

Abstract
Peptide binding to major histocompatibility complex (MHC) molecules is the most selective requisite for T-cell recognition. Therefore, prediction of peptide–MHC binding is the main basis for anticipating T-cell epitopes. A very popular and accurate method to predict peptide–MHC binding is based on motif-profiles and here we show how to make them using EPIMHC (http://imed.med.ucm.es/epimhc/). EPIMHC is a database of T-cell epitopes and MHC-binding peptides that unlike any related resource provides a framework for computational vaccinology. In this chapter, we describe how to derive peptide–MHC binding motif-profiles in EPIMHC and use them to predict peptide–MHC binding and T-cell epitopes. Moreover, we show evidence that customization of peptide–MHC binding predictors can lead to enhanced epitope predictions.
Key words: MHC, HLA, PSSM, T-cell epitope, Prediction
Introduction
T cells play a key role in fighting infectious agents such as pathogenic viruses, bacteria and parasites, as well as in cancer immune surveillance, eliminating tumoral cells. T cells respond to foreign peptide antigens (T-cell epitopes) bound the cell surface expressed major histocompatibility complex (MHC) molecules [1–4]. There are two main classes of MHC molecules, MHC class I (MHCI) and MHC class II (MHCII) that are in turn recognized by CD8+ T and CD4+ T cells, respectively [4]. In humans, MHC molecules are known as Human Leukocyte Antigens (HLAs) and are extremely polymorphic [5]. HLA polymorphisms are the basis for distinct peptide binding specificity of HLA allelic variants [5].
The relevance of T-cell epitopes for understanding disease pathology [6] and for epitope-based vaccines [7–10] has led to the identification of thousands of epitopes and MHC-peptide ligands from all kind of antigens. The availability of this vast amount of data has had two major intertwined consequences. On the one hand, it has given rise to comprehensive databases and resources to store the ever-increasing data. On the other hand, it has fueled the development of computational approaches for the prediction of T-cell epitopes.
Relevant examples of T-cell epitope and MHC-peptide ligand databases include SYFPEITHI [11], JenPep [12], and MHCBN [13], TEPIDAS [14], ImmuneEpitope Database [15], and EPIMHC [16]. These resources are instrumental for the analysis of peptide–MHC binding and T-cell epitope immunogenicity, primarily serving as source of data but also providing analytic and predictive tools. All these databases are based on relational databases and share a considerable amount of capabilities. Yet they also have unique features. Here we will work with EPIMHC [16], a highly curated database of T-cell epitopes and MHC-binding peptides that unlike any related resource enables tailored predictions of T-cell epitopes using custom-made peptide–MHC binding motif-profiles [17–19].
T-cell epitopes are determined by several molecular events [20–22], of which peptide–MHC binding is the most selective. Therefore, prediction of peptide–MHC binding is the main basis to anticipate T-cell epitopes [23]. Peptide–MHC binding predictions can be achieved through a great variety of methods [23], including peptide–MHC binding motif-profiles [17–19] which rank among the most successful and popular of them [24]. These motif-profiles consist of weighted position specific scoring matrices (PSSM) [25] created from sets of aligned peptide sequences known to bind to the relevant MHC molecules.
Prediction of T-cell epitopes using a large set of MHC-specific motif-profiles is readily available for free public use in at our RANKPEP site (http://imed.med.ucm.es/Tools/rankpep.html). We generated the peptide–MHC binding profiles available in RANKPEP from the largest non-redundant set of peptides that we could identify. In computational cross-validations, RANKPEP profiles exhibited a great performance [17]. However, they are not necessarily the best for all predictive matters. In fact there is no general consensus on what peptides should be included for peptide–MHC binding model building. Therefore, in this chapter, we illustrate how to use EPIMHC to derive custom-made peptide–MHC binding motif-profiles and produce tailored T-cell epitope predictions.
Materials
EPIMHC Database and Query Form
EPIMHC is a database with comprehensive information on MHC-restricted ligands and T-cell epitopes that are observed in real proteins from a great variety of sources including tumor antigens. Peptide data were collected from related databases [11, 26, 27] and the literature and was incorporated into the database upon computational curation (altered peptide ligands are not included). EPIMHC data is structured as a relational database with a set of related tables (Fig. 1). Entries in EPIMHC are unique with regard to the combination of two features: the sequence of the peptide and the MHC restriction element. Main annotations in EPIMHC include information on the ligand (sequence, length, MHC binding, T-cell activity, processing, protein source, protein name, and organisms), the MHC restriction element (CLASS, MHC molecule and MHC source), and publication reference. The “processing” field indicates whether the peptides are processed and presented from their protein sources in vivo (annotated as natural). “MHC binding” and “immunogenicity level” fields follow a qualitative annotation of four values (high, moderate, little, unknown). The immunogenicity level only applies to peptides with reported “T-cell activity.” Immunogenicity and MHC-binding binding levels were obtained from the literature and translated onto the indicated qualitative values as previously reported [26]. If no information on peptide–MHC binding and/or Immunogenicity level was found, then such fields were annotated as unknown.
Fig. 1.
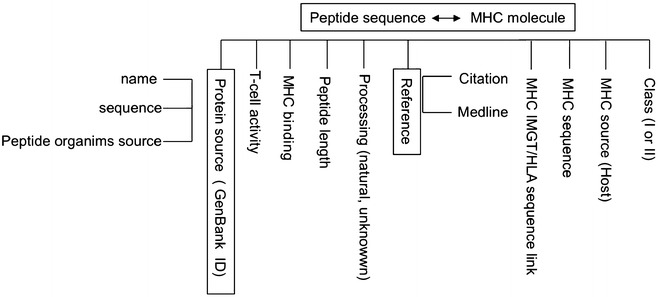
EPIMHC database structure
EPIMHC database is accessible online at http://imed.med.ucm.es/epimhc/ (Fig. 2) through an intuitive and user-friendly Web interface. This server allows for complex database queries, combining any annotation field, thanks to the underlying SQL language. For example, users can search for peptide ligands and/or epitopes that are restricted by one, various or all MHC molecules (left side of the screen), and restrict the search according to various criteria like length and source of the peptide (right of the screen). Also, any field of interest can be included in the search output. The EPIMHC search output will be described in detail in the Method section in the context of the generation of custom-made profiles.
Fig. 2.

EPIMHC database Web interface. EPIMHC resource is available for free public use at http://imed.med.ucm.es/epimhc/
Prediction of a Peptide–MHC Binding and T-Cell Epitopes Using Profiles
As mentioned, motif-profiles consist of weighted PSSM [25] created from a set of aligned peptide sequences known to bind to a given MHC molecule. In order to predict peptide–MHC binding and T-cell epitope using motif-profiles, we used a search algorithm known as RANKPEP [17, 18]. The algorithm uses the profile coefficients to score all possible peptide fragments in a protein with the width of the PSSM and ranks them by score. The width of a PSSM is given by the number of residue sites in a multiple sequence alignment. Although rank per se is insufficient to assess whether a peptide is a potential binder, we have shown that T-cell epitopes score among the top 2 % ranking peptides [17, 18]. Motif-profiles assume that peptide residues contribute independently to MHC binding. This assumption is well supported by experimental data, although there are reported instances in which the contribution of peptide residues to MHC-binding is influenced by neighboring residues [28].
RANKPEP is accessible online for public use at the site http://imed.med.ucm.es/Tools/rankpep.html (Fig. 3). Currently, 88 and 50 different MHCI and MHCII molecules, respectively, can be targeted for peptide binding predictions in RANKPEP using the relevant motif-profiles. The profiles available in RANKPEP have been derived from large sets of non-redundant peptide–MHC binders, without taking in consideration their T-cell activity and source. These sets can include self-peptides eluted from MHC molecules. The RANKPEP Web server is flexible, intuitive and combines several interesting features. Notably, users can upload their own motif-profiles, such as those generated using EPIMHC (see Subheading 3). A simplified version of the RANKPEP input form can also be launched from EPIMHC to facilitate tailored prediction of T-cell epitopes using custom-made profiles (see Subheading 3).
Fig. 3.

RANKPEP Web server. The figure depicts a screenshot of the RANKPEP interface with the option of uploading custom-made profiles highlighted. RANKPEP is available for free public use at http://imed.med.ucm.es/Tools/rankpep.html
Methods
In this section, we show a step-by-step guide to derive a specific peptide–MHC binding motif-profile in EPIMHC and produce tailored T-cell epitope predictions. In particular, we will target the prediction of A*0201-restricted CD8 T-cell epitopes from SARS coronavirus nucleoprotein (GI: 30173007). This protein contains 7 experimentally identified A*0201-restricted CD8 T-cell epitopes (Table 1) and we will use that knowledge to assess the predictive accuracy of various peptide–MHC binding motif-profiles.
Table 1.
Known A*0201-restricted CD8 T-cell epitopes in SARS nucleoprotein
| Epitope sequence | Location | References |
|---|---|---|
| ALNTPKDHI | 139–147 | [34] |
| LQLPQGTTL | 160–168 | [34] |
| LALLLLDRL | 220–228 | [34] |
| LLLDRLNQL | 223–231 | [34] |
| RLNQLESKV | 227–235 | [34] |
| GMSRIGMEV | 317–325 | [34] |
| ILLNKHIDA | 352–360 | [34] |
None of these epitopes has been used for profile building in EPIMHC
Peptide Selection and Motif-Profile Building
We will build a motif-profile from all 9-mer peptides that are annotated in EPIMHC to bind with high affinity to the human MHC I molecule HLA-A*0201 (A*0201) (see Notes 1–6). To this end, we first do a search in EPIMHC with the following selection criteria, leaving the remaining fields with default settings:
Select HLA-A*0201 in MHC SELECTION.
Select 9 in LENGTH (see Note 2).
Select high in PEPTIDE BINDING LEVEL.
In Fig. 4a we show a screen capture of the selection described. Upon submitting the search, we get 178 peptides (Fig. 4b). EPIMHC search results consist of a tabulated list, rows, of records fitting the search criteria. The table columns provide the information fields selected by the users in the query form. The default fields are those shown in Fig. 4b and include the MHC restriction element (MHC), the MHC class (I or II) (CLASS), the sequence of the peptide (SEQUENCE), the name of the source protein (PROTEIN SOURCE NAME), whether the peptide is an epitope (Yes or NO) (EPITOPE), T-cell activity level (EPITOPE LEVEL), the source organisms of the peptide (PEPTIDE SOURCE ORGANISMS), and length of the peptide (LENGTH). Clicking on the peptide sequence will show its location onto the relevant protein source. Also, one can retrieve the protein source record in NCBI by clicking on the relevant protein source names. Users can select any record by clicking on the record-checkbox or select all records by clicking on the option check all at the bottom of the result page and download the data in a variety of text formats from the relevant action buttons (FASTA Sequence, Table format, or Full Record).
Fig. 4.
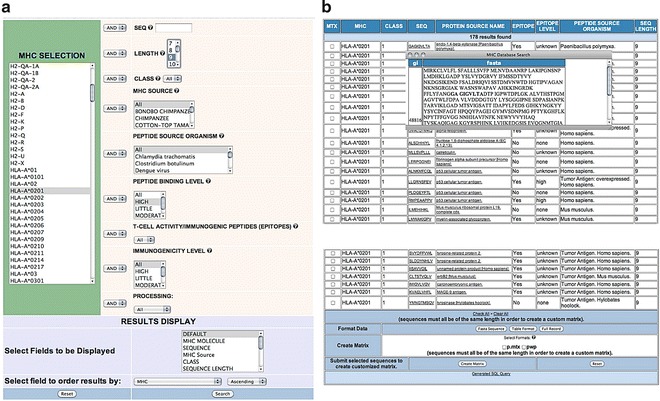
EPIMHC search example and output. The figure illustrates a search example in the EPIMHC resource for peptides binding to HLA-A*0201 with high affinity (a) and the result of that specific search (b)
Motif-profiles are built from peptide sequences selected in the output. Currently, EPIMHC can generate two types of profiles that are specified by clicking on either the p.mtx or the pwp checkboxes. The first one uses a branch proportional weighting method [29], while the second uses position-based weights [30]. To make a motif-profile incorporating position-based weights from all peptide records, we follow the next sequential steps (highlighted in Fig. 5a):
Click on check all (all peptides will be selected) (see Note 6).
Click the p.mtx check box.
Click the create matrix action button.
Fig. 5.
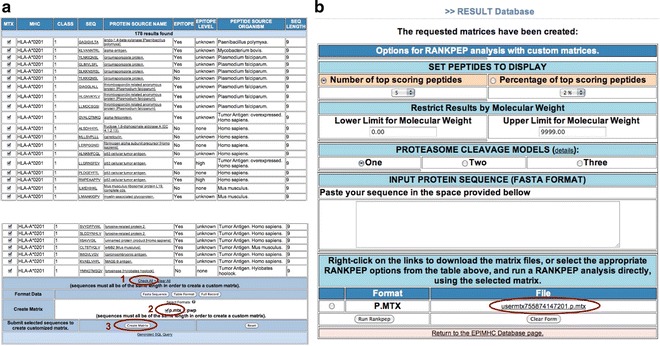
Profile-building using peptides selected in the EPIMHC output. (a) Figure highlights the steps that are needed (labeled 1, 2, and 3) to make a motif-profile with position-based weights from all peptides in the EPIMHC search output (b) RANKPEP form launched by EPIMHC with ready-to-use custom-made profile (highlighted)
Upon hitting the create matrix button, EPIMHC opens a simplified version of the RANKPEP Web interface that incorporates the custom-made motif-profile from the selected peptides (Fig. 5b). The profile appears under the File field of the form and can be downloaded with a mouse right click (PC) or by a mouse click-and-hold (MAC). The profile thus generated, shown in Fig. 6, has the format required by the MAST-motif search algorithm [31] and can be uploaded to the original RANKPEP Web server to produce custom predictions of peptide–MHC binding and T-cell epitopes. However, the RANKPEP interface launched by EPIMHC allows a more direct and simple way to achieve such a task (Fig. 5b). Under SET DISPLAY OPTIONS, users can select between two options to set the number of peptides to be returned by the algorithm: one is as a fixed number of top scoring peptides and the other as a percentage of top scoring peptides. Users can also restrict the peptides sorted by RANKPEP by molecular weight (MW) so that only peptides within a MW window will be returned. By default, MW filtering is not applied. The RANKPEP interface also provides three models for proteasomal cleavage predictions [17, 22]. By default, model one is selected. These models will be applied regardless of the class of the MHC targeted for predictions but the predictions are only meaningful for MHC I-restricted peptides (see Note 4).
Fig. 6.
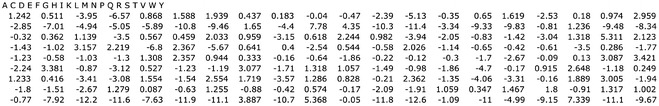
EPIMHC profile with position-based weights generated from 178 9-mer peptides binding to HLA-A*0201 with high affinity
Prediction of Peptide–MHC Binding and T-Cell Epitopes With EPIMHC Custom-Made Profiles
To target SARS nucleoprotein for T-cell epitope predictions using the RANKPEP form launched by EPIMHC with the custom-made profile we carry on as follows:
Set peptides to display to 2 % of top scoring peptides (see Note 5).
Paste the SARS nucleoprotein, FASTA format, in the text box INPUT section.
Click on the matrix check box.
Click on the action button Run Rankpep.
The indicated steps are highlighted in Fig. 7a and we describe next the RANKPEP output (Fig. 7b)
Fig. 7.
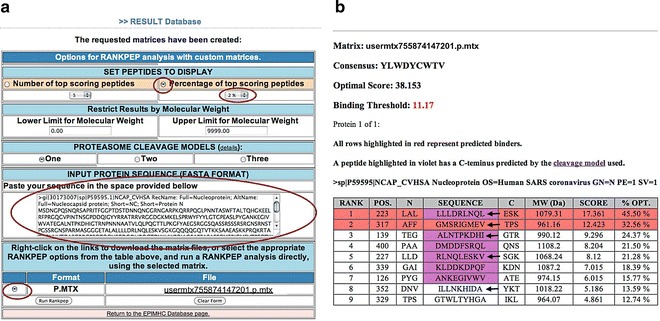
Tailored prediction of peptide–MHC binding using the RANKPEP form launched by EPIMHC (a) The figure illustrate the steps to predict HLA-A*0201-restricted peptides from SARS nucleoprotein. (b) RANKEP output showing the prediction results
The top part of the RANKPEP output shows the matrix (profile) used for the predictions, a consensus sequence that would reach the largest score, optimal (largest) score and a binding threshold (BT). The later is an important feature. Large scores lead to top ranking peptides and are indicative of peptide–MHC binding. However, rank per se is insufficient to know whether a given peptide will bind to a particular MHC molecule, e.g., scoring a single peptide. Therefore, EPIMHC provides a profile-specific BT that serves to identify the most confident peptide–MHC binders and T-cell epitopes as those with a score ≥ BT. The profile-specific BT provided by EPIMC is obtained by scoring all the peptides used to make the relevant profile matching the 90 percentile value of all peptide scores [18]. The next part in the RANKPEP output consists of a list of peptides from the input protein ranked by the scores obtained with the relevant profile. In our case, RANKPEP shows only 9 peptides from SARS nucleoprotein because we selected to display only the 2 % of top scoring peptides. For every peptide, RANKPEP shows its rank (RANK), location in the protein sequence (POS), sequence (SEQUENCE), three N-terminal (N) and C-terminal (C) flaking residues, score (SCORE), and relative score, in percentage, with regard to the optimum score (%OPT). Peptides whose scores are equal or greater than the BT score are highlighted in red, and those containing a C-terminal end predicted to be the result of proteasomal cleavage are shown in violet.
As we made a profile from peptides binding with high affinity to A*0201, we are predicting peptides from SARS nucleoprotein that bind to A*0201 and hence potential A*0201-restricted CD8 T-cell epitopes. In fact, in the results shown in Fig. 7b, it is possible to identify 5 out of the 7 known A*0201-restricted CD8 T-cell epitopes from SARS nucleoprotein (Table 2).
Table 2.
Description of custom-made profiles used in this study
| EPIMHC search selection | EPIMHC search result and profile building | ||||||
|---|---|---|---|---|---|---|---|
| Profile name | MHC molecule | PEP. binding level | PEP. source organism | T-cell activity | Length | Peptides | Method |
| Profile #1 | HLA-A*0201 | High | All | All | 9 | 178 | p.mtxb |
| Profile #2 | HLA-A*0201 | High | All | YES | 9 | 95 | p.mtxb |
| Profile #3 | HLA-A*0201 | High | Virusesa | All | 9 | 48 | p.mtxb |
| Profile #4 | HLA-A*0201 | High | Virusesa | YES | 9 | 32 | p.mtxb |
aThe viruses selected in EPIMHC were the following: Dengue virus, Epstein-Barr virus, Hepatitis B virus, Hepatitis C virus, Human immunodeficiency virus 1, Human immunodeficiency virus 1 OPT, Human immunodeficiency virus 2, Influenza A virus, Measles virus, and West Nile virus. None of the CD8 T-cell epitopes from SARS nucleoprotein are included in any of the peptide selections used for making these profiles
bPSSMs generated using position-based weights [30]
Comparison of CD8 T-Cell Epitope Predictions Using Various Custom-Made Profiles
The goodness of peptide–MHC binding and T-cell epitope predictions provided by any predictive model, including motif-profiles, is tied to the data used for model building [32, 33]. To demonstrate such influence, here we will compare the predictions of A*0201-restricted CD8 T-cell epitopes from SARS nucleoprotein that are obtained with 4 distinct motif-profiles, including that described in the previous section (hereafter profile #1). The specific peptide selections that give rise to the different profiles used in this section are detailed in Table 2. Briefly, all profile-motifs are generated from peptides binding with high affinity to A*0201. In addition, profile #3 and #4 only include peptides from viruses and profile #2 and #4 only include peptides known to display T-cell activity (they are epitopes). To evaluate the predictive performance of these profiles, we scored and ranked all peptides from SARS nucleoprotein and compared the ranking achieved by the known SARS nucleoprotein A*0201-restricted CD8 T-cell epitopes shown in Table 1. These results are summarized in Table 3. All four motif-profiles produce related results, ranking the known CD8 T-cell epitopes among the top scoring peptides of SARS nucleoprotein. This is expected as A*0201-restricted CD8 T-cell epitopes and the peptides used for model building have in common the ability to bind to A*0201. However, there are also differences in the results. Thus, only the profiles derived from viral peptides are capable of predicting the known A*0201-restricted CD8 T-cell epitopes from SARS nucleoprotein among the top 11 scoring peptides (Table 3). Judging from the dispersion of the ranks (Table 3), the best overall epitope predictions are obtained with a motif-profile derived from viral peptides with known T-cell activity (T-cell epitopes). It remains to be explored whether discarding peptide–MHC binders with no reported T-cell activity always improves the resulting T-cell epitope prediction models. It is important to note that none of the known epitopes used in these analyses have been used to derive any of the profiles. In fact, the current version of EPIMHC does not contain any SARS peptides at all.
Table 3.
Ranking and statistics of known SARS nucleoprotein A*0201-restricted CD8 T-cell epitopes using four custom-made motif-profiles
| Epitope | Profile #1 | Profile # 2 | Profile #3 | Profile # 4 |
|---|---|---|---|---|
| Alntpkdhi | 3 | 3 | 4 | 4 |
| GMSRIGMEV | 2 | 1 | 1 | 3 |
| Illnkhida | 8 | 7 | 5 | 5 |
| Lalllldrl | 34 | 11 | 30 | 11 |
| LLLDRLNQL | 1 | 2 | 9 | 2 |
| LQLPQGTTL | 10 | 13 | 7 | 10 |
| RLNQLESKV | 5 | 4 | 3 | 1 |
| Rank | Statistics | |||
| Median | 5 | 4 | 5 | 4 |
| Mean | 9 | 5.8 | 5.4 | 5.1 |
| Stdev | 11.3 | 4.5 | 9.8 | 3.9 |
| Range | 1–34 | 1–13 | 1–30 | 1–11 |
Peptide ranks are obtained after scoring all peptides in SARS nucleoprotein with the relevant motif-profiles and depict the peptide score relative to that of all remaining peptides. Rank 1 means that the peptide has the largest score of all peptides
In conclusion, profiles are very powerful at capturing nontrivial motifs and the results shown here support that epitope predictions can be improved using customized peptide–MHC binding profiles. EPIMHC is the only available resource readily suitable for that task.
Notes
In EPIMHC, users can make profiles from any peptide selection but the peptides must be related to some extend (e.g., binding to the same MHC) to produce profiles yielding meaningful predictions (see Subheading 3.1).
EPIMHC is better suited for making MHC I-specific profiles. Moreover, profiles can only be generated from peptides with the same length; otherwise EPIMHC returns an error (see Subheading 3.1). There are practical and structure-based reasons for this limitation as discussed by Reche et al. [18].
EPIMHC can also produce profiles from peptides that have been selected to bind to MHC II molecules, provided that they have the same length (see Subheading 3.1). However, as data availability is limited, we recommend using a motif discovery program such as MEME [31] for making peptide–MHC II binding profiles from peptides of any length as described in previous reports [17, 18].
The proteasomal cleavage predictions should not be taken in consideration when predicting peptide binding to MHC II molecules: the proteasome is not involved in class II antigen processing. We are working in correcting this inconsistency.
For RANKPEP to return all peptides in a given protein sorted by score, users need to make the following selections in the RANKPEP input form: first set peptides to display by number and then select 990 from the pull-down menu.
To generate profiles that are capable of capturing the relevant peptide–MHC binding feature, we suggest using a minimum of five peptides.
Acknowledgements
This work was supported by grant SAF2009-08103 from Ministerio de Ciencia e Innovación to PAR.
Contributor Information
Rajat K. De, Phone: +91-33-25753105, Email: rajat@isical.ac.in
Namrata Tomar, Phone: +91-33-25753105, Email: namrata.tomar@gmail.com.
Pedro Reche, Email: parecheg@med.ucm.es.
References
- 1.Zinkernagel M, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248:701–702. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 2.Margulies DH. Interactions of TCRs with MHC-peptide complexes: a quantitative basis for mechanistic models. Curr Opin Immunol. 1997;9:390–395. doi: 10.1016/S0952-7915(97)80086-6. [DOI] [PubMed] [Google Scholar]
- 3.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- 4.Wang J-H, Reinherz E. Structural basis of T cell recognition of peptides bound to MHC molecules. Mol Immunol. 2001;38:1039–1049. doi: 10.1016/S0161-5890(02)00033-0. [DOI] [PubMed] [Google Scholar]
- 5.Reche PA, Reinherz EL. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol. 2003;331:623–641. doi: 10.1016/S0022-2836(03)00750-2. [DOI] [PubMed] [Google Scholar]
- 6.Tchernev G, Orfanos CE. Antigen mimicry, epitope spreading and the pathogenesis of pemphigus. Tissue Antigens. 2006;68:280–286. doi: 10.1111/j.1399-0039.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- 7.Reche PA, Keskin DB, Hussey RE, Ancuta P, Gabuzda D, Reinherz EL. Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med Immunol. 2006;5:1. doi: 10.1186/1476-9433-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akdis CA, Akdis M, Blesken T, Wymann D, Alkan SS, Muller U, Blaser K. Epitope-specific T cell tolerance to phospholipase A2 in bee venom immunotherapy and recovery by IL-2 and IL-15 in vitro. J Clin Invest. 1996;98:1676–1683. doi: 10.1172/JCI118963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stienekemeier M, Falk K, Rotzschke O, Weishaupt A, Schneider C, Toyka KV, Gold R, Strominger JL. Vaccination, prevention, and treatment of experimental autoimmune neuritis (EAN) by an oligomerized T cell epitope. Proc Natl Acad Sci U S A. 2001;98:13872–13877. doi: 10.1073/pnas.241504598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazoura E, Apostolopoulos V. Insights into peptide-based vaccine design for cancer immunotherapy. Curr Med Chem. 2005;12:1481–1494. doi: 10.2174/0929867054039017. [DOI] [PubMed] [Google Scholar]
- 11.Rammensee HG, Bachmann J, Emmerich NPN, Bacho OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 12.Blythe MJ, Doytchinova IA, Flower D. JenPep: a database of quantitative functional peptide data for immunology. Bioinformatics. 2002;18:434–439. doi: 10.1093/bioinformatics/18.3.434. [DOI] [PubMed] [Google Scholar]
- 13.Bhasin M, Singh H, Raghava GP. MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics. 2003;19:665–666. doi: 10.1093/bioinformatics/btg055. [DOI] [PubMed] [Google Scholar]
- 14.Diez-Rivero CM, Garcia-Boronat M, Reche PA. Integrating T-cell epitope annotations with sequence and structural information using DAS. Bioinformation. 2008;3:156–158. doi: 10.6026/97320630003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, et al. Immune epitope database analysis resource (IEDB-AR) Nucleic Acids Res. 2008;36:W513–W518. doi: 10.1093/nar/gkn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reche PA, Zhang H, Glutting JP, Reinherz EL. EPIMHC: a curated database of MHC-binding peptides for customized computational vaccinology. Bioinformatics. 2005;21:2140–2141. doi: 10.1093/bioinformatics/bti269. [DOI] [PubMed] [Google Scholar]
- 17.Reche PA, Glutting J-P, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56:405–419. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 18.Reche PA, Glutting JP, Reinherz EL. Prediction of MHC class I binding peptides using profile motifs. Hum Immunol. 2002;63:701–709. doi: 10.1016/S0198-8859(02)00432-9. [DOI] [PubMed] [Google Scholar]
- 19.Reche PA, Reinherz EL. Prediction of peptide-MHC binding using profiles. Methods Mol Biol. 2007;409:185–200. doi: 10.1007/978-1-60327-118-9_13. [DOI] [PubMed] [Google Scholar]
- 20.Jensen PE. Recent advances in antigen processing and presentation. Nat Immunol. 2007;8:1041–1048. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- 21.Diez-Rivero CM, Chenlo B, Zuluaga P, Reche PA. Quantitative modeling of peptide binding to TAP using support vector machine. Proteins. 2010;78:63–72. doi: 10.1002/prot.22535. [DOI] [PubMed] [Google Scholar]
- 22.Diez-Rivero CM, Lafuente EM, Reche PA. Computational analysis and modeling of cleavage by the immunoproteasome and the constitutive proteasome. BMC Bioinformatics. 2010;11:479. doi: 10.1186/1471-2105-11-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lafuente EM, Reche PA. Prediction of MHC-peptide binding: a systematic and comprehensive overview. Curr Pharm Des. 2009;15:3209–3220. doi: 10.2174/138161209789105162. [DOI] [PubMed] [Google Scholar]
- 24.Gowthaman U, Agrewala JN. In silico tools for predicting peptides binding to HLA-class II molecules: more confusion than conclusion. J Proteome Res. 2008;7:154–163. doi: 10.1021/pr070527b. [DOI] [PubMed] [Google Scholar]
- 25.Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci U S A. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brusic V, Rudy G, Harrison LC. MHCPEP: a database of MHC-binding peptides. Nucleic Acids Res. 1994;22:3663–3665. doi: 10.1093/nar/22.17.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korber BTM, Moor JP, Brander C, Walker BD, Haynes BF, Koup R. HIV molecular immunology compendium. Los Alamos, NM: Los Alamos National Laboratory; 1998. [Google Scholar]
- 28.Peters B, Tong W, Sidney J, Sette A, Weng Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics. 2003;19:1765–1772. doi: 10.1093/bioinformatics/btg247. [DOI] [PubMed] [Google Scholar]
- 29.Thompson JD, Higgins DG, Gibson TJ. Improved sensitivity of profile searches through the use of sequence weights and gap excision. Comput Appl Biosci. 1994;10:19–29. doi: 10.1093/bioinformatics/10.1.19. [DOI] [PubMed] [Google Scholar]
- 30.Henikoff S, Henikoff JG. Position-based sequence weights. J Mol Biol. 1994;243:574–578. doi: 10.1016/0022-2836(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 31.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Naves E, Lafuente EM, Reche PA. Recognition of the ligand-type specificity of classical and non-classical MHC I proteins. FEBS Lett. 2011;585:3478–3484. doi: 10.1016/j.febslet.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Martínez-Naves E, Lafuente EM, Reche PA (2011) In: Liò P, Nicosia G, Stibor T (eds.), 10th International conference on artificial immune systems. Springer-Verlag Cambridge, England, Vol. 6825, 55–65
- 34.Cheung YK, Cheng SC, Sin FW, Chan KT, Xie Y. Induction of T-cell response by a DNA vaccine encoding a novel HLA-A*0201 severe acute respiratory syndrome coronavirus epitope. Vaccine. 2007;25:6070–6077. doi: 10.1016/j.vaccine.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]