

Abstract
Influenza is often complicated by bacterial pathogens that colonize the nasopharynx and invade the middle ear and/or lung epithelium. Incidence and pathogenicity of influenza-bacterial coinfections are multifactorial processes that involve various pathogenic virulence factors and host responses with distinct site- and strain-specific differences. Animal models and kinetic models have improved our understanding of how influenza viruses interact with their bacterial co-pathogens and the accompanying immune responses. Data from these models indicate that considerable alterations in epithelial surfaces and aberrant immune responses lead to severe inflammation, a key driver of bacterial acquisition and infection severity following influenza. However, further experimental and analytical studies are essential to determining the full mechanistic spectrum of different viral and bacterial strains and species and to finding new ways to prevent and treat influenza-associated bacterial coinfections. Here, we review recent advances regarding transmission and disease potential of influenza-associated bacterial infections and discuss the current gaps in knowledge.
Keywords: Influenza Virus, Respiratory Syncytial Virus, Alveolar Macrophage, Influenza Infection, Influenza Virus Infection
Introduction
Pneumonia is a leading cause of death in the United States and worldwide [(Centers for Disease Control Deaths and Mortality); World Health Organization]. Multiple respiratory viruses and bacteria can cause an infection that leads to severe pneumonia, and it is now recognized that a high proportion of community-acquired pneumonia is caused by coinfections. Pathogens including influenza viruses, parainfluenza viruses, respiratory syncytial virus (RSV), human metapneumovirus (HMPV), (pneumococcus), , and group A streptococcus (S. pyogenes, or GAS), and others, alone or in various combinations, cause millions of ambulatory care visits for pneumonia and thousands of deaths each year in the United States. The resulting economic burden is greater than 17 billion dollars (File and Marrie 2010). In addition, otitis media is the leading reason for visits to a pediatrician (2.4 million visits per year) (Centers for Disease Control Deaths and Mortality, Centers for Disease Control Estimated Burden of Acute Otits Externa)], further increasing the health care cost of these pathogens. Although otitis media has classically been considered a bacterial disease, an increasing amount of evidence suggests that viral infections are a common cause and a great deal of acute otitis media (AOM) results from coinfections with two or more pathogens (Heikkinen 2000).
Of the multiple viruses and bacteria that participate in coinfections of the lung and middle ear, one of the most important is the influenza virus. Although influenza is a major public health threat on its own, bacterial coinfections complicating influenza contribute greatly by exacerbating disease severity. Detailed descriptions of fatal cases date as far back as the eighteenth century (Laennec 1923) indicating that viral-bacterial coinfections have been recognized as being prevalent for hundreds of years. Since then, further study has taken place. The most infamous event was during the “Spanish Flu” pandemic in 1918–1919 where more than 95 % of the 50+ million deaths were complicated by a bacterial coinfection (Morens et al. 2008). Although significant improvements regarding health care have been made, new pathogenic strains emerge and complications from bacterial coinfections continue. Approximately 50–70 % of severe or fatal cases in the 1957 H2N2 and 1968 H3N2 pandemics and nearly one-third of those in the 2009 H1N1 pandemic had bacterial complications (Louria et al. 1959; Weinberger et al. 2011). Furthermore, when a bacterial coinfection was identified, mortality was high despite appropriate antibiotic use in the majority of cases (Domínguez-Cherit 2009; Kumar 2009; Jain et al. 2009; Palacios et al. 2009). Today, it is well recognized that bacterial pneumonia complicates disease initiated by respiratory pathogens like influenza viruses.
Pneumococcus remains the most frequently identified bacterial pathogen associated with influenza infections and the most common cause of community-acquired pneumonia (CAP) despite use of the pneumococcal conjugate vaccine (PCV) in children and adults (Nelson et al. 2008). However, over the last decade, S. aureus dominated influenza-associated childhood fatalities in the US and accounted for ~75 % of deaths from bacterial coinfections. S. aureus has likely become a more common cause of fulminant coinfections due to the emergence in some countries of the methicillin-resistant clonotypes USA300 and USA400 (MRSA). It is unclear why these strains are more likely lead to secondary pneumonia with influenza than other circulating strains. There is currently no vaccine for S. aureus. Group A streptococcus only occasionally complicates viral infections (Chaussee et al. 2011) and, when present, falls behind pneumococcus and S. aureus in prevalence.
There is little systematic surveillance of bacterial coinfections during seasonal influenza, but this continued threat to public health has led to increased research on the co-pathogenesis of pneumonia due to influenza viruses and bacterial pathogens [reviewed in (Short et al. 2012a; Bosch et al. 2013; Metzger and Sun 2013; McCullers 2014)]. This research has significantly improved the current state of knowledge of influenza coinfections through the use of animal models and, more recently, through the use of theoretical models. Key questions regarding transmission, invasion, and pathogenicity remain unanswered. Identifying how a bacterial coinfection renders mild influenza infections fatal is key to effectively combating pneumonia and preparing for future influenza pandemics.
Animal Models to Study Influenza-Bacterial Coinfections
During the 1918 influenza pandemic, the armed forces of several countries made detailed accounts of infectious disease-related illnesses since their efforts during World War I were severely impacted (Brundage and Shanks 2007; Shanks et al. 2010). This led to the first animal studies confirming that bacteria contribute to disease during influenza virus infections by using filtered and unfiltered human sputum (Wherry and Butterfield 1921). These experiments were followed in 1931 by Shope, who conducted controlled experiments in pigs with a swine influenza virus and suis (Shope 1931), and in the 1940s by Francis and Torregrosa, who used a mouse model with the mouse-adapted influenza A/Puerto Rico/8/1934 virus together with pneumococcus, S. aureus, or H. influenzae (Francis and de Torregrosa 1945). Since then, a variety of animal models have been used to study coinfections (Fig. 1).
Fig. 1.

Animal models of influenza-associated bacterial coinfections. Several different animal models have been used to study the effect that influenza viruses have on bacterial transmission and colonization and on invasive diseases, such as acute otitis media and pneumonia (Wherry and Butterfield 1921; Shope 1931; Francis and de Torregrosa 1945; Berendt et al. 1975; Rarey et al. 1987; Hajek et al. 1999; Hirano et al. 1999; McCullers and Rehg 2002; Okamoto et al. 2003; Seki et al. 2004; Peltola et al. 2006; Montgomery et al. 2008; Small et al. 2010; Diavatopoulos et al. 2010b; Lee et al. 2010; Jamieson et al. 2010; McCullers et al. 2010; Loving et al. 2010; Iverson et al. 2011; Kudva et al. 2011; Ayala et al. 2011; Chaussee et al. 2011; Short et al. 2011; Mina et al. 2013; McHugh et al. 2013; Redford et al. 2014)
The sequential viral-bacterial mouse model of pneumonia, which we characterized in detail in 2002, remains the most useful and well-defined system for investigating coinfections, particularly given the lack of comprehensive data from natural infections in humans. In the initial model, sublethal doses of PR8 and of a type 2 laboratory strain of pneumococcus (D39) reproducibly caused severe secondary bacterial pneumonia when given intranasally in BALB/c mice (McCullers and Rehg 2002). The influenza virus infection had to precede the bacterial challenge to observe synergistic disease. An interval of 3–14 days between inoculations with the organisms resulted in the most severe disease, and peak severity occurred when pneumococcus was given 7 days postviral infection. Simultaneous administration of the two pathogens had only additive effects on morbidity, rather than the synergistic effects observed during the sequential infection. This model was later improved by engineering pneumococcal strains to express luciferase, which allows for quantitative bioluminescent imaging to track progression of the infection in live mice (McCullers and Bartmess 2003).
Multiple strains of influenza, including the 2009 H1N1 pandemic virus, can prime mice for secondary pneumonia (Wanzeck et al. 2011), but the doses necessary to have comparable results differ in a strain-dependent fashion. In addition, other viruses (e.g., rhinovirus, adenovirus, coronavirus, parainfluenza virus, HMPV, and RSV) have been used within the same model framework (reviewed in (Bosch et al. 2013). A variety of clinical outcomes can be modeled with different pneumococcal strains, including pneumonia with and without bacteremia, sepsis with secondary seeding of organs leading to pneumonia, otitis media, and sinusitis (Peltola et al. 2005; McCullers et al. 2007; Smith et al. 2007). Furthermore, multiple bacterial species can synergize with influenza viruses to cause disease (Fig. 1).
The mouse model for influenza-bacterial coinfections has several limitations. For example, viruses that replicate well in mice are required to produce robust and reproducible effects, a limitation that affects the certainty with which conclusions can be extrapolated to humans. This is mitigated somewhat by using different species of mice, including the C57BL/6 strain, which behaves similarly to the BALB/c strain (Karlstrom et al. 2011), and the DBA/2 strain, which is highly permissive to a variety of human influenza strains (Alymova et al. 2011). In addition, a ferret model can be used to confirm results found using the mouse model or to answer questions about strain-related differences in pathogenesis since ferrets are susceptible to most human viruses and exhibit a disease course similar to humans (Peltola et al. 2006; McCullers et al. 2010).
Chinchillas and weanling ferrets can also be infected with a variety of pneumococcal strains (Hajek et al. 1999; McCullers et al. 2010), although the disease manifestations do not map precisely to the mouse model. There are limited data with other viruses and bacteria in the ferret model, but unpublished experience from our laboratory has shown that S. aureus will not cause respiratory infections in ferrets even when the animals are preinfected with influenza. Another limitation of the mouse model is the poor transmission potential for respiratory viruses or bacteria between mice, thus requiring the use of alternate models such as neonatal mice (Diavatopoulos et al. 2010a) or ferrets (McCullers et al. 2010) for transmission studies.
Early animal models of AOM utilized the chinchilla due to their large and accessible middle ear spaces (Hajek et al. 1999). These studies demonstrated that the greatest incidence of AOM occurred in animals receiving bacteria 4–8 days following influenza (Hajek et al. 1999), similar to the data concerning timing of pneumonia. More recently, juvenile and infant mouse models have been developed so that diseases of young adults and children, respectively, can be mimicked (McCullers et al. 2007; Diavatopoulos et al. 2010a). Similar models are used to investigate the effects that influenza viruses have on bacterial colonization (Tong et al. 2001; Nakamura et al. 2011).
Studying viral-bacterial interactions in animal models has significantly increased our knowledge about the transmission and pathogenicity of coinfections. However, age, gender, weight, and exposure to anesthesia all contribute to susceptibility to infection in animals in these models, so extreme care must be taken in pathogenesis studies to control all these variables. In addition, studies must carefully select pathogen strains, inoculum sizes, and the sequence and timing of infections since all influence the progression of bacterial pneumonia following influenza.
Effect of Influenza Virus Infection on Pneumococcal Transmission
Influenza viruses readily transmit from person to person via small or large respiratory droplets from a sneeze or cough. Successful transmission and infection typically begins 1 day prior to developing symptoms, which can last up to 7 days in adults and 21 days or more in children (World Health Organization Writing Group et al. 2006). While influenza viruses can spread by large droplets up to six feet away, pneumococcal transmission is thought to require close contact of individuals. Recent evidence, however, suggests that this distance can be lengthened if the individual is virus infected. In fact, epidemiological studies found connections between upper respiratory tract (URT) infections, likely of viral origin, and an increase in bacterial transmission and carriage prevalence (Gwaltney et al. 1975; García-Rodríguez and Fresnadillo Martínez 2002; Pettigrew et al. 2008; Murphy et al. 2009; Ansaldi et al. 2012).
Influenza virus’ impact on pneumococcal transmission was recently illustrated in the ferret model where transmission events and recipient acquisition increased while the distance necessary for successful bacterial acquisition decreased (McCullers et al. 2010). Both bacterial titers and disease severity intensified in the contact ferrets. This relationship was further examined in the infant mouse model, where influenza virus replication and nasopharyngeal bacterial growth were deemed essential for pneumococcal transmission between littermates (Diavatopoulos et al. 2010b; Short et al. 2012b).
These outcomes may not be seen with all influenza-pneumococcal pairings since all observed effects were both viral and bacterial strain dependent. For instance, H3N2 influenza viruses enhance pneumococcal sinusitis and AOM and induce bacterial colonization and disease more frequently than H1N1 or influenza B viruses (Peltola et al. 2006; Short et al. 2013b). Similarly, colonization and AOM development were greater with pneumococcal serotype 19F compared to serotype 7F (McCullers et al. 2010).
Although the precise mechanisms responsible for enhancing the transmission profile that influenza viruses provide pneumococci are currently unknown, it is likely due to an increase in pathogen density and frequency of secretion events (e.g., sneezing and coughing) in the infected individual combined with a decrease in immunity and resistance from natural barriers breaking down in the person who is newly exposed.
Mechanisms of Interaction Between Influenza Viruses and Bacterial Pathogens
During an influenza virus infection, the respiratory tract environment is primed for efficient bacterial invasion. Natural physiological barriers are compromised and a heightened state of inflammation is reached. Numerous factors dictate whether an individual develops a mild or serious infection. The time between exposure to the virus and the bacteria and the pathogen strain and inoculum size all influence influenza coinfection pathogenesis. In addition, many of the virulence factors expressed by each viral and bacterial pathogen act in strain-specific and site-specific manners and can favor different outcomes. The extensive, and growing, list of possible mechanisms (Fig. 2) emphasizes the need to understand how each interacts and how to effectively combat the disease.
Fig. 2.

Influenza-bacterial interaction during coinfections. Numerous alterations of the respiratory epithelium and host immune responses occur during influenza virus infection that predisposes a host to coinfection with bacterial pathogens. As influenza virus infects and kills host cells, epithelial surfaces become exposed and permissive to bacterial attachment. Physical barriers (e.g., mucociliary transport) are damaged, pathogen detection is decreased, anti-microbial peptides (AMPs) are downregulated, receptors are upregulated, virus production is enhanced, bacterial transepithelial migration is permitted, and repair mechanisms are lost. Several host responses are also dampened, altered, or removed. Alveolar macrophages, neutrophils, dendritic cells, and NK cells have altered cytokine profiles and become impaired and/or depleted. These changes result in a heightened inflammatory environment with decreased bacterial surveillance and eradication
Influenza Virus Effects on Physiologic Barriers to Bacterial Invasion
As an influenza virus infection progresses, respiratory tract damage accumulates and primes the damaged and undamaged areas for bacterial colonization due to disrupted mechanical clearance mechanisms and exposed receptors. Airway damage from overexuberant inflammatory responses and disruption of specific immune responses to viral pathogens leave the airways suitable for invasion by bacterial pathogens (reviewed in Short et al. 2012a; Bosch et al. 2013; Metzger and Sun 2013; McCullers 2014).
The host depends on the mucociliary apparatus in the lung and nasal passages to clear invading pathogens, but viral insults can damage the respiratory epithelium and inhibit this mode of removal (Pittet et al. 2010). Receptors [e.g., plate-activating factor receptor (PAFr) (Cundell et al. 1995; Miller et al. 2007)] permissive to attachment of bacterial invaders become exposed in these inflamed areas, as shown by autopsy studies in humans and in vivo infections in mice (Giles and Shuttleworth 1957; Oseasohn et al. 1959; Herzog et al. 1959; Plotkowski et al. 1986, 1993; Louie et al. 2009). Additional adhesion sites in the lung appear as the viral lesions begin to heal. Pneumococcus, H. influenzae, and S. aureus, in particular, all use bacterial adhesions to bind exposed laminin, type I and IV collagen, and fibrin/fibrinogen deposition in areas of incomplete healing (Fainstein et al. 1980). Injured or differentiating cells also provide new sites on apical receptors [e.g., asialylated glycans or integrins) for both S. aureus and Pseudomonas aeruginosa (reviewed in Puchelle et al. 2006).
This increased attachment within the lung, trachea, and nasopharyngeal surfaces may be mediated, at least in part, by viral neuraminidase (NA) activity (Hirano et al. 1999; Peltola et al. 2005), which facilitates bacterial adherence by exposing host cell receptors and providing decoy receptors when sialylated mucins are disrupted. Some bacteria, like pneumococcus, use their own NAs to access receptors and cleave sialic acids to avoid host defenses and prevent mucociliary clearance, replacing, or complementing antecedent viral infections (Camara et al. 1991).
Although decreased mucociliary transport impacts bacterial access to the middle ear, receptor-mediated mechanisms may not be as relevant in AOM. In neonatal mice infected with influenza virus, bacteria can be seen localizing to inflammatory infiltrates, rather than to the epithelium (Short et al. 2011), suggesting that different mechanisms are driving the enhanced bacterial replication. It has been hypothesized that viral-mediated destruction still has a role, but in the context of nutrient availability rather than receptor upregulation (Short et al. 2011).
The influenza glycoprotein hemagglutinin (HA), which binds sialic acid residues on host cell surfaces and aids viral internalization, has an indirect effect on influenza-pneumococcal synergy within the middle ear (Short et al. 2013b). The HA specificity is sufficient to produce differential viral replication and bacterial localization. Here, H3 viruses have higher replicative ability than H1 viruses, even across various NAs, but the effect is site specific and does not depend on cell tropisms (Short et al. 2013a). It is likely that similar mechanisms dictate each type of infection with both the HA and NA having specific roles, but the interactions remain complex. It does, however, help to explain the differential outcomes that different influenza strains have on pneumococcal coinfection.
Bacterial Invasion of the Nasopharynx and Middle Ear
Various bacterial species frequently colonize the nasopharynx and reach a balance with the mucosal immune response such that they are not harmful to the host. In this state, bacteria exist in either biofilms or move between intracellular and extracellular states. Most often, colonizing strains remain in the upper airways since movement to the lower respiratory tract is inhibited by physical barriers (e.g., mucociliary mechanisms) and by immune responses (e.g., resident immune cells, complement, and mucosal antibodies). In this manner, the breakdown of physical barriers and disruption of host responses can result in bacteria emerging from biofilms (Marks et al. 2013).
Most invasive infections and pneumonia occur within a short period of time after a new strain is acquired from the environment rather than from a long-term carriage isolate disseminating to other sites in the body. This is likely due to colonizing strains being limited by systemic immunity, such as pathogen-specific serum IgG, which prevents successful invasion of the lower airways but may tolerate carriage [reviewed in (McCullers et al. 2010)]. In the mouse model, bacteria are delivered directly to the lung, which mimics a direct-inhalation scenario. It is unclear whether this scenario is physiologically relevant in humans, or whether some period of colonization of the nasopharynx must occur prior to invasion and dissemination [reviewed in (McCullers 2014)]. The inoculum size and volume, and the length of anesthesia all influence how much bacteria reach the lower airways and thus the disease model being studied. Both the ferret and mouse AOM models, on the other hand, use colonization as a prerequisite for pneumonia or AOM, so a more natural infection for this site can be instigated.
Influenza viruses and live-attenuated influenza vaccines (LAIV) can result in prolonged bacterial colonization and enhanced bacterial replication within the nasopharynx in both mice and ferrets (Peltola et al. 2006; Nakamura et al. 2011; Short et al. 2012b; Mina et al. 2013). This may be mediated by type I interferon responses and bacterial toxins, such as pneumococcal pneumolysin (Nakamura et al. 2011). Nasopharyngeal colonization can result in bacterial migration to the middle ear via the Eustachian tube. In mice AOM models, animals become colonized with pneumococcus within 72 h after inoculation and can experience recurrent episodes when virus infected (McCullers et al. 2007). Of those that developed AOM (~70 %), resolution occurred within 48 h but colonization persisted for nearly 30 days (McCullers et al. 2007). In the chinchilla model, animals infected with influenza virus experience negative middle ear pressure and eardrum inflammation associated with epithelial damage and cellular and mucosal debris accumulation in the Eustachian tube (Giebink et al. 1987). Similarly, both virus-mediated inflammation and hearing loss are observed in neonatal mice and ferrets (Rarey et al. 1987; Short et al. 2011). However, in contrast to the findings in chinchillas, minimal Eustachian tube damage is observed and bacterial localization specificity suggests that invasion techniques observed in other models are unlikely to be relevant to bacterial AOM (Short et al. 2011). Nevertheless, the increased pathology may support bacterial replication in the middle ear.
Bacterial Invasion of the Lung
If bacteria are successful in migrating to the lungs, a combination of increasing bacterial burden and an accompanying, intense inflammatory response may result in the host developing pneumonia. The early stages of pneumonia are marked by capillary congestion and fluid in the alveolar regions, which provides a medium where pneumococci can readily grow [reviewed in (McCullers 2001)]. As blood vessels become permeable, inflammatory cells are allowed to enter the lung, receptors become upregulated and bacteria easily adhere to, invade, and kill epithelial cells. The combined effects result in significant inflammation, a hallmark of pneumonia.
During coinfections, the host is in a relative state of immune dysregulation with heightened inflammatory and anti-inflammatory responses [reviewed in (Short et al. 2012a; Bosch et al. 2013; Metzger and Sun 2013; McCullers 2014)] likely due to expression of various pathogenic factors. Bacterial cytotoxins, like the pneumococcal pneumolysin (Tuomanen et al. 1995; Kadioglu et al. 2008), S. aureus panton-valentine leukocidin (PVL) (Niemann et al. 2012) and B. pertussis toxin (PT) (Ayala et al. 2011), are known to influence host inflammation and may work in concert with viral cytotoxins. These bacterial factors may intensify the cell death and inflammatory signaling resulting from pores formed by the influenza cytotoxic protein, PB1-F2 (Chen et al. 2001).
The PB1-F2 protein of some influenza viruses increases pathologic effects by causing cell death, increasing viral replication, and altering inflammatory responses to primary viral infections and to bacterial coinfections (Conenello et al. 2007; McAuley et al. 2007, 2010a, b; Smith et al. 2011a, 2013). PB1-F2 can act in a proapoptotic fashion due to its mitochondrial targeting sequence and ability to form pores when interacting with membrane-based proteins (Chen et al. 2001; Gibbs et al. 2003; Chanturiya et al. 2004; Zamarin et al. 2005; Danishuddin et al. 2010; McAuley et al. 2010a). This likely results in the death of epithelial cells and immune cells, which may balance the high replicative ability and support rapid spread through cell monolayers thereby contributing to virulence in vivo (Zamarin et al. 2006; McAuley et al. 2010a, b; Smith et al. 2011a; Varga et al. 2011, 2012). Kinetic analyses suggest that this mechanism impacts viral loads during the later stages of the influenza infection, but is overshadowed by more prominent mechanisms during secondary bacterial infections (Smith et al. 2011a, 2013).
These cellular effects have been mapped to a specific set of amino acids in the C-terminal end of the protein, which are found in most of the early twentieth century H1N1 strains (McAuley et al. 2010a). Although rare (Hai et al. 2010), a serine at position 66 (i.e., ‘66S polymorphism’) impacts virulence with highly pathogenic strains with full-length PB1-F2s (e.g., 1918 H1N1, H5N1) but not less pathogenic strains with truncated PB1-F2s (e.g., 2009 H1N1) (Conenello et al. 2007, 2011; Hai et al. 2010; Varga et al. 2011, 2012). The 66S polymorphism facilitates binding of PB1-F2 to the mitochondrial antiviral-signaling (MAVS) protein adaptor protein and subsequent inhibition of interferon production (Varga et al. 2011, 2012). As a result, viral virulence in primary infection and secondary bacterial infection models is severely exacerbated.
The most relevant PB1-F2 mechanism may be its ability to modulate the immune response during influenza infections and coinfections. The high proinflammatory activity of PB1-F2 intensifies disease in animal models, particularly with respect to induction and severity of bacterial coinfections (McAuley et al. 2007, 2010a; Alymova et al. 2011; Weeks-Gorospe et al. 2012), and is marked by a large influx of immune cells and cytokine storm (Conenello et al. 2007; McAuley et al. 2007, 2010a). Pathogenic PB1-F2s, such as that from the 1918 pandemic strain, elevate neutrophils and macrophages and contribute to the pathologic tissue destruction observed during bacterial coinfections (McAuley et al. 2007). This is likely due to regulation of the type I interferon response (le Goffic et al. 2010; Conenello et al. 2011; Varga et al. 2011, 2012) and apoptotic monocytes infected with influenza (Chen et al. 2001; Gibbs et al. 2003; Zamarin et al. 2005). Specific molecular signatures that facilitate this inflammatory environment have been identified (McAuley et al. 2007, 2010b). Amino acids 62L, 75R, 79R, and 82L in the C-terminal portion of PB1-F2 of select strains are positively associated with inflammation and hypercytokinemia in infected animals and negatively correlated with survival. The precise mechanism and contribution of these signatures singly or in combination is unclear.
Pathogenicity of the 1918 H1N1 pandemic strain was likely impacted by possession all four inflammatory signatures together with the 66S polymorphism. While the 1957 H2N2 and 1968 H3N2 pandemic strains also had all four amino acids, both lacked the 66S polymorphism. Subsequent circulating strains became truncated in the H1N1 lineage around 1948 (McAuley et al. 2010a) and mutations in the H3N2 lineage resulted in loss of the inflammatory signatures by the 1980s. In fact, an antibacterial phenotype emerged in the H3N2 lineage such that viruses and invading bacteria compete via PB1-F2 expression rather than synergize (Alymova et al. 2011; Weeks-Gorospe et al. 2012). The expression of these signatures and the length of PB1-F2 proteins vary widely within influenza viruses. In general, viruses with only the 66S signature or a full set of the polymorphisms strongly support bacterial pathogens, while truncation results in an intermediate phenotype (Zell et al. 2007) and a single substitution at position 82 confers anti-inflammatory properties (Weeks-Gorospe et al. 2012). The range of phenotypes highlights the intricate and complicated nature of influenza infections and coinfections and links specific molecular signatures to pathogenicity. It is important to note that each of these effects was demonstrated in animal models using specific viruses [e.g., H3N2 (Alymova et al. 2011), swine H1N1, H1N2, H3N2 (Weeks-Gorospe et al. 2012)]. The H5N1 viruses are of particular interest for further study since they have little diversity in their PB1-F2, are typically full length, and are more likely to possess the full inflammatory panel with the exception of the 66S polymorphism (Smith and McCullers 2013).
Influenza Virus Effects on Host Immune Responses
A key prerequisite for bacterial invasion into respiratory epithelium is the induction of inflammation. Several immune responses are activated and act to control bacterial pathogens that invade the lung (Joyce et al. 2009; Koppe et al. 2012). A robust initial response is sufficient to immobilize bacterial invaders before full establishment and uncontrolled growth puts the host in a harmful inflammatory state. The degree of attack and the initial replicative ability within mice are dose-dependent and occur in the lung only when alveolar macrophages become overwhelmed with bacteria (Smith et al. 2011b).
For small inocula of bacteria, resident macrophages provide the first line of defense and result in rapid elimination of bacterial pathogens while maintaining homeostasis, which is represented by a low inflammatory state (Knapp et al. 2003; Dockrell et al. 2003 Smith et al. 2003, 2011b). However, given a large invasion or a compromised host state (e.g., influenza virus infected), bacterial outgrowth occurs and an inflammatory response is launched. Neutrophils appear first and are followed shortly by inflammatory macrophages (Jonsson et al. 1985; Fillion et al. 2001; Knapp et al. 2003). The inflammatory influx in the lungs results from bacterial recognition by antigen presenting cells (APCs), and subsequent cytokine and chemokine production. Bacterial phagocytosis by these cells is only efficient if ample complement proteins are available to opsonize the pathogen or if type-specific antibody is made available by B-cells.
Respiratory viruses compromise many aspects of the early detection and response to bacterial pathogens like pneumococcus or S. aureus [reviewed in (Short et al. 2012a; Bosch et al. 2013; Metzger and Sun 2013; McCullers 2014)]. Viruses and bacteria also activate many of the same cytokines, inflammatory cells, and pattern recognition receptors (e.g., TLR4) that can synergize during coinfections and generate inflammation (Navarini et al. 2006; Joyce et al. 2009; Karlstrom et al. 2011; Kuri et al. 2013). Interference of immune responses occurs through various manners, such as by viral expression of multifunctional proteins like the influenza virus NS-1 (Hale et al. 2008) and PB1-F2 (McAuley et al. 2007, 2010a). Depending on the stage of influenza, the innate, cellular, and anergic responses may differentially synergize.
Inflammation in Pneumonia
Since phagocytic cells are critical in creating a bactericidal environment, it is not surprising that these cells are impacted by viral and bacterial mechanisms when secondary infections occur. The activity of neutrophils and macrophages is dampened along with their cytokine production as natural killer (NK) cells become impaired during influenza virus infection and undergo additional suppression during coinfections as a result of heightened TNF-α expression (Small et al. 2010). Further functional suppression of these cells occurs when type I interferons (IFN-α,β) place epithelial cells in antiviral states and alter their chemotactic functions (Joyce et al. 2009; Shahangian et al. 2009; le Goffic et al. 2010; Conenello et al. 2011; Nakamura et al. 2011; Tian et al. 2012; Li et al. 2012). For instance, neutrophil chemoattractants KC and MIP-2 (Shahangian et al. 2009) and macrophage chemoattractant CCL2 (Nakamura et al. 2011) all become downregulated, which inhibits recruitment of immune cells leading to inefficient bacterial clearance. IFN-α,β may also decrease Th-17 cytokines IL-17, IL-22, and IL-23 during S. aureus coinfection, which increases inflammation and decreases viral and bacterial clearance (Kudva et al. 2011).
Production of interferon-γ increases during influenza resolution and can downregulate bacterial scavenger receptors (e.g., MARCO) on macrophages leaving phagocytic cells suppressed and cytokine profiles altered (Didierlaurent et al. 2008; Sun and Metzger 2008). Additional proinflammatory [e.g., IL-1β, TNF-α, IL-6, and IL-12 (Seki et al. 2004; Smith et al. 2007; Shahangian et al. 2009; Nakamura et al. 2011; McHugh et al. 2013)] and anti-inflammatory cytokines [e.g., IL-10 (van der Sluijs et al. 2004)] become inflated and further compound downstream events like macrophage and neutrophil recruitment and dendritic cell function during influenza-pneumococcal coinfection (Sun and Metzger 2008; Shahangian et al. 2009; Wu et al. 2011; Nakamura et al. 2011; Kuri et al. 2013). Even viral and bacterial pathogens themselves can induce apoptosis of phagocytic cells (Colamussi et al. 1999; Engelich et al. 2001; Kobayashi et al. 2003, 2010; McNamee and Harmsen 2006), thus leaving the infected areas severely damaged.
Many of these responses require time to activate, thus the susceptibility of a host to bacterial pathogens following influenza virus infection indicates an effect that may be fully realized during viral preinfection. Indeed, novel analyses of coinfection kinetics identified and detailed the dominant mechanism driving influenza-pneumococcal synergy as a direct viral-dependent reduction in bacterial phagocytosis by alveolar macrophages (Smith et al. 2013). We predicted that this phagocytosis is reduced by 85–90 % at day 7 of the influenza virus infection. We later confirmed that this was a major driver of influenza-pneumococcal synergy with a mouse model by labeling and tracking these cells before and during influenza infections (Ghoneim et al. 2013). Our experiments showed that the resident macrophage population declines as influenza progresses, suggesting that influenza virus directly depletes these cells rather than simply reducing their function. Furthermore, bacterial outgrowth correlated to the level of depletion, which offers new insight into why the timing of bacterial infection has a profound impact on disease outcome (Ghoneim et al. 2013).
As the vigorous antiviral inflammatory response begins to subside, a new state of innate immune activation that may alter responsiveness to new pathogenic insults is reached. The lung becomes repopulated with resident alveolar macrophages as recruited macrophages proliferate and differentiate. In an attempt to return the lung to homeostasis, wound-healing processes coordinate an anti-inflammatory response characterized by IL-10 (van der Sluijs et al. 2004; Hussell and Cavanagh 2009) and suppress pathogen recognition systems [reviewed in (Metzger and Sun 2013)].
During the recovery phase, the host becomes immunologically desensitized both locally and systemically (van der Sluijs et al. 2004; Didierlaurent et al. 2008), which can last for several weeks and prolongs the opportunity for bacterial invasion. The degree and length of this suppression is viral strain-dependent (Ludewick et al. 2011), and occurs through diverse mechanisms. For instance, alveolar macrophages with high expression of homeostatic moieties such as CD200R, a regulatory anti-inflammatory ligand (Barclay et al. 2002; Minas and Liversidge 2006; Snelgrove et al. 2008; Jiang-Shieh et al. 2010), become desensitized when expression of CD200 on apoptotic immune cells increases and open the airways to bacterial invasion (Goulding et al. 2011). In conjunction, absence of CD200R in mice inhibits bacterial outgrowth and prevents migration of bacteria to exogenous sites, such as the blood, in influenza-infected mice (Goulding et al. 2011). Elevated glucocorticoid levels also cause sustained immunosuppression, as was demonstrated in a model of Listeria monocytogenes coinfection (Jamieson et al. 2010).
Inflammation in Acute Otitis Media
The inflammatory nature of influenza-pneumococcal coinfections extends to middle ear invasions. Middle ear inflammation, regardless of pathogenic origin, is sufficient to induce AOM. Influenza viruses can initiate this inflammation (Abramson et al. 1981, 1982; Short et al. 2013b), cause hearing loss and instigate bacterial growth within the ear cavity in a viral strain-dependent, but cell tropism independent, manner (Short et al. 2013a).
Inflammation in AOM is characterized by an influx of neutrophils and expression of key proinflammatory genes (i.e., pro-IL-1β, IL-1α, and CXCL2) (Abramson et al. 1981, 1982; Short et al. 2011, 2013b). Chinchilla studies indicate that immunosuppression, rather than inflammation, is the key mechanism contributing to enhanced pneumococcal replication since influenza viruses can inhibit neutrophils and render clearance ineffective (Abramson et al. 1981, 1982). However, it has also been hypothesized that the enhanced bacterial growth results from nutrients becoming available in areas damaged by this response (Short et al. 2013b). More experiments are clearly necessary to elucidate the underlying relationship between viral replication and inflammation in AOM.
Bacterial Effects on Influenza Virus Clearance
The mechanisms discussed thus far have detailed how the virus affects host responses to invading bacteria. The relationship is somewhat complementary, however, since rebounds in viral load and reduced viral clearance are consistently observed in animal models (McCullers and Rehg 2002; Iverson et al. 2011; Weeks-Gorospe et al. 2012; Smith et al. 2013). The rapidity of the viral reply suggests a fast-acting mechanism, which may occur through direct interactions of the two pathogens, bacterial interference with antiviral immunity, or virulence factors synergizing (Smith et al. 2013). Kinetic studies suggest that pneumococci directly interact with influenza-infected epithelial cells to cause a sudden release of virus (Smith et al. 2013), but experimental studies have not been crafted to confirm this prediction.
The precise effects of bacterial virulence factors are likely type specific as S. aureus may be capable of cleaving influenza hemaggluttinin to enhance invasion of host cells and thus impact viral load (Tashiro et al. 1987). It is feasible that additional, although unknown, bacterial virulence factors have immune-modulatory effects and could interfere with viral clearance, such as T-cell-mediated infected cell clearance or other innate immune components. Determining if bacterial gene products from various species have similar effects during infection and, if so, how they complement viral factors is an important, but largely unexplored, area. Another interesting area for investigation in animal models is the idea that the respiratory and gastrointestinal microbiome affects the development of antiviral responses to pathogens (Ichinohe et al. 2011; Licciardi et al. 2012; Abt et al. 2012; Wang et al. 2013). Understanding how commensal species influence host immune status may help explain the heterogeneity in responses to pathogenic invasions.
Treatment and Prevention
The outcome of influenza virus coinfection is often severe despite appropriate vaccination and treatment. In addition, antiviral and antimicrobial resistance is increasing (Musher et al. 2002; Levy and Marshall 2004; Hayden 2006) and many treatment options have the potential to cause adverse effects on the host (McCullers and English 2008; Karlstrom et al. 2011). With the high prevalence of viral-bacterial coinfections in some situations, discovering treatment options that can prevent or treat both the influenza virus infection and the secondary bacterial infection are of utmost importance.
Antiviral Treatment
Several antiviral drugs targeted against various influenza virus components have been or are currently being developed [reviewed in (Hayden 2013)]. Neuraminidase inhibitors (NAIs) are one class of drugs that have become the pillar of influenza treatment in recent years. NAIs act to block virus from budding out of infected cells, thereby preventing the spread of virus to neighboring cells (Moscona 2005).
NAI therapy can prevent secondary bacterial pneumonia in animals infected with influenza. Mice given NAI treatment within 72 h postinfluenza infection or prophylactically experienced improved survival due to delayed development of and progression of pneumonia with NAI treatment, decreased viral loads, and reduced secondary bacterial infections. Treatment in the later stages of influenza virus infection (i.e., 5 days) reduces bacterial invasion without any impact on viral loads, signifying that mechanisms independent of replication inhibition are in play (McCullers 2005). Their action may directly, or indirectly, lessen the effects of viral virulence factors, prevent receptor exposure, reduce use of sialic acids as catabolic substrates, and/or activate immunological components in a viral strain-dependent manner [reviewed in (McCullers 2011)].
Although NAI resistance remains problematic and may be reduced with combination therapy, treatment with multiple NAIs can inhibit antiviral efficacy (Duval et al. 2010). NAIs in combination with antibiotics, on the other hand, can facilitate recovery from influenza and alter the coinfection pathogenesis in infected animals (McCullers 2004). Other antivirals [e.g., peramivir and laninamivir (NAIs), favipiravir (RNA polymerase inhibitor)] that are not yet licensed may provide benefit to coinfected hosts, but these have yet to be tested in animal coinfection models.
It is important to note that NAIs specifically target influenza virus NA and do not inhibit bacterial NAs with clinically relevant doses (Nishikawa et al. 2012). While NAIs may reduce incidence of bacterial pneumonia, and thus antibiotic requirements, NAs derived from invasive or commensal bacteria may antagonize their effectiveness (Nishikawa et al. 2012). Thus, NAI therapy could have differential bacterial-dependent effects as well.
Antibacterial Treatment
Unlike antivirals, which interrupt disease progression by preventing viral spread, antibiotics work to eliminate pathogens directly. Some antibiotics, however, kill bacteria through mechanisms that can have harmful repercussions. For instance, therapy with cell wall active agents (e.g., ampicillin), the mainstay of treatment of community-acquired pneumonia in children (Bradley et al. 2011), causes significant inflammation and lung injury in animal models (Karlström et al. 2009). The characteristic inflammation in secondary bacterial infections is due to immune cells responding to the release of bacterial components, such as cell wall components, during lysis (Karlstrom et al. 2011). Thus, alternative treatments that eliminate pathogens while preserving host integrity are desirable.
Antibiotics that reduce neutrophil influx or cytokines and thus circumvent the inflammatory tissue damage are beneficial in coinfected animals (Karlstrom et al. 2011; Liu et al. 2013). In particular, protein synthesis inhibitors (e.g., clindamycin) and macrolides (e.g., azithromycin) have anti-inflammatory properties in addition to bactericidal activity, thereby clinically curing mice with influenza-associated pneumococcal pneumonia (Karlström et al. 2009). Nevertheless, antibiotic treatment alone is suboptimal.
Anti-inflammatory agents, such as corticosteroids (e.g., dexamethasone), in conjunction with antibiotic therapy improve beta-lactam-induced immunopathology and mortality in animals with severe pneumonia. However, giving dexamethasone prophylactically during influenza infections negatively impacts adaptive immunity and results in reduced viral clearance (Ghoneim and McCullers 2013). Thus, use of immune-modulatory approaches may be best reserved for severe infections where inflammation is driving poor outcomes but avoided in primary viral infections where detrimental effects on the host response may result. Given the benefit of anti-inflammatory treatment and the importance of inflammation in viral-bacterial coinfections, better success may occur if specific inflammatory pathways or pathogenic factors are targeted [reviewed in (McCullers 2011)].
Vaccination
Vaccination remains fundamental to prevention of influenza and bacterial infections. Data from animal models indicate that vaccinating against influenza viruses effectively circumvents bacterial associated pneumonia (Huber et al. 2010; Chaussee et al. 2011; Mina et al. 2013) but may support colonization and replication in the URT (Mina et al. 2013). An important caveat of current vaccines against influenza viruses is that partial protection of related strains may not be sufficient to alleviate bacterial complications. On the other hand, antibacterial vaccines are important to block the specific bacteria being targeted and to prevent clinically severe influenza infections by reducing the coinfection component.
Vaccinating animals against some bacteria (e.g., pneumococcus, H. influenzae) prevents invasive diseases caused by these pathogens, but protection is limited to vaccine-specific types and efficacy may be lost in influenza virus infected animals (Mina et al. 2013). In addition, vaccines against other coinfecting bacteria, such as S. aureus, are not currently available. Interestingly, vaccination with a live-attenuated B. pertussis vaccine can protect against lethal challenges with influenza virus by controlling cytokine responses that lead to virus-mediated inflammation (Li et al. 2010). Although the vaccine has not yet been approved for use in humans, it is promising and may benefit as a prophylactic agent against infection with influenza viruses.
Kinetic Modeling
Unraveling the relationships between pathogen replication and interactions and the resulting airway alterations and inflammation that are driving coinfection host pathology and disease is complicated. Kinetic models are a robust means of analyzing experimental results and explaining biological phenomena without testing every scenario experimentally. They have proven valuable in the identification and characterization of mechanisms driving influenza virus infections, pneumococcal infections, and bacterial coinfection establishment and severity.
Modeling Influenza Virus Infections
A growing body of work modeling in vivo influenza virus infections has improved our knowledge about the viral life cycle, viral control by the host, pathogenic differences in strains, and efficacy of antiviral treatment [reviewed in (Smith and Ribeiro 2010; Beauchemin and Handel 2011; Smith and Perelson 2011)]. These models have characterized the spread of virus during early infection and yielded estimates of strain-specific viral infection and production rates, infected cell life spans, and infectious virus half-life, all of which are not amenable to experimental investigation.
Most of these studies model data from humans or large animals where only nasal wash titers are available and, thus, are restricted to studying nasopharyngeal infections (Baccam et al. 2006; Saenz et al. 2010; Canini and Carrat 2010; Pawelek et al. 2012). A few models, however, take advantage of data collected in the mouse model, including pathogen and immunological measurements, to study invasive lung infections (Handel et al. 2010; Miao et al. 2010; Smith et al. 2011a). Viral load dynamics can be accurately modeled using target-cell limitation (Fig. 3), through undefined mechanisms, as the primary means of viral control while excluding specific immune responses (Baccam et al. 2006; Smith et al. 2011a). It is important to note that these models do not discount the fact that immunological factors may drive influenza virus pathogenesis, but that this information can simply not be extracted from viral loads (Smith et al. 2010; Miao et al. 2011). Quantifying the effect that host factors have on viral replication has been restricted by the limited amount of data detailing the innate immune responses [reviewed in (Smith and Perelson 2011)]. As more data arise, new quantitative descriptions of influenza virus kinetics will be developed and will undoubtedly aid experimental interpretation.
Fig. 3.

Kinetics of influenza-pneumococcal coinfection. Model schematic and equations that result in the observed kinetics of influenza virus infection followed by pneumococcus given 7 days postinfluenza infection (Smith et al. 2013). During primary influenza, susceptible epithelial (target) cells (T) become infected at a rate βV per cell. Infected cells (I
1) first undergo an eclipse phase at rate k per cell prior to entering a state (I
2) in which virus is produced. Productively infected cells are lost, through apoptosis, viral cytopathic effects, or removal by immune cells, at a rate δ per cell. Virus (V) is produced at rate p per cell, which is significantly increased by bacterial presence (aP
z) (boxed), and cleared at rate c. Invading pneumococci (P) proliferate at maximum rate r with a tissue capacity K
p CFU/ml. Bacteria are cleared via phagocytosis by alveolar macrophages (MA) at rate γf per cell, which is significantly reduced by virus presence 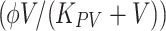 (boxed). With this kinetic description, viral titers increase exponentially, peak and begin to decline prior to bacterial invasion. Once bacteria are present, a viral rebound occurs and bacteria grow exponentially before reaching a maximum capacity. The potential increase in bacterial adherence to virus-infected cells and any accompanying cell death has little effect are excluded here
(boxed). With this kinetic description, viral titers increase exponentially, peak and begin to decline prior to bacterial invasion. Once bacteria are present, a viral rebound occurs and bacteria grow exponentially before reaching a maximum capacity. The potential increase in bacterial adherence to virus-infected cells and any accompanying cell death has little effect are excluded here
Modeling Bacterial Infections
Kinetic models depicting bacterial infections are an exciting new tool being used to study pathogenesis (Smith et al. 2011b). Capitalizing on data obtainable in the mouse model system, we characterized the dose-dependent innate immune control of a pneumococcal invasion and quantified the contributions of alveolar macrophages, neutrophils, inflammatory macrophages, cytokines, and damage to bacterial pathogenesis (Smith et al. 2011b).
Model analysis revealed the exact thresholds for bacterial establishment, growth, and eradication with alveolar macrophages playing a central role. The dose-dependent invasive ability of pneumococci was solely dependent on the number and phagocytic ability of resident macrophages initially present. Thus, any alterations to resident cells, such as death from an antecedent viral infection, would result in immediate pathogenic invasion. While the rapid neutrophil influx could facilitate bacterial removal, pneumococcal-induced neutrophil apoptosis hindered complete eradication. This process was also dependent on alveolar macrophages and whether they were engaged damage control rather than bacterial clearance. Inflammatory macrophages had little effect on clearance but still contributed to respiratory tract damage. Through this model, we successfully captured the biochemical, cellular, immunological interactions of pneumococci with the host and identify the critical processes driving pathogenesis.
Modeling Coinfections
Modeling the interactions of two pathogens requires the combination of previously developed single infection models. Thus far, the only model depicting a coinfection is one that we formulated for influenza-pneumococcal coinfection using the models discussed above (Fig. 3) (Smith et al. 2013). We quantitated the enhanced bacterial growth and viral rebound and evaluated prior hypotheses about the interaction between influenza, pneumococcus, and the host.
Careful model development and analyses showed that any enhanced bacterial adherence to epithelial cells, with respect to both invasion and cell death, was negligible compared to the viral-induced impairment of alveolar macrophages. While the mechanism for this prediction is not available through initial modeling efforts, it did pinpoint the process driving influenza-pneumococcal synergy that should be subject to further examination in the laboratory (Smith et al. 2013). In fact, the decrease in phagocytosis by alveolar macrophage was later determined to be a result of influenza virus directly killing these cells (Ghoneim et al. 2013). Remarkably, both our models (Smith et al. 2013) and experiments (Ghoneim et al. 2013) agreed that these cells diminished to 85–90 % of their baseline level within 7 days.
Receptor-mediated mechanisms may still drive the synergy, although not in the context of enhanced invasion. Our model predicts that bacterial interaction with virus-infected epithelial cells releases virus and thus increases viral loads postbacterial invasion. Bacterial proteases or NAs may liberate virus in the same manner as viral NA. S. aureus proteases can activate influenza virus HA cleavage and enhance viral invasion into host cells (Tashiro et al. 1987). Furthermore, some commensal bacteria [e.g., S. mitis (Nonaka et al. 1983; Beighton and Whiley 1990), certain S. pneumoniae (Scanlon et al. 1989), Actinomyces naeslundii and A. viscosus (Moncla and Braham 1989), Porphyromonas gingivalis (Moncla et al. 1990), and S. oralis (Homer et al. 1996)] that secrete NAs or exogenous NA can rescue influenza virus replication if viral NA is missing or inhibited (Liu and Air 1993; Hughes et al. 2000; Nishikawa et al. 2012). Thus, it is feasible that other NA possessing bacterial species can act in the manner predicted by our model. To uncover the underlying mechanism, a combination of in vitro and in vivo experiments with viruses and bacteria that exhibit differential expression of NA is necessary (Smith et al. 2013).
Concluding Remarks and Key Research Questions
It is becoming better appreciated that pneumonia is frequently caused by coinfecting pathogens. Viral-mediated mechanisms are also important in other invasive infections, such as otitis media. The underlying relationship between viral and bacterial density, inflammation, and the host microbiome during influenza coinfections is exceptionally complex. Even with the growing body of work detailing various aspects of viral-bacterial coinfections, determining the precise contributions of each interrelated factor is challenging. Furthermore, studying coinfections has become problematic due to the numerous site-, pathogen-, time-, and host-specific variations to consider. Thus, it is necessary to employ the next generation of analyses using a mixture of animal models and kinetic models with the goal of obtaining results translatable to infections in humans.
Some important areas for consideration include determining how different bacterial virulence factors leverage the environment set forth by influenza viruses to cause disease, how timing of sequential infections impacts each of the aforementioned mechanisms, how the synergistic relationship is facilitated by host genetics, and how each of these factors differ between the nasopharyngeal, middle ear, and lung niches and between different viral and bacterial species. Our understanding of coinfection biology should increase as new and different data emerge. With such data, treatment options suitable for clinical practice are permissive to investigation as we focus on preparation for the next influenza pandemic.
Acknowledgments
This work was supported by NIH grant AI100946 (AMS) and ALSAC (JAM). We thank Betsy Williford and Klo Spelshouse (SJCRH Biomedical Communications) for assistance with figure illustration.
Contributor Information
Richard W. Compans, Phone: +1(404)727-2015, Email: rcompan@emory.edu
Michael B. A. Oldstone, Phone: +1858-784-8054, Email: mbaobo@scripps.edu
Amber M. Smith, Email: amber.smith@stjude.org
References
- Abramson JS, Giebink GS, Mills EL, Quie PG. Polymorphonuclear leukocyte dysfunction during influenza virus infection in chinchillas. J Infect Dis. 1981;143:836–845. doi: 10.1093/infdis/143.6.836. [DOI] [PubMed] [Google Scholar]
- Abramson JS, Giebink GS, Quie PG. Influenza A virus-induced polymorphonuclear leukocyte dysfunction in the pathogenesis of experimental pneumococcal otitis media. Infect Immun. 1982;36:289–296. doi: 10.1128/iai.36.1.289-296.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abt MC, Osborne LC, Monticelli LA, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alymova IV, Green AM, van de Velde N, et al. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J Virol. 2011;85:12324–12333. doi: 10.1128/JVI.05872-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansaldi F, de Florentiis D, Parodi V, et al. Bacterial carriage and respiratory tract infections in subjects > or = 60 years during an influenza season: implications for the epidemiology of community acquired pneumonia and influenza vaccine effectiveness. J Prev Med Hyg. 2012;53:94–97. [PubMed] [Google Scholar]
- Ayala VI, Teijaro JR, Farber DL, et al. Bordetella pertussis infection exacerbates influenza virus infection through pertussis toxin-mediated suppression of innate immunity. PLoS ONE. 2011;6:e19016. doi: 10.1371/journal.pone.0019016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccam P, Beauchemin C, Macken CA, et al. Kinetics of influenza a virus infection in humans. J Virol. 2006;80:7590–7599. doi: 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23:285–290. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- Beauchemin CA, Handel A. A review of mathematical models of influenza A infections within a host or cell culture: lessons learned and challenges ahead. BMC Public Health. 2011;11:S7. doi: 10.1186/1471-2458-11-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beighton D, Whiley RA. Sialidase activity of the “Streptococcus milleri group” and other viridans group Streptococci. J Clin Microbiol. 1990;28:1431–1433. doi: 10.1128/jcm.28.6.1431-1433.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendt RF, Long GG, Walker JS. Influenza alone and in sequence with pneumonia due to Streptococcus pneumoniae in the squirrel monkey. J Infect Dis. 1975;132:689–693. doi: 10.1093/infdis/132.6.689. [DOI] [PubMed] [Google Scholar]
- Bosch AATM, Biesbroek G, Trzcinski K, et al. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 2013;9:e1003057. doi: 10.1371/journal.ppat.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JS, Byington CL, Shah SS, et al. The management of community-acquired pneumonia in infants and children older than 3 months of age: clinical practice guidelines by the pediatric infectious diseases society and the infectious diseases society of America. Clin Infect Dis. 2011;53:e25–e76. doi: 10.1093/cid/cir531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage JF, Shanks GD. What really happened during the 1918 influenza pandemic? The importance of bacterial secondary infections. J Infect Dis. 2007;196:1717–1718. doi: 10.1086/522355. [DOI] [PubMed] [Google Scholar]
- Camara M, Mitchell TJ, Andrew PW, Boulnois GJ. Streptococcus pneumoniae produces at least two distinct enzymes with neuraminidase activity: cloning and expression of a second neuraminidase gene in Escherichia coli. Infect Immun. 1991;59:2856–2858. doi: 10.1128/iai.59.8.2856-2858.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canini L, Carrat F. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J Virol. 2010;85:2764–2770. doi: 10.1128/JVI.01318-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control Deaths and Mortality
- Centers for Disease Control Estimated Burden of Acute Otits Externa
- Chanturiya AN, Basanez G, Schubert U, et al. PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol. 2004;78:6304–6312. doi: 10.1128/JVI.78.12.6304-6312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaussee MS, Sandbulte HR, Schuneman MJ, et al. Inactivated and live, attenuated influenza vaccines protect mice against influenza: Streptococcus pyogenes super-infections. Vaccine. 2011;29:3773–3781. doi: 10.1016/j.vaccine.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Calvo PA, Malide D, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Colamussi ML, White MR, Crouch E, Hartshorn KL. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood. 1999;93:2395–2403. [PubMed] [Google Scholar]
- Conenello GM, Tisoncik JR, Rosenzweig E, et al. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol. 2011;85:652–662. doi: 10.1128/JVI.01987-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conenello GM, Zamarin D, Perrone LA, et al. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:e141. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundell DR, Gerard NP, Gerard C, et al. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- Danishuddin M, Khan SN, Khan AU. Molecular interactions between mitochondrial membrane proteins and the C-terminal domain of PB1-F2: an in silico approach. J Mol Model. 2010;16:535–541. doi: 10.1007/s00894-009-0555-5. [DOI] [PubMed] [Google Scholar]
- Diavatopoulos DA, Short KR, Price JT, et al. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. 2010;24:1789–1798. doi: 10.1096/fj.09-146779. [DOI] [PubMed] [Google Scholar]
- Diavatopoulos DA, Short KR, Price JT, et al. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. 2010;24:1789–1798. doi: 10.1096/fj.09-146779. [DOI] [PubMed] [Google Scholar]
- Didierlaurent A, Goulding J, Patel S, et al. Sustained desensitization to bacterial toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockrell DH, Marriott HM, Prince LR, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol (Baltimore Md 1950) 2003;171:5380–5388. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- Domínguez-Cherit G. Critically ill patients with 2009 influenza A(H1N1) in Mexico. JAMA. 2009;302:1880. doi: 10.1001/jama.2009.1536. [DOI] [PubMed] [Google Scholar]
- Duval X, van der Werf S, Blanchon T, et al. Efficacy of Oseltamivir-Zanamivir combination compared to each monotherapy for seasonal influenza: a randomized placebo-controlled trial. PLoS Med. 2010;7:e1000362. doi: 10.1371/journal.pmed.1000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. 2001;69:50–56. [PubMed] [Google Scholar]
- Fainstein V, Musher DM, Cate TR. Bacterial adherence to pharyngeal cells during viral infection. J Infect Dis. 1980;141:172–176. doi: 10.1093/infdis/141.2.172. [DOI] [PubMed] [Google Scholar]
- File TM, Jr, Marrie TJ. Burden of community-acquired pneumonia in North American adults. Postgrad Med. 2010;122:130–141. doi: 10.3810/pgm.2010.03.2130. [DOI] [PubMed] [Google Scholar]
- Fillion I, Ouellet N, Simard M, et al. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol (Baltimore Md 1950) 2001;166:7353–7361. doi: 10.4049/jimmunol.166.12.7353. [DOI] [PubMed] [Google Scholar]
- Francis T, de Torregrosa MV. Combined infection of mice with H. influenzae and influenza virus by the intranasal route. J Infect Dis. 1945;76:70–77. [Google Scholar]
- García-Rodríguez JA, Fresnadillo Martínez MJ (2002) Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J Antimicrob Chemother 50 Suppl S2:59–73 [DOI] [PubMed]
- Ghoneim HE, McCullers JA. Adjunctive corticosteroid therapy improves lung immunopathology and survival during severe secondary pneumococcal pneumonia in mice. J Infect Dis. 2013 doi: 10.1093/infdis/jit653. [DOI] [PubMed] [Google Scholar]
- Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. 2013;191:1250–1259. doi: 10.4049/jimmunol.1300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JS, Malide D, Hornung F, et al. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003;77:7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebink G, Ripley M, Wright P. Eustachian tube histopathology during experimental influenza A virus infection in the chinchilla. Ann Otol Rhinol Laryngol. 1987;96:199–206. doi: 10.1177/000348948709600212. [DOI] [PubMed] [Google Scholar]
- Giles C, Shuttleworth EM. Postmortem findings in 46 influenza deaths. Lancet. 1957;273:1224–1225. doi: 10.1016/s0140-6736(57)90180-0. [DOI] [PubMed] [Google Scholar]
- le Goffic R, Bouguyon E, Chevalier C, et al. Influenza A virus protein PB1-F2 exacerbates IFN-expression of human respiratory epithelial cells. J Immunol. 2010;185:4812–4823. doi: 10.4049/jimmunol.0903952. [DOI] [PubMed] [Google Scholar]
- Goulding J, Godlee A, Vekaria S, et al. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. 2011;204:1086–1094. doi: 10.1093/infdis/jir467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwaltney JM, Sande MA, Austrian R, Hendley JO. Spread of Streptococcus pneumoniae in families. II. Relation of transfer of S. pneumoniae to incidence of colds and serum antibody. J Infect Dis. 1975;132:62–68. doi: 10.1093/infdis/132.1.62. [DOI] [PubMed] [Google Scholar]
- Hai R, Schmolke M, Varga ZT, et al. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J Virol. 2010;84:4442–4450. doi: 10.1128/JVI.02717-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek DM, Yuan Z, Quartey MK, Giebink GS (1999) Otitis media: the chinchilla model. In: Zak O, Sande MA (eds) Handbook of animal models of infection: experimental models in antimicrobial chemotherapy Academic, San Diego, CA, pp 389–401
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Handel A, Longini IM, Antia R. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J R Soc Interface. 2010;7:35–47. doi: 10.1098/rsif.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG. Antiviral resistance in influenza viruses—implications for management and pandemic response. N Engl J Med. 2006;354:785–788. doi: 10.1056/NEJMp068030. [DOI] [PubMed] [Google Scholar]
- Hayden FG. Newer influenza antivirals, biotherapeutics and combinations: novel influenza antivirals and therapeutics. Influenza Other Respir Viruses. 2013;7:63–75. doi: 10.1111/irv.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T. The role of respiratory viruses in otitis media. Vaccine. 2000;19:S51–S55. doi: 10.1016/s0264-410x(00)00278-4. [DOI] [PubMed] [Google Scholar]
- Herzog H, Staub H, Richterich R. Gas-analytical studies in severe pneumonia; observations during the 1957 influenza epidemic. Lancet. 1959;1:593–597. doi: 10.1016/s0140-6736(59)92351-7. [DOI] [PubMed] [Google Scholar]
- Hirano T, Kurono Y, Ichimiya I, et al. Effects of influenza A virus on lectin-binding patterns in murine nasopharyngeal mucosa and on bacterial colonization. Otolaryngol Head Neck Surg. 1999;121:616–621. doi: 10.1016/S0194-5998(99)70068-9. [DOI] [PubMed] [Google Scholar]
- Homer KA, Kelley S, Hawkes J, et al. Metabolism of glycoprotein-derived sialic acid and N-acetylglucosamine by Streptococcus oralis. Microbiol Read Engl. 1996;142(Pt 5):1221–1230. doi: 10.1099/13500872-142-5-1221. [DOI] [PubMed] [Google Scholar]
- Huber VC, Peltola V, Iverson AR, McCullers JA. Contribution of Vaccine-induced immunity toward either the HA or the NA component of influenza viruses limits secondary bacterial complications. J Virol. 2010;84:4105–4108. doi: 10.1128/JVI.02621-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MT, Matrosovich M, Rodgers ME, et al. Influenza A viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J Virol. 2000;74:5206–5212. doi: 10.1128/jvi.74.11.5206-5212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussell T, Cavanagh MM. The innate immune rheostat: influence on lung inflammatory disease and secondary bacterial pneumonia. Biochem Soc Trans. 2009;37:811. doi: 10.1042/BST0370811. [DOI] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Kumamoto Y, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Nat Acad Sci U.S.A. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson AR, Boyd KL, McAuley JL, et al. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis. 2011;203:880–888. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Kamimoto L, Bramley AM, et al. Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med. 2009;361:1935–1944. doi: 10.1056/NEJMoa0906695. [DOI] [PubMed] [Google Scholar]
- Jamieson AM, Yu S, Annicelli CH, Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe. 2010;7:103–114. doi: 10.1016/j.chom.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang-Shieh Y-F, Chien H-F, Chang C-Y, et al. Distribution and expression of CD200 in the rat respiratory system under normal and endotoxin-induced pathological conditions. J Anat. 2010;216:407–416. doi: 10.1111/j.1469-7580.2009.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson S, Musher DM, Chapman A, et al. Phagocytosis and killing of common bacterial pathogens of the lung by human alveolar macrophages. J Infect Dis. 1985;152:4–13. doi: 10.1093/infdis/152.1.4. [DOI] [PubMed] [Google Scholar]
- Joyce EA, Popper SJ, Falkow S. Streptococcus pneumoniae nasopharyngeal colonization induces type I interferons and interferon-induced gene expression. BMC Genomics. 2009;10:404. doi: 10.1186/1471-2164-10-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- Karlström Å, Boyd KL, English BK, McCullers JA. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J Infect Dis. 2009;199:311–319. doi: 10.1086/596051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstrom A, Heston SM, Boyd KL, et al. Toll-like receptor 2 mediates fatal immunopathology in mice during treatment of secondary pneumococcal pneumonia following influenza. J Infect Dis. 2011;204:1358–1366. doi: 10.1093/infdis/jir522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S, Leemans JC, Florquin S, et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2003;167:171–179. doi: 10.1164/rccm.200207-698OC. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J. Innate Immun. 2010;2:560–575. doi: 10.1159/000317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Braughton KR, Whitney AR, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Nat Acad Sci U.S.A. 2003;100:10948–10953. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system: Innate immune recognition of Streptococcus pneumoniae. Cell Microbiol. 2012;14:460–466. doi: 10.1111/j.1462-5822.2011.01746.x. [DOI] [PubMed] [Google Scholar]
- Kudva A, Scheller EV, Robinson KM, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol (Baltimore Md 1950) 2011;186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA. 2009;302:1872. doi: 10.1001/jama.2009.1496. [DOI] [PubMed] [Google Scholar]
- Kuri T, Smed Sörensen A, Thomas S, et al. Influenza A virus-mediated priming enhances cytokine secretion by human dendritic cells infected with Streptococcus pneumoniae: Influenza virus and pneumococcal co-infection of human DCs. Cell Microbiol. 2013;15:1385–1400. doi: 10.1111/cmi.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laennec RTH (1923) Translation of selected passages from De l’auscultation mediate. Bale & Danielsson, London
- Lee LN, Dias P, Han D, et al. A mouse model of lethal synergism between influenza virus and haemophilus influenzae. Am J Pathol. 2010;176:800–811. doi: 10.2353/ajpath.2010.090596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Li R, Lim A, Phoon MC, et al. Attenuated bordetella pertussis protects against highly pathogenic influenza a viruses by dampening the cytokine storm. J Virol. 2010;84:7105–7113. doi: 10.1128/JVI.02542-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J Virol. 2012;86:12304–12312. doi: 10.1128/JVI.01269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciardi PV, Toh ZQ, Dunne E, et al. Protecting against ppneumococcal disease: critical interactions between probiotics and the airway microbiome. PLoS Pathog. 2012;8:e1002652. doi: 10.1371/journal.ppat.1002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Air GM. Selection and characterization of a neuraminidase-minus mutant of influenza virus and its rescue by cloned neuraminidase genes. Virology. 1993;194:403–407. doi: 10.1006/viro.1993.1276. [DOI] [PubMed] [Google Scholar]
- Liu X, He Y, Xiao K, et al. Effect of linezolid on clinical severity and pulmonary cytokines in a murine model of influenza A and Staphylococcus aureus coinfection. PLoS ONE. 2013;8:e57483. doi: 10.1371/journal.pone.0057483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie J, Jean C, Chen TH, et al. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May-August 2009. Morb Mortal Wkly Rep. 2009;58:1071–1074. [PubMed] [Google Scholar]
- Louria DB, Blumenfeld HL, Ellis JT, et al. Studies on influenza in the pandemic of 1957-1958. II. Pulmonary Complications Of Influenza. J Clin Invest. 1959;38:213–265. doi: 10.1172/JCI103791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving CL, Brockmeier SL, Vincent AL, et al. Influenza virus coinfection with Bordetella bronchiseptica enhances bacterial colonization and host responses exacerbating pulmonary lesions. Microb Pathog. 2010;49:237–245. doi: 10.1016/j.micpath.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Ludewick HP, Aerts L, Hamelin M-E, Boivin G. Long-term impairment of Streptococcus pneumoniae lung clearance is observed after initial infection with influenza A virus but not human metapneumovirus in mice. J Gen Virol. 2011;92:1662–1665. doi: 10.1099/vir.0.030825-0. [DOI] [PubMed] [Google Scholar]
- Marks LR, Davidson BA, Knight PR, Hakansson AP (2013) Interkingdom signaling induces streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4:e00438–13. doi:10.1128/mBio.00438-13 [DOI] [PMC free article] [PubMed]
- McAuley JL, Chipuk JE, Boyd KL, et al. PB1-F2 proteins from H5N1 and 20th century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010;6:e1001014. doi: 10.1371/journal.ppat.1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Hornung F, Boyd KL, et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249. doi: 10.1016/j.chom.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Zhang K, McCullers JA. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol. 2010;84:558–564. doi: 10.1128/JVI.01785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA. Molecular pathogenesis of pneumococcal pneumonia. Front Biosci. 2001;6:d877. doi: 10.2741/mccullers. [DOI] [PubMed] [Google Scholar]
- McCullers JA. Antiviral therapy of influenza. Expert Opin Investig Drugs. 2005;14:305–312. doi: 10.1517/13543784.14.3.305. [DOI] [PubMed] [Google Scholar]
- McCullers JA. Preventing and treating secondary bacterial infections with antiviral agents. Antivir Ther. 2011;16:123–135. doi: 10.3851/IMP1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. 2014;12:252–262. doi: 10.1038/nrmicro3231. [DOI] [PubMed] [Google Scholar]
- McCullers JA. Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J Infect Dis. 2004;190:519–526. doi: 10.1086/421525. [DOI] [PubMed] [Google Scholar]
- McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J Infect Dis. 2003;187:1000–1009. doi: 10.1086/368163. [DOI] [PubMed] [Google Scholar]
- McCullers JA, English BK. Improving therapeutic strategies for secondary bacterial pneumonia following influenza. Future Microbiol. 2008;3:397–404. doi: 10.2217/17460913.3.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA, Karlström Å, Iverson AR, et al. Novel strategy to prevent otitis media caused by colonizing Streptococcus pneumoniae. PLoS Pathog. 2007;3:e28. doi: 10.1371/journal.ppat.0030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA, McAuley JL, Browall S, et al. Influenza enhances susceptibility to natural acquisition of and disease due to Streptococcus pneumoniae in ferrets. J Infect Dis. 2010;202:1287–1295. doi: 10.1086/656333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA, Rehg JE. Lethal synergism between influenza virus and streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- McHugh KJ, Mandalapu S, Kolls JK, et al. A novel outbred mouse model of 2009 pandemic influenza and bacterial co-infection severity. PLoS ONE. 2013;8:e82865. doi: 10.1371/journal.pone.0082865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74:6707–6721. doi: 10.1128/IAI.00789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol. 2013;191:2047–2052. doi: 10.4049/jimmunol.1301152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Hollenbaugh JA, Zand MS, et al. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J Virol. 2010;84:6687–6698. doi: 10.1128/JVI.00266-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Xia X, Perelson AS, Wu H. On identifiability of nonlinear ODE models and applications in viral dynamics. SIAM Rev. 2011;53:3–39. doi: 10.1137/090757009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ML, Gao G, Pestina T, et al. Hypersusceptibility to invasive pneumococcal infection in experimental sickle cell disease involves platelet-activating factor receptor. J Infect Dis. 2007;195:581–584. doi: 10.1086/510626. [DOI] [PubMed] [Google Scholar]
- Mina MJ, Klugman KP, McCullers JA. Liveattenuated influenza vaccine, but not pneumococcal conjugate vaccine, protects against increased density and duration of pneumococcal carriage after influenza infection in pneumococcal colonized mice. J Infect Dis. 2013;208:1281–1285. doi: 10.1093/infdis/jit317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minas K, Liversidge J. Is the CD200/CD200 receptor interaction more than just a myeloid cell inhibitory signal? Crit Rev Immunol. 2006;26:213–230. doi: 10.1615/critrevimmunol.v26.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncla BJ, Braham P. Detection of sialidase (neuraminidase) activity in Actinomyces species by using 2’-(4-methylumbelliferyl) alpha-D-N-acetylneuraminic acid in a filter paper spot test. J Clin Microbiol. 1989;27:182–184. doi: 10.1128/jcm.27.1.182-184.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncla BJ, Braham P, Hillier SL. Sialidase (neuraminidase) activity among gram-negative anaerobic and capnophilic bacteria. J Clin Microbiol. 1990;28:422–425. doi: 10.1128/jcm.28.3.422-425.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery CP, Boyle-Vavra S, Adem PV, et al. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J Infect Dis. 2008;198:561–570. doi: 10.1086/590157. [DOI] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- Murphy TF, Bakaletz LO, Smeesters PR. Microbial interactions in the respiratory tract. Pediatr Infect Dis J. 2009;28:S121–S126. doi: 10.1097/INF.0b013e3181b6d7ec. [DOI] [PubMed] [Google Scholar]
- Musher DM, Dowell ME, Shortridge VD, et al. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. N Engl J Med. 2002;346:630–631. doi: 10.1056/NEJM200202213460820. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. 2011;121:3657–3665. doi: 10.1172/JCI57762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarini AA, Recher M, Lang KS, et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc Nat Acad Sci U.S.A. 2006;103:15535–15539. doi: 10.1073/pnas.0607325103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JC, Jackson M, Yu O, et al. Impact of the introduction of pneumococcal conjugate vaccine on rates of communityacquired pneumonia in children and adults. Vaccine. 2008;26:4947–4954. doi: 10.1016/j.vaccine.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Niemann S, Ehrhardt C, Medina E, et al. Combined action of influenza virus and Staphylococcus aureus panton-valentine leukocidin provokes severe lung epithelium damage. J Infect Dis. 2012;206:1138–1148. doi: 10.1093/infdis/jis468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Shimizu K, Tanaka T, et al. Bacterial neuraminidase rescues influenza virus replication from inhibition by a neuraminidase inhibitor. PLoS ONE. 2012;7:e45371. doi: 10.1371/journal.pone.0045371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Ishikawa Y, Otsuka M, et al. Purification and some properties of neuraminidase isolated from the culture medium of oral bacterium Streptococcus mitis ATCC 9811. J Dent Res. 1983;62:792–797. doi: 10.1177/00220345830620070301. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Kawabata S, Nakagawa I, et al. Influenza A virus-infected hosts boost an invasive type of Streptococcus pyogenes infection in mice. J Virol. 2003;77:4104–4112. doi: 10.1128/JVI.77.7.4104-4112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oseasohn R, Adelson L, Kaji M. Clinicopathologic study of thirty-three fatal cases of Asian influenza. N Engl J Med. 1959;260:509–518. doi: 10.1056/NEJM195903122601101. [DOI] [PubMed] [Google Scholar]
- Palacios G, Hornig M, Cisterna D, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS ONE. 2009;4:e8540. doi: 10.1371/journal.pone.0008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelek KA, Huynh GT, Quinlivan M, et al. Modeling within-host dynamics of influenza virus infection including immune responses. PLoS Comput Biol. 2012;8:e1002588. doi: 10.1371/journal.pcbi.1002588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola VT, Boyd KL, McAuley JL, et al. Bacterial sinusitis and otitis media following influenza virus infection in ferrets. Infect Immun. 2006;74:2562–2567. doi: 10.1128/IAI.74.5.2562-2567.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola VT, Murti KG, McCullers JA. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J Infect Dis. 2005;192:249–257. doi: 10.1086/430954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew MM, Gent JF, Revai K, et al. Microbial interactions during upper respiratory tract infections. Emerg Infect Dis. 2008;14:1584–1591. doi: 10.3201/eid1410.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet LA, Hall-Stoodley L, Rutkowski MR, Harmsen AG. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2010;42:450–460. doi: 10.1165/rcmb.2007-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkowski MC, Bajolet-Laudinat O, Puchelle E. Cellular and molecular mechanisms of bacterial adhesion to respiratory mucosa. Eur Respir J. 1993;6:903–916. [PubMed] [Google Scholar]
- Plotkowski MC, Puchelle E, Beck G, et al. Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am Rev Respir Dis. 1986;134:1040–1044. doi: 10.1164/arrd.1986.134.5.1040. [DOI] [PubMed] [Google Scholar]
- Puchelle E, Zahm J-M, Tournier J-M, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:726–733. doi: 10.1513/pats.200605-126SF. [DOI] [PubMed] [Google Scholar]
- Rarey KE, DeLacure MA, Sandridge SA, Small PA. Effect of upper respiratory infection on hearing in the ferret model. Am J Otolaryngol. 1987;8:161–170. doi: 10.1016/s0196-0709(87)80040-6. [DOI] [PubMed] [Google Scholar]
- Redford PS, Mayer-Barber KD, McNab FW, et al. Influenza A virus impairs control of mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis. 2014;209:270–274. doi: 10.1093/infdis/jit424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz RA, Quinlivan M, Elton D, et al. Dynamics of influenza virus infection and pathology. J Virol. 2010;84:3974–3983. doi: 10.1128/JVI.02078-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon KL, Diven WF, Glew RH. Purification and properties of Streptococcus pneumoniae neuraminidase. Enzyme. 1989;41:143–150. doi: 10.1159/000469069. [DOI] [PubMed] [Google Scholar]
- Seki M, Yanagihara K, Higashiyama Y, et al. Immunokinetics in severe pneumonia due to influenza virus and bacteria coinfection in mice. Eur Respir J. 2004;24:143–149. doi: 10.1183/09031936.04.00126103. [DOI] [PubMed] [Google Scholar]
- Shahangian A, Chow EK, Tian X, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks GD, Mackenzie A, McLaughlin R, et al. Mortality risk factors during the 1918-1919 influenza pandemic in the Australian army. J Infect Dis. 2010;201:1880–1889. doi: 10.1086/652868. [DOI] [PubMed] [Google Scholar]
- Shope RE. Swine influenza: I. Experimental transmission and pathology. J Exp Med. 1931;54:349–359. doi: 10.1084/jem.54.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Diavatopoulos DA, Thornton R, et al. Influenza virus induces bacterial and nonbacterial otitis media. J Infect Dis. 2011;204:1857–1865. doi: 10.1093/infdis/jir618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Habets MN, Hermans PW, Diavatopoulos DA. Interactions between Streptococcus pneumoniae and influenza virus: a mutually beneficial relationship? Future Microbiol. 2012;7:609–624. doi: 10.2217/fmb.12.29. [DOI] [PubMed] [Google Scholar]
- Short KR, Habets MN, Payne J, et al. Influenza A virusinduced bacterial otitis media is independent of virus tropism for α2,6-linked sialic acid. Virol J. 2013;10:128. doi: 10.1186/1743-422X-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Reading PC, Brown LE, et al. Influenza-induced inflammation drives pneumococcal otitis media. Infect Immun. 2013;81:645–652. doi: 10.1128/IAI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Reading PC, Wang N et al (2012b) Increased nasopharyngeal bacterial titers and local inflammation facilitate transmission of Streptococcus pneumoniae. mBio 3:e00255–12. doi:10.1128/mBio.00255-12 [DOI] [PMC free article] [PubMed]
- van der Sluijs KF, van Elden LJR, Nijhuis M, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol (Baltimore Md 1950) 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- Small C-L, Shaler CR, McCormick S, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. 2010;184:2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
- Smith AM, Adler FR, McAuley JL, et al. Effect of 1918 PB1-F2 expression on influenza A virus infection kinetics. PLoS Comput Biol. 2011;7:e1001081. doi: 10.1371/journal.pcbi.1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Adler FR, Perelson AS. An accurate two-phase approximate solution to an acute viral infection model. J Math Biol. 2010;60:711–726. doi: 10.1007/s00285-009-0281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Adler FR, Ribeiro RM, et al. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 2013;9:e1003238. doi: 10.1371/journal.ppat.1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, McCullers JA. Molecular signatures of virulence in the PB1-F2 proteins of H5N1 influenza viruses. Virus Res. 2013;178:146–150. doi: 10.1016/j.virusres.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, McCullers JA, Adler FR. Mathematical model of a three-stage innate immune response to a pneumococcal lung infection. J Theor Biol. 2011;276:106–116. doi: 10.1016/j.jtbi.2011.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Perelson AS. Influenza A virus infection kinetics: quantitative data and models. Wiley Interdiscip Rev Syst Biol Med. 2011;3:429–445. doi: 10.1002/wsbm.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Ribeiro RM. Modeling the viral dynamics of influenza A virus infection. Crit Rev Immunol. 2010;30:291–298. doi: 10.1615/critrevimmunol.v30.i3.60. [DOI] [PubMed] [Google Scholar]
- Smith MW, Schmidt JE, Rehg JE, et al. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp Med. 2007;57:82–89. [PMC free article] [PubMed] [Google Scholar]
- Snelgrove RJ, Goulding J, Didierlaurent AM, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. 2008;9:1074–1083. doi: 10.1038/ni.1637. [DOI] [PubMed] [Google Scholar]
- Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- Tashiro M, Ciborowski P, Klenk H-D, et al. Role of Staphylococcus protease in the development of influenza pneumonia. Nature. 1987;325:536–537. doi: 10.1038/325536a0. [DOI] [PubMed] [Google Scholar]
- Tian X, Xu F, Lung WY, et al. Poly I: C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS ONE. 2012;7:e41879. doi: 10.1371/journal.pone.0041879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong HH, Weiser JN, James MA, DeMaria TF. Effect of influenza A virus infection on nasopharyngeal colonization and otitis media induced by transparent or opaque phenotype variants of Streptococcus pneumoniae in the chinchilla model. Infect Immun. 2001;69:602–606. doi: 10.1128/IAI.69.1.602-606.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomanen EI, Austrian R, Masure HR. Pathogenesis of pneumococcal infection. N Engl J Med. 1995;332:1280–1284. doi: 10.1056/NEJM199505113321907. [DOI] [PubMed] [Google Scholar]
- Varga ZT, Grant A, Manicassamy B, Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga ZT, Ramos I, Hai R, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li F, Sun R, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat Commun. 2013 doi: 10.1038/ncomms3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanzeck K, Boyd KL, McCullers JA. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am J Respir Crit Care Med. 2011;183:767–773. doi: 10.1164/rccm.201007-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks-Gorospe JN, Hurtig HR, Iverson AR, et al. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with gram-positive respiratory pathogens. J Virol. 2012;86:9035–9043. doi: 10.1128/JVI.00369-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DM, Simonsen L, Jordan R, et al. Impact of the 2009 influenza pandemic on pneumococcal pneumonia hospitalizations in the United States. J Infect Dis. 2011;205:458–465. doi: 10.1093/infdis/jir749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry WB, Butterfield CT. Inhalation experiments on influenza and pneumonia, and on the importance of spray-borne bacteria in respiratory infections. Public Health Rep (1896–1970) 1921;36:1443. [Google Scholar]
- World Health Organization Global Burden of Disease
- World Health Organization Writing Group. Bell D, Nicoll A, et al. Non-pharmaceutical interventions for pandemic influenza, international measures. Emerg Infect Dis. 2006;12:81–87. doi: 10.3201/eid1201.051370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Mao H, Ling M-T, et al. Successive influenza virus infection and Streptococcus pneumoniae stimulation alter human dendritic cell function. BMC Infect Dis. 2011;11:201. doi: 10.1186/1471-2334-11-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, García-Sastre A, Xiao X, et al. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell R, Krumbholz A, Eitner A, et al. Prevalence of PB1-F2 of influenza A viruses. J Gen Virol. 2007;88:536–546. doi: 10.1099/vir.0.82378-0. [DOI] [PubMed] [Google Scholar]