

Abstract
Transgenics are powerful mouse models to understand the biological functions of genes. This chapter gives a short overview of the requirements and considerations in designing a transgene. In addition, potential important choices that have to be made in advance for the successful designing and generating a transgenic mouse model are discussed. Methods for DNA purification for microinjection are also provided in this chapter.
Key words: Transgenic, Gene expression, Promoter, DNA construct
Introduction
The application of transgenesis has increased exponentially since its introduction in the early 1980s and is still one of the most powerful methods to study gene function. As approaches to solve scientific problems became more complex, transgene design evolved alongside. At this moment, the versatility in strategies and applications of transgenic animal models are both staggering and exciting at the same time. An attempt to give a fully comprehensive overview of all variations in transgene design described in the scientific press would be an illusion and exceed the aim of this chapter. Nevertheless, the strategy to generate a transgenic animal model (i.e., design of a transgene) warrants special attention to ensure the highest chances for success. Therefore, this chapter provides a concise overview of the elementary requirements of a transgenic construct and some considerations in transgene design. In addition, a number of aspects are discussed that may influence choices early on in the process of designing and generating a transgenic mouse model.
The first choice in transgene design concerns the donor species (origin) of the transgene DNA and the biological properties of the transgene (see Subheading 1.1). Secondly, transgenes, including elements that control their expression, may either be fully derived from a native genomic locus or be assembled from genomic DNA or copy DNA (cDNA) and (heterologous) regulatory elements (see Subheading 1.2). In addition, a range of regulatory systems offers a certain degree of control over transgene expression (see Subheadings 1.3 and 1.4). Size constraints, inherent to particular cloning systems, may limit the use of native regulatory elements: if a transgene becomes too large for regular plasmid or cosmid-based vectors, or when genetic complementation is desired (e.g., with DNA fragments spanning large genomic deletions), one can switch to systems that allow cloning of very large DNA segments (see Subheading 1.3; 10.1007/978-1-60761-974-1_9). A number of frequently encountered drawbacks are worth paying extra attention to; these are summarized in the notes.
Since the purity of the microinjected DNA is the very first determinant of success, detailed protocols are provided for DNA purification methods of conventionally sized (i.e., at maximum 20 kb) transgenes (see Subheading 2). For the purification of large DNA segments, the reader is referred to 10.1007/978-1-60761-974-1_9.
Origin of the Transgene
In considering transgenic technology, the choice of origin of the transgene, i.e., the species the transgene originates from, is an important one. The origin of a transgene may range from prokaryotes (e.g., reporter genes such as ß-galactosidase) to worms or flies and higher eukaryotes like man. Human DNA is applied most widely to generate transgenes in experimental biomedical research. This choice offers several advantages. First, from a biomedical viewpoint, many known genetic disorders in humans have been mapped and extensively characterized at the molecular level: mutations or deletions have often been identified and mutant alleles are readily available (1–7). This makes it possible to generate transgenic models with “diseased” alleles and study structure–function relationships in the context of common (mouse) alleles. Second, despite structural divergence, most genes have been well conserved between mouse and man. This may become an important issue when, for instance, the biological activity of the (trans)gene product is dependent on protein–protein interactions or homodimerization. Such interactions may no longer occur between proteins originating from species that have diverged too much during evolution. It is possible that the (trans)gene product alters expression of the mouse homolog, either at a transcriptional, translational, or posttranslational (stability) level. Needless to say, these aspects are important, since they may all affect the outcome of experiments. Third, at a practical level, screening for founder mice generated by pronuclear microinjection, may be difficult when the transgene is also derived from the mouse; the same holds for expressional analysis; such analyses would require quantitation at the DNA and mRNA level, respectively. Structural differences between human and mouse genes make it possible to screen for transgeneity relatively easy. Primary sequence differences, often concentrated in noncoding regions (introns), provide a convenient way to discern between the transgene and the endogenous mouse gene by simple restriction endonucleases analysis. Sequence differences are not necessarily confined to noncoding regions but may also occur in coding regions (exons): the mRNA transcribed from the human transgene and that from the endogenous murine counterpart may differ in size and/or nucleotide composition. The latter may become useful in case size similarity hampers straightforward interpretation. If gene products differ at the amino acid level, Western analysis also presents a means to discriminate between endogenous and transgene-related expression, provided antisera are available that specifically detect the (trans)gene product. Alternatively, discrimination of the transgene-encoded protein can be easily accomplished by adding an epitope to the proteins’ amino acid sequence, such as a synthetic Flag-, multiple Histidine (His), protein-derived tags (i.e., Hemagglutinin (HA), c-myc) or virus-based tag (i.e., V5, PY – polyoma). In addition, follow-up studies such as immunoprecipitation, immunohistochemistry, flow cytometry, chromatin immunoprecipitation (ChIP assay) and protein purification, may significantly benefit from such tags, in case the availability of immunological tools against the transgene product is limiting. Importantly, the effect of tag addition should be evaluated in vitro prior to embarking on in vivo experiments, as the addition of small peptide tags may affect protein function in vivo.
In summary, there is a wide choice in the origin of the DNA used to construct a transgene. As holds for the choice of expressional control (see Subheading 1.3), the choice of transgene origin is mostly determined by the aim of the experimental model itself. For biomedical studies, the use of human transgenes may be preferred, if not obligatory. If the study of gene function is the aim and overexpression is the experimental approach, a human gene may simply offer a practical solution for screening purposes. In addition, an alternative approach to generate transgenic mice is discussed in 10.1007/978-1-60761-974-1_10.
Intron-Exon Boundaries
Whereas cDNA-based expression vectors on average work fine in vitro and designing a transgenic construct using cDNA may seem straightforward, cDNA-based transgenes often function in vivo, but expression levels are frequently low, and such transgenes are often silenced. It appears important to preserve the intron–exon boundaries at least to some extent in a transgene. The native intron–exon structure of a gene need not be preserved in its entirety though. If the size of a genomic DNA transgene is too large for conventional cloning techniques, combining cDNA sequences with a few genomic intron–exon boundaries may circumvent this problem (see Fig. 1). Inclusion of only one generic intron in a transgene has been shown to augment transgene expression significantly (8, 9).
Fig. 1.
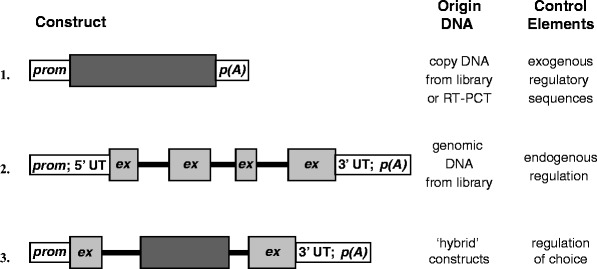
Schematic overview of basic construct design. The basis of the above scheme is the use of eukaryotic coding sequences. The origin of the coding sequences is indicated in the figure. Since regulatory regions are usually not cloned along with cDNA, these have to be provided “separately” and are most often not endogenous (1). Endogenous regulatory elements may be included when the DNA originated from a genomic clone (2). The experimenter has a certain degree of freedom to tailor transgene design to specific requirements ((3); see also Fig. 3). The example depicts a transgene constructed in part of genomic and cDNA sequences. Choice of cDNA and or genomic DNA-based transgenes is discussed in Subheading 1.2. Prom promoter sequences, ex exon, p(A) poly(A+) signal, 5′ and 3′ UT 5′ and 3′ untranslated regions, the thin black lines represent introns (2 and 3).
It appears that the origin of the intron need not necessarily be same as that of the (trans)gene of interest, but may in fact be heterologous or even a hybrid of sequences from different origins. Often, (part of) the first (noncoding) exon attached to a promoter is used in combination with coding sequences within a transgene; care should be taken that transgene translation starts at the intended ATG, and not in upstream heterologous exon sequences (see Subheading 1.3). Moreover, the effect of including introns in a transgene seems independent of its position within the transcriptional unit, although 3′-positioned splice acceptor and donor sequences have been known to result in aberrant splicing products. These observations suggest that recognition and processing by spliceosomes is instrumental in the observed upregulation of transgene expression. In addition, some endogenous introns appear to harbor regulatory elements with structural and functional similarities to enhancers, Locus control regions (LCRs), or Matrix attachment regions (MARs) (see Subheading 2.2) which direct transgene expression in a position-independent or cell type-specific fashion (9–17).
Endogenous Regulatory Elements; Transgene Size
The choice of regulatory elements that drive transgene expression is broad (Fig. 2), and is primarily determined by the aim of the model. However, in all instances, a number of indispensable elements that control gene expression need to be included in a transgene.
Fig. 2.

Regulatory sequences in transgene design. Depending on the nature of the animal model and its specific application, there are numerous choices in as far as the regulation of transgene expression is concerned; such regulatory control may comprise more than a promoter only (see Subheading 1.3). (1) Eukaryotic regulatory sequences may be derived from the gene of interest, i.e., autologous (see Subheading 1.3) or from a different gene. (2) The required expression profile may be systemic or tissue specific (see Subheadings 1.3 and 1.4); alternatively (over)expression in all tissues may be achieved with more general promoters. (3) Finally, specific animal models or embryonic lethality may dictate the need for an inducible expression system (see Subheading 1.4). Regulatory sequences of viral origin are widely used to drive transgene expression and frequently confer tissue-specific expression characteristics to a transgene.
The promoter, the region of DNA at which gene expression is initiated by binding of the RNA polymerase transcriptional machinery, is the most basic and essential element controlling gene expression. The promoter region should comprise a Kozak/ATG sequence at which transgene translation commences (see Subheading 1.2; (18)). If the expression pattern of a transgene needs to parallel that of the endogenous mouse gene, one needs to include native regulatory elements. Regulatory elements can be included that augment transgene expression, such as enhancers, which typically act in an orientation-independent manner. MARs, scaffold attachment regions (SARs), and chromosomal insulators are believed to insulate (trans)gene expression from the influences of surrounding chromatin (15). LCRs confer position-independent and copy number-dependent expressional characteristics to a transgene. In addition, LCRs provide transgene expression at physiological levels, often with a cell lineage-specific enhancer activity. The application of LCRs in transgenesis is discussed in detail elsewhere (reviewed in (15)). The advantage of including such elements in transgenes is obvious: whereas transgenes with “minimal” promoters may become inactive by the insertion into transcriptionally silent chromatin, transgenes carrying, for instance, LCRs will not. However, not all endogenous loci contain such elements and most often their position relative to coding regions within the locus is not known. If faithful reproduction of the endogenous expression profile is required (see also Subheading 1.4), without actual knowledge of the position of regulatory element within a transgene, there is obvious advantage in using large DNA segments as transgenes (see Subheading 1.3).
In the early days of transgenesis, it was often difficult to obtain faithful transgene expression patterns, i.e., which parallel expression of their endogenous counterparts, for a number of reasons (e.g., lack of knowledge in regard to nature and location of regulatory sequences of a locus; size restrictions of cloning systems). The use of a full-length relatively small mammalian gene (i.e., 15–20 kb), including 5′ and 3′ and internal regulatory regions, may yield faithful transgene expression patterns. In such a fortunate situation, not only coding sequences, but also cell lineage-specific and other regulatory elements are located within or close to the intron–exon structure of a locus. However, the exact location of elements that exert transcriptional control over a (trans)gene of interest need not always be known and often there may be many kb removed from the actual transcriptional start site. For a number of applications, like genetic complementation of large deletions and gene therapy, it is imperative to include such regulatory features in a transgene (19–21). Fortunately, when transgenes become too large (i.e., up to 100–150 kb) for “conventional” plasmid-based cloning, a number of modern cloning techniques have overcome this hurdle: one needs to resort to cloning systems employing P1 artificial chromosomes (PAC), yeast artificial chromosomes (YAC), or bacterial artificial chromosomes (BAC) (see 10.1007/978-1-60761-974-1_9). In addition, other cloning methodologies have been described to generate plasmids for biotechnology purposes, such as recombineering (see 10.1007/978-1-60761-974-1_11). In principle, any gene can be cloned into these systems. Exceedingly large YACs (>500 kb) are transferred into embryonic stem cells first and via this route used to generate transgenic mice (see 10.1007/978-1-60761-974-1_9). An obvious and important advantage of using large stretches of genomic DNA is that with these systems the chances of obtaining cell lineage-specific, integration site-independent, and copy number-dependent expression characteristics are greatly improved (22, 23).
Heterologous Regulatory Elements
If overexpression or ectopic expression is required, general type heterologous promoters (e.g., such as widely applied viral promoters) and/or enhancers are widely used. The use of heterologous and autologous (i.e., endogenous to the gene in interest) regulatory elements is often combined (see for example Figs. 1 and 3). The very first transgenic mouse models generated made use of the general-type metallothioneine promoter (pMT) (24, 26–28). This promoter was used to control expression of human, rat, or viral transgenes and, although showing a relatively high level of basal expression, proved to be further inducible with glucocorticoids, heavy metals, or bacterial endotoxin (LPS) (29, 30). The use of heterologous promoters and other regulatory elements has become wide spread, and as indicated before, is determined primarily by the aim of the animal model (see Fig. 2). To ensure global and ubiquitous expression of a particular transgene, general-type promoters, such as those derived from histones, ß-actin, or housekeeping genes (e.g., phosphoglycerate kinase (PGK)), but also viral promoters are often applied. Needless to say, if a heterologous promoter is chosen to drive transgene expression, one should adhere to those promoters that have been proven to function in vivo, or thoroughly test the novel system first. In the latter case, the experimenter should realize that in vitro expression characteristics of novel promoters may be very different than those in vivo. It is therefore strongly recommended to test novel promoters in vivo before use in a transgenic animal model.
Fig. 3.
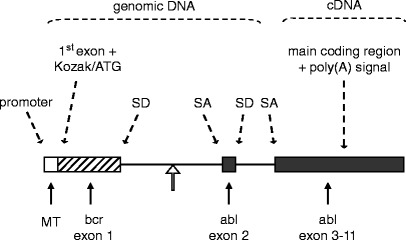
A textbook example of transgene design. The transgene depicted was constructed to study the development of Philadelphia-positive childhood leukemia in a mouse model. The Philadelphia chromosome, the hallmark of this clinical type of leukemia, results from a reciprocal translocation between chromosomes 9 and 22. As a result, the BCR locus and the ABL locus become joined. The genomic ABL locus itself is more than 200 kb in size, the first intron spanning approximately 175 kb. Breakpoints within the ABL locus are known to occur relatively 5′ within the first intron. The transgene harbors a heterologous (see Subheading 1.4) general-type metallothioneine promoter (24). The first BCR exon plus part of the first intron, which is fused (open arrow) to a short part of the first ABL introns and the second ABL exon, provide a splice donor (SD) and a splice acceptor (SA) site and preserve a simple but truthful mammalian intron–exon structure (see Subheading 1.2); an additional SD/SA pair comes from the ABL exon 2/intron 2 and intron 2/exon 3 boundaries. The main body of ABL exons, which spans about 32 kb, was cloned into the transgene as a cDNA segment. In this configuration, the transgene spans a mere 10 kb. Above illustration is adapted after (25).
If tissue-restricted expression patterns are required, specific promoters that confer this selectivity are chosen. In order to determine the minimal requirements for tissue-restricted expression of a particular promoter, it needs to be dissected at the molecular level. Classical promoter studies can be applied in vitro and in vivo to map the elements present within and surrounding a gene of interest. The promoter is tested in vivo by fusing it to reporter genes, such as ß-galactosidase, CAT, luciferase, or GFP. This approach is standardly used to examine spatio-temporal expression patterns and tissue specificity of native promoter sequences in vivo.
To bypass embryonic lethality of a transgene or to study the effects of tissue restricted transgene (over)expression, tissue-specific, inducible, or combinations of these regulatory systems (binary transgenic systems) may be employed. Several binary transgenic systems have been developed, often employing prokaryotic expression control systems (31–38). A number of exciting applications of transgenic technology are described in.
Materials
Transgene Release, DNA Preparation
Qiagen Gel Extraction kit.
Restriction Endonuclease(s) of choice.
Spermidine.
Ethidium bromide (10,000× stock = 5 mg/ml in sterile water).
Electrophoresis buffer: TAE: 0.04 M Tris-acetate, 1 mM EDTA or TBE: 0.09 M Tris-borate, pH 8.0, 2 mM EDTA.
Agarose.
Microinjection buffer: 10 mM Tris–HCl, pH 7.4, 0.1 mM EDTA (filter (0.2 μm) sterilized).
Laboratory Equipment
Thermoblock, adjustable temperature control.
Microcentrifuge capable of 12,000 × g.
Electrophoresis equipment.
Spectrophotometer.
Optional
-
5.
Elutip-D minicolumns (Schleicher & Schuell).
-
6.
3 ml and 5 ml polypropylene syringes.
-
7.
Disposable filters (Schleicher & Schuell; Uniflo 0.45 μ).
-
8.
1 M Tris–HCl pH 7.4.
-
9.
0.5 M EDTA, pH 8.0.
-
10.
5 M Sodium chloride, TC (tissue culture) grade.
-
11.
Elutip buff. I: 20 mM Tris–HCl pH 7.4, 1 mM EDTA, 0.2 M NaCl TC-grade.
-
12.
Elutip buff. II: 20 mM Tris–HCl pH 7.4, 1 mM EDTA, 1.0 M NaCl TC-grade.
-
13.
Ice cold 100% EtOH, 70% EtOH.
Methods
Transgene Release, DNA Preparation
The transgene is released from vector sequences by restriction endonuclease (REN) digestion. Removal of prokaryotic sequences should be carried out as completely as possible (see Note 2). Depending on the materials present on location, several methods can be pursued to extract the linearized transgene from agarose gels. Commercially available agarose gel extraction kit, such Qiagen gel extraction kit, may be used. Although optional, some laboratories apply Elutip-D columns for subsequent purification of DNA for microinjection, with very satisfactory and consistent results (see Subheading 3.1.4; Note 8).
Transgene Release
For regular-sized transgenes, 15 μg transgene DNA is sufficient to purify for microinjection. For practical reasons, we usually isolate up to 50 μg DNA. Vector sequences are typically removed in a relatively large reaction volume (i.e., 400–600 μl), in the presence of spermidine to ensure full digestion at all restriction sites.
Run REN digestion-check (10–15 μl digested DNA) and stop the reaction by adding 2 μl of 0.5 M EDTA. At this stage, the DNA may be mixed with loading buffer and directly loaded onto a preparative agarose gel.
Transgene and vector sequences are separated by electrophoresis. Fragments are excised from agarose gels (see Note 7). DNA isolation is carried out according to manufacturer’s instructions. Elute DNA with microinjection buffer. Optionally perform an additional DNA purification (see Subheading 3.2).
DNA Purification (seeNote6)
Adjust DNA solution to solute concentrations of elutip buffer I: to 100 μl DNA in TE-buffer add 9.0 μl 1 M Tris–HCl, pH 7.4, 0.8 μl 0.5 M EDTA, 20 μl 5 M NaCl, 370 μl dH2O.
Cut off Elutip-D column just below the white matrix and activate column with 1–2 ml elutip buffer II (5 ml syringe).
Wash column with 5 ml elutip buffer I.
Attach filter between syringe and Elutip.
Load column with 400 μl DNA slowly (drop wise).
Wash both filter and column with 2–3 ml buffer I; remove buffer completely (i.e., flush air through) discard filter.
Rinse sterile Eppendorf tube with sterile water to remove dust.
Elute DNA with 400 μl elutip buffer II, very slowly.
Add 1 ml 100% Ethanol; no extra Sodium acetate is added, no coprecipitants are added (see Note 9).
Precipitate at −20°C, overnight, or 15–20 min on dry ice.
Pellet-precipitated DNA at maximum speed for 30 min, 4°C.
Wash pellet twice with 70% Ethanol.
Remove Ethanol with drawn-out Pasteur pipet and air dry briefly.
Dissolve pellet in 50 μl filtered (0.2 μm) special TE buffer for microinjection: 10 mM Tris–HCl, pH 7.4, 0.1 mM EDTA.
Run concentration check; make a microinjection solution of 1–5 DNA ng/μl (keep stock at −20°C). Freezing aliquots of the working solution is recommended (i.e., prevents repeated freezing and thawing).
Notes
Aside from promoter choice, the overall structure of a transgene has considerable influence on its activity. In eukaryotes, foreign DNA sequences (e.g., viral DNA, transgenes) are often methylated and inactivated as part of a host defense system against transcription of potentially harmful genes (39–41). Although the exact mechanisms by which (trans)genes are transcriptionally silenced are not clear on all aspects, the presence of prokaryotic sequences (i.e., plasmid) should be minimized in a transgenic construct (the strategy (i.e., restriction sites) by which the transgene is released from plasmid sequences is a crucial part of the transgene design). In addition, it appears that the presence of bona fide intron–exon structures in a transgene circumvents some of these problems (see Subheading 1.2).
Expression of the transgene may be low in several independent animal lines or not detectable at all. Although this may indicate a flaw in the design of the construct, it is possible, that the (trans)gene product causes embryonic death: transgene (over)expression interferes with normal embryogenesis. In the latter case, the use of different regulatory sequences should be considered (see Subheadings 1.3 and 1.4). To rule out poor construct design, it is strongly recommended to evaluate the biological activity (i.e., basic expression) of a transgene and the size of the transgenic mRNA in cultured cells (e.g., by transient transfection or electroporation and subsequent Northern analysis).
High copy number insertions are often associated with a significant decrease in transgene expression as a result of silencing (42–44), most likely because these are perceived as repeats in the mammalian genome. The presence of regulatory elements that confer position independent and copy number dependent transgene expression, may circumvent this problem. Occasionally, insertion as inverted repeats may also affect transgene activity (29).
An inserted transgene may affect the expression of (nearby) endogenous genes, which influences the phenotype in an unforeseen manner. Transgene insertion may cause haplo-insufficiency, or when integrated in an imprinted locus or in a gene on the X-chromosome, it may even cause a null-mutant phenotype, entirely unrelated to the intended model. To ascribe a certain phenotype to transgene activity, it is therefore imperative to include more than one independent transgenic line in the studies, as is standardly done for ES cell-mediated genetic manipulation in mice.
Although without doubt more involving than conventional transgenesis, it is possible to study the behavior of recessive mutations in mice, by overexpression at supra-physiological levels (18). Alternatively, a (conditional) knockin for the mutation may be generated, or the transgenic lines may be backcrossed to a (conditional) knockout for the endogenous gene (19).
In our experience, the slightest impurities will have a serious impact on the efficiency with which transgenic founders are generated. Often times, residual Ethidium Bromide is a source of trouble. Traces Ethidium Bromide in a DNA preparation is easily detected on an agarose gel from which Ethidium Bromide has been omitted. If DNA is detectable in this fashion, the preparation should be reextracted with Phenol-chloroform-isoamylalcohol a number of times.
Manipulation of DNA, that is to be used to generate transgenic animals via pronuclear injection, should be done with utmost care: shield DNA as much as possible from UV-light (i.e., day light, UV-light box) when in the presence of Ethidium Bromide, since DNA can be damaged and mutated as a result. Working surfaces and equipment (UV tray; scalpels) are clean when isolating fragments from agarose gels.
Since DNA for microinjection needs to be extremely pure, it is not precipitated with coprecipitants. Moreover, some coprecipitants, like Dextran T-500 (Pharmacia), for instance, are toxic to zygotes.
Elutip-D purification is a convenient method to purify DNA for pronuclear injection (see 10.1007/978-1-60761-974-1_2). Yields are not very high (loading capacity column), but the DNA obtained is ultrapure. As an alternative to Elutip-D purification, dialysis against TE-buffer for microinjection is often used. However, care should be taken that the materials used are absolutely free of soap and other contaminants, since these substances have a strongly negative effect on the survival of injected fertilized eggs.
Contributor Information
Marten H. Hofker, Phone: + 31-50-3637932/5777, FAX: + 31-50-3638971, Email: m.h.hofker@med.umcg.nl
Jan van Deursen, Phone: 507-284-2524, FAX: 507- 266-0824, Email: vandeursen.jan@mayo.edu.
References
- 1.Bates GP, Mangiarini L, Mahal A, Davies SW. Transgenic models of Huntington’s disease. Hum Mol Genet. 1997;6:1633–1637. doi: 10.1093/hmg/6.10.1633. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/S0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 3.Gramolini AO, Jasmin BJ. Duchenne muscular dystrophy and the neuromuscular junction: the utrophin link. Bioessays. 1997;19:747–750. doi: 10.1002/bies.950190903. [DOI] [PubMed] [Google Scholar]
- 4.Erickson RP. Mouse models of human genetic disease: which mouse is more like a man? Bioessays. 1996;18:993–998. doi: 10.1002/bies.950181209. [DOI] [PubMed] [Google Scholar]
- 5.Hsiao K, Prusiner SB. Molecular genetics and transgenic model of Gertsmann-Straussler-Scheinker disease. Alzheimer Dis Assoc Disord. 1991;5:155–162. doi: 10.1097/00002093-199100530-00001. [DOI] [PubMed] [Google Scholar]
- 6.Leavitt BR, Wellington CL, Hayden MR. Recent insights into the molecular pathogenesis of Huntington disease. Semin Neurol. 1999;19:385–395. doi: 10.1055/s-2008-1040853. [DOI] [PubMed] [Google Scholar]
- 7.Mehler MF. Brain dystrophin, neurogenetics and mental retardation. Brain Res Brain Res Rev. 2000;32:277–307. doi: 10.1016/S0165-0173(99)00090-9. [DOI] [PubMed] [Google Scholar]
- 8.Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aronow BJ, Silbiger RN, Dusing MR, Stock JL, et al. Functional analysis of the human adenosine deaminase gene thymic regulatory region and its ability to generate position-independent transgene expression. Mol Cell Biol. 1992;12:4170–4185. doi: 10.1128/mcb.12.9.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, et al. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 11.Brooks AR, Nagy BP, Taylor S, Simonet WS, et al. Sequences containing the second-intron enhancer are essential for transcription of the human apolipoprotein B gene in the livers of transgenic mice. Mol Cell Biol. 1994;14:2243–2256. doi: 10.1128/MCB.14.4.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charron G, Guy LG, Bazinet M, Julien JP. Multiple neuron-specific enhancers in the gene coding for the human neurofilament light chain. J Biol Chem. 1995;270:30604–30610. doi: 10.1074/jbc.270.51.30604. [DOI] [PubMed] [Google Scholar]
- 13.Dale TC, Krnacik MJ, Schmidhauser C, Yang CL, et al. High-level expression of the rat whey acidic protein gene is mediated by elements in the promoter and 3′ untranslated region. Mol Cell Biol. 1992;12:905–914. doi: 10.1128/mcb.12.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krnacik MJ, Li S, Liao J, Rosen JM. Position-independent expression of whey acidic protein transgenes. J Biol Chem. 1995;270:11119–11129. doi: 10.1074/jbc.270.19.11119. [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Harju S, Peterson KR. Locus control regions: coming of age at a decade plus. Trends Genet. 1999;15:403–408. doi: 10.1016/S0168-9525(99)01780-1. [DOI] [PubMed] [Google Scholar]
- 16.Liska DJ, Reed MJ, Sage EH, Bornstein P. Cell-specific expression of alpha 1(I) collagen-hGH minigenes in transgenic mice. J Cell Biol. 1994;125:695–704. doi: 10.1083/jcb.125.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman L, Parr B, Lendahl U, Cunningham M, et al. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 18.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 19.Manson AL, Trezise AE, MacVinish LJ, Kasschau KD, et al. Complementation of null CF mice with a human CFTR YAC transgene. EMBO J. 1997;16:4238–4249. doi: 10.1093/emboj/16.14.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun T, Jayatilake D, Afink GB, Ataliotis P, et al. A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFRalpha in prenatal lung growth. Development. 2000;127:4519–4529. doi: 10.1242/dev.127.21.4519. [DOI] [PubMed] [Google Scholar]
- 21.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/S0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 22.Schedl A, Montoliu L, Kelsey G, Schutz G. A yeast artificial chromosome covering the tyrosinase gene confers copy number-dependent expression in transgenic mice. Nature. 1993;362:258–261. doi: 10.1038/362258a0. [DOI] [PubMed] [Google Scholar]
- 23.Vassaux G, Manson AL, Huxley C. Copy number-dependent expression of a YAC-cloned human CFTR gene in a human epithelial cell line. Gene Ther. 1997;4:618–623. doi: 10.1038/sj.gt.3300442. [DOI] [PubMed] [Google Scholar]
- 24.Palmiter RD, Norstedt G, Gelinas RE, Hammer RE, et al. Metallothionein-human GH fusion genes stimulate growth of mice. Science. 1983;222:809–814. doi: 10.1126/science.6356363. [DOI] [PubMed] [Google Scholar]
- 25.Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, et al. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 26.Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, et al. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature. 1982;300:611–615. doi: 10.1038/300611a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brinster RL, Palmiter RD. Transgenic mice containing growth hormone fusion genes. Philos Trans R Soc Lond B Biol Sci. 1984;307:309–312. doi: 10.1098/rstb.1984.0132. [DOI] [PubMed] [Google Scholar]
- 28.Brinster RL, Chen HY, Trumbauer M, Senear AW, et al. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell. 1981;27:223–231. doi: 10.1016/0092-8674(81)90376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmiter RD, Wilkie TM, Chen HY, Brinster RL. Transmission distortion and mosaicism in an unusual transgenic mouse pedigree. Cell. 1984;36:869–877. doi: 10.1016/0092-8674(84)90036-9. [DOI] [PubMed] [Google Scholar]
- 30.Ghoshal K, Jacob ST. Regulation of metallothionein gene expression. Prog Nucleic Acid Res Mol Biol. 2000;66:357–384. doi: 10.1016/S0079-6603(00)66034-8. [DOI] [PubMed] [Google Scholar]
- 31.Fishman GI. Timing is everything in life: conditional transgene expression in the cardiovascular system. Circ Res. 1998;82:837–844. doi: 10.1161/01.res.82.8.837. [DOI] [PubMed] [Google Scholar]
- 32.Chin L, DePinho RA. Flipping the oncogene switch: illumination of tumor maintenance and regression. Trends Genet. 2000;16:147–150. doi: 10.1016/S0168-9525(99)01968-X. [DOI] [PubMed] [Google Scholar]
- 33.Gardner DP, Byrne GW, Ruddle FH, Kappen C. Spatial and temporal regulation of a lacZ reporter transgene in a binary transgenic mouse system. Transgenic Res. 1996;5:37–48. doi: 10.1007/BF01979920. [DOI] [PubMed] [Google Scholar]
- 34.Furth PA, St Onge L, Boger H, Gruss P, et al. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci U S A. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaisser F. Inducible gene expression and gene modification in transgenic mice. J Am Soc Nephrol. 2000;11(Suppl 16):S95–S100. [PubMed] [Google Scholar]
- 36.St-Onge L, Furth PA, Gruss P. Temporal control of the Cre recombinase in transgenic mice by a tetracycline responsive promoter. Nucleic Acids Res. 1996;24:3875–3877. doi: 10.1093/nar/24.19.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shockett P, Difilippantonio M, Hellman N, Schatz DG. A modified tetracycline-regulated system provides autoregulatory, inducible gene expression in cultured cells and transgenic mice. Proc Natl Acad Sci U S A. 1995;92:6522–6526. doi: 10.1073/pnas.92.14.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao B, Magdaleno S, Chua S, Wang YL, et al. Transgenic mouse models for lung cancer. Exp Lung Res. 2000;26:567–579. doi: 10.1080/01902140150216675. [DOI] [PubMed] [Google Scholar]
- 39.Scrable H, Stambrook PJ. A genetic program for deletion of foreign DNA from the mammalian genome. Mutat Res. 1999;429:225–237. doi: 10.1016/S0027-5107(99)00114-1. [DOI] [PubMed] [Google Scholar]
- 40.Doerfler W. Patterns of DNA methylation – evolutionary vestiges of foreign DNA inactivation as a host defense mechanism. A proposal. Biol Chem Hoppe Seyler. 1991;372:557–564. doi: 10.1515/bchm3.1991.372.2.557. [DOI] [PubMed] [Google Scholar]
- 41.Clark AJ, Harold G, Yull FE. Mammalian cDNA and prokaryotic reporter sequences silence adjacent transgenes in transgenic mice. Nucleic Acids Res. 1997;25:1009–1014. doi: 10.1093/nar/25.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin DI, Whitelaw E. The vagaries of variegating transgenes. Bioessays. 1996;18:919–923. doi: 10.1002/bies.950181111. [DOI] [PubMed] [Google Scholar]
- 43.Henikoff S. Conspiracy of silence among repeated transgenes. Bioessays. 1998;20:532–535. doi: 10.1002/(SICI)1521-1878(199807)20:7<532::AID-BIES3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 44.Garrick D, Fiering S, Martin DI, Whitelaw E. Repeat-induced gene silencing in mammals. Nat Genet. 1998;18:56–59. doi: 10.1038/ng0198-56. [DOI] [PubMed] [Google Scholar]