

Abstract
Proteases play an important role in health and disease of the lung. In the normal lungs, proteases maintain their homeostatic functions that regulate processes like its regeneration and repair. Dysregulation of proteases–antiproteases balance is crucial in the manifestation of different types of lung diseases. Chronic inflammatory lung pathologies are associated with a marked increase in protease activities. Thus, in addition to protease activities, inhibition of anti-proteolytic control mechanisms are also important for effective microbial infection and inflammation in the lung. Herein, we briefly summarize the role of different proteases and to some extent antiproteases in regulating a variety of lung diseases.
Keywords: Protease, Antiprotease, Lung diseases, Infection, Inflammation
Introduction
The lung possesses a large number of anti-inflammatory components [1], which fight against microbial infections.
Serine, cysteine, aspartic, and metalloproteases are the principal classes of protease present in the human lung. A good number of evidence suggest that neutrophil serine proteases (NSPs) such as elastase, proteinase 3 (PR3), cathepsin G (CatG), and matrix metalloproteases (MMPs) are major pathogenic determinants of chronic inflammatory lung disorders [1]. The lung proteases act in concert with the proteases of invading microbes, inactivate antiproteases, and antimicrobial compounds and thereby play a pivotal role in different types of lung diseases including chronic obstructive pulmonary disease (COPD), asthma, acute respiratory distress syndrome (ARDS), influenza, and cancer [1].
The lung proteases can either intracellularly or extracellularly regulate processes such as tissue remodeling, mucin production, neutrophil chemotaxis, and microbial destruction. Additionally, they regulate infection and inflammation in the lung, for example neutrophil elastase (NE), a serine protease, which plays critical role in the progression of a variety of lung diseases. It can regulate activities of CatB and MMP-2 in alveolar macrophages [2] and also activates proMMP-2, MMP-7, and MMP-9 [3–5], indicating that NE may act as a proinflammatory mediator. In some cases, NE regulates important signaling pathways that modulate innate immunity [6, 7]. NE’s multiple roles characterize it as a decisive factor controlling many aspects of infection and inflammation in the lung.
Pulmonary Hypertension
Pulmonary hypertension (PAH) occurs due to elevation of pulmonary artery pressure and if it is prolonged, then right ventricular failure may occur with subsequent fatality [8]. PAH often leads to secondary complications of many pulmonary disorders such as COPD, asthma and chronic bronchitis bronchopulmonary displasia, cystic fibrosis, chronic bronchitis, and emphysema [9, 10].
Serine Protease and Pulmonary Hypertension
The role of oxidants such as hydrogen peroxide, hydroperoxides, superoxide, and peroxynitrite in producing PAH is now well established [11–15]. Administration of the oxidant, tert-butylhydroperoxide (tert-buOOH) to the perfusate of isolated rabbit lungs causes pulmonary vasospasm [16]. Oxidant-induced pulmonary vasoconstriction can be blocked by the cyclooxygenase or thromboxane synthase inhibitor, indomethacin and is closely correlated with the thromboxane level in the effluent perfusate [13, 16–19], suggesting a critical role of thromboxane in pulmonary vasoconstriction. On the other hand, TMB-8, an intracellular Ca2+ ([Ca2+]i) antagonists, has been shown to prevent oxidant-mediated pulmonary vasoconstriction [13]. Thus, oxidant-mediated PAH is triggered by an increase in [Ca2+]i. In many occasions PAH occur due to an increase in [Ca2+]i caused by stimulants such as thromboxane A2 and endothelin-1 that generates oxidants. Oxidant and Ca2+ ionophore-mediated pulmonary hypertension has been observed to be inhibited by serine protease inhibitors, for example, aprotinin [19–21].
The mechanism by which oxidants stimulate production of the arachidonic acid (AA) metabolites has gained considerable interest. A report by Chakraborti et al. [22] indicated that oxidants, e.g., tert-buOOH stimulation of pulmonary artery endothelial and smooth muscle cells caused a marked increase in phospholipase A2 (PLA2) activity with subsequent generation of AA. Mepacrine, an inhibitor of PLA2 inhibits tert-buOOH-induced increase in PLA2 activity, thromboxane B2 production, and PAH [19, 22]. Some investigators, considering the analogy of activation of pancreatic phospholipase A2 [23], suggested that a serine protease might be involved in regulating PLA2 activity [24]. Chakraborti et al. [25, 26] demonstrated that oxidant-mediated activation of PLA2 activity in pulmonary endothelial and smooth muscle cells occur with the involvement of proteolytically activated protein kinase Cα (PKCα). They have also demonstrated that oxidants caused increase in [Ca2+]i in pulmonary endothelial and smooth muscle cells can activate an aprotinin sensitive protease having mol mass of ~43 kDa [26]. The protease then proteolytically activates PKCα resulting in stimulation of cPLA2 (Fig. 1), which generates thromboxane and that has been observed to be important in producing PAH [25, 26]. Oxidants elicit an increase in [Ca2+]i due to proteolytic activation of PKC-δ by MMP-2 resulting in phosphorylation of a pertussis toxin sensitive protein (Gi) leading to inhibition of Na+ dependent Ca2+ uptake (Na+/Ca2+ exchanger) in the endoplasmic reticulum (ER) [27–31] (Fig. 2), whereas the role of cell membrane for an increase in [Ca2+]i has been observed to be due to phosphorylation of G i via proteolytically activated PKCα by an aprotinin sensitive serine protease leading to inhibition of Na+ dependent Ca2+ efflux (Na+/Ca2+ exchanger) (Fig. 1) in pulmonary vascular cells [24–26].
Fig. 1.
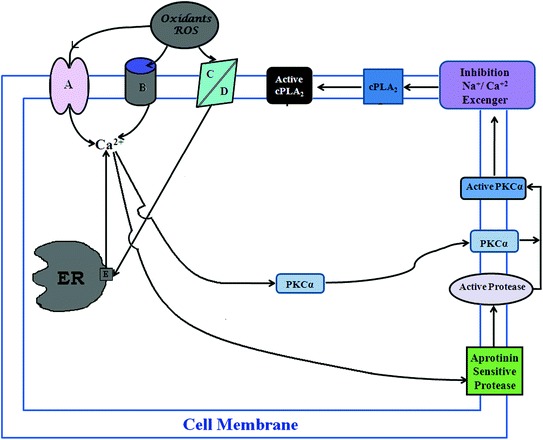
Schematic representation of the underlying mechanism associated with oxidant (reactive oxygen species: ROS)-mediated cPLA2 activation in pulmonary vascular endothelial and smooth muscle cells. A calcium channels; B membrane bound Ca2+ stores; C anion channels; D diffusion; E inhibition of Na+ dependent Ca2+ uptake
Fig. 2.
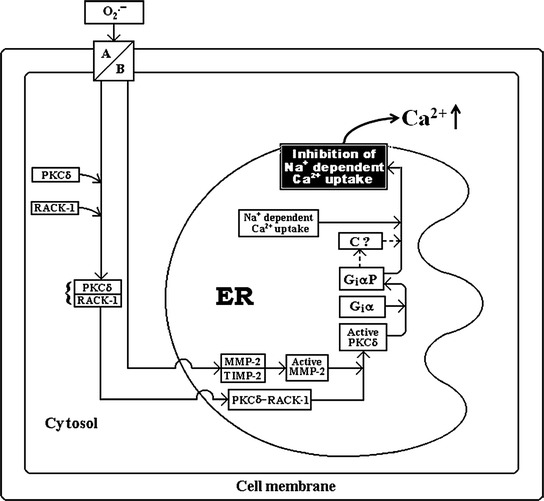
Schematic representation of the underlying mechanism of oxidant (reactive oxygen species: ROS) triggered inhibition of Na+ dependent Ca2+ uptake in bovine pulmonary smooth muscle ER resulting in an increase in [Ca2+]i. A Anion channel; B diffusion; MMP-2 matrix metalloprotease-2; TIMP-2 tissue inhibitor of metalloprotease-2; PKCδ protein kinase C delta; RACK-1 receptor for activated C kinase-1; Giα inhibitory G protein α subunit; GiαP phosphorylated Giα; C Na+-K+-ATPase/Na+-H+ exchanger. (Taken from Chakraborti et al. (2005) Mol Cell Biochem. 280: 107–117 with permission)
In many systems, Ca2+-ATPase represents only about 1% of the total proteins [32]. In heart sarcolemmal vesicles, Ca2+ uptake via NCX produces maximum transport velocity and that has been demonstrated to be about 30-fold up than that elicited by the sarcoplasmic reticulum {S(ER)} Ca2+ pump system [33]. In addition to the Ca2+ pump, Na+ dependent Ca2+ uptake system is an important mechanism to sequester Ca2+ in the ER of pulmonary vascular cells [29–33]. A decrease in Ca2+ sequestration by proteolytic inhibition of Na+ dependent Ca2+ uptake has been observed to measure duration of free [Ca2+]i transient, which eventually produces vasoconstriction [34, 35]. In different systems, Na+/Ca2+ exchanger controls the contractility of smooth muscle cells [34, 35]. For example, contractile dysregulation in the myocardium could be related with activation of proteases [36]. Thus, the role of cell membrane associated aprotinin sensitive protease and ER MMP-2 on Na+/Ca2+ exchange in pulmonary artery endothelial and smooth muscle cells under oxidant triggered condition is an important mechanism for the pathological manifestation of pulmonary vasoconstriction [24–26, 29–31].
MMPs and Pulmonary Hypertension
PAH is characterized by persistent vasoconstriction and remodeling of pulmonary vasculature associated with activation of proteases, for instance, MMPs [34]. Remodeling of pulmonary artery is associated with an alteration of extracellular matrix (ECM) turnover with concomitant change in ECM proteins level. In PAH, dysregulation of ECM turnover has been suggested to play an important role in the pathological remodeling process [35, 36]. ECM degradation occurs by different proteases of which matrix metalloproteases (MMPs) has been shown to play the crucial role [37, 38]. Of the MMPs, MMP-2, and MMP-9 are able to cleave basement membrane associated type IV collagen, which increase remodeling of the pulmonary vasculature in PAH [39]. Given the potency of MMPs, its activity is tightly regulated at the transcriptional and post-translational level, where the tissue inhibitors of MMP (TIMPs) play a pivotal role [40, 41].
IL-1, a potent endogenously generated inducer of PAH, elicits its effect via an increase in the level of TGF and TNF in pulmonary smooth muscle cells. TGF causes an increase in the expression of the 92 kDa proMMP-9 and 72 kDa proMMP-2 mRNAs, while TNF triggers activation of proMMP-9 and proMMP-2 [42].
MMP-2 is produced upon activation of proMMP-2 by a variety of stimuli under different pathophysiological conditions. It has been observed that the activation of proMMP-2 occurs at the cell membrane. Interaction between MT1-MMP and TIMP-2 is an important phenomenon in the activation of proMMP-2. The MT1MMP-TIMP2 associates with proMMP-2 and forms a trimolecular complex, which triggers the activation of proMMP-2 and subsequently generates MMP-2 [41, 42]. The activation of proMMP-2 in pulmonary artery smooth muscle cells occur with the involvement of protein kinase C-α dependent and NF-кB-MT1MMP-mediated signaling mechanism. TNF-α augments mRNA and protein expression of MT1MMP, while the expression level of TIMP-2 diminishes. The increase in TNF-α leads to IKK activation, IB phosphorylation and degradation, and subsequently activation of NF-кB. Upon activation, NF-кB binds to the MT1-MMP promoter, thereby enhancing its expression and subsequently increases proMMP-2 level in association with TIMP-2 that is modulated by protein kinase C-α at the cell membrane [41, 42] (Fig. 3). This indicates therapeutic potentiality of PKC inhibitors in ameliorating the PAH, where activation of proMMP-2 is an important phenomenon.
Fig. 3.
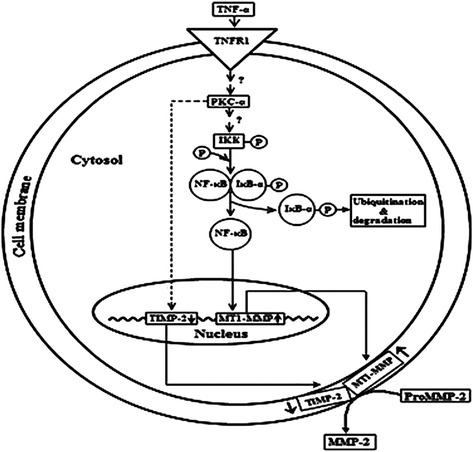
Schematic representation of TNFα-induced proMMP-2 activation in the SMCs. TNFα binds to cell surface receptor TNFR1. Upon binding, TNFα induces PKCα activation, which subsequently activates IKK by phosphorylation. Activated IKK then phosphorylates IkB-α, which upon phosphorylation is ubiquitinated and degraded in the cytosol. The free NF-кB then translocates to the nucleus and increases the expression of MT1-MMP, which then accumulates on the cell surface. PKC-α, on the other hand, also down regulates TIMP-2 expression by mechanism that is currently unknown. (Taken from Roy et al. (2013) J Biochem 153:289–302 with permission)
ProMMP-9 activation by TNF-α has been observed to occur with the involvement of an aprotinin sensitive serine protease [42]. TNF-α was shown to inhibit aprotinin and TIMP-1 mRNA and protein expression, which trigger activation of proMMP-2 resulting in the stimulation of MMP-2 (Fig. 4). Under IL-1β stimulation, the aprotinin sensitive protease was not activated, although a discernible inhibition of TIMP-2 mRNA and protein expression were triggered by TNF-α [42]. Thus, IL-1-induced stimulation of the two progelatinases occurs via different mechanisms.
Fig. 4.
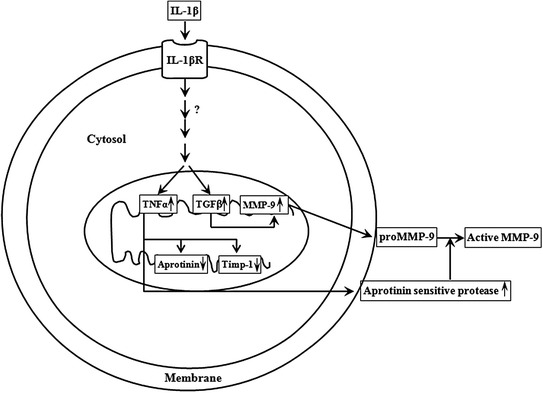
Schematic representation of proMMP-9 activation by an aprotinin sensitive protease during IL-1β stimulation of pulmonary artery smooth muscle cells. IL-1β treatment to the cells stimulates TGF-β and TNF-α. TGF-β stimulates expression of proMMP-9, while TNF-α activates proMMP-9 via an increase in a ~43 kDa aprotinin sensitive protease concomitant with the down regulation of aprotinin and TIMP-1 expression
Influenza
Influenza viruses are highly infectious and trigger acute respiratory diseases with significant morbidity and mortality in humans and other animals [43–46].
Influenza viruses can be classified as A, B, or C. Influenza virus A, found in humans and other mammals and birds, played a nefarious role in causing the three twentieth century major influenza outbreak and also the influenza outbreak of swine origin that occurred in the recent past [47]. Many of the influenza A-related mortalities are attributable to secondary bacterial pneumonia [48, 49].
Haemagglutinin (HA) protein contributes critically to influenza virus-mediated pathogenicity. HA of influenza virus binds to sialic acid containing cell surface receptors. HA upon cleavage by a good number of host of protease(s) forms HA1 and HA2 subunits and that has been fused with host cell membrane, which subsequently initiates the infection process [49–52]. In most cases, the cleavage site of HA of avian and mammalian influenza viruses is a single arginine, albeit a single lysine amino acid has also been observed at the cleavage site in some cases. Cleavage can occur extracellularly by trypsin [53, 54] and proteases such as plasmin [55–57], tryptase of bronchiolar epithelial and mast cells [58], and also by bacterial proteases [59–61].
Several other proteases expressed in the lung are also able to facilitate influenza virus spread. Böttcher et al. [62] demonstrated that TMPRSS2 and TMPRSS11D, transmembrane serine proteases, (a.k.a. human airway trypsin-like protease: HAT) activate the influenza viruses H1N1 (A/Memphis/14/96), H2N9 (A/Mallard/Alberta/205/98), and H3N2 (A/Texas/6/96) upon cleavage of haemagglutinin (HA) and contribute to the high pathogenicity of these influenza viruses in the lung [63]. In addition to HAT, TMPRSS-2 and -4, the granzymes (Gzm) such as GzmA, GzmB, and GzmE are known to play a key role in the process of cleavage of 1918 H1N1 HA as a part of the progression of the influenza disease [62, 63].
The sites of virus replication in the microenvironments of respiratory tract represent complex extracellular proteases (such as trypsin and tryptase), which activate a family of receptors called protease activated receptors (PARs) [64, 65] and that play an important role in both virus replication and innate immune response [58, 66]. Four PARs (PAR1-4) are known to be activated by different proteases. After cleavage of the receptor(s) by proteases, the newly released amino terminal sequence binds and internally activates the receptor [67]. In the airways of IAV-infected mice, an increase in the level PAR2 upon IFNγ-mediated modulation has been shown to play a crucial role in influenza pathology [68, 69].
Multiple serine protease activities are implicated in mediating influenza virus infection. Inhibition of influenza A virus infection in cultured lung epithelial cells by serine protease inhibitors, for example, aprotinin markedly protects mice from infection [70]. Another serine protease inhibitor, camostat has also been shown to possess anti-influenza (Taiwan/1/86) virus pathology [71].
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) is associated with the pathological manifestations of emphysema and chronic bronchitis. Emphysema is characterized by a marked destruction of the alveolar septa with concomitant decrease in lung plasticity and that results in gas trapping leading to a marked decrease in pulmonary oxygenation in the lung. Chronic bronchitis usually occurs with inflamed and thickened airways along with an increase in mucus production by the cells in the airways, which leads to a marked increase in cough and difficulty in breathing. Noxious particles present in cigarette smoke and automobile exhaustion have been observed to be an important causative agent of COPD [72, 73].
In the early 1960s, proteases have been shown to produce lung lesions in experimental animals similar to human emphysema. Initial studies in this scenario included metalloproteinases, papain and subsequently serine proteases, for example, porcine pancreatic elastase [74].
Serine Proteases and COPD
The identification of α1-PI (an endogenous serine protease inhibitor) and subsequent research confirmed that a strong association exists between the development of emphysema and inherited deficiency of the inhibitor [75]. A critical role of serine proteases have been established in the pathobiology of emphysema. Preliminary studies in this genetic condition of inherited α1-PI deficiency indicated that chronic bronchitis is associated with the early onset of the disease [76, 77].
The lung epithelium of normal individuals is protected from the detrimental effects of neutrophil serine proteases (NSPs) by a battery of antiproteases. Large quantities of NSPs (~20-fold w.r.t. normal subjects) released by neutrophils in acute and chronic inflammatory conditions overpowered antiprotease activities, leading to uncontrolled proteolysis and subsequently lung damage [74–76].
Alpha-1-PI deficiency increases activities of different enzymes secreted by activated neutrophils such as NE, cathepsin G (CatG), and proteinase 3 (PR3), all of which are capable of damaging different components of the ECM such as collagen, laminin, fibrillin, and elastin. However, several evidences suggest that it is the destruction of lung elastin that is important in causing emphysema, which generates COPD pathophysiology in animal model systems [75, 76].
Human neutrophil elastase (HNE), by destroying elastin, plays a critical role in the development of pulmonary emphysema [76, 77]. HNE has many other biological activities. For example, it stimulates mucin production [78], activates MMPs [5], inactivates TIMPs [79], and generates neutrophil chemotactic elastin-derived fragments [80, 81]. PR3 and CatG, two elastase homologues secreted in massive quantities from neutrophils at inflammatory sites, have also been shown to have proinflammatory functions acting through various mechanisms [80–82]. The most abundant is the α1-PI, which targets preferentially HNE. Secretory leukoprotease inhibitor (SLPI), an inhibitor of HNE and CatG, but not of PR3, has been shown to control excess proteolysis in the upper airways [83]. Elafin, derived from trappin-2 (pre-elafin) [84], is an NSP inhibitor that controls the activities of HNE and PR3 [85]. Other NSP inhibitors including 1-antichymotrypsin and monocyte/NE inhibitor (MNEI) were found to play relatively minor role as protease inhibitors [86, 87]. NSPs, therefore, could prove useful as therapeutic target for a number of inflammatory lung diseases.
There are differences in pathological manifestations among smoking and non-smoking α1-PI deficient COPD patients. The smokers with COPD frequently show pulmonary emphysema and bronchitis, while patients of the latter category often show emphysema without bronchitis. Although the chronic smokers usually suffer from the antiprotease inactivation in both the airways and respiratory units as a result of oxidant-mediated inactivation of α1-PI, the non-smoking α1-PI deficient individuals possess diminished antiprotease content primarily in respiring units, which are free of mucus glands and depends upon α1-PI for antiprotease defence [88].
Oxidative stress induced by cigarette smoke in COPD patients may promote the inflammatory state by recruiting additional neutrophils and upregulating the inflammatory transcription factor, NF-кB and neutralizing TIMPs in addition to α1-PI and SLPI [89].
Urokinase Plasminogen Activator and COPD
In COPD patients, an increase in urokinase plasminogen activator (uPA) level in the airway epithelial and alveolar cells, and lung macrophages cause destruction of small airways and alveolus of the lung [90]. Urokinase plasminogen activator receptor (uPAR), in addition to functioning as a protease receptor, mediates intracellular signaling [91]. An increase in uPAR level in the macrophages has been observed in patients with COPD, which suggests the critical role of uPAR in inflammation and tissue remodeling including parenchymal destruction and fibrosis of small airways [92].
MMPs and COPD
MMPs are known to induce morphological changes in the lung that are prevalent in COPD. Several MMPs are known to play important roles in the pathogenesis of COPD [93]. Lung parenchyma and inflammatory cells such as neutrophils and macrophages are the major sources of MMPs in patients with COPD [94].
MMP-12 (a.k.a. macrophage elastase) is known to play an important role in COPD pathogenesis. A marked increase in MMP-12 expression in alveolar macrophages is associated with smoking associated emphysema [95]. In mouse, deletion of MMP-12 gene prevents cigarette smoke-induced inflammation, neutrophil influx, and emphysema in the lung [96, 97]. Genetic analysis of human COPD patients demonstrated that the common serine (codon 357) of the MMP-12 gene plays a crucial role in the pathological manifestations of matrix degradation, which has been observed to be related with the severity of the disease [98, 99].
Analysis of COPD lung tissue indicated an increase in the activity of MMP-1 and MMP-8, but not MMP-13 [100, 101]. An increase in MMP-1 activity was found in type II pneumocytes in patients with emphysema, but not in normal control subjects [101]. Neutrophil-derived MMP-8 levels were markedly increased in patients with COPD in comparison to the normal subjects [101]. Prominent increase in MMP-2 and MMP-9 expression has been observed in the lung of COPD patients [102]. During interleukin-10 (IL-10)-mediated airflow obstruction, an imbalance between MMP-9 and TIMP-1 results in an increase in MMP-9 activity was found in an animal model system [103].
Acrolein, a component of cigarette smoke‚ has been shown to initiate cleavage of proMMP-9, thereby producing active MMP-9. However, MMP-9 knockout mice do not completely inhibit cigarette smoke-induced emphysema, suggesting that other MMPs also play role in COPD pathogenesis [103]. Importantly, MMP-14, the membrane-type MMP (MT1-MMP), has been observed to be induced by acrolein which upon increase in mucin production leads to COPD [104] (Fig. 5).
Fig. 5.
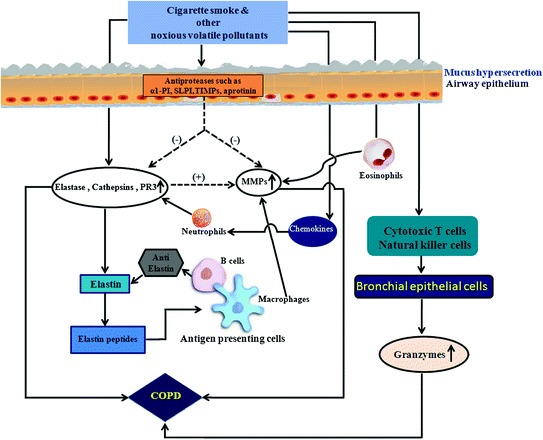
Cigarette smoke and other noxious volatile components-mediated dysregulation of protease–antiprotease balance resulting in an increase in protease activity for pathological manifestation of COPD
Lung Fibrosis
Lung fibrosis (a.k.a. interstitial lung diseases) is a chronic disorder, exemplified by a marked increase in matrix degradation and intra-alveolar fibrosis leading to dyspenea, impaired oxygen transfer and alveolar collapse [105, 106]. Lung fibrosis occurs in the alveolar space and interstitium and is characterized by a widespread accumulation of differentiated fibroblasts (i.e., myofibroblasts) and ECM components.
Fibrotic disorders in the lung are associated with dysregulation of proteolytic activities. A considerable number of reports have suggested the involvement of cathepsins in this scenario. Enhanced proteolytic processing of CatB was observed in the lungs during an increase in TGF-β, suggesting that CatB may participate in fibrogenesis [107].
Microarray studies have revealed that in addition to NE, MMP-7 (a.k.a. matrilysin) is an important COPD marker. MMP-7 degrades decorin, the extracellular proteoglycan, which subsequently releases decorin-bound transforming growth factor-β (TGF-β) [108] and that subsequently contributes to TGF-β activation, which is known as a critical marker of COPD [109].
Silicosis
An increase in the activity of MMP-2, MMP-9, and stromelysin has been demonstrated in alveolar macrophages from silica-treated rats, which contribute to extracellular matrix (ECM) and basement membrane (BM) degradation [110]. Administration of silica particles to mice causes upregulation of cathepsin K (CatK) expression and activity in silicotic lung homogenates compared to control lungs. Lung fibroblasts and macrophages were known as the main CatK-producing cells. Expression of CatK is inversely correlated to the level of TGF-β1, suggesting a protective role of CatK during silicotic process [111]. Mature active CatB, -H, -K, -L, and -S were identified in the broncho alveolar lavage fluids (BALFs) of patients suffering from silicosis. Among them, CatH has been observed to be the most abundant aminopeptidase, while CatB and CatL were mostly found thiol dependant endoproteases. Importantly, an increase in cathepsins/inhibitors ratio has been shown to favor uncontrolled proteolysis during silicosis [110, 111].
Cystic Fibrosis
In cystic fibrosis (CF), a marked increase in the activities of proteases could damage the airway architecture and that contributes to progressive bronchiectasis, a condition where the bronchial tubes of lungs are permanently damaged and enlarged due to infection in the bronchi [112, 113]. CF is an autosomal recessive genetic disorder caused by loss of expression or functional mutations to the cystic fibrosis transmembrane conductance regulator (CFTR) [114, 115]. CF affects multiple organs, albeit the pathology associated with CF appears to be due to its effect on the respiratory system. Non-functional CFTR channels in CF patients prevent the regulation of chloride and sodium ions across epithelial membranes leading to an increase in dehydrated mucus secretions in the lungs [112–116].
The key immune cell mediators seen in CF patients are polymorphonuclear neutrophils [115]. Upon recruitment, activated neutrophils release a wide variety of proteases, which induce inflammatory response and subsequently tissue damage [115, 116].
The impairment of mucociliary clearance mainly revolves around the interactions between NE and mucins. Mucins are a family of highly glycosylated proteins produced by epithelial cells and are the main components of the mucus found clogging of the airways in CF patients [117, 118]. NE has been shown to regulate the mucins via activation of TNF—converting enzyme, which upregulates their expression via epidermal growth factor receptor (EGFR) pathway [118–120]. CF patients cannot efficiently clear mucus due to damage by proteases to the cilia structures in the lungs and, therefore, are highly susceptible to chronic bacterial infections [121–123].
MMPs were found to play a crucial role in CF pathogenesis [124]. MMP levels are increased in the BAL of CF individuals [125]. MMPs produce proline-glycine-proline (PGP), a neutrophil chemoattractant derived from extracellular matrix, which regulates the immune response during CF [126].
A marked increase in NE has been shown in CF lung, which causes airway remodeling by degrading ECM proteins such as elastin and fibronectin [127]. The resulting alteration of airway epithelial cell membrane by NE induces neutrophil-mediated inflammation upon increase in the expression of proinflammatory cytokines, for example, IL-8, which results in neutrophil-mediated inflammation by upregulating the proinflammatory MMPs, CatG, and PR3 leading to tissue damage in the CF lung [6].
Asthma and Allergy
Asthma
Asthma is a chronic inflammatory disease of the airways and its occurence and propagation is on the rise. The number of patients with asthma is estimated to attain a staggering figure of 100 million globally by 2025 [128]. Generally, asthma is triggered by the activation of adaptive immune response that upon inducing the lung triggers mucus production, increased IgE level, airway remodeling, and airway hyperactivity [129].
Manifestation of Asthma is characterized by acute inflammatory response and airway obstruction [130]. In both acute and chronic asthma, proinflammatory cells, including neutrophils, eosinophils, mast cells and macrophages enters into the lung tissue [131, 132]. These proinflammatory cells secrete a variety of extracellular proteases of which serine proteases and MMPs are important as these enzymes play prominent role in asthma pathogenesis [133–139].
Plasminogen can be converted to the active enzyme plasmin by tissue type plasminogen activator (PA) or urokinase-type PA (u-PA). Tissue PA and u-PA are associated with the dissolution of fibrin and also in the degradation of ECM components [140]. Plasminogen activator inhibitor-1 (PAI-1) is a major inhibitor of tissue type plasminogen activator and u-PA, and thereby contributes to matrix formation by preventing matrix degradation. Mast cells (MCs) in the airways of patients with asthma are crucial in initiating allergic inflammation [141]. MCs and bronchial epithelial cells (BECs) are the major source of plasminogen activator inhibitor (PAI-1). The interactions between the BECs and the MCs are important in maintaining persistent inflammation and structural changes in asthma [142].
IgE-mediated inflammation is well known for the pathogenesis of asthma. MCs-derived TGF-β upon cross-linking with IgE receptor enhances PAI-1 production in BECs. This increase in the production of PAI-1 has been suggested to play a critical role in the development of fibrosis that occurs adjacent to the epithelium [143]. Conceivably, drugs that inhibit activation of MCs, for example, by anti-IgE may prove useful in preventing airway remodeling in asthma.
Serine Protease and MMP in Asthma Pathophysiology
Proteolytic enzymes including NE and MMP-9 play important roles in tissue remodeling and repair in the airways [144]. The proteolytic enzymes levels are increased in asthma, which occurs due to an imbalance in the protease–antiprotease system.
In neutrophilic asthma, high levels of active NE and proMMP-9 were observed, whereas only a small amount of MMP-9 has been observed to be active. However, eosinophilic asthma was characterized with high level of active MMP-9 without free elastase. Thus, a differential profile of protease activity has been observed in asthma. A deficiency of antiproteases may explain the differential enzyme activity observed in eosinophilic and neutrophilic asthma. An increase in the level of MMP-9 bound to TIMP-1 in subjects with neutrophilic asthma in comparison to the eosinophilic asthma has been observed. This could explain about the presence of low level of active MMP-9 in neutrophilic asthma. This relative deficiency of TIMP-1 in subjects with eosinophilic asthma in comparison to neutrophilic asthma may explain about the high level of active MMP-9 that exists in the sputum of subjects in this group [144–146].
Alpha1-PI level has been observed to be increased in neutrophilic asthma, but its function was impaired leading to a marked increase in free elastase (NE) activity. Proteolytic inactivation of α1PI may lead to a form, which acts as an activator of neutrophils and that could result in superoxide (O2 . −) production [146]. However, role of proteolytic enzymes in specific inflammatory phenotypes of asthma is not clearly known. In contrast, IL-8 (a potent chemoattractant and an activator of neutrophils) plays an important role in eosinophilic asthma [6]. NE can induce production of IL-8 and its potency has been observed to be elevated upon proteolytic processing by MMP-9 [147].
Patients with persistent inflammatory asthma elicit more non-eosinophilic asthma progression than that of the eosinophilic asthma. These exacerbations are not prevented by corticosteroid treatment [148]. COPD and neutrophilic asthmatic patients generally show chronic airway inflammation associated with a marked airway neutrophilia, which are not discernibly responsive to inhaled corticosteroids [148, 149].
Cytokines and Asthma
Inflammation in asthma has been observed to be mediated by a specific subclass of T-lymphocytes referred primarily to Th2 lymphocytes, which causes inflammation and remodeling via secretion of specific cytokines [150].
Cytokines, for example, IL-11 play primary role in mediating asthma pathophysiology through its receptor (IL-11R). ADAM-10, a matrix metalloprotease, can release the IL-11R ectodomain upon cleavage of IL-11 receptor. Serine proteases such as NE and PR3 can also cleave the IL-11R. The resulting truncated soluble IL-11R (sIL-11R) activates the inflammatory cells. Thus, IL-11 signaling pathology proceeds upon proteolytic cleavage of its receptor [151].
An increase in the numbers of apoptotic airway epithelial cells in COPD has been observed to be associated with secondary necrosis [152, 153]. In severe asthma, conditions associated with increased airway neutrophilia, tissue damage and an increase in apoptosis of airway epithelial and smooth muscle cells have also been demonstrated [154]. Granzymes, a family of serine protease, have a repute to initiate immune-mediated cell death. Cytotoxic T cells and natural killer (NK) via granzyme-mediated pathway induces apoptosis of target cells, e.g., bronchial epithelial cells. Granzymes play critical roles in a variety of age-related chronic inflammatory diseases. There are five human granzymes identified so far in the lungs. These are granzyme A (GzmA-tryptase), granzyme B (GzmB-aspase), granzyme H (GzmH-chymase), granzyme K (GzmK-tryptase), and granzyme M (GzmM-metase). Granzymes, especially granzyme B and perforin, are stored in secretory granules of cytotoxic cells, and are released into the intercellular space following adhesion to the target cells. In presence of Ca2+, perforin pores in the cell membrane enable entry of granzyme B and subsequently induces caspase-dependent apoptosis [155], which may be an important mechanism of lung injury in asthma.
Allergy
Many aeroallergens like house dust mites and fungal allergens associated proteases play important role in asthma pathophysiology. Epidemiological studies suggested that sensitivity to fungal allergens could be an important cause of allergic asthma [156].
Alternaria Alternate and Asthma Severity
The fungus Alternaria alternate has been observed to cause asthma under certain circumstances [157, 158]. The allergen of the fungus possesses intrinsic proteolytic activities and that upon activating protease activated receptors (PARs) play a prominent role in mediating allergic airway diseases. Interleukin-33 (IL-33) has been observed to be associated with the development of allergic asthma [159]. IL-33 expression in the lung was found to be elevated in the asthmatic subjects and the asthma severity could be positively correlated with the IL-33 expression in the airways [160, 161].
Alternaria driven release of IL-33 occurs with the involvement of a serine protease specific to this aeroallergen. The Alternaria serine proteases cause marked inflammation because of the capacity of the serine protease to drive IL-33 release, which in turn induces rapid onset of asthma exacerbations. Thus, targeting the protease—IL-33 signaling axis could prove useful as a therapeutic measure in this kind of asthma pathogenesis [162].
Aeroallergenicity of Acanthamoeba
The free living amoeba, Acanthamoeba trophozoite, is found in human airway cavities and possesses high protease activities, which can elicit allergic airway inflammation [163]. Intranasal inoculation of A. trophozoite or its excretory secretory (ES) proteins in mice have been shown to elicit allergic airway inflammation. ES proteins with strong protease activities stimulate dendritic cells and also able to enhance the differentiation of early T cells into mature IL-4 secreting T cells. Treatment of ES proteins in the protease activated receptor (PAR-2) knockout mouse showed inhibition of lung airway inflammation and Th2 immune responses with lower IgE level compared with the normal mouse. This suggests a role of PAR-2 in the aeroallergenicity of Acanthamoeba allergens [163].
Seasonal Rhinitis and Asthma
Allergic diseases like seasonal rhinitis and asthma are generally result from exposure to airborne pollens. Asthmatic patients allergic to pollen have been observed to develop a chronic inflammation of the airways leading to bronchial obstruction and hyper responsiveness. Upon inhalation, pollen grains release a wide variety of allergens with protease activities, which may act as inflammatory mediators and subsequently pathogenesis of respiratory allergies. The proteases were known to inactivate lung regulatory neuropeptides, for example, substance P and vasoactive intestinal peptide (VIP) [164] leading to dysregulation of the contraction-relaxation rhythm of the respiratory airways. The inhaled allergens were processed by dendritic cells (DCs) present at the subepithelial regions, and then present allergen peptides to native T-lymphocytes for stimulation of IgE production. Some pollen allergens exhibit proteases such as aminopeptidase and trypsin-like serine protease activities, which can cleave proteins from junctional complexes between epithelial cells [165]. A 98 kDa aminopeptidase of Parietaria judaica, for instance, has been shown to cause detachment of A549 human alveolar epithelial cells by degrading intercellular adhesion proteins from tight junctions and adherens junctions [165, 166]. Pollen proteases can induce degradation of cell junction proteins such as occluding, claudin-1 and E-cadherin, thereby help allergens to cross the epithelial barrier and contact with DCs for intensifying immune response [164–166].
Organic Dust Allergy
Organic dusts made for agriculture may lead to airway inflammation, which may cause sinusitis and chronic bronchitis to workers in agricultural industries [167–170]. Workers in livestock industries working in concentrated animal feeding operations (CAFOs) are susceptible to chronic airway diseases [171]. Extracts of dust collected from CAFOs are potent stimulators of lung inflammatory responses. Hog dust extract (HDE) contains active proteases, which play a critical role in lung inflammatory processes [172, 173].
Epidermal growth factor receptor (EGFR) signaling has been shown to play an important role in the proinflammatory response of bronchial epithelial cells (BECs) to HDE [172]. The proinflammatory effect of HDE has been suggested to be due to the proteolytic activation of PARs, especially PAR-2 [173]. In lung epithelial cells, actions of fungal, cockroach and dust mite allergen proteases are mediated by the cleavage and activation of protease activated receptor-2 (PAR-2) [174–176]. However, PAR-1 is unable to mediate the effects of these proteases. PARS play an important role in bronchial fibroblast proliferation; epithelial cell wound healing and hypersecretion of mucus [177, 178]. By inhibiting HDE proteases or abrogating activation of epithelial cells, PAR-2 could inhibit HDE-induced inflammatory indexes in bronchial epithelial cells (BECs) and subsequently inhibition of the allergic response [179]. Thus, targeting the protease activity of organic dusts made for agricultural usage and other air borne dusts may prove useful as a strategy for preventing airway inflammation in agricultural workers, who are generally exposed to dusty agricultural environments.
Cockroach Allergy
Bernton and Brown [180] first observed skin rashes upon exposure of cockroach over the skin of allergic patients. Subsequently, a considerable number of evidence have confirmed that exposure to cockroach can induce allergy. Proteases associated with cockroach can produce bleb formation to the skin, which could play a critical role in the development of allergic disease [181–183].
Cockroach allergens such as saliva, feces, cast skins, and dead bodies contain serine protease activities, however, feces (frass) was found to be the prominent source of allergens, which contains serine protease activity [184, 185]. Sensitization of wild type mice to German cockroach frass (GC frass) has been shown to increase allergic hyperactivity (AHR) due to a marked increase in serum IgE and also production of cytokines such as IL-13, IL-4, IL-5, and IL-17 [186]. Sensitization of PAR-2 deficient mice with GC frass, however, did not show a discernible increase in allergic airway inflammation indicating a role of PAR-2 in mediating allergic airway inflammation [185].
Lung Cancer
Lung cancer is one the most prevalent and lethal diseases worldwide. Despite recent advances in chemotherapy, the molecular basis of its progression to a metastatic disease remains unclear.
Exposure of bronchial epithelial cells of smokers produce oxidants and reactive oxygen species (ROS) with consequent cellular responses to activate NF-кB and other transcription factors that regulate inflammation-related genes and initiates several signaling pathways depending on both genetic and epigenetic factors and manifest COPD and lung cancer [187, 188].
MMPs and Lung Cancer
Lung cancers are of two major types: (i) small cell lung carcinoma (LC); and (ii) non-small cell lung carcinoma (NSCLC). ECM and basement membrane components are proteolytically cleaved by MMPs. These components play a pivotal role in cancer progression. Lung cancers express high levels of MMPs. MMP-7 and MMP-9 expressions were found to be markedly high in NSCLC in comparison to the normal tissue [189]. MMP-1 and MMP-3 promoter polymorphisms are associated in modifying susceptibility to NSCLC and also to an increase in the risk of lymphatic metastasis of these tumors [190]. MMP-2 and MMP-9 activities have been found to be associated with an increase in tumor spread [191].
Treatment of mice with CH1104I (dual inhibitor of MMP-2 and -9) has been shown to markedly inhibit metastasis of lung carcinoma, which suggests that inhibition of MMP-2 and -9 could significantly inhibit tumor invasion and metastasis [192]. MMP-2 and -9 expressions may have prognostic implications in patients with NSCLC [193]. Over expression of MMP-1 has been observed to induce the formation of lung metastases [194, 195].
High Temperature Requirement A (HtrA) Serine Protease and Lung Cancer
HtrA (a.k.a. DegP) is a heat shock-induced serine protease that is active in the periplasm of Escherichia coli. Homologues of HtrA were found in a wide range of bacteria and in eukaryotes. Till date, four human homologues of the bacterial serine protease HtrA have been described, which are named as HtrA-1, -2, -3, and -4 [196, 197]. They have a variety of functions including cancer [198].
HtrA1 has been observed to be downregulated in lung cancers [198]. In human cancer cells, HtrA1 over expression prevents cancer cell growth and proliferation suggesting HtrA1 as a tumor suppressor. A modest expression of HtrA1 has been observed in primary tumors and lymph node metastases. However, the exact functions of HtrA1 in cancer are mostly unknown. A previous report suggested that HtrA1 elicits its function by inhibiting TGF-β pathway [199]. The role of TGF-β in cancer progression is well documented [200]. Accordingly, the TGF-β signaling pathway has been considered as both a tumor suppressor and promoter pathway of tumor progression and invasion. It, therefore, seems probable that activation of TGF-β signaling pathway occurs when HtrA1 is downregulated, thereby contributing to the cancer progression. Alternatively, over expression of HtrA1 has been shown to induce apoptosis [201]. Thus, loss of HtrA1 expression alters regulation of apoptosis and could lead to cancer progression [201].
HtrA1 degrades tubulin by disrupting microtubules (MTs), which suggest that HtrA1 could play an important role in regulating MT and tubulin stability and MT-associated cellular functions [245]. HtrA1 also regulates cell migration and offers a potential role in regulating MT organization associated with cell migration. However, the exact mechanism(s) by which HtrA1 regulates cell migration is currently unclear. Active HtrA1 upon removal of N-terminal Kazal-type trypsin inhibitory domains contributes to cell death through caspase dependent, as well caspase-independent mechanisms [202].
The role of HtrA2 protease in stress responses and apoptosis in lung cells has been established and a previous report suggested its involvement in cisplatin-induced death of renal cells [203]. Over expression of HtrA2 by cisplatin has been observed to follow the release of HtrA2 from mitochondria to the cytosol and it degrades anti-apoptotic proteins. This mechanism seems to be obligatory to trigger mitochondrial permeabilization for HtrA2 to participate in cell death [203, 204]. Therefore, HtrA2 plays a vital role in programmed cell death upon eliminating the caspase inhibitory activity of apoptosis [205, 206]. However, the detail mechanism(s) by which HtrA2 regulates lung cancer is currently unknown.
Smoking is a critical factor for lung cancers and the HtrA3 has been shown to be associated with smoking-induced lung cancer. HtrA3 expression has been found to be markedly downregulated in lung cancer cell lines and also primary lung tumors isolated from heavy smokers [207]. HtrA3, in contrast to the steady HtrA1 and HtrA2 expression, has been identified as a probable target for cigarette smoke-induced changes in normal human bronchial epithelial cells [207]. It has also been suggested that cigarette smoke-induced methylation of HtrA3 may play an important role in the etiology of smoking-linked lung cancer [207].
Studies on HtrA3 exon in lung cancer cell lines indicates that it possesses core xenobiotic response element (XRE) consensus sequence, 5-TNGCGTG-3 and is a target for methylation at CpG. XRE is located in the promoter region of the genes involved in metabolizing xenobiotic carcinogens, mainly aryl hydrocarbons from cigarette smoke and environmental pollutants, for instance, automobile exhaust [208, 209]. These compounds upregulate the gene products of XRE via aryl hydrocarbon receptors and multitude of other transcription factors [210, 211]. The degree of methylation of HtAr3 is similar when studied in A549 and H157 lung epithelial cells, when treated with NNK (Nicotine-derived nitrosamine ketone), a.k.a 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, an important tobacco-specific nitrosamines, which play key role in carcinogenesis [208]. NNK suppressed the expression of HtAr3, whereas 5-azo-dc has been shown to induce it [208]. Differential expressivity of HtAr3 may be controlled by other cross-talking mechanisms such as histone deacetylation, micro RNA activation, loss of heterozygosity and genetic mutation. Along with that HTAr3, HtAr1, and HtAr2 are also believed to be upregulated by xenobiotic stress [208, 212]. However, more research is needed to ascertain them as therapeutic targets pertaining to lung cancer for epigenetic therapies as well as prognosis to forecast tumor response.
Tuberculosis
Upregulation of CatG, but not NE, has been shown to induce cell death of activated Mycobacterium tuberculosis infected macrophages. The substrate specificity of CatG and NE is distinct. CatG cleaves the C-terminus of aromatic or positively charged amino acid residues, while NE cleaves the C-terminus of small hydrophobic amino acid residues [213, 214]. Enhanced necrosis in infected macrophages by CatG may result from proteolysis of specific target sequences, which are currently unknown. It has been shown that serpinb3a inhibition of CatG is necessary to prevent necrosis induced by IFN-γ in M. tuberculosis infected macrophages [214].
Role of matrix metalloproteinases (MMPs) as an important mediator of tissue destructive response in TB has now been clearly known [215, 216]. MMPs can cleave ECM components [217]. In humans, MMP-1 cleaves fibrillar components (types I and III collagen) of ECM. The level of MMP-3, the activator of MMP-1 is high in respiratory secretions of TB patients than control subjects [216–218]. In rabbits, MMP-1, -3, -7, -12, and -13 expressions are elevated in granulomatous and cavitary pathologies in human respiratory secretions observed in vivo model systems of TB [218–220]. In a human monocyte infection model, a marked reduction in TIMP-3 has been observed to be correlated with TB pathogenesis [221]. In a mice model, TIMP-3 has been observed to be associated with cavity formation and subsequently ECM degradation, which are important for TB pathogenesis [221, 222].
Acute Lung Injury
Severe acute respiratory distress syndrome (SARS) was identified in 2003, which triggered death of thousands of people worldwide [223, 224]. A new type of coronavirus has been identified and found responsible for the SARS, which produces pneumonia and associated high fever and severe dyspnea and subsequently acute respiratory distress syndrome (ARDS) followed by death due to acute lung injury (ALI) [224, 225]. ARDS is characterized by accumulation of inflammatory cells and severe hypoxia that leads to pulmonary edema [226].
Renin-angiotensin system (RAS) has been observed to play a critical role in SARS. In animal studies, a prominent role of angiotensin converting enzyme (ACE) in the pathogenesis of ARDS has been suggested [227, 228]. ACE2, a homologue of ACE, was shown to be a key regulator for coronovirus infection that produces SARS. ACE2 has been shown to be expressed in the lungs of both healthy and diseased humans, and it protects against SARS-induced ALI [229–231]. Therefore, it seems conceivable that ACE2 might prove a novel therapeutic target for SARS-coronovirus-induced ARDS that develops in emerging lung infectious diseases including influenza [231].
Elafin and ALI
Elafin, a serine protease inhibitor having mol wt of 6 kDa, found in lung secretions. Elafin is formed by proteolytic cleavage of its precursor protein, trappin-2 [232]. The antiprotease activity of elafin is located in the C-terminal domain having specificity for NE and proteinase 3. The N-terminus transglutaminase substrate binding motif (GQDPVK) of elafin cross-links with extracellular matrix proteins [233, 234].
In ALI, the protease–antiprotease balance alters in favor of proteases leading to an increase in protease activity, and this protease burden can produce pulmonary edema [235]. A decrease in plasma elafin level has been observed to be correlated with altered elafin gene expression and seems a critical component for an increase in acute respiratory distress syndrome (ARDS) [236–238].
In ALI patients, the 20S proteasome was observed to be markedly higher compared with normal subjects [239], but elafin level was shown to be decreased with consequent proteolytic degradation of antiproteases by the 20S proteasome in lung patients with ALI. This decrease may contribute to an increase in NE activity in ALI regulation and expression; however, its biological role in the lung is currently unknown. The cleavage of elafin by 20S proteosome suggests that the increment of antiprotease levels in ALI patients could prove clinically beneficial in attenuating uncontrolled activity of NE. Elafin’s multifunctional properties could prove useful as the therapeutic target for ALI [239–241].
Elane and ALI
Elane has a potential catalytic activity to hydrolyze elastin. Under physiological conditions, lungs are protected from this enzyme by endogenous inhibitors such as α1-PI, α2-macroglobulin, and SLPI. However, in the course of ALI, the balance between elane and its endogenous inhibitors is disregulated in favor of the enzyme [242–244] leading to massive infiltration of neutrophils into the lungs and subsequently tissue injury. Therefore, peptidic and non-peptidic elane inhibitors may prove useful for treating ALI associated with systemic inflammation [245, 246].
Age-Related Pulmonary Diseases
Granzymes, especially granzyme A and granzyme B, are the most abundant granzymes involving the membrane perforating molecule, perforin, which induce cell death [247]. Perforin facilitates granzymes entry into the target cell and that subsequently induces cell death [248]. Granzyme A has originally been thought to induce caspase-independent cell death; however, recent findings suggest that granzyme A may be involved in immune regulation of age-related lung disorders [249, 250]. In contrast, granzyme B induces apoptosis through caspase-dependent and -independent pathways [251]. An increase in granzyme A and B activities is known to promote generation of proinflammatory cytokines. ECM degradation and formation of autoantigens may exacerbate the inflammatory response [251]. Chronic inflammation is a hallmark of age-related cardiovascular and lung diseases. Thus, granzyme A and B may serve as important agent in promoting a positive feedback cycle that may be common to many persistent age-related disorders [251, 252]. However, role of other granzymes such as GzmH, GzmK, and GzmM in age-related ARDS are currently unknown.
Aspergillosis
Invasive pulmonary aspergillosis (IPA) elicited by the filamentous members of the genus Aspergillus can have devastating role in immune compromised individuals [253, 254]. Aspergillus fumigatus is responsible for the majority of IPA infections. Normal individuals are at little risk because of the effectiveness of their lung defences. However, inhalation of conidia becomes life threatening to subjects having weak immunity, which could allow the conidia to germinate into invasive hyphae in the lung [255].
The populations at greatest risk for IPA are patients with cancer, solid organ transplants, bone marrow transplants and those with advanced AIDS [256]. A. fumigatus secretes proteases like an alkaline serine protease (ALP), a metalloprotease (Mep), an aspartic protease (Pep) and a prolyl endopeptidase in the lungs [257–260]. A. fumigates secreted proteases are expressed in the lung during infection [261, 262].
A. fumigates binds to ECM proteins with the involvement of polysaccharides and glycoproteins of the conidial cell wall [263]. Several agents secreted from fungus such as proteases and toxins have been shown to influence its infection in host lung tissue [263].
Systemic Sclerosis
Systemic sclerosis (SSc) is an autoimmune disease, which could occur due to vascular injuries and fibrosis in skin and certain internal organs [264]. Some cytokines and growth factors like transforming growth factor-β (TGF-β) have been observed to stimulate fibroblast proliferation [264]. The disintegrin and metalloprotease-12 (ADAM-12) possess the extracellular cell binding functions. ADAM-12 is expressed in two alternative forms: (i) membrane-anchored form (ADAM12-L); and (ii) a short secreted form (ADAM12-S). An increase in the level of serum ADAM12-S level plays an important role in the pathological events of diffuse cutaneous systemic sclerosis (dcSSc) [264, 265].
ADAM-12 plays a critical role in fibrotic process. ADAM12-S has the ability to degrade physiological substrates such as the ECM substrates: fibronectin, type IV collagen [265] and also insulin-like growth factor binding protein (IGFBPs) [266, 267]. Degradation of IGFBPs augments the association between insulin-like growth factors, for example, IGF-I and its receptors. IGF-I down regulates collagenase activity with consequent increase in collagen production [268], which indicates that IGF-I could be an important mediator in the progression of fibrosis. Additionally, ADAM-12 has been observed to be upregulated in chronic wound suggesting that ADAM-12 could be related to the fibrotic process [269].
Bronchopulmonary Dysplasia
Bronchopulmonary dysplasia (BPD) usually occurs in prematurely born infants. Due to deficiency of lung development, BPD patients require prolonged medical ventilation for oxygen. BPD causes an increase in morbidity and mortality in preterm infants. BPD is characterized by chronic inflammation, alveolar hypoplasia and respiratory infections [270, 271]. In an animal model of BPD, a significant high elastase activity along with excessive proteolytic degradation of the elastic fibers due to a marked decrease in the levels of endogenous protease inhibitors are usually observed in the lung secretion [272]. Additionally, a discernible increase in mRNA and protein expression and also activities of cathepsins-B, -H, -K, -L, and -S have been observed in new borne BPD tracheal aspirates [273]. BAL fluid of newborn preterm infants with BPD showed elevation of MMP-9 and a decrease in free TIMP-1 level [274, 275].
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis, a rare and progressive lung disease, usually affects women of pre-menopausal age. This disease is characterized by the infiltration of smooth muscle cells that express contractile proteins, for example, desmin. Immunohistochemical studies have demonstrated a strong expression of CatK restricted to lymphangioleiomyomatosis cells. CatK has been suggested as a marker for diagnosis of lymphangioleiomyomatosis [276].
Bronchiolitis Obliterans Syndrome
Bronchiolitis obliterans syndrome (BOS) is a complication, which usually occurs during chronic rejection of lung transplant patients. Elevated levels of MMP-8 and MMP-9 were observed in obliterative bronchiolitis patients after a few years of lung transplantation [277]. A marked elevation in gelatinase activity was found in BAL fluid from BOS patients that could be due to MMP-9 secretion by local neutrophils [278]. In lung transplant model, inhibition of matrix metalloproteases in the donor and recipient, respectively, before lung harvest and after lung transplantation have been shown to improve oxygenation and markedly decreased PMN leukocyte influx into the isograft [279]. Both MMP-8 and MMP-9 deficient mice were observed to be protected from BOS as evidenced by a marked decrease in neutrophil influx and collagen deposition [280, 281].
MMPs and TIMPs were suggested to play a crucial role important role during lung allograft rejection. While TIMP-1 and TIMP-2 over expression have not been observed to elicit consistent effect on the level of cytokines or rejection pathology, MMP inhibition via systemic administration of MMP inhibitors were shown to reduce lung allograft rejection [282].
Lung Surfactant Proteins and Proteases
Lung surfactants are a mixture of lipids and proteins complex, which form a thin film in the lung alveoli and that plays a vital role in respiratory function especially gas exchange [283]. Additionally, the surfactant also shows the first line of innate immune defence in the lung. Its mode of action appears to lie in the inhibition of microbial infectivity and attenuation of inflammatory responses [284]. SP-A, SP-B, SP-C, and SP-D are the major surfactant proteins, which elicit important roles to trigger immune response in the lung [285–289]. SP-A has been demonstrated to be an important surfactant component having relevant functional immune response during Staphylococcus aureus infection [290].
Surfactant protein D (SP-D) is an important target of numerous proteases present in the CF lung. Host defence appears to be impaired due to proteolysis of SP-D and may contribute to the supportive lung disease in CF. SP-D, a glycoprotein of the collecting family, is produced and secreted by alveolar type II cells and non-ciliated bronchial epithelial cells [291].
SP-D has been observed to be protective against a wide variety of pathogens such as Pseudomonas aeruginosa, Haemophilus influenza, and A. fumigates [291–293]. Upon binding with SP-D, these pathogens trigger their agglutination, enhanced killing and clearance [294, 295]. SP-D has been shown to cause a number of secretions, which present larger protection in the body. An important characteristic of CF is chronic neutrophil-mediated inflammation in the airways mainly with an increase in the levels of HLE and PR3 [296–298]. Proteolytic degradation of some proteins such as SP-A and SP-D was found in BALF of CF patients [299].
Proteases have been observed to modulate surfactant activity in addition to its action on mucus proteins. Secretory proteases of P. aeruginosa can degrade SP-A and SP-B from lipid–protein complexes [300]. Purified elastase or secretory protease IV of P. aeruginosa supernatants have also been shown to degrade SP-A and SP-D [301–303]. The protease IV-mediated degradation of SP-A and SP-D may cause a discernible loss of bacterial aggregation or increase bacterial phagocytosis by alveolar macrophages [304]. NSPs like NE, PR3 and CatG can cleave the surfactant proteins [305]. These proteases may also cleave SP-D within the conserved sub-region of the C-terminal lectin domain with the generation of a ~35-kDa fragment, which decreases bacterial aggregation and mannan binding of SP-D [306]. P. aeruginosa elastase digestion of SP-D has also been shown to produce the 35-kDa fragment that retain the N-terminal collagen tail, albeit devoid of functional C-terminal globular lectin domain, which consequently elicit loss of innate immune functions [300, 303, 304].
In CF, COPD and asthma, like chronic lung airway diseases, an increase in the epidermal growth factor receptor (EGFR) could be the mechanism for mucus production [307, 308]. EGFR phosphorylation has been observed to activate mitogen activated protein kinases (MAPKs)-dependent signaling pathways, which in turn stimulates MMPs, for example, ADAM-17 and also NSPs leading to the production of mucins [309–312].
Human airway trypsin-like protease (HAT) has been observed to enhance the synthesis of mucus glycoconjugates in airway epithelial cells [313]. HAT is a natural ligand for PAR-2 present in bronchial cells [314] and HAT-dependent upregulation of mucin genes have also been shown to occur via PAR-EGFR signaling pathway [313].
Particulate Matter
Particulate matter (PM) having diameter of about 10 nm (PM10) is a complex mixture of metals, polycyclic aromatic hydrocarbons, nitrates, sulfates and other chemicals [315], where traffic and industrial activities have an important impact on that composition. Adverse effects of PM10, especially on alveolar epithelia were related to inflammation triggered by phagocytic cells upon PM10 internalization [316]. An immediate response is to augment generation of cytokines and chemokines such as IL-1, IL-6, and IL-8, TNF-α [317, 318].
Airborne PM10 is a risk factor for the development of a variety of lung diseases including cancer [315, 316]. In vitro, treatment of PM10 induces an increase in MMP-2 and MMP-9 activities, which causes ECM degradation during acute lung injury [319]. PM10 was found to be responsible for lung diseases such as tuberculosis [320], emphysema [321] and COPD [322]. In A549 lung epithelial cells, PM10 causes a marked decrease in E-cadherin/β-catenin expression and subsequently induces potentially invasive characteristics and thereby could contribute to cancer development [323].
Conclusion and Future Perspective
In order to fight against infections, the lung is orchestrated with different antiproteases and anti-inflammatory components. In pulmonary diseases like COPD and asthma, the balance between host proteases and their secreted endogenous inhibitors shift toward the proteases. Proteases such as NSPs, MMPs, and cathepsins are known to act along with the bacterial proteases and play important role in the manifestation of a variety of pulmonary diseases. Thus, agents that restore lung protease–antiprotease balance by upregulating endogenous protease inhibitors and/or down regulating host protease activities, appear important to control excessive inflammatory responses in the lung.
Inflammatory cells, which are rich in oxidants and proteases cause proteolytic inactivation of protein inhibitors of proteases. SLPI and trappin-2/elafin are relatively stable; even though they may be cleaved and inactivated by proteases, for example, by cathepsins at their N-terminal end, albeit it does not affect their inhibitory potency [324]. In contrast, cleavage of elafin by P. aeruginosa proteases may inactivate its antiprotease activity [325]. To overcome the unwanted proteolysis of antiproteases, encapsulations of protease inhibitors within liposomes have been suggested [326]. Aerosol delivery of liposomes entrapped antiproteases may prove useful since it has several features such as sustained release and relatively high loading capacities.
NE has been chosen as a target for inhibition by synthetic compounds because it is a widely recognized serine protease, which has been shown to be associated with a variety of lung diseases including CF [327]. DX-890, a small protein inhibitor of NE, has been observed to be tolerable in rats and humans after phase 1 clinical trial [328]. This compound was shown to be involved in IL-8 release from CF neutrophils and to reduce neutrophil transmigration through the epithelial barrier. Outcome of phase 3 clinical trials will reveal the usefulness of DX-890 as a therapeutic measure for a variety of lung diseases.
In a guinea pig model system, the dual MMP9/MMP12 inhibitor, AZ11557272 was found to be protective toward cigarette smoke-induced emphysema [329]. This compound markedly reduces number of inflammatory cells in bronchoalveolar lavage (BAL) fluid and has also been shown to decrease smoke-induced air space enlargement, which suggests that MMP-2 and MMP-12 could be the potential targets for therapeutic intervention in COPD. AS112108, another dual MMP-9/MMP-12 inhibitor, has been shown to inhibit the early inflammatory responses with a decrease in neutrophil numbers [330]. AS111793, a selective MMP-12 inhibitor, elicited dose-dependent inhibition in the levels of neutrophils and macrophages in bronchoalveolar lavage (BAL) fluid, and also on the concentration of several inflammation markers that express after cigarette smoke exposure [331]. However, it did not inhibit lung inflammation observed by lipopolysaccharide (LPS). Understanding the mechanism of these antiproteases will eventually lead us to gain an insight into the basic biochemical mechanisms that regulate COPD.
MMP-1 has been considered as one of the target proteases in lung cancer [332]. The generation of MMP-1 deficient mouse model suggested pro-tumorigenic role of the enzyme [333]. Further studies on MMP-1 and other MMPs in knockout mice will be required to evaluate the functional redundancy and relative relevance of MMP-1 and other MMPs in cell proliferation, regulation of inflammatory cells, and different stages of cancer progression.
Acknowledgements
This work was supported by DST-SERB (Govt. of India) and also the DST-PURSE programme of the University of Kalyani.
Contributor Information
Sajal Chakraborti, Phone: +91919831228224, FAX: +91913325828282, Email: saj_chakra@rediffmail.com.
Tapati Chakraborti, Email: t_chakraborti@yahoo.com.
Naranjan S. Dhalla, Phone: +11204-235-3421, Email: nsdhalla@sbrc.ca
Sajal Chakraborti, Email: saj_chakra@rediffmail.com.
References
- 1.Lecaille F, Lalmanach G, Andrault PM. Antimicrobial proteins and peptides in human lung diseases: a friend and foe partnership with host proteases. Biochimie. 2016;122:151–168. doi: 10.1016/j.biochi.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Geraghty P, Rogan MP, Greene CM, et al. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. J Immunol. 2007;178:5871–5878. doi: 10.4049/jimmunol.178.9.5871. [DOI] [PubMed] [Google Scholar]
- 3.Imai K, Yokohama Y, Nakanishi I, et al. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells. Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J Biol Chem. 1995;270:6691–6697. doi: 10.1074/jbc.270.12.6691. [DOI] [PubMed] [Google Scholar]
- 4.Ferry G, Lonchampt M, Pennel L, et al. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS Lett. 1997;402:111–115. doi: 10.1016/S0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- 5.Shamamian P, Schwartz JD, Pocock BJ, et al. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura H, Yoshimura K, McElvaney NG, et al. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest. 1992;89:1478–1484. doi: 10.1172/JCI115738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benarafa C, Priebe GP, Remold-O’Donnell E. The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J Exp Med. 2007;204:1901–1909. doi: 10.1084/jem.20070494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 9.Perkett EA, Lyons RM, Moses HL, et al. Transforming growth factor-beta activity in sheep lung lymph during the development of pulmonary hypertension. J Clin Invest. 1990;86:1459–1464. doi: 10.1172/JCI114862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyrick B, Reid L. Pulmonary hypertension. Anatomic and physiologic correlates. Clin Chest Med. 1983;4:199–217. [PubMed] [Google Scholar]
- 11.Archer SL, Nelson DP, Weir EK. Detection of activated O2 species in vitro and in rat lungs by chemiluminescence. J Appl Physiol. 1989;67:1912–1921. doi: 10.1152/jappl.1989.67.5.1912. [DOI] [PubMed] [Google Scholar]
- 12.Farrukh IS, Sciuto AM, Spannhake EW, et al. Leukotriene D4 increases pulmonary vascular permeability and pressure by different mechanisms in the rabbit. Am Rev Respir Dis. 1986;134:229–232. doi: 10.1164/arrd.1986.134.2.229. [DOI] [PubMed] [Google Scholar]
- 13.Farrukh IS, Michael JR, Summer WR, et al. Thromboxane-induced pulmonary vasoconstriction: involvement of calcium. J Appl Physiol. 1985;58:34–44. doi: 10.1152/jappl.1985.58.1.34. [DOI] [PubMed] [Google Scholar]
- 14.Freeman BA, Topolosky MK, Crapo JD. Hyperoxia increases oxygen radical production in rat lung homogenates. Arch Biochem Biophys. 1982;216:477–484. doi: 10.1016/0003-9861(82)90236-3. [DOI] [PubMed] [Google Scholar]
- 15.Gurtner GH, Michael JR, Farrukh IS, et al. Mechanism of hyperoxia-induced pulmonary vascular paralysis: effect of antioxidant pretreatment. J Appl Physiol. 1985;59:953–958. doi: 10.1152/jappl.1985.59.3.953. [DOI] [PubMed] [Google Scholar]
- 16.Farrukh IS, Michael JR, Peters SP, et al. The role of cyclooxygenase and lipoxygenase mediators in oxidant-induced lung injury. Am Rev Respir Dis. 1988;137:1343–1349. doi: 10.1164/ajrccm/137.6.1343. [DOI] [PubMed] [Google Scholar]
- 17.Feddersen CO, Chang S, Czartalomna J, et al. Arachidonic acid causes cyclooxygenase-dependent and -independent pulmonary vasodilation. J Appl Physiol. 1990;68:1799–1808. doi: 10.1152/jappl.1990.68.5.1799. [DOI] [PubMed] [Google Scholar]
- 18.Gurtner GH, Knoblauch A, Smith PL, et al. Oxidant- and lipid-induced pulmonary vasoconstriction mediated by arachidonic acid metabolites. J Appl Physiol. 1983;55:949–954. doi: 10.1152/jappl.1983.55.3.949. [DOI] [PubMed] [Google Scholar]
- 19.Gurtner GH, Burke-Wolin T. Interactions of oxidant stress and vascular reactivity. Am J Physiol. 1991;260:L207–L211. doi: 10.1152/ajplung.1991.260.4.L207. [DOI] [PubMed] [Google Scholar]
- 20.Seeger W, Wolf H, Graubert E, et al. Influence of aprotinin and gabexate mesilate on arachidonic acid release by the Ca-ionophore A 23187 in the lung. Adv Exp Med Biol. 1983;156:553–567. [PubMed] [Google Scholar]
- 21.White RP. Pharmacodynamic effects of tosyl-arginine methyl ester (TAME) on isolated human arteries. Gen Pharmacol. 1988;19:387–392. doi: 10.1016/0306-3623(88)90034-1. [DOI] [PubMed] [Google Scholar]
- 22.Chakraborti S, Gurtner GH, Michael JR. Oxidant-mediated activation of phospholipase A2 in pulmonary endothelium. Am J Physiol. 1989;257:L430–L437. doi: 10.1152/ajplung.1989.257.6.L430. [DOI] [PubMed] [Google Scholar]
- 23.de Haas GH, Postema NM, Nieuwenhuizen W, van Deenen LL. Purification and properties of an anionic zymogen of phospholipase A from porcine pancreas. Biochim Biophys Acta. 1968;159(1):118–129. doi: 10.1016/0005-2744(68)90249-0. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborti S, Roy S, Mandal A, et al. Role of PKCα-p38MAPK-Giα axis in NADPH oxidase derived O2—mediated activation of cPLA2 under U46619 stimulation in pulmonary artery smooth muscle cells. Arch Biochem Biophys. 2012;523:169–180. doi: 10.1016/j.abb.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborti T, Das S, Chakraborti S. Proteolytic activation of protein kinase Calpha by peroxynitrite in stimulating cytosolic phospholipase A2 in pulmonary endothelium: involvement of a pertussis toxin sensitive protein. Biochemistry (USA) 2005;44:5246–5257. doi: 10.1021/bi0477889. [DOI] [PubMed] [Google Scholar]
- 26.Chakraborti S, Chowdhury A, Chakraborti T. Cross-talk between p(38)MAPK and G iα in regulating cPLA 2 activity by ET-1 in pulmonary smooth muscle cells. Mol Cell Biochem. 2015;400:107–123. doi: 10.1007/s11010-014-2267-0. [DOI] [PubMed] [Google Scholar]
- 27.Chakraborti S, Mandal A, Das S, Chakraborti T. Inhibition of Na+/Ca2+ exchanger by peroxynitrite in microsomes of pulmonary smooth muscle: role of matrix metalloproteinase-2. Biochim Biophys Acta. 2004;1671:70–78. doi: 10.1016/j.bbagen.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborti S, Mandal A, Das S, Chakraborti T. Role of MMP-2 in PKCdelta-mediated inhibition of Na+ dependent Ca2+ uptake in microsomes of pulmonary smooth muscle: involvement of a pertussis toxin sensitive protein. Mol Cell Biochem. 2005;280:107–117. doi: 10.1007/s11010-005-8237-9. [DOI] [PubMed] [Google Scholar]
- 29.Chakraborti T, Ghosh SK, Michael JR, et al. Role of an aprotinin-sensitive protease in the activation of Ca2+-ATPase by superoxide radical O2−∙ in microsomes of pulmonary vascular smooth muscle. Biochem J. 1996;317:885–890. doi: 10.1042/bj3170885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh SK, Chakraborti T, Michael JR, et al. Oxidant-mediated proteolytic activation of Ca2+-ATPase in microsomes of pulmonary smooth muscle. FEBS Lett. 1996;387:171–174. doi: 10.1016/0014-5793(96)00471-1. [DOI] [PubMed] [Google Scholar]
- 31.Chakraborti T, Das S, Mandal M, et al. Role of Ca2+ dependent metalloprotease-2 in stimulating Ca2+ ATPase activity under peroxynitrite treatment in bovine pulmonary artery smooth muscle cell membrane. IUBMB Life. 2002;53:167–173. doi: 10.1080/15216540212337. [DOI] [PubMed] [Google Scholar]
- 32.Bouchard RA, Bose D. Contribution of sarcolemmal sodium-calcium exchange and intracellular calcium release to force development in isolated canine ventricular muscle. J Gen Physiol. 1992;99:931–960. doi: 10.1085/jgp.99.6.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blaustein MP. Sodium/calcium exchange and the control of contractility in cardiac muscle and vascular smooth muscle. J Cardiovasc Pharmacol. 1988;5:S56–S68. doi: 10.1097/00005344-198806125-00011. [DOI] [PubMed] [Google Scholar]
- 34.Vieillard-Baron A, Frisdal E, Raffestin B, et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer limits monocrotaline-induced pulmonary vascular remodeling in rats. Hum Gene Ther. 2003;14:861–869. doi: 10.1089/104303403765701150. [DOI] [PubMed] [Google Scholar]
- 35.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Ann Rev Physiol. 1997;59:89–144. doi: 10.1146/annurev.physiol.59.1.89. [DOI] [PubMed] [Google Scholar]
- 36.Stamenkovic I. Extracellular matrix remodelling: the role of matrix metalloproteinases. J Pathol. 2003;200:448–464. doi: 10.1002/path.1400. [DOI] [PubMed] [Google Scholar]
- 37.Woessner JF., Jr Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–2154. [PubMed] [Google Scholar]
- 38.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/A:1026028303196. [DOI] [PubMed] [Google Scholar]
- 39.Mandal M, Das S, Chakraborti T, Mandal A, Chakraborti S. Identification, purification and partial characterization of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) in bovine pulmonary artery smooth muscle. Mol Cell Biochem. 2003;254:145–155. doi: 10.1023/A:1027312913250. [DOI] [PubMed] [Google Scholar]
- 40.Mandal M, Mandal A, Das S, Chakraborti T, Chakraborti S. Identification, purification and partial characterization of tissue inhibitor of matrix metalloproteinase-2 in bovine pulmonary artery smooth muscle. Mol Cell Biochem. 2003;254:275–287. doi: 10.1023/A:1027389602772. [DOI] [PubMed] [Google Scholar]
- 41.Roy S, Chakraborti T, Chowdhury A, Chakraborti S. Role of PKC-α in NF-κB-MT1-MMP-mediated activation of proMMP-2 by TNF-α in pulmonary artery smooth muscle cells. J Biochem. 2013;153:289–302. doi: 10.1093/jb/mvs150. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Samanta K, Chakraborti T, et al. Role of TGF-β1 and TNF-α in IL-1β mediated activation of proMMP-9 in pulmonary artery smooth muscle cells: involvement of an aprotinin sensitive protease. Arch Biochem Biophys. 2011;513:61–69. doi: 10.1016/j.abb.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 43.Palese P. Influenza: old and new threats. Nat Med. 2004;10:S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- 44.Yewdell J, Garcia-Sastre A. Influenza virus still surprises. Curr Opin Microbiol. 2002;5:414–418. doi: 10.1016/S1369-5274(02)00346-6. [DOI] [PubMed] [Google Scholar]
- 45.Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol. 2005;3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 46.Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanquer J, Blanquer R, Borra’s R, et al. Aetiology of community acquired pneumonia in Valencia, Spain: a multicentre prospective study. Thorax. 1991;46:508–511. doi: 10.1136/thx.46.7.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lauderdale TL, Chang FY, Ben RJ, et al. Etiology of community acquired pneumonia among adult patients requiring hospitalization in Taiwan. Respir Med. 2005;99:1079–1086. doi: 10.1016/j.rmed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 49.Garten W, Klenk HD (2008) Cleavage activation of the influenza virus hemagglutinin and its role in pathogenesis. In Avian influenza: monographs in virology, vol 27. Karger, Basel, Switzerland
- 50.Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994;2:39–43. doi: 10.1016/0966-842X(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 51.Steinhauer DA. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 1999;258:1–20. doi: 10.1006/viro.1999.9716. [DOI] [PubMed] [Google Scholar]
- 52.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 53.Klenk HD, Rott R, Orlich M, et al. Activation of influenza A viruses by trypsin treatment. Virology. 1975;68:426–439. doi: 10.1016/0042-6822(75)90284-6. [DOI] [PubMed] [Google Scholar]
- 54.Lazarowitz SG, Choppin PW. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology. 1975;68:440–454. doi: 10.1016/0042-6822(75)90285-8. [DOI] [PubMed] [Google Scholar]
- 55.Goto H, Kawaoka Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci U S A. 1998;95:10224–10228. doi: 10.1073/pnas.95.17.10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazarowitz SG, Goldberg AR, Choppin PW. Proteolytic cleavage by plasmin of the HA polypeptide of influenza virus: host cell activation of serum plasminogen. Virology. 1973;56:172–180. doi: 10.1016/0042-6822(73)90296-1. [DOI] [PubMed] [Google Scholar]
- 57.LeBouder F, Morello E, Rimmelzwaan GF, et al. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J Virol. 2008;82:6820–6828. doi: 10.1128/JVI.00246-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kido H, Okumura Y, Yamada H, et al. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des. 2007;13:405–414. doi: 10.2174/138161207780162971. [DOI] [PubMed] [Google Scholar]
- 59.Tashiro M, Ciborowski P, Klenk HD, et al. Role of Staphylococcus protease in the development of influenza pneumonia. Nature. 1987;325:536–537. doi: 10.1038/325536a0. [DOI] [PubMed] [Google Scholar]
- 60.Scheiblauer H, Reinacher M, Tashiro M, et al. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis. 1992;166:783–791. doi: 10.1093/infdis/166.4.783. [DOI] [PubMed] [Google Scholar]
- 61.Bahgat MM, Błazejewska P, Schughart K. Inhibition of lung serine proteases in mice: a potentially new approach to control influenza infection. Virol J. 2011;8:27. doi: 10.1186/1743-422X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Böttcher E, Matrosovich T, Beyerle M, et al. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chaipan C, Kobasa D, Bertram S, et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. 2009;83:3200–3211. doi: 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steinhoff M, Buddenkotte J, Shpacovitch V, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 65.Vergnolle N. Proteinase-activated receptors (PARs) in infection and inflammation in the gut. Int J Biochem Cell Biol. 2008;40:1219–1227. doi: 10.1016/j.biocel.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 66.Riteau B, de Vaureix C, Lefevre F. Trypsin increases pseudorabies virus production through activation of the ERK signalling pathway. J Gen Virol. 2006;87:1109–1112. doi: 10.1099/vir.0.81609-0. [DOI] [PubMed] [Google Scholar]
- 67.Chignard M, Pidard D. Neutrophil and pathogen proteinases versus proteinase-activated receptor-2 lung epithelial cells: more terminators than activators. Am J Respir Cell Mol Biol. 2006;34:394–398. doi: 10.1165/rcmb.2005-0250TR. [DOI] [PubMed] [Google Scholar]
- 68.Lan RS, Stewart GA, Goldie RG, et al. Altered expression and in vivo lung function of protease-activated receptors during influenza A virus infection in mice. Am J Physiol. 2004;286:L388–L398. doi: 10.1152/ajplung.00286.2003. [DOI] [PubMed] [Google Scholar]
- 69.Khoufache K, LeBouder F, Morello E, et al. Protective role for protease-activated receptor-2 against influenza virus pathogenesis via an IFN-gamma-dependent pathway. J Immunol. 2009;182:7795–7802. doi: 10.4049/jimmunol.0803743. [DOI] [PubMed] [Google Scholar]
- 70.Ovcharenko AV, Zhirnov OP. Aprotinin aerosol treatment of influenza and paramyxovirus bronchopneumonia of mice. Antiviral Res. 1994;23:107–118. doi: 10.1016/0166-3542(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 71.Lee MG, Kim KH, Park KY, et al. Evaluation of anti-influenza effects of camostat in mice infected with non-adapted human influenza viruses. Arch Virol. 1996;141:1979–1989. doi: 10.1007/BF01718208. [DOI] [PubMed] [Google Scholar]
- 72.Manzano-Leon N, Quintana R, Sanchez B. Variation in the composition and in vitro proinflammatory effect of urban particulate matter from different sites. J Biochem Mol Toxicol. 2013;27:87–97. doi: 10.1002/jbt.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heijink IH, de Bruin HG, Dennebos R et al (2016) Cigarette smoke-induced epithelial expression of WNT-5B: implications for COPD. Eur Respir J ERJ-01541-2015 [DOI] [PubMed]
- 74.Chadwick D, Goode JA (eds) (2001) Chronic obstructive pulmonary disease: pathogenesis to treatment. Novartis Foundation Symposium 234, vol 234. Novartis Foundation. ISBN: 0-471-49437-2
- 75.Stockley JA, Stockley RA. Pulmonary physiology of chronic obstructive pulmonary disease, cystic fibrosis, and alpha-1 antitrypsin deficiency. Ann Am Thorac Soc. 2016;2:S118–S122. doi: 10.1513/AnnalsATS.201504-224KV. [DOI] [PubMed] [Google Scholar]
- 76.Zuo L, Pannell BK, Zhou T, et al. Historical role of alpha-1-antitrypsin deficiency in respiratory and hepatic complications. Gene. 2016;589:118–122. doi: 10.1016/j.gene.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 77.Ganrot PO, Laurell CB, Eriksson S. Obstructive lung disease and trypsin inhibitors in α1-antitrypsin deficiency. Scand J Clin Lab Invest. 1967;19:205–208. doi: 10.3109/00365516709090627. [DOI] [PubMed] [Google Scholar]
- 78.Kueppers F, Briscoe WA, Bearn AG. Hereditary deficiency of serum α-l-antitrypsin. Science. 1964;146:1678–1679. doi: 10.1126/science.146.3652.1678. [DOI] [PubMed] [Google Scholar]
- 79.Okada Y, Watanabe S, Nakanishi I, et al. Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinases. FEBS Lett. 1988;229:157–160. doi: 10.1016/0014-5793(88)80817-2. [DOI] [PubMed] [Google Scholar]
- 80.Hunninghake GW, Davidson JM, Rennard S. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science. 1981;212:925–927. doi: 10.1126/science.7233186. [DOI] [PubMed] [Google Scholar]
- 81.Kessenbrock K, Dau T, Jenne DE. Tailor-made inflammation: how neutrophil serine proteases modulate the inflammatory response. J Mol Med. 2010;89:23–28. doi: 10.1007/s00109-010-0677-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Korkmaz B, Horwitz MS, Jenne DE, et al. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. 2011;62:726–759. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pham CT. Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol. 2008;40:1317–1333. doi: 10.1016/j.biocel.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kramps JA, Franken C, Dijkman JH. ELISA for quantitative measurement of low-molecular-weight bronchial protease inhibitor in human sputum. Am Rev Respir Dis. 1984;129:959–963. doi: 10.1164/arrd.1984.129.6.959. [DOI] [PubMed] [Google Scholar]
- 85.Vogelmeier C, Hubbard RC, Fells GA, et al. Anti-neutrophil elastase defense of the normal human respiratory epithelial surface provided by the secretory leukoprotease inhibitor. J Clin Invest. 1991;87:482–488. doi: 10.1172/JCI115021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schalkwijk J, Wiedow O, Hirose S. The trappin gene family: proteins defined by an N-terminal transglutaminase substrate domain and a C-terminal four-disulphide core. Biochem J. 1999;340:569–577. doi: 10.1042/bj3400569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hochstrasser K, Albrecht GJ, Schonberger OL, et al. An elastase-specific inhibitor from human bronchial mucus: isolation and characterization. Hoppe-Seyler’s Z Physiol Chem. 1981;362:1369–1375. doi: 10.1515/bchm2.1981.362.2.1369. [DOI] [PubMed] [Google Scholar]
- 88.Takubo Y, Guerassimov A, Ghezzo H, et al. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med. 2002;166:1596–1603. doi: 10.1164/rccm.2202001. [DOI] [PubMed] [Google Scholar]
- 89.Janoff A, Carp H, Lee DK. Inactivation of alpha 1-proteinase inhibitor and bronchial mucous proteinase inhibitor by cigarette smoke in vitro and in vivo. Bull Eur Physiopathol Respir. 1980;16:321–340. doi: 10.1016/b978-0-08-027379-2.50034-x. [DOI] [PubMed] [Google Scholar]
- 90.Simon RH, Gross TJ, Edwards JA, et al. Fibrin degradation by rat pulmonary alveolar epithelial cells. Am J Physiol. 1992;262:L482–L488. doi: 10.1152/ajplung.1992.262.4.L482. [DOI] [PubMed] [Google Scholar]
- 91.Blasi F, Carmeliet P. uPAR: a versatile signaling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Xiao W, Jiang Y, et al. Levels of components of the urokinase-type plasminogen activator system are related to chronic obstructive pulmonary disease parenchymal destruction and airway remodelling. J Int Med Res. 2012;40:976–985. doi: 10.1177/147323001204000316. [DOI] [PubMed] [Google Scholar]
- 93.Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax. 2016;71:126–132. doi: 10.1136/thoraxjnl-2015-207428. [DOI] [PubMed] [Google Scholar]
- 94.Navratilova Z, Kolek V, Petrek M (2016) Matrix metalloproteinases and their inhibitors in chronic obstructive pulmonary disease. Arch Immunol Ther Exp (Warsz) 64:177–193 [DOI] [PubMed]
- 95.Churg A, Wang RD, Tai H, et al. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 96.Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert Opin Ther Targets. 2009;13:287–295. doi: 10.1517/14728220902751632. [DOI] [PubMed] [Google Scholar]
- 97.Aida Y, Shibata Y, Abe S, et al. Inhibition of elastase-pulmonary emphysema in dominant-negative MafB transgenic mice. Int J Biol Sci. 2014;10:882–894. doi: 10.7150/ijbs.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haq I, Lowrey GE, Kalsheker N, et al. Matrix metalloproteinase-12 (MMP-12) SNP affects MMP activity, lung macrophage infiltration and protects against emphysema in COPD. Thorax. 2011;66:970–976. doi: 10.1136/thx.2011.159087. [DOI] [PubMed] [Google Scholar]
- 99.Navratilova Z, Zatloukal J, Kriegova E, et al. Simultaneous up-regulation of matrix metalloproteinases 1, 2, 3, 7, 8, 9 and tissue inhibitors of metalloproteinases 1, 4 in serum of patients with chronic obstructive pulmonary disease. Respirology. 2012;17:1006–1012. doi: 10.1111/j.1440-1843.2012.02197.x. [DOI] [PubMed] [Google Scholar]
- 100.Raulo SM, Sorsa TA, Kiili MT, et al. Evaluation of collagenase activity, matrix metalloproteinase-8, and matrix metalloproteinase-13 in horses with chronic obstructive pulmonary disease. Am J Vet Res. 2001;62:1142–1148. doi: 10.2460/ajvr.2001.62.1142. [DOI] [PubMed] [Google Scholar]
- 101.Roderick JT, Cheryl LF, Laura M, et al. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am J Respir Cell Mol Biol. 2006;35:289–297. doi: 10.1165/rcmb.2005-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deshmukh HS, Shaver C, Case LM, et al. Acrolein-activated matrix metalloproteinase-9 contributes to persistent mucin production. Am J Respir Cell Mol Biol. 2008;38:446–454. doi: 10.1165/rcmb.2006-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lim S, Roche N, Oliver BG, et al. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med. 2000;162:1355–1360. doi: 10.1164/ajrccm.162.4.9910097. [DOI] [PubMed] [Google Scholar]
- 104.Deshmukh HS, McLachlan A, Atkinson JJ, et al. Matrix metalloproteinase-14 mediates a phenotypic shift in the airways to increase mucin production. Am J Respir Crit Care Med. 2009;180:834–845. doi: 10.1164/rccm.200903-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145–150. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 106.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 107.Moles A, Tarrats N, Fernández-Checa JC, et al. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology. 2009;49:1297–1307. doi: 10.1002/hep.22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Imai K, Hiramatsu A, Fukushima D, et al. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J. 1997;322:809–814. doi: 10.1042/bj3220809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van den Brûle S, Misson P, Bühling F, et al. Overexpression of cathepsin K during silica-induced lung fibrosis and control by TGF-beta. Respir Res. 2005;6:84. doi: 10.1186/1465-9921-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Scabilloni JF, Wang L, Antonini JM, et al. Matrix metalloproteinase induction in fibrosis and fibrotic nodule formation due to silica inhalation. Am J Physiol Lung Cell Mol Physiol. 2005;288:L709–L717. doi: 10.1152/ajplung.00034.2004. [DOI] [PubMed] [Google Scholar]
- 111.Perdereau C, Godat E, Maurel MC, et al. Cysteine cathepsins in human silicotic bronchoalveolar lavage fluids. Biochim Biophys Acta. 2006;1762:351–356. doi: 10.1016/j.bbadis.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 112.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 113.Gibson GJ, Loddenkemper R, Lundbäck B. Respiratory health and disease in Europe: the new European Lung White Book. Eur Respir J. 2013;42:559–563. doi: 10.1183/09031936.00105513. [DOI] [PubMed] [Google Scholar]
- 114.Tilly BC, Winter MC, Ostedgaard LS, et al. Cyclic AMP-dependent protein kinase activation of cystic fibrosis transmembrane conductance regulator chloride channels in planar lipid bilayers. J Biol Chem. 1992;267:9470–9473. [PubMed] [Google Scholar]
- 115.Downey DG, Bell SC, Elborn JS (2009) Neutrophils in cystic fibrosis. Thorax 64:81–88 [DOI] [PubMed]
- 116.Kreda SM, Davis CW, Rose MC. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb Perspect Med. 2012;2:a009589. doi: 10.1101/cshperspect.a009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Voynow JA, Rubin BK. Mucins, mucus, and sputum. Chest. 2009;135:505–512. doi: 10.1378/chest.08-0412. [DOI] [PubMed] [Google Scholar]
- 118.Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production inhuman airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J Immunol. 2005;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- 119.Song JS, Cho KS, Yoon HK, et al. Neutrophil elastase causes MUC5AC mucin synthesis via EGF receptor, ERK and NF-kB pathways in A549 cells. Korean J Intern Med. 2005;20:275–283. doi: 10.3904/kjim.2005.20.4.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol. 2002;26:447–452. doi: 10.1165/ajrcmb.26.4.4473. [DOI] [PubMed] [Google Scholar]
- 121.Amitani R, Wilson R, Rutman A. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am J Respir Cell Mol Biol. 1991;4:26–32. doi: 10.1165/ajrcmb/4.1.26. [DOI] [PubMed] [Google Scholar]
- 122.Mall MA. Role of cilia, mucus, and airway surface liquid in mucociliary dysfunction: lessons from mouse models. J Aerosol Med Pulm Drug Deliv. 2008;21:13–24. doi: 10.1089/jamp.2007.0659. [DOI] [PubMed] [Google Scholar]
- 123.Åstrand AB, Hemmerling M, Root J, et al. Linking increased airway hydration, ciliary beating, and mucociliary clearance through ENaC inhibition. Am J Physiol Lung Cell Mol Physiol. 2015;308:L22–L32. doi: 10.1152/ajplung.00163.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gaggar A, Hector A, Bratcher PE, et al. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur Respir J. 2011;38:721–727. doi: 10.1183/09031936.00173210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gaggar A, Li Y, Weathington N, et al. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am J Physiol Lung Cell Mol Physiol. 2007;293:L96–L104. doi: 10.1152/ajplung.00492.2006. [DOI] [PubMed] [Google Scholar]
- 126.Gaggar A, Jackson PL, Noerager BD, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matthew S, Twigg SB, Philip L, et al. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediat Inflamm. 2015;2015:10. doi: 10.1155/2015/293053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Braman SS. The global burden of asthma. Chest. 2006;130:4S–12S. doi: 10.1378/chest.130.1_suppl.4S. [DOI] [PubMed] [Google Scholar]
- 130.Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 131.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 132.Agrawal DK, Shao Z. Pathogenesis of allergic airway inflammation. Curr Allergy Asthma Rep. 2010;10:39–48. doi: 10.1007/s11882-009-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J Allergy Clin Immunol. 2008;121:560–570. doi: 10.1016/j.jaci.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cataldo D, Munaut C, Noel A, et al. MMP2 and MMP9 linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 2000;123:259–267. doi: 10.1159/000024452. [DOI] [PubMed] [Google Scholar]
- 135.Corry DB, Rishi K, Kanellis J, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2 deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greenlee KJ, Corry DB, Engler DA, et al. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. 2006;177:7312–7321. doi: 10.4049/jimmunol.177.10.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87:69–98. doi: 10.1152/physrev.00022.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McMillan SJ, Kearley J, Campbell JD. Matrix metalloproteinase9 deficiency results in enhanced allergen induced airway inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 139.Tsai YS, Tseng YT, Chen PS, et al. Protective effects of elafin against adult asthma. Allergy Asthma Proc. 2016;37:15–34. doi: 10.2500/aap.2016.37.3932. [DOI] [PubMed] [Google Scholar]
- 140.Rijken DC, Sakharov DV. Basic principles in thrombolysis: regulatory role of plasminogen. Thromb Res. 2001;103:S41–S49. doi: 10.1016/S0049-3848(01)00296-1. [DOI] [PubMed] [Google Scholar]
- 141.Pesci A, Foresi A, Bertorelli G, et al. Histochemical characteristics and degranulation of mast cells in epithelium and lamina propria of bronchial biopsies from asthmatic and normal subjects. Am Rev Respir Dis. 1993;147:684–689. doi: 10.1164/ajrccm/147.3.684. [DOI] [PubMed] [Google Scholar]
- 142.Cho SH, Tam SW, Demissie-Sanders S, et al. Production of plasminogen activator inhibitor-1 by human mast cells and its possible role in asthma. J Immunol. 2000;165:3154–3161. doi: 10.4049/jimmunol.165.6.3154. [DOI] [PubMed] [Google Scholar]
- 143.Cho SH, Lee SH, Kato A, et al. Cross-talk between mast cells and bronchial epithelial cells in plasminogen activator inhibitor-1 production via transforming growth factor β1. Am J Respir Cell Mol Biol. 2015;52:88–95. doi: 10.1165/rcmb.2013-0399OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vignola AM, Riccobono L, Mirabella A, et al. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlated with airflow obstruction in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1998;158:1945–1950. doi: 10.1164/ajrccm.158.6.9803014. [DOI] [PubMed] [Google Scholar]
- 145.Mautino G, Henriquet C, Gougat C, et al. Increased expression of tissue inhibitor of metalloproteinase-1 and loss of correlation with matrix metalloproteinase-9 by macrophages in asthma. Lab Invest. 1999;79:39–47. [PubMed] [Google Scholar]
- 146.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma. Chest. 2001;119:1329–1336. doi: 10.1378/chest.119.5.1329. [DOI] [PubMed] [Google Scholar]
- 147.Van Den Steen PE, Proost P, Wuyts A, et al. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO—and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- 148.George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis. 2016;7:34–51. doi: 10.1177/2040622315609251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol. 2001;54:577–589. doi: 10.1136/jcp.54.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hodge S, Hodge G, Holmes M, et al. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J. 2005;25:447–454. doi: 10.1183/09031936.05.00077604. [DOI] [PubMed] [Google Scholar]
- 151.Lokau J, Nitz R, Agthe M, et al. Proteolytic cleavage governs interleukin-11 trans signaling. Cell Rep. 2016;14:1761–1773. doi: 10.1016/j.celrep.2016.01.053. [DOI] [PubMed] [Google Scholar]
- 152.Trautmann A, Kruger K, Akdis M, et al. Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol. 2005;138:142–150. doi: 10.1159/000088436. [DOI] [PubMed] [Google Scholar]
- 153.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol. 2007;4:419–429. [PubMed] [Google Scholar]
- 154.Solarewicz-Madejek K, Basinski TM, Crameri R, et al. T cells and eosinophils in bronchial smooth muscle cell death in asthma. Clin Exp Allergy. 2009;39:845–855. doi: 10.1111/j.1365-2222.2009.03244.x. [DOI] [PubMed] [Google Scholar]
- 155.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3:361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 156.Lopez M, Salvaggio JE. Mold-sensitive asthma. Clin Rev Allergy. 1985;3:183–196. doi: 10.1007/BF02992982. [DOI] [PubMed] [Google Scholar]
- 157.Agarwal R. Severe asthma with fungal sensitization. Curr Allergy Asthma Rep. 2011;11:403–413. doi: 10.1007/s11882-011-0217-4. [DOI] [PubMed] [Google Scholar]
- 158.Downs SH, Mitakakis TZ, Marks GB, et al. Clinical importance of Alternaria exposure in children. Am J Respir Crit Care Med. 2001;164:455–459. doi: 10.1164/ajrccm.164.3.2008042. [DOI] [PubMed] [Google Scholar]
- 159.Kouzaki H, Iijima K, Kobayashi T, et al. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Prefontaine D, Nadigel J, Chouiali F, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 161.Prefontaine D, Lajoie-Kadoch S, Foley S, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–5103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 162.Snelgrove RJ, Gregory LG, Peiró T, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583–592. doi: 10.1016/j.jaci.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park MK, Cho MK, Kang SA, et al. Acanthamoeba protease activity promotes allergic airway inflammation via protease-activated receptor 2. PLoS ONE. 2014;9:e92726. doi: 10.1371/journal.pone.0092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Widmer F, Hayes PJ, Whittaker RG, et al. Substrate preference profiles of proteases released by allergenic pollens. Clin Exp Allergy. 2000;30:571–576. doi: 10.1046/j.1365-2222.2000.00784.x. [DOI] [PubMed] [Google Scholar]
- 165.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 166.Matheson N, Schmidt J, Travis J. Isolation and properties of an angiotensin II-cleaving peptidase from mesquite pollen. Am J Respir Cell Mol Biol. 1995;12:441–448. doi: 10.1165/ajrcmb.12.4.7695924. [DOI] [PubMed] [Google Scholar]
- 167.Medical Section of the American Lung Association The official conference report of the American Thoracic Society and Approved by the ATS board of Directors. Respiratory health hazards in agriculture. Am J Respir Crit Care Med. 1998;158:S1–S76. doi: 10.1164/ajrccm.158.supplement_1.rccm1585s1. [DOI] [PubMed] [Google Scholar]
- 168.Kirkhorn SR, Garry VF. Agricultural lung diseases. Environ Health Perspect. 2000;108:705–712. doi: 10.1289/ehp.00108s4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Langley RL. Consequences of respiratory exposures in the farm environment. N C Med J. 2011;72:477–480. [PubMed] [Google Scholar]
- 170.Poole JA, Romberger DJ. Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol. 2012;12:126–132. doi: 10.1097/ACI.0b013e3283511d0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.May S, Romberger DJ, Poole JA. Respiratory health effects of large animal farming environments. J Toxicol Environ Health B Crit Rev. 2012;15:524–541. doi: 10.1080/10937404.2012.744288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dodmane PR, Schulte NA, Heires AJ, et al. Airway epithelial epidermal growth factor receptor mediates hogbarn dust-induced cytokine release but not Ca2 response. Am J Respir Cell Mol Biol. 2011;45:882–888. doi: 10.1165/rcmb.2010-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Romberger DJ, Heires AJ, Nordgren TM, et al. Proteases in agricultural dust induce lung inflammation through PAR-1 and PAR-2 activation. Am J Physiol Lung Cell Mol Physiol. 2015;309:L388–L399. doi: 10.1152/ajplung.00025.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kouzaki H, O’Grady SM, Lawrence CB, et al. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol. 2009;183:1427–1434. doi: 10.4049/jimmunol.0900904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Page K, Strunk VS, Hershenson MB. Cockroach proteases increase IL-8 expression in human bronchial epithelial cells via activation of protease activated receptor (PAR)-2 and extracellular-signal-regulated kinase. J Allergy Clin Immunol. 2003;112:1112–1118. doi: 10.1016/j.jaci.2003.08.050. [DOI] [PubMed] [Google Scholar]
- 176.Post S, Heijink IH, Petersen AH, et al. Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS ONE. 2014;9:e91206. doi: 10.1371/journal.pone.0091206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liu C, Li Q, Zhou X, et al. Human airway trypsin-like protease induces mucin5AC hypersecretion via a protease activated receptor 2-mediated pathway in human airway epithelial cells. Arch Biochem Biophys. 2013;535:234–240. doi: 10.1016/j.abb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 178.Matsushima R, Takahashi A, Nakaya Y, et al. Human airway trypsin-like protease stimulates human bronchial fibroblast proliferation in a protease-activated receptor-2-dependent pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290:L385–L395. doi: 10.1152/ajplung.00098.2005. [DOI] [PubMed] [Google Scholar]
- 179.Asokananthan N, Graham PT, Stewart DJ, et al. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Derp 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol. 2002;169:4572–4578. doi: 10.4049/jimmunol.169.8.4572. [DOI] [PubMed] [Google Scholar]
- 180.Bernton H, Brown H. Insect allergy-preliminary studies of the cockroach. J Allergy. 1964;35:506–513. doi: 10.1016/0021-8707(64)90082-6. [DOI] [PubMed] [Google Scholar]
- 181.Arruda LK, Vailes LD, Ferriani VPL, et al. Cockroach allergens and asthma. Curr Rev Allergy Clin Immunol. 2001;107:419–428. doi: 10.1067/mai.2001.112854. [DOI] [PubMed] [Google Scholar]
- 182.Crain EF, Walter M, O’Connor GT, et al. Home and allergic characteristics of children with asthma in seven U.S. urban communities and design of an environmental intervention: the inner-city asthma study. Env Health Persp. 2002;110:939–945. doi: 10.1289/ehp.02110939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bhat RK, Page K, Tan A, et al. German cockroach extract increases bronchial epithelial cell interleukin-8 expression. Clin Exp Allergy. 2003;33:35–42. doi: 10.1046/j.1365-2222.2002.01481.x. [DOI] [PubMed] [Google Scholar]
- 184.Hughes VS, Page K. German cockroach frass proteases cleave promatrix metalloproteinase-9. Exp Lung Res. 2007;33:135–150. doi: 10.1080/01902140701356561. [DOI] [PubMed] [Google Scholar]
- 185.Page K, Ledford JR, Zhou P, et al. Mucosal sensitization to German cockroach involves protease-activated receptor-2. Respir Res. 2010;11:62. doi: 10.1186/1465-9921-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Page K. Role of cockroach proteases in allergic disease. Curr Allergy Asthma Rep. 2012;12:448–455. doi: 10.1007/s11882-012-0276-1. [DOI] [PubMed] [Google Scholar]
- 187.Young RP, Hopkins RJ. How the genetics of lung cancer may overlap with COPD. Respirology. 2011;16:1047–1055. doi: 10.1111/j.1440-1843.2011.02019.x. [DOI] [PubMed] [Google Scholar]
- 188.Brzóska K, Bartłomiejczyk T, Sochanowicz B, et al. Matrix metalloproteinase 3 polymorphisms as a potential marker of enhanced susceptibility to lung cancer in chronic obstructive pulmonary disease subjects. Ann Agric Environ Med. 2014;21:546–551. doi: 10.5604/12321966.1120599. [DOI] [PubMed] [Google Scholar]
- 189.Safranek J, Pesta M, Holubec L, et al. Expression of MMP-7, MMP-9, TIMP-1 and TIMP-2 mRNA in lung tissue of patients with non-small cell lung cancer (NSCLC) and benign pulmonary disease. Anticancer Res. 2009;29:2513–2517. [PubMed] [Google Scholar]
- 190.Fang S, Jin X, Wang R, et al. Polymorphisms in the MMP1 and MMP3 promoter and non-small cell lung carcinoma in North China. Carcinogenesis. 2005;26:481–486. doi: 10.1093/carcin/bgh327. [DOI] [PubMed] [Google Scholar]
- 191.Turpeenniemi-Hujanen T. Gelatinases (MMP-2 and-9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87:287–297. doi: 10.1016/j.biochi.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 192.Chen MH, Cui SX, Cheng YN, et al. Galloyl cyclic-imide derivative CH1104I inhibits tumour invasion through suppressing matrix metalloproteinase activity. Anticancer Drugs. 2008;19:957–965. doi: 10.1097/CAD.0b013e328313e15b. [DOI] [PubMed] [Google Scholar]
- 193.Qian Q, Wang Q, Zhan P, et al. The role of matrix metalloproteinase 2 on the survival of patients with non-small cell lung cancer: a systematic review with meta-analysis. Cancer Invest. 2010;28:661–669. doi: 10.3109/07357901003735634. [DOI] [PubMed] [Google Scholar]
- 194.Pulukuri SM, Rao JS. Matrix metalloproteinase-1 promotes prostate tumour growth and metastasis. Int J Oncol. 2008;32:757–765. [PMC free article] [PubMed] [Google Scholar]
- 195.Houghton AM, Grisolano JL, Baumann ML, et al. Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res. 2006;66:6149–6155. doi: 10.1158/0008-5472.CAN-04-0297. [DOI] [PubMed] [Google Scholar]
- 196.Nie GY, Hampton A, Li Y, et al. Identification and cloning of two isoforms of human high-temperature requirement factor A3 (HtrA3), characterization of its genomic structure and comparison of its tissue distribution with HtrA1 and HtrA2. Biochem J. 2003;371:39–48. doi: 10.1042/bj20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell. 2002;10:443–455. doi: 10.1016/S1097-2765(02)00658-5. [DOI] [PubMed] [Google Scholar]
- 198.Esposito V, Campioni M, De Luca A, et al. Analysis of HtrA1 serine protease expression in human lung cancer. Anticancer Res. 2006;26:3455–3459. [PubMed] [Google Scholar]
- 199.Oka C, Tsujimoto R, Kajikawa M, et al. HtrA1 serine protease inhibits signaling mediated by TGF, family proteins. Development. 2004;131:1041–1053. doi: 10.1242/dev.00999. [DOI] [PubMed] [Google Scholar]
- 200.Hirotaka O, Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 2002;21:7421–7434. doi: 10.1038/sj.onc.1205802. [DOI] [PubMed] [Google Scholar]
- 201.Shen ZT, Shen JS, Ji XQ et al (2016) TGF-β1 -rs1982073 polymorphism contributes to radiation pneumonitis in lung cancer patients: a meta-analysis. J Cell Mol Med. doi:10.1111/jcmm.12933 [DOI] [PMC free article] [PubMed]
- 202.Chien J, Ota T, Aletti G, et al. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol. 2009;2009:4177–4187. doi: 10.1128/MCB.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cilenti L, Kyriazis GA, Soundarapandian MM, et al. Omi/HtrA2 protease mediates cisplatin-induced cell death in renal cells. Am J Physiol Renal Physiol. 2005;288:371–379. doi: 10.1152/ajprenal.00154.2004. [DOI] [PubMed] [Google Scholar]
- 204.Egger L, Schneider J, Rhême C, et al. Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ. 2003;10:1188–1203. doi: 10.1038/sj.cdd.4401288. [DOI] [PubMed] [Google Scholar]
- 205.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 206.Wolf BB, Green DR. Apoptosis: letting slip the dogs of war. Curr Biol. 2002;5:R177–R179. doi: 10.1016/S0960-9822(02)00736-4. [DOI] [PubMed] [Google Scholar]
- 207.Jorgensen ED, Dozmorov I, Frank MB, et al. Global gene expression analysis of human bronchial epithelial cells treated with tobacco condensates. Cell Cycle. 2004;3:1154–1168. doi: 10.4161/cc.3.9.1078. [DOI] [PubMed] [Google Scholar]
- 208.Beleford D, Liu Z, Rattan R, et al. Methylation induced gene silencing of HtrA3 in smoking-related lung cancer. Clin Cancer Res. 2010;16:398–409. doi: 10.1158/1078-0432.CCR-09-1677. [DOI] [PubMed] [Google Scholar]
- 209.Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 2005;338:311–317. doi: 10.1016/j.bbrc.2005.08.162. [DOI] [PubMed] [Google Scholar]
- 210.Safe S, Wang F, Porter W, et al. Ah receptor agonists as endocrine disruptors: antiestrogenic activity and mechanisms. Toxicol Lett. 1998;102:343–347. doi: 10.1016/S0378-4274(98)00331-2. [DOI] [PubMed] [Google Scholar]
- 211.Hockings JK, Thorne PA, Kemp MQ, et al. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006;66:2224–2232. doi: 10.1158/0008-5472.CAN-05-1619. [DOI] [PubMed] [Google Scholar]
- 212.Chien J, Campioni M, Shridhar V, et al. HtrA serine proteases as potential therapeutic targets in cancer. Curr Cancer Drug Targets. 2009;9:451–468. doi: 10.2174/156800909788486704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie. 2008;90:227–242. doi: 10.1016/j.biochi.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 214.Reece ST, Loddenkemper C, Askew DJ, et al. Serine protease activity contributes to control of Mycobacterium tuberculosis in hypoxic lung granulomas in mice. J Clin Invest. 2010;120:3365–3376. doi: 10.1172/JCI42796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Elkington P, Shiomi T, Breen R, et al. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest. 2011;121:1827–1833. doi: 10.1172/JCI45666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Walker NF, Clark SO, Oni T, et al. Doxycycline and HIV infection suppress tuberculosis-induced matrix metalloproteinases. Am J Resp Crit Care Med. 2012;185:989–997. doi: 10.1164/rccm.201110-1769OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Elkington PT, Armiento JM, Friedland JS. Tuberculosis immunopathology: the neglected role of extracellular matrix destruction. Sci Transl Med. 2011;3:71–76. doi: 10.1126/scitranslmed.3001847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Seddon J, Kasprowicz V, Walker NF, et al. Procollagen III N terminal propeptide and desmosine are released by matrix destruction in pulmonary tuberculosis. J Infect Dis. 2013;208:1571–1579. doi: 10.1093/infdis/jit343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ugarte-Gil CA, Elkington P, Gilman RH, et al. Induced sputum MMP-1, -3 and -8 concentrations during treatment of tuberculosis. PLoS ONE. 2013;8:e61333. doi: 10.1371/journal.pone.0061333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Singh S, Saraiva L, Elkington PT, et al. Regulation of matrix metalloproteinase-1, -3, and -9 in Mycobacterium tuberculosis-dependent respiratory networks by the rapamycin-sensitive PI3K/p70(S6K) cascade. FASEB J. 2014;28:85–93. doi: 10.1096/fj.13-235507. [DOI] [PubMed] [Google Scholar]
- 221.Leco KJ, Waterhouse P, Sanchez OH, et al. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3) J Clin Invest. 2001;108:817–829. doi: 10.1172/JCI200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kübler A, Luna B, Larsson C, et al. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J Pathol. 2015;235:431–444. doi: 10.1002/path.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Peiris JS, Yuen KY, Osterhaus AD, et al. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 224.Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hudson LD, Milberg JA, Anardi D, et al. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 226.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 227.Raiden S, Nahmod K, Nahmod V, et al. Nonpeptide antagonists of AT1 receptor for angiotensin II delay the onset of acute respiratory distress syndrome. J Pharmacol Exp Ther. 2002;303:45–51. doi: 10.1124/jpet.102.037382. [DOI] [PubMed] [Google Scholar]
- 228.Marshall RP, Webb S, Bellingan GJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166:646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
- 229.Jerng JS, Yu CJ, Wang HC, et al. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit Care Med. 2006;34:1001–1006. doi: 10.1097/01.CCM.0000206107.92476.39. [DOI] [PubMed] [Google Scholar]
- 230.Hamming I, Timens W, Bulthuis ML, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Guyot N, Zani ML, Berger P, et al. Proteolytic susceptibility of the serine protease inhibitor trappin-2 (pre-elafin): evidence for tryptase-mediated generation of elafin. Biol Chem. 2005;386:391–399. doi: 10.1515/BC.2005.047. [DOI] [PubMed] [Google Scholar]
- 233.Molhuizen HO, Alkemade HA, Zeeuwen PL, et al. SKALP/elafin: an elastase inhibitor from cultured human keratinocytes. Purification, cDNA sequence, and evidence for transglutaminase cross-linking. J Biol Chem. 1993;268:12028–12032. [PubMed] [Google Scholar]
- 234.Nara K, Ito S, Ito T, et al. Elastase inhibitor elafin is a new type of proteinase inhibitor which has a transglutaminase-mediated anchoring sequence termed ‘cementoin’. J Biochem. 1994;115:441–448. doi: 10.1093/oxfordjournals.jbchem.a124357. [DOI] [PubMed] [Google Scholar]
- 235.Petty TL. Protease mechanisms in the pathogenesis of acute lung injury. Ann N Y Acad Sci. 1991;624:267–277. doi: 10.1111/j.1749-6632.1991.tb17025.x. [DOI] [PubMed] [Google Scholar]
- 236.Wang Z, Beach D, Su L, et al. A genome-wide expression analysis in blood identifies pre-elafin as a biomarker in ARDS. Am J Respir Cell Mol Biol. 2008;38:724–732. doi: 10.1165/rcmb.2007-0354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wang Z, Chen F, Zhai R, et al. Plasma neutrophil elastase and elafin imbalance is associated with acute respiratory distress syndrome (ARDS) development. PLoS ONE. 2009;4:e4380. doi: 10.1371/journal.pone.0004380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tejera P, Wang Z, Zhai R, et al. Genetic polymorphisms of peptidase inhibitor 3 (elafin) are associated with ARDS. Am J Respir Cell Mol Biol. 2009;41:696–704. doi: 10.1165/rcmb.2008-0410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sixt SU, Adamzik M, Spyrka D, et al. Alveolar extracellular 20S proteasome in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2009;179:1098–1106. doi: 10.1164/rccm.200802-199OC. [DOI] [PubMed] [Google Scholar]
- 240.Roth GA, Moser B, Krenn C, et al. Heightened levels of circulating 20S proteasome in critically ill patients. Eur J Clin Invest. 2005;35:399–403. doi: 10.1111/j.1365-2362.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 241.Kerrin A, Weldon S, Chung AHK, et al. Proteolytic cleavage of elafin by 20S proteasome may contribute to inflammation in acute lung injury. Thorax. 2013;68:315–321. doi: 10.1136/thoraxjnl-2012-202536. [DOI] [PubMed] [Google Scholar]
- 242.Merritt TA, Cochrane CG, Holcomb K, et al. Elastase and alpha 1-proteinase inhibitor activity in tracheal aspirates during respiratory distress syndrome. Role of inflammation in the pathogenesis of bronchopulmonary dysplasia. Clin Invest. 1983;72:656–666. doi: 10.1172/JCI111015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Janoff A. Elastases and emphysema. Current assessment of the protease antiprotease hypothesis. Am Rev Respir Dis. 1985;132:417–433. doi: 10.1164/arrd.1985.132.2.417. [DOI] [PubMed] [Google Scholar]
- 244.Taggart CC, Greene CM, Carroll TP, et al. Elastolytic proteases: inflammation resolution and dysregulation in chronic infective lung disease. Am J Respir Crit Care Med. 2005;171:1070–1076. doi: 10.1164/rccm.200407-881PP. [DOI] [PubMed] [Google Scholar]
- 245.Kawabata K, Suzuki M, Sugitani M, et al. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem Biophys Res Commun. 1991;177:814–820. doi: 10.1016/0006-291X(91)91862-7. [DOI] [PubMed] [Google Scholar]
- 246.Inoue Y, Omodani T, Shiratake R, et al. Development of a highly water-soluble peptide-based human neutrophil elastase inhibitor; AE-3763 for treatment of acute organ injury. Bioorg Med Chem. 2009;17:7477–7486. doi: 10.1016/j.bmc.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 247.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways ofprogrammed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Boivin WA, Cooper DM, Hiebert PR, et al. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest. 2009;89:1195–1220. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Metkar SS, Menaa C, Pardo J, et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. 2008;29:720–733. doi: 10.1016/j.immuni.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 250.Hendel A, Hiebert PR, Boivin WA, et al. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ. 2010;17:596–606. doi: 10.1038/cdd.2010.5. [DOI] [PubMed] [Google Scholar]
- 251.Pardo J, Aguilo JI, Anel A, et al. The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes Infect. 2009;11:452–459. doi: 10.1016/j.micinf.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 252.García-Laorden MI, Stroo I, Blok DC, et al. Granzymes A and B regulate the local inflammatory response during Klebsiella pneumoniae pneumonia. J Innate Immun. 2016;8:258–268. doi: 10.1159/000443401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Latge JP. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev. 1999;12:310–350. doi: 10.1128/cmr.12.2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Segal BJ. Aspergillosis. N Engl J Med. 2009;360:1870–1884. doi: 10.1056/NEJMra0808853. [DOI] [PubMed] [Google Scholar]
- 255.Woodcock AA, Steel N, Moore CB, et al. Fungal contamination of bedding. Allergy. 2006;61:140–142. doi: 10.1111/j.1398-9995.2005.00941.x. [DOI] [PubMed] [Google Scholar]
- 256.Moutaouakil M, Monod M, Prevost MC, et al. Identification of the 33-kDa alkaline protease of Aspergillus fumigates in vitro and in vivo. J Med Microbiol. 1993;39:393–399. doi: 10.1099/00222615-39-5-393. [DOI] [PubMed] [Google Scholar]
- 257.Kunert J, Kopecek P, et al. Multiple forms of the serine proteinase ALP of Aspergillus fumigatus. Mycoses. 2000;43:339–347. doi: 10.1046/j.1439-0507.2000.00586.x. [DOI] [PubMed] [Google Scholar]
- 258.Monod M, Paris S, Sanglard D, et al. Isolation and characterization of a secreted metalloprotease of Aspergillus fumigatus. Infect Immun. 1993;61:4099–5104. doi: 10.1128/iai.61.10.4099-4104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Reichard U, Eiffert H, Ruchel R. Purification and characterization of an extracellular aspartic proteinase of Aspergillus fumigatus. J Med Vet Mycol. 1994;32:427–436. doi: 10.1080/02681219480000581. [DOI] [PubMed] [Google Scholar]
- 260.Kolattukudy PE, Lee JD, Rogers LM, et al. Evidence for possible involvement of an elastolytic serine protease in aspergillosis. Infect Immun. 1993;61:2357–2368. doi: 10.1128/iai.61.6.2357-2368.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Markaryan A, Morozova I, Yu H, et al. Purification and characterization of an elastinolytic metalloprotease from Aspergillus fumigatus and immunoelectron microscopic evidence of secretion of this enzyme by the fungus invading the murine lung. Infect Immun. 1994;62:2149–2157. doi: 10.1128/iai.62.6.2149-2157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.McDonagh A, Fedorova ND, Crabtree J, et al. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 2008;4:e1000154. doi: 10.1371/journal.ppat.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Bouchara JP, Sanchez M, Esnault K, et al. Interactions between Aspergillus fumigatus and host matrix protein. Contrib Microbiol. 1999;2:167–181. doi: 10.1159/000060293. [DOI] [PubMed] [Google Scholar]
- 264.Asano Y. Future treatments in systemic sclerosis. J Dermatol. 2010;37:54–70. doi: 10.1111/j.1346-8138.2009.00758.x. [DOI] [PubMed] [Google Scholar]
- 265.Roy R, Wewer UM, Zurakowski D, et al. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem. 2004;279:51323–51330. doi: 10.1074/jbc.M409565200. [DOI] [PubMed] [Google Scholar]
- 266.Shi Z, Xu W, Loechel F, et al. ADAM 12, a disintegrin metalloprotease, interacts with insulin-like growth factor-binding protein-3. J Biol Chem. 2000;275:18574–18580. doi: 10.1074/jbc.M002172200. [DOI] [PubMed] [Google Scholar]
- 267.Loechel F, Fox JW, Murphy G, et al. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278:511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- 268.Jones JI, Clemmons DR, et al. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 269.Harsha A, Stojadinovic O, Brem H, et al. ADAM12: a potential target for the treatment of chronic wounds. J Mol Med (Berl) 2008;86:961–969. doi: 10.1007/s00109-008-0353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol. 2003;8:73–81. doi: 10.1016/S1084-2756(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 271.Madurga A, Mizíková I, Ruiz-Camp J, et al. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2013;305:893–905. doi: 10.1152/ajplung.00267.2013. [DOI] [PubMed] [Google Scholar]
- 272.Bruce MC, Schuyler M, Martin RJ, et al. Risk factors for the degradation of lung elastic fibers in the ventilated neonate. Implications for impaired lung development in bronchopulmonary dysplasia. Am Rev Respir Dis. 1992;146:204–212. doi: 10.1164/ajrccm/146.1.204. [DOI] [PubMed] [Google Scholar]
- 273.Altiok O, Yasumatsu R, Bingol-Karakoc G, et al. Imbalance between cysteine proteases and inhibitors in a baboon model of bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2006;173:318–326. doi: 10.1164/rccm.200503-425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Davies PL, Spiller OB, Beeton ML, et al. Relationship of proteinases and proteinase inhibitors with microbial presence in chronic lung disease of prematurity. Thorax. 2010;65:246–251. doi: 10.1136/thx.2009.116061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Bry K, Hogmalm A, Backstrom E. Mechanisms of inflammatory lung injury in the neonate: lessons from a transgenicmouse model of bronchopulmonary dysplasia. Semin Perinatol. 2010;34:211–221. doi: 10.1053/j.semperi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 276.Chilosi M, Pea M, Martignoni G, et al. Cathepsin-k expression in pulmonary lymphangioleiomyomatosis. Mod Pathol. 2009;22:161–166. doi: 10.1038/modpathol.2008.189. [DOI] [PubMed] [Google Scholar]
- 277.Smith GN, Jr, Mickler EA, Payne KK, et al. Lung transplant metalloproteinase levels are elevated prior to bronchiolitis obliterans syndrome. Am J Transplant. 2007;7:1856–1861. doi: 10.1111/j.1600-6143.2007.01850.x. [DOI] [PubMed] [Google Scholar]
- 278.Riise GC, Ericson P, Bozinovski S, et al. Increased net gelatinase but not serine protease activity in bronchiolitis obliterans syndrome. J Heart Lung Transplant. 2010;29:800–807. doi: 10.1016/j.healun.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 279.Iwata T, Chiyo M, Yoshida S, et al. Lung transplant ischemia reperfusion injury: metalloprotease inhibition down-regulates exposure of type V collagen, growth-related oncogene-induced neutrophil chemotaxis, and tumour necrosis factor-a expression. Transplantation. 2008;85:417–426. doi: 10.1097/TP.0b013e31815e91b6. [DOI] [PubMed] [Google Scholar]
- 280.Khatwa UA, Kleibrink BE, Shapiro SD, et al. MMP-8 promotes polymorphonuclear cell migration through collagen barriers in obliterative bronchiolitis. J Leukocyte Biol. 2010;87:69–77. doi: 10.1189/jlb.0509361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Fernandez FG, Campbell LG, Liu W, et al. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am J Transplant. 2005;5:671–683. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]
- 282.Yoshida S, Iwata T, Chiyo M, et al. Metalloproteinase inhibition has differential effects on alloimmunity, autoimmunity, and histopathology in the transplanted lung. Transplantation. 2007;83:799–808. doi: 10.1097/01.tp.0000258600.05531.5d. [DOI] [PubMed] [Google Scholar]
- 283.King RJ, Clements JA. Surface active materials from dog lung. I. Method of isolation. Am J Physiol. 1972;223:707–714. doi: 10.1152/ajplegacy.1972.223.3.707. [DOI] [PubMed] [Google Scholar]
- 284.LeVine AM, Kurak KE, Wright JR, et al. Surfactant protein-a binds group b streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-a-deficient mice. Am J Respir Cell Mol Biol. 1999;20:279–286. doi: 10.1165/ajrcmb.20.2.3303. [DOI] [PubMed] [Google Scholar]
- 285.Crouch E, Hartshorn K, Ofek I. Collectins and pulmonary innate immunity. Immunol Rev. 2000;173:52–65. doi: 10.1034/j.1600-065X.2000.917311.x. [DOI] [PubMed] [Google Scholar]
- 286.Hartshorn KL, Crouch E, White MR, et al. Pulmonary surfactant proteins a and d enhance neutrophil uptake of bacteria. Am J Physiol. 1998;274:L958–L969. doi: 10.1152/ajplung.1998.274.6.L958. [DOI] [PubMed] [Google Scholar]
- 287.Sano H, Sohma H, Muta T, et al. Pulmonary surfactant protein a modulates the cellular response to smooth and rough lipopolysaccharides by interaction with cd14. J Immunol. 1999;163:387–395. [PubMed] [Google Scholar]
- 288.Garcia-Verdugo I, Descamps D, Chiganard M, et al. Protease/anti-protease net work and modulation of mucus production and surfactant activity. Biochimie. 2010;92:1608–1617. doi: 10.1016/j.biochi.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 289.Murakami S, Iwaki D, Mitsuzawa H, et al. Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor-alpha secretion in u937 cells and alveolar macrophages by direct interaction with toll-like receptor 2. J Biol Chem. 2002;277:6830–6837. doi: 10.1074/jbc.M106671200. [DOI] [PubMed] [Google Scholar]
- 290.Kantyka T, Pyrc K, Gruca M, et al. Staphylococcus aureus proteases degrade lung surfactant protein A potentially impairing innate immunity of the lung. J Innate Immun. 2013;5:251–260. doi: 10.1159/000345417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Reid KBM. Interactions of surfactant protein D with pathogens, allergens and phagocytes. Biochem Biophys Acta. 1998;1408:290–295. doi: 10.1016/s0925-4439(98)00074-x. [DOI] [PubMed] [Google Scholar]
- 292.Restrepo CI, Dong Q, Savov J, et al. SP-D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol. 1999;21:576–585. doi: 10.1165/ajrcmb.21.5.3334. [DOI] [PubMed] [Google Scholar]
- 293.Crouch EC. Structure, biological properties, and expression of surfactant protein D (SP-D) Biochem Biophys Acta. 1998;1408:278–289. doi: 10.1016/s0925-4439(98)00073-8. [DOI] [PubMed] [Google Scholar]
- 294.Kuan SF, Rust K, Crouch EC. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest. 1992;90:97–106. doi: 10.1172/JCI115861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Madan T, Eggleton P, Kishore U, et al. Binding of pulmonary surfactant protein A and D to Aspergillus fumigatus conidia enhance phagocytosis and killing by human neutrophils and alveolar macrophages. Infect Immun. 1997;65:3171–3179. doi: 10.1128/iai.65.8.3171-3179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Persson A, Chang D, Crouch E. Surfactant protein D is a divalent cation-dependent carbohydrate-binding protein. J Biol Chem. 1990;265:5755–5760. [PubMed] [Google Scholar]
- 297.Pikaar JC, Voorhut WF, van Golde LM, et al. Opsonic activities of surfactant protein A and D in phagocytosis of gram-negative bacterias by alveolar macrophages. J Infect Dis. 1995;172:481–489. doi: 10.1093/infdis/172.2.481. [DOI] [PubMed] [Google Scholar]
- 298.Birrer P, McElvaney G, Rüdelberg A, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;151:207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- 299.von Bredow C, Birrer P, Griese M. Degradation of surfactant protein A and other bronchoalveolar lavage fluid proteins in patients with cystic fibrosis. Eur Respir J. 2001;17:716–722. doi: 10.1183/09031936.01.17407160. [DOI] [PubMed] [Google Scholar]
- 300.Beatty AL, Malloy JL, Wright JR. Pseudomonas aeruginosa degrades pulmonary surfactant and increases conversion in vitro. Am J Respir Cell Mol Biol. 2005;32:128–134. doi: 10.1165/rcmb.2004-0276OC. [DOI] [PubMed] [Google Scholar]
- 301.Malloy JL, Veldhuizen RA, Thibodeaux BA, et al. Pseudomonas aeruginosa protease IV degrades surfactant proteins and inhibits surfactant host defense and biophysical functions. Am J Physiol Lung Cell Mol Physiol. 2005;288:409–418. doi: 10.1152/ajplung.00322.2004. [DOI] [PubMed] [Google Scholar]
- 302.Alcorn JF, Wright JR. Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J Biol Chem. 2004;279:30871–30879. doi: 10.1074/jbc.M400796200. [DOI] [PubMed] [Google Scholar]
- 303.Mariencheck WI, Alcorn JF, Palmer SM, et al. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol. 2003;28:528–537. doi: 10.1165/rcmb.2002-0141OC. [DOI] [PubMed] [Google Scholar]
- 304.Hirche TO, Crouch EC, Espinola M, et al. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem. 2004;279:27688–27698. doi: 10.1074/jbc.M402936200. [DOI] [PubMed] [Google Scholar]
- 305.Liau DF, Yin NX, Huang J, et al. Effects of human polymorphonuclear leukocyte elastase upon surfactant proteins in vitro. Biochim Biophys Acta. 1996;1302:117–128. doi: 10.1016/0005-2760(96)00042-2. [DOI] [PubMed] [Google Scholar]
- 306.Griese M, Wiesener A, Lottspeich F, et al. Limited proteolysis of surfactant protein D causes a loss of its calcium-dependent lectin functions. Biochim Biophys Acta. 2003;1638:157–163. doi: 10.1016/S0925-4439(03)00063-2. [DOI] [PubMed] [Google Scholar]
- 307.Burgel PR, Nadel JA, et al. Roles of epidermal growth factor receptor activation in epithelial cell repair and mucin production in airway epithelium. Thorax. 2004;59:992–996. doi: 10.1136/thx.2003.018879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Burgel PR, Nadel JA, et al. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respir. 2008;J32:1068–1081. doi: 10.1183/09031936.00172007. [DOI] [PubMed] [Google Scholar]
- 309.Hewson CA, Edbrooke MR, Johnston SL, et al. PMA induces the MUC5AC respiratory mucin in human bronchial epithelial cells, via PKC, EGF/TGF-alpha, Ras/Raf, MEK, ERK and Sp1-dependent mechanisms. J Mol Biol. 2004;344:683–695. doi: 10.1016/j.jmb.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 310.Wu DY, Wu R, Chen Y, et al. PMA stimulates MUC5B gene expression through an Sp1-based mechanism in airway epithelial cells. Am J Respir Cell Mol Biol. 2007;37:589–597. doi: 10.1165/rcmb.2007-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lundgren JD, Rieves RD, Mullol J, et al. The effect of neutrophil proteinase enzymes on the release of mucus from feline and human airway cultures. Respir Med. 1994;88:511–518. doi: 10.1016/S0954-6111(05)80333-6. [DOI] [PubMed] [Google Scholar]
- 312.Kohri K, Ueki IF, Nadel JA, et al. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol. 2002;283:531–540. doi: 10.1152/ajplung.00455.2001. [DOI] [PubMed] [Google Scholar]
- 313.Chokki M, Yamamura S, Eguchi H, et al. Human airway trypsin-like protease increases mucin gene expression in airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:470–478. doi: 10.1165/rcmb.2003-0199OC. [DOI] [PubMed] [Google Scholar]
- 314.Miki M, Nakamura Y, Takahashi A, et al. Effect of human airway trypsin-like protease on intracellular free Ca2+ concentration in human bronchial epithelial cells. J Med Invest. 2003;50:95–107. [PubMed] [Google Scholar]
- 315.Englert N. Fine particles and human health—a review of epidemiological studies. Toxicol Lett. 2004;149:235–242. doi: 10.1016/j.toxlet.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 316.Mukae H, Hogg JC, English D, et al. Phagocytosis of particulate air pollutants by human alveolar macrophages stimulates the bone marrow. Am J Physiol Lung Cell Mol Physiol. 2000;279:L924–L931. doi: 10.1152/ajplung.2000.279.5.L924. [DOI] [PubMed] [Google Scholar]
- 317.van Eeden SF, Tan WC, Suwa T, et al. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)) Am J Respir Crit Care Med. 2001;164:826–830. doi: 10.1164/ajrccm.164.5.2010160. [DOI] [PubMed] [Google Scholar]
- 318.Sun J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct. 2010;2010:985132. doi: 10.1155/2010/985132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Wang J, Zhang H, Su C, et al. Dexamethasone ameliorates H2S-induced acute lung injury by alleviating matrix metalloproteinase-2 and -9 expression. PLoS ONE. 2014;9:e94701. doi: 10.1371/journal.pone.0094701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Ong CW, Elkington PT, Friedland JS. Tuberculosis, pulmonary cavitation and matrix metalloproteinases. Am J Respir Crit Care Med. 2014;190:9–18. doi: 10.1164/rccm.201311-2106PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur Respir J. 2014;43:82–91. doi: 10.1183/09031936.00174612. [DOI] [PubMed] [Google Scholar]
- 322.Kumar M, Phougat N, Ruhil S, et al. Genomics of chronic obstructive pulmonary disease (COPD); exploring the SNPs of protease–antiprotease pathway. Curr Genomics. 2013;14:204–213. doi: 10.2174/1389202911314030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Morales-Bárcenas R, Chirino YI, Sánchez-Pérez Y, et al. Particulate matter (PM10) induces metalloprotease activity and invasion in airway epithelial cells. Toxicol Lett. 2015;237:167–173. doi: 10.1016/j.toxlet.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 324.Taggart CC, Lowe GJ, Greene CM, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276:33345–33352. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- 325.Quinn DJ, Weldon S, Taggart CC. Antiproteases as therapeutics to target inflammation in cystic fibrosis. Open Respir Med J. 2010;4:20–31. doi: 10.2174/1874306401004010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Gibbons A, McElvaney NG, Cryan SA. A dry powder formulation of liposome-encapsulated recombinant secretory leukocyte protease inhibitor (rSLPI) for inhalation: preparation and characterisation. AAPS Pharm Sci Tech. 2010;11:1411–1421. doi: 10.1208/s12249-010-9500-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Delacourt C, erigault SH, Delclaux C, et al. Protection against acute lung injury by intravenous or intratracheal pretreatment with EPI-HNE-4, a new potent neutrophil elastase inhibitor. Am J Respir Cell Mol Biol. 2002;26:290–297. doi: 10.1165/ajrcmb.26.3.4611. [DOI] [PubMed] [Google Scholar]
- 328.Dunlevy FK, Martin SL, de Courcey F, et al. Anti-inflammatory effects of DX-890, a human neutrophil elastase inhibitor. J Cyst Fibros. 2012;11:300–304. doi: 10.1016/j.jcf.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 329.Churg A, Wang R, Wang X, et al. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. 2007;62:706–713. doi: 10.1136/thx.2006.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.le Quémen C, Lagente V, Guénon I, Muzio V, Gillon J-Y, Boichot E (2008) Anti-inflammatory properties of MMP inhibitors in experimental models of chronic obstructive pulmonary disease and lung inflammation. In: Lagente V, Boichot E (eds) Matrix metalloproteinases in tissue remodelling and inflammation. Springer, New York, pp 57–69
- 331.Le Quément C, Guénon I, Gillon JY, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol. 2008;154:1206–1215. doi: 10.1038/bjp.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Jawad MU, Garamszegi N, Garamszegi SP, et al. Matrix metalloproteinase 1: role in sarcoma biology. PLoS ONE. 2010;5:e14250. doi: 10.1371/journal.pone.0014250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Fanjul-Fernández M, Folgueras AR, Fueyo A, et al. Matrix metalloproteinase MMP-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J Biol Chem. 2013;288:14647–14656. doi: 10.1074/jbc.M112.439893. [DOI] [PMC free article] [PubMed] [Google Scholar]