

Abstract
Approximately 10,000 l of air and 8,000 l of blood transit the respiratory system each day driven by small pressure gradients developed in response to rhythmic contraction and relaxation of striated muscle under both voluntary and involuntary control of the central nervous system. Matching of air- and blood flow results from central and local reflexes responding to both internal and external stimuli and subsequently controlling the pumps, as well as the smooth muscle in walls of the airways and blood vessels. A wide range of neural and immune mechanisms protect the lungs against environmental insults, and many are adaptive in nature, resulting in memory that increases sensitivity and responsiveness upon repeated exposure to stimuli. In over 10 % of the population, the responses to environmental stimuli become pathological, resulting in excessive sensitivity and aberrant responses to both specific and nonspecific stimuli, and culminate in physical remodeling of the airways and lungs. Prevention, definitive diagnosis, and effective treatment of the disorders require a better understanding of the mechanisms underlying excessive responses to environmental stimuli.
Keywords: Pulmonary anatomy and physiology, Innervations of the respiratory tract, Airway remodeling
An average adult at rest moves over 10,000 l of air through the respiratory tract and exchanges over 400 l of O2 and CO2 across the alveolar surface each day. Toxins and pathogens in the ambient air can damage the lung tissue, interfere with respiratory mechanics, and limit gas exchange. Both passive and active processes protect the lungs against environmental insults by filtering the inspired air; altering airflow and blood flow to limit alveolar and systemic exposure; capturing, killing, and rapidly clearing pathogens; and efficiently repairing damaged tissues to restore the mucosal barrier. Some of the defense mechanisms are adaptive, increasing the sensitivity of the system and intensity of responses to future insults. Dysregulation of defense pathways can increase sensitivity to nonspecific stimuli and lead to responses that are out of proportion to the insult. This chapter provides a brief overview of the anatomy and physiology of the respiratory tract in relation to development of hypersensitivity and hyperresponsiveness of the respiratory tract.
Functional Anatomy of the Respiratory Tract
Flow of air through the respiratory tract is driven by small pressure gradients, and both the work of breathing and partitioning of airflow to different regions of the lungs are impacted by the resistance to airflow in each segment of the airways. The resistance to laminar flow of air through a rigid tube is related to its length and diameter. But most airways are deformable, not rigid; and are affected by mechanical forces and pressures impinging on the wall. Muscle, blood vessels, and glands in the airway wall are under local and reflex control, and actively respond to changes in the internal and external environments.
The respiratory tract is divided into two broad divisions: the upper and lower airways. The upper airways consist of the extrathoracic structures from the nares and mouth down to, and including, the larynx. Being extrathoracic, these airways are impacted by barometric pressure impinging on their external surface. The lower airways include the extrathoracic and intrathoracic portions of the trachea; the extrapulmonary and intrapulmonary bronchi; and the bronchioles, alveolar ducts, and alveoli, which are intrathoracic and intrapulmonary. Intrathoracic extrapulmonary airways are impacted by pressure that develops in the pleural space, which is normally negative, but becomes positive during forced expiration. Intrapulmonary airways are surrounded by alveoli, and their diameter is affected by changes in tension in the lung tissue impinging on their surface, as well as pressure of the alveolar gas, which oscillates from negative to positive during the respiratory cycle. Airways proximal to the respiratory bronchioles lack alveoli and are therefore classified as conducting airways, whereas the respiratory bronchioles, alveolar ducts, and alveoli constitute the gas exchange region of the lungs. With the exception of the pharynx, the walls of the airways become less rigid distally, with the walls of the upper airways containing bone and cartilage, the large lower airways containing cartilage rings or plates, and the small lower airways lacking bone and cartilage.
The nose and mouth represent two parallel pathways by which air enters and leaves the system. The nasal cavity and nasopharynx have a large surface area and tortuous path of airflow that is conducive to filtering, warming, and humidifying ambient air. The nares open into a vestibule, which leads to a constricted region called the nasal valve (Rhee and Kimbell 2012). Inferior, middle, and superior turbinates posterior to the nasal valve protrude into the airstream from each side of the central septum, increasing the surface area for exchange of heat and moisture. An olfactory region with a high density of olfactory receptors is located high in the nasal cavity. The majority of air passes through the nasal cavity along the floor, between the hard palate and inferior turbinate, and is forced to make a near 90° bend in the nasopharynx. Air flowing through the upper regions of the nasal cavity is forced to flow around the turbinates, but enters the nasopharynx at a less acute angle (Kelly et al. 2000). Both pathways are conducive to removal of large particles by impaction with the airway surface. Airflow through the nose is laminar at velocities up to 200 ml/s, which includes resting conditions; but the velocity is not uniform throughout the nose (Kelly et al. 2000). The olfactory region has a relatively low airflow velocity, which is presumed to protect the delicate olfactory receptors as well as keep the air in contact with the receptors for a longer period of time, allowing for detection of scents within the airstream (Kelly et al. 2000). At flow rates greater than 200 ml/s, turbulence develops, which increases resistance and work of breathing. The nasal cavity has fairly rigid walls, and the main factors that impact resistance to airflow in this region are physical obstructions such as polyps and septal deviation, swelling of the mucosa, and accumulation of secretions in the airway lumen.
The pharynx is posterior to the turbinates and separated into the nasopharynx, oropharynx, and laryngopharynx. The nasopharynx passes over the soft palate before opening into the oropharynx. Right and left Eustachian tubes open into the nasopharynx and connect the middle ear with the upper airway. In contrast to the nasal cavity, the pharynx in humans is deformable. The hyoid bone and soft tissue comprising the airway wall in this region are not directly attached to the surrounding skeletal structures, making the wall of the pharynx deformable (Dempsey et al. 2010; Susarla et al. 2010). In humans, there is a relatively large distance between the soft palate and epiglottis, and the face is flattened, resulting in a long, narrow oropharynx upon which the tongue encroaches. Another unique feature is that adipose tissue accumulates in the surrounding tissue and can apply pressure to the outside of the airway, affecting lumen diameter. As discussed later, all of these factors predispose the pharynx to collapsing late in expiration and during inspiration. The tendency to collapse is offset by two factors, neural reflexes that activate pharyngeal muscles and stiffen the wall, and by traction developed in the pharyngeal wall as the lung inflates (Dempsey et al. 2010; Susarla et al. 2010).
The epiglottis and larynx play major roles in protecting the lower airways. The epiglottis deflects solids and liquids traveling through the oropharynx airway from entering the larynx, directing them down the esophagus. The larynx has a high density of receptors for diverse chemical entities, that when stimulated evoke a cough reflex that expels solids and liquids from airway.
Humans have two lungs, which are divided into three lobes on the right—superior, middle, and inferior—and two lobes on the left—superior and inferior. The inner surface of the chest wall and outer surface of the lung are covered by parietal and visceral pleura, respectively, which are separated by a thin fluid-filled space. Anatomical and functional tracer studies indicate that drainage of the pleural space is via the superior mediastinal, intercostals, paraesophageal, and intra-abdominal lymph nodes, with the superior mediastinal lymph nodes being the sentinel nodes (Parungo et al. 2004, 2005).
The trachea, bronchi, and terminal bronchioles are conducting airways that lack alveoli. The trachea extends from the larynx in the cervical region to the carina in the thoracic cavity. Like the larynx, the carina is sensitive to tactile stimulation, which evokes cough. The wall of the trachea is supported by 15–20 C-shaped rings of cartilage interspersed with soft tissue. The posterior membrane connecting the ends of cartilaginous rings contains smooth muscle that can shorten the distance between the ends of the cartilage, decreasing the diameter of the trachea. Smooth muscle is also located between the cartilaginous rings allowing at least a small degree of active regulation of tracheal length. Shortening of the trachea can reduce resistance to airflow by both decreasing the length of the airway and increasing traction on the pharynx, stiffening the wall and opposing collapse.
In contrast to the trachea, the bronchial wall is supported by plates of cartilage with a higher degree of deformability than the tracheal rings. The bronchioles lack cartilage, with the wall consisting of epithelium attached to its basement membrane, a lamina propria, and a smooth muscle layer. Alveolar septa attach to the outside of the bronchioles, and changes in alveolar pressure and tension in the lung tissue have large effects on the diameter of the bronchioles. The bronchioles branch into alveolar ducts that terminate in alveolar sacs.
Dichotomous branching of the airways leads to an increase in the number of airways in parallel. The diameter of each airway decreases; but the aggregate, or total cross-sectional diameter of the airspace increases distally, which has two prominent functional impacts. First, the velocity of airflow diminishes, decreasing turbulence. Second, total resistance to airflow decreases exponentially with the increase in the effective/aggregate radius of the airways. Only about 2 % of the total airway resistance is accounted for by the bronchioles and alveolar ducts; and likewise only a small decrease in lumen pressure occurs along normal distal airways during exhalation. The relatively small pressure drop along the intrapulmonary airways minimizes the tendency for dynamic compression of the airways induced by the differential between a positive alveolar pressure surrounding the airways and reduced luminal pressure due the resistive drop during exhalation. Partial obstruction of distal airways due to contraction of smooth muscle or accumulation of secretions in the airway lumen leads to increased resistance and a greater-than-normal decrease in luminal pressure, predisposing the airways to close earlier than normal during exhalation.
The airways terminate in alveolar sacs, which are polygonal structures covered largely by a simple squamous Type 1 alveolar epithelial cells. Cuboidal alveolar Type II cells that produce and secrete surface active alveolar surfactant localize to the corners of the alveoli. The barrier between the blood in the capillaries and the air in the each alveolus is less than 1 μm thick. The thinness of the barrier is in part due to the alveolar epithelium and alveolar capillary endothelium sharing the same basement membrane, thereby eliminating excess interstitial tissue. This extremely thin barrier facilitates rapid diffusion of gas between the alveolar air and blood while maintaining an intact barrier between the external and internal environments.
Collagen and elastin fibers run through the lung tissue, interconnecting proteoglycans and glycosaminoglycans of the ground substance with the cells making up the lung surface, alveolar walls, blood vessels, and airways. This fibrous matrix results in interdependence of lung structures (i.e., airways, blood vessels, alveoli) (Faffe and Zin 2009). The elastin fibers are largely responsible for the elastance of the lung tissue and its tendency to return to its original shape after being deformed. Collagen fibers are stiffer than elastin fibers, and at low lung volumes are thought to be slack. As lung volumes increase, collagen fibers become taut and provide tissue rigidity and opposition to over-stretching of the lung tissue.
In addition to the tissue properties that affect expansion and contraction of the lungs, the air–liquid interface at the alveolar surface impacts lung mechanics and function. Surface tension pulls on surface of curved air–liquid interfaces, tending to reduce the surface area of the interface. The pressure developed by surface tension adds to the tissue recoil force, facilitating exhalation but resisting lung expansion. The pressure developed by surface tension promotes the transfer of air from alveoli with small radii into larger alveoli, which can result in collapse of small alveoli and development of atelectasis if surface tension is too great. Surface tension also induces a pressure differential between fluid in the interstitial compartment and the airway surface, which acts to pull water out of capillaries and the alveolar wall and into the airspace, leading to alveolar edema.
Surface tension in normal lungs is reduced by surfactant released from Type II pneumocytes. Surfactant is comprised of phospholipid, mainly dipalmitoylphosphatidylcholine (DPPC), neutral lipids including cholesterol, and proteins (Perez-Gil and Weaver 2010). When released from Type II cells, the lipids form a complex tubular structure, tubular myelin, in the liquid layer on the alveolar surface. During lung inflation, surfactant apoproteins B and C (SP-B and SP-C) facilitate the insertion of the lipid into the interfacial film to maintain a relatively uniform concentration of the surface-active material at the interface. Compression of the surface during exhalation leads to regeneration of the tubular myelin. The insertion of the hydrophilic heads of the phospholipid molecules into the water layer is thought to disrupt the intermolecular attraction of the water molecules that give rise to surface tension.
Both SP-B and SP-C are produced as proproteins in Type II cells. SP-B is also produced by Clara cells, but unlike Type II cells that process the proprotein to produce mature SP-B, Clara cells cannot produce the mature protein. Of the two proteins, SP-B appears to be of greater importance on an acute basis, and a 75 % reduction in SP-B levels in the airspaces is fatal (Perez-Gil and Weaver 2010). In contrast, SP-C deficiency leads to chronic, progressive lung disease in both humans and animals (Perez-Gil and Weaver 2010).
Two other surfactant-associated proteins, SP-A and SP-D, belong the collectin family of proteins with collagen-like lectin domains. SP-A has some ability to facilitate formation of tubular myelin and adsorption of surfactant on the alveolar surface, but also enhances killing, aggregation, and phagocytosis of microorganisms (Orgeig et al. 2010). The antimicrobial activity of SP-A is a result of direct opsonization of pathogens, as well as upregulation of pattern recognition receptors on phagocytes that mediate uptake and elimination of the foreign bodies. SP-D has similar functional properties to SP-A, but in addition, modulates macrophage, dendritic cell, and T-cell functions (Orgeig et al. 2010; Forbes and Haczku 2010). Deficiency in SP-D leads to increased numbers of myeloid dendritic cells in the lungs that promote Th2 responses. Treatment with recombinant SP-D can reduce allergen-induced eosinophilic inflammation and antigen-specific IgE production (Orgeig et al. 2010).
Separate circulations supply blood to the nose, larynx, trachea, and lungs. The nasal mucosa is perfused by the sphenopalatine branches of the maxillary artery, the anterior and posterior ethmoidal branches of the ophthalmic artery, and the superior labial artery, a branch of the facial artery (Osborn 1978). The larynx is perfused by the superior and inferior branches of the laryngeal artery, which stem from superior and inferior thyroid arteries, and run to the larynx alongside the superior and inferior laryngeal nerves (Weir 1997). Blood flow to the trachea is via the inferior thyroid, supreme intercostal, subclavian, internal mammary, innominate, and bronchial arteries. The branching from these arteries connects with one another to form longitudinal tracheal anastomoses that give rise to intercartilaginous submucosal capillary plexuses (Salassa et al. 1977). The bronchial circulation provides oxygenated blood to the bronchi and walls of the intrapulmonary airways, and the pulmonary circulation delivers mixed venous blood to the alveolar capillaries for gas exchange.
An extensive lymphatic system drains the lungs and thoracic cavity (Gray 2000; Carati and Gannon 2006). The lymph vessels are divided into parietal and visceral components. Plexuses of parietal lymphatic vessels drain into sternal, intercostals, diaphragmatic, and axillary nodes. The visceral lymph nodes are located along the mediastinum in associated with the esophagus, large veins and arteries, and pericardium and along the trachea and bronchi. Drainage of the lungs is via two plexuses: superficial and deep. The superficial plexus is located beneath the visceral pleura and drains to nodes at the hilus. The deep plexus consists of vessels surrounding airways and blood vessels. In the bronchioles, there is a single plexus, but in the bronchi there are two plexuses, submucosal and peribronchial. The deep plexus drains into tracheobronchial nodes. The lymphatic vessels terminate at the alveolar ducts, and fluid from the alveolar wall moves to the vessels in response to the “pumping” action of respiration.
Airway Musculature
Muscles of the upper airways are striated under autonomous, and in some cases, conscious control. Pharyngeal muscles are separated into four muscle groups based on the structures they control: the tongue, palate, hyoid bone, and wall of the pharynx (Edwards and White 2011). The extrinsic muscles of the tongue include the genioglossus, styloglossus, and hypoglossus. The genioglossus pulls the tongue forward and down, keeping the tongue from occluding the oropharynx. The genioglossus exhibits phasic activity during inspiration, tonic activity during expiration, and responds to hypoxia, hypercapnia, and negative pressure in the larynx, indicating that it is controlled in least in part through the respiratory centers in the brain stem. Both the styloglossus and hypoglossus retract the tongue. When activated in concert with the genioglossus muscle, they help stiffen the tongue and oppose collapse of the oropharynx.
The palate is controlled by five muscles: the tensor palatini, levator palatini, palatoglossus, palatopharyngeus, and muscular uvula. The tensor palatine stiffens the soft palate. The levator palatini and palatoglossus raise and lower the soft palate, respectively, either closing or opening the nasal and oral air passages, respectively. The palatopharyngeus lifts and moves the soft palate anteriorly, and the muscular uvula raises the uvula. The geniohyoid, mylohyoid, thyrohyoid, sternohyoid, stylohyoid, and omohyoid manipulate the hyoid bone, moving it anteriorly and caudally, dilating the upper airway. The pharyngeal constrictor muscles assist in swallowing and constrict the airway at normal and elevated lung volumes (Edwards and White 2011).
At the level of the larynx, the muscle in the airway wall transitions to a smooth muscle phenotype. Smooth muscle is found throughout the airways, from the trachea to alveolar ducts.
Innervation
Upper Airways
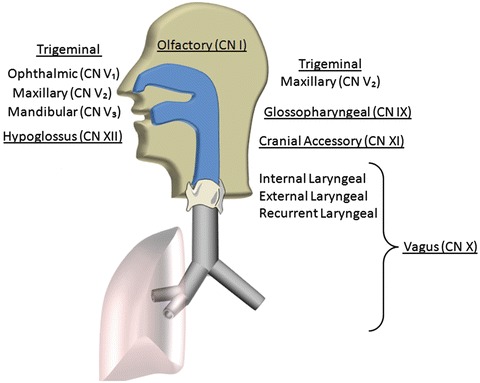
The upper airways are innervated by sensory, motor, parasympathetic, and sympathetic neurons (Fig. 2.1a) (Sarin et al. 2006; Yoshida et al. 2000; Undem et al. 1999). Olfaction is carried in cranial nerve I (CN I), and taste is carried in the facial and glossopharyngeal nerves (CN VII and IX). Other sensory modalities are from the mouth, nasal cavity, and nasopharynx are transmitted in the trigeminal nerve (CN V) via the ophthalmic (V1), maxillary (V2), and mandibular (V3) branches. A pharyngeal plexus innervated by the glossopharyngeal (CN IX), vagus (CN X), cranial accessory (CN XI) nerves conveys sensation from the lower aspect of the nasopharynx, the oropharynx, and supralaryngeal pharynx. Sensation from the larynx is carried in the internal, external, and recurrent laryngeal branches of the vagus nerve.
Fig. 2.1.
Cranial nerves innervating the upper and lower airways
The soft palate and walls of the pharynx contain both constrictor and dilator muscles that move contents of the pharynx to the esophagus, close the nasopharynx and larynx during swallowing, and stabilize the airway during normal breathing. Branchial motor neurons originating in the trigeminal motor nucleus pass through the trigeminal nerve, trigeminal ganglion, and mandibular nerves to innervate the tensor veli palatine, which stiffens the soft palate. The vagus nerve (CN X) innervates the remaining muscles of the soft palate (levator veli palatine, palatoglossus, palatopharyngeus, musculus uvulae), the major constrictor muscles in the pharyngeal wall (superior, middle, and inferior constrictors), the salpingopharyngeus muscle that draws the larynx upward during swallowing, and the muscle of the larynx. The glossopharyngeal nerve (CN IX) innervates the stylopharyngeus, which also draws the larynx upward, dilating the pharynx. Motor control of the tongue is mainly via the hypoglossal nerve (CN XII), except for the palatoglossus muscle that is innervated from the vagus nerve via the pharyngeal plexus, as mentioned above.
Autonomic innervation of the upper airways is from the deep petrosal, facial, and vagus nerves. Parasympathetic innervation of the nose is via the facial nerve. Preganglionic parasympathetic fibers originate in facial and superior salivatory nuclei, exit the brain stem in the facial nerve (CN VII), and branch into the greater superior petrosal nerve. The greater superior petrosal nerve merges with the deep petrosal nerve to form the Vidian nerve that leads to the pyterygopalatine or sphenopalatine, ganglion. Postganglionic fibers from the spenopalatine ganglion distribute to the surface epithelium, glands, arteries, and veins in the nasal mucosa. Acetylcholine (Ach) is the major preganglionic and postganglionic transmitter, although vasoactive intestinal peptide (VIP), peptide histidine methionine, peptide histidine valine, secretoneurin, and nitric oxide have also been associated with cholinergic neurons (Sarin et al. 2006). Parasympathetic output induces serous and mucous secretions and vasodilation.
Preganglionic sympathetic fibers innervating the nose arise from the thoracic and lumbar regions of the spinal cord and are carried to the superior cervical ganglion via the vagosympathetic trunk. Postganglionic sympathetic fibers from the superior cervical ganglion destined for the nasal cavity and nasopharynx are carried in the deep petrosal nerve, which merges with the greater petrosal nerve to form the Vidian nerve (Sarin et al. 2006). The fibers pass through the sphenopalatine ganglion and are distributed to the mucosa with fibers from the trigeminal nerve. Postganglionic sympathetic fibers innervating the pharyngeal plexus derive from direct gray rami communicans from the superior ganglion. Sympathetic fibers primarily innervate the mucosal vasculature, inducing vasoconstriction. Activation of α1 and α2 adrenoceptors or neuropeptide Y receptors on arterioles and venous sinusoids leads to vasoconstriction, lower mucosal blood flow, and less pooling of blood in the mucosal vasculature (Sarin et al. 2006). Adrenergic stimulation has been associated with secretion by glands, but sympathetic innervation of glands is sparse (Undem et al. 1999).
Lower Airways
The lungs are innervated by parasympathetic cholinergic and parasympathetic noncholinergic neurons carried in the vagus nerve (CN X), and sympathetic adrenergic and sympathetic nonadrenergic neurons carried in spinal nerves between T1 and T6 (Fig. 2.1b) (Lee and Pisarri 2001; Canning 2006). Parasympathetic innervation of the lungs is conserved across species, whereas sympathetic innervation exhibits significant species variation. The majority of sympathetic postganglionic efferent fibers originate in the superior cervical ganglion and thoracic ganglia. Human airway smooth muscle exhibits little sympathetic innervation, whereas other species exhibit significant sympathetic-mediated constriction and relaxation effects based on whether α- or β- adrenoceptors are expressed on the postsynaptic membrane (Canning 2006). Likewise, in humans, a convincing role for sympathetic control of gland and epithelial secretions has not been demonstrated (Rogers 2001).
Parasympathetic innervation of the lungs includes afferent and efferent neurons. Preganglionic parasympathetic efferent neurons emanate from dorsal motor nuclei of the vagus or nucleus ambiguus. Postganglionic fibers originate in plexuses within the airway wall associated with mucosa, submucosa, smooth muscle, peri-tracheal, and peribronchial layers (Wine 2007). Afferent fibers exiting the lungs travel through the vagus nerve to the nodose and intracranial jugular ganglia, which in turn have outputs to the nucleus tractus solitarius. Afferent neurons include myelinated Aδ and nonmyelinated C fibers. Aδ fibers exhibit an organized pattern of terminals with attachments to the mucosal extracellular matrix making them responsive to mechanical deformation of the airway wall (Canning 2011). The Aδ mechano-sensitive fibers can be separated into slowly adapting (SAR) and rapidly adapting (RAR) functional phenotypes, with SARs being associated with regulatory inflation and deflation reflexes (e.g., Hering-Breuer); and RARs being polymodal and associated with defensive reflexes such as cough and sneeze (Widdicombe 1982). Lung afferent C-fibers are small, nonmyelinated fibers that terminate in the airway wall, vasculature, and parenchyma. Similar to nociceptors in other tissues, these fibers are polymodal and are typically classified as chemoreceptors (Lee and Pisarri 2001). C fibers in the lungs are classified into two groups, bronchial and pulmonary, based on the circulation that perfuses the tissues they innervate.
In addition to conventional sympathetic adrenergic and parasympathetic cholinergic fibers, the upper and lower airways are replete with neurons that release nonadrenergic noncholinergic transmitters, either in concert with or independent of acetylcholine and norepinephrine. The noncholinergic nonadrenergic (NANC) neurons were originally referred to as a discrete subset (Rogers 2001), but in the last decade there has been increasing reference to these neurons as subtypes of sympathetic and parasympathetic neurons (Canning 2006).
Although less developed than in the gut, the neural network within the airway wall is extensive and exhibits some capacity to control airway functions without efferent input from the central nervous system. One mechanism by which this occurs is antidromic conduction of action potential in branches of afferent nerve fibers. Action potentials generated at sensory nerve terminals in the airway mucosa can be conducted along branches of the afferent neuron leading to the surface epithelium, glands, and blood vessels within the airway wall. Release of neurotransmitters, particularly NANC transmitters, results in intrinsic control of airway functions, initiating responses or modulating centrally mediated responses. Axon reflexes are common in the airways of rats and guinea pigs, but their overall importance in humans is a matter of debate. While direct empirical evidence of the importance of intrinsic control of airway tone and secretory activity is lacking, the fact that airways in transplanted lungs can function relatively normally for years without external innervations argues in favor of some intrinsic mechanism for maintaining airway patency and protecting the airways from environmental insult (Wine 2007) (Fig. 2.2).
Fig. 2.2.
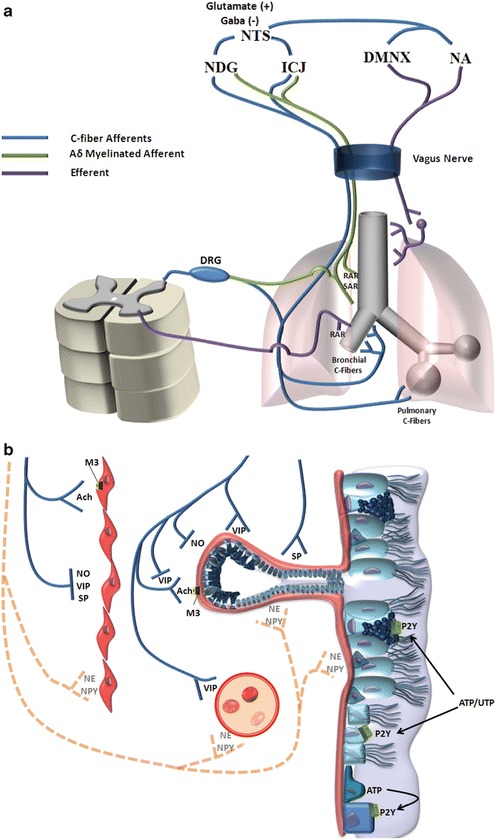
Innervation of the lower airways. (a) Distribution of autonomic fibers. (b) End organ innervations within the airway mucosa. Dashed lines represent sympathetic fibers, which have minimal activity in humans. RAR rapidly adapting receptor, SAR slowly adapting receptor, DRG dorsal root ganglion, NDG nodose ganglion, ICJ intracranial jugular ganglion, NTS nucleus tractus solitarius, DMNX dorsal motor nucleus of cranial nerve X, NA nucleus ambiguus, M3 muscarinic receptor subtype 3, Ach acetylcholine, NO nitric oxide, VIP vasoactive intestinal peptide, SP substance P, NE norepinephrine, NPY neuropeptide Y, P2Y purinergic receptor 2Y
Mechanical, Thermal, and Chemical Receptors
Whereas SAR’s respond primarily to mechanical deformation, RAR and C-fibers are polymodal, responding to diverse stimuli including temperature, acidity, and osmolarity. Transient receptor potential cation channels (TRP channels) are involved in transduction of environmental stimuli into physiologically relevant cellular responses. TRP channels are express on numerous cell types within the airway wall, including neurons (Fig. 2.3).
Fig. 2.3.
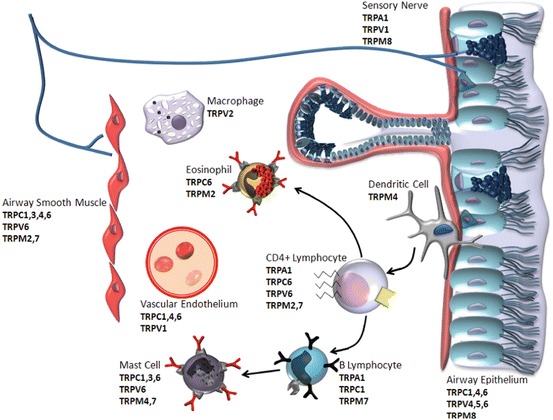
Expression of transient receptor potential (TRP) cation channel subtypes in the structural and inflammatory cells of the airway mucosa. TRP subtypes are defined in the text
TRP channels were first described in relation to phospholipase C (PLC)-dependent phototransduction in Drosophila (Hardie and Minke 1995), and later as a family of mammalian proteins involved in capacitative calcium entry induced by diverse stimuli via PLC-dependent and -independent mechanisms (Birnbaumer et al. 1996; Zhu et al. 1996). In 2001, TRPs were implicated in hypoxic vasoconstriction (McDaniel et al. 2001; 2002), bronchoconstriction, and bronchial smooth muscle proliferation (Sweeney et al. 2002); and by 2003 were recognized as potential targets in diverse environmental and inflammatory lung diseases (Li et al. 2003). From a more fundamental standpoint, they constitute a critical interface between the environment and the lungs, transducing changes in temperature, osmolality, pressure, stretch, pH, and chemical stimuli into transmembrane cation fluxes, membrane potential changes, and intracellular second messenger signals. In addition to responding to environmental cues, changes in intracellular second messengers originating from activation of other signaling cascades can modulate TRP function, including their sensitivity to primary stimuli (Moran et al. 2011).
The TRP family has 28 members subdivided into six subfamilies on the basis of sequence homology and chemical activation: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), and TRPML (mucolipin). Activation of TRP can lead to depolarization due to enhanced cation conductance, elevation of intracellular Ca2+ concentration, or hyperpolarization when the channels are co-expressed with BKCa K+ channels (Moran et al. 2011; Kim et al. 2009). TRPs are expressed by many of the major cell types involved in asthma and COPD, and data indicate that TRP channels are involved in osmotic sensing, modulation of vascular permeability, mucociliary clearance, and inflammation (Colsoul et al. 2009). In most cases, in vivo studies confirming physiological and pathophysiological roles for these receptors are lacking, but in the case of TRPA1, TRPC6, TRPV1, TRPV4, and TRPM4 in vivo validation is emerging (Banner et al. 2011). TRPA1 is activated by diverse chemical stimuli, including changes in pH, and cold. TRPV1 is activated by heat, acid, and high chemical stimuli, but at higher concentrations than those known to activate TRPA1. TRPC6 is expressed in vascular smooth muscle and endothelium, and its activity is affected by mechanical stimuli, including stretch. TRPC6 has been linked to hypoxic vasoconstriction in pulmonary vasculature (Weissmann et al. 2006) and plays a role in formation of pulmonary edema following pulmonary ischemia and reperfusion, as well as podocyte formation in kidney cells (Kim et al. 2009). TRPV4 has also been linked to changes in vascular permeability and formation of pulmonary edema induced by high vascular pressure, airway inflation pressures, and tidal volumes (Banner et al. 2011).
TRP channels are expressed on cells involved in innate and adaptive immunity, and have been implicated in inflammatory responses to environmental stimuli. TRPA1 and TRPC6 have been associated with enhanced allergic inflammation (Caceres et al. 2009; Sel et al. 2008). In contrast, TRPV1 has been reported to protect against allergic sensitization to aeroallergens but not sensitization to system allergens (Mori et al. 2011). Differential effects of TRPV1 that are dependent on the route of sensitization is an interesting observation in that neutrophilic inflammation and airway hyperresponsiveness induced by LPS and allergen have also been shown to be affected by the route of sensitization (Wilson et al. 2009), raising the question of whether TRPV plays a role in differentiating between immune responses originating systemically and at a mucosal barrier. TRPM4 has been associated with down-regulation of IgE-mediated mast cell activation in vivo (Banner et al. 2011).
Reflexes
For this discussion, reflexes in the respiratory system will be separated into two categories, regulatory and defensive. Regulatory reflexes are homeostatic in nature. Physiological parameters are monitored, and ventilation and perfusion are modulated to maintain the composition of arterial blood while maximizing efficiency of breathing and gas exchange. The major physiological parameters monitored in regulatory reflexes are mechanical deformation of airways and blood vessels, CO2 via pH, and arterial partial pressure O2 (). In contrast, defensive reflexes respond to environmental stimuli to protect cardiopulmonary function. Composition, or quality, of the air is monitored; and ventilation, perfusion, and mucosal functions are modulated to minimize exposure of tissues to injurious, or detrimental factors. The major parameters inducing defensive reflexes are changes in tactile sensation, temperature, acidity, and molecular structures at the mucosal surface.
Regulatory Reflexes
Maintenance of O2, CO2, and pH levels is a primary function of the respiratory system. The basic reflexes linking changes in O2, CO2, and pH to ventilation are described in textbooks of physiology and will not be reiterated here; but changes in sensitivity to CO2 in relation to respiratory drive and sleep apnea and reflex control of the airways by O2 and CO2 deserve consideration. Under normal conditions, hypercapnia and hypoxia are primary stimuli of the respiratory central pattern generator (CPG) in the brainstem. During sleep in normal individuals, the sensitivity to CO2 decreases, resulting in an elevation of by 3–6 mmHg (Dempsey et al. 2010; Skatrud and Dempsey 1983). In parallel, the apnea threshold, or the below which an individual will be apneic, increases in parallel and remains 4–5 mmHg lower than the steady state . Normal individuals retain sensitivity to small changes in CO2 during sleep and respond with changes in ventilation that maintain within a narrow range and above the apnea threshold (Naughton 2010). So, remains high but stable. In contrast, patients with sleep apnea do not exhibit elevated during sleep or a reduction in the apnea threshold, resulting in a smaller difference between the steady state during sleep and apnea threshold (Naughton 2010; Xie et al. 2002). Thus, small decreases in lead to apnea and unstable breathing.
During inspiration, negative pressure develops in the airway lumen, acting to collapse the airway and restrict airflow, particularly in the extrathoracic airways that are surrounded by barometric pressure. Reflexes exist to both preempt and respond to collapse of the upper airways. Reflexes that preempt closure of the upper airways originate in the CPG. Many of the muscles of the upper airways receive input from the CPG, which synchronizes their activity with inspiration and expiration and modifies their activity in response to respiratory stimulants and depressants, such as hypoxia, hypercapnia, and opiates. The genioglossus is the upper airway muscle that is best studied in this regard. The genioglossus contracts 50–100 ms before the beginning of inspiration, pulling the tongue forward and down, away from the pharynx, presumably in preparation for the development of negative airway pressure that would act to pull the tongue back into the oropharynx (Edwards and White 2011).
In addition to extrinsic regulation by the CPG, muscles in the pharynx, and the lower airways as well, respond to local deformation of airway and lung tissue leading to reactive regulation of airway diameter, respiratory drive, and cardiac function. The effect of mechanical deformation is observed both within and across regions of the airways. The processes have been defined best in the lower airways, but are also known to be present in the upper airways. Both the active development of compressing transmural pressure during inspiration and passive compression by surrounding tissues tends to collapse the oropharynx. Collapse of the pharyngeal wall is opposed by active and passive forces (Susarla et al. 2010). Negative pressure that develops in the pharynx during inspiration activates mechanoreceptors in the pharyngeal wall, inducing reflex contraction of constrictor muscles that stabilize the wall and oppose collapse. Simultaneously, expansion of the lungs increases traction on the upper airways, stiffening and opposing collapse of the pharynx. Conversely, positive pressure developed during exhalation induces activation of pharyngeal dilator muscles. Simultaneously, loss of traction on the pharynx as the lung deflates leads to progressive loss of tension in the pharyngeal wall during exhalation. The combination of a high level of pharyngeal dilator activity and compression by fat deposits in the wall of the pharynx has been implicated in collapse of the oropharynx late in expiration in obstructive sleep apnea (Susarla et al. 2010).
Pressure changes in the upper airways also affect ventilation (Widdicombe 2011). In adults, negative pressure in the pharynx, particularly in response to nasal obstruction, inhibits respiratory drive and increases expiratory and inspiratory time, and positive pressure has a small stimulatory effect on breathing frequency. Reflex stabilization of the upper airways by positive pressure, and reversing the decrease in respiratory drive associated with negative pressure may underlie the effectiveness of continuous positive airway pressure in alleviating sleep apnea (Widdicombe 2011).
Of the three classes of sensory fibers found in the airways—SAR, RAR, and C-fibers—SAR neurons are most closely associated with regulatory reflexes. Activation leads to tachycardia, relaxation of airway smooth muscle, inhibition of inspiratory drive, and prolongation of expiration (Kubin et al. 2006; Coleridge and Coleridge 1994; Hlastala and Berger 2001). Tachycardia increases cardiac output to accommodate the increase in venous return associated with the lower thoracic pressure and to better match the volumes of blood and air in the lungs. Relaxation of airway smooth muscle allows the airways to expand in concert with the increase in recoil force within the lung tissue. Inhibition of inspiratory drive opposes overinflation, and prolongation of expiration insures that inspiratory and expiratory volumes are matched.
Defensive Reflexes
Defensive reflexes mediate changes in mucociliary clearance, sneezing and coughing to actively clear inhaled agents from the airways. Reflex changes in ventilation and perfusion limit distribution of inspired agents throughout the lungs and to other organs that could be damaged due to exposure to the toxin. Compared to SAR fibers involved in regulatory reflexes, RAR neurons, and C-fibers exhibit greater sensitivity to tactile stimulation, pH, and chemical agents; and play major roles in defensive reflexes. RAR neurons respond to mechanical deformation but also to inhaled irritants (e.g., smoke and ammonia (Kubin et al. 2006)) and low osmolarity solutions (Coleridge and Coleridge 1994). C-fibers are less sensitive to mechanical deformation; but highly sensitive to pH changes, chemicals (e.g., ozone and capsaicin), high osmolarity solutions, and temperature changes (Kubin et al. 2006; Coleridge and Coleridge 1994). Response of the RAR irritant receptors is associated with cough, bronchoconstriction, and mucus secretion (Hlastala and Berger 2001). C-fiber activation is associated with cough, rapid shallow breathing and apnea, bronchoconstriction, mucus secretion, bradycardia, and hypotension (Kubin et al. 2006; Coleridge and Coleridge 1994; Hlastala and Berger 2001).
Under normal conditions, SAR neurons, RAR neurons, and C-fiber neurons are distinct subpopulations physiologically and anatomically. The respond to unique stimuli, exhibit unique temporal responses to stimuli, and terminate in separate regions within the NTS. Yet, outputs from the NTS converge on brainstem and spinal neurons controlling ventilation, cardiac function, airway diameter, salivary and mucus gland secretion, and pain, leading to significant overlap in efferent responses to SAR, RAR, and C-fiber activation (Kubin et al. 2006). Furthermore, physiological differences between the subclasses become blurred in inflammation, which can change the expression patterns of TRP channels in the cells, increasing the diversity of stimuli that activate each neuronal subtype.
Exposure of the nose to diverse environmental stimuli leads to sneezing, mucus secretion, itching, nasal congestion, and nasal obstruction (Tran et al. 2011). Each of these responses can be induced locally, but there is clear evidence of reflex involvement in sneezing, nasal secretion, and congestion (Sarin et al. 2006). Sneezing is induced by irritants or tactile stimulation of the nasal mucosa, but also by exposure of the eyes to bright light or exposure of areas of the skin to sudden changes in temperature will induce sneezing. Rhinorrhea, or excessive nasal secretions, can be induced by local exposure to histamine, allergens, cold air, hypertonic solutions, the TRPV1 activator capsaicin, or bradykinin; but it can also be induced in the contralateral nostril (Sarin et al. 2006). The reflex nature of the response is evident from the fact that the contralateral response is eliminated by cutting the Vidian nerve. Nasal congestion occurs in response to vasodilation of mucosal blood vessels and pooling of blood in the mucosa. Congestion can be induced by exposure of skin at different sites on the body to changes in temperature. The reflex nature of congestion is further indicated by the observation that congestion can cycle from one nostril to the other (Sarin et al. 2006). This nasal cycle can be disrupted by inhibition of either sympathetic or parasympathetic outflow, leading to the idea that the cycling results from oscillating imbalance in parasympathetic-mediated vasodilation and sympathetic-mediated vasoconstriction.
Cough is both a protective mechanism for clearing pathogens, chemicals, and debris from the airways and a chronic, often debilitating, manifestation of infection and inflammation. The role of cough in lung defense is emphasized by the fact that loss of the cough reflex is accompanied by an increase in the incidence of pneumonia (Duarte and Myers 2012). Interestingly, the cough reflex is lost following denervation of the lower airways during lung transplant surgery, but is then partially restored in some cases (Duarte and Myers 2012). The restoration indicates that at least a part of the cough reflex is intrinsic to the lungs and does not require input from the central nervous system.
The cough reflex can be initiated via mechanical or chemical stimuli through parallel vagal afferent pathways. At least two activation pathways exist, one that is activated by capsaicin and inhibited under anesthesia, and another that is not activated by capsaicin and is retained under anesthesia. The former pathway is linked to bronchopulmonary C-fibers (Canning 2011). C-fibers express the transient receptor potential cation channels, TRPV1, which endows them responsiveness to capsaicin, and TRPA1, which is activated by acrolein in cigarette smoke, allyl isothiocyanate, and nicotine (Banner et al. 2011). They terminate in the mucosa, submucosa, parenchyma, and vasculature from the larynx to the peripheral lung. Cough induced by chemical activation of C-fibers is often paroxysmal.
Cough can also be initiated by receptors in the larynx, trachea, and large bronchi that activate Aδ fibers. In contrast to cough initiated by C-fibers, cough associated with Aδ fibers is rarely paroxysmal and is observed under anesthesia. Over 60 years ago, JG Widdicombe described vagal afferent fibers that were insensitive to mechanical stimulation of the airway surface, adapted rapidly to lung inflation, and could induce cough (Widdicombe 1954a, b). This initial description has led to the idea that RAR fibers are cough receptors, but Canning cautions against this interpretation (Canning 2010, 2011). Aδ fibers are not a homogenous population, but rather there are multiple subpopulations that differ in peripheral termination sites, sensitivity to mechanical stimuli, and reflexes they activate. In some cases, activation of RAR fibers and cough can be dissociated, indicating that the cough receptor that activate Aδ fibers are separate entities from RARs. Specifically, stimuli that activate RAR fibers—hyperventilation, breathing against a closed glottis, and bronchoconstriction induced by histamine, neurokinin A, and leukotrienes—do not elicit cough (Canning 2010). The cough receptors do not express the capsaicin receptor, TRPV1, but respond to protons and therefore may express other acid-sensitive ion channels. More definitive identification of cough receptors has been elusive, possibly due to plasticity in the pathway. Undem and colleagues have demonstrated that aeroallergen challenge induces TRPV1 expression in Aδ fibers (Lieu et al. 2012). The aeroallergen-induced change in phenotype increases the range of stimuli that lead to neural activation.
In addition to cough, irritation in the upper airways can induce apnea, bradycardia, and changes in peripheral vascular resistance (Widdicombe 2011). The diving reflex stimulated by exposure of the face and nasal mucosa to cold water is a well-recognized example of a cardiopulmonary reflex originating in the face and upper airways. The apnea and bradycardia induced by immersion in cold water can be so intense as to be fatal. The diving reflex is inhibited by interrupting conduction in the trigeminal nerve and can be mimicked by stimulating the ophthalmic branch (V1) of the trigeminal nerve. The systemic vascular responses to immersion in cold water is species dependent, but in humans exposing the nose to cold water leads to peripheral vasoconstriction and peripheral hypertension, but retention of coronary and cerebral blood flow. The diving reflex also entails closure of the larynx, reducing the risk of aspirating water.
An aspiration reflex is associated with mechanical stimulation of the nose, nasopharynx, and oropharynx. The reflex facilitates clearance of inhaled particles to the esophagus and can be mimicked by stimulation of the glossopharyngeal nerve. The aspiration reflex is closely associated with sniffing induced by some odors, but is unique in the fact that it is elicited by mechanical deformation. The aspiration reflex exhibits species diversity and relatively weak in humans.
Similar to the nose and pharynx, the larynx also exhibits a high density of mechanical, irritant, and chemical receptors. When stimulated, these receptors induce apnea, hypertension, bradycardia, bronchoconstriction, and cough. As would be expected from the diversity of sensory nerve endings, the stimuli affecting the larynx are diverse and include mechanical deformation, ammonia, SO2, smoke, 10–30 % CO2, and osmolarity (Widdicombe 2011). In the case of osmolarity, changes in the concentration of permeant ions, particularly chloride, is a primary stimulus for receptor activation.
Deposition and Clearance of Airborne Particles
Deposition
Airborne particles are removed from inspired air by the impaction, interception, sedimentation, and diffusion. Impaction is the process by which inertia causes the particle to collide with the wall of the airway where the airstream deviates from a straight path. Particles with diameters greater than 10 μm (e.g., pollens, coarse particulate from mining and grinding activities) are deposit largely in the upper airways due to inertial impaction. Deflection of the inspired air around the turbinates and through the bend in the nasopharynx causes large particles with high inertia to impact with the front edge of the turbinates and posterior wall of the nasopharynx. The degree of impaction is related to the velocity of airflow, and as airflow rates decrease more large particles escape the upper airways and enter the lower airways, depositing in the trachea-bronchial region (Nakorn Tippayawong and Det Damrongsak 2003; Heyder 2004). Large particles escaping the upper airways continue to impact the airway wall at branch points/bifurcations in the lower airways. Interception is a term use to describe deposition resulting from a particle brushing against the wall of the airway and being trapped in the mucus, airway surface liquid, glycocalyx, or cell membranes. Fibers have a particularly high probability of being intercepted by the airways wall. Sedimentation refers to the settling of particles out of the airstream as velocity of airflow decreases. High rates of sedimentation are observed in the distal bronchi and bronchioles, where airflow velocity decreases due to an exponential increase in cross-sectional diameter of the airways. Particles that escape impaction, interception, and sedimentation can come in contact with and deposit on the airway wall via diffusion. Particles with aerodynamic diameters less than 10 μm (e.g., particles from combustion) are carried in the airstream to the lower airways and are considered to be respirable; and particles less than approximately 1 μm are carried into the alveoli; but the composition and surface characteristics of the particles greatly affect their deposition pattern. Hydrophilic particles tend to increase in size as inspired air is humidified, resulting in greater rates of deposition than predicted on their original dimensions (Heyder 2004). Likewise, aggregation of dispersed particles increases deposition within the lungs and consolidation of particles within the airways. Hydrophobic particles exhibit less deposition in the larger airways than expected based on aerodynamic diameter, but greater deposition in distal regions where the hydrophobicity of alveolar surfactant can facilitate their interaction with the airway surface (Heyder 2004).
Mucociliary Clearance
Particles depositing on the airway surface become entrapped in mucus gel layer floating on the airway epithelium. The airway surface is covered by a 7 μm film of electrolyte, the periciliary layer, that supports an overlying layer of mucus that is 7–70 μm thick and composed of electrolyte and gel-forming mucin glycoproteins (Fahy and Dickey 2010; Tarran et al. 2006). Electrolyte and mucins are secreted by both the glands and surface epithelium. Glands are found from the nose through the bronchi, and the density of glands decreases with increasing airway generation. In the large airways, 95 % of the mucus is secreted from submucosal glands, and only 5 % is derived from the surface epithelium, but in airways smaller than 2 mm, nonciliated cells in the surface epithelium are the only source of mucin glycoproteins (Trout et al. 2001).
The structure of mucus glands was described in 1969 by Meyrick and colleagues from serial thin sectioning of the airway wall (Meyrick et al. 1969), and subsequent studies defined the functional attributes of different regions of the glands (Wine 2007). The prominent portion of the gland observed in histological sections consists of mucous tubules lined by eosinophilic secretory cells. The mucous tubules are flanked by tubules lined with electrolyte secreting epithelial cells. Distally, the serous tubules secrete electrolyte that hydrates the mucus and moves it towards the airway lumen. Proximal to the mucous tubule is the collecting duct, where electrolyte content of the mucus can be adjusted. The gland opens to the airway through a ciliated duct that propels the mucus onto the airway surface.
Mucin glycoproteins impart the gel-like character to mucus. MUCAC and MUC5B are the major polymer-forming mucins in the airways (Fahy and Dickey 2010). MUC5AC is expressed mainly in large proximal airways by the superficial epithelial cells. MUC5B is expressed in glands throughout the proximal airways and by secretory cells in the surface epithelium in both proximal and distal airways. Mucins are stored in secretory cells in a condensed form and hydrated extracellularly following exocytosis (Fahy and Dickey 2010). The mucin glycans provide a substrate which can bind microbial proteins and trap inhaled particles, thereby limit further penetration of the mucosal barrier (Fahy and Dickey 2010). In addition, the gel acts as a sieve with a pore size of approximately 500 nm.
Electrolyte homeostasis in the airways represents a balance in at least three processes: passive transepithelial diffusion of salt and water, transepithelial Na+ absorption mediated by basolateral Na/K ATPase and apical Na+ channels (ENaC), and transepithelial secretion of Cl− and HCO3 − via the cystic fibrosis transmembrane regulator (CFTR) and calcium-activated Cl− channels (CaCC) (Wine 2007; Tarran et al. 2006). CFTR activity is regulated by cAMP, and the importance of this pathway in electrolyte balance is highlighted by the airway dehydration associated with cystic fibrosis. ATP may also play a role in regulating the depth of the periciliary and mucus layers. ATP is released by the epithelium into the airway lumen and acts through P2Y receptors to stimulate airway epithelial Cl− secretion via CaCC and inhibit airway epithelial Na+ absorption via ENaC (Tarran et al. 2006). Breakdown of ATP to adenosine and subsequent activation of A2b receptors on the apical epithelial surface can regulate CFTR and ENaC secondarily (Tarran et al. 2006). ATP also stimulates surfactant release from Type II pneumocytes and thereby reduces surface tension (Mishra et al. 2011). Surface tension is associated with alveolar edema, and in this context, ATP would reduce the formation of alveolar edema.
The role of purinergic regulation in lung function is well recognized in the context of cystic fibrosis and may also be relevant to asthma. ATP functions in recruitment and activation of dendritic cells, neutrophils, and eosinophils (Mortaz et al. 2010; Willart and Lambrecht 2009; Muller et al. 2010); and can promote allergic responses through the innate immune system by inducing airway epithelial cells to secrete IL-33, which promotes Th2 responses (Kouzaki et al. 2011). Single nucleotide polymorphisms of purinergic receptors are associated with FEV1, FEV1/FVC, airway hyperresponsiveness, and bronchodilator responses; and the associations are affected by house dust mite exposure (Bunyavanich et al. 2012).
Airway Resistance
The nose accounts for approximately 50 % of the total resistance to airflow in the airways and therefore also accounts for an equivalent proportional decrease in lumenal pressure as air flows between the nares and alveoli. The configuration of the respiratory system leads to region-specific closure of airways during inspiration and expiration. Respiratory muscles act on the chest wall and diaphragm to change the pressure in the pleural space and alveoli, which subsequently drive airflow. The nose accounts for approximately 50 % of the total resistance to airflow through the respiratory system during nasal breathing. The nasal resistance lowers the pressure in the pharynx during inspiration, predisposing the pharynx to collapse during inspiration (Ferris et al. 1964). In contrast, the restriction opposes collapse of the distal airways during exhalation by providing a backpressure in the airway lumen. By comparison, the intrathoracic extrapulmonary airways are surrounded by pleural pressure, which remains negative relative to barometric pressure during quiet breathing. Thus, during normal resting breathing, pleural pressure opposes closure of intrathoracic airways during inspiration. During forced exhalation, pleural pressure can be positive, which tends to compress the intrathoracic extrapulmonary airways. Likewise, intrapulmonary airways are surrounded by alveoli, and development of positive alveolar pressure during exhalation acts as a compressive force. Under normal conditions, the resistance of the distal airways is low, leading to minimal decrease in the luminal pressure, minimizing the pressure differential across the wall of the intrapulmonary airways. Alveolar septa impinge upon the walls of the intrapulmonary airways, and stretch of the septa opposes collapse of the airways, particularly at high lung volumes. Increase in the resistance of the distal airways due to smooth muscle contraction, accumulation of secretions in the airway lumen, or destruction of lung tissue that reduces elastic recoil leads a premature drop in the luminal pressure and dynamic compression of the airways during exhalation (Irvin and Bates 2009).
The collagen and elastin network combined with contractile elements, surface tension, surface-active material, and fluid dynamics result in complex responses of the lungs to applied pressures. The resulting viscoelastic properties of the lungs give rise to tissue resistance that is dependent on breathing frequency, increasing at low frequencies and decreasing at higher frequencies. At respiratory rates of 12–24 bpm, tissue resistance can account for 40 % of the resistance to breathing (Faffe and Zin 2009). The role of tissue resistance in responses to allergen challenge in mice is well established (Irvin and Bates 2009), but less is known about its role in humans. Readers wanting to delve deeper into lung mechanics and models of lung behavior are directed to the excellent review by Faffe and colleagues (Faffe and Zin 2009) as well as the work of Jason Bates (Bates and Lutchen 2005).
Control of Airway Smooth Muscle
Airway smooth muscle exhibits basal tone; and numerous hormones, neurotransmitters, inflammatory mediators, environmental agents, and therapeutics alter the airway smooth muscle contractile activity. Two primary factors influencing the level of bronchomotor tone under basal conditions is parasympathetic cholinergic stimulation of contraction via M3 receptors on the smooth muscle, and parasympathetic noncholinergic fibers induce bronchodilation via VIP, Substance P, and NO (Canning 2006). While there is abundant expression of VIP in the trachea and bronchi (Groneberg et al. 2006; Kraneveld and Nijkamp 2001; Groneberg et al. 2001), there is a lack of definitive evidence for expression of the known VIP receptors—PAC, VPAC1, and VPAC2—on airway smooth muscle, yet VPAC2 agonists induce bronchodilation both in vivo and in vitro (Tannu et al. 2010). So, the mechanism by which VIP induces smooth muscle relaxation is unclear. Hormonally and therapeutically, bronchodilation via β-adrenergic agonists and elevation of cAMP is well documented.
In humans, SP and NKA contract large airways via activation of NK2 receptors (Kraneveld and Nijkamp 2001). NK1 agonists are without effect. SP and NKA also contract small airways, but the effects are mediated by thromboxane and A2 and NK1 receptors (Kraneveld and Nijkamp 2001).
Mast cell- and basophil-derived mediators, including histamine and cysteinyl leukotrienes, are potent bronchoconstrictors, and bidirectional interaction exists between mast cells and parasympathetic afferents (Kraneveld and Nijkamp 2001). NANC nerve terminals in the airway wall express histamine receptors capable of increasing sensitivity of the neurons to stimuli, lowering the stimulus threshold for release of neuropeptides. In human mast cells, SP can down-regulate FcεRI and the response to IgE (McCary et al. 2010). In vivo, down-regulation of FcεRI would reduce acute bronchoconstriction in response to allergen, which would be in opposition to bronchoconstriction induced by direct effect of SP binding to NK2 receptors airway smooth muscle. Reciprocally, mast cell lines have been shown to express NK1 and NK2 receptors, and SP modulates mast cell activity (Kraneveld and Nijkamp 2001). Similarly, both nerves and mast cells have been shown to secrete and respond to nerve growth factor (NGF) (Kraneveld and Nijkamp 2001). Cholinergic, adrenergic, and nonadrenergic noncholinergic neurotransmitters regulate secretions in the airways. Adrenergic stimulation induces electrolyte secretion onto the airway surface. VIP induces low rates of secretion from glands, and acetylcholine induces high rates of glandular secretion (Wine 2007).
Control of Mucus Secretion
Control of electrolyte and mucin secretion varies across species and is still not completely understood. In humans, acetylcholine from parasympathetic cholinergic efferent fibers acts on muscarinic M3 receptors to induce both mucin and electrolyte secretion (Rogers 2001; Wine 2007). At least a portion of the Ach-induced electrolyte secretion is secondary to Ca2+-induced Cl− transport that is independent of the cystic CFTR (Wine 2007). Ach-dependent regulation of mucus secretion is thought to be the major pathway controlling reflex stimulation of mucus secretion through the autonomic motor nuclei.
VIP constitutes a second major control pathway in human airways that parallels and modulates Ach-induced mucus secretion. VIP acting on VIP type 1 receptors (VPAC1R) expressed on gland and surface epithelial cells stimulates electrolyte and mucin secretion (Kim et al. 2011; Miotto et al. 2004). Unlike Ach-induced electrolyte secretion, VIP stimulates HCO3 − secretion that is dependent on CFTR and therefore is impaired in cystic fibrosis (Wine 2007). In addition to the epithelial expression, VIP receptors are also found presynaptically on parasympathetic cholinergic efferent fibers. Activation of these receptors inhibits Ach-induced mucus secretion, and this thought to be the dominant effect of VIP on submucosal glands (Rogers 2001).
Inflammation alters expression of VIP receptors, and it is unclear whether the alteration contributes to, or compensates for increased mucus production. Consistent with this paradigm, in a rat model of airway inflammation, VIP expression in the airway wall was negatively correlated with ozone-induced changes in mucin secretion (Li et al. 2009). In humans, VIP and VIP receptors expression are increased in the nasal mucosa of allergic rhinitis subjects (Kim et al. 2011; Park and Christman 2006) and bronchial mucosa of smokers with chronic bronchitis (Miotto et al. 2004). It is unknown if the increased expression observed in human tissues is related to inhibitory activity at cholinergic synapses, or is associated with NANC stimulation that synergizes with the cholinergic stimulation.
In some species, sensory efferent/axon reflex control of glandular secretion is common and involves CGRP, NKA, and/or SP (Rogers 2001). As stated above, sensory efferent innervation is not common. There is scant evidence of CGRP, NKA, or NKB expression in human airways, but SP is expressed, along with NK1, NK2, and NK3 receptors (Canning 2006). Interestingly, in 2007, Wine and colleagues demonstrated a direct effect of SP on volume secretion by human bronchial glands, the effect was 10 % of what was observed in porcine glands stimulated with SP and even less and less than 10 % of the secretion rate observed in human glands stimulated with carbachol (Choi et al. 2007). SP was synergized with VIP, but not carbachol, and was shown to involve electrolyte secretion through a CFTR-dependent pathway.
In contrast to glands, secretion of mucins from goblet cells in the surface epithelia is reported to be controlled primarily by ATP and UTP in the airway surface liquid acting on apical P2Y purinoceptors (Davis and Dickey 2008). Bronchioles are normally devoid of goblet cells, and the major nonciliated secretory cell in this region is the Clara cell. Under normal condition, Clara cells secrete mucin glycoproteins in a constitutive fashion, with secretion rates matching production, and no accumulation of glycoproteins in secretory granules. During inflammation, production of mucin glycoproteins increases, and mucin granules appear in the cells, resulting in a goblet cell phenotype.
Airway Inflammation
The inflammatory response to inhaled pathogens is highly variable and affected by the nature of the agent, its concentration in inspired air, duration and periodicity of exposure, and expression of comorbid conditions. A summary of common inflammatory cells in the lungs and some of their interactions are illustrated in Fig. 2.4. Innate and adaptive responses drive the inflammation, leading to redundancy in the pathways leading to the inflammatory response. Epithelial cells, dendritic cells, macrophages, mast cells, natural killer (NK) cells, and nerves respond to pathogens in the airways and environmental stimuli, altering airway function, mounting defense responses, and initiating inflammation. Interaction with T and B lymphocytes leads to adaptive immune responses, immunologic memory, and efficiency of subsequent responses. There is a tendency in reviews and discussions in this field to focus on pro-inflammatory mechanisms, but it should be noted that counter-regulatory mechanisms are in place that limit the inflammatory response and thereby protect the tissues from intrinsic damage.
Fig. 2.4.
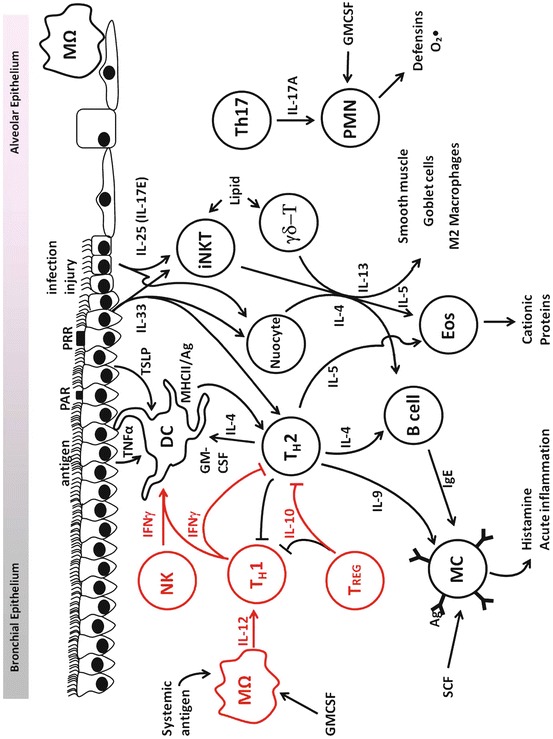
Overview of cells and cytokines implicated in airway inflammation underlying asthma. Red indicates pathways that down-regulate TH2-driven inflammation. PAR protease-activated receptor, PRR pattern recognition receptor, MΩ macrophage, NK natural killer cell, DC dendritic cell, Th T-helper lymphocyte, iNKT invariant natural killer T lymphocyte, γδ-T γδ-T lymphocyte, PMN polymorphonuclear lymphocyte, T reg regulatory T lymphocyte, B cell B lymphocyte, MC mast cell, Eos eosinophil
Macrophages are found in the alveolar, interstitial, and intravascular compartments. The alveolar macrophage exists at the air–tissue interface and acutely responds to environmental stimuli. They stain brightly for nonspecific esterase. Upon activation they secrete reactive oxygen species (ROS) and cytokines, and exhibit Fc-independent phagocytosis (Lohmann-Matthes et al. 1994). Interstitial macrophages exhibit less secretion of ROS and cytokines, but have a greater capacity for antigen presentation. Intravascular macrophages are thought to engulf material then transit from the lung and into the blood. They are distinguished from monocytes by a greater capacity for phagocytosis (Lohmann-Matthes et al. 1994). Dendritic cells are similar to interstitial macrophages, but these cells have even greater capacity for antigen presentation.
Inhaled particles activate macrophages via cell surface receptors (scavenger receptors and pattern recognition receptors (PRRs)) that recognize common structures that are shared by broad classes of pathogen (pathogen-associated molecular patterns or PAMPs). Scavenger receptors represent a diverse family of molecules capable of binding inorganic molecules such as silica and titanium, as well as organic molecules such as low density lipoprotein (Murphy et al. 2005). Eight classes of scavenger receptors have been described and are designated A through H. Receptors classes A, B, D, and E, specifically SR-AI/II, MARCO, CD36, CD68, and LOX-1, have been implicated in responses to environmental pathogens (Thakur et al. 2008; Yatera et al. 2011; Lewis et al. 2009). PRRs recognize PAMPs including mannans in the yeast cell wall, bacterial-derived formylated peptides, lipopolysaccharides, and lipoteichoic acids; and PRR–PAMP interaction be direct or may depend on intermediate or adapter molecules. Toll-like receptors (TLRs) bind PAMPs directly. TLR2 and 4 are expressed by alveolar macrophages and mediate many of the responses to pathogens (Xiang et al. 2010; Xiang and Fan. 2010). TLRs also recognize damage-associated molecular patterns (DAMPs), leading macrophages to respond to tissue injury following environmental exposures and inflammatory responses (Xiang et al. 2010). Particles and pathogens may also bind indirectly to macrophages following opsonization with immunoglobulins, complement, or collectins including mannose binding protein and surfactant apoproteins A and D. Opsonized pathogens bind to macrophage via Fc (FcγRI, II, and III; FcεRI; and FcαRI), complement receptors (CR1, 3, 4), or collectin receptors (SP-R210, cC1qR, CD14, SRCL) (Selman et al. 2008; Aderem and Underhill 1999; Grubor et al. 2006).
Macrophages play a central role in dust-induced occupational diseases, such as silicosis, asbestosis, and berylliosis. Activation by particles leads to phagocytosis and secretion of ROI (reactive oxygen intermediates such as superoxide, hydrogen peroxide, hydroxyradicals) and RNI (reactive nitrogen intermediates like nitric oxide and peroxynitrite). Activation also alters immune and inflammatory responses via secretion of cytokines (e.g., TNF-α, IL-12, TGF-β) and chemoattractants (e.g., CCL17 and CCL22 act on TH2 cells, and CCL11 attracts eosinophils). The specific stimulus controls the secretory products that are released and the ultimate physiological and pathological responses of the macrophage.
Dendritic cells are found throughout the respiratory tract and can be separated into at least five subtypes based on cell surface markers and origin: conventional, plasmacytoid, monocyte-derived inflammatory, innate killer, and alveolar dendritic cells (Hammad and Lambrecht 2011; Lambrecht and Hammad 2009). Dendritic cells play a central role in both initiating immune responses to allergens, microbes, and viruses and maintaining balance within the immune system. Accordingly, different subsets have specialized, and occasionally opposing effects on immune responses. For example, dendritic cells of myeloid origin, including conventional and inflammatory dendritic cells, actively promote TH1 and TH2 immune responses against foreign antigens (Hammad and Lambrecht 2011; Hammad et al. 2010). In contrast, some plasmacytoid dendritic cells have anti-inflammatory activity that leads to apoptosis of eosinophils and lymphocytes through expression of programmed death 1 ligand (PD-1L) (Kool et al. 2009).
In normal lungs, immature dendritic cells express high levels of PRRs, but low levels of accessory molecules necessary for binding and activating naïve T-lymphocytes (CD40, CD54, CD58, CD80, and CD86). Immature dendritic cells are thereby poised to take up and process antigens, but are inefficient in presenting antigen to lymphocytes. In the presence of inflammatory mediators including GM-CSF, IL-4, CD40L, TNF-α, IL-1β, IL-6, and thymic stromal lymphopoietin (TSLP), DAMPs, PAMPs, or specific ligation with T-helper cells, immature dendritic cells differentiate and express high levels of MHCII and the accessory molecules necessary for efficient antigen presentation. Exposure to antigen and inflammatory mediators, which normally occurs in peripheral tissues, causes the dendritic cell to migrate to the draining lymph nodes where the mature dendritic cell then presents antigens with high efficiency in the T cell zones of the secondary lymphoid tissues. Maturation factors for dendritic cells are prevalent in the mucosa in many diseases, particularly in allergic airways disease, and result in recruitment of monocytes that differentiate into mature dendritic cells (Ganesh et al. 2009; Blanchfield and Mannie 2010; Hamilton and Anderson 2004; Ritz et al. 2002; Soruri and Zwirner 2005).
Dendritic cells process and present both peptide and non-peptide antigens, and the type of antigen affects dendritic cell maturation. Viral- and cell-associated antigens induce maturation to a phenotype characterized by expression of CD70 and Notch Ligand Delta and secretion of IL-12, which promotes development of TH1 cells that are thought to mediate antiviral immunity and promote cell-mediated diseases such as Sarcoidosis (Parisinos 2011). In contrast, soluble peptide allergens induce dendritic cells, particularly myeloid dendritic cells, to mature into a TH2-promoting phenotype thought to play a critical role in allergic respiratory diseases (Tsoumakidou et al. 2008). Interestingly, viral infections are a primary trigger for asthmatic reactions, so the dichotomy between the effects of viral and allergenic antigens is not absolute. Viral exposure has been shown to induce the expression of the high affinity IgE receptor (FcεRI) on murine dendritic cells, and activation of the receptor leads to expression of CCL28, a chemoattractant for T and B lymphocytes (Holtzman et al. 2009; Grayson et al. 2007).
Lymphocytes comprise a relatively small percentage of the inflammatory cells in environmental lung diseases compared to macrophages, PMNs, and eosinophils (<10 % of the cells in bronchoalveolar lavage fluid); but are thought to play crucial regulatory roles. All three subtypes (T cells, B cells, and natural killer (NK) cells) are found in the lungs following environmental exposures.
T cells express the CD3 cell surface marker, which is a subunit of the T cell receptor. They are subdivided into CD4+ (T helper (TH), CD4+ T regulatory T (Treg), γδ T, and invariant Natural Killer T-lymphocytes (iNKT)) and CD8+ (cytotoxic TC)) cells. TH cells are further divided into effector and memory TH1, TH2, TH9, and TH17 cells. TH1 cells secrete IFN-γ and promote cellular immunity. TH2 cells secrete IL-4, IL-5, IL-6, IL-9, IL-10, and IL-13 that support humoral immune responses. IL-4 is a key cytokine in needed for differentiation of the TH2 subset and immunoglobulin class switching to the IgE isotype. IL-5 is a primary growth factor for eosinophils, and IL-13 induces smooth muscle and goblet cell growth. IL-6 is a growth factor for T, B, and mast cells. IL-9 also promotes mast cell growth. IL-10 promotes B cell growth, but inhibits TH1 and TH2 responses. TH17 cells secrete IL-17A and F, which promote neutrophilic inflammation. iNKT cells can be activated by glycolipid bound to the MHC-class-I-like cell surface protein, CD1d, leading to secretion of IL-4, IL-5, and IL-13 and TH2-like inflammation (Meyer et al. 2008; Iwamura and Nakayama 2010). iNKT cells are responsive to IL-25 and IL-33, which are expressed by epithelial cells in response to activation of PRRs. iNKT cells may thereby act as effector cells for epithelial cell-initiated responses to diverse environmental stimuli, helping to explain the preponderance of the Th2 response in the allergic lungs. IL-25 is also secreted by mast cells and TH2 lymphocytes. This reciprocal activation of iNKT, mast cells, and TH2 lymphocytes represents a point of synergism between the adaptive and innate immune systems (Iwamura and Nakayama 2010). iNKT cells are relatively resistant to corticosteroids and may contribute to steroid-resistance in asthma. γδ T cells also respond to lipid antigens presented in the context of CD1d on the surface of DCs (Lloyd and Hessel 2010). Some γδ T cells can express cytokines associated with asthma, including IL-5 and IL-13 (Robinson 2010) and IL-17A (Murdoch and Lloyd 2010). However, γδ T cells may not be purely pro-inflammatory and have been associated with resolution of allergic responses in mice (Murdoch and Lloyd 2010).
Treg cells secrete the immunosuppressive cytokines TGF-β and IL-10 and are thought to play an important role in moderating pulmonary allergic sensitivity and immune responsives (Nouri-Aria and Durham 2008; Ray et al. 2010; Wan and Flavell 2007). TGF-β is also implicated in wound healing and tissue remodeling. These cells are therefore capable of down-regulating inflammation and promoting repair of lung tissue. In the lungs, an excessive repair response however may result in fibrosis.
CD8 TC bind to the non-variable region of MHCI stabilizing the interaction between the TC cells and cells that express viral antigens in the context of MHCI, positioning the TC cells to release perforin, granzymes, and granulysin that kill the virus-infected cells. TC cells differentiate into subsets that are functionally similar to those described for TH cells, including TC1, TC2, TC17, FOXP3+, γδ+, effector, and memory cells. In a TH2 environment, TC cells are induced to produce IL-5, augmenting the allergic response and providing a potential mechanism for virus-induced asthma exacerbations (Coyle et al. 1995).
NK cells are CD3 negative cells that lyse cells expressing abnormal levels of MHCI. NK cells play diverse roles in allergic asthma. They can produce large amounts of IFN-γ, and thereby affect DC maturation. They express MHC II, and in the presence of costimulatory molecules present the antigens to TH2 cells (Korsgren et al. 1999), but depletion does not impair antibody responses to antigen (Hanna et al. 2004). Consistent with secretion of IFN-γ, NK cells attenuate ovalbumin-induced airway responses in mice (Wang et al. 1998) and are required for timely resolution of OVA-induced airway inflammation, possibly via Resolvin E1 binding to its receptor, CMKLR1, on NK cells (Matsubara et al. 2007).
Multiple subpopulations of NK cells have been described based on cell surface markers, cytolytic capacity, and cytokine secretion. IL-12 induces a cytolytic phenotype (Haworth et al. 2011). IL-18 also induces differentiation of NK cells towards a phenotype of low cytolytic activity and robust IFN-γ secretion (Moretta et al. 2006). Exposure to IL-4 inhibits development of a cytolytic phenotype (Agaugue et al. 2008), and a subpopulation of NK cells has been described, NK2 cell, that secrete TH2 cytokines and promote humoral immunity (Moretta et al. 2006). Thus, depending on the environment, NK cells can modulate antigen presentation and development of effector cells.
Polymorphonuclear leukocytes (PMNs) are prominent in the airways of smokers and are among the first cell types to infiltrate the lung during the airway inflammatory response to allergen. PMNs are also prominent following acute lung injury and in the lungs of subjects who die of status asthmaticus (Deniz et al. 2002). In COPD, PMN numbers correlate with impairment of pulmonary function (Cowburn et al. 2008; Perl et al. 2011). PMNs are activated and recruited to the lungs by diverse signals, including IL-8 acting on the CXCR1 and CXCR2 chemokine receptors and leukotriene-B4 (LTB4), TNF-α, IL-1β, IL-6, and MCP-1 (Bathoorn et al. 2008). Circulating PMN binds to pulmonary capillary endothelial cells via selectins (P-, E-, and L-selectin) and β2-integrins. The majority of transendothelial migration of neutrophils is via active endothelial engulfment triggered by engagement of ICAM1 (Perl et al. 2011; Bathoorn et al. 2008). In the tissues, PMNs migrate to the site of inflammation by phosphoinositide 3-kinase-dependent chemotaxis. Circulating PMNs have a relatively short lifespan (6–12 h), but apoptosis can be inhibited and lifespan extended at sites of inflammation by lipid activators, TNF, IL-1, IL-6, IL-8, GM-CSF, G-CSF, and hypoxia; thereby extending their longevity in the tissues (Cowburn et al. 2008; van Buul et al. 2007). Activated neutrophils secrete elastase, cathepsins, myeloperoxidase, and ROS; all of which can lead to further tissue damage. Macrophages play a role in down-regulating neutrophilic inflammation by phagocytosis of apoptotic neutrophils and secretion IL-10 and TGF-β (Perl et al. 2011). Impairment of macrophages in COPD may contribute to chronic neutrophilic inflammation.
Anti-inflammatory lipid mediators acting on the PMNs, endothelium, and epithelium inhibit PMN migration and promote resolution of neutrophilic inflammation (Cowburn et al. 2008). Five categories of anti-inflammatory mediators have been described: lipoxins, resolvins, protectins, cyclopentenones, and polyisoprenyl phosphates. These mediators reduce PMN migration and inhibit release of pro-inflammatory mediators while increasing apoptosis and clearance by macrophages (Haworth et al. 2011; Campbell et al. 2007; Haworth and Levy 2007). These mediators increase expression of anti-adhesion molecules on epithelium and consequently promote epithelial clearance of PMN (Haworth and Levy 2007).
Eosinophils are commonly associated with allergic asthma although their number can vary widely between subtypes of patients (Campbell et al. 2007). Eosinophils are also prevalent in 20–40 % of COPD patients (Welte and Groneberg 2006; Akuthota et al. 2011). Whether eosinophils are bystanders or effectors in these diseases is a matter of debate (Saha and Brightling 2006). IL-5 is a primary eosinophil growth factor, and complement C5a and platelet activating factor are major chemoattractants (Akuthota et al. 2011; Sugita et al. 2003). Other cytokines that are active in eosinophil recruitment include IL-2, IL-3, GM-CSF, eotaxin, and RANTES (Resnick and Weller 1993). Vascular cell adhesion molecule 1 (VCAM-1) is upregulated on endothelial cells by IL-4t does not efficiently interact with neutrophils, which may be why eosinophils accumulate in allergic airways to a greater degree than neutrophils (Resnick and Weller 1993). When activated, eosinophils release cationic proteins, leukotrienes, and ROS (Resnick and Weller 1993). In addition, eosinophils contain over 30 different cytokines that are released in response to specific stimulation (Sugita et al. 2003; Spencer et al. 2009). Eosinophils augment the TH2 response to allergen by production of IL-4 and IL-5 (Spencer et al. 2009). They also inhibit growth of respiratory syncytial virus (Akuthota et al. 2011).
Mast cells respond to environmental stimuli through TRPs, PRRs, crosslinking of FcεRI by allergen and allergen-specific IgE. Mast cells originate from a myeloid progenitor that is distinct from the granulocyte and macrophage progenitor. Immature precursor cells are released from the bone marrow and circulate to peripheral tissues where they mature into effector cells. The mechanisms underlying recruitment of mast cell precursors to the lungs have not been fully defined but appear to involve interactions of integrins α4β1 and α4β7 with VCAM-1 in response to LTB4, PGE2, IL-9, and other signals from NKT and Treg cells (Akuthota et al. 2011). In the mucosa, mast cells mature in response to stem cell factor (SCF) produced by the epithelium (Collington et al. 2011; Weller et al. 2011), acting through the c-kit receptor (Wen et al. 1996). When mature, mast cells respond to PAMPs, DAMPs, environmental stimuli, and allergen-specific IgE by immediate release of preformed mediators, including histamine, serotonin, and serine proteases, followed by synthesis and release of cysteinyl leukotrienes (LTC4, D4, E4, and F4), LTB4, thromboxane, and platelet activating factor. Mast cells also release pro-inflammatory cytokines including TNF-α, IL-4, IL-5, IL-6, and IL-13, which augment and prolong mucosal inflammation (Collington et al. 2011).
Mast cell products have widespread effects in the lungs. Histamine induces smooth muscle contraction, increases endothelial permeability, stimulates epithelial secretory activity, and modulates afferent nerve activity. Histamine stimulates or inhibits nerve activity depending on the balance of H1and H3 histamine receptor ligation (Weller et al. 2011). Leukotriene receptors are found on diverse structural and inflammatory cells types and regulate global aspects of airway and vascular function, as well as immunity and inflammation (Weller et al. 2011; Groneberg et al. 2004). LTB4 is a potent chemoattractant and activator of neutrophils, eosinophils, macrophages, T cells, and DCs (Okunishi and Peters-Golden 2011). Mast cells have been implicated in myocardial remodeling and fibrosis (Okunishi and Peters-Golden 2011) and pulmonary responses to silica and nanoparticles (Levick et al. 2011).
Epithelial cells have emerged as important participants in inflammatory responses to environmental stimuli. The epithelium expresses a wide array of receptors to monitor inspired air for pathogens and toxins, including protease-activated receptors (PARs), TLRs, nucleotide-binding oligomerization domain [NOD]-like receptors, and TRPs. Activation leads to release of cytotoxic proteins, collectins, and ROS that contribute to killing and clearance of microbes (Brown et al. 2007; Shannahan et al. 2012). Epithelial cells modulate immune responses through release of cytokines that act on dendritic cells and T cells. For example, GM-CSF and TSLP induce dendritic cells to promote TH2 immune responses. IFN-α and -β promote TH1 responses, and IFN-λ (IL-28 and IL-29) promotes Treg growth (Schleimer et al. 2007). IL-33 production by epithelial cells promotes mast cell activation and TH2 cell differentiation, and IL-25 activates iNKT cells to produce TH2 cytokines. Recent studies by McKenzie indicate that IL-25 also induces nuocytes that produce TH2 cytokines (Schleimer et al. 2007). IL-6 production results in the differentiation of antibody-secreting B cells. Ligation of CD40 on epithelial cells can initiate inflammatory cell recruitment via release of RANTES, MCP, CCL28, and IL-8. Epithelial cells can also regulate survival of immune cells via expression of PD-L1, PD-L2, and Fas ligand (Barlow and McKenzie 2011; Neill et al. 2010).
In addition to its roles in immunity and inflammation, the airway epithelium is thought to play an important role in mucosal repair and tissue remodeling through the release of pro-fibrotic mediators and enzymes (Schleimer et al. 2007). The epithelium may play an even more direct role in fibrosis via epithelial–mesenchymal transition (EMT). EMT is a form of epithelial plasticity in which epithelial cells redifferentiate into migratory fibroblastoid cells. It is well recognized that epithelial damage leads to release of TGF-β, epidermal growth factor (EGF), and fibroblast growth factors (FGFs) that facilitate wound healing and promote fibrosis. TGF-β down-regulates E-cadherin and tight junctions between the epithelial cells and upregulates mesenchymal markers (Hackett. 2012). To date, most of the work in this area has been in mice, and definitive evidence of whether EMT plays a role in human airways is lacking.
Neurons interact with inflammatory and immune cells in a bidirectional fashion. Afferent nerves express receptors for inflammatory mediators. One prominent example mentioned above is H1 and H3 histamine receptors on afferent parasympathetic neurons (Johnson et al. 2011). Another is LTB4 acting on TRPV1 channels (Groneberg et al. 2004).
Work supporting development of cholinergic antagonists for use in COPD has led to documentation of muscarinic receptors on diverse inflammatory cells that mediate acetylcholine-induced proliferation and chemotaxis (Hwang et al. 2000). At least some of the acetylcholine effects are thought to be due to increased secretion of LTB4. Interestingly, while the clinical use of cholinergic antagonists is increasing, definitive proof of a direct role for acetylcholine in airway inflammation is lacking (Kistemaker et al. 2012).
In contrast to acetylcholine, a strong case is emerging for a role of VIP in immune and inflammatory processes. Two subtypes of VIP receptors with opposing activity have been described, VPAC1 and VPAC2. VPAC1 activation is immunosuppressive, causing down-regulation of both cytotoxic and allergic responses (Kistemaker et al. 2012). VIP induces generation of Treg cells in the spleen of mice (Voice et al. 2002), which may account for some of its immune suppression. VPAC1 is expressed constitutively, and long-acting VIP analogues are being developed as means to suppress inflammation in allergic asthma and COPD (Szema et al. 2011). VPAC1 has also been associated with enhanced Th17 responses, and the impact of VIP-VPAC1 effects in disease has yet to be defined (Onoue et al. 2010, 2011; Misaka et al. 2010).
In contrast to VPAC1, VPAC2 expression is induced following activation of the TCR and is associated with enhanced IgE antibody responses and heightened cutaneous reactions (Yadav et al. 2008). Through VPAC2, VIP augments co-stimulation via B7, inhibits apoptosis of activated TH2 cells, and inhibits IL-12 production, which has the effect of skewing the TH1/TH2 balance towards TH2 immunity (Voice et al. 2002). The net effect of VPAC2 activation is augmentation of TH2 effector and memory functions in peripheral tissues exposed to allergen.
There is also some evidence that tachykinins modulate inflammatory processes. Alveolar macrophages express NK1 receptors, and exposure to substance P increases metalloproteinase, TNF-α, and IL-1β secretion (Delgado and Ganea 2000; Delgado et al. 2000; Goetzl et al. 2008). Knockout of preprotachykinin A gene attenuates LPS-induced airway hyperreactivity and airway inflammation (Xu et al. 2008, 2011; Xu and Xu 2010). As of 2010, clinical trials with NK receptor antagonists had not validated a role of tachykinins in asthma (Helyes et al. 2010). Expression of NK1 receptors is upregulated by cigarette smoke, which may account for increased susceptibility of smokers to infection, fibrosis, cancer, and emphysema (Ramalho et al. 2011).
Counter Regulation
Numerous negative feedback pathways limit the duration and magnitude of inflammatory responses (Xu et al. 2011; Xu and Xu 2010). Detailed discussion of these mechanisms is beyond the scope of this chapter, but a few examples are noteworthy because they portray a general paradigm. Similar to traditional negative feedback loops in the endocrine system, some mediators released during immune activation act on inhibitory receptors to restrict further cellular activation. Notable examples are nitric oxide, prostaglandins, and catabolites of tryptophan. Prostaglandins produced by the cyclooxygenase pathway interact with heterotrimeric G protein-coupled receptors to stimulate adenylate cyclase formation of cAMP. Nitric oxide produced by nitric oxide synthetase acts through guanylate cyclase to increase cGMP, which in certain cases inhibits immune and inflammatory processes (Bosnjak et al. 2011; Saxon et al. 2008; Cook and Bochner 2010; Bjorgo et al. 2011; Bopp et al. 2009; Baumer et al. 2007; Heijink and Van Oosterhout 2006; Mitkiewicz et al. 2011). Similarly, activation of indoleamine 2,3-dioxygenase and subsequent release of tryptophan metabolites can stimulate aryl hydrocarbon receptors that promote tolerance to aeroallergens (Taher et al. 2008). Inhibitory Fc receptors that bind the Fc domain of IgG, such as FcγRIIB, are expressed on B cells and mast cells, and when activated reduce cellular activation and cytokine production (Taher et al. 2008). Upregulation and activation of decoy cytokine receptors, such as IL-13R α2, can have potent inhibitory and negative feedback consequences, thereby opposing the inflammatory activity of cytokines (Saxon et al. 2008; Karra and Levi-Schaffer 2011; Smith and Clatworthy 2010; Zhu et al. 2010).
In addition to receptors that respond to soluble mediators, there is a plethora of inhibitory receptors that respond to ligands expressed on the surface of activated immune cells. CTLA-4 is expressed on the surface of activated T cells and inhibits CD28 binding to B7 on antigen presenting cells. Expression of CTLA4 limits co-stimulation required for T-cell activation (Mentink-Kane and Wynn 2004). PD-1 acts in a similar fashion. PD-1 is expressed on activated T and B cells, and one of its ligands, PDL-2 is expressed on myeloid dendritic cells and macrophages. Expression of PD-1 provides negative feedback to further activation of T and B cells (Chen and Shi 2006; Bour-Jordan et al. 2011). CD22 and CD72 are co-inhibitory receptors on B cells. Their ligands are expressed on diverse cell types, but when activated simultaneously the B cell is inhibited. Similarly, CD200R is expressed on dendritic cells and macrophages, and CD200 is more widely expressed on epithelium, T cells, and B cells (Chen and Shi 2006; Singh et al. 2011; Okazaki and Honjo 2006). CD200 interaction with CD200R is thought to maintain balance in the lungs between protection and immunopathology (Minas and Liversidge 2006; Gorczynski 2005; Barclay et al. 2002).
TGF-β is a pro-fibrotic growth factor produced by activated monocytes, pulmonary macrophages, eosinophils, neutrophils, airway epithelial cells, smooth muscle cells, mast cells, lung fibroblasts, and T Treg cells (Holt and Strickland 2008). TGF-β is a strong stimulating factor for epithelial cell differentiation and fibroblast secretion of extracellular matrix proteins, contributing to the airway remodeling observed in chronic pulmonary allergic disease pathology. TGF-β can be pro-inflammatory when acting in concert with cytokines such as IL-1, IL-6, IL-23, or IL-4 by augmenting differentiation of TH17 and TH2 cells. TGF-β has pronounced anti-inflammatory activity by inhibiting IgE production, mast cell proliferation, T cell, B cell, NK cell, and cytotoxic lymphocyte function (Commins et al. 2010; Chung and Barnes 1999) or anti-inflammatory role by inhibiting IgE production, mast cell proliferation, T cell, B cell, NK cell, and cytotoxic lymphocyte functions (Chung and Barnes 1999). TGF-β is a primary cytokine responsible for the differentiation of CD4+ CD25+ T regulatory lymphocytes (Treg cells) that release the anti-inflammatory cytokine IL-10. Reduction in the numbers of Treg cells in hypersensitivity disorders may allow activation of APC and effector cells to promote inflammation. However, the role of Treg cells in pulmonary inflammation may be more complicated because some subsets of Treg cells, particularly the inducible iTreg subset exhibits a high level of plasticity, and can transform into IL-17 and IFN-γ producing effector cells to drive inflammation (Akdis and Akdis 2009). Thus, whether Treg cells act to promote or attenuate inflammation could depend on the local environment.
IL-10, originally named “cytokine synthesis inhibitor factor”, is produced by TH0, TH1, TH2, TReg, TC cells, mast cells, and activated monocytes, macrophages, and dendritic cells. IL-10 decreases TH1 and TH2 cytokine production, suppresses monocyte and macrophage activity, inhibits production of ROI and RNI, and down-regulates secretion of pro-inflammatory cytokines. However, IL-10 is pleiotropic, and while its dominant effect is suppression of adaptive immune responses, it also promotes growth of mast cells, T cells, and induces T cell growth in certain circumstances by induction of the IL-2 receptors (Vock et al. 2010).
Osteopontin (OPN) is a TH1 cytokine with emerging potential for affecting allergic responses. OPN appears to augment primary sensitization, but inhibits responses to secondary antigen exposure (Chung and Barnes 1999; Vock et al. 2010). The complex responses to OPN may be due to different domains on the protein, as ligation of different epitopes with antibodies leads to different effects (Xanthou et al. 2007).
Remodeling
Repeated exposure of the lungs to irritants and pathogens leads to a constellation of morphological and physiological changes manifested throughout the respiratory tract. The changes manifested in an individual may vary depending on the stimulus, exposure history, genetic and epigenetic background of the individual, and coincident pathogenic conditions or comorbidities.
Irritants and allergens often induce remodeling of the airway wall typically associated with asthma. Smooth muscle hypertrophy, subepithelial collagen deposition, and edema are observed throughout the airways. IL-4, -9, -13, -17, -23, and -25 induce growth of mucin secreting cells in the surface epithelium and submucosal glands (Konno et al. 2011). In mild asthma, the surface epithelium is mainly affected (Turner and Jones 2009; Zuhdi Alimam et al. 2000; Dabbagh et al. 1999), but with increased severity of the disease, submucosal glands enlarge (Ordonez et al. 2001). Small airways that are normally devoid of goblet cells are characterized by Clara cells that differentiate into a mucin-secreting cell phenotype (Dunnill et al. 1969). Corticosteroids are effective in inhibiting mucus hypersecretion in asthma, thereby revealing a major role of inflammatory mediators in mucus hypersecretion (Davis and Dickey 2008).
Deposition of collagens I, III, and V, fibronectin, tenascin, lumican, and biglycan are increased in asthma (Barnes 2002). Increased synthesis is paralleled by an altered balance in metalloproteinase (MMP)-9 and tissue inhibitor of metalloproteinase (TIMP) (Bergeron et al. 2009) resulting in altered ECM turnover and thickening of the lamina reticularis (Suzuki et al. 2001). Degradation of cartilage in the airway wall can also be observed in asthma (Al-Muhsen et al. 2011).
The number and diameter of blood vessels in the airway wall are increased in asthma (Haraguchi et al. 1999; Noble et al. 2002). Epithelial cells, eosinophils, lymphocytes, mast cells, basophils, macrophages, endothelial cells, and smooth muscle cells secrete and angiogenic factors that can drive increase in vascularity, including vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), tryptase, chymase, IL-8, TGF-β, TNF-α, NGF, and angiogenin.
Although not well documented in humans, neural remodeling is likely to occur in asthma. In rhesus macaques, postnatal exposure to house dust mite and ozone reduced neural projections to the epithelium and increased lateral projections to the basal lamina (Dunnill et al. 1969). One year after exposure, nerve density in the airway wall was increased, and projections to the epithelium were abnormal (Larsen et al. 2012; Larson et al. 2004). In rodents, allergic aeroallergens and irritants such as ozone stimulate release of neurotrophins, such as NGF, that promote neural growth within the airway mucosa (Kajekar et al. 2007). Ovalbumin sensitization induces TRPV1 expression in airway Aδ afferent fibers in guinea pigs, which could increase the diversity of stimuli that activate these fibers and lead to cough in allergic asthma (Joos et al. 2000; Ito et al. 2008; Hunter et al. 2011). Another indication of altered neural activity in allergic asthma is that inhibition of TRPA1 inhibits ovalbumin-induced hyperresponsiveness in rats (Lieu et al. 2012).
Summary
The function of the respiratory system is based on bulk flow of air in response to small pressure gradients developed by respiratory muscle contraction coupled with passive diffusion of gases between inspired air and circulating blood of the alveolar capillaries in response to differential gas pressures. Extensive mechanisms exist to protect the lungs against environmental insults, and many of these processes are adaptive in nature, resulting in memory that increases sensitivity and responsiveness to antigenic stimuli. Adaptations are observed locally and systemically and involve responses by epithelial, nerve, muscle, and immune cells. In over 10 % of the population, the responses to environmental stimuli become pathological and result in excessive sensitivity and aberrant responses to both specific and nonspecific stimuli, and culminate in a progressive and deleterious remodeling of the airways and lungs. Environmental diseases, such as extrinsic asthma, rhinitis, and allergy are increasing and represent major health issues around the world. A better understanding of the mechanisms underlying responses to environmental factors is required for more effective prevention and treatment of these disorders.
Contributor Information
William J. Meggs, Email: MEGGSW@ecu.edu
Michael R. Van Scott, Email: VANSCOTTMI@ecu.edu
References
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Agaugue S, Marcenaro E, Ferranti B, Moretta L, Moretta A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood. 2008;112(5):1776–83. doi: 10.1182/blood-2008-02-135871. [DOI] [PubMed] [Google Scholar]
- Akdis CA, Akdis M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J Allergy Clin Immunol. 2009;123(4):735–46. doi: 10.1016/j.jaci.2009.02.030. [DOI] [PubMed] [Google Scholar]
- Akuthota P, Xenakis JJ, Weller PF. Eosinophils: offenders or general bystanders in allergic airway disease and pulmonary immunity? J Innate Immun. 2011;3(2):113–9. doi: 10.1159/000323433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011;128(3):451–62. doi: 10.1016/j.jaci.2011.04.047. [DOI] [PubMed] [Google Scholar]
- Banner KH, Igney F, Poll C. TRP channels: emerging targets for respiratory disease. Pharmacol Ther. 2011;130(3):371–84. doi: 10.1016/j.pharmthera.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23(6):285–90. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- Barlow JL, McKenzie AN. Nuocytes: expanding the innate cell repertoire in type-2 immunity. J Leukoc Biol. 2011;90(5):867–74. doi: 10.1189/jlb.0311160. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Current and future therapies for airway mucus hypersecretion. Novartis Found Symp. 2002;248:237–49. [PubMed] [Google Scholar]
- Bates JH, Lutchen KR. The interface between measurement and modeling of peripheral lung mechanics. Respir Physiol Neurobiol. 2005;148(1–2):153–64. doi: 10.1016/j.resp.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Bathoorn E, Kerstjens H, Postma D, Timens W, MacNee W. Airways inflammation and treatment during acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2008;3(2):217–29. doi: 10.2147/copd.s1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumer W, Hoppmann J, Rundfeldt C, Kietzmann M. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets. 2007;6(1):17–26. doi: 10.2174/187152807780077318. [DOI] [PubMed] [Google Scholar]
- Bergeron C, Al-Ramli W, Hamid Q. Remodeling in asthma. Proc Am Thorac Soc. 2009;6(3):301–5. doi: 10.1513/pats.200808-089RM. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proc Natl Acad Sci USA. 1996;93(26):15195–202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorgo E, Moltu K, Tasken K. Phosphodiesterases as targets for modulating T-cell responses. Handb Exp Pharmacol. 2011;204(204):345–63. doi: 10.1007/978-3-642-17969-3_15. [DOI] [PubMed] [Google Scholar]
- Blanchfield JL, Mannie MD. A GMCSF-neuroantigen fusion protein is a potent tolerogen in experimental autoimmune encephalomyelitis (EAE) that is associated with efficient targeting of neuroantigen to APC. J Leukoc Biol. 2010;87(3):509–21. doi: 10.1189/jlb.0709520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, Schild H, Buhl R, Schmitt E, Taube C. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol. 2009;182(7):4017–24. doi: 10.4049/jimmunol.0803310. [DOI] [PubMed] [Google Scholar]
- Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM. Treatment of allergic asthma: modulation of Th2 cells and their responses. Respir Res. 2011;12:114. doi: 10.1186/1465-9921-12-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev. 2011;241(1):180–205. doi: 10.1111/j.1600-065X.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Swindle EJ, Kushnir-Sukhov NM, Holian A, Metcalfe DD. Silica-directed mast cell activation is enhanced by scavenger receptors. Am J Respir Cell Mol Biol. 2007;36(1):43–52. doi: 10.1165/rcmb.2006-0197OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunyavanich S, Boyce JA, Raby BA, Weiss ST. Gene-by-environment effect of house dust mite on purinergic receptor P2Y12 (P2RY12) and lung function in children with asthma. Clin Exp Allergy. 2012;42(2):229–37. doi: 10.1111/j.1365-2222.2011.03874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D’Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, Homer RJ, Cohn L, Huang X, Moran MM, Jordt SE. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci USA. 2009;106(22):9099–104. doi: 10.1073/pnas.0900591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EL, Louis NA, Tomassetti SE, Canny GO, Arita M, Serhan CN, Colgan SP. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 2007;21(12):3162–70. doi: 10.1096/fj.07-8473com. [DOI] [PubMed] [Google Scholar]
- Canning BJ. Functional implications of the multiple afferent pathways regulating cough. Pulm Pharmacol Ther. 2011;24(3):295–9. doi: 10.1016/j.pupt.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Canning BJ. Afferent nerves regulating the cough reflex: mechanisms and mediators of cough in disease. Otolaryngol Clin North Am. 2010;43(1):15–25. doi: 10.1016/j.otc.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning BJ. Reflex regulation of airway smooth muscle tone. J Appl Physiol. 2006;101(3):971–85. doi: 10.1152/japplphysiol.00313.2006. [DOI] [PubMed] [Google Scholar]
- Carati CJ, Gannon BJ. Lymphatic system. In: Anonymous, editor. Encyclopedia of respiratory medicine. New York: Elsevier; 2006. p. 643. [Google Scholar]
- Chen YQ, Shi HZ. CD28/CTLA-4–CD80/CD86 and ICOS–B7RP–1 costimulatory pathway in bronchial asthma. Allergy. 2006;61(1):15–26. doi: 10.1111/j.1398-9995.2006.01008.x. [DOI] [PubMed] [Google Scholar]
- Choi JY, Joo NS, Krouse ME, Wu JV, Robbins RC, Ianowski JP, Hanrahan JW, Wine JJ. Synergistic airway gland mucus secretion in response to vasoactive intestinal peptide and carbachol is lost in cystic fibrosis. J Clin Invest. 2007;117(10):3118–27. doi: 10.1172/JCI31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KF, Barnes PJ. Cytokines in asthma. Thorax. 1999;54(9):825–57. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleridge HM, Coleridge JC. Pulmonary reflexes: neural mechanisms of pulmonary defense. Annu Rev Physiol. 1994;56:69–91. doi: 10.1146/annurev.ph.56.030194.000441. [DOI] [PubMed] [Google Scholar]
- Collington SJ, Williams TJ, Weller CL. Mechanisms underlying the localisation of mast cells in tissues. Trends Immunol. 2011;32(10):478–85. doi: 10.1016/j.it.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Colsoul B, Nilius B, Vennekens R. On the putative role of transient receptor potential cation channels in asthma. Clin Exp Allergy. 2009;39(10):1456–66. doi: 10.1111/j.1365-2222.2009.03315.x. [DOI] [PubMed] [Google Scholar]
- Commins SP, Borish L, Steinke JW. Immunologic messenger molecules: cytokines, interferons, and chemokines. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S53–72. doi: 10.1016/j.jaci.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Cook ML, Bochner BS. Update on biological therapeutics for asthma. World Allergy Organiz J. 2010;3(6):188–94. doi: 10.1097/WOX.0b013e3181e5ec5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest. 2008;134(3):606–12. doi: 10.1378/chest.08-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle AJ, Erard F, Bertrand C, Walti S, Pircher H, Le Gros G. Virus-specific CD8+ cells can switch to interleukin 5 production and induce airway eosinophilia. J Exp Med. 1995;181(3):1229–33. doi: 10.1084/jem.181.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. 1999;162(10):6233–7. [PubMed] [Google Scholar]
- Davis CW, Dickey BF. Regulated airway goblet cell mucin secretion. Annu Rev Physiol. 2008;70:487–512. doi: 10.1146/annurev.physiol.70.113006.100638. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. VIP and PACAP inhibit activation induced apoptosis in T lymphocytes. Ann N Y Acad Sci. 2000;921:55–67. doi: 10.1111/j.1749-6632.2000.tb06951.x. [DOI] [PubMed] [Google Scholar]
- Delgado M, Leceta J, Sun W, Gomariz RP, Ganea D. VIP and PACAP induce shift to a Th2 response by upregulating B7.2 expression. Ann N Y Acad Sci. 2000;921:68–78. doi: 10.1111/j.1749-6632.2000.tb06952.x. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90(1):47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz G, Akdis M, Aktas E, Blaser K, Akdis CA. Human NK1 and NK2 subsets determined by purification of IFN-gamma-secreting and IFN-gamma-nonsecreting NK cells. Eur J Immunol. 2002;32(3):879–84. doi: 10.1002/1521-4141(200203)32:3<879::AID-IMMU879>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Duarte AG, Myers AC. Cough reflex in lung transplant recipients. Lung. 2012;190(1):23–7. doi: 10.1007/s00408-011-9352-x. [DOI] [PubMed] [Google Scholar]
- Dunnill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, in chronic bronchitis, and in emphysema. Thorax. 1969;24(2):176–9. doi: 10.1136/thx.24.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, White DP. Control of the pharyngeal musculature during wakefulness and sleep: implications in normal controls and sleep apnea. Head Neck. 2011;33(Suppl 1):S37–45. doi: 10.1002/hed.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faffe DS, Zin WA. Lung parenchymal mechanics in health and disease. Physiol Rev. 2009;89(3):759–75. doi: 10.1152/physrev.00019.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–47. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris BG, Jr, J M, LH O. Partitioning of respiratory flow resistance in man. J Appl Physiol. 1964;19:653–8. doi: 10.1152/jappl.1964.19.4.653. [DOI] [PubMed] [Google Scholar]
- Forbes LR, Haczku A. SP-D and regulation of the pulmonary innate immune system in allergic airway changes. Clin Exp Allergy. 2010;40(4):547–62. doi: 10.1111/j.1365-2222.2010.03483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c+CD8a–dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol. 2009;21(3):269–82. doi: 10.1093/intimm/dxn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzl EJ, Chan RC, Yadav M. Diverse mechanisms and consequences of immunoadoption of neuromediator systems. Ann N Y Acad Sci. 2008;1144:56–60. doi: 10.1196/annals.1418.008. [DOI] [PubMed] [Google Scholar]
- Gorczynski RM. CD200 and its receptors as targets for immunoregulation. Curr Opin Investig Drugs. 2005;6(5):483–8. [PubMed] [Google Scholar]
- Gray H. Anatomy of the human body. New York: Bartelby.com, Inc.; 2000. p. 1396. [Google Scholar]
- Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, Battaile JT, Alevy Y, Yan L, Agapov E, Kim EY, Holtzman MJ. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. 2007;204(11):2759–69. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groneberg DA, Hartmann P, Dinh QT, Fischer A. Expression and distribution of vasoactive intestinal polypeptide receptor VPAC(2) mRNA in human airways. Lab Invest. 2001;81(5):749–55. doi: 10.1038/labinvest.3780283. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Quarcoo D, Frossard N, Fischer A. Neurogenic mechanisms in bronchial inflammatory diseases. Allergy. 2004;59(11):1139–52. doi: 10.1111/j.1398-9995.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Rabe KF, Fischer A. Novel concepts of neuropeptide-based drug therapy: vasoactive intestinal polypeptide and its receptors. Eur J Pharmacol. 2006;533(1–3):182–94. doi: 10.1016/j.ejphar.2005.12.055. [DOI] [PubMed] [Google Scholar]
- Grubor B, Meyerholz DK, Ackermann MR. Collectins and cationic antimicrobial peptides of the respiratory epithelia. Vet Pathol. 2006;43(5):595–612. doi: 10.1354/vp.43-5-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12(1):53–9. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- Hamilton JA, Anderson GP. GM-CSF biology. Growth Factors. 2004;22(4):225–31. doi: 10.1080/08977190412331279881. [DOI] [PubMed] [Google Scholar]
- Hammad H, Lambrecht BN. Dendritic cells and airway epithelial cells at the interface between innate and adaptive immune responses. Allergy. 2011;66(5):579–87. doi: 10.1111/j.1398-9995.2010.02528.x. [DOI] [PubMed] [Google Scholar]
- Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M, Muskens F, Lambrecht BN. Inflammatory dendritic cells—not basophils—are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207(10):2097–111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Gonen-Gross T, Fitchett J, Rowe T, Daniels M, Arnon TI, Gazit R, Joseph A, Schjetne KW, Steinle A, Porgador A, Mevorach D, Goldman-Wohl D, Yagel S, LaBarre MJ, Buckner JH, Mandelboim O. Novel APC-like properties of human NK cells directly regulate T cell activation. J Clin Invest. 2004;114(11):1612–23. doi: 10.1172/JCI22787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Haraguchi M, Shimura S, Shirato K. Morphometric analysis of bronchial cartilage in chronic obstructive pulmonary disease and bronchial asthma. Am J Respir Crit Care Med. 1999;159(3):1005–13. doi: 10.1164/ajrccm.159.3.9712144. [DOI] [PubMed] [Google Scholar]
- Hardie RC, Minke B. Phosphoinositide-mediated phototransduction in Drosophila photoreceptors: the role of Ca2+ and trp. Cell Calcium. 1995;18(4):256–74. doi: 10.1016/0143-4160(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Haworth O, Cernadas M, Levy BD. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol. 2011;186(11):6129–35. doi: 10.4049/jimmunol.1004007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur Respir J. 2007;30(5):980–92. doi: 10.1183/09031936.00005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink IH, Van Oosterhout AJ. Strategies for targeting T-cells in allergic diseases and asthma. Pharmacol Ther. 2006;112(2):489–500. doi: 10.1016/j.pharmthera.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Helyes Z, Elekes K, Sandor K, Szitter I, Kereskai L, Pinter E, Kemeny A, Szolcsanyi J, McLaughlin L, Vasiliou S, Kipar A, Zimmer A, Hunt SP, Stewart JP, Quinn JP. Involvement of preprotachykinin A gene-encoded peptides and the neurokinin 1 receptor in endotoxin-induced murine airway inflammation. Neuropeptides. 2010;44(5):399–406. doi: 10.1016/j.npep.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Heyder J. Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proc Am Thorac Soc. 2004;1(4):315–20. doi: 10.1513/pats.200409-046TA. [DOI] [PubMed] [Google Scholar]
- Hlastala MP, Berger AJ. Physiology of respiration. New York: Oxford University Press; 2001. [Google Scholar]
- Holt PG, Strickland DH. The CD200-CD200R axis in local control of lung inflammation. Nat Immunol. 2008;9(9):1011–3. doi: 10.1038/ni0908-1011. [DOI] [PubMed] [Google Scholar]
- Holtzman MJ, Byers DE, Benoit LA, Battaile JT, You Y, Agapov E, Park C, Grayson MH, Kim EY, Patel AC. Immune pathways for translating viral infection into chronic airway disease. Adv Immunol. 2009;102:245–76. doi: 10.1016/S0065-2776(09)01205-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DD, Carrell-Jacks LA, Batchelor TP, Dey RD. Role of nerve growth factor in ozone-induced neural responses in early postnatal airway development. Am J Respir Cell Mol Biol. 2011;45(2):359–65. doi: 10.1165/rcmb.2010-0345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA. 2000;97(11):6155–60. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin CG, Bates JH. Physiologic dysfunction of the asthmatic lung: what's going on down there, anyway? Proc Am Thorac Soc. 2009;6(3):306–11. doi: 10.1513/pats.200808-091RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito W, Tanimoto M, Ono K, Mizuno S, Yoshida A, Koga H, Fuchimoto Y, Kondo N, Tanimoto Y, Kiura K, Matsumoto K, Kataoka M, Nakamura T, Gelfand EW, Kanehiro A. Growth factors temporally associate with airway responsiveness and inflammation in allergen-exposed mice. Int Arch Allergy Immunol. 2008;145(4):324–39. doi: 10.1159/000110891. [DOI] [PubMed] [Google Scholar]
- Iwamura C, Nakayama T. Role of NKT cells in allergic asthma. Curr Opin Immunol. 2010;22(6):807–13. doi: 10.1016/j.coi.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One. 2011;6(1):e16175. doi: 10.1371/journal.pone.0016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joos GF, Germonpre PR, Pauwels RA. Role of tachykinins in asthma. Allergy. 2000;55(4):321–37. doi: 10.1034/j.1398-9995.2000.00112.x. [DOI] [PubMed] [Google Scholar]
- Kajekar R, Pieczarka EM, Smiley-Jewell SM, Schelegle ES, Fanucchi MV, Plopper CG. Early postnatal exposure to allergen and ozone leads to hyperinnervation of the pulmonary epithelium. Respir Physiol Neurobiol. 2007;155(1):55–63. doi: 10.1016/j.resp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Karra L, Levi-Schaffer F. Down-regulation of mast cell responses through ITIM containing inhibitory receptors. Adv Exp Med Biol. 2011;716:143–59. doi: 10.1007/978-1-4419-9533-9_9. [DOI] [PubMed] [Google Scholar]
- Kelly JT, Prasad AK, Wexler AS. Detailed flow patterns in the nasal cavity. J Appl Physiol. 2000;89(1):323–37. doi: 10.1152/jappl.2000.89.1.323. [DOI] [PubMed] [Google Scholar]
- Kim DH, Park IH, Cho JS, Lee YM, Choi H, Lee HM. Alterations of vasoactive intestinal polypeptide receptors in allergic rhinitis. Am J Rhinol Allergy. 2011;25(1):e44–7. doi: 10.2500/ajra.2011.25.3568. [DOI] [PubMed] [Google Scholar]
- Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol. 2009;75(3):466–77. doi: 10.1124/mol.108.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistemaker LE, Oenema TA, Meurs H, Gosens R. Regulation of airway inflammation and remodeling by muscarinic receptors: Perspectives on anticholinergic therapy in asthma and COPD. Life Sci. 2012;91(21–22):1126–33. doi: 10.1016/j.lfs.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Konno S, Kurokawa M, Uede T, Nishimura M, Huang SK. Role of osteopontin, a multifunctional protein, in allergy and asthma. Clin Exp Allergy. 2011;41(10):1360–6. doi: 10.1111/j.1365-2222.2011.03775.x. [DOI] [PubMed] [Google Scholar]
- Kool M, van Nimwegen M, Willart MA, Muskens F, Boon L, Smit JJ, Coyle A, Clausen BE, Hoogsteden HC, Lambrecht BN, Hammad H. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol. 2009;183(2):1074–82. doi: 10.4049/jimmunol.0900471. [DOI] [PubMed] [Google Scholar]
- Korsgren M, Persson CG, Sundler F, Bjerke T, Hansson T, Chambers BJ, Hong S, Van Kaer L, Ljunggren HG, Korsgren O. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med. 1999;189(3):553–62. doi: 10.1084/jem.189.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186(7):4375–87. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraneveld AD, Nijkamp FP. Tachykinins and neuro-immune interactions in asthma. Int Immunopharmacol. 2001;1(9–10):1629–50. doi: 10.1016/s1567-5769(01)00099-6. [DOI] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. 2006;101(2):618–27. doi: 10.1152/japplphysiol.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31(3):412–24. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Larsen JM, Steen-Jensen DB, Laursen JM, Sondergaard JN, Musavian HS, Butt TM, Brix S. Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS One. 2012;7(2):e31976. doi: 10.1371/journal.pone.0031976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson SD, Schelegle ES, Walby WF, Gershwin LJ, Fanuccihi MV, Evans MJ, Joad JP, Tarkington BK, Hyde DM, Plopper CG. Postnatal remodeling of the neural components of the epithelial-mesenchymal trophic unit in the proximal airways of infant rhesus monkeys exposed to ozone and allergen. Toxicol Appl Pharmacol. 2004;194(3):211–20. doi: 10.1016/j.taap.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Lee LY, Pisarri TE. Afferent properties and reflex functions of bronchopulmonary C-fibers. Respir Physiol. 2001;125(1–2):47–65. doi: 10.1016/s0034-5687(00)00204-8. [DOI] [PubMed] [Google Scholar]
- Levick SP, Melendez GC, Plante E, McLarty JL, Brower GL, Janicki JS. Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res. 2011;89(1):12–9. doi: 10.1093/cvr/cvq272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S, Singh D, Evans CE. Cyclic hydrostatic pressure and cotton particles stimulate synthesis by human lung macrophages of cytokines in vitro. Respir Res. 2009;10:44. doi: 10.1186/1465-9921-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Westwick J, Poll C. Transient receptor potential (TRP) channels as potential drug targets in respiratory disease. Cell Calcium. 2003;33(5–6):551–8. doi: 10.1016/s0143-4160(03)00060-5. [DOI] [PubMed] [Google Scholar]
- Li X, Wang XM, Zhang JS. Relevance of vasoactive intestinal peptide and total bronchial mucin in rat lung. Sheng Li Xue Bao. 2009;61(6):539–43. [PubMed] [Google Scholar]
- Lieu TM, Myers AC, Meeker S, Undem BJ. TRPV1 induction in airway vagal low-threshold mechanosensory neurons by allergen challenge and neurotrophic factors. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L941–8. doi: 10.1152/ajplung.00366.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10(12):838–48. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J. 1994;7(9):1678–89. [PubMed] [Google Scholar]
- Matsubara S, Takeda K, Kodama T, Joetham A, Miyahara N, Koya T, Swasey CH, Okamoto M, Dakhama A, Gelfand EW. IL-2 and IL-18 attenuation of airway hyperresponsiveness requires STAT4, IFN-gamma, and natural killer cells. Am J Respir Cell Mol Biol. 2007;36(3):324–32. doi: 10.1165/rcmb.2006-0231OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCary C, Tancowny BP, Catalli A, Grammer LC, Harris KE, Schleimer RP, Kulka M. Substance P downregulates expression of the high affinity IgE receptor (FcepsilonRI) by human mast cells. J Neuroimmunol. 2010;220(1–2):17–24. doi: 10.1016/j.jneuroim.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca(2+) entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol. 2001;280(5):L870–80. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- Mentink-Kane MM, Wynn TA. Opposing roles for IL-13 and IL-13 receptor alpha 2 in health and disease. Immunol Rev. 2004;202:191–202. doi: 10.1111/j.0105-2896.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- Meyer EH, DeKruyff RH, Umetsu DT. T cells and NKT cells in the pathogenesis of asthma. Annu Rev Med. 2008;59:281–92. doi: 10.1146/annurev.med.59.061506.154139. [DOI] [PubMed] [Google Scholar]
- Meyrick B, Sturgess JM, Reid L. A reconstruction of the duct system and secretory tubules of the human bronchial submucosal gland. Thorax. 1969;24(6):729–36. doi: 10.1136/thx.24.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minas K, Liversidge J. Is the CD200/CD200 receptor interaction more than just a myeloid cell inhibitory signal? Crit Rev Immunol. 2006;26(3):213–30. doi: 10.1615/critrevimmunol.v26.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto D, Boschetto P, Bononi I, Zeni E, Cavallesco G, Fabbri LM, Mapp CE. Vasoactive intestinal peptide receptors in the airways of smokers with chronic bronchitis. Eur Respir J. 2004;24(6):958–63. doi: 10.1183/09031936.04.10031504. [DOI] [PubMed] [Google Scholar]
- Misaka S, Aoki Y, Karaki S, Kuwahara A, Mizumoto T, Onoue S, Yamada S. Inhalable powder formulation of a stabilized vasoactive intestinal peptide (VIP) derivative: anti-inflammatory effect in experimental asthmatic rats. Peptides. 2010;31(1):72–8. doi: 10.1016/j.peptides.2009.09.032. [DOI] [PubMed] [Google Scholar]
- Mishra A, Chintagari NR, Guo Y, Weng T, Su L, Liu L. Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J Cell Sci. 2011;124(Pt 4):657–68. doi: 10.1242/jcs.066977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitkiewicz M, Kuropatwa M, Kurowska E, Gorczyca WA. Different effects of soluble and particulate guanylyl cyclases on expression of inflammatory cytokines in rat peripheral blood mononuclear cells. Immunobiology. 2011;216(3):423–30. doi: 10.1016/j.imbio.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10(8):601–20. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Moretta L, Ferlazzo G, Bottino C, Vitale M, Pende D, Mingari MC, Moretta A. Effector and regulatory events during natural killer-dendritic cell interactions. Immunol Rev. 2006;214:219–28. doi: 10.1111/j.1600-065X.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- Mori T, Saito K, Ohki Y, Arakawa H, Tominaga M, Tokuyama K. Lack of transient receptor potential vanilloid-1 enhances Th2-biased immune response of the airways in mice receiving intranasal, but not intraperitoneal, sensitization. Int Arch Allergy Immunol. 2011;156(3):305–12. doi: 10.1159/000323889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortaz E, Folkerts G, Nijkamp FP, Henricks PA. ATP and the pathogenesis of COPD. Eur J Pharmacol. 2010;638(1–3):1–4. doi: 10.1016/j.ejphar.2010.04.019. [DOI] [PubMed] [Google Scholar]
- Muller T, Robaye B, Vieira RP, Ferrari D, Grimm M, Jakob T, Martin SF, Di Virgilio F, Boeynaems JM, Virchow JC, Idzko M. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy. 2010;65(12):1545–53. doi: 10.1111/j.1398-9995.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- Murdoch JR, Lloyd CM. Resolution of allergic airway inflammation and airway hyperreactivity is mediated by IL-17-producing {gamma}{delta}T cells. Am J Respir Crit Care Med. 2010;182(4):464–76. doi: 10.1164/rccm.200911-1775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182(1):1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Tippayawong N, Damrongsak D. Prediction of particle deposition in human respiratory system. Thammasat Int J Sci Technol. 2003;8(2):65–71. [Google Scholar]
- Naughton MT. Loop gain in apnea: gaining control or controlling the gain? Am J Respir Crit Care Med. 2010;181(2):103–5. doi: 10.1164/rccm.200909-1449ED. [DOI] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble PB, Turner DJ, Mitchell HW. Relationship of airway narrowing, compliance, and cartilage in isolated bronchial segments. J Appl Physiol. 2002;92(3):1119–24. doi: 10.1152/japplphysiol.00662.2001. [DOI] [PubMed] [Google Scholar]
- Nouri-Aria KT, Durham SR. Regulatory T cells and allergic disease. Inflamm Allergy Drug Targets. 2008;7(4):237–52. doi: 10.2174/187152808786848405. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Okunishi K, Peters-Golden M. Leukotrienes and airway inflammation. Biochim Biophys Acta. 2011;1810(11):1096–102. doi: 10.1016/j.bbagen.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue S, Aoki Y, Matsui T, Kojo Y, Misaka S, Mizumoto T, Yamada S. Formulation design and in vivo evaluation of dry powder inhalation system of new vasoactive intestinal peptide derivative ([R(15, 20, 21), L(17), A(24,25), des-N(28)]-VIP-GRR) in experimental asthma/COPD model rats. Int J Pharm. 2011;410(1–2):54–60. doi: 10.1016/j.ijpharm.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Onoue S, Misaka S, Aoki Y, Karaki S, Kuwahara A, Ohide A, Mizumoto T, Yamada S. Inhalable powder formulation of vasoactive intestinal peptide derivative, [R15,20,21, L17]-VIP-GRR, attenuated neutrophilic airway inflammation in cigarette smoke-exposed rats. Eur J Pharm Sci. 2010;41(3–4):508–14. doi: 10.1016/j.ejps.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Ordonez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y, Novikov A, Dolganov G, Fahy JV. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. 2001;163(2):517–23. doi: 10.1164/ajrccm.163.2.2004039. [DOI] [PubMed] [Google Scholar]
- Orgeig S, Hiemstra PS, Veldhuizen EJ, Casals C, Clark HW, Haczku A, Knudsen L, Possmayer F. Recent advances in alveolar biology: evolution and function of alveolar proteins. Respir Physiol Neurobiol. 2010;173(Suppl):S43–54. doi: 10.1016/j.resp.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn AG. The nasal arteries. AJR Am J Roentgenol. 1978;130(1):89–97. doi: 10.2214/ajr.130.1.89. [DOI] [PubMed] [Google Scholar]
- Parisinos CA. Sarcoidosis is a Th1/Th17 multisystem disorder: wider implications. Thorax. 2011;66(11):1011–2. doi: 10.1136/thoraxjnl-2011-200290. [DOI] [PubMed] [Google Scholar]
- Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L797–805. doi: 10.1152/ajplung.00513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parungo CP, Colson YL, Kim SW, Kim S, Cohn LH, Bawendi MG, Frangioni JV. Sentinel lymph node mapping of the pleural space. Chest. 2005;127(5):1799–804. doi: 10.1378/chest.127.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parungo CP, Ohnishi S, De Grand AM, Laurence RG, Soltesz EG, Colson YL, Kang PM, Mihaljevic T, Cohn LH, Frangioni JV. In vivo optical imaging of pleural space drainage to lymph nodes of prognostic significance. Ann Surg Oncol. 2004;11(12):1085–92. doi: 10.1245/ASO.2004.03.054. [DOI] [PubMed] [Google Scholar]
- Perez-Gil J, Weaver TE. Pulmonary surfactant pathophysiology: current models and open questions. Physiology (Bethesda) 2010;25(3):132–41. doi: 10.1152/physiol.00006.2010. [DOI] [PubMed] [Google Scholar]
- Perl M, Lomas-Neira J, Venet F, Chung CS, Ayala A. Pathogenesis of indirect (secondary) acute lung injury. Expert Rev Respir Med. 2011;5(1):115–26. doi: 10.1586/ers.10.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raemdonck K, de Alba J, Birrell MA, Grace M, Maher SA, Irvin CG, Fozard JR, O'Byrne PM, Belvisi MG. A role for sensory nerves in the late asthmatic response. Thorax. 2012;67(1):19–25. doi: 10.1136/thoraxjnl-2011-200365. [DOI] [PubMed] [Google Scholar]
- Ramalho R, Soares R, Couto N, Moreira A. Tachykinin receptors antagonism for asthma: a systematic review. BMC Pulm Med. 2011;11:41. doi: 10.1186/1471-2466-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P. Regulatory T cells in many flavors control asthma. Mucosal Immunol. 2010;3(3):216–29. doi: 10.1038/mi.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick MB, Weller PF. Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol. 1993;8(4):349–55. doi: 10.1165/ajrcmb/8.4.349. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Kimbell JS. The nasal valve dilemma: the narrow straw vs the weak wall. Arch Facial Plast Surg. 2012;14(1):9–10. doi: 10.1001/archfacial.2011.1450. [DOI] [PubMed] [Google Scholar]
- Ritz SA, Stampfli MR, Davies DE, Holgate ST, Jordana M. On the generation of allergic airway diseases: from GM-CSF to Kyoto. Trends Immunol. 2002;23(8):396–402. doi: 10.1016/s1471-4906(02)02278-0. [DOI] [PubMed] [Google Scholar]
- Robinson DS. The role of the T cell in asthma. J Allergy Clin Immunol. 2010;126(6):1081–91. doi: 10.1016/j.jaci.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol. 2001;125(1–2):129–44. doi: 10.1016/s0034-5687(00)00209-7. [DOI] [PubMed] [Google Scholar]
- Saha S, Brightling CE. Eosinophilic airway inflammation in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(1):39–47. doi: 10.2147/copd.2006.1.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salassa JR, Pearson BW, Payne WS. Gross and microscopical blood supply of the trachea. Ann Thorac Surg. 1977;24(2):100–7. doi: 10.1016/s0003-4975(10)63716-2. [DOI] [PubMed] [Google Scholar]
- Sarin S, Undem B, Sanico A, Togias A. The role of the nervous system in rhinitis. J Allergy Clin Immunol. 2006;118(5):999–1016. doi: 10.1016/j.jaci.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Saxon A, Kepley C, Zhang K. “Accentuate the negative, eliminate the positive”: engineering allergy therapeutics to block allergic reactivity through negative signaling. J Allergy Clin Immunol. 2008;121(2):320–5. doi: 10.1016/j.jaci.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120(6):1279–84. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sel S, Rost BR, Yildirim AO, Sel B, Kalwa H, Fehrenbach H, Renz H, Gudermann T, Dietrich A. Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin Exp Allergy. 2008;38(9):1548–58. doi: 10.1111/j.1365-2222.2008.03043.x. [DOI] [PubMed] [Google Scholar]
- Selman L, Skjodt K, Nielsen O, Floridon C, Holmskov U, Hansen S. Expression and tissue localization of collectin placenta 1 (CL-P1, SRCL) in human tissues. Mol Immunol. 2008;45(11):3278–88. doi: 10.1016/j.molimm.2008.02.018. [DOI] [PubMed] [Google Scholar]
- Shannahan JH, Kodavanti UP, Brown JM. Manufactured and airborne nanoparticle cardiopulmonary interactions: a review of mechanisms and the possible contribution of mast cells. Inhal Toxicol. 2012;24(5):320–39. doi: 10.3109/08958378.2012.668229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Stock P, Akbari O. Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy. 2011;66(2):155–62. doi: 10.1111/j.1398-9995.2010.02458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol. 1983;55(3):813–22. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10(5):328–43. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruri A, Zwirner J. Dendritic cells: limited potential in immunotherapy. Int J Biochem Cell Biol. 2005;37(2):241–5. doi: 10.1016/j.biocel.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Spencer LA, Szela CT, Perez SA, Kirchhoffer CL, Neves JS, Radke AL, Weller PF. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol. 2009;85(1):117–23. doi: 10.1189/jlb.0108058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita M, Kuribayashi K, Nakagomi T, Miyata S, Matsuyama T, Kitada O. Allergic bronchial asthma: airway inflammation and hyperresponsiveness. Intern Med. 2003;42(8):636–43. doi: 10.2169/internalmedicine.42.636. [DOI] [PubMed] [Google Scholar]
- Susarla SM, Thomas RJ, Abramson ZR, Kaban LB. Biomechanics of the upper airway: changing concepts in the pathogenesis of obstructive sleep apnea. Int J Oral Maxillofac Surg. 2010;39(12):1149–59. doi: 10.1016/j.ijom.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Kato T, Miyazaki Y, Iwata M, Noda Y, Takagi K, Nakashima N, Torii K. Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in sputum from patients with bronchial asthma. J Asthma. 2001;38(6):477–84. doi: 10.1081/jas-100105868. [DOI] [PubMed] [Google Scholar]
- Sweeney M, McDaniel SS, Platoshyn O, Zhang S, Yu Y, Lapp BR, Zhao Y, Thistlethwaite PA, Yuan JX. Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J Appl Physiol. 2002;92(4):1594–602. doi: 10.1152/japplphysiol.00722.2001. [DOI] [PubMed] [Google Scholar]
- Szema AM, Hamidi SA, Golightly MG, Rueb TP, Chen JJ. VIP Regulates the Development & Proliferation of Treg in vivo in spleen. Allergy Asthma Clin Immunol. 2011;7:19. doi: 10.1186/1710-1492-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher YA, Piavaux BJ, Gras R, van Esch BC, Hofman GA, Bloksma N, Henricks PA, van Oosterhout AJ. Indoleamine 2,3-dioxygenase-dependent tryptophan metabolites contribute to tolerance induction during allergen immunotherapy in a mouse model. J Allergy Clin Immunol. 2008;121(4):983–91.e2. doi: 10.1016/j.jaci.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Tannu SA, Renzetti LM, Tare N, Ventre JD, Lavelle D, Lin TA, Morschauser A, Paciorek J, Bolin DR, Michel H, Singer L, Hargaden M, Knowles I, Gardiner P, Cazzola M, Calzetta L, Matera MG, Hicks A. Dual bronchodilatory and pulmonary anti-inflammatory activity of RO5024118, a novel agonist at vasoactive intestinal peptide VPAC2 receptors. Br J Pharmacol. 2010;161(6):1329–42. doi: 10.1111/j.1476-5381.2010.00975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol. 2006;68:543–61. doi: 10.1146/annurev.physiol.68.072304.112754. [DOI] [PubMed] [Google Scholar]
- Thakur SA, Hamilton RF, Jr, Holian A. Role of scavenger receptor a family in lung inflammation from exposure to environmental particles. J Immunotoxicol. 2008;5(2):151–7. doi: 10.1080/15476910802085863. [DOI] [PubMed] [Google Scholar]
- Tran NP, Vickery J, Blaiss MS. Management of rhinitis: allergic and non-allergic. Allergy Asthma Immunol Res. 2011;3(3):148–56. doi: 10.4168/aair.2011.3.3.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trout L, Corboz MR, Ballard ST. Mechanism of substance P-induced liquid secretion across bronchial epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L639–45. doi: 10.1152/ajplung.2001.281.3.L639. [DOI] [PubMed] [Google Scholar]
- Tsoumakidou M, Demedts IK, Brusselle GG, Jeffery PK. Dendritic cells in chronic obstructive pulmonary disease: new players in an old game. Am J Respir Crit Care Med. 2008;177(11):1180–6. doi: 10.1164/rccm.200711-1727PP. [DOI] [PubMed] [Google Scholar]
- Turner J, Jones CE. Regulation of mucin expression in respiratory diseases. Biochem Soc Trans. 2009;37(Pt 4):877–81. doi: 10.1042/BST0370877. [DOI] [PubMed] [Google Scholar]
- Undem BJ, McAlexander M, Hunter DD. Neurobiology of the upper and lower airways. Allergy. 1999;54(Suppl 57):81–93. doi: 10.1111/j.1398-9995.1999.tb04409.x. [DOI] [PubMed] [Google Scholar]
- van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178(7):1279–93. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vock C, Hauber HP, Wegmann M. The other T helper cells in asthma pathogenesis. J Allergy (Cairo) 2010;2010:519298. doi: 10.1155/2010/519298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voice JK, Dorsam G, Chan RC, Grinninger C, Kong Y, Goetzl EJ. Immunoeffector and immunoregulatory activities of vasoactive intestinal peptide. Regul Pept. 2002;109(1–3):199–208. doi: 10.1016/s0167-0115(02)00182-9. [DOI] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Regulatory T cells, transforming growth factor-beta, and immune suppression. Proc Am Thorac Soc. 2007;4(3):271–6. doi: 10.1513/pats.200701-020AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ellison CA, Gartner JG, HayGlass KT. Natural killer cell depletion fails to influence initial CD4 T cell commitment in vivo in exogenous antigen-stimulated cytokine and antibody responses. J Immunol. 1998;160(3):1098–105. [PubMed] [Google Scholar]
- Weir N. Anatomy of the larynx and tracheobronchial tree. In: Kerr AG, editor. Scott-Brown’s otolaryngology. 6. Oxford: Butterworth-Heinemann; 1997. pp. 13–4. [Google Scholar]
- Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos Y, Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci USA. 2006;103(50):19093–8. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller CL, Collington SJ, Williams T, Lamb JR. Mast cells in health and disease. Clin Sci (Lond) 2011;120(11):473–84. doi: 10.1042/CS20100459. [DOI] [PubMed] [Google Scholar]
- Welte T, Groneberg DA. Asthma and COPD. Exp Toxicol Pathol. 2006;57(Suppl 2):35–40. doi: 10.1016/j.etp.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Wen LP, Fahrni JA, Matsui S, Rosen GD. Airway epithelial cells produce stem cell factor. Biochim Biophys Acta. 1996;1314(3):183–6. doi: 10.1016/s0167-4889(96)00138-3. [DOI] [PubMed] [Google Scholar]
- Widdicombe JG. Pulmonary and respiratory tract receptors. J Exp Biol. 1982;100:41–57. doi: 10.1242/jeb.100.1.41. [DOI] [PubMed] [Google Scholar]
- Widdicombe JG. Receptors in the trachea and bronchi of the cat. J Physiol. 1954;123(1):71–104. doi: 10.1113/jphysiol.1954.sp005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdicombe JG. Respiratory reflexes from the trachea and bronchi of the cat. J Physiol. 1954;123(1):55–70. doi: 10.1113/jphysiol.1954.sp005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdicombe JG. Reflexes from the upper respiratory tract. In: Anonymous, editor. Comprehensive physiology. New Jersey: John Wiley & Sons Inc.; 2011. [Google Scholar]
- Willart MA, Lambrecht BN. The danger within: endogenous danger signals, atopy and asthma. Clin Exp Allergy. 2009;39(1):12–9. doi: 10.1111/j.1365-2222.2008.03118.x. [DOI] [PubMed] [Google Scholar]
- Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180(8):720–30. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine JJ. Parasympathetic control of airway submucosal glands: central reflexes and the airway intrinsic nervous system. Auton Neurosci. 2007;133(1):35–54. doi: 10.1016/j.autneu.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthou G, Alissafi T, Semitekolou M, Simoes DC, Economidou E, Gaga M, Lambrecht BN, Lloyd CM, Panoutsakopoulou V. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat Med. 2007;13(5):570–8. doi: 10.1038/nm1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med. 2010;16(1–2):69–82. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Fan J, Fan J. Association of Toll-like receptor signaling and reactive oxygen species: a potential therapeutic target for posttrauma acute lung injury. Mediators Inflamm. 2010;2010:916425. doi: 10.1155/2010/916425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165(9):1245–50. doi: 10.1164/rccm.200110-022OC. [DOI] [PubMed] [Google Scholar]
- Xu J, Xu F. Role of neurogenic substance P in overexpression of alveolar macrophages’ neurokinin 1 receptor in mice exposed to cigarette smoke. Exp Lung Res. 2010;36(4):243–54. doi: 10.3109/01902140903398275. [DOI] [PubMed] [Google Scholar]
- Xu J, Xu F, Barrett E. Metalloelastase in lungs and alveolar macrophages is modulated by extracellular substance P in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L162–70. doi: 10.1152/ajplung.00282.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xu F, Lin Y. Cigarette smoke synergizes lipopolysaccharide-induced interleukin-1beta and tumor necrosis factor-alpha secretion from macrophages via substance P-mediated nuclear factor-kappaB activation. Am J Respir Cell Mol Biol. 2011;44(3):302–8. doi: 10.1165/rcmb.2009-0288OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Rosenbaum J, Goetzl EJ. Cutting edge: vasoactive intestinal peptide (VIP) induces differentiation of Th17 cells with a distinctive cytokine profile. J Immunol. 2008;180(5):2772–6. doi: 10.4049/jimmunol.180.5.2772. [DOI] [PubMed] [Google Scholar]
- Yatera K, Morimoto Y, Kim HN, Myojo T, Mukae H. Foam cell formation of alveolar macrophages in Clara cell ablated mice inhaling crystalline silica. Inhal Toxicol. 2011;23(12):736–44. doi: 10.3109/08958378.2011.608741. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Tanaka Y, Hirano M, Nakashima T. Sensory innervation of the pharynx and larynx. Am J Med. 2000;108(Suppl 4a):51S–61. doi: 10.1016/s0002-9343(99)00342-3. [DOI] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85(5):661–71. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Oh SY, Cho YS, Zhang L, Kim YK, Zheng T. Tyrosine phosphatase SHP-1 in allergic and anaphylactic inflammation. Immunol Res. 2010;47(1–3):3–13. doi: 10.1007/s12026-009-8134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhdi Alimam M, Piazza FM, Selby DM, Letwin N, Huang L, Rose MC. Muc-5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol. 2000;22(3):253–60. doi: 10.1165/ajrcmb.22.3.3768. [DOI] [PubMed] [Google Scholar]