect particle size, which in turn determines the degree to which particles can penetrate the lungs. Highly respirable aerosols produced with a homogenous size distribution, commonly associated with sophisticated biological weapons, target the lower airways and alveolar spaces of the lung. Naturally-produced infectious aerosols, on the other hand, tend to be heterogeneous with respect to size, and will deposit throughout the respiratory tract. These physical differences not only dictate the degree of mucosal exposure associated with an aerosol, but may also impact the nature of the ensuing disease.
The network of food production, processing, and distribution in the US is vast and potentially vulnerable to deliberate contamination with toxins or infectious agents (Sobel and Watson 2009). The vulnerability of the food supply at the national level is evidenced by the fact that food-borne outbreaks caused by a single source of Salmonella or shiga toxin-producing E. coli O157:H7 (STEC) are not uncommon. For example, just the past two years, two Salmonella outbreaks have accounted for more than 2,000 illnesses (and nine deaths) in 44 states and certain provinces in Canada (Maki 2009). The first Salmonella outbreak occurred between April and August 2008 and was linked to contaminated peppers (and possibly tomatoes) that originated in Mexico and were subsequently processed in the southwest US. The second outbreak, due to S. typhimurium, occurred between October 2008 and March 2009, and was traced to a single peanut processing plant in Georgia (www.cdc.gov/salmonella). The actual number of cases associated with these outbreaks is likely to be 20–30 times greater than the number of cases reported (Maki 2009). Contamination of water supplies has had a similar impact on public health. In 1993, an outbreak of waterborne cryptosporidium in Milwaukee, WI affected an estimated 403,000 people (Leclerc et al. 2002). Enteric infections also pose a significant threat at the local level, in which a food or water source directly accessible to consumers is deliberately adulterated. The best example of such an incident occurred in 1984 in The Dalles, OR, where members of a religious commune intentionally contaminated salad bars at public restaurants with S. typhimurium (Torok et al. 1997). That incident resulted in more than 700 individuals contracting gastroenteritis.
Assessing Degrees of Mucosal Involvement
From the perspective of vaccine development for biodefense, it is important to differentiate between biological agents that elicit mucosal infections and pathophysiology, and those agents that simply exploit mucosal tissues as a means to gain access to the systemic compartment. In the former case, mucosal immunity is likely to be essential in preventing and clearing infections. Therefore, vaccines against these agents must truly involve mucosa-associated lymphoid tissues. In situations where the mucosa functions solely as a port of entry, systemic immunity is likely to be sufficient to control infection. One such example is anthrax. While B. anthracis spores are highly infectious by aerosol, the vegetative bacteria generally do not proliferate locally. Rather, following inhalation, the bacteria disseminate systemically via the lymphatics and circulatory system. Once within the systemic compartment, B. anthracis germinates and produces two toxins which account for the lethality associated with infection (Leppla et al. 2002). Protective immunity to B. anthracis is associated primarily with anti-toxin serum IgG antibodies. Mucosal defense is of little (if any) importance in controlling anthrax. On the other hand, mucosal immunity is likely to be important in controlling infections caused by two other Category A bacterial pathogens, notably Y. pestis and F. tularensis, which cause mucosal and systemic complications following inhalation (Metzger et al. 2007).
For many of the Category A–C agents, transmission via the respiratory tract is considered an “unnatural” route of infection and the actual involvement of the mucosa in the pathogenesis of infection may not be currently known. As a consequence, initial host interaction and the subsequent pathophysiology will not necessarily coincide with established clinical outcomes associated with infection by the natural route. In addition, there may be no clinical data that adequately defines aerosol-related disease or how it differs from the natural route of infection. An example of one such agent is Staphylococcal enterotoxin B (SEB). As a member of the superantigen family of toxins, SEB forms “bridges” between Major Histocompatibility Class II molecules on antigen presenting cells and T cell receptors on specific subsets of CD4+ and CD8+ T cells. As a consequence of SEB binding, T cells release massive amounts of proinflammatory cytokines and undergo hyper-proliferation which ultimately results in their depletion (Kappler et al. 1989; White et al. 1989). Staphylococcal enterotoxin B is clinically associated with food poisoning, as ingestion of microgram quantities of the toxin are sufficient to evoke violent vomiting, diarrhea, fever, and, in severe cases, lethal shock (Bergdoll 1983). Despite being classified as an enterotoxin, SEB is extremely pathogenic following aerosol challenge. Rhesus monkeys administered SEB as an aerosol suffered from vomiting and diarrhea within hours, and died approximately two days later (Tseng et al. 1993; Mattix et al. 1995). Postmortem analysis indicated that the animals likely succumbed to severe pulmonary edema triggered by SEB-mediated T cell proliferation in the respiratory mucosa (Mattix et al. 1995).
In the case of alphaviruses, the pathogenesis associated with infection is highly dependent on the route of exposure, at least in experimental animal models. It is hypothesized that exposure to aerosols induces disease directly via the olfactory bulb, whereas experimental infection via injection generally causes a disseminated viremia prior to nervous system engagement and encephalitis. These different manifestations of clinical symptoms and outcome following different routes of exposure have been demonstrated for the Venezuelan and Eastern equine encephalitis viruses (Ryzhikov et al. 1991). Correspondingly, immunity to the alphaviruses may depend on the route of exposure. While serum IgG antibodies may be effective for neutralizing virus following exposure via subcutaneous injection (similar to a natural exposure), these antibodies may not control viral spread following aerosol challenge. Cell-mediated immunity (CMI) rather than humoral immunity may be more important following aerosol challenge. Thus, induction of secretory antibodies at the respiratory mucosal barrier may not necessarily be an important aspect of rational vaccine design and development against these viruses (Pratt et al. 2003).
A final group of biothreat agents are those which are broadly toxic and potentially lethal irrespective of the route of exposure. In this case, systemic immunity may suffice to protect against lethality but may not prevent localized tissue damage in the mucosa. One such example is ricin toxin, which elicits both local and systemic inflammation and cell death following injection, inhalation or ingestion (Wilhelmsen and Pitt 1996; Mantis 2005; Yoder et al. 2007). Ricin is a bipartite toxin capable of intoxicating all known cell types. The ricin toxin B subunit (RTB) is a lectin with specificity for β-1,3 linked galactose and N-acetylgalactosamine residues. The A subunit (RTA) is an RNA N-glycosidase whose substrate is a conserved adenine residue within the so-called sarcin/ricin loop of eukaryotic 28S ribosomal RNA. Monkeys exposed to lethal doses of ricin by aerosol suffered peribronchovascular edema, mixed inflammatory cell infiltrates, and widespread necrosis in the airways and alveoli (Wilhelmsen and Pitt 1996). Death occurred 36–48 h post exposure. In a rodent model, serum IgG was sufficient to prevent death of animals after a lethal aerosol challenge, but not sufficient to prevent toxin-induced lung lesions (Griffiths et al. 1995). Although these studies need to be confirmed in a nonhuman primate model, the data suggest that vaccine development strategies must aim at eliciting both systemic and mucosal immunity to confer complete protection against certain select agents such as ricin.
Inherent Challenges in Development of Mucosal Vaccines for Biodefense
The development of vaccines is, in general, an extremely challenging and expensive undertaking. It is estimated that a single vaccine takes 10–15 years to reach licensure and at a cost exceeding hundreds of million of dollars (Levine 2006). The development of vaccines for biodefense faces additional hurdles, including the following:
(i) The requirement for specialized containment facilities for biothreat research and animal studies, especially aerosol challenges.
BSL-3 facilities are absolutely required to perform research using fully virulent strains of the CDC Category A agents (e.g., B. anthracis, Y. pestis, and F. tularensis). Smallpox requires BSL-4 containment; this virus is held by only two laboratories in the world. Most Category B agents can be used safely under BSL-2 conditions, but generally not in aerosolized form. Ricin, abrin, SEB, epsilon toxin, for example, are 10–1,000 fold more toxic via inhalation when compared to ingestion or transdermal exposure, and therefore, require BSL3 facilities for animal challenge studies (Mantis 2005).
In an effort to enable research on Category A and B priority pathogens, NIAID has established a national network of regional (RBL) and national (NBL) biodefense laboratories (Fig. 1) as part of the Regional Centers of Excellence (RCEs) for Biodefense and Emerging Infectious Diseases. These RCEs provide services and resources to the scientific community in all aspects of biodefense research, including BSL3 containment laboratory access, preclinical development activities, expertise in immunological, proteomic and genomic techniques, high throughput small molecule screening, and aerobiology facilities (Fig. 2). As of 2008, there were six RBLs scattered throughout the US, with seven more under construction. In addition, there were two NBLs nearing completion: one at the University of Texas Medical Branch at Galveston, and one at Boston University Medical Center. The RBLs and NBLs are designed to serve as regional and national centers for collaborative research on pathogenesis, therapeutics, diagnostics, and vaccines.
Fig. 1.
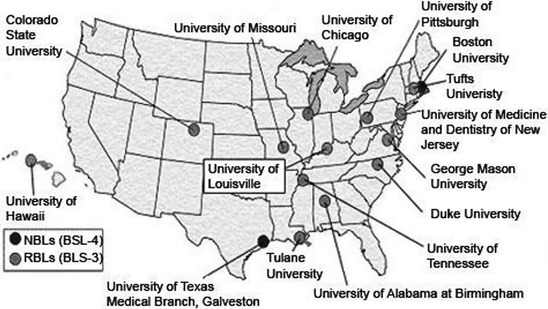
Locations of NIAID-Sponsored Regional and National Biodefense Laboratories in the United States. Image from www3.niaid.nih.gov/LabsAndResources/resources/dmid/NBL_RBL/site.htm
Fig. 2.

Class III biological safety cabinets for biodefense research available at RBLs. The NIAID-sponsored RBLs provide full containment for infectious aerosol challenge studies with primates, as well as instrumentation for specialized bioaerosol characterization studies
(ii) Lack of established animal models, especially involving mucosal challenge. While immunity to many Category A–B agents has been studied for decades, in most cases these models have involved systemic not mucosal exposure. Many of the animal models that have been developed are limited to rodents; there are very few large animal models (rabbit, nonhuman primate) available for efficacy/evaluation studies. Even fewer models exist that appropriately describe the pathophysiology in terms of cellular/molecular mechanisms of the disease process. The lack of relevant animal models represents a major developmental hurdle for testing and comparing biodefense vaccines and therapeutics.
(iii) Limited (or no) access to clinical samples. The natural incidence of infection by most Category A and many Category B select agents is low or so sporadic that obtaining clinical samples for research purposes is not technically feasible. For example, in 2006 there were 20 cases of food borne botulism, 95 cases of tularemia, 17 cases of plague and 1 incident of anthrax (www.cdc.gov/mmWR/PDF/wk/mm5553). With these infection rates, it is virtually impossible to obtain sufficient numbers of clinical samples from individuals at specific time points following infection, or to correlate clinical outcomes with observations made in animal models.
(iv) Vaccine efficacy trials may not be feasible. In general, Phase II-III clinical efficacy trials of candidate vaccines for biothreat agents are not feasible or ethical. In recognition of this issue, the Food and Drug Administration (FDA) has implemented the so-called “animal rule” which enables candidate biodefense vaccines to proceed towards licensure based on efficacy studies in relevant animal models (Crawford 2002; Sullivan et al. 2009). However, according to the FDA, the animal models must mimic the pathogenesis of human disease, and the defined end point(s) of efficacy studies must correlate with the desired effects in humans. This stipulation is somewhat of a “catch-22”, considering that the human response to many select agents is not known (see above).
(v) Sole procurer of biodefense vaccines is US government. Vaccine development is driven in large part by market forces (Levine 2006). In the biodefense realm, the sole agency responsible for the purchase of vaccines is the US government, specifically the Biomedical Advanced Research and Development Authority (BARDA) (Trull et al. 2007). The BARDA budget is largely devoted to end stage acquisition, not development. Therefore, the costs of vaccine development must be covered by private investment or NIAID grants/contracts, which are highly competitive.
(vi) Vaccines must have unusually long shelf lives. It is anticipated that most biodefense vaccines will be administered to limited and specific populations of individuals (e.g. emergency responders, healthcare providers, politicians). The public at large will receive such vaccines only in the event of a local, regional or national health emergency. Therefore, following acquisition by BARDA (see above), most vaccines will simply be stockpiled. From the perspective of vaccine development, this poses significant challenges, as the vaccine formulations must be impervious (possibility over periods of years) to chemical inactivation, protein unfolding, denaturation and aggregation.
Yersinia pestis as a Case Study in Biodefense Vaccine Development
The causative agent of plague, Yersinia pestis, represents a hallmark pathogen for which both mucosal and systemic immunity is essential for protection. The Y. pestis organism is a gram-negative bacillus that can be transmitted by flea-bite or by aerosol. Depending upon the mode of transmission and success of infection, three forms of the disease may manifest: bubonic, septicemic, and/or pneumonic plague. Upon flea-bite, the bacterium is introduced into the bloodstream and eventually seeds the nearest lymph nodes. The bacterium is ingested by nonactivated macrophages that are unable to control the growth of the organism. Inflammation produced in response to bacterial proliferation causes a characteristic swelling, or bubo, the classical feature of bubonic plague. Eventually, bacteria can disseminate throughout the bloodstream leading to septicemic plague and the colonization of additional sites. Pneumonic plague can result from dissemination to the lung alveoli or from the inhalation of aerosolized organisms. Pneumonic plague has a very rapid onset (1–3 days). It is highly contagious and approaches a 100% fatality rate if left untreated. Host immune responses ultimately fail to control the growth and dissemination of the organism. Without early antibiotic treatment, death can result from either pneumonia or endotoxin-induced septicemic shock (Cornelius et al. 2007). Due to the intrinsic virulence of Y. pestis, its ease of transmission by aerosol, and the lack of a vaccine, this bacterium poses a significant threat to biodefense and is classified as a Category A priority pathogen.
The extensive research aimed at producing an effective vaccine against plague has revealed the significant contribution of mucosal immunity in protection against the respiratory form of the disease. It has long been recognized that serum antibodies generated against whole-killed Y. pestis can prevent bubonic and septicemic forms of the disease (Smiley 2008). However, the inability of serum antibody to prevent pneumonic plague led to the hypothesis that local mucosal immunity in the lung may be essential for protection. This has been substantiated in numerous prime/boost immunization studies with recombinant capsule F1 and low calcium response (Lcr) V subunit antigens using murine models of bubonic and pneumonic plague. Parenteral vaccination with rF1-V can protect mice against subcutaneous and aerosol challenge with Y. pestis (Titball and Williamson 2001). In this instance, the protection is attributed to induction of systemic F1-V specific IgG which transudate into the lung to protect against inhaled bacteria (Williamson et al. 1997). The additional presence of antigen-specific IgA in pulmonary fluids may further contribute to protection in the respiratory tract. Survival against aerosolized Y. pestis was enhanced by increasing the nasal boost dosage of rF1-V, and both serum and pulmonary antibody titers to V antigen were the best predictors of outcome (Reed and Martinez 2006). This is consistent with previous studies demonstrating that vaccination with F1 or V alone is sufficient for protection in mice against both bubonic and pneumonic plague, while vaccination with the F1-V combination provides additive protection (Titball and Williamson 2001).
In the context of a biological attack by aerosol, it is likely that systemic immunity is equally important as mucosal immunity in preventing the septicemic stage from developing after inhalation of Y. pestis. Mucosal immunity appears to be essential for preventing pulmonary inflammation and pneumonia, while systemic immunity is required for preventing bacterial dissemination and septicemia. This is evidenced by vaccine studies in mice that report rapid, fulminating disease and endotoxin-induced death in sham-vaccinated animals challenged with aerosolized Y. pestis (Reed and Martinez 2006). In vaccinated animals that survive the same challenge dose, both systemic and mucosal antibody titers correlate with protection. In contrast, vaccinated animals that eventually succumb to the disease display no evidence of bacterial dissemination from the lung but develop lethal pneumonia (Reed and Martinez 2006). This could be explained by the induction of a systemic immune response in the absence of a mucosal response in these animals. The importance of systemic antibody is also exemplified by the demonstration that passive transfer of F1-V mouse antisera can protect recipient normal or SCID mice from bubonic and pneumonic challenge with Y. pestis (Motin et al. 1994; Anderson et al. 1996; Green et al. 1999). However, B-cell deficient mice vaccinated with live Y. pestis are protected against pneumonic plague unless they are additionally depleted of CD4+ and CD8+ T cells, IFN-γ, or TNF-α (Parent et al. 2005). Recent studies with nonhuman primates also suggest that F1-V antibodies alone may not solely correlate with protection against pneumonic plague (Smiley 2008). Thus, induction of cellular immunity may be critical in the development of an effective vaccine against plague.
Due to the likely importance of cellular immunity in preventing pneumonic plague, there is renewed interest in the development of live-attenuated Y. pestis strains as vaccines (Smiley 2008). Whether such bacterial strains could be safely utilized as mucosal vaccine formulations is debatable. Future subunit vaccines for Y. pestis will likely utilize recombinant F1-V as the major antigenic components since no other vaccine candidates have yielded better immunogenicity and protection (Titball and Williamson 2001). However, the incorporation of additional T cell antigens may be required to achieve complete and durable immunity.
Conclusion
A contentious issue within the biomedical community has been the commitment of disproportionate amounts of the US NIH budget to support biodefense research (Trull et al. 2007). Opponents have argued that devotion of monies to pursue diagnostics, therapeutics and vaccines against relatively “obscure” pathogens and toxins has been detrimental to more basic research programs aimed at preventing infectious diseases of immediate global importance. However, biodefense research should significantly impact overall worldwide health in several ways.
First, the development of vaccines, diagnostics, and therapeutics for Category A–C agents, which are generally most infectious/toxic in the respiratory and/or gastrointestinal tract, has required more basic research in mucosal innate and adaptive immunology, which will enhance our understanding of host defense against a variety of mucosal pathogens. Second, the testing of potential vaccines or therapeutics at international field sites where food- and water-borne diseases are endemic is likely to reduce deaths caused by enteric pathogens such as S. dysenteriae 1 and shiga toxin-producing E. coli. Finally, the development of vaccines for HIV, tuberculosis, and other mucosal diseases that currently cause great mortality will benefit from the identification of novel adjuvants (Guy 2007), new particle delivery platforms (Bramwell et al. 2005), and improved live-attenuated oral delivery vehicles (Galen et al. 2009).
In conclusion, the development of vaccines and other countermeasures against the diverse microbial pathogens and toxins that have been deemed potential biothreats by public health organizations is a daunting challenge for the scientific community. Certainly, the development of vaccines against the entire list of biothreat agents is neither necessary nor realistic, but efforts towards this end should reveal new and fundamentally significant insights into innate and adaptive immunity in the mucosae, and they will undoubtedly have beneficial implications for combating infectious diseases as a whole.
Acknowledgments
The authors would like to thank members of their respective laboratories for helpful discussions. NJM and CJR would like to acknowledge Dr. Robert Brey (Soligenix, Inc.) for insight into the reality of vaccine development. This work was supported by NIH grants U54AI057158, U01AI070624, U01AI075509.
Contributor Information
Pamela A. Kozlowski, Email: pkozlo@lsuhsc.edu
N. J. Mantis, Email: nmantis@wadsworth.org
References
- Anderson GW, Jr, Leary SE, Williamson ED, et al. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis. Infect Immun. 1996;64:4580–4585. doi: 10.1128/iai.64.11.4580-4585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artenstein AW. New generation smallpox vaccines: a review of preclinical and clinical data. Rev Med Virol. 2008;18:217–231. doi: 10.1002/rmv.571. [DOI] [PubMed] [Google Scholar]
- Bergdoll MS. Enterotoxins. In: Easmon CSF, Adlam C, editors. Staphylococci and staphylococcal infections, vol 2. New York: Academic Press; 1983. [Google Scholar]
- Bramwell VW, Eyles JE, Oya Alpar H. Particulate delivery systems for biodefense subunit vaccines. Adv Drug Deliv Rev. 2005;57:1247–1265. doi: 10.1016/j.addr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Cornelius C, Quenee L, Anderson D, et al. Protective immunity against plague. Adv Exp Med Biol. 2007;603:415–424. doi: 10.1007/978-0-387-72124-8_38. [DOI] [PubMed] [Google Scholar]
- Crawford LM. New drug and biological drug. Products; evidence needed to demonstrate effectiveness of new drugs when human efficacies studies are not ethical or feasible. Federal Register. 2002;67:37988–37998. [PubMed] [Google Scholar]
- Galen JE, Pasetti MF, Tennant S, et al. Salmonella enterica serovar typhi live vector vaccines finally come of age. Immunol Cell Biol. 2009;87:400–412. doi: 10.1038/icb.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M, Rogers D, Russell P, et al. The SCID/Beige mouse as a model to investigate protection against Yersinia pestis. FEMS Immunol Med Microbiol. 1999;23:107–113. doi: 10.1111/j.1574-695X.1999.tb01229.x. [DOI] [PubMed] [Google Scholar]
- Griffiths GD, Lindsay CD, Allenby AC, et al. Protection against inhalation toxicity of ricin and abrin by immunisation. Hum Exp Toxicol. 1995;14:155–164. doi: 10.1177/096032719501400201. [DOI] [PubMed] [Google Scholar]
- Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007;5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- Henderson DA. The looming threat of bioterrorism. Science. 1999;283:1279–1282. doi: 10.1126/science.283.5406.1279. [DOI] [PubMed] [Google Scholar]
- Kappler J, Kotzin B, Herron L, et al. V beta-specific stimulation of human T cells by staphylococcal toxins. Science. 1989;244:811–813. doi: 10.1126/science.2524876. [DOI] [PubMed] [Google Scholar]
- Kortepeter MG, Cieslak TJ, Eitzen EM. Bioterrorism. J Environ Health. 2001;63:21–24. [PubMed] [Google Scholar]
- Leclerc H, Schwartzbrod L, Dei-Cas E. Microbial agents associated with waterborne diseases. Crit Rev Microbiol. 2002;28:371–409. doi: 10.1080/1040-840291046768. [DOI] [PubMed] [Google Scholar]
- Leppla SH, Robbins JB, Schneerson R, et al. Development of an improved vaccine for anthrax. J Clin Invest. 2002;110:141–144. doi: 10.1172/JCI16204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MM. Enteric infections and the vaccines to counter them: future directions. Vaccine. 2006;24:3865–3873. doi: 10.1016/j.vaccine.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Maki DG. Coming to grips with foodborne infection–peanut butter, peppers, and nationwide salmonella outbreaks. N Engl J Med. 2009;360:949–953. doi: 10.1056/NEJMp0806575. [DOI] [PubMed] [Google Scholar]
- Mantis NJ. Vaccines against the category B toxins: Staphylococcal enterotoxin B, epsilon toxin and ricin. Adv Drug Deliv Rev. 2005;57:1424–1439. doi: 10.1016/j.addr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Mattix ME, Hunt RE, Wilhelmsen CL, et al. Aerosolized staphylococcal enterotoxin B-induced pulmonary lesions in rhesus monkeys (Macaca mulatta) Toxicol Pathol. 1995;23:262–268. doi: 10.1177/019262339502300304. [DOI] [PubMed] [Google Scholar]
- Metzger DW, Bakshi CS, Kirimanjeswara G. Mucosal immunopathogenesis of Francisella tularensis. Ann NY Acad Sci. 2007;1105:266–283. doi: 10.1196/annals.1409.007. [DOI] [PubMed] [Google Scholar]
- Motin VL, Nakajima R, Smirnov GB, et al. Passive immunity to yersiniae mediated by anti-recombinant V antigen and protein A–V antigen fusion peptide. Infect Immun. 1994;62:4192–4201. doi: 10.1128/iai.62.10.4192-4201.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent MA, Berggren KN, Kummer LW, et al. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun. 2005;73:7304–7310. doi: 10.1128/IAI.73.11.7304-7310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WD, Davis NL, Johnston RE, et al. Genetically engineered, live attenuated vaccines for Venezuelan equine encephalitis: testing in animal models. Vaccine. 2003;21:3854–3862. doi: 10.1016/S0264-410X(03)00328-1. [DOI] [PubMed] [Google Scholar]
- Reed DS, Martinez MJ. Respiratory immunity is an important component of protection elicited by subunit vaccination against pneumonic plague. Vaccine. 2006;24:2283–2289. doi: 10.1016/j.vaccine.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Rotz LD, Khan AS, Lillibridge SR, et al. Public health assessment of potential biological terrorism agents. Emerg Infect Dis. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryzhikov AB, Tkacheva NV, Sergeev AN, et al. Venezuelan equine encephalitis virus propagation in the olfactory tract of normal and immunized mice. Biomed Sci. 1991;2:607–614. [PubMed] [Google Scholar]
- Smiley ST. Immune defense against pneumonic plague. Immunol Rev. 2008;225:256–271. doi: 10.1111/j.1600-065X.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel J, Watson J. Intentional terrorist contamination of food and water. In: Lutwick L, Lutwick S, editors. Beyond anthrax: the weaponization of infectious diseases. New York: Springer; 2009. [Google Scholar]
- Sobel J, Khan AS, Swerdlow DL. Threat of a biological terrorist attack on the US food supply: the CDC perspective. Lancet. 2002;359:874–880. doi: 10.1016/S0140-6736(02)07947-3. [DOI] [PubMed] [Google Scholar]
- Sullivan NJ, Martin JE, Graham BS, et al. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat Rev Microbiol. 2009;7:393–400. doi: 10.1038/nrmicro2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titball RW, Williamson ED. Vaccination against bubonic and pneumonic plague. Vaccine. 2001;19:4175–4184. doi: 10.1016/S0264-410X(01)00163-3. [DOI] [PubMed] [Google Scholar]
- Torok TJ, Tauxe RV, Wise RP, et al. A large community outbreak of salmonellosis caused by intentional contamination of restaurant salad bars. JAMA. 1997;278:389–395. doi: 10.1001/jama.1997.03550050051033. [DOI] [PubMed] [Google Scholar]
- Trull MC, du Laney TV, Dibner MD. Turning biodefense dollars into products. Nat Biotechnol. 2007;25:179–184. doi: 10.1038/nbt0207-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng J, Komisar JL, Chen JY, et al. Immunity and responses of circulating leukocytes and lymphocytes in monkeys to aerosolized staphylococcal enterotoxin B. Infect Immun. 1993;61:391–398. doi: 10.1128/iai.61.2.391-398.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Herman A, Pullen AM, et al. The V beta-specific superantigen staphylococcal enterotoxin B: stimulation of mature T cells and clonal deletion in neonatal mice. Cell. 1989;56:27–35. doi: 10.1016/0092-8674(89)90980-X. [DOI] [PubMed] [Google Scholar]
- Wilhelmsen CL, Pitt ML. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet Pathol. 1996;33:296–302. doi: 10.1177/030098589603300306. [DOI] [PubMed] [Google Scholar]
- Williamson ED, Eley SM, Stagg AJ, et al. A sub-unit vaccine elicits IgG in serum, spleen cell cultures and bronchial washings and protects immunized animals against pneumonic plague. Vaccine. 1997;15:1079–1084. doi: 10.1016/S0264-410X(96)00303-9. [DOI] [PubMed] [Google Scholar]
- Yoder JM, Aslam RU, Mantis NJ. Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect Immun. 2007;75:1745–1750. doi: 10.1128/IAI.01528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]