

Abstract
Self-replicating RNA derived from the genomes of positive strand RNA viruses represents a powerful tool for both molecular studies on virus biology and approaches to novel safe and effective vaccines. The following chapter summarizes the principles how such RNAs can be established and used for design of vaccines. Due to the large variety of strategies needed to circumvent specific pitfalls in the design of such constructs the technical details of the experiments are not described here but can be found in the cited literature.
Keywords: Self-replicating RNA, Positive strand RNA virus, Alphavirus, Flavivirus, Pestivirus, Classical swine fever virus
Introduction
The story of self-replicating RNA started with the recognition of the infectious nature of some viral RNA genomes in the 1950s and 1960s [1–7]. The evidence that naked RNA upon introduction into cells is able to promote a full replication cycle including release of infectious virus particles represented the starting point for a new era of research on RNA virus molecular biology and its application. Due to the technical difficulties, RNA is not amenable to site specific manipulation so that reverse genetics systems for RNA viruses always rely on a c DNA intermediate [8, 9]. First successful approaches towards recovery of replicating viruses from cloned c DNA were published for positive-strand RNA viruses relying on transfection of plasmid DNA containing a virus derived c DNA insert [10]. Soon afterwards, in vitro transcription and transfection of viral genome-like RNA was described leading to recovery of infectious progeny virus [11, 12].
Positive-strand RNA viruses were the first RNA viruses amenable to direct genetic manipulation due to their simple strategy of gene expression and replication [13]. The genomic RNA (vRNA) represents an mRNA able to govern the production of all viral proteins necessary for the initiation of virus replication. Products of the first round of translation of the viral genomic RNA assemble into a replicase complex that polymerizes a minus strand complementary to the genome (c RNA) as a template for the synthesis of additional mRNA molecules. Thus, for all positive- strand RNA viruses the components of the replicase complex have to be translated directly from the genomic RNA. Viral polypeptides not required for RNA replication, which mainly constitute structural proteins, can either also be translated from the genomic RNA or from one or more subgenomic mRNAs transcribed from the negative sense c RNA template, depending on the specific type of virus. Genomes of members of the group using the former expression strategy contain one long open reading frame (ORF). Translation of this RNA leads to a polyprotein that is co-translationally and posttranslationally processed by viral and host cellular proteases. The members of the families and belong to this first group (Fig. 1). The second group comprises the families , , , and . These viruses are characterized by the subgenomic RNAs used for expression of part of their genes (Fig. 1). In contrast to the first group, the replicase genes of these viruses are located in the 5′ part of the genome upstream of the structural genes. For all of these viruses the subgenomic RNAs are 3′ co-terminal with the genomic RNA.
Fig. 1.
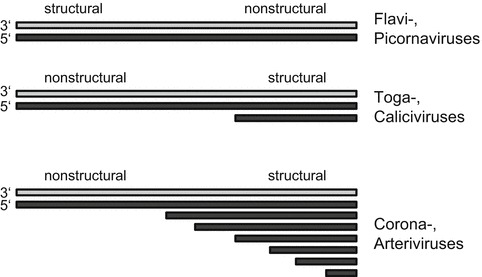
Genome structures and gene expression strategies of different positive strand RNA viruses. Schematic representation of the RNA species found in cells infected with the indicated viruses. For flaviviruses and picornaviruses, only RNA of genome size is generated. The RNA with positive polarity (genome orientation) is translated into one polyprotein that is subsequently processed into the viral proteins. Togaviruses and caliciviruses transcribe one RNA of subgenomic length encoding the structural proteins. Coronaviruses and arteriviruses use multiple subgenomic mRNAs for expression of structural and accessory proteins. RNA in coding orientation (mRNA sense) is represented by black bars whereas grey bars symbolize negative strand intermediates of viral genome replication. The location of structural and nonstructural genes in the viral genomes is indicated
The possibility to recover self-replicating viral RNA from cloned c DNA sequences opened a window to sophisticated studies on the mechanisms of RNA virus replication. Moreover, this knowledge was crucial for establishment of rationally attenuated viruses as well as development of strategies for use of self-replicating RNA expressing foreign genes for vaccine purposes and other applications. In this chapter, we present the technical principles used for establishment of self-replicating RNA and selected examples for its application in the context of vaccine development.
Methods for Establishment of Self-Replicating RNAs
Basic Strategies: A Historical Review
Due to the greater instability of (single stranded) RNA versus DNA and the wealth of techniques for DNA manipulation in contrast to the difficulties of direct RNA manipulation recombinant virus systems are based on DNA constructs, even in the case of RNA viruses where these systems rely on c DNA of the viral RNA. Due to the infectious nature of the positive strand RNA virus genome reverse genetics systems for positive strand RNA viruses need not be much more complicated than to be a way to deliver genome-like RNA into cells for successful replication of said RNA and for virus recovery. The history of reverse genetic systems for positive strand RNA viruses highlights the pitfalls that may be encountered in the design of a reverse genetic system and show solutions how to circumvent these difficulties. Some of these difficulties are covalently linked to the genome structures found in different positive strand RNA virus families. The genome can be capped or linked to a so-called VPg-protein or contain a naked 5′ end. The 3′ end can form loop structures or be a poly-A tail as would be expected for mRNAs. Depending on the virus the correct 5′ and 3′ end is very important as they can be crucial for replication and/or translation, or the production of subgenomic RNA (Fig. 1).
The first infectious c DNA clone of a eukaryotic virus was a c DNA clone for poliovirus [10]. This construct had the complete c DNA-sequence including a 37 residue poly (A) tail in the plasmid pBR322 and yielded infectious virus particles upon transfection in mammalian cells. This first construct did not contain a dedicated promoter to ensure the transcription of viral RNA, but nevertheless led to enough RNA expression for virus recovery. Three years later, the performance of poliovirus c DNA clones could be significantly ameliorated through the introduction of SV40 transcription and replication signals and transfection of the resulting construct into cells expressing the SV40 large T antigen [14], thus ensuring replication of the DNA-plasmid in eukaryotic cells leading to a higher yield of viral RNA and recovered virus (Fig. 2, left part). For other picornaviruses, cloning the c DNA into a bacterial plasmid was not enough to establish constructs leading to the production of infectious virus progeny. Indeed, the first c DNA clone for rhinovirus type 14 failed to produce infectious viral particles, but addition of an SP6 bacteriophage promoter upstream of the c DNA combined with in vitro transcription of the c DNA, produced RNA that led to infectious progeny upon transfection into cells [12] (Fig. 2, middle). An equivalent approach was also used in the reverse genetics system for brome mosaic virus, which consists of three plasmids containing the c DNAs to the three viral genomic RNAs immediately downstream of a λ-phage promoter to drive in vitro transcription. Combined transfection of the three in vitro- transcribed RNAs led to virus infection in plants [11].
Fig. 2.

Different strategies to generate reverse genetic systems for positive strand RNA viruses. Upper part: Viral RNA can either be obtained from purified virus particles or from infected cells trough total RNA extraction. c DNA of the viral genome can be generated using a specific primer complementary to the 3′ end of the viral genome if the sequence is known, oligo-d(T) primers for polyadenylated genomes or random priming in case of unknown sequences. RNA can also be used in high throughput sequencing approaches to determine the viral genomic sequence including the genomic ends. Middle part: To obtain efficient reverse genetics systems the c DNA needs to be cloned downstream of promoter sequences. This can either be a RNA polymerase II promoter if the vRNA shall be transcribed in the nucleus of transfected cells, or bacteriophage promoters like T7 for in vitro transcription. When possible, the c DNA is assembled in one full length construct (left). Alternatively, the c DNA can be cloned in fragments into different plasmids to avoid instability or to break down large genomes to sizes that are more amenable to manipulation (right). Lower part: From full length plasmids containing a eukaryotic promoter vRNA will be transcribed by the cellular machinery upon transfection of the c DNA construct. After export of the RNA into the cytoplasm its translation will provide the viral proteins necessary for replication (left). Full length plasmids with bacteriophage promoters are linearized before RNA synthesis via run-off in vitro transcription (middle). When the viral c DNA is cloned in several fragments, the complete c DNA needs first to be assembled into a full length c DNA template by in vitro ligation to obtain a template for in vitro transcription (right). The resulting RNA is transfected into cells where it is translated. In all cases translation of the RNA within transfected cells generates the viral replicase proteins that are necessary and sufficient to initiate virus replication and production of viral particles
Another problem encountered in the generation of reverse genetic systems was the fact that some plasmids containing viral c DNA were unstable in and/or induced cytotoxicity. In many cases, cytotoxicity or instability of the viral c DNA could be countered successfully by use of low copy plasmids with for example P15A origins of replication restricting the plasmid copy number to 1 or very few per cell. This approach was successful in all of the first infectious clones for pestiviruses (ncpBVDV, cpBVDV, and CSFV) [15–18] but failed in case of yellow fever virus ( YFV) . This problem led to the development of a strategy using two or more plasmids, each of which contained a different part of the virus-derived c DNA. The first YFV infectious c DNA clone (17D vaccine strain, first flavivirus infectious clone at all) consisted of two separated fragments corresponding to the 5′ and 3′ half of the genome, respectively. Infectious RNA was generated through in vitro ligation of the two fragments followed by in vitro transcription [19] (Fig. 2 right part).
Correct 5′ end 3′ ends of the viral genome are often very important for the success of a reverse genetics system, as many viruses rely on special structures at their termini for replication and/or translation. All systems described above used restriction enzyme sites introduced directly downstream of the viral c DNA to linearize the plasmids before run-off in vitro transcription to obtain RNA 3′ ends identical or as similar as possible to those of the viral genome. With regard to the 5′ end of the RNA the use of bacteriophage promoters (mostly T7 or SP6) allowed to transcribe RNA with a marginally modified or even the desired start since only a 5′ G residue is necessary for efficient transcription by these enzymes. All infectious c DNA clones for members of the were established with a T7 promoter directly upstream of the genomic c DNA and a blunt cutting restriction enzyme with a recognition site that directly overlaps the 3′ end of the genome to allow run-off in vitro transcription resulting in RNA identical to viral genomic RNA [15–19].
As an alternative to in vitro ligation of c DNA fragments an interesting approach based on reconstitution of full length viral genomic RNA via intracellular RNA recombination has been developed. RNA recombination is a naturally occurring process and very widespread in RNA viruses. It gives rise to new virus variants such as the cytopathic biotype of pestiviruses [20–24]. Recombination of RNA of positive strand RNA viruses that replicate in the cytoplasm of infected cells, is different from DNA recombination or cellular RNA splicing, in which dedicated cellular machinery joins the ends of the respective nucleic acids. Recombination of cytoplasmic RNA is thought to occur either through template switching by the RNA-dependent polymerase during genome replication or through breakage of the RNA and joining with other RNA ends [22]. Several experiments with pestivirus and poliovirus mutants have shown that RNA recombination can happen independently of active RNA replication [25, 26]. In these experiments RNA fragments that each encoded only part of the RNA-dependent RNA polymerase were co-transfected into cells and were sufficient to lead to the recovery of infectious virus. The fact that intracellular recombination of viral RNA occurs rather frequently has been used as a tool to manipulate viral genomes not (yet) accessible to reverse genetics systems by c DNA clones or similar approaches via recombination of (mutated) genome fragments [27–30].
The above mentioned instability of viral full length c DNA clones is in part dependent on the size of the viral genome. The first c DNA clones were established for members of the with genome sizes of about 7.5 kb [10, 12, 14]. Members of the have genome sizes of 9.5–14 kb. Coronaviruses have the largest RNA genomes and therefore remained inaccessible to reverse genetic systems based on c DNA clones for a long time. Instead, targeted mutagenesis was achieved through recombination of transfected in vitro- transcribed RNA representing only a part of the viral genome and full length viral RNA in infected cells [27–29]. It took 19 years from the first infectious full length c DNA clone of a picornavirus to a full length infectious c DNA clone of a member of the [31]. The latter construct was for the transmissible gastroenteritis virus ( TGEV) and used a bacterial artificial chromosome ( BAC) to propagate the large virus derived c DNA with low copy number, as parts of the genome were toxic to the bacteria. Furthermore, this c DNA clone contained the TGEV sequence downstream of a cytomegalovirus immediate early promoter and upon transfection of the DNA into cells, viral RNA was produced by the cellular RNA polymerase II, which then led to the production of infectious viral particles. The same year, a second c DNA system for TGEV was published using five separate plasmids which together contained the full length genome and needed to be assembled through in vitro ligation before RNA synthesis [32]. Yet another approach followed a year later for the avian coronavirus infectious bronchitis virus in which the genomic c DNA was inserted into the genome of vaccinia virus, a large DNA virus from the family Poxviridae [33]. However, also strategies based on the use of RNA recombination are still employed for establishment of recombinant coronaviruses [30].
Methods to generate the long viral c DNA have changed in the last 35 years. The first approaches were based on c DNA libraries made from purified virion RNA or RNA of infected cells [10, 15–19, 34–36]. Later, full length PCR amplification of viral genomes became feasible through the availability of proofreading polymerases that allowed generation of an infectious clone after a single round of reverse transcription, followed by long-range PCR [37, 38]. With the rapid development of nucleic acid synthesis and high throughput sequencing it is now possible to generate c DNA clones through synthesis of the corresponding DNA sequences simply with the knowledge of the sequence. This was first demonstrated once again with poliovirus, but recently a c DNA clone system based on synthetic plasmids was published for the coronavirus porcine epidemic diarrhea virus [39, 40].
Road Map to Recovery of Self-Replicating RNA
Development of a strategy for establishment of a reverse genetics system for a new virus, which allows generation of self-replicating RNA and recovery of recombinant virus, requires knowledge on the molecular biology of this virus and a variety of considerations with regard to the final aim of the approach. The first step will usually be the determination of the sequence of the viral genome including the correct 5′ and 3′ ends. The latter information can be obtained by so-called RACE technology (rapid amplification of c DNA ends), PCR based systems that nowadays are provided by different commercial suppliers. The knowledge of the sequence will provide the necessary information on the genome organization which helps to understand the gene expression strategy of the virus. An important question in this context concerns the mechanism promoting initiation of translation and replication of the viral genomic RNA. As described above, translation of the genome is necessary to provide the components of the replicase that starts genome replication and thereby initiates the viral life cycle. Positive strand RNA viruses have developed a variety of strategies to ensure initiation of translation of their RNA [41–43]. In most systems, the infectious c DNA construct can be designed in a way that cis-acting structures important for translation and replication of the genome-like RNA derived from the construct will be equivalent to what is found in the viral genome. There are, however, special cases providing problems. Caliciviruses have a protein called VPg covalently bound to 5′ end of the viral RNA, which functions as a substitute for the cap structure driving translation initiation in eukaryotic mRNAs. This protein is most likely also crucial for the RNA to be accepted as a substrate for RNA replication but cannot be easily linked to in vitro- transcribed viral RNA. Replacing VPg by a standard cap structure was found to be sufficient for translation and initiation of replication of the in vitro- transcribed RNA, but with quite low efficiency [36, 44, 45].
Similarly, the 3′ end of the viral RNA is important for successful recovery of self-replicating RNA. Many viruses contain a poly(A) tail at the 3′ end and thereby mimic the structure of a standard eukaryotic mRNA ensuring efficient translation. The poly(A) tail should also be important for replication of the viral RNA since it is the sequence at which transcription has to start during minus strand synthesis. Other viral genomes contain no poly(A) but for example specific secondary structures representing important cis-acting elements for both translation and RNA replication. As a general rule, any virus with a genome containing a poly(A) tail should also have such a sequence in its infectious c DNA construct, whereas viruses without a poly(A) tail can be expected to be very sensitive to changes in the sequence at their genomic 3′ end, so that steps should be undertaken to ensure generation of the correct genomic end during transcription.
When the necessary information on the viral genome and strategy of gene expression are available the next point to be decided is where and how transcription of the c DNA construct should occur. For the majority of reverse genetics systems for positive strand RNA viruses the genome-like RNA is generated in vitro and subsequently introduced into cells via transfection. This strategy is characterized by some methodical advantages, especially the simple generation of correct end sequences through use of bacteriophage RNA polymerases and “run-off” transcription. The transcription procedure was improved over the years so that highly efficient kits yielding large amounts of RNA became commercially available. The most common promoters used in in vitro transcription systems are the phage promoters T7 and SP6. These can be placed immediately upstream of the c DNA sequence ensuring a correct 5′ end of the resulting RNA. To obtain capped transcripts either a cap analog (like m7G(5′)ppp(5′)G) has to be included in the in vitro transcription reaction or the RNA needs to be capped after the in vitro transcription (using vaccinia virus derived capping systems). If the genomic RNA should contain a poly-A tail this needs to be either included in the template construct or added after transcription using a poly(A) polymerase . The second choice adds yet another step to the generation of the RNA and thus might reduce yield.
The above mentioned ways to introduction of a cap structure into in vitro-transcribed RNA work with only rather low efficiency. Thus, the alternative strategy relying on transfection of the plasmid DNA followed by in vivo transcription of the genome-like RNA can be advantageous when the production of capped transcripts is necessary, since RNA produced in transfected cells is 5′ capped and 3′ polyadenylated by cellular enzymes. A problem with this approach is the relatively high chance of further post-transcriptional modification of the RNA like splicing, which could abrogate any infectivity. To obtain correct genomic ends for non-polyadenylated viruses with this approach ribozyme sequences such as the hepatitis delta ribozyme can be added at the ends, which will cleave themselves off and leave the correct terminus [46]. In fact, reverse genetics systems for positive strand RNA viruses using direct transfection of DNA into cells are much rarer than in vitro transcription based approaches.
An interesting alternative combining features of the in vitro transcription system with the advantages of DNA transfection is based on helper viruses like the vaccinia virus MVA- T7. Cells infected with the latter virus contain bacteriophage T7 RNA polymerase expressed by MVA- T7 which upon introduction of plasmid constructs with T7 promoters will transcribe the desired RNA in the cytoplasm of the cell which avoids nuclear location and the danger of unwanted splicing of the RNA product. The defined start site of T7 based transcription allows for an easy production of the correct 5′ end just as during in vitro transcription. Insertion of ribozyme sequences into the plasmid can ensure the formation of the desired defined 3′ end of the transcript. Since vaccinia virus replicates in the cytoplasm it expresses enzymes that cap and polyadenylate its own transcripts efficiently, which is also true for the T7 transcripts. The final result is the efficient production of a capped and polyadenylated transcript with correct ends within the cell which can lead to superior performance compared to in vitro transcription/ transfection of RNA [45].
As mentioned before instability of viral sequences in E. coli while propagating the c DNA plasmids can be countered by different measures. It is preferable to use low copy plasmids or BACs to minimize the amount of plasmids with toxic sequences in the bacteria. Moreover, BACs can carry large inserts and thus are suitable for every positive strand RNA virus genome including those of coronaviruses. Sequences that seem to be deleterious for the propagation in E. coli can be disrupted by strategically placed intron sequences, if virus recovery is achieved via plasmid transfection into cells and intracellular RNA synthesis through RNA polymerase II. The intron will be spliced out of the produced RNA regenerating the viral sequence within the cells. This approach was employed in the production of a TGEV infectious clone [47].
Taken together, the establishment of systems for generation of self-replicating RNAs and recovery of infectious recombinant positive strand RNA viruses are in principle straight forward today but have to pay attention to the individual features of the respective system and the aims to be achieved. Depending on the system, more rarely used strategies like RNA recombination based generation of mutants might show up as the feasible solution. In fact, system specific problems had to be solved during development of almost any reverse genetics system that is routinely used now but the available repertoire of possible solutions for such problems will facilitate such approaches in the future.
Use of Self-Replicating RNA for Vaccine Purposes
The development of techniques allowing the recovery of self-replicating viral RNA from c DNA was not only a milestone for basic research on RNA virus biology but also opened a door to new approaches towards modified live vaccines against viral diseases. In contrast to the traditional ways relying for attenuation on elaborate passaging of viruses in tissue culture cells or unusual host animals, reverse genetics systems allow for defined mutagenesis and rational attenuation.
Vaccines Based on Full Length Viral RNA
Pestiviruses represent a good example for different approaches towards live attenuated viral vaccines. Members of the genus are economically important pathogens of farm animals that are grouped in the family together with their closest relatives, the hepaciviruses. Most important pestiviruses are the classical swine fever virus ( CSFV) and the bovine viral diarrhea virus ( BVDV) [48]. All members of the Flaviviridae are enveloped viruses with positive strand RNA genomes containing one long open reading frame. The economic impact of pestiviruses results at least in part from causing a wide range of pregnancy disorders and persistent infection due to their ability to cross the placenta in pregnant animals [48]. Persistently infected animals represent an important reservoir for virus spread. Vaccination represents a feasible means to interrupt the cycle of transmission as long as the vaccines do not only prevent disease but also fetal transmission of the pathogens. To fulfill the latter demand pestivirus vaccines have to be very potent.
The so-called CSFV C-strain represents an example of a successful pestivirus vaccine. It is a traditional modified live vaccine that was attenuated via serial passages in rabbit cells resulting in a very safe and efficient vaccine virus with so far undefined basis of attenuation. The latter is also true for different live BVDV strains used for vaccination in various countries worldwide. As an important disadvantage of these vaccines the attenuated viruses can still cross the placenta and infect the fetus in pregnant animals which in case of the conventional live BVDV vaccines can even lead to abortion. Using a reverse genetics approach we were able to establish a BVDV mutant with defined genomic deletions of nonessential sequences that knocked out two viral factors interfering with the host’s type 1 interferon response without significantly impairing viral replication [49]. As a consequence of these changes affecting viral mechanisms blocking the innate immune response to BVDV infection not only complete attenuation of the mutant virus was observed but also the inability to infect the fetus in pregnant animals, the prerequisite for pregnancy disorders and persistent infection.
Another approach based on deletion of nonessential sequences was described for coronaviruses. Members of the family represent important pathogens of man and animals among which SARS and MERS coronavirus (SARS-CoV and MERS-CoV) are best known [50, 51]. As outlined above, coronaviruses have by far the largest known RNA genomes which encode not only essential but also some nonessential accessory proteins. Deletion of five of the eight group-specific ORFs (ORF3a, OF3b, ORF6, ORF7a, and ORF7b), either alone or in various combinations, from the SARS-CoV genomic RNA did not result in clear indications for attenuation in a mouse model. In contrast, a viable SARS-CoV mutant lacking the sequence coding for the E protein (ORF4) was recovered that exhibited reduced virulence in two animal models probably by enhanced response of the immune response to the infection [52–56]. E represents one of the membrane bound structural proteins of the virus and is involved in virion assembly and release. Such deletion mutants are still being characterized and improved but might provide a basis for the development of coronavirus vaccines in the future.
Not only deletion of sequences but also exchange of genomic fragments between related viruses is easily done via reverse genetics and can lead to attenuation and other desired features. As an example, a chimeric pestivirus was established as a vaccine against classical swine fever (Fig. 3). This concept was based on the replacement of the region coding for the major envelope protein E2 of a BVDV genome by the corresponding sequence of CSFV. The resulting virus CP7_E2alf was only able to infect pigs and thus displayed the tropism of CSFV. The chimeric virus was fully attenuated but nevertheless induced strong protective immunity [57–59]. As an important further advantage, the configuration of this chimera allows for serologic differentiation between vaccinated animals and those having been infected by a CSFV field virus, a feature of major importance for control and eradication programs in veterinary medicine.
Fig. 3.

Generation of a chimeric viral genome from two parental RNAs. On the level of a c DNA construct, one protein-coding sequence is replaced by the corresponding gene of the other virus (principle used for the pestivirus vaccine CP7_E2alf [58]). internal ribosome entry site
Similar approaches as for CP7_E2alf were also used for members of the genus in the family . The first approach employed the yellow fever virus ( YFV) vaccine strain 17D, a virus developed in 1936 by empirical passage. YFV 17D is a very effective and safe vaccine that was found to highly trigger the innate immune response which helps driving the adaptive immune response to long lasting protective immunity [60–63]. Therefore, YFV 17D was chosen as backbone for a chimeric Japanese encephalitis/yellow fever virus vaccine (ChimeriVax™-JE) in which the surface protein prM/E coding region of YFV was replaced by the corresponding but modified JEV sequence resulting in a safe and effective vaccine launched by the end of 2012 (trade name IMOJEV™) [64]. Similar constructs in the 17D background were established with prM-E sequences from West Nile virus or the four dengue virus (DENV) genotypes and tested as vaccine candidates [65–69]. Also chimeric DENV composed of sequences from two different DENV genotypes and encompassing attenuating mutations were established and tested, as well as chimeras of DENV with tick-borne encephalitis virus sequences [70, 71].
The chimeric approach has also been followed in vaccine trials in alphaviruses, another group of positive strand RNA viruses belonging to the family . Low virulent Sindbis virus provided the backbone for these approaches that used exchange of the complete structural protein coding regions with sequences from highly virulent alphaviruses like Eastern or Western or Venezuelan equine encephalitis virus (EEE or WEE or VEE). The chimerization process itself led to significant attenuation of the resulting viruses that were found to be highly immunogenic (for review, see ref. 72). Nevertheless, the safety issue is a major concern in such approaches since biomarkers for the attenuation of Sindbis virus are not known and small animal models for testing virulence in most cases not adequate to evaluate attenuation in humans. Further research is needed to fully evaluate these vaccine candidates.
It has to be stressed that all the approaches described above employ self-replicating RNAs that represent either full length viral genomes or such RNA with deletion of nonessential sequences. Accordingly, these constructs allow the recovery of infectious virus particles. As presented above, introduction of the in vitro-transcribed recombinant RNA into a cell via transfection starts its autonomous replication leading to release of infectious viruses that after amplification in tissue culture serve as vaccine. Upon administration, the immune response is triggered because the vaccine virus mimics all steps of a field virus infection but without induction of significant symptoms.
Replicons as Vaccines
The use of fully replication competent recombinant viruses bears a certain risk of reversion to virulence. Depending on the type of attenuating mutations this risk can be significant or only theoretically relevant as for viruses containing more than one deletion. Introduction of deletions into RNA virus genomes can lead to recovery of attenuated viruses in some special cases but will in most cases result in RNAs that are no longer able to promote the generation of infectious progeny. As long as the deletions do not concern the sequences responsible for replication of the RNA, such mutant RNAs will behave as replicons that amplify autonomously when introduced into a cell and lead to translation of significant amounts of the encoded proteins. A typical replicon approach is based on deletion of sequences coding for one or more structural components of the virus (Fig. 4). Such replicons were important tools for research on RNA replication of for example pestiviruses and hepaciviruses [73, 74]. For pestiviruses, replicons have also been tested in vaccination approaches [75]. In all cases, essential sequences were deleted from the genomes so that the vaccine candidates need complementation in trans by stably transfected cells providing the missing factors. Infection of a host organism with the virus particles secreted from these complementing cells represents a dead-end since no infectious virus can be released from non-complementing cells. Thus, these vaccines cross the border between live attenuated viruses and killed vaccines exhibiting safety features at least very close to killed vaccines. The big advantage over killed vaccines is the ability to express viral proteins within cells leading to MHC presentation of viral peptides and activation of a T-cell response in addition to the humoral response.
Fig. 4.

Genome structure of a replicon. Deletion of a structural protein- coding region from the viral genome (represented by a dotted line) without interrupting the reading frame results in an RNA able to replicate autonomously within cells but deficient in production of infectious progeny
An interesting example of a further developed replicon approach is found in flaviviruses with the so-called pseudo-infectious vaccines. Integration of deletions of different length into the capsid protein coding region of the viral genomes results in autonomously replicating RNAs that are no longer able to promote the generation of infectious virus particles [72, 76, 77]. However, cells harboring these replicons will secrete large amounts of immunogenic prM/E particles. Propagation of such replicons in stably transfected cells providing the missing C protein in trans leads to virus-like particles able to conduct a single round of infection with highly efficient establishment of cells producing the prM/E particles. As a further approach, a DNA based vaccination has been developed that relies on two separate plasmids, one containing a c DNA representing the capsid-deleted viral genome and another expressing the missing capsid protein [72]. Cells that have taken up both plasmids will not only translate and present viral sequences but will also release infectious virus particles that can infect further cells leading to an enhanced stimulus of the immune system. Again, chimeric approaches with replicon backbones derived from one flavivirus species and prM/E coding sequences from another species have been tested successfully [72].
Self-Replicating RNAs as Vectors for Expression of Foreign Genes
The chimeric systems described above represent a special case of a more general approach towards vaccines based on self-replicating RNA that contain foreign sequences. Similar to the chimeric constructs mentioned before the replacement of viral protein-coding sequences by foreign genes can be used to express the desired proteins for immunization. In contrast to the chimeric viruses with a structural protein exchange between closely related viruses, such constructs will usually not yield autonomously propagating infectious viruses but replicons harboring non related sequences instead of the original structural proteins. Alternatively, the foreign sequences can be inserted into the viral genome as additional information without loss of essential viral functions so that fully replication-competent recombinant viruses can be produced. A prerequisite for the successful establishment of such self-replicating RNAs expressing foreign genes is the development of a strategy for integration and expression of the latter sequences without disturbing the autonomous replication of the RNA. Due to the fact that positive strand RNA viruses use expression strategies based on translation of polyproteins and subsequent proteolytic processing, the integration of foreign sequences into a viral open reading frame has to be combined with a specific processing step. A common approach avoiding fusion of significant numbers of unwanted residues to the proteins of interest is to place the foreign sequence at the 5′ end of the viral ORF and insert the foot and mouth disease virus (FMDV) 2A- coding region between the foreign sequence and the viral polyprotein (Fig. 5). FMDV 2A is a short peptide of 18 amino acids that is able to induce an irregular stop and restart of translation [78, 79]. In fact, 2A provokes an interruption of the polyprotein translation at its own carboxy terminus leading to release of an upstream protein fragment with 2A at its C-terminus and restart of translation with the proline following 2A in the polyprotein, so that the viral proteins following downstream are free of any added residues. An elegant approach avoiding any fusion of unwanted residues relies on the establishment of bicistronic RNAs. In such constructs the foreign sequence is usually also placed at the 5′ end of the ORF with a stop codon at the desired end of the translated region. Instead of a protein coding region ensuring processing of proteins an internal ribosome entry site ( IRES) is integrated between foreign sequence and the viral ORF (Fig. 6) [74, 80]. The foreign sequence is expressed using the strategy that initiates translation of the viral proteins in the wt virus. Its translation terminates at a stop codon at the 3′ end. The IRES recruits ribosomes to the start site at the 5′ end of the viral ORF and thereby promotes translation of the proteins necessary for replication of the recombinant RNA. An alternative arrangement has been published for BVDV, in which IRES and foreign sequence are placed in the 3′ NTR (Fig. 6) [81].
Fig. 5.

Expression of a foreign protein from a viral genome via the viral polyprotein. The foreign sequence is inserted at the 5′ end of the viral ORF followed by a sequence coding for the FMDV 2A that promotes the separation of the foreign protein from the viral polyprotein during translation of the recombinant RNA
Fig. 6.

Schematic representation of bicistronic self-replicating RNAs. On top, a standard positive strand virus genome with a single long open reading frame is shown (similar to RNA of picornaviruses or pestiviruses). Foreign sequences can be inserted together with a second IRES (internal ribosome entry site) instead of structural protein-coding sequences (bicistronic replicon—middle) or as insertion (bicistronic virus, bottom panel)
Similarly, viruses like alphaviruses that use subgenomic RNAs for expression of their structural proteins can be adapted to expression of foreign sequences with an approach relying on the standard genome organization and expression strategy of the viruses. The viral RNA contains promoter sequences that recruit the viral RNA polymerase to internal sites of the minus strand RNA replication intermediate and start transcription of a mRNA of subgenomic length [82, 83]. Replacing the viral structural protein coding sequence downstream of this internal promoter with the desired foreign sequence will lead to a replicon that transcribes an mRNA coding for the foreign protein (Fig. 7). Alternatively, the subgenomic RNA promoter can be duplicated and inserted together with the desired foreign sequence as an additional cistron into the viral genomic RNA thereby preserving its ability to drive the generation of infectious replication competent virus particles. Based on these principles, a variety of vaccine strategies has been developed [71].
Fig. 7.
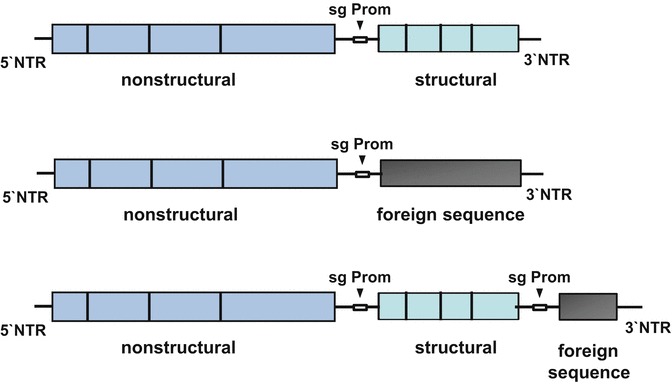
Strategies to express foreign sequences in alphaviruses. On top, a standard alphavirus genome is shown. It contains two open reading frames, the second of which is expressed from as subgenomic mRNA transcribed under control of the subgenomic promoter (sg Prom). Replacement of the structural protein coding second ORF by a foreign sequence will lead to a replicon expressing the desired protein (middle), whereas insertion of the foreign sequence downstream of a duplicated sg promoter generates a recombinant virus expressing the desired sequence via a second sg mRNA (bottom). See also Fig. 1
As a matter of fact, basically all approaches using self-replicating RNA for vaccination employ packaging of the RNA into virions or virus-like particles. The reason for this preference over naked or stabilized RNA is based on the extraordinary performance of the viral infection machinery resulting in highly efficient delivery of the RNA into cells. Since self-replicating RNAs derived from viral genomes exhibit the intrinsic property for specific packaging into viral particles, the use of this strategy is easy and straight forward. It has, however, to be mentioned that many virus particles display quite limited stability so that approaches relying on stabilized RNA could well be advantageous in certain situations especially when a cold chain during transport and delivery cannot be provided. An interesting opportunity for the future could be a vaccine formulation containing the RNA genome of a fully replication competent attenuated virus in stabilized form so that infectious virus is generated in the vaccinee upon administration. This approach would combine the superior resistance of stabilized RNA with the efficacy of a modified live viral vaccine.
Contributor Information
Thomas Kramps, Phone: +494949-176-7257-4818, Email: thkramps@gmail.com.
Knut Elbers, Phone: +4949+49 (6132) 77-143060, FAX: +4949+49 (6132) 77-2509, Email: nut.elbers@boehringer-ingelheim.com.
Birke Andrea Tews, Email: Birke.Tews@fli.de.
Gregor Meyers, Email: gregor.meyers@fli.de.
References
- 1.Colter JS, Bird HH, Brown RA. Infectivity of ribonucleic acid from Ehrlich ascites tumour cells infected with Mengo encephalitis. Nature. 1957;179(4565):859–860. doi: 10.1038/179859a0. [DOI] [PubMed] [Google Scholar]
- 2.Colter JS, Bird HH, Moyer AW, Brown RA. Infectivity of ribonucleic acid isolated from virus-infected tissues. Virology. 1957;4(3):522–532. doi: 10.1016/0042-6822(57)90084-3. [DOI] [PubMed] [Google Scholar]
- 3.Sprunt K, Redman WM, Alexander HE. Infectious ribonucleic acid derived from enteroviruses. Proc Soc Exp Biol Med Soc Exp Biol Med. 1959;101:604–608. doi: 10.3181/00379727-101-25033. [DOI] [PubMed] [Google Scholar]
- 4.Holland JJ, Hoyer BH, Mc LL, Syverton JT. Enteroviral ribonucleic acid. I. Recovery from virus and assimilation by cells. J Exp Med. 1960;112:821–839. doi: 10.1084/jem.112.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holland JJ, Mc LL, Hoyer BH, Syverton JT. Enteroviral ribonucleic acid. II. Biological, physical, and chemical studies. J Exp Med. 1960;112:841–864. doi: 10.1084/jem.112.5.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ketler A, Hamparian VV, Hilleman MR. Characterization and classification of ECHO 28-rhinovirus-coryzavirus agents. Proc Soc Exp Biol Med Soc Exp Biol Med. 1962;110:821–831. doi: 10.3181/00379727-110-27662. [DOI] [PubMed] [Google Scholar]
- 7.Dimmock NJ. Biophysical studies of a rhinovirus. Extraction and assay of infectious ribonucleic acid. Nature. 1966;209(5025):792–794. doi: 10.1038/209792a0. [DOI] [PubMed] [Google Scholar]
- 8.Mogler MA, Kamrud KI. RNA-based viral vectors. Expert Rev Vaccines. 2015;14(2):283–312. doi: 10.1586/14760584.2015.979798. [DOI] [PubMed] [Google Scholar]
- 9.Conzelmann KK, Meyers G. Genetic engineering of animal RNA viruses. Trends Microbiol. 1996;4(10):386–393. doi: 10.1016/0966-842X(96)10062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Racaniello VR, Baltimore D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science. 1981;214(4523):916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- 11.Ahlquist P, French R, Janda M, Loesch-Fries LS. Multicomponent RNA plant virus infection derived from cloned viral cDNA. Proc Natl Acad Sci U S A. 1984;81(22):7066–7070. doi: 10.1073/pnas.81.22.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizutani S, Colonno RJ. In vitro synthesis of an infectious RNA from cDNA clones of human rhinovirus type 14. J Virol. 1985;56(2):628–632. doi: 10.1128/jvi.56.2.628-632.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baltimore D. Expression of animal virus genomes. Bacteriol Rev. 1971;35(3):235–241. doi: 10.1128/br.35.3.235-241.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semler BL, Dorner AJ, Wimmer E. Production of infectious poliovirus from cloned cDNA is dramatically increased by SV40 transcription and replication signals. Nucleic Acids Res. 1984;12(12):5123–5141. doi: 10.1093/nar/12.12.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moormann RJ, van Gennip HG, Miedema GK, Hulst MM, van Rijn PA. Infectious RNA transcribed from an engineered full-length cDNA template of the genome of a pestivirus. J Virol. 1996;70(2):763–770. doi: 10.1128/jvi.70.2.763-770.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyers G, Tautz N, Becher P, Thiel HJ, Kümmerer BM. Recovery of cytopathogenic and noncytopathogenic bovine viral diarrhea viruses from cDNA constructs. J Virol. 1996;70(12):8606–8613. doi: 10.1128/jvi.70.12.8606-8613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyers G, Thiel HJ, Rümenapf T. Classical swine fever virus: recovery of infectious viruses from cDNA constructs and generation of recombinant cytopathogenic defective interfering particles. J Virol. 1996;70(3):1588–1595. doi: 10.1128/jvi.70.3.1588-1595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruggli N, Tratschin JD, Mittelholzer C, Hofmann MA. Nucleotide sequence of classical swine fever virus strain Alfort/187 and transcription of infectious RNA from stably cloned full-length cDNA. J Virol. 1996;70(6):3478–3487. doi: 10.1128/jvi.70.6.3478-3487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1989;1(3):285–296. [PubMed] [Google Scholar]
- 20.Becher P, Orlich M, König M, Thiel HJ (1999) Nonhomologous RNA recombination in bovine viral diarrhea virus: molecular characterization of a variety of subgenomic RNAs isolated during an outbreak of fatal mucosal disease. J Virol 73(7):5646–5653 [DOI] [PMC free article] [PubMed]
- 21.Becher P, Orlich M, Thiel HJ. RNA recombination between persisting pestivirus and a vaccine strain: generation of cytopathogenic virus and induction of lethal disease. J Virol. 2001;75(14):6256–6264. doi: 10.1128/JVI.75.14.6256-6264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becher P, Tautz N. RNA recombination in pestiviruses: cellular RNA sequences in viral genomes highlight the role of host factors for viral persistence and lethal disease. RNA Biol. 2011;8(2):216–224. doi: 10.4161/rna.8.2.14514. [DOI] [PubMed] [Google Scholar]
- 23.Meyers G, Rümenapf T, Thiel HJ. Ubiquitin in a togavirus. Nature. 1989;341:491. doi: 10.1038/341491a0. [DOI] [PubMed] [Google Scholar]
- 24.Meyers G, Thiel HJ. Molecular characterization of pestiviruses. Adv Virus Res. 1996;47:53–117. doi: 10.1016/S0065-3527(08)60734-4. [DOI] [PubMed] [Google Scholar]
- 25.Gallei A, Pankraz A, Thiel HJ, Becher P. RNA recombination in vivo in the absence of viral replication. J Virol. 2004;78(12):6271–6281. doi: 10.1128/JVI.78.12.6271-6281.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gmyl AP. Nonreplicative homologous RNA recombination: promiscuous joining of RNA pieces? RNA. 2003;9(10):1221–1231. doi: 10.1261/rna.5111803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koetzner CA, Parker MM, Ricard CS, Sturman LS, Masters PS. Repair and mutagenesis of the genome of a deletion mutant of the coronavirus mouse hepatitis virus by targeted RNA recombination. J Virol. 1992;66(4):1841–1848. doi: 10.1128/jvi.66.4.1841-1848.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao CL, Lai MMC. RNA recombination in a coronavirus - recombination between viral genomic RNA and transfected RNA fragments. J Virol. 1992;66(10):6117–6124. doi: 10.1128/jvi.66.10.6117-6124.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Most RG, Heijnen L, Spaan WJ, de Groot RJ. Homologous RNA recombination allows efficient introduction of site-specific mutations into the genome of coronavirus MHV-A59 via synthetic co-replicating RNAs. Nucleic Acids Res. 1992;20(13):3375–3381. doi: 10.1093/nar/20.13.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masters PS, Rottier PJ. Coronavirus reverse genetics by targeted RNA recombination. Curr Top Microbiol Immunol. 2005;287:133–159. doi: 10.1007/3-540-26765-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almazan F, Gonzalez JM, Penzes Z, Izeta A, Calvo E, Plana-Duran J, Enjuanes L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A. 2000;97(10):5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yount B, Curtis KM, Baric RS. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J Virol. 2000;74(22):10600–10611. doi: 10.1128/JVI.74.22.10600-10611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casais R, Thiel V, Siddell SG, Cavanagh D, Britton P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J Virol. 2001;75(24):12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Racaniello VR, Baltimore D. Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc Natl Acad Sci U S A. 1981;78(8):4887–4891. doi: 10.1073/pnas.78.8.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rice CM, Levis R, Strauss JH, Huang HV. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J Virol. 1987;61(12):3809–3819. doi: 10.1128/jvi.61.12.3809-3819.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sosnovtsev S, Green KY. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology. 1995;210(2):383–390. doi: 10.1006/viro.1995.1354. [DOI] [PubMed] [Google Scholar]
- 37.Chumakov KM. PCR engineering of viral quasispecies: a new method to preserve and manipulate genetic diversity of RNA virus populations. J Virol. 1996;70(10):7331–7334. doi: 10.1128/jvi.70.10.7331-7334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen TB, Reimann I, Hoffmann B, Depner K, Uttenthal A, Beer M. Direct recovery of infectious pestivirus from a full-length RT-PCR amplicon. J Virol Methods. 2008;149(2):330–333. doi: 10.1016/j.jviromet.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 39.Cello J, Paul AV, Wimmer E. Chemical synthesis of poliovirus cDNA: generation of infectious virus in the absence of natural template. Science. 2002;297(5583):1016–1018. doi: 10.1126/science.1072266. [DOI] [PubMed] [Google Scholar]
- 40.Beall A, Yount B, Lin CM, Hou Y, Wang Q, Saif L, Baric R. Characterization of a pathogenic full-length cDNA clone and transmission model for porcine epidemic diarrhea virus strain PC22A. MBio. 2016;7(1):e01451–15. doi: 10.1128/mBio.01451-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bushell M, Sarnow P. Hijacking the translation apparatus by RNA viruses. J Cell Biol. 2002;158(3):395–399. doi: 10.1083/jcb.200205044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firth AE, Brierley I. Non-canonical translation in RNA viruses. J Gen Virol. 2012;93(Pt 7):1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Lastra M, Ramdohr P, Letelier A, Vallejos M, Vera-Otarola J, Valiente-Echeverria F. Translation initiation of viral mRNAs. Rev Med Virol. 2010;20(3):177–195. doi: 10.1002/rmv.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sosnovtsev SV, Belliot G, Chang KO, Onwudiwe O, Green KY. Feline calicivirus VP2 is essential for the production of infectious virions. J Virol. 2005;79(7):4012–4024. doi: 10.1128/JVI.79.7.4012-4024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thumfart JO, Meyers G. Feline calicivirus: recovery of wild-type and recombinant viruses after transfection of cRNA or cDNA constructs. J Virol. 2002;76(12):6398–6407. doi: 10.1128/JVI.76.12.6398-6407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnell MJ, Mebatsion T, Conzelmann KK. Infectious rabies viruses from cloned cDNA. EMBO J. 1994;13(18):4195–4203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez JM, Penzes Z, Almazan F, Calvo E, Enjuanes L. Stabilization of a full-length infectious cDNA clone of transmissible gastroenteritis coronavirus by insertion of an intron. J Virol. 2002;76(9):4655–4661. doi: 10.1128/JVI.76.9.4655-4661.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tautz N, Tews BA, Meyers G. The molecular biology of pestiviruses. Adv Virus Res. 2015;93:47–160. doi: 10.1016/bs.aivir.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Meyers G, Ege A, Fetzer C, von Freyburg M, Elbers K, Carr V, Prentice H, Charleston B, Schürmann EM. Bovine viral diarrhoea virus: prevention of persistent foetal infection by a combination of two mutations affecting the Erns RNase and the Npro protease. J Virol. 2007;81(7):3327–3338. doi: 10.1128/JVI.02372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol. 2004;7(4):412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, Denison MR, Davis N, Baric RS. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol. 2005;79(23):14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeDiego ML, Alvarez E, Almazan F, Rejas MT, Lamirande E, Roberts A, Shieh WJ, Zaki SR, Subbarao K, Enjuanes L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J Virol. 2007;81(4):1701–1713. doi: 10.1128/JVI.01467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dediego ML, Pewe L, Alvarez E, Rejas MT, Perlman S, Enjuanes L. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology. 2008;376(2):379–389. doi: 10.1016/j.virol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamirande EW, DeDiego ML, Roberts A, Jackson JP, Alvarez E, Sheahan T, Shieh WJ, Zaki SR, Baric R, Enjuanes L, Subbarao K. A live attenuated severe acute respiratory syndrome coronavirus is immunogenic and efficacious in golden Syrian hamsters. J Virol. 2008;82(15):7721–7724. doi: 10.1128/JVI.00304-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Netland J, DeDiego ML, Zhao J, Fett C, Alvarez E, Nieto-Torres JL, Enjuanes L, Perlman S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology. 2010;399(1):120–128. doi: 10.1016/j.virol.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beer M, Reimann I, Hoffmann B, Depner K. Novel marker vaccines against classical swine fever. Vaccine. 2007;25(30):5665–5670. doi: 10.1016/j.vaccine.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 58.Koenig P, Lange E, Reimann I, Beer M. CP7_E2alf: a safe and efficient marker vaccine strain for oral immunisation of wild boar against Classical swine fever virus (CSFV) Vaccine. 2007;25(17):3391–3399. doi: 10.1016/j.vaccine.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 59.Leifer I, Lange E, Reimann I, Blome S, Juanola S, Duran JP, Beer M. Modified live marker vaccine candidate CP7_E2alf provides early onset of protection against lethal challenge infection with classical swine fever virus after both intramuscular and oral immunization. Vaccine. 2009;27(47):6522–6529. doi: 10.1016/j.vaccine.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 60.Gaucher D, Therrien R, Kettaf N, Angermann BR, Boucher G, Filali-Mouhim A, Moser JM, Mehta RS, Drake DR, 3rd, Castro E, Akondy R, Rinfret A, Yassine-Diab B, Said EA, Chouikh Y, Cameron MJ, Clum R, Kelvin D, Somogyi R, Greller LD, Balderas RS, Wilkinson P, Pantaleo G, Tartaglia J, Haddad EK, Sekaly RP. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205(13):3119–3131. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203(2):413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009;10(1):116–125. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Querec TD, Pulendran B. Understanding the role of innate immunity in the mechanism of action of the live attenuated Yellow Fever Vaccine 17D. Adv Exp Med Biol. 2007;590:43–53. doi: 10.1007/978-0-387-34814-8_3. [DOI] [PubMed] [Google Scholar]
- 64.Monath TP, Guirakhoo F, Nichols R, Yoksan S, Schrader R, Murphy C, Blum P, Woodward S, McCarthy K, Mathis D, Johnson C, Bedford P. Chimeric live, attenuated vaccine against Japanese encephalitis (ChimeriVax-JE): phase 2 clinical trials for safety and immunogenicity, effect of vaccine dose and schedule, and memory response to challenge with inactivated Japanese encephalitis antigen. J Infect Dis. 2003;188(8):1213–1230. doi: 10.1086/378356. [DOI] [PubMed] [Google Scholar]
- 65.Guirakhoo F, Kitchener S, Morrison D, Forrat R, McCarthy K, Nichols R, Yoksan S, Duan X, Ermak TH, Kanesa-Thasan N, Bedford P, Lang J, Quentin-Millet MJ, Monath TP. Live attenuated chimeric yellow fever dengue type 2 (ChimeriVax-DEN2) vaccine: phase I clinical trial for safety and immunogenicity: effect of yellow fever pre-immunity in induction of cross neutralizing antibody responses to all 4 dengue serotypes. Hum Vaccines. 2006;2(2):60–67. doi: 10.4161/hv.2.2.2555. [DOI] [PubMed] [Google Scholar]
- 66.Guirakhoo F, Weltzin R, Chambers TJ, Zhang ZX, Soike K, Ratterree M, Arroyo J, Georgakopoulos K, Catalan J, Monath TP. Recombinant chimeric yellow fever-dengue type 2 virus is immunogenic and protective in nonhuman primates. J Virol. 2000;74(12):5477–5485. doi: 10.1128/JVI.74.12.5477-5485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monath TP, Arroyo J, Miller C, Guirakhoo F. West Nile virus vaccine. Curr Drug Targets Infect Disord. 2001;1(1):37–50. doi: 10.2174/1568005013343254. [DOI] [PubMed] [Google Scholar]
- 68.Monath TP, Liu J, Kanesa-Thasan N, Myers GA, Nichols R, Deary A, McCarthy K, Johnson C, Ermak T, Shin S, Arroyo J, Guirakhoo F, Kennedy JS, Ennis FA, Green S, Bedford P. A live, attenuated recombinant West Nile virus vaccine. Proc Natl Acad Sci U S A. 2006;103(17):6694–6699. doi: 10.1073/pnas.0601932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pugachev KV, Guirakhoo F, Trent DW, Monath TP. Traditional and novel approaches to flavivirus vaccines. Int J Parasitol. 2003;33(5-6):567–582. doi: 10.1016/S0020-7519(03)00063-8. [DOI] [PubMed] [Google Scholar]
- 70.Pletnev AG, Bray M, Lai CJ. Chimeric tick-borne encephalitis and dengue type 4 viruses: effects of mutations on neurovirulence in mice. J Virol. 1993;67(8):4956–4963. doi: 10.1128/jvi.67.8.4956-4963.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Monath TP. Recombinant, chimeric, live, attenuated vaccines against Flaviviruses and Alphaviruses. In: Dormitzer PR, Mandel CW, Rappuoli R, editors. Replicating vaccines: a new generation. Basel: Springer; 2011. pp. 349–438. [Google Scholar]
- 72.Roby JA, Hall RA, Khromykh AA. Nucleic acid-based infections and pseudo-infectious flavivirus vaccines. In: Dormitzer PR, Mandel CW, Rappuoli R, editors. Replicating vaccines: a new generation. Basel: Springer; 2011. pp. 299–320. [Google Scholar]
- 73.Behrens SE, Grassmann CW, Thiel HJ, Meyers G, Tautz N. Characterization of an autonomous subgenomic pestivirus RNA replicon. J Virol. 1998;72(3):2364–2372. doi: 10.1128/jvi.72.3.2364-2372.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lohmann V, Körner F, Koch JO, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 75.Reimann I, Semmler I, Beer M. Packaged replicons of bovine viral diarrhea virus are capable of inducing a protective immune response. Virology. 2007;366(2):377–386. doi: 10.1016/j.virol.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 76.Kofler RM, Aberle JH, Aberle SW, Allison SL, Heinz FX, Mandl CW. Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc Natl Acad Sci U S A. 2004;101(7):1951–1956. doi: 10.1073/pnas.0307145101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mandl CW. Flavivirus immunization with capsid-deletion mutants: basics, benefits, and barriers. Viral Immunol. 2004;17(4):461–472. doi: 10.1089/vim.2004.17.461. [DOI] [PubMed] [Google Scholar]
- 78.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol. 2001;82(Pt 5):1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 79.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol. 2001;82(Pt 5):1013–1025. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- 80.Tautz N, Harada T, Kaiser A, Rinck G, Behrens SE, Thiel HJ. Establishment and characterization of cytopathogenic and noncytopathogenic pestivirus replicons. J Virol. 1999;73(11):9422–9432. doi: 10.1128/jvi.73.11.9422-9432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baroth M, Peters Y, Schönbrunner ER, Behrens SE (2010) Stable recombinants of bovine viral diarrhea virus containing a hepatitis C virus insert. J Gen Virol 91(Pt 5):1213–1217. doi:10.1099/vir.0.016998-0 [DOI] [PubMed]
- 82.Agapov EV, Frolov I, Lindenbach BD, Pragai BM, Schlesinger S, Rice CM. Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc Natl Acad Sci U S A. 1998;95:12989–12994. doi: 10.1073/pnas.95.22.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]