

Abstract
The surface envelope protein of any virus is major determinant of the host cell that is infected and as a result a major determinant of viral pathogenesis. Retroviruses have a single surface protein named Env. It is a trimer of heterodimers and is responsible for binding to the host cell receptor and mediating fusion between the viral and host membranes. In this review we will discuss the history of the discovery of the avian leukosis virus (ALV) and human immunodeficiency virus type 1 (HIV-1) Env proteins and their receptor specificity, comparing the many differences but having some similarities. Much of the progress in these fields has relied on viral genetics and genetic polymorphisms in the host population. A special feature of HIV-1 is that its persistent infection in its human host, to the point of depleting its favorite target cells, allows the virus to evolve new entry phenotypes to expand its host range into several new cell types. This variety of entry phenotypes has led to confusion in the field leading to the major form of entry phenotype of HIV-1 being overlooked until recently. Thus an important part of this story is the description and naming of the most abundant entry form of the virus: R5 T cell-tropic HIV-1.
Introduction
Retroviruses are sloppy. One can marvel at the regularity of the surface envelope protein of flaviviruses and alphaviruses where the proteins outside of the membrane are organized with the icosahedral shell inside of the membrane. One can envy the high density of envelope proteins that decorate the surface of coronaviruses and influenza virus. While some retroviruses show-off a high density of their surface envelope protein (Env), for others inclusion of the Env protein almost seems like an afterthought, given the apparent low density of Env on the virus surface (Zhu et al. 2006; Martin et al. 2016). In the case of HIV-1, the Env protein appears on the surface of the cell and if not quickly incorporated into a budding virion it is cycled back off the surface (Rowell et al. 1995; Sauter et al. 1996), presumably to avoid marking the cell as infected, a biologic example of “use it or lose it.”
All retroviruses encode an Env protein, representing a primordial strategy for how at least some viral particles are able to fuse their membranes with the target cellular membrane. There are cellular proteins that can fuse membranes, for example the SNAREs, but the details of how they accomplish this are sufficiently different that it is hard to argue they are the origin of the viral envelope proteins like retroviruses use. Ironically it is easier to make the opposite claim. The cellular protein syncytin 1 is responsible for fusing cells to make a multicellular placenta in primates. Syncytin 1 is derived from an endogenous retrovirus whose env gene is now developmentally regulated. When expressed it fuses adjacent cells in the same way a viral membrane is fused to the cellular membrane; similar capture events appear to have happened in other eutherian mammals (Cornelis et al. 2013). The current lack of a true cellular protein that acts like the retroviral Env protein, and the fact that the retroviral Env protein functions in the same way as the influenza viral protein (and the surface protein of other distant viruses too), points to an early evolution of this type of membrane fusion capacity in enveloped viruses with this gene evolving with viruses as they generated different lineages. It is also likely that this gene has been passed among different viral lineages followed by distinctive evolution within the lineages to retain the basic fusion mechanism but with greatly varying sequence.
There are several universal points about this class of viral entry proteins (Fig. 1). First, they are type 1 transmembrane (TM) proteins with an N-terminal signal sequence that threads them into the endoplasmic reticulum and a stop-transfer sequence that stays in the membrane near the C terminus of the protein. This gives a luminal/outside N terminus of the protein and leaves the C terminus “inside” of the cell, which later becomes the inside of the virus particle when it buds from the cell taking some of the cellular membrane as its envelope. Second, there is an obligatory trimerization of the protein to stabilize its structure. Third, each subunit of the trimer is cleaved by a host protein (furin or furin-like) in the late Golgi compartment to create an extracellular component and a TM component, both of which are retained in the trimer complex and with each other. Fourth, the extracellular component (called SU for surface component in retrovirus nomenclature) of the cleaved protein is responsible for interacting with the host receptor(s), while the TM component exposes a hydrophobic stretch of amino acids (fusion peptide) at its new N terminus that is able to insert into the host membrane to facilitate fusion of host and viral membranes. There is a signal transduction event between the SU and TM components of the protein that occurs when the SU component binds its receptor thus defining the moment when fusion to the host cell must occur. There are diverse host cell receptors for viruses with this type of surface envelope protein, and the protein domains that bind the receptor and transduce the signal have lost any semblance of a common ancestor. And fifth, in contrast to the diversity of SU protein function the fusion mechanism is highly conserved, in function if not in sequence. Insertion of the TM N-terminal fusion peptide into the host cell membrane is linked to the formation of a proximal trimer of a heptad repeat. This is followed by a more distal heptad repeat that folds up on the N-terminal heptad repeat to form a hairpin. Both of these heptad repeats (hr) are in the extracellular domain of TM such that the N-terminal repeat (hr1) is adjacent to the fusion peptide inserted into the host membrane and the C-terminal heptad repeat (hr2) is adjacent to the viral membrane-spanning portion of TM; the juxtaposition of the heptad repeats in the hairpin brings the two membranes together to promote fusion, bringing the inside of the virus particle into the inside of the target host cell.
Fig. 1.
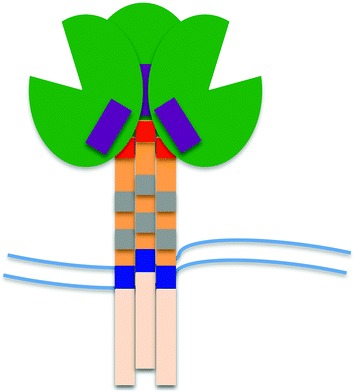
The retroviral Env protein. The viral Env protein exists as a trimer of heterodimers (SU/TM) embedded in the viral membrane (parallel wavy lines). The TM/transmembrane protein is orange with the lighter shade of orange indicating the cytoplasmic domain, the dark blue portion indicating the membrane-spanning domain, the gray regions representing the heptad repeats (hr1 and hr2) involved in the formation of the six helix bundle, and the red portion indicating the N-terminal fusion peptide. The green region represents the receptor binding/surface/SU protein, with the cleft indicating the receptor-binding region. The purple box indicates the region for HIV-1 that rearranges after binding CD4 to form the coreceptor binding site
There are several other curious points to mention about viruses that use this type of viral entry protein. First, the somewhat loose relationship between the TM viral envelope protein and the viral capsid inside of the viral envelope allows other proteins to be incorporated as TM proteins into the virus particle. Cellular proteins can be incorporated and there is an ongoing interest as to whether such proteins can alter the biology of the virus particle. Second, other viral proteins can be incorporated into the virus envelope which now gives the virus a host range specificity not encoded in its own genome, a phenomenon known as pseudotyping. Third, the virus-producing cell still expresses the cellular receptor. Since the virus does not what its envelope proteins interacting with the host cell receptor on a cell that is already infected, viruses use a variety of strategies of lowering the amount of receptor on the surface of the infected cell and regulating the fusogenicity of the envelope protein until the virus particle has budded. The overall effect of either down-regulating the receptor or tying up the receptor with newly synthesized Env protein prevents the infected cell from getting super-infected (usually), a phenomenon known as interference.
In this review we will focus on the SU domain of two very different retroviruses: avian leukosis virus (ALV) and human immunodeficiency virus type 1 (HIV-1). We will examine what sequence evolution can tell us about the proteins, we will look at their receptors and the conformational changes induced by binding to the receptor, and we will explore how these proteins can change host range to target different cell types. Some commonalities will emerge, but it will also be striking how different these two groups of viruses have solved the challenge of host cell targeting.
Avian Leukosis Virus (ALV)
It was around 1900 that enough evidence was accumulating to make the concept of a virus tenable. At that time most of the evidence was based on identifying a transmissible disease-causing agent that could be passed through a filter that would retain bacteria. There was an ongoing discussion as to whether these filterable agents could be very very small bacteria (but still independently living), toxins, or viruses that could only replicate within a host cell. As viruses became synonymous with filterable agents the list of viral diseases grew. There were no tools to measure a virus directly so disease was the only readout initially. Thus Ellermann and Bang described the first ALV-associated disease with the description of avian leukemia in 1908. Within a few years Peyton Rous would get credit for describing the first tumor-causing virus, Rous sarcoma virus, in 1910, for which he would get the Nobel Prize in 1966.
There was of course an underlying biology of these viruses that could not have been guessed in 1910, with genetic heterogeneity in the chickens defining susceptibility and in the virus changing receptors, and even endogenous viruses in the chicken genome. These phenomena were all revealed slowly, initially as unexpected observations that had to be reproduced enough so that a description could be formulated that could then be used to support suggestions of underlying mechanisms.
History of Viruses and Susceptibility
When disease is the only readout progress is slow. In a retrospective published by Harry Rubin in 2011, he reviewed the role new assays played in developing an understanding of the viruses, not just the disease (Rubin 2011). In 1938 Keogh showed that (tumor-like) lesions could be produced on the chorioallantoic membrane (CAM) of a chicken egg providing a semi-quantitative alternative to titering virus in chickens. It took another 20 years until Prince discovered that the high number of false negatives (i.e., eggs that gave no lesions) was a genetic trait of the chicken strains used, defining a sensitive and resistant allele for the virus used, and the first measure of the viral receptor. It was also around this time that Howard Temin, then a graduate student collaborating with Harry Rubin, perfected the focus-forming assay with an agar overlay to allow titration of the transforming activity of RSV. Each egg could be turned into multiple plates of chicken embryo fibroblasts (CEFs) and read much easier than counting lesions on CAM.
With a new, more powerful, assay the genetics of the virus started to come into focus. A subset of embryos that should have given cells that were susceptible to transformation by RSV were in fact resistant, defining a resistance-inducing factor (RIF). For a number of years avian leukosis viruses had been studied for their natural infection of chickens, and it turned out that the RIF was the result of the occasional embryo that was already infected with an ALV. This revealed the phenomenon of interference, the mechanism being that when a cell is already expressing a viral envelope protein it removes or engages its normal receptor precluding infection with a new virus with the same receptor specificity.
It had been 50 years of research and experimental passaging of Rous sarcoma virus by this time so it should not be surprising that different virus stocks had acquired different properties and even different passenger viruses. RSV is unique in that it can be propagated as a replication competent virus, in contrast to most other acutely transforming retroviruses that carry deletion of part of the viral genome with the acquisition of the cellular oncogene. But RSV will delete the v-src gene to give rise to a non-acutely transforming, replication competent Rous-associated virus (RAV), again essentially a standard ALV. However, the isolation of the first RAV (RAV-1) with its associated interference properties was followed by the isolation of a second RAV (RAV-2) with distinct interference properties. The clear implication was that these two viruses used different host receptors. Furthermore, the isolation of defective forms of the transforming virus component (defective for replication) allowed rescue of the transforming genome with any ALV helper virus, with the transforming component now having the cell infectivity properties of the helper virus (i.e., a pseudotype). The combination of cell susceptibility to infection and transformation, and the ability to test viruses for the use of the same or different receptors through interference led to the identification of virus subgroups, specifically subgroups A, B, C, D, and E (with J coming later). Conversely it was possible to identify the genetic loci for susceptibility in chickens, the presumed receptors which were named tva, tvb, and tvc (with tvb serving a receptor for subgroups D and E ALV). Thus great strides in the genetics of the virus and the host were accomplished using the focus-forming assay. These insights into the biology of the virus and the host set the stage for the coming tools of cloning and sequencing.
The Viral Env Gene/Protein
The development of electron microscopy led to the ability to “see” viruses and it became apparent that viruses had complex structures. Advances in growing and purifying the virus, and in protein analysis allowed for the identification of proteins associated with the virus particle, and presumably encoded by the viral genome. In the early 1970s a small group of investigators, including Peter Vogt, had characterized virion proteins for RSV, including virion-associated glycoproteins (Duesberg et al. 1970; Robinson et al. 1970). By 1971 the combination of EM, virus purification, radioactive labeling of proteins, and protease treatment was used to show that it was the virion glycoprotein that was on the exterior of the viral envelope (Rifkin and Compans 1971). With viral genetics providing recombinants with altered host range it became possible to map the location of the glycoprotein within the viral genome using the pre-sequencing tool of assessing patterns of RNase T1-resistant oligonucleotides displayed using 2D electrophoresis, placing the env gene at the 3′ end of the ALV genome (Joho et al. 1975; Coffin and Billiter 1976; Wang et al. 1976).
Sequencing, Cloning, and Sequencing
In February of 1975 The Asilomar Conference on Recombinant DNA was held to consider the risks and rewards of using recombinant DNA tools and cloning. With guidelines in place a new era of biology began. Virologists had to become at least mediocre bacteriologists capable of growing phage and plasmids and isolating biological clones. Those up to the task were rewarded with a new view of genes and genomes. With the availability of unlimited amounts of cloned DNA, sequencing tools quickly followed and gene and inferred protein sequences were revealed. Since DNA is relatively homogeneous in its chemical properties, in contrast to its information properties, virtually anything could be cloned with the order determined by the investigator’s interest. For those interested in viruses with RNA genomes the challenge was greater, while for retrovirologists most went after the DNA form of the viral genome. However, in an early tour de force, virtually the entire RSV genome was sequenced as cDNA products that were made using random primers, purified reverse transcriptase, and viral genomic RNA (Schwartz et al. 1983). The authors noted in their 1983 paper that “at the time this project was initiated, molecular cloning of RSV was prohibited.” The placement of the env gene, upstream of the v-src gene, was proven by comparing the amino-terminal protein sequences of the SU (gp85) and TM (gp37) Env protein subunits and placing them on the nucleotide sequence (Hunter et al. 1983). The strain of RSV that was sequenced carried a subgroup C env gene providing the first view of the sequence of this protein.
One of the funny things about looking at sequences is that you learn some things from looking at the first sequence of a gene, but you learn a whole lot of information that was not available by looking at the first sequence when you get to look at the second sequence, hopefully a similar but not identical sequence. Specifically you learn about which regions are identical (or nearly identical) and which are different. For evolutionary differences there are two considerations: first is a relatively uninteresting evolutionary drift that occurs with evolutionary distance; more interesting are the sequences that rapidly evolve due to strong selective pressure where the differences can be linked to changes in biological function. Thus the second and third sequences reported, for subgroup B and E env genes, revealed conserved and variable regions that could only be interpreted as the protein framework and two regions of variability as determinants of receptor specificity, named host range 1 and 2 or hr 1 and 2 (Dorner et al. 1985). The analysis of subgroups A and D env genes (Bova et al. 1986, 1988) reinforced this idea.
Receptors
The molecular cloning of receptors was a difficult endeavor. In cloning the viral genome hybridization probes made as radioactive cDNA from viral genomic RNA could be used to find the desired clones. However, even though the receptors had been characterized genetically and named (tva, tvb, etc.), the only assay for the receptors was a biological assay. After an effort of several years the first ALV receptor was cloned for subgroup A viruses and identified as being related to the low density lipoprotein receptor (Bates et al. 1993). Next the receptor for subgroup B and D viruses was cloned and recognized as a member of the TNFR protein family (Brojatsch et al. 1996), then the receptor for subgroup E was identified as another member of the TNFR family (Adkins et al. 2001), and finally the subgroup C receptor was identified as a member of the butyrophilin protein family (Elleder et al. 2005). The lesson learned is that viruses pick out receptors using a logic that as yet escapes us. The polymorphisms in the receptor/susceptibility loci, in part expanded in the population by breeding but still from naturally existing alleles, suggests selection against having functional receptors by the host. This is analogous to the presence of xenotropic MLV endogenous viruses, i.e., an endogenous virus that can not infect its host (or that the host lost the functional receptor after the endogenous provirus was fixed). Since evolution is not functionally blind, it must be the case that there is a primary receptor and at least a low level of interaction with other surface proteins, as different from each other as they are, to allow the evolution of different subgroups with different receptor specificities. The ultimate “receptor switch” then provides the strong selective pressure for rapid sequence change in the hr1 and hr2 regions.
Subgroup J, Biology in Real Time
Humans are overly egocentric such that we do not readily conceive of the next thing until it happens as a surprise. There will always be new viral variants that appear with a slightly different constellation of properties that allow the next epidemic. An unknown primate lentivirus was percolating along in chimpanzees and we became aware of it only when it turned into a worldwide epidemic in humans. Thus we should not be surprised that just as we were getting comfortable with the biology of ALV, and ways to avoid it in chicken flocks, a new subgroup appeared, subgroup J. As reviewed in Payne and Nair (2012) a new pathogenic subgroup of ALV was identified in the late 1980s, subsequently shown to be a recombinant between ALV and the env gene of an endogenous retroviral allele. The receptor for this virus was cloned and shown to be a Na+/H+ exchanger type 1 protein (Chai and Bates 2006), thus adding to the seemingly non sequitur list of cellular proteins capable of serving as receptors.
HIV-1 Env Proteins: Still Trying to Get It Right
HIV-1 was discovered around the time other retroviral genomes were just coming into the hands of cloners. Given its novelty as the second human retrovirus, and certainly the scarier of the two, the understanding of the framework of its biology occurred quickly. An important early observation was that CD4 on the surface of T cells was a receptor important for cell entry, demonstrated by blocking viral infection with an anti-CD4 antibody (Dalgleish et al. 1984; Klatzmann et al. 1984). This is a fundamental property of HIV-1 as no CD4-independent virus has ever been isolated, although it has been possible to select for a CD4-independent SIV in cell culture (Swanstrom et al. 2016). However, after identifying CD4 as a receptor for HIV-1, the entry field took a confusing twist that is still confounding us today. In a way that was analogous to some chickens/eggs/CEFs being resistant to infection (for lack of a functional receptor), certain strains of HIV-1 could not grow on cell lines that clearly expressed CD4, even though all viral isolates could grow in PBMCs. Thirty years later we are still trying to overcome the initial interpretation of these results that now need to be updated.
The identification of CD4 as a receptor and the use of transformed T cell lines that express CD4 (usually derived from a leukemia) make perfect sense. The ability of some strains of virus to grow in the T cell lines earned them the name T cell-tropic. Those that failed to grow had to be something else. Macrophages express a low level of CD4 and can support a low level of entry for most isolates and overt replication for some isolates. This was enough for the isolates that could not grow in the T cell lines to earn the name macrophage-tropic. Thus HIV-1 isolates fell into these two nice categories, T cell-tropic and macrophage-tropic. Unfortunately both names are inappropriate for what they were trying to describe.
This problem of misidentification became exacerbated with the important discovery that HIV-1, unlike most of the other viruses we know about, has a second receptor now called the co-receptor. A coreceptor was identified for one of the “T cell-tropic” viruses which was the chemokine receptor CXCR4 (Feng et al. 1996). By analogy the “macrophage-tropic” viruses should use a similar molecule, which was quickly identified as the chemokine receptor CCR5 (Alkhatib et al. 1996; Choe et al. 1996; Deng et al. 1996; Doranz et al. 1996; Dragic et al. 1996). Viruses using these coreceptors were named X4 and R5 viruses and the dual entry phenotype of HIV-1 was further engrained as X4 T cell-tropic and R5 macrophage-tropic. If the names were inappropriate before this only made it worse. In fact there are three types of HIV-1 whose entry phenotypes are most accurately described as a jumble of these two inappropriate names.
The missing piece in this story was apparent to a few investigators who came to understand that the “R5 macrophage-tropic” viruses were not homogeneous. Some were effective at using a low density of CD4 while most were inefficient at entering cells that displayed a low density of CD4 (Kabat et al. 1994; Platt et al. 1998). Furthermore, the viruses capable of using a low density of CD4 were most reliably found in the brain late in infection (Gorry et al. 2002; Peters et al. 2004; Dunfee et al. 2006; Martin-Garcia et al. 2006; Peters et al. 2006; Duenas-Decamp et al. 2009; Schnell et al. 2011). Since macrophages have a low density of CD4 (CD4 has no function in macrophages) (Lee et al. 1999; Joseph et al. 2014), it is a small leap to suggest that the viruses that can use a low density of CD4 to enter cells are in fact the ones that have evolved to become macrophage-tropic. We used a cell line (Affinofile cells) designed to allow regulated control of CD4 levels (Johnston et al. 2009) to explore these issues at length. After analyzing between 100 and 200 env genes cloned from a variety of sources, we came to the conclusion that R5 macrophage-tropic viruses are rare (see for example Ping et al. 2013). This left the majority of R5 viruses as distinct from R5 macrophage-tropic viruses and without a name.
As discussed later, X4 viruses evolve from R5 viruses so the default version of HIV-1 has to be an R5 virus. But if most of these isolates do not enter macrophages efficiently then where are these viruses growing? Of course the answer is T cells and they are rightly called T cell-tropic, or more descriptively R5 T cell-tropic. However, we started this discussion with the idea that only X4 viruses were growing in T cells, or more accurately T cell lines. For those who are good at puzzles the answer is probably clear. The CD4+ T cells lines used to grow HIV-1 in the early days expressed CXCR4 but not CCR5, a point that at the time was meaningless since the concept of the coreceptor did not exist. However, just as chickens, eggs, and CEFs can lack functional receptors for certain subgroups of ALV, these cell lines were heterogeneous for expression of the coreceptors, although in general most of these transformed cell lines express CXCR4 and not CCR5. The better analogy comes with the observation that about 10% of northern Europeans carry a CCR5 allele with an inactivating mutation, meaning that about 1% of this population is resistant to infection by the most common form of HIV-1, i.e., R5 T cell-tropic virus (Dean et al. 1996; Samson et al. 1996; Liu et al. 1996). Such individuals can be infected with an X4 T cell-tropic virus (Theodorou et al. 1997; Michael et al. 1998), although transmission of this virus is rare. It should be noted that when we have examined X4 viruses where the env genes were isolated without tissue culture passage we find that they require a high density of CD4 so that they are still appropriately called X4 T cell-tropic (M. Bednar and R.S., in preparation).
To summarize HIV-1 entry phenotypes thus far: there are three entry phenotypes, R5 T cell-tropic, R5 macrophage-tropic, and X4 T cell-tropic. The default form of HIV-1 is R5 T cell-tropic, using the CCR5 coreceptor and requiring a high density of CD4 for entry. X4 T cell-tropic viruses evolve late in disease with a switch in coreceptor use. In an analogous way macrophage-tropic viruses also evolve late in disease (in a T cell-poor environment) and gain the ability to enter cells with a low density of CD4 more efficiently. Thus the vexing legacy of the early entry work is that in naming two types of entry phenotypes the one that was excluded is the one that is actually the predominant form of HIV-1: R5 T cell-tropic.
The Two Evolutionary Variants of R5 T Cell-Tropic Viruses
The transmitted form of HIV-1 and the form that is found in the blood throughout most of the infection is the R5 T cell-tropic form of HIV-1 (using CCR5 as the coreceptor and requiring a high density of CD4 for efficient entry) (Ochsenbauer et al. 2012; Ping et al. 2013). That means that the other forms must evolve from the R5 T cell-tropic form (Fig. 2). With the discovery of coreceptors it was possible to make the link with the CXCR4-using version of HIV-1 as appearing late in infection (Connor et al. 1997; Brumme et al. 2005; Moyle et al. 2005). As noted above for the evolution of different subgroups of ALV (probably on a much longer time scale), the R5 T cell-tropic version of HIV-1 must interact at least a little bit with CXCR4 to allow that evolutionary pathway to occur. At the extreme, the adaptation is so complete that the virus losses the ability to interact with CCR5, however most viruses never get that far in their evolution so the typical virus seen is termed dual-tropic, meaning it can use both CXCR4 and CCR5. We have observed that these dual-tropic viruses have a reduced affinity for CCR5, as measured by increased sensitivity to a CCR5 antagonist, suggesting that the dual-tropic viruses are more X4 than R5 (M. Bednar and R.S., in preparation). We have also observed that X4 viruses require a high density of CD4 for efficient entry, retaining their X4 T cell-tropic moniker. Since X4 viruses evolve late in disease we can speculate that either they require an immunodeficient host to evolve or that they evolve when the preferred target cells, CD4+ CCR5+ T cells, become limiting. One theory is that CD4+ CXCR4+ naive T cells support the replication of X4 viruses (Ribeiro et al. 2006).
Fig. 2.
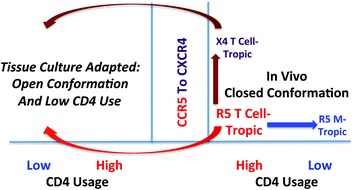
Pathways for the evolution of the HIV-1 Env protein entry phenotype. The major entry phenotype form for HIV-1 is the R5 T cell-tropic form. It uses CCR5 as the coreceptor, but requires a high density of CD4, as is found on T cells, for efficient entry. In vivo it evolves to switch coreceptor to use CXCR4. Alternatively, it can evolve to use a low density of CD4 to enter cells such as macrophages, which have a density of CD4 about 25-fold lower than that found on T cells. Also in vivo, these viruses are found in a closed conformation, i.e., resistant to neutralization, especially to antibodies targeting the epitopes exposed after binding CD4. In cell culture the virus follows another evolutionary pathway in which an open conformation is generated allowing the use of a low density of CD4 for entry. Presumably this enables more rapid entry under culture conditions. This can happen for both the X4 form of the virus and the R5 T cell-tropic form of the virus and should be considered an artifact of tissue culture adaptation
The second evolutionary variant of HIV-1 is of course the macrophage-tropic variant. Where the change in coreceptor use is more dramatic and easy to measure, the change in CD4 use is less dramatic and harder to measure (Joseph et al. 2014; Arrildt et al. 2015), accounting for much of the confusion about this phenotype. These viruses evolve in a T cell-poor environment, especially in the CNS (Sturdevant et al. 2015). In addition to their ability to efficiently enter cells with a low density of CD4 there is a tightly linked phenotype of increased sensitivity to neutralization by soluble CD4 (Arrildt et al. 2015). It appears these viruses are primed to undergo the fusion conformation cascade with fewer interactions with CD4.
A Fourth Entry Phenotype Is an Artifact of Tissue Culture Adaptation
It is common practice in studying a virus to grow the virus in tissue culture. For most of the viral functions this is probably not a bad idea, at least in the short term. However, given that many attenuated viral vaccines were developed simply by passaging an isolate in culture we have to acknowledge that passage in culture does put the virus under a very different selective pressure compared to what it experiences in vivo. The strongest selective pressure in cell culture is likely to occur on the viral entry phenotype. There is no wrong cell to infect given that the culture is largely homogeneous, so viral replication in cell culture becomes a race to see who can enter cells most quickly. HIV-1 seems to be especially susceptible to a serious cell culture artifact that is as yet not fully appreciated.
As noted above the HIV-1 Env protein goes through a conformational change when binding the CD4 receptor (McDougal et al. 1986; White et al. 2010; Munro et al. 2014). For most viruses this would lead to insertion of the fusion peptide followed by membrane fusion. However, for HIV-1 there is an extra step. The first conformational change is to create and expose the CCR5 binding site. Binding to CCR5 then triggers insertion of the fusion peptide, formation of the six helix bundle, and membrane fusion between the host and viral membranes. Thus the HIV-1 Env protein trimer must transition through a lot of conformational space to carry out its job. When examining the antibodies from people infected with HIV-1 there seem to be abundant antibodies to epitopes that are created after binding to CD4. This is likely the selective pressure that keeps the trimer in a “closed” conformation where these epitopes are either covered or not yet even formed. The transient exposure of these epitopes after CD4 engagement leaves too little space at the surface of the cell for these antibodies to be effective (Labrijn et al. 2003). However, in cell culture no such antibody selective pressure exists.
An important observation was the realization that passage of HIV-1 in tissue culture led to a highly neutralization sensitive form of the Env protein where it was now sensitive to these CD4 binding-induced epitopes (Moore et al. 1995). The conformation associated with this state has become known as the “open” conformation. The important thing to realize is that a virus with its Env protein in an open conformation is a tissue culture artifact, such viruses are not found in vivo (Mascola et al. 1996; Harris et al. 2011). In the race to grow the fastest in cell culture the incorporation of mutations that dispense with the closed conformation and allow the virus to skip most of the conformational change induced by binding CD4 are selected, although these viruses are still CD4-dependent (Fig. 2).
The next idea we will discuss is partly data-driven and partly prediction. Tissue culture adaptation has another phenotype that causes confusion with macrophage tropism; like macrophage-tropic viruses, tissue culture-adapted viruses are also able to use a low density of CD4 to enter cells (Kabat et al. 1994). The important distinction to make is that tissue culture adaptation selects for an open conformation while macrophage tropism does not. There do seem to be some structural changes in the Env protein associated with macrophage tropism, but these do not go as far as representing the tissue culture-adapted open conformation (Arrildt et al. 2015). This is an important distinction to make because these two forms appear to share the feature of being able to use CD4 at a low density. We would predict that the pathway to low CD4 use in the context of tissue culture adaptation is distinct from the more relevant pathway that the virus uses in vivo to become macrophage-tropic. In developing a relevant understanding of macrophage tropism it will be important to rigorously avoid confounding information that comes from tissue culture-adapted viruses.
Acknowledgements
Our own work is supported by the National Institutes of Health. We thank many lab members who contributed to the ideas in this review. We also thank Adrienne Swanstrom for editorial assistance.
Personal Perspective from RS
I first met Peter Vogt in 1975. I was a new postdoctoral fellow in the Bishop/Varmus lab at UCSF where we participated in the “West Coast RNA Tumor Virus Meeting.” It was a group that met several times each year with somewhat changing characters but with a core of Peter’s lab, Mike’s and Harold’s lab, Peter Duesberg’s lab, and Steve Martin’s lab. I joined this group just after the discovery of v-src sequences in the normal vertebrate genome. The race was on to genetically define new viral oncogenes and to test their host cell origin, and to try to understand what the functions of the protein products were and how they could change to induce cellular transformation and cancer. At least from my perspective Peter Vogt was the one who brought the biology of these viruses to this larger group, providing the starting point for the important molecular understanding of the transforming genes that followed. Peter has been a full partner in this field, fittingly spending these last years searching for small molecule inhibitors of the proteins encoded in the same transforming genes he helped discover.
Peter’s personal story of how he became an academic researcher and his long successful career are inspiring but not mine to tell. In my time with Peter I always found him to be incredibly thoughtful and generous. He is truly a gentle man. He has a deep-seated love of art that led him to be a painter in his own right. I have two Peter Vogt paintings, one of a rose demonstrating an eye for capturing simple beauty. The other is a street scene from Pasadena where he lived while on the faculty at USC Medical School. Pasadena played an important role for me in my extended family when I was young so to be able to capture part of that memory as seen through Peter’s eyes is a special treat. True to Peter’s nature, both of these paintings (and many others) were donated to USC to allow them to be sold to raise funds for graduate education.
I think Peter enjoys people in general and he is good spirits. I stand with a large group of people who have seen Peter as a role model for scientific excellence and personal humility. I have always enjoyed his company given the endlessly interesting things he pursues. One of the things I have noticed over the years is that PIs frequently have their personality reflected through the people in their lab. Thus it is easy to point to many of Peter’s trainees as scientifically rigorous and genuinely nice people. I am sure Klaus and Eric take pride in being able to share some of their feelings toward Peter by creating this volume in his honor.
I met Eric Hunter when he was a postdoctoral fellow with Peter, Eric forever being slightly older than I am. It is a friendship that has lasted over 40 years. When Eric asked if I would contribute a chapter there was really only one answer. In 1990 Peter approached me to coedit CTMI volume 157 “Retroviruses: Strategies of Replication.” In trying to round out my own list of authors I asked Eric to write a chapter on “Retrovirus Envelope Glycoproteins” which he agreed to do with me as a coauthor. Now the shoe is on the other foot, but as I look back on that old chapter it is clear that we did not bring the breadth to the topic that (mostly) Eric and I managed as younger writers.
When I met Peter it was a time when young people were joining the field with the hope of getting to work on their own new oncogene. Alas, some of us were just virologists. It is fun to use the miracle of PubMed to be reminded of Peter’s work before I met him. The foundational work that set the stage for the discovery of cellular oncogenes also provided the underlying biology for some of the molecular studies I did. The introduction of molecular cloning and sequencing allowed us to ask how the sequences of the ALV strains differed given their host range differences. A few years later another generation of postdocs, Paul Bates and John Young, would build on the same biology to clone the cellular receptors for some of these strains to provide a complete view of how the virus chooses which cells to infect.
Along the way the oncogene field and the retrovirus field largely split. The next retrovirus challenge made itself known with the discovery of HIV. I have taken as our charge in this new review to revisit the biology of viral entry for the avian retroviruses that brought us v-src, v-myc, v-myb, etc., and today threaten new ALV pandemics. We have paired those lessons with the ones learned from HIV-1 infecting human cells. It is perhaps an odd pairing but simply reflects the twists and turns of a typical scientist following related biological phenomena. Finally, this effort is offered with affection and admiration to one of my heroes.
Contributor Information
Eric Hunter, Phone: +11+1-404-727-8587, Email: ehunte4@emory.edu.
Klaus Bister, Phone: +4343512 507 57500, Email: klaus.bister@uibk.ac.at.
Ronald Swanstrom, Email: ron_swanstrom@med.unc.edu.
References
- Adkins HB, Blacklow SC, Young JA (2001) Two functionally distinct forms of a retroviral receptor explain the nonreciprocal receptor interference among subgroups B, D, and E avian leukosis viruses. J Virol 75(8):3520–3526. doi:10.1128/JVI.75.8.3520-3526.2001 [DOI] [PMC free article] [PubMed]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272(5270):1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Arrildt KT, LaBranche CC, Joseph SB, Dukhovlinova EN, Graham WD, Ping L-H, Schnell G, Sturdevant CB, Kincer LP, Mallewa M, Heyderman RS, Van Rie A, Cohen MS, Spudich S, Price RW, Montefiori DC, Swanstrom R (2015) Phenotypic correlates of HIV-1 macrophage tropism. J Virol 89(22):11294–11311. doi:10.1128/jvi.00946-15 [DOI] [PMC free article] [PubMed]
- Bates P, Young JA, Varmus HE (1993) A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell 74(6):1043–1051 [DOI] [PubMed]
- Bova CA, Manfredi JP, Swanstrom R (1986) Env genes of avian retroviruses: nucleotide sequence and molecular recombinants define host range determinants. Virology 152(2):343–354 [DOI] [PubMed]
- Bova CA, Olsen JC, Swanstrom R (1988) The avian retrovirus env gene family: molecular analysis of host range and antigenic variants. J Virol 62(1):75–83 [DOI] [PMC free article] [PubMed]
- Brojatsch J, Naughton J, Rolls MM, Zingler K, Young JA (1996) CAR1, a TNFR-related protein, is a cellular receptor for cytopathic avian leukosis-sarcoma viruses and mediates apoptosis. Cell 87(5):845–855 [DOI] [PubMed]
- Brumme ZL, Goodrich J, Mayer HB, Brumme CJ, Henrick BM, Wynhoven B, Asselin JJ, Cheung PK, Hogg RS, Montaner JS, Harrigan PR. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J Infect Dis. 2005;192(3):466–474. doi: 10.1086/431519. [DOI] [PubMed] [Google Scholar]
- Chai N, Bates P (2006) Na+/H+ exchanger type 1 is a receptor for pathogenic subgroup J avian leukosis virus. Proc Natl Acad Sci U S A 103 (14):5531–5536. doi:10.1073/pnas.0509785103 [DOI] [PMC free article] [PubMed]
- Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85(7):1135–1148. doi: 10.1016/S0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- Coffin JM, Billeter MA (1976) A physical map of the Rous sarcoma virus genome. J Mol Biol 100(3):293–318 [DOI] [PubMed]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR (1997) Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J Exp Med 185(4):621–628 [DOI] [PMC free article] [PubMed]
- Cornelis G, Heidmann O, Degrelle SA, Vernochet C, Lavialle C, Letzelter C, Bernard-Stoecklin S, Hassanin A, Mulot B, Guillomot M, Hue I, Heidmann T, Dupressoir A (2013) Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc Natl Acad Sci U S A 110(9):E828–E837. doi:10.1073/pnas.1215787110 [DOI] [PMC free article] [PubMed]
- Dalgleish AG, Beverley PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA (1984) The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312(5996):763−767. doi:10.1038/312763a0 [DOI] [PubMed]
- Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, Donfield S, Vlahov D, Kaslow R, Saah A, Rinaldo C, Detels R, Obrien SJ (1996) Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273(5283):1856–1862. doi:10.1126/science.273.5283.1856 [DOI] [PubMed]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85(7):1149–1158. doi: 10.1016/S0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- Dorner AJ, Stoye JP, Coffin JM (1985) Molecular basis of host range variation in avian retroviruses. J Virol 53(1):32–39 [DOI] [PMC free article] [PubMed]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381(6584):667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Duenas-Decamp MJ, Peters PJ, Burton D, Clapham PR (2009) Determinants flanking the CD4 binding loop modulate macrophage tropism of human immunodeficiency virus type 1 R5 envelopes. J Virol 83(6):2575–2583. doi:10.1128/jvi.02133-08 [DOI] [PMC free article] [PubMed]
- Duesberg PH, Martin GS, Vogt PK (1970) Glycoprotein components of avian and murine RNA tumor viruses. Virology 41(4):631–646 [DOI] [PubMed]
- Dunfee RL, Thomas ER, Gorry PR, Wang JB, Taylor J, Kunstman K, Wolinsky SM, Gabuzda D (2006) The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc Natl Acad Sci U S A 103(41):15160–15165. doi:10.1073/pnas.0605513103 [DOI] [PMC free article] [PubMed]
- Elleder D, Stepanets V, Melder DC, Senigl F, Geryk J, Pajer P, Plachy J, Hejnar J, Svoboda J, Federspiel MJ (2005) The receptor for the subgroup C avian sarcoma and leukosis viruses, Tvc, is related to mammalian butyrophilins, members of the immunoglobulin superfamily. J Virol 79(16):10408–10419. doi:10.1128/JVI.79.16.10408-10419.2005 [DOI] [PMC free article] [PubMed]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Taylor J, Holm GH, Mehle A, Morgan T, Cayabyab M, Farzan M, Wang H, Bell JE, Kunstman K, Moore JP, Wolinsky SM, Gabuzda D (2002) Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. J Virol 76(12):6277–6292. doi:10.1128/jvi.76.12.6277-6292.2002 [DOI] [PMC free article] [PubMed]
- Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang Y, Depetris R, Marozsan AJ, Sanders RW, Klasse PJ, Milne JLS, Wilson IA, Olson WC, Moore JP, Subramaniam S (2011) Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci U S A 108(28):11440–11445. doi:10.1073/pnas.1101414108 [DOI] [PMC free article] [PubMed]
- Hunter E, Hill E, Hardwick M, Bhown A, Schwartz DE, Tizard R (1983) Complete sequence of the Rous sarcoma virus env gene: identification of structural and functional regions of its product. J Virol 46(3):920–936 [DOI] [PMC free article] [PubMed]
- Johnston SH, Lobritz MA, Nguyen S, Lassen K, Delair S, Posta F, Bryson YJ, Arts EJ, Chou T, Lee B. A quantitative affinity-profiling system that reveals distinct CD4/CCR5 usage patterns among human immunodeficiency virus type 1 and simian immunodeficiency virus strains. J Virol. 2009;83(21):11016–11026. doi: 10.1128/JVI.01242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho RH, Billeter MA, Weissmann C (1975) Mapping of biological functions on RNA of avian tumor viruses: location of regions required for transformation and determination of host range. Proc Natl Acad Sci U S A 72(12):4772–4776 [DOI] [PMC free article] [PubMed]
- Joseph SB, Arrildt KT, Swanstrom AE, Schnell G, Lee B, Hoxie JA, Swanstrom R. Quantification of entry phenotypes of macrophage-tropic HIV-1 across a wide range of CD4 densities. J Virol. 2014;88(4):1858–1869. doi: 10.1128/JVI.02477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat D, Kozak SL, Wehrly K, Chesebro B (1994) Differences in CD4 dependence for infectivity of laboratory-adapted and primary patient isolates of human immunodeficiency virus type 1. J Virol 68(4):2570–2577 [DOI] [PMC free article] [PubMed]
- Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L (1984) T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312(5996):767–768 [DOI] [PubMed]
- Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, Venturi M, Petropoulos CJ, Wrin T, Dimitrov DS, Robinson J, Kwong PD, Wyatt RT, Sodroski J, Burton DR (2003) Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol 77(19):10557–10565 [DOI] [PMC free article] [PubMed]
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW (1999) Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A 96(9):5215–5220 [DOI] [PMC free article] [PubMed]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR (1996) Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86(3):367–377. doi:10.1016/s0092-8674(00)80110-5 [DOI] [PubMed]
- Martin JL, Cao S, Maldonado JO, Zhang W, Mansky LM (2016) Distinct particle morphologies revealed through comparative parallel analyses of retrovirus-like particles. J Virol 90(18):8074–8084. doi:10.1128/JVI.00666-16 [DOI] [PMC free article] [PubMed]
- Martin-Garcia J, Cao W, Varela-Rohena A, Plassmeyer ML, Gonzalez-Scarano F (2006) HIV-1 tropism for the central nervous system: brain-derived envelope glycoproteins with lower CD4 dependence and reduced sensitivity to a fusion inhibitor. Virology 346(1):169–179 [DOI] [PubMed]
- Mascola JR, Snyder SW, Weislow OS, Belay SM, Belshe RB, Schwartz DH, Clements ML, Dolin R, Graham BS, Gorse GJ, Keefer MC, McElrath MJ, Walker MC, Wagner KF, McNeil JG, McCutchan FE, Burke DS (1996) Immunization with Envelope Subunit Vaccine Products Elicits Neutralizing Antibodies against Laboratory-Adapted but Not Primary Isolates of Human Immunodeficiency Virus Type 1. J Infect Dis 173(2):340–348 [DOI] [PubMed]
- McDougal JS, Nicholson JK, Cross GD, Cort SP, Kennedy MS, Mawle AC (1986) Binding of the human retrovirus HTLV-III/LAV/ARV/HIV to the CD4 (T4) molecule: conformation dependence, epitope mapping, antibody inhibition, and potential for idiotypic mimicry. J Immunol 137(9):2937–2944 [PubMed]
- Michael NL, Nelson JA, KewalRamani VN, Chang G, O’Brien SJ, Mascola JR, Volsky B, Louder M, White GC, 2nd, Littman DR, Swanstrom R, O’Brien TR (1998) Exclusive and persistent use of the entry coreceptor CXCR4 by human immunodeficiency virus type 1 from a subject homozygous for CCR5 delta32. J Virol 72(7):6040–6047 [DOI] [PMC free article] [PubMed]
- Moore JP, Cao YZ, Qing L, Sattentau QJ, Pyati J, Koduri R, Robinson J, Barbas CF, Burton DR, Ho DD (1995) Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J Virol 69(1):101–109 [DOI] [PMC free article] [PubMed]
- Moyle GJ, Wildfire A, Mandalia S, Mayer H, Goodrich J, Whitcomb J, Gazzard BG. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J Infect Dis. 2005;191(6):866–872. doi: 10.1086/428096. [DOI] [PubMed] [Google Scholar]
- Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, Kwong PD, Blanchard SC, Mothes W (2014) Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346(6210):759–763. doi:10.1126/science.1254426 [DOI] [PMC free article] [PubMed]
- Ochsenbauer C, Edmonds TG, Ding HT, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC (2012) Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol 86(5):2715–2728 [DOI] [PMC free article] [PubMed]
- Payne LN, Nair V (2012) The long view: 40 years of avian leukosis research. Avian Pathol 41(1):11–19. doi:10.1080/03079457.2011.646237 [DOI] [PubMed]
- Peters PJ, Bhattacharya J, Hibbitts S, Dittmar MT, Simmons G, Bell J, Simmonds P, Clapham PR (2004) Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J Virol 78(13):6915–6926. doi:10.1128/jvi.78.13.6915-6926.2004 [DOI] [PMC free article] [PubMed]
- Peters PJ, Sullivan WM, Duenas-Decamp MJ, Bhattacharya J, Ankghuambom C, Brown R, Luzuriaga K, Bell J, Simmonds P, Ball J, Clapham PR (2006) Non-macrophage-tropic human immunodeficiency virus type 1 R5 envelopes predominate in blood, lymph nodes, and semen: Implications for transmission and pathogenesis. J Virol 80(13):6324–6332. doi:10.1128/jvi.02328-05 [DOI] [PMC free article] [PubMed]
- Ping LH, Joseph SB, Anderson JA, Abrahams MR, Salazar-Gonzalez JF, Kincer LP, Treurnicht FK, Arney L, Ojeda S, Zhang M, Keys J, Potter EL, Chu H, Moore P, Salazar MG, Iyer S, Jabara C, Kirchherr J, Mapanje C, Ngandu N, Seoighe C, Hoffman I, Gao F, Tang Y, Labranche C, Lee B, Saville A, Vermeulen M, Fiscus S, Morris L, Karim SA, Haynes BF, Shaw GM, Korber BT, Hahn BH, Cohen MS, Montefiori D, Williamson C, Swanstrom R (2013) Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J Virol 87(13):7218–7233. doi:10.1128/JVI.03577-12 [DOI] [PMC free article] [PubMed]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72(4):2855–2864 [DOI] [PMC free article] [PubMed]
- Ribeiro RM, Hazenberg MD, Perelson AS, Davenport MP (2006) Naive and memory cell turnover as drivers of CCR5-to-CXCR4 tropism switch in human immunodeficiency virus type 1: implications for therapy. J Virol 80(2):802–809. doi:10.1128/JVI.80.2.802-809.2006 [DOI] [PMC free article] [PubMed]
- Rifkin D, Compans RW (1971) Identification of the spike proteins of Rous sarcoma virus. Virology 46(2):485–489 [DOI] [PubMed]
- Robinson WS, Hung P, Robinson HL, Ralph DD (1970) Proteins of avian tumor viruses with different coat antigens. J Virol 6(5):695–698 [DOI] [PMC free article] [PubMed]
- Rowell JF, Stanhope PE, Siliciano RF (1995) Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J Immunol 155(1):473–488 [PubMed]
- Rubin H (2011) The early history of tumor virology: Rous, RIF, and RAV. Proc Natl Acad Sci U S A 108(35):14389–14396. doi:10.1073/pnas.1108655108 [DOI] [PMC free article] [PubMed]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi YJ, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M (1996) Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382(6593):722–725. doi:10.1038/382722a0 [DOI] [PubMed]
- Sauter MM, Pelchen-Matthews A, Bron R, Marsh M, LaBranche CC, Vance PJ, Romano J, Haggarty BS, Hart TK, Lee WM, Hoxie JA (1996) An internalization signal in the simian immunodeficiency virus transmembrane protein cytoplasmic domain modulates expression of envelope glycoproteins on the cell surface. J Cell Biol 132(5):795–811 [DOI] [PMC free article] [PubMed]
- Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog. 2011;7(10):e1002286. doi: 10.1371/journal.ppat.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DE, Tizard R, Gilbert W (1983) Nucleotide sequence of Rous sarcoma virus. Cell 32(3):853–869 [DOI] [PubMed]
- Sturdevant CB, Joseph SB, Schnell G, Price RW, Swanstrom R, Spudich S (2015) Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. Plos Pathog 11(3):e1004720. doi:10.1371/journal.ppat.1004720 [DOI] [PMC free article] [PubMed]
- Swanstrom AE, Haggarty B, Jordan APO, Romano J, Leslie GJ, Aye PP, Marx PA, Lackner AA, Del Prete GQ, Robinson JE, Betts MR, Montefiori DC, LaBranche CC, Hoxie JA (2016) Derivation and characterization of a CD4-independent, non-CD4-tropic simian immunodeficiency virus. J Virol 90(10):4966–4980. doi:10.1128/jvi.02851-15 [DOI] [PMC free article] [PubMed]
- Theodorou I, Meyer L, Magierowska M, Katlama C, Rouzioux C. HIV-1 infection in an individual homozygous for CCR5 delta 32. Seroco Study Group. Lancet. 1997;349(9060):1219–1220. doi: 10.1016/S0140-6736(05)62411-7. [DOI] [PubMed] [Google Scholar]
- Wang LH, Duesberg P, Mellon P, Vogt PK (1976) Distribution of envelope-specific and sarcoma-specific nucleotide sequences from different parents in the RNAs of avian tumor virus recombinants. Proc Natl Acad Sci U S A 73(4):1073–1077 [DOI] [PMC free article] [PubMed]
- White TA, Bartesaghi A, Borgnia MJ, Meyerson JR, de la Cruz MJ, Bess JW, Nandwani R, Hoxie JA, Lifson JD, Milne JLS (2010) Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS pathog 6(12):e1001249 [DOI] [PMC free article] [PubMed]
- Zhu P, Liu J, Bess J, Jr., Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441(7095):847–852. doi:10.1038/nature04817 [DOI] [PubMed]