

Abstract
One avenue for inferring the function of a protein is to learn what proteins it may bind to in the cell. Among the various methodologies, one way for doing so is to affinity select peptide ligands from a phage-displayed combinatorial peptide library and then to examine if the proteins that carry such peptide sequences interact with the target protein in the cell. With the protocols described in this chapter, a laboratory with skills in microbiology, molecular biology, and protein biochemistry can readily identify peptides in the library that bind selectively, and with micromolar affinity, to a given target protein on the time scale of 2 months. To illustrate this approach, we use a library of bacteriophage M13 particles, which display 12-mer combinatorial peptides, to affinity select different peptide ligands for two different targets, the SH3 domain of the human Lyn protein tyrosine kinase and a segment of the yeast serine/threonine protein kinase Cbk1. The binding properties of the selected peptide ligands are then dissected by sequence alignment, Kunkel mutagenesis, and alanine scanning. Finally, the peptide ligands can be used to predict cellular interacting proteins and serve as the starting point for drug discovery.
Key words: Affinity selections, Cbk1 protein kinase, Kunkel mutagenesis, Phage ELISA, Sequence LOGO, Src homology 3 (SH3) domain
Introduction
Very often in research projects, there is interest in mapping the protein–protein interactions of a protein of interest as a way of understanding its function in the cell or virus. While a variety of techniques exist for this purpose, such as yeast two-hybrid screening, mass spectrometry, and protein complementation assays, another approach is the use of phage-displayed combinatorial peptide libraries. In such an approach, one takes a purified, recombinant protein and isolates peptide ligands to it through affinity selection (Fig. 1). Interestingly, the phage-displayed peptides bind at “hot spots” for molecular interaction and very often share the same primary structure as short regions within cellular interacting proteins. Several examples where this approach has proven useful include such targets as protein interacting domains [1, 2].
Fig. 1.
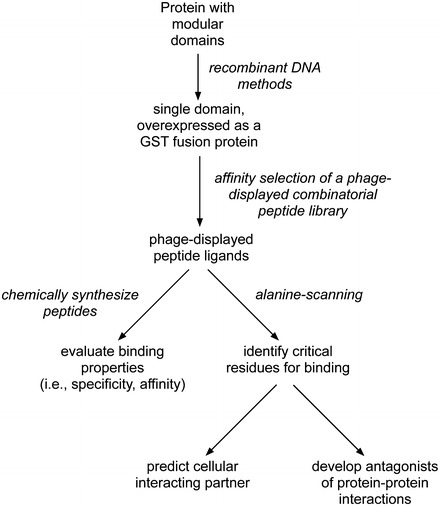
A general workflow diagram for isolating and characterizing the peptide ligands to protein domains or fragments using phage display methods. In principle, coding regions of any protein domain of interest can be converted into recombinant DNA and expressed as a fusion protein to a partner such as glutathione S-transferase (GST). With a soluble protein in hand, one can perform affinity selections with phage-displayed combinatorial peptide libraries to identify its peptide ligands. The binding properties of those ligands can be then further characterized through affinity and specificity measurements. To assess the importance of each residue in the peptide ligand, a mutagenic analysis known as alanine scanning can be performed on the recombinant DNA. With that knowledge, potential interacting partners of the protein of interest can be predicted [14]. Finally, improving affinity and specificity of selected ligands through directed evolution may lead to the development of antagonists, which could be used as inhibitors of specific cellular interaction for proof-of-principle experiments of drug development
To demonstrate the utility of this approach, we describe its application to two targets, human protein tyrosine kinase, Lyn, and the yeast serine/threonine-protein kinase Cbk1. Lyn was first discovered as a viral oncogene [3] and later appreciated to be a proto-oncogene in humans. It is a non-receptor protein tyrosine kinase, which is a member of the Src family of proteins. It has a modular architecture: a Src homology 3 (SH3) domain, a Src homology 2 (SH2) domain, several linker regions, and a catalytic domain that phosphorylate tyrosines in proteins [4]. The SH3 domain plays a role in mediating protein–protein interactions and has been described to bind proline-rich motifs in proteins. The Cbk1 belongs to a large family of NDR/LATS protein kinases, which is conserved across eukaryotes and includes members such as myotonic dystrophy kinase [5]. Cbk1 plays a role in controlling cell separation after cytokinesis, cell integrity, and polarized growth in Saccharomyces cerevisiae [6]. To date, only a few substrates of Cbk1 have been reported. One of them, Ace2, a transcription factor that is activated by Cbk1 via three phosphorylation sites [7], is responsible for transcriptional control of enzymes required for septum degradation after cytokinesis [6]. The other, Ssd1, is an RNA-binding protein that suppresses translation of certain mRNAs and which loses activity when phosphorylated by Cbk1 [8].
We chose these two proteins as targets in parallel affinity selection experiments to illustrate that the same phage-displayed combinatorial peptide library can yield very different peptide ligands to different targets. The Lyn SH3 domain has previously been used in affinity selection experiments, and so it served as a positive control. The Cbk1 protein had not been used previously in affinity selection of combinatorial peptide libraries. We also show that the Kunkel mutagenesis in combination with alanine scanning and ELISA are very simple and expedient methods for evaluating the contribution of certain amino acids in the peptide ligands for binding to their targets.
Materials
Prepare using deionized water (diH2O). Autoclave or filter sterilize and store at room temperature, unless indicated otherwise.
Reagents
Phosphate buffered saline (PBS): 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4.
Phosphate buffered saline containing 0.1 % Tween (PBST).
Wash buffer 1: PBS (store at 4 °C).
Wash buffer 2: PBS containing 0.1 % Tween (store at 4 °C).
Blocking buffer 1: PBS containing 3 % (w/v) bovine serum albumin (BSA) (store at 4 °C).
Blocking buffer 2: PBS containing 3 % (w/v) skim milk (SM) (prepare before use).
Purified glutathione S-transferase (GST) fusion target.
Glutathione S-transferase (GST), purified protein, 1 mg/mL in PBS.
Glutathione Magnetic Beads (Pierce Chemical Co., catalog # 88821).
Combinatorial phage display library (e.g., Ph.D.™ 12-mer Phage Display Peptide Library, New England BioLabs).
Acid elution buffer: 50 mM glycine–HCl: 0.375 g glycine per 100 mL of deionized water (diH2O), pH 2.
Neutralization buffer: 2 M Tris base: 24.22 g Tris base per 100 mL of diH2O, pH 10 adjusted with NaOH.
Escherichia coli strain TG1 (genotype: supE thi-1 D(lac-proAB) hsdD5[F′ traD36 proAB + lacIq lacZDM15]) (Lucigen; see Note 1).
YT medium, 2×: 10 g tryptone, 10 g yeast extract, 5 g NaCl per 1 L of diH2O.
Agar powder.
YT, 2× agar plates (10 cm): 2 × YT medium, agar powder 1.5 % (w/v).
YT, 2× top agar: 2 × YT, agar powder 0.8 % (w/v).
Anti-M13 antibody conjugated to horseradish peroxidase (HRP) (GE Healthcare), diluted 1:5,000 in PBST.
2,2-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS), 10 mg tablets (Pierce Chemical Co.).
Sodium citrate, 50 mM: 10.5 g citrate monohydrate per 1 L of diH2O, pH 4 adjusted with citric acid, filter sterilized (store at 4 °C).
Chromogenic substrate solution: 1 tablet of ABTS, 10 mL of 50 mM sodium citrate, 100 μL of 3 % H2O2 (prepare immediately before use).
Isopropyl-β-d-thiogalactopyranoside (IPTG), 100 mM: 0.238 g IPTG per 10 mL of diH2O, filter sterilized (store at −20 °C).
5-bromo-4-chloro-3-indolyl-beta-d-galactoside (X-Gal), 2 % (w/v): 100 mg X-Gal per 5 mL dimethyl sulfoxide (DMSO), keep in light-impermeable tubes (store at −20 °C) (MP Biomedicals).
A modified M13 bacteriophage vector containing an amber stop codon, as described by Scholle et al. [9] in gene III.
Phage precipitation solution, 24 % polyethylene glycol (PEG) 8000 (w/v), 3 M NaCl: 240 g PEG, 175.3 g NaCl per 1 L of diH2O, filter sterilized.
Escherichia coli strain SS320 (MC1061F′) (genotype: hsdR mcrB araD139 (araABC-leu)7679 lacX74 galUgalK rpsL thi F′[proAB + lacIqlacZ M15 Tn10 (tetr)]) (Lucigen).
T4 polynucleotide kinase (New England BioLabs).
T4 DNA ligase (New England BioLabs).
T7 DNA polymerase (New England BioLabs).
TM buffer (10×), 500 mM Tris–HCl, 100 mM MgCl2: 0.606 g Tris base, 0.246 g MgCl2 per 10 mL of diH2O, pH 7.5 adjusted with HCl, filter sterilized (store at −20 °C).
Dithiothreitol (DTT), 100 mM: 15.45 g of DTT in 100 mL of diH2O, filter sterilized (store at −20 °C).
ATP, 10 mM (New England BioLabs).
dNTP, 10 mM (New England BioLabs).
SOC medium: 20 g tryptone, 5 g yeast extract, 0.5 g NaCl, 2.5 mL of 1 M KCl, 10 mL of 1 M MgCl2, 10 mL of 1 M MgSO4, and 20 mL of 1 M glucose (added after autoclaving the media).
Equipments
Magnetic separator (accommodating 1.5 and 2 mL Eppendorf Tubes).
Rotating shaker (accommodating 1.5 and 2 mL Eppendorf Tubes).
Multichannel pipette 20–200 and 200–1,000 μL.
Filtered tips 20, 200, and 1,000 μL.
Nunc MaxiSorp flat-bottom 96 well plates (Thermo Fisher Scientific, Inc.).
Round bottom 96 DeepWell 2 mL plate, polypropylene, sterilized (Thermo Fisher Scientific, Inc.).
Square top round bottom 24 deep-well 10 mL plate, polypropylene, sterilized (GE Healthcare).
SealPlate® adhesive sealing film for microplate (EXCEL Scientific, Inc.).
AeraSeal™ breathable sealing film for microplates (EXCEL Scientific, Inc.).
Centrifuge with a deep well plate swing-bucket rotor.
General Methods
Concentrations of proteins were determined by measuring the absorbance of samples at 280 nanometer (nm) wavelength, with a NanoDrop ND-1000 UV spectrophotometer (Thermo Fisher Scientific, Inc.).
Washing steps for microtiter plate in enzyme-linked immunosorbent assay (ELISA) were carried on ELx405™ plate washer (BioTek®) using PBST.
ELISA read outs were detected by measuring absorbance at 405 nm on POLARstar OPTIMA microplate reader (BMG Labtech).
Double-stranded DNA samples were isolated with the Wizard® Plus SV Minipreps DNA Purification System (Promega Corp.).
Single-stranded DNA samples were isolated with the QIAprep Spin M13 purification kit (Qiagen).
Covalently closed, circular double-stranded M13 DNA, synthesized via Kunkel mutagenesis [9, 10], was purified with QIAquick PCR Purification Kit (Qiagen).
Concentrations of DNA samples were determined using NanoDrop ND-1000 UV spectrophotometer (Thermo Fisher Scientific, Inc.) and measuring the optical absorbance at 260 nm.
The following steps: phosphorylation of oligonucleotides, annealing to the template, and synthesis of covalently closed circular double-stranded DNA (cccDNA) were performed using DNA Engine Dyad® thermal cycler (Bio-Rad).
Methods
In described methods, affinity selections with phage-displayed combinatorial peptide libraries (Figs. 2 and 3) were employed to identify peptide ligands that bind to human Lyn SH3 domain and yeast Cbk1 protein kinase. In general, once peptide ligands have been identified for a protein target, an interacting motif can be deduced via sequence alignment (Logo, Fig. 4) of the primary structures of the displayed binding peptides. However, to assess the functionality of this motif, amino acid replacements of the critical residues are generally necessary. While this is possible through the chemical synthesis of variant peptides, it is more expedient to use mutagenesis techniques, such as alanine scanning (Fig. 5, [11]).
Fig. 2.
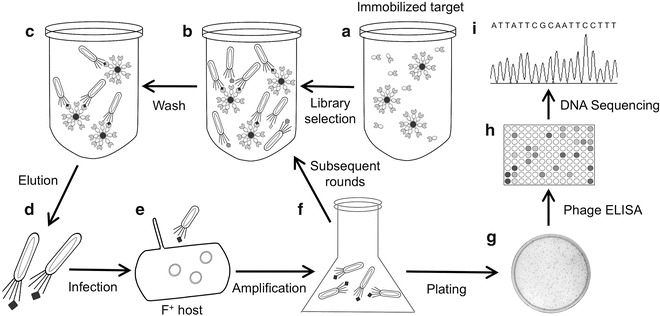
Schematic representation of affinity selection process with phage-displayed combinatorial peptide libraries. (A) The target protein is first immobilized on beads via its fusion partner or biotin–streptavidin interaction (e.g., GST fusion and glutathione beads or biotinylated target and streptavidin-coated magnetic bead). (B ) Next, the phage-displayed library is incubated with the target. (C ) The beads are washed to remove any unbound phage, and (D ) the bound virions are then eluted. (E ) A bacterial host, which carries an F pilus (F+), is infected with the eluted virions and (F ) grown overnight to amplify potential “binders,” which are then used in subsequent rounds of affinity selections. Two to three additional rounds of selections are usually performed to complete the process. (G ) In the final step, the cells, which have been infected with the eluted phage, are plated, and (H ) single clones are tested via a phage enzyme-linked immunosorbent assay (ELISA). (I ) DNA isolated from binding clones is then analyzed to identify the predicted primary structures of the peptide ligands
Fig. 3.
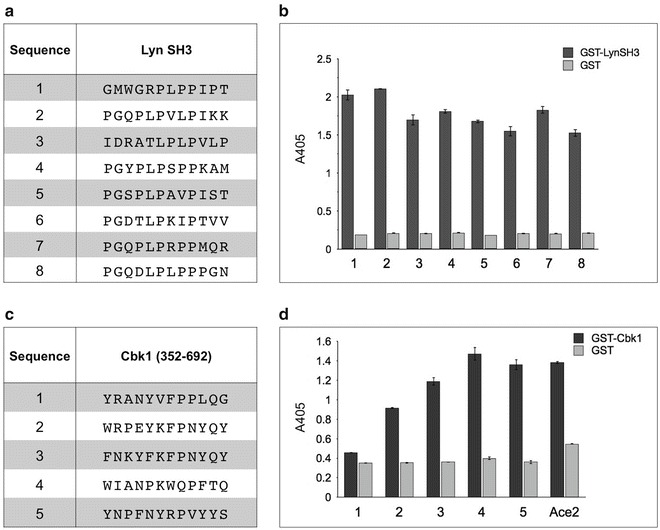
Analysis of peptides isolated against two targets, GST fusions to the SH3 domain of human Lyn tyrosine protein kinase, Lyn SH3 (aa 63–123), and to the yeast NDR/LATS family protein kinase, Cbk1 (aa 352–692). Purified GST-Cbk1 (aa 352–692) protein was a gift from Dr. Eric Weiss. (a, c) All clones were isolated from a 12-mer NNK combinatorial peptide library ANL7 [9], following three rounds of affinity selection, as described in Subheading 3.1. (b, d) Binding of each clone was analyzed via phage ELISA (as described in Subheading 3.2). Wells of microtiter plate were coated with 800 ng of the target. Following blocking with 3 % skim milk in PBS, peptide-displaying phage supernatants were incubated with the target, and binding of the peptides to the target detected via an anti-phage antibody conjugated to horseradish peroxidase (anti-M13-HRP). All peptide ligands were assayed either in duplicate, for Lyn SH3, or triplicate, for Cbk1 (aa 352–692). (d) The Ace2 peptide sequence (unpublished data, Dr. Eric Weiss) was derived from the Ace2 transcription factor, which is a known interacting partner of Cbk1 [6]
Fig. 4.

Sequence logos generated using an online application, WebLogo3, available at http://weblogo.berkeley.edu/. (a) Alignment of Lyn SH3 binding sequences revealed the well-known PxxP motif [15, 16] that can be further classified as x−5 x−4 x−3 x−2 L P x1 x2 P, with preference for proline at position −5 and −2, glycine at position −4, and hydrophobic residues at position +2. (b) Alignment of Cbk1 (aa 352–692) binding sequences (1–3; Fig. 3c) revealed a known Y/FxFP docking motif to Cbk1 [17]. Also, it should be noted that sequence 3 (Fig. 3c) contains the FKFP motif, which is present in Ssd1, a known Cbk1 substrate [8, 17]. Our findings clearly indicate that potential binding partners of certain targets can be deduced via selections with phage-displayed peptide libraries. As only three sequences were used for alignment, any additional amino acid preference analysis could be biased. (c) Further characterization of the isolated Y/FxFP motif showed preference to positively charged residues, R and K, at the “x” position. Other allowed residues at that position included V, M, T, Q, when at least one positively charged residue was observed outside the motif, or C, when a second cysteine was present following the motif. All sequences incorporated in the LOGO (32 isolates, data not shown) were isolated by screening phage-displayed focused library. The library was synthesized via Kunkel mutagenesis (as described in Subheading 3.3.1) by inserting Ssd1 fragment, TEQS xxx FP xxx, with six randomized positions (where x = NNK, N = A, C, T, or G, K = G or T) into the SAM vector [9] at N-terminus of gene III. The oligonucleotide used for library construction was 5′ TCT CGA AGG TCT AGA GAC GTC MNN MNN MNN AGG GAA MNN MNN MNN TGA CTG TTC GGT AGG CCT ACT CGA GGA GTG 3′ (IDT DNA, Coralville, Iowa), where underlined sequence represents nucleotides complementary to the wild-type SAM vector [9]. Ssd1 sequence used for library construction was obtained from Dr. Eric Weiss, NU (unpublished data)
Fig. 5.
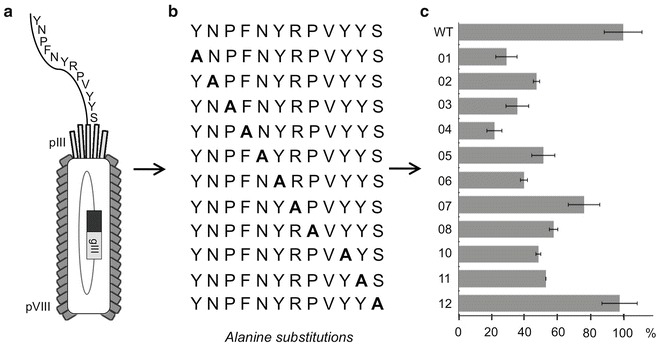
Alanine scanning. (a) The peptide sequence, NH2-YNPFNYRPVYYS-COOH, isolated against GST fusion to Cbk1 (aa 352–692) (Fig. 3c) has been fused to the N-terminus of the minor coat protein, pIII, in the bacteriophage M13 genome, via Kunkel mutagenesis, as described in Subheading 3.3.1. The fusion was generated using the SAM phage vector. The oligonucleotide used for generation of the wild-type clone fusion was 5′ TCT CGA AGG TCT AGA GAC GTC AGA GTA GTA AAC CGG ACG GTA GTT GAA CGG GTT GTA AGG CCT ACT CGA GGA GTG 3′ (IDT DNA, Coralville, Iowa), where underlined sequence represents nucleotides complementary to wild-type SAM vector. The fusion facilitates pentavalent display of the peptide; however, for the simplicity of the figure, only one displayed copy was shown. (b) Alanine substitutions were generated via Kunkel mutagenesis (as described in Subheading 3.3.1) to determine which residues are involved in the interaction with Cbk1 protein. Alanine mutants have been generated using the SAM phage vector and analogous oligonucleotides (as described for the WT clone, a). For unknown technical reasons, clone V9A failed to be generated. (c) All mutant phage-displayed peptides were analyzed via phage ELISA. The results suggested that residues Y1, P3, F4, and Y6 contribute the most to peptide bioreactivity
Screens for Peptide Ligands Using Glutathione Magnetic Beads and GST Fusion Proteins
For the purpose of this selection protocol, the target domain was overexpressed in Escherichia coli in the form of a glutathione S-transferase (GST) fusion protein. Recombinant protein can be prepared via commercially available pGEX vectors (GE Healthcare). Figure 2 presents a schematic diagram of the selection protocol utilizing glutathione-conjugated magnetic beads. To maintain optimum sterility and eliminate carryover, only filtered tips should be used throughout the selection procedure.
Add 20 μL of glutathione magnetic bead slurry (5 μL of settled beads) into 2 mL Eppendorf Tube, and wash twice with 600 μL of ice-cold wash buffer #1 (PBS) on magnetic separator. Remove the supernatant.
Resuspend the beads in 600 μL of wash buffer #1 with 20 μg of the GST fusion protein. Incubate at 4 °C for 1 h on the rotating shaker (see Note 2).
Remove the supernatant. Resuspend in 800 μL of ice-cold blocking buffer #1 (PBS with 3 % BSA), and incubate at 4 °C for at least 3–4 h on the rotating shaker (see Note 3).
Remove the supernatant. Resuspend in 200 μL of blocking buffer #1. Add 50 μg of GST protein to remove potential GST-binding phage clones and 20 library equivalents (e.g., if the library size is 1010 clones and the titer 1013 pfu/mL use 20 μL). Bring the volume to 600 μL with ice-cold wash buffer #1. Incubate at 4 °C for 1 h on the rotating shaker.
Remove the supernatant. Wash three times with 800 μL of ice-cold wash buffer #2 (PBST). Remove supernatant, add 800 μL of ice-cold wash buffer #1, and transfer into a new 2 mL Eppendorf Tube to eliminate any plastic-bound phage.
Repeat the washing step two more times with wash buffer #1. Remove the supernatant.
Elute the phage with 200 μL of elution buffer. Incubate for 10 min at room temperature, and gently mix the content every couple minutes.
Place the tube on magnetic separator, and transfer the eluted phage supernatant into a new 1.5 mL Eppendorf Tube containing 12 μL of neutralization buffer, and mix well.
To amplify the phage for subsequent round of selection, in a culture tube, mix 150 μL of eluted phage with 300 μL of fresh mid-log TG1 cells. Incubate for 10 min at 37 °C without shaking. Add 5 mL of 2 × YT medium, and incubate overnight at 37 °C in a shaking incubator (approximately 240 rpm). Keep remaining phage at 4 °C as a stock of selection-isolated phage clones (see Note 4).
To perform two additional rounds of selection, repeat steps 4 through 9. Spin down the overnight cultures to pellet the cells. Use 200 μL of fresh overnight-amplified phage supernatant (step 4).
(Optional). A quality control (polyclonal) enzyme-linked immunosorbent assay (ELISA) can be performed to verify if the subsequent steps of selection were successful. Use the remaining overnight-amplified phage supernatant from round 2 and 3 and follow steps 1–10 in Subheading 3.2.
Following the final round of selection, perform serial dilutions of the eluted phage in the range from 10−2 to 10−6 and mix 10 μL of each dilution (final range: 10−4–10−8), with 400 μL of fresh mid-log TG1 cells. Incubate for 10 min at 37 °C without shaking (see Note 5).
If blue-white screening can be performed, add 45 μL of 100 mM IPTG and 80 μL of 2 % X-Gal.
Immediately before plating, add 4 mL of 2 × YT top agar (0.8 %), gently swirl, and pour over prewarmed 2 × YT agar plates. Incubate overnight at 37 °C.
To test individual phage clones, aliquot 50 μL of mid-log TG1 cells into each well on a sterile deep-well round bottom polypropylene 96-well plate.
From a plate with well-separated plaques, using 200 μL pipette tips, transfer single phage clones into the wells of the plate. Incubate for 10 min at 37 °C without shaking (see Note 6).
Using a multichannel pipette, add 500 μL of 2 × YT medium and incubate overnight at 37 °C in a shaking incubator (approximately 240 rpm).
Use phage supernatant to perform phage ELISA (Subheading 3.2). Keep at least 50 μL of the supernatant at 4 °C as phage stock.
Conformation of Binding Activity of Isolated Peptides via Phage ELISA
Coat 96-well microtiter plates with the target protein and GST as negative control. Use approximately 250 ng of protein per well (70–100 μL per well), diluted in PBS. Incubate for 1–2 h at room temperature or overnight at 4 °C.
Wash the plates with 200 μL of PBST using automated plate washer or manually with multichannel pipette.
Fill the plates with 250 μL of blocking buffer #2. Incubate for at least 1 h at room temperature or overnight at 4 °C.
Wash the plates with 200 μL of PBST one more time.
Add phage supernatant (same per well volume as the target protein) and incubate 1–2 h at room temperature.
Wash the plates with 200 μL of PBST three more times.
Add anti-M13 HRP-conjugated antibody diluted 1:5,000 in PBST (same per well volume as the target protein), and incubate 30 min to 1 h at room temperature.
Wash the plates with 200 μL of PBST three more times.
Add chromogenic substrate solution (Subheading 2.1, item 21) (same per well volume as the target protein), and incubate for 10–30 min.
Quantify the results by measuring the optical absorbance at 405 nm, using a microtiter plate spectrophotometer.
Characterization of Isolated Peptides
All binding phage clones isolated via ELISA are amplified and their binding regions subsequently identified by DNA sequencing (see Note 7). If the selection generates enough unique isolates, the binding motif of the target can be predicted by sequence alignment known as LOGO plot (Fig. 4). With that knowledge, one can attempt to better define the known interactions and to predict new substrates of the target. To further assess the importance of each residue in the motif, mutagenic analysis known as alanine scanning can be performed (Fig. 5, [11]). In this method, each residue is replaced, one by one, by alanine (or by glycine if originally occupied by alanine). One way to generate all the variants is to incorporate them into M13 phage genome via Kunkel mutagenesis (described in Subheading 3.3.1) [10, 12, 13]. Once the phage-displayed mutant pool is generated, the effect of the substitutions on their binding to the target is evaluated by phage ELISA (described in Subheading 3.2).
Kunkel Mutagenesis
For the purpose of this protocol, a modified M13 phage vector containing an amber stop codon has to be first obtained [9]. In the vector constructed by Scholle et al., an amber stop codon, TAG, has been placed at the N-terminus of the coding region of gene III, following the signal sequence of protein III (pIII). That modification eliminates the need for generation of uracil-containing single-stranded DNA (U-ssDNA) template, using E. coli strain CJ236 (dut − ung −). A mutagenic oligonucleotide, containing both complementary and exogenous DNA fragments, is then annealed to the ssDNA template, with the exogenous fragment looping over the TAG-containing vector region. Once the double-stranded DNA (dsDNA) is synthesized from the ssDNA template, it is electroporated into non-suppressor E. coli strain (e.g., SS320). If the TAG triplet-containing region has not been replaced by the exogenous fragment, the phage will not be propagated as the minor coat protein, pIII, cannot be translated. The wild-type phage can be propagated via the suppressor E. coli strain such as TG1 cells. The amber stop codon is then translated into glutamine. The use of TAG-containing template facilitates nearly 100 % recombination efficiency [9].
To prepare ssDNA template, infect 30 mL of fresh mid-log TG1 cells, grown in 2 × YT medium, with appropriate phage particles (see Note 8). Incubate for 1 h at 37 °C without shaking. Mix the culture gently from time to time to resuspend settling cells. Incubate overnight at 20–25 °C for approximately 20 h in a shaking incubator with a speed not exceeding 250 rpm; recommended is 200 rpm [12].
Prepare a cell-free supernatant by centrifugation and syringe filtration with 0.22 μm filter.
Concentrate the phage by mixing the cell-free phage supernatant with phage precipitation solution (Subheading 2.1, item 25) at 5:1 ration (i.e., 1 mL of solution per 5 mL of phage supernatant), and incubate at room temperature for approximately 20 min.
Pellet the phage by centrifugation and resuspend at room temperature PBS (see Note 9).
To extract the ssDNA template, process the sample using QIAprep Spin M13 kit.
Phosphorylate oligonucleotides (26.4 pmol each, 1.32 μL of 20 μM oligonucleotide) by mixing 5 units of T4 polynucleotide kinase, 1 μL of 10× TM buffer, 1 μL of 10 mM ATP, and 0.5 μL of 100 mM DTT in a final volume to 10 μL, adjusted with diH2O. Incubate for 1 h at 37 °C using thermal cycler.
To anneal to the template, mix phosphorylated oligonucleotides with ssDNA template at the molar ratio of 3:1 (oligonucleotide:template), and add 5 μL of 10× TM buffer in a 50 μL reaction adjusted with diH2O. Incubate the mixture at 90 °C for 2 min, and then decrease the temperature to 25 °C at a rate of 1 °C per min using thermal cycler.
To synthesize the covalently closed circular double-stranded DNA product, add the following to the reaction: 3 μL of 10 mM ATP, 3 μL of 100 mM DTT, 5 μL of dNTPs, 8 Weiss units of T4 DNA ligase, and 8 units of T7 DNA polymerase. Incubate at 20 °C for 30 min using thermal cycler.
Analyze the in vitro synthesized dsDNA by resolving on 1 % agarose gel.
Purify the dsDNA with the QIAquick PCR Purification Kit and elute using diH2O.
In a prechilled 0.2 cm cuvette, mix SS320 electrocompetent cells and 100 ng to 1 μg of dsDNA. Electroporate at 2,400 V using an electroporator. Allow the cells to recover in 1 mL of prewarmed SOC medium, and incubate at 37 °C in a shaking incubator (150 rpm), for 30 min to 1 h.
To isolate single recombinant clones, mix 1 and 10 μL of transformation reaction with 400 μL of fresh mid-log SS320. Incubate for 10 min at 37 °C without shaking.
Immediately before plating, add 4 mL of 2 × YT top agar (0.8 %), gently swirl, and pour over prewarmed 2 × YT agar plates. Incubate overnight at 37 °C.
Amplify phage from single plaques and process the DNA from the virions in the culture supernatant for sequencing (see Note 7).
Alanine Scanning of the Phage-Displayed Peptide
Generate the phage-displayed wild-type “binder” and all desired alanine substitutions following Kunkel mutagenesis protocol, as described in Subheading 3.3.1.
Identify the desired recombinant clones via sequencing (see Note 7).
Perform phage ELISA, as described in Subheading 3.2, to evaluate the effect of the alanine substitutions on the binding to the target.
Notes
Prior to selection procedure, multiple single aliquots (100 μL) of TG1 cells in 16 % glycerol can be prepared and stored at −80 °C. To assure TG1 infectivity, the cells are first streaked on M9 plates (per 1 L, 8 g sodium phosphate heptahydrate, 3 g potassium phosphate monobasic, 0.5 g sodium chloride, 1 g ammonium chloride, 0.24 g magnesium sulfate, 4 g glucose, 11.1 mg calcium chloride, and 15 g agar). Following 48 h incubation at 37 °C, singles colonies are picked and grown till mid-log phase. Aliquots of 100 μL mid-log TG1 containing 16 % glycerol are flash frozen in liquid nitrogen and stored at −80 °C. To prepare fresh culture, single aliquot of TG1 cells is added to 5–10 mL of 2 × YT and grown at 37 °C till mid-log phase.
If low quantities of target are available, a small amount can be used to coat the beads (e.g., 10 μg of protein and 10 μL of slurry, respectively). Also, decreasing the amount of used target can facilitate isolation of higher affinity clones.
For convenience, target-bound beads can be prepared for the entire selection procedure and stored at 4 °C in the blocking buffer #1 (PBS containing 3 % BSA (w/v)) for up to a week.
As long as no contamination is introduced, phage particles can be stored in a culture medium at 4 °C for many years.
Since the phage titer, recovered after final round of selection, depends on many controllable and uncontrollable factors such as stringency of the selections (i.e., number of washes, amount of target used) or type and structure of the target, from our experience, we recommend to cover a wide dilution range from at least 10−4–10−8.
If filtered tips are used for phage transfer, it is convenient to briefly rinse and remove the tips with multichannel pipette.
In order to identify DNA sequences of potential “binders,” the phage is first amplified by infecting 200 μL of TG1 cells (from a mid-log phase culture) with 10 μL of phage supernatant (or single plaque) and incubated overnight in a shaking incubator (240 rpm), in 5 mL final volume. The pelleted cells are then used for isolation of dsDNA that consists of replicative form of phage DNA. Subsequently, the samples are analyzed via sequencing. Remaining phage supernatant can be stored at 4 °C (see Note 4).
In order to isolate high quantity of ssDNA template, large amount of phage particles has to be first collected. Amplification of phage by infecting cells with just a single plaque may result in low quantity of template. Thus, we suggest a two-step preparation process, where the PEG-precipitated concentrated virions are used to infect the fresh mid-log cells.
To remove any polyethylene glycol (PEG) residues from the surface of the tube prior to phage reconstitution, it is recommended to first briefly rinse the side of the tube with PBS, avoiding the phage pellet. Usually 1–3 mL of room temperature PBS is used to resuspend the pelleted virions.
Closing Remarks
One of the major advantages of affinity selection of phage-displayed combinatorial peptide libraries is the potential for rapid discovery of peptide ligands to a target protein. It is relatively straightforward to use homemade or commercially purchased libraries to isolate peptide ligands to a given target and then deduce a binding motif. A peptide with a consensus motif can then be used to study the biology of the target (i.e., predict cellular interacting partner, solve the three-dimensional structure of the peptide and target in a complex, and to inhibit the target in cells). However, when only one or a few peptide ligands are isolated to a target, it is difficult to know a priori what residues in the phage-displayed peptide ligand contribute to binding. While one can explore the binding parameters of the peptide sequence through chemical synthesis of peptides with systematic amino acid replacements across the primary structure, we present an alternative, faster, and easier approach involving Kunkel mutagenesis, alanine scanning, and ELISA. We find that by coupling these three techniques, one can readily determine at first pass many important aspects of the binding interaction.
Acknowledgments
This work was funded by the Chicago Biomedical Consortium, with support from the Searle Funds at the Chicago Community Trust. We would like to thank Mr. Kyle Schneider, Dr. Brian Yeh, and Dr. Eric Weiss (NU) for providing the GST-Cbk1 DNA constructs and purified GST-Cbk1 fusion protein. We are grateful to Dr. Michael Kierny and Dr. Renhua Huang (UIC) for their helpful comments on this manuscript.
Contributor Information
Ratmir Derda, Phone: +1780 492 8370, Email: ratmir@ualberta.ca.
Brian K. Kay, Email: bkay@uic.edu
References
- 1.Deshayes K. Exploring protein - protein interactions using peptide libraries displayed on phage. In: Sidhu SS, editor. Phage display in biotechnology and drug discovery. Boca Raton, FL: CRC Press; 2005. pp. 55–282. [Google Scholar]
- 2.Kokoszka ME, Han Z, Karatan E, Kay BK (2015) Mapping intracellular protein networks. In: Sidhu SS and Geyer CR (eds) Phage display in biotechnology and drug discovery, 2nd (ed.). CRC, New York, NY, pp 229–248
- 3.Yamanashi Y, Fukushige S, Semba K, Sukegawa J, Miyajima N, Matsubara K, Yamamoto T, Toyoshima K. The yes-related cellular gene lyn encodes a possible tyrosine kinase similar to p56lck. Mol Cell Biol. 1987;7:237–243. doi: 10.1128/mcb.7.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784:56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Racki WJ, Bécam AM, Nasr F, Herbert CJ. Cbk1p, a protein similar to the human myotonic dystrophy kinase, is essential for normal morphogenesis in Saccharomyces cerevisiae. EMBO J. 2000;19:4524–4532. doi: 10.1093/emboj/19.17.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss EL, Kurischko C, Zhang C, Shokat K, Drubin DG, Luca FC. The Saccharomyces cerevisiae Mob2p-Cbk1p kinase complex promotes polarized growth and acts with the mitotic exit network to facilitate daughter cell-specific localization of Ace2p transcription factor. J Cell Biol. 2002;158:885–900. doi: 10.1083/jcb.200203094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazanka E, Alexander J, Yeh BJ, Charoenpong P, Lowery DM, Yaffe M, Weiss EL. The NDR/LATS family kinase Cbk1 directly controls transcriptional asymmetry. PLoS Biol. 2008;6:e203. doi: 10.1371/journal.pbio.0060203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen JM, Wanless AG, Seidel CW, Weiss EL. Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr Biol. 2009;19:2114–2120. doi: 10.1016/j.cub.2009.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scholle MD, Kehoe JW, Kay BK. Efficient construction of a large collection of phage-displayed combinatorial peptide libraries. Comb Chem High Throughput Screen. 2005;8:545–551. doi: 10.2174/1386207054867337. [DOI] [PubMed] [Google Scholar]
- 10.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-C. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 12.Huang R, Fang P, Kay BK. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods. 2012;58:10–17. doi: 10.1016/j.ymeth.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kay BK, Kasanov J, Knight S, Kurakin A. Convergent evolution with combinatorial peptides. FEBS Lett. 2000;480:55–62. doi: 10.1016/S0014-5793(00)01778-6. [DOI] [PubMed] [Google Scholar]
- 15.Cesareni G, Panni S, Nardelli G, Castagnoli L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 2002;256:38–44. doi: 10.1016/S0014-5793(01)03307-5. [DOI] [PubMed] [Google Scholar]
- 16.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quilliam LA, Kay BK. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCy, Crk, and Grb2. Proc Natl Acad Sci U S A. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen Ba AN, Yeh BJ, van Dyk D, Davidson AR, Andrews BJ, Weiss EL, Moses AM. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci Signal. 2012;5:rs1. doi: 10.1126/scisignal.2002515. [DOI] [PMC free article] [PubMed] [Google Scholar]