

Abstract
Central nervous system (CNS) infections—i.e., infections involving the brain (cerebrum and cerebellum), spinal cord, optic nerves, and their covering membranes—are medical emergencies that are associated with substantial morbidity, mortality, or long-term sequelae that may have catastrophic implications for the quality of life of affected individuals. Acute CNS infections that warrant neurointensive care (ICU) admission fall broadly into three categories—meningitis, encephalitis, and abscesses—and generally result from blood-borne spread of the respective microorganisms. Other causes of CNS infections include head trauma resulting in fractures at the base of the skull or the cribriform plate that can lead to an opening between the CNS and the sinuses, mastoid, the middle ear, or the nasopharynx. Extrinsic contamination of the CNS can occur intraoperatively during neurosurgical procedures. Also, implanted medical devices or adjunct hardware (e.g., shunts, ventriculostomies, or external drainage tubes) and congenital malformations (e.g., spina bifida or sinus tracts) can become colonized and serve as sources or foci of infection. Viruses, such as rabies, herpes simplex virus, or polioviruses, can spread to the CNS via intraneural pathways resulting in encephalitis. If infection occurs at sites (e.g., middle ear or mastoid) contiguous with the CNS, infection may spread directly into the CNS causing brain abscesses; alternatively, the organism may reach the CNS indirectly via venous drainage or the sheaths of cranial and spinal nerves. Abscesses also may become localized in the subdural or epidural spaces. Meningitis results if bacteria spread directly from an abscess to the subarachnoid space. CNS abscesses may be a result of pyogenic meningitis or from septic emboli associated with endocarditis, lung abscess, or other serious purulent infections. Breaches of the blood–brain barrier (BBB) can result in CNS infections. Causes of such breaches include damage (e.g., microhemorrhage or necrosis of surrounding tissue) to the BBB; mechanical obstruction of microvessels by parasitized red blood cells, leukocytes, or platelets; overproduction of cytokines that degrade tight junction proteins; or microbe-specific interactions with the BBB that facilitate transcellular passage of the microorganism. The microorganisms that cause CNS infections include a wide range of bacteria, mycobacteria, yeasts, fungi, viruses, spirochaetes (e.g., neurosyphilis), and parasites (e.g., cerebral malaria and strongyloidiasis). The clinical picture of the various infections can be nonspecific or characterized by distinct, recognizable clinical syndromes. At some juncture, individuals with severe acute CNS infections require critical care management that warrants neuro-ICU admission. The implications for CNS infections are serious and complex and include the increased human and material resources necessary to manage very sick patients, the difficulties in triaging patients with vague or mild symptoms, and ascertaining the precise cause and degree of CNS involvement at the time of admission to the neuro-ICU. This chapter addresses a wide range of severe CNS infections that are better managed in the neuro-ICU. Topics covered include the medical epidemiology of the respective CNS infection; discussions of the relevant neuroanatomy and blood supply (essential for understanding the pathogenesis of CNS infections) and pathophysiology; symptoms and signs; diagnostic procedures, including essential neuroimaging studies; therapeutic options, including empirical therapy where indicated; and the perennial issue of the utility and effectiveness of steroid therapy for certain CNS infections. Finally, therapeutic options and alternatives are discussed, including the choices of antimicrobial agents best able to cross the BBB, supportive therapy, and prognosis.
Keywords: Acute bacterial meningitis, Amoebic meningoencephalitis, Aseptic meningitis, Aspergillus infections of the CNS, Bartonella (cat-scratch disease) CNS infection, Blastomycosis of the CNS, Brain abscess, Candida CNS infections, Cat-scratch fever, Cerebral malaria, Cerebritis, CNS complications of strongyloidiasis, CNS infections caused by rapidly growing mycobacteria, CNS complications of Rocky Mountain spotted fever, CNS mucormycosis, CNS mycoses, CNS zygomycosis, Coccidioidal meningitis, Cryptococcus meningitis, Cytomegalovirus encephalitis, Dengue, Echinococcus involvement of the CNS, External ventricular drainage infections, Fungal CNS infections, Fungal meningitis, Histoplasma CNS infections, HIV encephalopathy, Ehrlichiosis of the CNS, Lyme disease, Mycobacterium tuberculosis infections of the CNS, Neurocysticercosis, Neurosyphilis, Parasitic infections of the CNS, Progressive multifocal leukoencephalopathy, Pyogenic bacterial abscesses of the CNS, Rhinocerebral mucormycosis, Rickettsial diseases of the CNS, Spinal epidural abscess, Spinal tuberculosis, Steroids use in CNS infections, Therapy of CNS infections, Vertebral osteomyelitis, Viral meningitis, Whipple’s disease of the CNS
Introduction
Acute infections of the central nervous system (CNS) are medical emergencies that if not addressed promptly result in significant mortality or long-term sequelae that have catastrophic implications for the quality of life of affected individuals. To fully understand the pathogenesis, clinical implications, and management of CNS infections, some knowledge of applied neuroanatomy is essential.
The CNS is defined by the brain (cerebrum and cerebellum), spinal cord, optic nerves, and their covering membranes. These structures are protected within the rigid confines of the skull and spinal canal of the vertebral column. The cerebral cortex (the outermost, gray tissue layer of the cerebrum) and the spinal cord are covered by three layers of continuous protective tissue called the meninges. The innermost meningeal layer that directly overlies the cerebral cortex is called the pia mater. The middle and outermost layers are known as the arachnoid and dura mater, respectively. The dura mater forms several intracranial compartments, including sinuses for venous drainage. Parts of the arachnoid—the arachnoid villi—project into these sinuses. The subpial space is continuous with the Virchow-Robin spaces. These two spaces transmit penetrating vessels to and from the brain parenchyma and do not connect with the subarachnoid space. The subarachnoid space lies between the pia mater and the arachnoid and is continuous in nature; the subdural space lies between the arachnoid and the dura mater. The epidural space lies between the dura and the skull. Certain infections can access the subpial and Virchow-Robin spaces, while most do not. Infections within the epidural spaces are usually caused by direct extension from adjacent infections and the infection remains in close proximity to the inciting source. Subdural infections are often associated with an extracerebral source, but these infections can spread widely within the subdural compartment well away from the inciting source. It is not uncommon to develop serous subdural effusions in bacterial meningitis. Subarachnoid infections are most often caused by hematogenous dissemination of organisms or viruses.
Cerebrospinal fluid (CSF) is continuously produced by the choroid plexuses within the four ventricles of the brain. CSF fills the lateral and third ventricles and circulates to the fourth ventricle, which lies between the cerebellum and the midbrain. CSF flows from the fourth ventricle into the subarachnoid space, where it bathes the entire CNS, and drains via the arachnoid villi into the superior sagittal sinus in the dura mater, where it is resorbed into the bloodstream.
A working knowledge of the blood supply is also essential for understanding the pathogenesis of CNS infections. The capillary supply to the brain and spinal cord is unique—the outermost layers of endothelial cells are fused together. These specialized brain microvascular endothelial cells constitute the blood–brain barrier, which separates the brain and the meninges from the circulating blood and impedes the influx of microorganisms, toxic agents, and most other compounds, while regulating the flow of essential nutrients and molecules for normal neural function. Thus, pathogens that breach the blood–brain barrier can cause CNS infections. Causes of such breaches include damage (e.g., microhemorrhage or necrosis of surrounding tissue) to the barrier; mechanical obstruction of microvessels by parasitized red blood cells, leukocytes, or platelets; overproduction of cytokines that degrade tight junction proteins; or microbe-specific interactions with the blood–brain barrier that facilitate transcellular passage of the microorganism (e.g., Escherichia coli, mycobacteria, and spirochaetes). The therapeutic implications are obvious—to be effective, antimicrobials prescribed for CNS infections must be able to cross the blood–brain barrier.
Routes of Infection
Acute CNS infections fall broadly into three categories—meningitis, encephalitis, and abscesses—and generally result from blood-borne spread of the respective microorganisms. Bacteremia or viremia can result from infection at sites adjacent or contiguous to the CNS, such as the mastoid, sinuses, or middle ear, or from primary infections at more remote anatomic sites (e.g., lungs, heart, skin, gastrointestinal tract, or kidney). In children the most common predisposing conditions are sinus or middle ear infection, which lead to transient bacteremia and hematogenous seeding of the CNS [1–3]. Bacterial infections of the paranasal and otomastoid sinuses often produce phlebothrombosis of adjacent draining cortical (cerebral) veins. This thrombotic process can extend into regional dural sinuses. The phlebothrombosis becomes thrombophlebitis offering a direct route of transmission from the infected sinus to the adjacent extra axial spaces or to the brain along cortical venous drainage pathways. Recognition of the venous involvement is essential, since the venous obstruction can produce intra axial brain swelling which may obscure the extra axial infection source contributing to misleading interpretations on brain and spinal imaging studies. The common relationship between paranasal sinus and otomastoid causes of intracranial infection reinforces the need for the clinician and radiologic imager to be well versed in both head and neck and paraspinous anatomy.
In patients with bacteremia or viremia, the organism, upon entering the venous sinuses, may cross the blood–brain barrier, penetrate the dura and arachnoid, and gain access to the subarachnoid space, thereby causing infection of the CSF and further dissemination of the infection throughout this anatomic space. Fractures at the base of the skull or the cribriform plate can lead to an opening between the CNS and the sinuses, mastoid, the middle ear, or the nasopharynx. Since these sites are all contiguous with the upper respiratory tract, CSF leaks occurring at any of these sites may enable respiratory flora to track back up into the subarachnoid space. Extrinsic contamination of the CNS can occur intraoperatively during neurosurgical procedures and, further, implanted medical devices or adjunct hardware (e.g., shunts, ventriculostomies, or external drainage tubes) can become colonized and serve as foci of infection. Congenital malformations, such as spina bifida or sinus tracts, can become colonized and serve as sources of infection. Viruses, such as rabies, herpes simplex virus (HSV), or polioviruses, can spread to the CNS via intraneural pathways resulting in encephalitis.
If infection occurs at sites (e.g., middle ear or mastoid) contiguous with the CNS, infection may spread directly into the CNS causing brain abscesses; alternatively, the organism may reach the CNS indirectly via venous drainage or the sheaths of cranial and spinal nerves. Abscesses also may become localized in the subdural or epidural spaces. Meningitis results if bacteria spread directly from an abscess to the subarachnoid space. CNS abscesses may be a result of pyogenic meningitis or from septic emboli associated with endocarditis, lung abscess, or other serious purulent infections, such as those caused by the Streptococcus milleri group.
Acute Bacterial Meningitis
Bacterial meningitis, a serious brain infection, can develop rapidly into a life-threatening infection even in previously healthy children or adults. Bacteria that cause meningitis enter the bloodstream, are carried toward the brain, and somehow manage to cross the defensive line of the blood–brain barrier outlined previously.
Definition
Bacterial meningitis can be defined as an inflammatory response to pyogenic bacterial invasion of the pia mater, the arachnoid membranes, and surrounding the CNS. This infection typically involves the entire length of the neuraxis including the brain (cerebrum, cerebellum), spinal cord, optic nerves, and their covering membranes because of the continuous nature of the subarachnoid space. Pyogenic meningitis is associated with a marked, acute inflammatory exudate; non-pyogenic microorganisms (e.g., mycobacteria or spirochetes like Leptospira spp.) are less commonly implicated. Clinically, onset is acute with development of headache, fever, irritability, and stiff neck with or without focal neurological signs over hours to a few days. Although the overall annual incidence of pyogenic bacterial meningitis in the United States is decreasing, the outcome is invariably fatal if left untreated [1, 4].
Epidemiology
The common causes of bacterial meningitis in the United States include Streptococcus pneumoniae, Neisseria meningitidis, group B Streptococcus spp., Listeria monocytogenes, and Haemophilus influenzae. Collectively, these agents account for over 95 % of cases (Table 22.1). The age distribution, predisposing conditions, and fatality rates for the most common agents are summarized in Fig. 22.1 and Table 22.1.
Table 22.1.
Causes of bacterial meningitis and overall case fatality rate according to organism
| Organism | % of total cases | Incidence | Predisposing conditions | Case fatality rate (%) |
|---|---|---|---|---|
| Streptococcus pneumoniae | 47 | 1.1 | Otitis media; sinusitis; alcoholism; cirrhosis; pneumococcal pneumonia; immunosuppression; skull fracture; CSF leak; myeloma; sickle cell disease | 21 |
| Neisseria meningitidis | 25 | 0.6 | “Closed” institutional setting; lack of specific antibody; complement deficiencies | 3 |
| Group B Streptococcus | 12 | 0.3 | Neonatal period; colonized mothers; preterm labor; prolonged rupture of membranes; intrapartum fever | 7 |
| Listeria monocytogenes | 8 | 0.2 | Neonatal period; immunosuppression; age; alcoholism/cirrhosis | 15 |
| Haemophilus influenzae | 7 | 0.2 | Lack of antibody to polysaccharide capsule; preceding otitis media | 6 |
Adapted with permission of the Massachusetts Medical Society from Schuchat et al. [1]
Fig. 22.1.
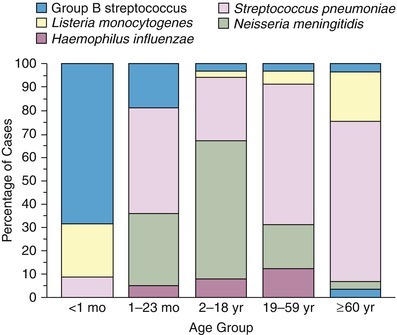
Pathogenic agents of bacterial meningitis according to age group (Used with permission of the Massachusetts Medical Society from Schuchat et al. [1])
Streptococcus pneumoniae
Of the three predominant organisms (S. pneumoniae, N. meningitidis, and H. influenzae) most often implicated in community-associated meningitis in the United States, S. pneumoniae is the most common and affects all age groups, except infants in the immediate neonatal period. The risk of pneumococcal meningitis varies with age but is significantly higher in infants than in young children and adults. Over the age of 70 years, the incidence rises again to approximately double the average incidence for young and middle-aged adults. In adults the major predisposing factors for infection include alcoholism, splenectomy, human immunodeficiency virus (HIV) infection, other acquired immunodeficiency conditions, diabetes, other underlying chronic conditions (e.g., chronic renal disease), and prior viral respiratory infection. Pneumococcal meningitis is the most common form of recurrent meningitis in patients who have CSF leaks. S. pneumoniae is spread by respiratory transmission in the general population and results in colonization of the nasopharynx with rates commonly in the range of 5–10 % of healthy adults. During the wintertime, carriage rates can rise to 20–30 % in certain populations depending on age and other factors. Overcrowding in day-care centers, military barracks, and prisons is often implicated in the transmission.
N. meningitidis
This is an aerobic, gram-negative diplococcus that colonizes the mucosal surface of the nasopharynx. The main mode of transmission is via direct contact with large droplet respiratory secretions from patients or asymptomatic carriers; humans are the only host. Invasive disease caused by this organism occurs in three clinical forms: meningitis (50 % of cases), blood infection (30 %), and pneumonia (10 %); other forms account for the remainder (10 %) of the cases. N. meningitidis has now become the leading cause of bacterial meningitis in the United States with an estimated annual incidence of approximately 0.5–1.5 cases per 100,000 population and at least tenfold higher in less-developed countries [1]. Persons at risk include household contacts of infected patients, military recruits, college freshmen who live in dormitories, microbiologists who work with isolates of N. meningitidis, persons traveling to a country where meningococcal disease is epidemic or highly endemic, and patients without spleens or with terminal complement component deficiencies. Infants less than 1 year of age and adolescents ages 16–21 years have higher rates of disease than other age groups, although infection can occur in all age groups including the elderly.
Strains of N. meningitidis are characterized according to the serologic recognition of polysaccharide epitopes on their capsule and outer membrane and are classified into serogroups A, B, C, W135, and Y. In the United States, strains from serogroups B, C, and Y cause the majority (45 %) of infections, whereas in less-developed countries serogroups A and C predominate; serogroup A has also been implicated in epidemics in sub-Saharan Africa.
H. influenzae
This is a small, pleomorphic, aerobic or facultative anaerobic gram-negative coccobacilli that is classified according to six serologically distinct antigenic types based on capsular polysaccharides a–f. Of these, only H. influenzae type b (Hib) is pathogenic. Although the nonencapsulated form of H. influenzae is a common inhabitant of the upper respiratory tract of healthy humans and causes localized infection (e.g., conjunctivitis or otitis media in children) without bacteremia, the more virulent encapsulated Hib serotype causes more invasive disease and is an important cause of meningitis or epiglottitis. Hib meningitis is relatively uncommon during the first 2 months of life probably because of the presence of passively transferred maternal antibodies. But by the fourth month of life, children lose these antibodies and become at risk of Hib meningitis. The occurrence of H. influenzae meningitis is directly associated with the presence and development of type-specific anticapsular antibodies to polyribosylribitol phosphate. Whether vaccine-induced or occurring naturally, presence of these antibodies is directly related to protection from invasive H. influenzae infection [5]. Various clinical studies have shown these antibodies to be opsonic and bactericidal against H. influenzae in vitro and protective in vivo. In the pre-vaccine era, colonization with nontypable strains of H. influenzae led to the development of cross-reacting antibodies that were largely protective against infection caused by type b strains. Before the introduction of the Hib conjugate polysaccharide capsular vaccine in 1986, H. influenzae was the most common cause of acute bacterial meningitis in children under the age of 5, with as many as 1 in 200 children acquiring invasive H. influenzae infection, including epiglottitis, septicemia, arthritis, and soft tissue infections in addition to meningitis. However, by 1995, the prevalence of meningitis caused by Hib had fallen 55 % [6]. Risk factors for Hib meningitis include malignancy, chronic renal disease, sickle cell disease, immunoglobulin dyscrasias, HIV infections, and cystic fibrosis.
Listeria monocytogenes
This is a facultative anaerobic, intracellular gram-positive bacillus that remains a significant cause of neonatal meningitis in the USA. Although the mode of transmission to humans is generally fecal-oral, transmission of the pathogen to the neonate generally occurs at the time of birth in mothers who had asymptomatic colonization of the genital or gastrointestinal tract prior to delivery. Domestic and wild animals are the main reservoirs for L. monocytogenes, but as the prevalence of stool carriage of this organism among asymptomatic adults is about 1 %, humans remain a small but still significant reservoir.
Generally, healthy adults exposed to the organism do not become ill, unless exposed to high numbers of infectious organisms, such as during an outbreak. L. monocytogenes causes meningitis most often in adults with depressed cell-mediated immunity, patients on steroids or other immunosuppressive agents, HIV-infected individuals, transplant recipients, and other vulnerable populations, such as persons with diabetes, alcohol abuse, chronic liver disease, renal disease requiring hemodialysis, or persons greater than 60 years of age. Outbreaks of L. monocytogenes meningitis in the United States and elsewhere have been associated with unpasteurized milk products, such as Swiss or feta cheese, undercooked chicken or hot dogs, seafood, vegetables, or coleslaw.
Other Microorganisms
Gram-negative microorganisms are important causes of acute bacterial meningitis. Persons with diabetes, a history of alcohol abuse, hepatic cirrhosis, or chronic urinary tract infections are particularly susceptible. E. coli with the K1 capsular polysaccharide antigen accounts for a majority of the cases of gram-negative meningitis in the newborn [7]. Carriage rates of the E. coli K1 serotypes vary in different populations but range from 7 to 38 % in women of childbearing age and may be as high as 50 % in nursing personnel [8–10]. In a study of 231 children presenting with bacterial meningitis to the emergency department during the era of widespread heptavalent conjugate pneumococcal vaccination, E. coli was implicated in 4 % and other gram-negative bacilli in 3.0 % [11, 12]. Other gram-negative organisms such as Klebsiella spp., Enterobacter spp., Pseudomonas spp., Citrobacter spp., and Salmonella spp. may also cause meningitis in the neonatal period, with an epidemiology similar to that of the E. coli K1 strains. Gram-negative bacillary meningitis still carries a worse prognosis than meningitis with a gram-positive organism [13]. Beyond the neonatal period, the vast majority of cases of gram-negative meningitis occur in the inpatient setting, especially following neurosurgery (e.g., craniotomy) and during placement of devices, such as ventriculostomy tubes, spinal surgery, or in patients who have suffered head trauma [14].
Group B Streptococcus (Streptococcus agalactiae) is the single most frequent cause of neonatal meningitis. This organism has been cultured from vaginal secretions in 30–40 % of women prior to delivery. During pregnancy, labor, and delivery, the microorganism can be transmitted to amniotic fluid or the newborn may become colonized as it passes through the birth canal. Transmission to the infant during delivery can result in neonatal meningitis within the first week of life. Alternatively, the organism may be acquired within the first few days after birth from adult contacts—relatives or hospital personnel—and meningitis may develop during the first 1–2 months after birth even though the infant might have appeared healthy at the time of delivery. While the probability of transmission from mothers colonized with S. agalactiae to neonates delivered vaginally is approximately 50 %, only 2 % of colonized neonates go on to develop invasive group B streptococcal disease. Group B streptococci produce polysaccharide capsules that manifest nine antigenic serotypes (types Ia–VIII). The type III group is responsible for the vast majority of neonatal meningitis; virulence factors (e.g., those causing production of higher levels of neuraminidase) have been described as an explanation for this.
Almost any microorganism that crosses the blood–brain barrier can cause acute meningitis. Other bacterial agents include group A streptococci, non-pneumococcal alpha hemolytic streptococci, Neisseria gonorrhea, Salmonella species, Flavobacterium meningosepticum, non-influenzae Haemophilus species, and even Bacillus anthracis (anthrax). Although S. aureus and Staphylococcus epidermidis are rarely implicated as causes of primary bacterial meningitis, these organisms are relatively common causes of bacterial meningitis following trauma, in situ CSF shunts, or neurosurgical procedures. Other microorganisms, such as Mycobacterium spp., Nocardia spp., yeasts and fungi (e.g., Coccidioides immitis, Histoplasma capsulatum, or Cryptococcus neoformans), Treponema spp., Brucella spp., Leptospira spp., or Toxoplasma gondii, can also produce meningitis. However, with the exception of Leptospira spp., these microorganisms tend to produce a more chronic form of meningitis and would not be considered agents of acute bacterial meningitis in the first instance [15]. For example, Mycobacterium spp., C. immitis, H. capsulatum, or C. neoformans would more likely produce chronic granulomatous inflammatory changes rather than acute pyogenic infections.
Pathogenesis of Meningeal Invasion
Colonization of the respiratory tract or nasopharynx is the critical first step preceding infection caused by the three microorganisms (S. pneumoniae, N. meningitides, and H. influenzae) most commonly associated with community-acquired meningitis. Biologically, colonization is mediated by affinity of these organisms for the nasopharyngeal mucosa. Colonization is facilitated by attachment and adherence of the microorganisms to cell surface receptors on nasopharyngeal epithelial cells, enabling them to replicate in the upper airway for prolonged periods. All three pathogens, typically, may colonize the upper airway without producing symptoms. Both host susceptibility and pathogen-specific factors (e.g., virulence and pathogenicity) are critical in the development of invasive disease. Many of these factors, though identified and characterized, are still not fully understood. For example, splenectomy definitely predisposes the affected person to invasive disease by S. pneumoniae, while it appears to have relatively little effect on the occurrence of invasive N. meningitidis disease, despite the fact that both are encapsulated organisms.
Adherence to nasopharyngeal mucosa is mediated by fimbriae or pili, in the case of gram-negative organisms. The pili of N. meningitidis are filamentous glycoproteins attached to the bacterial surface, traverse the polysaccharide capsule, and extend beyond the surface of the bacterium, where they can bind to specific receptors on nasopharyngeal cells, in this instance the CD4+ receptor [16, 17]. After receptor binding, further interaction with the host cell is established by certain outer membrane proteins on N. meningitidis, designated Opa and Opc [18]. Binding of the outer membrane proteins to specific receptors promotes engulfment of the N. meningitidis by the epithelial cells followed by transportation of the bacteria across the cell in membrane-bound vacuoles to the intravascular space; organisms also gain access to the intravascular space by creating separations in the tight junctions of columnar epithelial cells. N. meningitidis also possess other outer membrane proteins that function as IgA proteases, which can specifically degrade the surface IgA antibodies on epithelial cells, further enhancing the probability of invasive disease [19]. Once the mucosal barrier has been breached, the development of meningococcal disease is dependent upon the survival of the organism in the bloodstream.
Here, the most important virulence factor for survival of meningococci is the polysaccharide capsule that protects the organism against complement-mediated phagocytosis by neutrophils in the reticular endothelial system [20, 21]. Host defense is clearly determined by the existing humoral antibody to specific polysaccharide capsular types and the cellular responses of the innate immune system.
Protective IgG antibody for meningococcal disease is acquired through maternal transmission and is protective for the first few months after birth. Colonization by nonpathogenic Neisseria species and a possibly cross-reacting gram-negative organism such as E. coli K1 induces protective antibodies. Antibodies protect by promoting optimization of phagocytosis through opsonization and specific lysis via complement activation. For this reason, patients who are deficient in complement factor C5 are particularly susceptible to repeated invasive infections by N. meningitidis. In fact, individuals with an inherited deficiency of any of the terminal components of complement C5, C6, C7, and C8 have a greater risk of invasive disease [22].
Virulence factors for the invasion of S. pneumoniae seem to be primarily a function of the capsular polysaccharide type. There are at least 93 known capsular serotypes of S. pneumoniae with the various serotypes having different propensities for producing disease or developing antibiotic resistance [23, 24].
Colonization of the upper airway by H. influenzae is also mediated by fimbrial attachment to epithelial cells. Alpha fimbriae enhance the binding to the anterior nasopharynx and β fimbriae facilitate binding to the posterior ciliated nasopharyngeal cells [25]. Although H. influenzae type b strains that lack fimbriae generally are unable to colonize the nasopharynx, isolates from CSF do not express fimbria, suggesting that while the presence of fimbriae on H. influenzae is important for colonization of and attachment to nasopharyngeal mucosa, it does not appear play a significant role in the pathogenesis of meningitis [26, 27].
N. meningitis, S. pneumoniae, H. influenzae type b and other pathogens are capable of invading the CNS and infecting the meninges due to the incorporation of virulence factors [28]. The chain of events that ultimately lead to invasion of the subarachnoid space by these pathogens includes a cascade of events involving nasopharyngeal or middle ear colonization, bloodstream dissemination of the respective pathogen, crossing of the blood–brain and blood-CSF barriers, and finally entrance and survival of the implicated pathogen into the subarachnoid space and subsequent infection [28]. Bacteria migrate through the brain microvascular endothelial cells in enclosed vacuoles via a mechanism that is dependent on F-actin. Thus, transport through the cell appears to be dependent on cytoskeletal rearrangement involving both microfilaments and microtubules. Because of the blood–brain barrier, immunoglobulin and complement protein levels and leukocytes are significantly lower in CSF than in serum and interstitial fluid. Thus, in the early phase of infection involving the subarachnoid space, bacterial replication proceeds virtually unchecked by host defense mechanisms. Although the major host response to the invasion of the subarachnoid space by pathogenic microorganisms is a rapid influx of polymorphonuclear leukocytes, opsonization of bacteria and subsequent phagocytosis by neutrophils are hindered by the relative paucity of complement and immunoglobulins and the intrinsic fluid nature of CSF which is less facilitating to phagocytosis as compared to solid tissues. In addition, leukocyte proteases derived from the initial influx of leukocytes degrade whatever complement components are present in the CSF [29–31].
Lipopolysaccharide (LPS) molecules from gram-negative bacteria are known to be extremely potent in the development of inflammation, and intracisternal injection of purified LPS from H. influenzae also elicits a strong inflammatory response [31, 32]. The mechanism by which LPS and other bacterial cell wall components (e.g., teichoic acid and peptidoglycans from S. pneumoniae) act to stimulate inflammation is probably through the induction of inflammatory cytokines such as Interleukin 1 (IL-1) or tumor necrosis factor (TNF) [29, 32]. In vitro studies with LPS and with IL-1 and TNF show that incubation with endothelial cell monolayers leads to a rapid, transient increase in the expression of the intercellular adhesion molecules (ICAM-1 and ICAM-2) as well as the selectin molecules such as ELAM-1. As a result, neutrophils are able to bind to CNS vascular endothelial cells at vastly increased rates and then subsequently migrate by diapedesis into the subarachnoid space. At the same time, adherence of neutrophils to the capillary endothelium increases the permeability of the blood vessels enabling more protein leakage into the CSF and subarachnoid space, adding to the inflammatory exudate. Figure 22.2a, b shows the dense inflammatory infiltrate with neutrophils, lymphocytes, and macrophages seen in a patient with purulent meningitis caused by S. pneumoniae. IL-1 and TNF, in turn, stimulate leukocytes and other categories of inflammatory cells to produce and secrete a host of other proinflammatory cytokines, proteolytic enzymes, free radicals, and nitric oxide. The end result is edema of the surrounding tissues, cell injury, and tissue necrosis. Infiltration of the walls of small arteries and cortical veins also leads to a vasculitis with intimal thickening, narrowing and occlusion of small arteries, thrombophlebitis of the cortical veins, and thrombosis of the major venous sinuses, leading to ischemia and infarction of brain tissues.
Fig. 22.2.
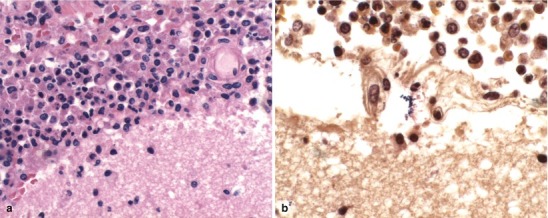
(a) Acute purulent meningitis caused by the Streptococcus pneumoniae. Leptomeninges expanded by a dense necroinflammatory infiltrate with neutrophils, lymphocytes, and macrophages (H&E 40×). (b) Gram stain of exudate specimens showing gram-positive diplococci (Both courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Inflammation of the pia mater and arachnoid affects glucose transport into the CSF resulting in a net lowering of CSF glucose levels. The pathophysiologic consequences of this intense neutrophil response in the subarachnoid space, tissue edema, and vasculitis account for most, if not all, of the serious clinical and pathologic consequences of meningitis, such as the increased permeability of the blood–brain barrier, increased intracranial pressure (ICP), hydrocephalus, and reduced cerebral blood flow, leading to cerebral hypoxia and death [32].
The ICP is raised via several mechanisms. First, vasogenic cerebral edema is caused by the increased permeability of the blood–brain barrier, which is a direct result of inflammatory bacterial products or the inflammatory cytokines released in response to these products. Second, the alterations in brain cellular membranes lead to cytotoxic cerebral edema resulting from increased intracellular water content, potassium leakage, and a shift in brain metabolism to anaerobic glycolysis with increased lactate production. And third, as a result of the inflammation in the subarachnoid space, there is decreased ability to reabsorb CSF, which leads to interstitial edema in brain parenchyma. All three mechanisms contribute to increased ICP pressure, which in turn may precipitate transtentorial brain herniation.
Clinical Manifestations
In the typical clinical presentation of meningitis in adults, fever, headache, and stiff neck predominate although there might be varying degrees of altered consciousness. Though vomiting is a common symptom generally attributed to raised ICP, it is also a well-recognized manifestation of the effects of the inflammatory process on the midbrain. The nature of the presentation of meningitis depends on the underlying microorganism responsible for the infection. For example, in pneumococcal meningitis (S. pneumoniae is the most common etiologic agent implicated in adult bacterial meningitis cases), the patient might have had pneumonia and bloodstream dissemination before progression to meningitis. Thus, the patient might have presented with chills and rigors or symptoms of upper respiratory tract infection, bronchitis, or pneumonia several days before the actual onset of meningitis symptoms. In the classic series by Carpenter and Petersdorf, approximately 27 % of patients had a sudden onset of headache, confusion, lethargy, and alteration of consciousness within the first 24 h before admission to hospital [33]. In contrast, 53 % presented with a more slowly progressive course over 1–7 days.
In a review of 493 episodes of meningitis, Durand and colleagues found that 95 % of the patients with acquired meningitis had a temperature greater than 37.7 °C on admission, while neck stiffness was present in 88 %. Only 22 % were alert, 51 % were confused or lethargic, and 22 % were responsive only to pain. Within the first 24–48 h of onset, 29 % had suffered focal seizures or had exhibited focal neurologic signs [34]. The most common predisposing factors for acute meningitis included pneumonia, sinusitis, otitis media, alcoholism, diabetes, or immunosuppression associated with conditions, such as malignancy, connective tissue diseases, sickle cell disease, diabetes, organ transplantation, splenectomy, dialysis, or steroids and other immunosuppressive therapy. For patients with some of these underlying conditions, the clinical presentation of meningitis may not be classic because of alteration of the immune response, with the diagnosis only being made upon further investigation of altered sensorium, persistent headache, or new onset seizures or neurological symptoms or signs. The presentation of patients with meningococcal meningitis may be similar to those with pneumococcal meningitis; clinically, one might not be able to distinguish between the two. However, meningococcal meningitis is part of the spectrum of N. meningitidis sepsis and the manifestations of meningococcal septicemia may precede the meningitis by 12–24 h. Signs indicative of underlying sepsis may dominate the clinical presentation. The initial presentation in meningococcemia may be completely nonspecific, with the patient simply complaining of feeling unwell but without overt symptoms or signs of meningitis. The clinical condition of these patients may progress to irreversible shock and death before the development or obvious manifestation of meningitis.
Early in meningococcemia, the patient may exhibit a subtle petechial rash (Fig. 22.3) that precedes progression to fulminant disseminated intravascular coagulation (DIC) and development of a more severe, prodigious purpuric rash leading to necrosis of the fingers and toes (Fig. 22.3). Purpura fulminans in the patient with meningococcemia is classically associated with hemorrhagic necrosis of the adrenal gland—the Waterhouse-Friderichsen syndrome. Thus, the clinical manifestation of meningococcal meningitis depends on the relative degree of meningococcemia and shock, as well as the severity of meningitis.
Fig. 22.3.
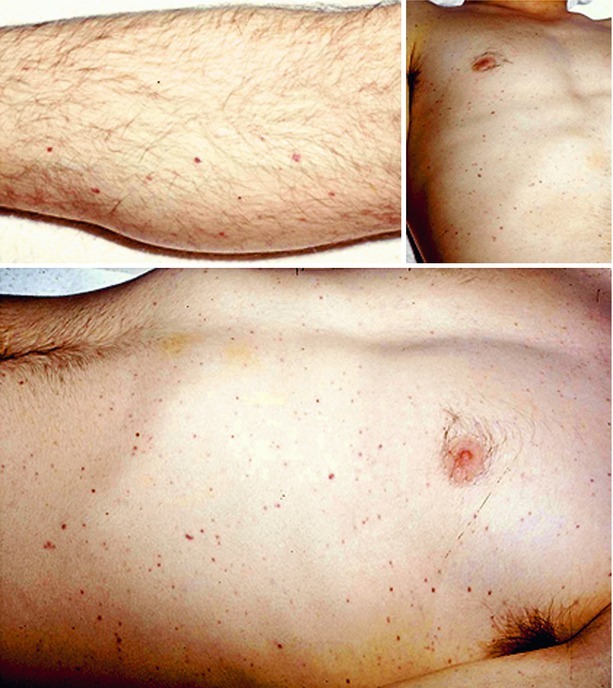
Petechial rash associated with meningococcal meningitis
It is important to recognize that the presentation of meningitis in the elderly and the very young may be subtler or more insidious compared with young adults and children. For example, in a review of 54 cases, Gorse and coworkers found that confusion was a predominating symptom in presentation among the elderly compared with the younger age group, and pneumonia was also more likely to be present in the older age group [35]. Typical symptoms and signs are also less commonly reported in the elderly since these patients often have cervical rigidity due to osteoarthritis, cervical spondylosis, or existing cerebrovascular disease. In addition, there may be hypertonicity of the neck muscles in conditions like Parkinson’s disease. Among the elderly, the meningitis itself may progress more rapidly, and patients are more likely to present in coma when compared with younger patient populations. With the development of coma, nuchal rigidity may be markedly less pronounced. Thus, when meningitis is suspected in the elderly, true nuchal rigidity has to be distinguished by careful physical examination. The absence of fever does not rule out the diagnosis of meningitis in the elderly patient.
In children, the presentation of meningitis is fundamentally similar to that in young and middle-aged adults, although nonspecific symptoms, such as irritability, nausea and vomiting, respiratory symptoms, and photophobia are more common in children. In neonates and infants, meningitis may present simply as fever or irritability; generally, there is a tendency for fever to be higher in children as compared to that in adults. The classic physical signs of meningeal inflammation or irritation described in medical textbooks are the Brudzinski’s and Kernig’s signs [36, 37]. Although Brudzinski originally described several signs of inflammation of the meninges, the best known of these is the so-called “nape of the neck” sign—the classic Brudzinski’s sign. This sign is elicited by flexing the neck forward. The stretching of the meninges induced by this movement results in involuntary flexion of the hips and knees. Kernig’s sign is elicited with the patient in the supine position and the thigh flexed on the abdomen with the knee flexed at a 45° angle. Upon passive extension of the leg in the presence of meningeal irritation, the patient resists extension with complaints of lower back and hamstring pain. Kernig’s and Brudzinski’s signs are neither sensitive nor specific indicators of meningitis and are potentially elicited in only about 50 % of children and only 5 % of adults with acute bacterial meningitis.
Diagnosis
An acute CNS infection is a medical emergency and bacterial meningitis may have to be differentiated from aseptic meningitis, encephalitis, brain abscess, subdural empyema, or noninfectious conditions affecting the CNS. Differentiation from encephalitis can be difficult and initially is made on clinical grounds. The classic features of meningitis (headache, neck stiffness, photophobia, fever, and vomiting) are often absent in neonates, patients who are immunocompromised—including persons with HIV infection—alcoholics, or the elderly. In encephalitis, altered state of consciousness, confusion, convulsions, and obtundation predominate. As the level of consciousness declines in patients with meningitis or encephalitis, differentiation between the two may only be possible through laboratory and radiographic findings. Because acute bacterial meningitis is a medical emergency, therapy should be implemented on clinical grounds without waiting for proof by laboratory or radiographic studies.
Fever and altered mental status with or without meningismus may occur in a variety of systemic infections as well as noninfectious conditions. For example, Rocky Mountain spotted fever (RMSF) can present with fever, shock, and a petechial rash (Fig. 22.4a–c), which must be differentiated from the rash associated with early meningococcemia (Fig. 22.3). Meningococcal disease may initially present simply as meningococcemia with shock and skin rash with minimal or absent meningeal signs. Other infections that present with headache and fever include brain abscess, influenza, leptospirosis, dengue, typhoid, parameningeal infections, or Q fever. Noninfectious, organic conditions, such as subarachnoid hemorrhage, acute hemorrhagic or ischemic strokes, cerebral venous sinus thrombosis, autoimmune disorders (e.g., temporal arteritis), neuroleptic malignant syndrome, status epilepticus, or toxic encephalopathies of various causes, can present precipitously with severe headache and fever, or nuchal rigidity.
Fig. 22.4.
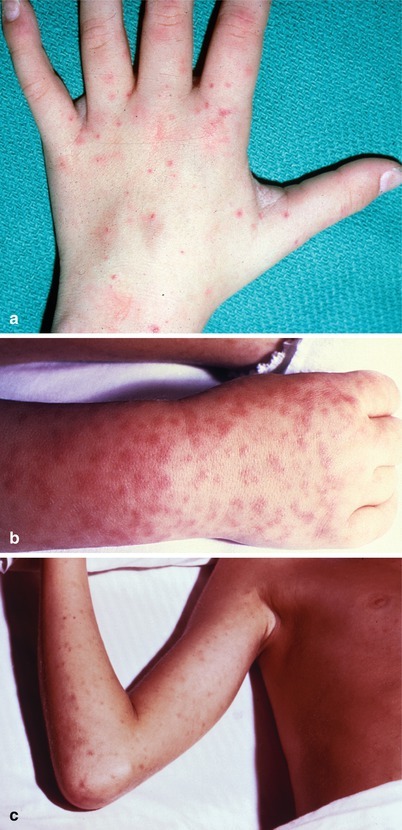
(a–c) Rocky Mountain spotted fever rash (a: Courtesy of Daniel J. Sexton, MD, Duke University Medical Center; b, c: Courtesy of the Centers for Disease Control and Prevention)
Lumbar Puncture and CSF Analysis
A lumbar puncture and analysis of the CSF facilitate the diagnosis of meningitis and other conditions affecting the CNS. However, the decision to perform a lumbar puncture on a patient with meningitis at presentation is precluded by the presence of raised ICP, which increases the risk of uncal, midbrain, medullary, or cerebellar tonsillar herniation after the procedure, leading to irreversible brain injury or death. Cerebral herniation occurs in about 5 % of patients with acute bacterial meningitis, accounting for about 30 % of the mortality [38]. Of note, the role of a CT scan is primarily to ascertain whether a space-occupying lesion is present; a CT scan cannot rule out the presence of increased ICP. Clinical signs suggestive of impending herniation include deteriorating level of consciousness, brainstem signs (including pupillary changes, decorticate posturing, or irregular respirations), a very recent seizure, absent oculocephalic reflexes, or papilledema. Lumbar puncture should be delayed in such patients, even for those with a normal CT scan, until preventive measures can be implemented to decrease ICP [38–42]. Other contraindications to immediate lumbar puncture include septic or hemodynamic shock, cardiorespiratory failure, presence of predisposing conditions for parameningeal abscesses (e.g., sinusitis, chronic ear discharge, or suppurative lung disease), bleeding disorders, and infection or loss of skin (e.g., burns) over the lumbar spine.
If the clinical picture is suggestive of bacterial meningitis or other intracranial infection and the patient is critically ill, especially if there is a rash or altered mental status, blood cultures should be drawn immediately and intravenous antimicrobial therapy initiated without delay. If the patient is not critically ill, one is certainly justified in withholding antimicrobial therapy until radiographic studies and lumbar puncture can be performed. If raised ICP is suspected and no focal lesions are defined by radiographic studies, one might consider intravenous infusion of mannitol (1 g/kg body weight) to reduce cerebral edema followed by a lumbar puncture after an interval of about 20 min. In addition to mannitol infusion, elective intubation and mechanical ventilation of the patient may be considered prior to the lumbar puncture procedure. Under these conditions, and using a 22-gauge needle, lumbar puncture can be performed without a significantly increased risk of herniation.
After insertion of the needle, the opening CSF pressure should be measured with the patient in the supine position. Normal opening pressure ranges from 1 to 10 cm H2O in young children, 6–20 cm H2O after 8 years of age, and up to 25 cm H2O in obese patients [43]. The level should fluctuate with respiration and can be elevated by the Valsalva maneuver. If the CSF pressure is measured again at the end of the procedure after appropriate volumes of CSF have been obtained and has dropped to zero, the possibility of a complete CSF block should be considered. CSF is normally crystal clear and colorless, not unlike a fine gin. A minimum of 200 white blood cells or 400 red blood cells/mm3 is necessary to impart turbidity to the fluid. CSF will appear reddish if more than 6,000 red blood cells/mm3 are present [43].
Xanthochromia is a yellow, orange, or pink discoloration of the CSF and is caused by the lysis of red blood cells resulting in hemoglobin breakdown to oxyhemoglobin, methemoglobin, and bilirubin. Discoloration begins after RBCs have been in spinal fluid for about 2 h. In cases of subarachnoid hemorrhage, xanthochromia occurs within 2–4 h after the initial cerebral bleed; xanthochromia may develop in vitro if the CSF specimen contains increased numbers of red blood cells and is not centrifuged immediately upon arrival in the laboratory. Xanthochromia also occurs when CSF protein concentrations are greater than 150 mg/dL. The macroscopic appearance of CSF only enables a diagnostic path for purulent versus aseptic meningitis; however, appearance alone is not sufficient to make a specific diagnosis of bacterial or viral meningitis.
In adults and children older than the neonatal age group, normal CSF generally contains less than 5 white cells/mm3, usually small lymphocytes. In neonates, CSF may contain up to 25–30 white cells/mm3 with up to 60 % neutrophils; this falls after a few days to the range of 8–9 white cells/mm3 [8]. Because red cells may be present in spinal fluid as a result of subarachnoid hemorrhage or through a traumatic tap, it is important to note whether red tinged or bloody CSF clears as sequential specimen tubes of CSF are obtained. Such clearing suggests a traumatic tap and can be documented in the laboratory by counting the red cells in successive tubes. CSF cells should be counted in the laboratory within 1–2 h of collection; further delays may result in a false low cell count because of cell lysis or adherence of cells to the walls of the specimen tube.
Glucose enters the CSF by transport through the choroid plexus and capillary endothelium in the subarachnoid space. CSF glucose levels are therefore a function of both active transport of glucose into the CNS and its rate of consumption within the CNS. CSF glucose levels in normal subjects are, on average, 60–70 % of the blood glucose levels. However, a study by Skipper and Davis showed the CSF to serum glucose ratio was accurate when the serum glucose was between 89 and 115 mg/dL [44]. For blood glucose levels greater than 125 mg/dL, the ratio was less than 60 %; for blood glucose levels greater than 192 mg/dL, the ratio fell to 50 % even among normal patients with no evidence of meningitis.
CSF protein levels are generally less than 40 mg/dL due to the exclusion of larger proteins by the blood–brain barrier. When the barrier breaks down during meningitis, CSF protein tends to rise and increases with duration of disease prior to initiation of therapy. Protein levels in newborn infants are significantly higher compared to older children and adults, averaging 90 mg/dL with normal levels up to 170 mg/dL. Extremely high levels (more than 1 g/dL) of CSF protein are suggestive of a CSF spinal block. Elevation of CSF protein on its own, however, is not specific for any specific type of meningitis.
Table 22.2 summarizes the CSF characteristics (macroscopic appearance, white cell count range and differential, protein and glucose levels) typically encountered in meningitis caused by various classes of organisms. In general, high CSF white cell counts are found in bacterial meningitis, where levels may be greater than 10,000 cells/mm3 with 95 % polymorphonuclear leukocytes (PMNs). Typically, WBC count in bacterial meningitis ranges between 500 and 5,000 cells/mm3, CSF glucose less than 40 mg/dL, and protein levels in the 100–500 mg/dL range. It is important to recognize that a predominance of PMNs may occur early in viral meningitis, within the first 24–48 h, but this gradually shifts to a mononuclear predominance over the next 8 h if the lumbar puncture is repeated [45, 46]. In patients with meningitis caused by L. monocytogenes, the organism grows and survives within the host cell cytoplasm, thereby stimulating a monocytic CSF response; in infants there may be a monocytic predominance.
Table 22.2.
CSF findings in acute and chronic meningitis and other CNS infectious conditions
| Type of infection | Macroscopic appearance | Cells | Protein (mg/dL) | Glucose (mg/dL) | Other tests |
|---|---|---|---|---|---|
| Normal | Clear | <5 lymphocytes/mm3 | 15–45 | 50–75 | Negative test results |
| Bacterial meningitis (S. pneumoniae; N. meningitides; L. monocytogenes) | Cloudy or turbid | Increased (commonly > 200) | >100 | Reduced (<40) | Gram stain, bacterial culture, and antigen tests may be positive |
| Typically >90 % PMNs | |||||
| Can be normal in meningococcemia | |||||
| Viral meningitis (enteroviruses; herpes simplex; arboviral encephalitis) | Clear or rarely opalescent | Increased | Usually <100 | Normal | Gram stain, bacterial culture, and antigen tests negative |
| May have PMN predominance early in the course of infection; converts to lymphocytic predominance within 12–24 h | PCR for HSV, VZV, arboviruses, and enteroviruses may be positive | ||||
| Fungal meningitis (cryptococcus; histoplasmosis; coccidioidomycosis) | Cloudy or turbid | >100 (<50 %) | 100–900 | < 40 | Cryptococcus can be diagnosed from India ink preps, antigen tests, or culture; PCR |
| Usual range 100–400 usually lymphocytic predominance | |||||
| May be normal in cryptococcal meningitis | |||||
| Tuberculous meningitis | Cloudy or turbid | Increased | 100–900 | <40 | Acid-fast bacilli occasionally seen on CSF smear stained with Kinyoun or Ziehl-Neelsen stains |
| Typically >100 | |||||
| Usual range 100–400 | |||||
| PMN early but converts to lymphocytic predominance | |||||
| Parameningeal infections (sinusitis; epidural abscess; paraspinous abscess) | Clear | <100 (<50 %) | Increased | Normal | |
| Occasionally PMN predominance | |||||
| If rupture into CSF, like acute meningitis |
Adapted with permission from Rand et al. [521]
Once CSF specimens are obtained, a gram stain should be performed immediately in patients with suspected bacterial meningitis followed by plating on solid culture media. Centrifugation of CSF improves the yield for both gram stain smears and culture. In general, a CSF concentration greater than 103 organisms/mL is required in order for organisms to be identified on light microscopy of the gram stain. With lower concentrations, there are simply too few organisms to detect by direct microscopy. Approximately 75 % of patients with acute bacterial meningitis will have a positive gram stain, and this percentage may drop to about 50 % among patients who have received significant doses of prior antimicrobial therapy. Generally, the gram stain is positive in 90 % of untreated patients with pneumococcal meningitis, 86 % of patients with meningitis due to H. influenzae, and approximately 75 % of cases due to N. meningitidis [47]. Among children, the overall sensitivity of gram stain to detect bacterial meningitis is 67 %. Moreover, most children without bacterial meningitis have negative gram stain with a negative predictive value of 99.9 %. Thus, CSF gram stain is useful in evaluating children for empiric therapy of bacterial meningitis [48].
In addition to gram stain, a number of other rapid diagnostic tests have been developed over the past 20 years for diagnosis of acute bacterial meningitis. In the 1970s counter immunoelectrophoresis (CIE) was used for direct detection of bacterial polysaccharide antigens; this test is quite insensitive and is no longer in use. Agglutination tests are commercially available for H. influenza, S. pneumoniae, and five serotypes of N. meningitidis. However, the sensitivity and specificity of these tests are no better than that of the gram stain, and they provide no additional diagnostic yield above and beyond the gram stain and the clinical picture and rarely influence the decision to treat empirically [49, 50]. Therefore, they are not currently recommended in the diagnosis of acute bacterial meningitis upon initial presentation. The underlying problem with these tests is that they are not sensitive and specific enough to establish a diagnosis upon which to initiate appropriate therapy. For example, if a patient is sick enough to be admitted to the hospital, and found to have a low CSF glucose level and raised CSF white blood count, one would still initiate empiric antimicrobial and supportive therapy even if the agglutination tests are negative.
Miscellaneous Testing
C-reactive protein (CRP) can be measured in CSF and, when greater than 100 μg/mL, may be useful in differentiating bacterial from viral meningitis [51]. Extensive literature exists describing the application of real-time polymerase chain reaction (PCR) for detection and quantification of various bacterial and viral pathogens in CSF of patients with a putative diagnosis of bacterial meningitis. Real-time PCR is faster and more sensitive than previous technologies. However, this technique is expensive and not readily available in most hospital laboratories. Moreover, in clinical practice, physicians are likely to initiate empirical antimicrobial therapy anyway after requesting testing by conventional methods, especially for patients with typical CNS symptoms and signs. In this case, rapid diagnostic testing using PCR assays is likely to not make a difference in the clinical the clinical decision making and medical management of the patient.
Imaging Studies
Neuroimaging plays little role in the diagnosis of acute bacterial meningitis except as indicated earlier to rule out the presence of mass lesions and raised ICP, which might increase the risk of herniation when lumbar puncture is performed. The major value of CT and MRI scans in patients with acute bacterial meningitis is in the investigation of complications, such as cerebral infarction, vasculitis, abscess, or hydrocephalus. Figure 22.5a–d shows the typical neuroradiological appearances of the brain in patients with meningitis caused by L. monocytogenes, N. meningitidis, S. pneumoniae, and M. tuberculosis. In patients with prolonged fever of 10 days duration or longer, up to 25 % may have a subdural effusion (Fig. 22.6). In some cases this may progress to a subdural empyema, which may account for the prolonged fever (Fig. 22.7). Cortical infarction is a common complication of bacterial meningitis and usually results from vasospasm of cerebral vasculature or vasculitis associated with the meningitis itself. The MRI scan is more sensitive than CT imaging in detecting cerebritis and cortical infarction. Cerebritis is an early complication that may occur during the first 4 days (Fig. 22.8). Early necrotic regions filled with polymorphonuclear cells, lymphocytes, and plasma cells and with ill-defined parenchymal swelling characterize cerebritis. In late cerebritis (4–8 days), central necrosis increases, there is vascular proliferation and more inflammatory cells, and suppurative foci begin to breakdown and become encapsulated.
Fig. 22.5.
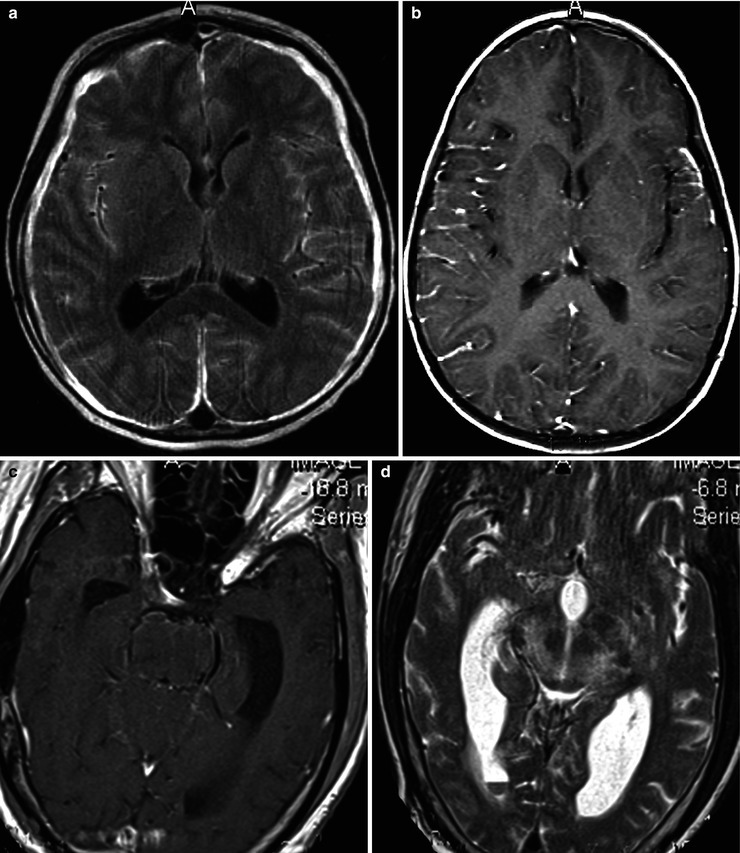
(a, b) Acute meningitis; images include fluid-sensitive, FLAIR T2-w sequence (a) and post-contrast T1-w (b) axial sequences. Findings in acute meningitis are frequently subtle especially viral meningitis. The FLAIR sequence. A presents normal CSF within the ventricles and sulci as hypointense (dark) relative to brain. With pial inflammation there is leak of proteinaceous fluid into the subpial and subdural spaces. This highly proteinaceous fluid is hyperintense (bright relative to brain) and thus becomes visualized on fluid-sensitive MRI sequences. Serous subdural effusions are often present as well, which are imaged as high-intensity fluid outside the brain, as in this case, but without contrast enhancement along their surfaces. These types of fluid collections are considered noninfective and they typically clear spontaneously after medical treatment. The contrasted MRI (b) demonstrates pial hyperemia along the right lateral cerebral convexity (compare to left side). On the left there is thickening of the pia probably early subpial empyema. (c, d) Listeria rhombencephalitis; images include post-contrast T1-w section (c) and a T2-w sequence (d). When the distribution of the inflammatory process involves mainly the upper brain stem, as in this instance, it is described as rhombencephalitis. Rhombencephalitis is uncommon but is one the manifestations of Listeria-based meningitis, as in this case. Note there is relatively little abnormal enhancement in this case of Listeria infection. If there is thick obvious enhancement in a similar distribution, findings would be more consistent with a granulomatous infection, as in fungal or tuberculous meningitis. Similar findings can also be part of noninfectious granulomatous pial disease, as in neurosarcoidosis and non-Langerhans histiocytosis; thus, tissue confirmation is usually necessary
Fig. 22.6.
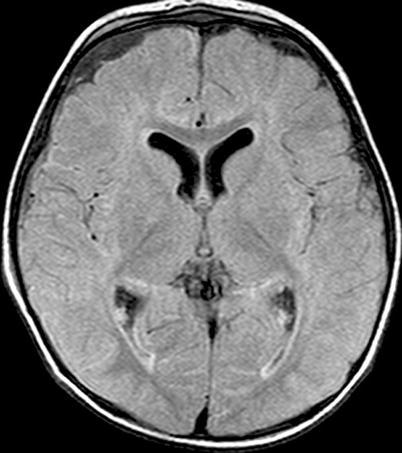
Active meningitis and secondary subdural effusions. This FLAIR sequence, which emphasizes tissue edema, but deemphasizes bulk CSF signal, shows increased signal along the trigones of the lateral ventricular surfaces indicative of ependymitis, plus increased signal along the pial surfaces indicative of meningitis, plus minimal ventriculomegaly. All of these findings commonly occur in acute meningitis. Additionally, there are small bifrontal extra axial fluid collections without any signal along their margins which are consistent with likely sterile subdural effusions. The fluid signal is minimally higher than CSF within the lateral ventricles indicating elevated CSF protein, a feature common to reactive subdural effusions
Fig. 22.7.
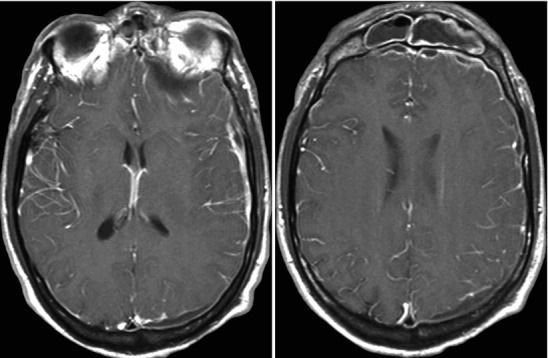
Acute frontal sinusitis with secondary subdural empyema; images include contiguous post-contrast mid-convexity axial MRI sections. This case illustrates the spread pattern of subdural empyema. The source of the infection is the frontal sinus. Once the infection accesses the subdural space it can spread widely within the intracranial compartment. In this instance, it continues all the way to the occipital region. These multicentric pockets of subdural empyema are often sequestered requiring multiple surgical drains. Thus, it is imperative that the full extent of the subdural empyema is appreciated
Fig. 22.8.
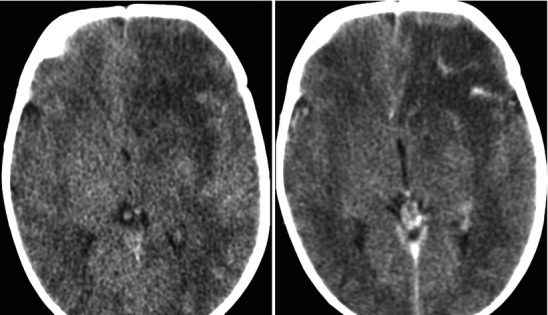
Acute left frontal lobe bacterial cerebritis; images include pre and post-contrast CT sections sagittal projection lower thoracic area. The early phase of brain infection (early cerebritis) demonstrates nonspecific cerebral edema and poorly defined contrast enhancement. There is frequently reactive pial hyperemia. In later stages the cerebritis will organize into early then mature stages of brain abscess
Treatment
The pathophysiology of the blood–brain barrier is of critical importance in determining the choice of antimicrobials for the treatment of acute bacterial meningitis. The penetration of the blood–brain barrier is a function of both the properties (e.g., lipid solubility, molecular size, and molecular structure) of the antimicrobial itself and the degree or extent of the inflammation of the meninges. For example, chloramphenicol, which is highly lipid soluble, will readily penetrate uninflamed meninges. Fortunately, in inflamed meninges therapeutic concentrations of penicillins, cephalosporins, and vancomycin can be achieved for treatment of the vast majority of cases of bacterial meningitis. Because only the free, unbound portion of an antimicrobial agent is capable of crossing the blood–brain barrier, the degree of protein binding of the antimicrobial in the patient serum is critical in determining how much of the agent eventually gets through to the CSF. With increased concentrations of protein in the CSF, protein binding becomes a significant factor in the effectiveness those antimicrobials that are highly protein bound.
The penetration of aminoglycosides is generally so poor that they are of little value in the treatment of acute meningitis when given intravenously, although they may be useful intrathecally. The penetration of the third-generation cephalosporins (e.g., ceftriaxone and cefotaxime) is significantly better than that of the first- and second-generation cephalosporins. Quinolones, tetracyclines, and macrolides do not penetrate the blood–brain barrier sufficiently to be useful first-line agents in the treatment of meningitis, whereas sulfa agents (e.g., trimethoprim-sulfa) and vancomycin, in the presence of inflamed meninges, may reach sufficient concentrations to be of therapeutic value.
Treatment regimens for acute bacterial meningitis in children above the age of 3 months and in adults up to the age of 50 is geared to treating the most common pathogens: N. meningitides, S. pneumoniae, and, less commonly, H. influenzae. The prevalence of penicillin-resistant S. pneumoniae has risen so that more than 50 % of strains may be resistant or exhibit intermediate resistance to penicillin in some parts of the United States and other countries. By definition, fully susceptible pneumococci are susceptible to penicillin at less than 0.1 μg/mL; intermediate susceptibility is defined by an MIC less than or equal to 2 μg/mL, whereas fully resistant S. pneumoniae are defined by MICs greater than or equal to 4 μg/mL. Among penicillin-resistant strains, resistance to the third-generation cephalosporins, including cefotaxime and ceftriaxone, has been increasing; resistance to ceftriaxone as high as 35 % has been documented for S. pneumoniae isolates in some areas [52–55].
Once a clinical diagnosis of acute bacterial meningitis is suspected or made, institution of antimicrobial therapy should be immediate. If clinical evaluation raises a suspicion of raised intracranial pressure, or if the patient manifests signs of papilledema or focal neurological deficits, blood should be drawn for culture and baseline testing (e.g., white blood count and glucose), and empiric antimicrobial therapy initiated before the patient is sent off for imaging studies of the brain. Choice of empirical antimicrobial therapy is dictated by the age of the patient, vaccine status, and whether bacterial meningitis was acquired in the community or within the healthcare setting.
For community-associated meningitis, the microorganisms most commonly implicated are S. pneumoniae, N. meningitidis, Listeria spp., other Streptococcus spp., S. aureus, and H. influenzae. Healthcare-associated meningitis usually follows neurosurgical procedures, such as craniotomy, placement of ventriculostomy tubes, or deep brain stimulation of the brain for Parkinson’s disease; pathogens most commonly implicated in healthcare settings include gram-negative microorganisms (e.g., Enterobacteriaceae and non-fermenters), S. aureus, and Streptococcus spp.
Initial empirical therapy for community-acquired meningitis: for adult and children 3 months–50 years, ceftriaxone or cefotaxime can be given, especially if risk factors (e.g., CSF leak, pneumonia, or sinusitis) for S. pneumoniae meningitis are present. If the patient is very sick or if gram-positive cocci are seen on CSF microscopy, vancomycin should be added to the therapeutic regimen to cover for penicillin-resistant S. pneumoniae in a dose of 2–3 g/day given every 8–12 h in adults and at 60 mg/kg for children in four divided doses, until it is known that the penicillin MIC is less than 0.1 mg/mL For patients with a history of idiosyncratic reactions to penicillin or cephalosporins, vancomycin is recommended, although chloramphenicol may be used in adults. For patients with a history of severe penicillin allergy, chloramphenicol at a dose of 4–6 g/day in four divided doses for adults should be given in place of the third-generation cephalosporin together with vancomycin. In adults more than 60 years of age, patients with chronic alcoholism, immunosuppression, or other debilitating conditions, the possibility of L. monocytogenes meningitis should be considered. Empirical therapy to cover L. monocytogenes includes addition of maximal doses of ampicillin (12 g daily dosed every 4 h) to the cephalosporin or vancomycin regimen until culture results for blood or CSF become available. For patients with penicillin allergy, the use of chloramphenicol, imipenem, or trimethoprim/sulfamethoxazole can be considered as an alternative to cover for Listeria until culture results are available. Therapy may need to be broadened depending on the results of the gram stain. In cases where gram-negative diplococci are seen, it is probably prudent to wait until culture results confirm N. meningitidis before narrowing the bacterial coverage to penicillin because of the possibility that the gram stain might have been misinterpreted. Treatment of the most common etiologic agents of acute bacterial meningitis is summarized in Table 22.3.
Table 22.3.
Therapy of acute bacterial meningitis
| Empirical treatment for patients with suspected meningitis but negative gram stain or culture | ||
| Age group | Likely organisms | Empiric regimen |
| Preterm to <1 month | Group B Streptococcus; Escherichia coli; Listeria sp. | Ampicillin 100 mg/kg IV q6h plus cefotaxime 50 mg/kg IV q6h or ampicillin 100 mg/kg IV q6h plus gentamicin 2.5 mg/kg IV q8h |
| 1 month–50 years | Streptococcus pneumoniae; Neisseria meningitidis; Haemophilus influenzae; Listeria sp. | Adult: ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q4-6 h plus vancomycin 15 mg/kg q6–8 h |
| Child: ceftriaxone 100 mg/kg/day IV (doses given q12h) or cefotaxime 200–300 mg/kg/day IV (doses given q6h) plus vancomycin 60 mg/kg/day IV (doses given q6h) | ||
| >50 years | S. pneumoniae; N. meningitidis; H. influenzae; Listeria sp.; aerobic gram-negative microorganisms | Ampicillin 2 g IV q4h plus ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q6h plus vancomycin 15 mg.kg IV q8–12 h |
| Trauma: skull fracture | S. pneumoniae; H. influenzae; Group B Streptococcus | Vancomycin 15 mg.kg IV q8–12 h plus ceftriaxone 2 g IV q12h |
| Trauma: penetrating | Staphylococcus aureus, coagulase-negative staphylococcus, Enterobacteriaceae, Pseudomonas spp. | Vancomycin 15 mg.kg IV q8–12 h plus cefepime 2 g IV q8h |
| Meningitis associated with shunts | S. aureus, coagulase-negative staphylococcus, Enterobacteriaceae, Pseudomonas spp. | Vancomycin 15 mg.kg IV q8–12 h plus cefepime 2 g IV q8h |
| Neurosurgery (e.g., craniotomy) | S. aureus, coagulase-negative staphylococcus, Enterobacteriaceae, Pseudomonas spp. | Vancomycin 15 mg.kg IV q8–12 h plus cefepime 2 g IV q8h |
| Therapy for patients with acute bacterial meningitis (suggested by gram stain or culture)—by microorganism | ||
| Microorganism | Treatment | Duration of therapy (days) |
| Streptococcus pneumoniae | ||
| Penicillin-susceptible isolate (MIC <0.1 μg/mL) | Adults: penicillin G 4 million units IV q4h or ampicillin 2 g IV q4–6 h | 10–14 |
| Children: 250,000–400,000 U/kg IV q4–6 h | ||
| Severe penicillin allergy: substitute cephalosporin agent with chloramphenicol 75–100 mg/kg/day in 4 divided doses | ||
| Isolate with intermediate (MIC = 0.1–1μg/mL) susceptibility to penicillin | Ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q4–6 h | |
| Isolate resistant (≥2μg/mL) to penicillin | Ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q4–6 h plus vancomycin 15 mg/kg q6–8 h | |
| Neisseria meningitidis | Adults: penicillin G 4 million units IV q4h or ampicillin 2 g IV q4–6 h or ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q4–6 h. Penicillin allergy: as for S. pneumoniae above | 7 |
| Children: penicillin G 250,000–400,000 U/kg IV q4–6 h. Penicillin allergy: substitute with chloramphenicol 75–100 mg/kg/day in 4 divided doses | ||
| Haemophilus influenzae | ||
| Beta-lactamase positive | Ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q6h | 7 |
| Beta-lactamase negative | Ampicillin 2 g IV q q4–6 h | |
| Group B Streptococcus (Streptococcus agalactiae) | ||
| Suspected/empiric | Preterm: ampicillin 200–300 mg/kg/day IV in 3 divided doses plus cefotaxime | 14–21 |
| Infants ≤7 days: ampicillin 200–300 mg/kg/day IV in 3 divided doses plus an aminoglycoside, adjusted for age and birth weight (BW), i.e., gentamicin 2.5 mg/kg IV q12h; 2.5 mg/kg IV q8–12 h if BW <2,000 g; 2.5 mg/kg IV q8h if BW >2,000 g | ||
| Infants >7 days: ampicillin 300 mg/kg/day iv in 4–6 doses/day plus an aminoglycoside, adjusted for age and BW, i.e., gentamicin 2.5 mg/kg IV q8–12 h if BW <2,000 g; 2.5 mg/kg IV q8h if BW >2,000 g | ||
| Intraventricular treatment not recommended | ||
| Known | Adults: penicillin G 4 million units IV q4h plus gentamicin 3–5 mg/kg IV daily, divided q8h | 14–21 |
| Infants ≤7 days: penicillin G 250,000–450,000 U/kg/day IV in 3 divided doses | ||
| Infants >7 days: penicillin G 450,000 U/kg/day IV | ||
| Listeria monocytogenes | ||
| Infants ≤7 days: ampicillin 200–300 mg/kg/day IV in 3 divided doses plus an aminoglycoside, adjusted for age and BW, i.e., gentamicin 2.5 mg/kg IV q12h if BW <2,000 g; 2.5 mg/kg IVq12h if BW >2,000 g | 21 or longer | |
| Infants >7 days: ampicillin 300 mg/kg/day IV in 4–6 doses/day plus an aminoglycoside, adjusted for age and BW, i.e., gentamicin 2.5 mg/kg IV q8–12 h if BW <2,000 g; 2.5 mg/kg IV q8h if BW >2,000 g | ||
| Adults >50, alcoholism, or other risk factors: ampicillin 2 g IV q4h plus ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q6h plus gentamicin 2 mg IV loading dose, then 1.7 mg/kg q8h plus dexamethasone 0.4 mg/kg IV q12 h x 2 | ||
| Penicillin allergy: trimethoprim/sulfamethoxazole | ||
| Consider stopping gentamicin after 1 week | ||
| Where ampicillin is suggested, amoxicillin may be used | ||
| Pseudomonas aeruginosa | Ceftazidime 1 g IV q8h or cefepime 2 g IV q8h plus gentamicin 3–5 mg/kg IV daily divided q8h | 21 |
| Enterobacteriaceae (e.g., Escherichia coli) | Ceftriaxone 2 g IV q12h or cefotaxime 2 g IV q4–6 h plus gentamicin 3–5mh/kg IV daily divided q8h | 21 |
The utility of adjunctive therapy with dexamethasone in the treatment of acute bacterial meningitis remains controversial. The use of dexamethasone as an adjunct to therapy in acute bacterial meningitis is complex. It has been shown clearly in animal models and in patient studies that dexamethasone reduces the level of inflammation and reduces the levels of the inflammatory cytokines IL1 beta and tumor necrosis factor alpha [56]. However, in an animal model, administration of dexamethasone together with vancomycin reduced the penetration of vancomycin into the CSF by 29 % and lowered the rate of bacterial clearance during the first 6 h in animals who received an intermediate dose of vancomycin. Animals that received a higher dose had therapeutic peaks maintained despite steroid use, suggesting that the anti-inflammatory effect of the steroids, which reduce entry of antibiotics into the CSF, may be overcome to some extent by increasing the dose [57]. In animal studies of experimental pneumococcal meningitis, an antibiotic-induced secondary inflammatory response in the CSF was demonstrated only in animals with high initial CSF bacterial concentrations; these effects were modulated by dexamethasone therapy [58].
Human studies of the use of dexamethasone have clearly shown that there is a reduction in severe hearing loss in patients who have H. influenza type b meningitis and there is a similar reduction in overall neurologic complications although perhaps not as significant. In children with meningitis due to S. pneumoniae, there also appears to be a significant reduction in long-term hearing loss [59]. Major side effects from dexamethasone include secondary fever and a small incidence of gastrointestinal bleeding which is probably negligible if treatment is limited to 2 days but increases up to 3 % in patients who received 4 or more days of treatment or more.
In summary, dexamethasone probably should be used as an adjunct in children at a dose of 0.4 mg/kg IV every 12 h for no more than 2 days and probably should be given just before, or at the time of, the first antibiotic dose to block any increase in any inflammatory cytokine production following initial bacterial lysis. A more recent study has shown that adjunctive dexamethasone in the treatment of acute bacterial meningitis in adults does not appear to significantly reduce death or neurological disability and concludes that the benefit of adjunctive dexamethasone for all or any subgroup of patients with bacterial meningitis remains unproven [60].
The duration of treatment of bacterial meningitis is based on empiric observation. In general, the minimum duration treatment is 7 days as long as the patient is afebrile for the last 4–5 days. Treatment of S. pneumoniae generally takes longer than H. influenzae and N. meningitidis and may be extended to 10–14 days, depending on the patient’s response. Meningitis following trauma and neurosurgical procedures is discussed elsewhere.
Complications
Elevated ICP is a result of cerebral edema due to acute bacterial meningitis and should be anticipated. Clinical manifestations of raised ICP include bradycardia, hypertension, altered mental status, drowsiness, obtundation and coma, third cranial nerve palsies, including unilateral or bilateral dilated, poorly reactive or nonreactive pupils, abnormal ocular movement, abnormal respiration, or decerebrate posturing. Papilledema is relatively uncommon and as such is an unreliable sign of raised ICP as it may take several hours to develop after the ICP has increased. Signs of herniation may supersede those of increased pressure and include unequal, dilated, or nonreactive pupils, dysconjugate eye movements, decorticate and decerebrate posturing, and bradycardia with abnormal respiratory patterns.
Patients who are awake and alert can be monitored closely. Patients who are obtunded or comatose, or who manifest other signs of increased ICP may well benefit from ICP monitoring. Pressures exceeding 20 mmHg should be treated and some studies suggest that even pressures greater than 15 mmHg may benefit from treatment [61]. An indication for treating at lower pressure levels is the phenomenon of “plateau waves” (Fig. 22.9), which are large elevations in pressure that occur spontaneously, or due to changes in cerebral blood flow, small shifts in intracranial blood volume resulting from hypoxia, fever, or otherwise innocuous events like tracheal suctioning. When these waves develop on a background of already increased ICP, herniation and irreversible brain stem injury may ensue [61, 62].
Fig. 22.9.
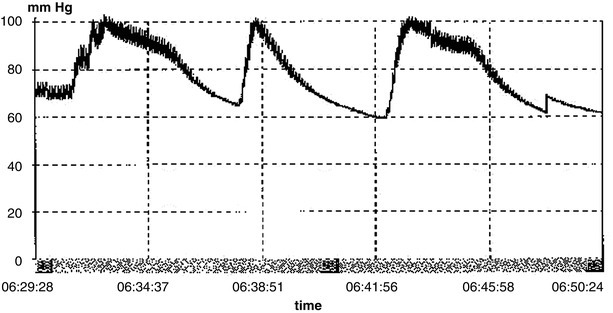
Plateau waves: characterized by a sudden rapid elevation of intracranial pressure to 50–100 mmHg for 5–20 min. After a sustained period of elevation, the termination of the wave is characterized by a rapid decrease of ICP. These waves are thought to be caused by changes in cerebral blood flow
The treatment of increased ICP includes elevation of the head of the bed to 30° above the horizontal to facilitate venous drainage, intubation, and hyperventilation to reduce and maintain the arterial PaCO2 concentrations to levels between 27 and 30 mmHg. Hypertonic osmotic agents, such as mannitol or hypertonic saline infusions, play a vital role in the reduction of elevated intracranial pressure and treatment of cerebral edema in patients with CNS infections [63–68]. Both mannitol and hypertonic saline reduce cerebral edema in many clinical syndromes [63–67]. However, recent data suggest that hypertonic saline appears to achieve a greater reduction in ICP than other osmotic agents [63, 65]. The object in using osmotic agents is to achieve a sustained reduction in intracranial pressure by modifying the modes and rates of administration of the respective osmotic agent [63–67].
Phenobarbital therapy may be considered if raised ICP remains uncontrolled by the foregoing interventions. Caution is advised in the use of hyperventilation to lower arterial PaCO2 concentrations because overly vigorous treatment may cause these values to fall below 25 mmHg, running the risk of further reductions in cerebral blood flow causing cerebral ischemia. The dose of mannitol in children is 0.5–2.0 g/kg infused over 30 min and repeated as necessary; in adults the usual dose is 0.25–1 g/kg bolus injection and 0.25 g/kg every 2–3 h as needed. Mannitol and hypertonic saline act as hyperosmolar agents and remain almost entirely within the intravascular space, producing an osmotic gradient that shifts intracranial fluid into this space. Serum osmolality should be frequently checked and kept between 315 and 320 mOsm/L [61–63, 67–71].
Dexamethasone has been used to reduce intracranial swelling in other settings primarily because of its effectiveness in vasogenic cerebral edema. Various clinical studies support the use of adjunctive dexamethasone in infants or children with H. influenza type b meningitis to reduce the risk of neurologic and audiologic complications, especially in those with raised ICP or coma. High-dose barbiturates may be helpful when other methods have failed to control increased ICP. Barbiturates decrease the CNS metabolic demand for oxygen thereby decreasing cerebral blood flow which, in turn, causes a fall in ICP. Phenobarbital is given at an initial dose of 5–10 mg/kg, at a rate of 1 mg/kg per minute followed by 1–3 mg/kg/h [61, 62]. Such therapies require regular ICP measurements and monitoring of cerebral electrical activity with electroencephalography. Phenobarbital is given until the ICP falls to levels 20 mmHg or until approximately 90 % burst suppression on the EEG (i.e., nine out of the ten screens of the EEG are flat) has been achieved. Serum phenobarbital concentrations should be kept within the range of 20–40 μg/mL. Pentobarbital is preferred because of its relatively short half-life (24 h) versus phenobarbital with a relatively longer half-life. Side effects of high-dose barbiturate therapy include cardiac depression with arrhythmias and hypotension, thus mandating invasive hemodynamic monitoring in these patients.
Seizures
Seizures occur in approximately 30–40 % of children and adults with acute bacterial meningitis within the first few days of illness. If not treated, seizures may progress to status epilepticus, which in turn can lead to anoxic damage of the temporal lobe, cerebellum, and thalamus. The principles of therapy are to control seizure activity quickly and definitively. To initiate therapy, short-acting anticonvulsants, such as lorazepam or diazepam, are administered followed by a long-acting agent like phenytoin. Lorazepam is given IV in doses of 1–4 mg in adults, and 0.05 mg/kg in children. Phenytoin is given IV at a dose of 18–20 mg/kg and at a rate of no more than 50 mg/min. The rate should be decreased if signs of toxicity, such as hypotension or a prolonged QT interval, develop. If phenytoin is not successful in controlling seizure activity, intubation and treatment with IV phenobarbital may be necessary. Patients must be watched and monitored carefully for signs of toxicity, such as hypotension and respiratory depression. Phenobarbital should be given IV at a rate of 100 mg/min until seizure activity stops, up to an initial dose of 20 mg/kg.
In children, the rate should be decreased to 30 mg/min. Should these measures fail to control seizures, general anesthesia and additional phenobarbital therapy may have to be considered.
Vaccination for Meningitis
N. meningitidis became a leading cause of bacterial meningitis in the United States after dramatic reductions in the incidence of S. pneumoniae and H. influenzae type b infections had been achieved as a result of using conjugate vaccines [1, 72, 73]. However, since 2000, meningococcal disease incidence has decreased and incidence for serogroups C and Y, which represent the majority of cases of vaccine-preventable meningococcal disease, is at historic lows. In 2005, a quadrivalent meningococcal polysaccharide-protein conjugate vaccine (MCV4) was licensed for use among persons aged 11–55 years, and during the same year, the Advisory Committee on Immunization Practices (ACIP) recommended routine vaccination with 1 dose of MCV4 for persons aged 11–12 years, persons entering high school (i.e., at approximately age 15 years) if not previously vaccinated with MCV4, and other persons at increased risk for meningococcal disease, including college freshmen living in dormitories [74]. In 2010, ACIP approved updated recommendations for the use of quadrivalent (serogroups A, C, Y, and W-135) meningococcal conjugate vaccines in adolescents and persons at high risk for meningococcal disease [75, 76]. The vaccine contains immunogenic polysaccharide capsular material from serogroups A, C, Y, and W-135. The vaccine has few side effects and is believed to be protective for at least 3–5 years.
Persons at increased risk for severe pneumococcal disease include those who are immunocompromised, asplenic or splenectomized, or patients with chronic illness such as chronic cardiovascular disease (e.g., congestive heart failure or cardiomyopathies), chronic pulmonary disease, diabetes mellitus, alcoholism, chronic liver disease (cirrhosis), or CSF leaks. CDC has updated recommendations from ACIP for prevention of invasive pneumococcal disease (i.e., bacteremia, meningitis, or infection of other normally sterile sites) through use of the 23-valent pneumococcal polysaccharide vaccine among all adults aged greater than or equal to 65 years and those adults aged 19–64 years with underlying medical conditions that put them at greater risk for serious pneumococcal infection [77].
Viral Meningitis and Encephalitis
Viral CNS infections may be classified as exogenous due to infection with a viral agent acquired outside the host or endogenous due to reactivation of viruses that have remained latent in the host. The majority of viral CNS infections are caused by exogenously acquired enteroviruses (Coxsackie virus A and B, echovirus, polio virus), arboviruses, and, less commonly, by HSV, mumps virus, varicella-zoster virus (VZV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), adenovirus, human immunodeficiency virus (HIV), West Nile virus (WNV), rabies virus, or lymphocytic choriomeningitis virus. HSV encephalitis is unique in that it may occur as part of the primary infection or be seen in patients in whom the infection has been latent for many years. CNS infections due to the other herpes viruses, such as EBV, VZV, or CMV occasionally may be seen as part of the primary infection but may also occur as reactivated infections in patients who are immunosuppressed or HIV-infected.
Epidemiology
Meningitis and meningoencephalitis are the most common viral CNS infections encountered in the United States. The overwhelming majority of these infections are caused by enteroviruses, which produce disease in outbreaks occurring mainly during the summer months, but may occur during May to October in warmer parts of the United States. While virtually all of the various serotypes of echovirus and Coxsackie virus can produce meningitis and meningoencephalitis, in addition to other syndromes, the 15 most commonly noted enteroviruses in the United States during 1970–2005 accounted for 83.5 % of CDC reports with known serotype [78]. The five most commonly reported serotypes (echoviruses 9, 11, 30, and 6 and Coxsackie virus B5) in descending order of frequency accounted for approximately half (48 %) of all reports [78]. CSF was the most common specimen type. The epidemiologic pattern is one in which certain strains, such as echovirus 30 or echovirus 9, cause disease endemically, while other strains occur in sporadic outbreaks varying from year to year in different regions. Enteroviruses are transmitted from person to person by the fecal-oral route and their activity tends to be increased in areas of overcrowding, poverty, and generally poor hygienic conditions.
Arboviruses account for the majority of epidemic cases of encephalitis. Their occurrence follows an identical seasonal distribution to that of viral meningitis and meningoencephalitis associated with enteroviruses. However, the mode of transmission is completely different. Arboviruses are spread by the bite of infected mosquitoes, which are part of a complex cycle of enzootic transmission between birds, mosquitoes, and small mammals. The epidemiology of these diseases may be affected in part by prevention efforts from the public health authorities. For example, many states maintain surveillance systems that include testing of mosquitoes for the presence of virus, as well as sentinel chicken flocks to determine arbovirus activity. Such efforts lead to early recognition of an outbreak and warnings by public health authorities for the population to take precautions such as insect repellants, wearing long sleeve shirts, and avoiding outdoor activity in the early evening hours when transmission is most likely to occur. In addition, mosquito control activities may contribute to reduction in rates of infection.
In August of 1999, an outbreak of encephalitis was detected in the borough of Queens, New York City: 62 patients were confirmed infected with an agent identified as the arbovirus West Nile virus (WNV); seven eventually died. This followed a massive die-off among birds, particularly crows that had been observed during the month before the outbreak. Most of those affected with serious illness were elderly, although one patient was 29 years old [79–81]. In 2003, there were over 8,000 cases of WNV infections reported to the CDC with 199 deaths; most of these cases involved the CNS. During the ensuing decade, WNV occurrence started to spread westward across the continental United States, and by the end of 2004 approximately 1 in 400 blood donors were thought to be infected with WNV (CDC data).
Although rabies is rare among humans in the United States, potential exposures to rabid animals lead to between 16,000 and 39,000 persons receiving post rabies exposure prophylaxis each year [82]. Since the 1950s, the incidence of rabies in domestic animals has declined dramatically because of immunization of dogs and other domestic animals. Unlike the situation in developing countries, wild animals are the most important potential source of rabies for both humans and domestic animals in the United States; most reported cases of rabies occur among raccoons, skunks, and foxes and various species of bats [82]. During 2010, the number of rabies cases—both in animals and humans—reported in the United States fell 8 % compared with the previous year [83].
Pathogenesis
Viral infection of the CNS occurs via two distinct routes: hematogenous and neuronal. Enteroviruses and arboviruses are carried to the CNS via the blood stream, while HSV and the rabies virus are carried to the CNS via nerve cells themselves. Because viruses must replicate intracellularly, the ability to cause disease is largely determined by whether viral surface proteins can attach to specific receptors on specific cells in affected tissues—i.e., tissue trophism. An example of viral tissue tropism being determined by the combination of viral surface proteins and specific tissue receptors is that of the binding of the HIV GP120 to the CD4+ receptor on T4 lymphocytes. Other tissues with HIV tropism include monocytes and derived cells (macrophages), Langerhans cells, glial cells, and dendritic cells, all of which express the CD4+ receptor. Cells that do not express this receptor generally do not become infected with HIV [84]. So important are these surface binding sites for their respective cellular receptors that several viruses such as rhinovirus, influenza virus, and poliovirus have evolved “sophisticated” molecular mechanisms to protect these sites from the host immune response.
Enteroviruses are transmitted in human populations largely through fecal-oral transmission. These viruses survive stomach acid, replicate in the intestine, and an initial viremia leads to infection of multiple organs within the body. A secondary viremia from these sources can lead to CNS involvement. The prompt production of antibody disrupts this second viremia and prevents invasion of the CNS. In the case of arboviruses, humans typically become infected when an infectious mosquito pierces the host epidermis to take a blood meal, depositing virus principally in the extravascular tissue although direct inoculation into the bloodstream can occur. Local replication is followed by viremia, and brain involvement is probably determined by viral tropism and the rapidity of the host immune response.
For CNS infections that occur following a viremia, invasion of the brain involves attachment of the virus to the endothelial cells, presumably via specific receptors. Following invasion, an acute inflammatory reaction occurs with a perivascular distribution within the brain parenchyma and varying degrees of involvement of the meninges, depending on the infecting viral agent. The perivascular inflammatory response is predominately mononuclear although polymorphonuclear leukocytes may be seen. Infection of neural cells results in degenerative changes and phagocytosis by tissue macrophages or microglial cells. Some pathologic features are unique to certain viruses: for example, cerebral atrophy and production of multinucleated giant cells and multiple nodules of infected microglia are seen in the white matter in patients with HIV encephalitis (Fig. 22.10), or the characteristic features of multinucleation, nuclear molding, chromatin margination, ground glass nuclei, and Cowdry type A intranuclear inclusion bodies seen in HSV infections (Fig. 22.11), with extensive asymmetrical necrosis in the temporal lobes, in the insulae, and in the cingulate gyri, typically seen in neuroimaging studies (Fig. 22.12a, b) [85]. In the case of rabies, histopathologic evidence of rabies encephalomyelitis (inflammation) in brain tissue and meninges includes mononuclear infiltration, perivascular cuffing of lymphocytes or polymorphonuclear cells, lymphocytic foci, Babes nodules consisting of glial cells, and the pathognomonic Negri body—an intracytoplasmic inclusion body within which the virus can be identified (Fig. 22.13a, b).
Fig. 22.10.
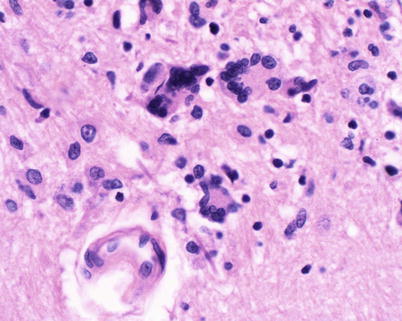
Primary HIV infection of the CNS. The pathology specimen is from the brain of a 25-year-old male with recently diagnosed HIV. The patient developed pneumonia and died of respiratory failure. The figure shows perivascular inflammation with multiple giant cells involving a small vessel in the pons (H&E, 40×) (Courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Fig. 22.11.
Herpes encephalitis (autopsy case: 32-year-old male patient with end-stage AIDS). Figure shows herpesvirus-infected neurons with marginated chromatin and glassy, smudged nuclei. H&E 60× (Courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Fig. 22.12.
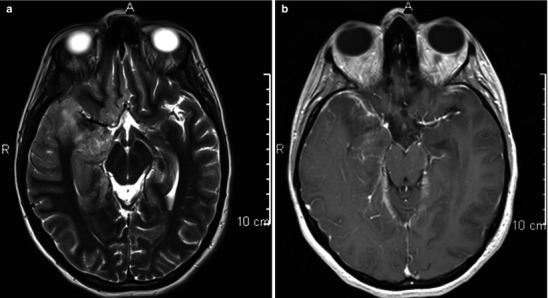
(a, b) Herpes (HSV-1) encephalitis; images include low convexity axial T2-w sequence (a) and post-contrast T1-w sequence in the same brain section level. The T2-w image (a) illustrates cytogenic edema distributed not only within the anterior and mesial right temporal lobe but also within the frontotemporal association bundle and basifrontal cortex. It involves gray and white matter. There is less obvious change on the left. This pattern, in the right clinical context, is typical of HSV-1 cerebritis. It has a differential of tumoral gliomatosis, but the latter typically has a more prolonged presenting clinical course. The post-contrast T1-w image (b) demonstrates pial and perivascular enhancement. HSV-1 is an angiophilic organism which can produce a necrotizing intrinsic angiitis which can cause subarachnoid hemorrhage, although not in this case
Fig. 22.13.
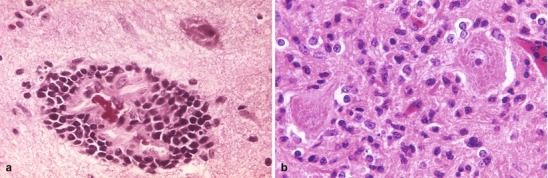
(a) This micrograph depicts the histopathologic changes associated with rabies encephalitis prepared using an H&E stain. Note the perivascular cuffing due to the perivascular accumulation of inflammatory cell infiltrates (i.e., lymphocytes and polymorphonuclear leukocytes). Figure (b) is a photomicrograph of H&E stained brain tissue from a rabies encephalitis patient displaying the pathognomonic finding of Negri bodies within the neuronal cytoplasm (Both: Courtesy of the Centers for Disease Control and Prevention)
Some viral infections, most notably HSV and rabies, spread to the CNS via a neuronal route. In the case of HSV, the distribution involves the medial part of the temporal lobe bilaterally with one temporal lobe generally much more involved than the other. Autopsy studies carried out on patients who died during active HSV encephalitis show the presence of virus in the olfactory bulbs, olfactory tracts, and the tracts of the limbic system which end in the hippocampus, amygdala, insula, cingulate gyrus, and olfactory cortex [86]. Thus, the virus appears to gain access to the CNS from the nasal mucosa to the olfactory bulbs and olfactory tracts, although the mechanism by which the virus does this remains unknown. About two-thirds of cases of HSV encephalitis in adults and older children occur in patients who have antibody to the virus at the time of infection. Many of these patients have a history of cold sores dating back 20–30 years. In the other one-third of patients, antibody to HSV is lacking at the time of onset of symptoms indicating that the encephalitis is likely part of the primary infection. Approximately 90–95 % of the cases of HSV encephalitis in older children and adults are due to HSV type I, with the remaining 5–10 % due to HSV type II. Neonatal herpes appears to be different, in that 90–95 % of these cases are due to HSV type II acquired from maternal or other sources at the time of birth. Infection of the CNS in neonates generally follows systemic viremic spread; however, there is no temporal lobe localization.
Rabies infection may result from contact with saliva or other secretions from infected animals as well as the animal bite itself. Rabies replicates initially at the local site of inoculation, and for this reason emergency preventive measures, such as thorough cleansing of the wound and infiltration with human rabies immunoglobulin, can be effective in preventing infection with this viral agent. In the process of local replication, the rabies virus invades the nerve sheaths and is transported via nerve cells to the CNS. The rapidity of the rabies virus in reaching the CNS is a function of the distance of the nerve endings from the CNS. Thus, bites on the lower extremities may take months to produce symptoms in the CNS, whereas bites in the face may reach the CNS within weeks. It is also important to recognize that the initial incident may be forgotten because of the length of time of the onset of CNS symptoms after the initial infecting event or because the inoculation may be unapparent as has been reported for bats [87]. Therefore, a high index of suspicion for rabies is essential when managing patients with encephalitis of unknown cause, especially in patients who exhibit signs of hyperirritability.
Clinical Manifestations
Viral meningitis is an acute illness characterized by fever, headache, stiff neck, photophobia, and varying degrees of nonspecific symptoms such as malaise, myalgia, nausea, vomiting, abdominal pain, or diarrhea. Generally, neither disturbance of mental status nor abnormal neurological signs are characteristic of viral meningitis. The presence of obtundation, disorientation, seizures, or localized neurologic signs should suggest brain parenchymal involvement and a diagnosis of encephalitis or meningoencephalitis. Neck stiffness is generally less severe compared with bacterial meningitis. Clinical clues to an enteroviral etiology include presence of a viral exanthem, pleuropericarditis, pleurodynia, painful oropharyngeal ulcers, or peripheral vesicular lesions suggestive of hand, foot, and mouth disease. The CSF in patients with viral meningitis is usually clear with an elevated white cell count; polymorphonuclear leukocytes may predominate within the first 24–48 h, although this number rarely exceeds 80 % of total CSF white cells. CSF protein is generally mildly elevated, and the glucose is normal with the occasional exception in about 10–20 % of patients with mumps and less often in patients with enterovirus or HSV infection. In viral meningitis, the CSF gram stain will be negative and routine CSF cultures will not yield bacterial growth. When the initial CSF analysis shows over 50 % polymorphonuclear leukocytes, it is not uncommon for clinicians to repeat the lumbar puncture over the next 12–24 h to determine whether there is a shift towards a lymphocytic predominance, as one would expect in viral meningitis.
A parameningeal infectious focus (e.g., brain abscess) will characteristically be associated with a CSF pleocytosis, a likely elevated protein, and a normal glucose. Thus, a patient with an epidural abscess or a brain abscess could present with mild headache, fever, and a CSF picture identical to that of viral meningitis. Some cases of sphenoid or frontal sinusitis may feature a CSF pleocytosis. Patients with fungal and tuberculous meningitis may also present with headache, fever, and stiff neck, but in general the clinical course is longer than an acute viral meningitis. The CSF in patients with fungal or tuberculous meningitis generally has a relatively low glucose level, usually less than 40 mg/dL.
The CSF in patients with cryptococcal meningitis may be completely normal. Fever, headache, and nonspecific CSF findings can also be seen in other infections, such as syphilis, ehrlichiosis, and noninfectious conditions, such as sarcoidosis, Behcet’s disease, systemic lupus erythematosus, vasculitis, or uveoparotitis.
Viruses other than enteroviruses can also produce aseptic meningitis. For example, HSV type II produces very typical aseptic meningitis with low-grade fever, headache, stiff neck, and photophobia as part of primary genital herpes infection. Therefore, it is important to question the sexually active patient about a history of genital herpetic lesions and to perform a pelvic examination in women where indicated. Aseptic meningitis can also be seen as part of the syndrome of primary HIV infection. Certain strains of leptospirosis will typically present with aseptic meningitis; however, most cases present in conjunction with systemic disease and severe involvement of other organs such as lung, liver, and kidney. Lymphocytic choriomeningitis virus (LCMV) belongs to the family Arenavirus and is found globally [88]. LCMV meningitis is typically associated with exposure to rodents, such as common house mouse (Mus musculus and M. domesticus), hamsters, and guinea pigs. Transmission occurs by inhalation (aerosol and droplet), fomites, or direct contact with excreta or blood from infected rodents. The incubation period is 1–2 weeks; symptoms are nonspecific and include fever, chills, myalgia, headache, photophobia, anorexia, pharyngitis, and cough. CNS invasion is seen only in a few patients either after an initial febrile illness. During the neurologic phase, patients acquire aseptic meningitis and peripheral leukocytosis. CSF leukocyte cell counts are often greater than 1,000 cells /μL, and glucose levels are low. LCMV meningitis may be associated with an ascending paralysis, transverse myelitis, or encephalitis; overall case fatality is less than 1 % [88].
The laboratory diagnosis of viral meningitis is generally one of exclusion. Viral cultures of CSF that yield growth of enteroviruses are diagnostic. However, these are positive in only 30–50 % of cases. PCR has become available for the diagnosis of enteroviral meningitis and these PCR platforms correlate very well with the results of viral culture. Unfortunately, PCR testing may not be routinely available at clinical microbiology laboratories for various reasons, including cost cutting measures and lack of trained personnel. Sending specimens to reference laboratories or highly specialized research laboratories is an alternative, but results generally will not be available during the acute phase of the patient’s illness. In the case of HSV meningitis, the diagnosis is confirmed if HSV is cultured from CSF or detected by PCR. For aseptic meningitis associated with systemic infections, such as leptospirosis, syphilis, or ehrlichiosis, diagnosis can be confirmed through standard serologic testing generally available at state public health laboratories or reference laboratories.
Because there are over 75 different enterovirus serotypes, testing is only possible for a subset of these. In addition there is tremendous overlap in the serologic response between the different serotypes such that seroconversion to one or more enterovirus serotype can occur. Moreover, since there is no specific therapy, expensive laboratory testing is unlikely to affect patient outcome. For these reasons, serologic studies for the diagnosis of enterovirus infections are generally not indicated.
A common diagnostic misconception is the usefulness of CSF antibodies. With the exception of the venereal disease research laboratory (VDRL) test, which indicates active CNS syphilis, and the ratio of measles antibodies in CSF to serum levels in extremely rare cases of subacute sclerosing panencephalitis (SSPE), routine CSF antibody testing does not affect patient outcomes. In general, infectious viral agents can more readily be diagnosed from serum rather than CSF studies. Tests for common viral infections and preferred diagnostic methods are outlined in Table 22.4.
Table 22.4.
Laboratory diagnosis of selected viral diseases
| Virus | CNS disease | Serology | Viral culture | Direct antigen | PCR/other |
|---|---|---|---|---|---|
| Herpes viruses | |||||
| Herpes simplex | Temporal lobe encephalitis | About 2/3 seropositive on admission, not helpful in Dx | Almost always negative in CSF, throat, etc. | None for CSF | ≥ 90 % sensitive on CSF—reference labs and some academic medical centers only |
| FA can be done on brain biopsy | |||||
| Varicella | Encephalitis in HIV patients | Not helpful diagnostically | Usually negative in CSF, throat, etc. | None for CSF | Reference labs only |
| FA can be done on brain biopsy | MRI may be suggestive | ||||
| Cytomegalovirus | Encephalitis in HIV patients | Almost always positive | Urine, throat, blood may be positive—consistent with but not proof of encephalitis | Antigenemia test on blood, positive test consistent with but not proof of encephalitis | If CSF positive, encephalitis likely, but asymptomatic HIV patients may also be positive |
| MRI may be suggestive | |||||
| Epstein-Barr Virus | Rare encephalitis in mononucleosis | Monospot test good presumptive test, may be negative in up to 20 % in 1st week | Not available | Not available | Reference labs only, not needed for diagnosis |
| VCA-IgG and IgM, positive IgM virtually diagnostic | |||||
| Human herpes virus 6 and 7 (HHV 6 and 7) | Seizures, encephalitis in 1–3 year olds | Reference labs only | Reference labs only | Not available | Reference labs only |
| Respiratory viruses | |||||
| Influenza A and B | Parainfluenza occasionally, others rarely cause encephalitis | Not useful | NP swabs; throat washings | Direct antigen ELISA available for RSV (excellent sensitivity), and Influenza (moderately sensitive) | Not available |
| Parainfluenza 1–3 | Cultures via bronchoscopy | ||||
| Adenovirus | Excellent sensitivity, diagnostic if positive | ||||
| Respiratory syncytial virus (RSV) | |||||
| Enterovirus | |||||
| Coxsackie | Summertime outbreaks of meningitis, meningoencephalitis | Not useful: too many serotypes and too much cross-reactivity | Send stool, throat, CSF | None | Available in reference labs, picks up most serotypes, may take a week to get results |
| Echovirus | If throat or CSF culture positive—diagnostically definitive | ||||
| If stool culture positive—presumptive (enteroviruses may be shed in stool for weeks) | |||||
| Arboviruses | |||||
| St. Louis encephalitis | Summertime outbreaks of encephalitis | Diagnostic, if positive | Generally not available | Not available | Not available |
| California encephalitis | Serum is preferred specimen; CSF not helpful | ||||
| Western equine | Done in reference laboratory and public health labs | ||||
| Eastern equine | |||||
| La Crosse | |||||
| West Nile virus | |||||
| HIV | Encephalopathy | ELISA | Research labs only | P 24 antigen in serum | Reference labs, many tertiary medical centers, used for following treatment |
| Confirm with Western Blot | |||||
| Lymphocytic choriomeningitis virus | Meningitis | Reference laboratory | Not available | Not available | Not available |
| JC virus | Progressive multifocal leukoencephalopathy | Reference laboratory | Not available | Not available | Available in reference laboratories |
| Mostly in HIV & other immunocompromised patients | Diagnostic if positive on CSF | ||||
| MRI highly suggestive | |||||
| Brain biopsy definitive | |||||
| Rabies | Encephalitis | Reference or state public health labs | Research labs only | Direct fluorescent antibody staining of hair follicles in skin biopsy from nape of neck above the hairline—50 % positive in 1st week, higher later | RT-PCR may be available in reference labs, state public health labs. Send saliva, CSF, or tissue |
| Antibody is generally undetectable before day 6, 50 % by day 8, and 100 % by day 15 | |||||
Adapted with permission from Rand et al. [521]
Finally, certain drugs such as sulfa and nonsteroidal anti-inflammatory agents can produce acute syndromes of aseptic meningitis (Table 22.5). Other noninfectious factors have been associated with an aseptic meningitis picture, including intravenous immunoglobulin, intrathecal administration of drugs, receipt of certain vaccines, malignancies, autoimmune conditions, and connective tissue disorders (Table 22.5) [89].
Table 22.5.
Nonviral causes of aseptic meningitis and encephalomyelitis
| Infective causes | |
| Ehrlichia spp. | Rocky Mountain spotted fever |
| Bacterial endocarditis | Brain abscess/cerebritis |
| Staphylococcus aureus | Treponema pallidum meningitis |
| Lyme disease | Leptospirosis |
| Mycoplasma pneumoniae | Listeria monocytogenes (rhomboid encephalitis) |
| Typhus | Legionella spp. |
| Cat-scratch disease | Nocardia spp. |
| Mycobacterium tuberculosis meningitis | Cryptococcus neoformans meningitis |
| Histoplasmosis | Coccidioidomycosis |
| Cerebral amebiasis | Cerebral malaria |
| Trypanosomiasis | Cerebral abscess and other parameningeal infections |
| Partially treated bacterial meningitis | CNS cysts (e.g., craniopharyngioma, dermoid/epidermoid) |
| Noninfective medical conditions | |
| Malignancy, especially lymphoma | Behçet’s disease |
| Still’s Disease | Systemic lupus erythematosus |
| Chronic subdural hematoma | Vasculitis |
| Intrathecal injections | Neurosurgery-related procedures |
| Drugs | |
| Certain nonsteroidal anti-inflammatory drugs | |
| Antimicrobial agents, including trimethoprim, cephalosporins, penicillin, amoxicillin, isoniazid, ciprofloxacin, metronidazole, and sulfonamides | |
| Carbamazepine | |
| Ranitidine, famotidine | |
| Azathioprine | |
| Sulfasalazine | |
| Indinavir | |
| Vaccines: measles, mumps, rubella (MMR), alone or in combination | |
| Monoclonal antibodies (e.g., muromonab-CD3) [OKT3] that targets the CD3 receptor | |
| Intravenous immunoglobulin | |
Adapted with permission from Rand et al. [521]
The hallmark of the presentation of viral encephalitis is fever, headache, confusion, drowsiness, and convulsions. Prominent signs include an altered level of consciousness with or without focal neurologic signs or meningism (i.e., the triad of neck stiffness, photophobia, and headache) in the setting of an acute febrile illness. By and large, the differential diagnosis is the same as for acute bacterial or viral meningitis. Varying degrees of nuchal rigidity can be present in patients with encephalitis. Lumbar puncture yields CSF with a picture similar to that of viral meningitis. HSV, VZV, arboviruses, and enteroviruses can now be diagnosed by PCR analyses of CSF specimens. MRI imaging is a useful test for ascertaining HSV encephalitis and for differentiating post infectious encephalomyelitis from viral encephalitis. It is important to recognize that some patients with arbovirus encephalitis, particularly Eastern equine encephalitis and West Nile virus encephalitis, may have focal lesions by MRI (Fig. 22.14). However, the distribution of these lesions is not consistent and differs from the pattern seen on neuroimaging studies of HSV encephalitis (Fig. 22.12a, b). Diagnosis of arbovirus encephalitis can almost always be made by serological testing as antibody titers to all of the common arboviruses are generally present by the time the patient presents with symptoms. Because there is only a very low background frequency of these antibodies in the general healthy population, a positive arbovirus serology can be accepted as clinically definitive of exposure or infection.
Fig. 22.14.
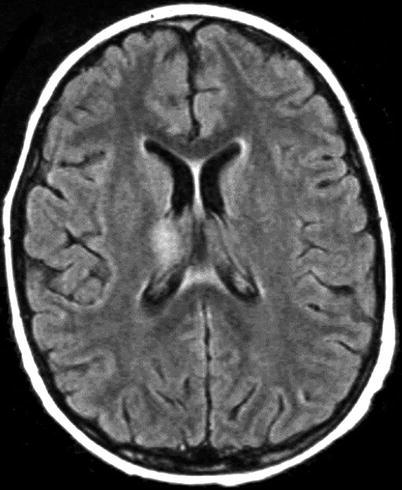
West Nile viral thalamic encephalitis T2-w mid-convexity brain section. Arbovirus (West Nile virus in this example) encephalitis typically has multicentric areas of cytogenic edema, which typically involve gray matter both in the cortex and in the central nuclear structures. The lesions are usually multicentric but not symmetric between hemispheres. In this example cytogenic edema is evident in the right thalamus and in the splenium of the corpus callosum. The splenial abnormality can be from the virus but may also occur following recent seizure activity
The diagnosis of HSV encephalitis is of critical importance because of the effectiveness of acyclovir in improving patient outcomes. HSV is the most common cause of sporadic encephalitis. Patients with HSV type I encephalitis generally present with a 3- to 5-day history of fever, headache, and focal signs, including dysphasia and personality changes that may progress to obtundation and coma; the latter can occur abruptly and may be associated with the onset of seizures. HSV encephalitis can occur in any age group from childhood through old age at any time of the year—i.e., there is no characteristic seasonal variation pattern in occurrence. The incidence is estimated at 1 in 250,000 to 1 in 500,000 people per year; HSV encephalitis accounts for approximately 10–20 % of viral encephalitides in the United States.
A diagnosis of HSV encephalitis is supported by MRI findings showing bilateral temporal lobe involvement that is generally asymmetrical (Fig. 22.12a, b). CT imaging may reveal frontotemporal changes in HSV encephalitis. In untreated patients, this may progress to hemorrhagic lesions; swelling of the more severely affected temporal lobe may result in a mass effect sufficient to produce a shift of the midline structures and tentorial herniation. PCR for HSV in the spinal fluid is almost uniformly positive in patients with HSV encephalitis, despite the fact that in 1–4 % of cases, one cannot grow the virus from CSF or specimens from other anatomic sites in patients [90, 91]. The use of PCR in combination with the detection of a specific intrathecal antibody response to HSV currently represents the most reliable strategy for the diagnosis and monitoring of the treatment of adult patients with HSV encephalitis [90–93]. Serologic diagnosis is not particularly helpful early on, although, in general, all patients with HSV encephalitis will show a significant rise in HSV titer both in the spinal fluid and serum; seroconversion can be ascertained in patients with primary HSV infection. EEG studies may reveal focal features.
Conditions that mimic encephalitis include brain abscess, subdural empyema, cerebritis due to Listeria spp., mycoplasma, fungal infections, tuberculosis, cryptococcus, rickettsia, toxoplasmosis, mucor, and agents frequently associated with bacterial meningitis, such as pneumococcus and meningococcus (Fig. 22.5c, d) [94, 95]. Influenza A virus can cause encephalitis during epidemics; children are particularly affected. Tumors, subdural hematomas, CNS lupus, adrenal leukodystrophy, acute strokes, neuroleptic malignant syndrome, or Reye’s syndrome can mimic the symptoms and signs of encephalitis.
Encephalitis can sometimes occur as part of systemic infection with common viruses that do not normally produce encephalitis. For example, EBV infection can, on occasion, present with seizures and even coma; these patients generally recover completely. CNS involvement with toxoplasmosis, lymphoma, VZV, and CMV in HIV-infected patients can also mimic the features of encephalitis. Table 22.5 lists nonviral infections that may resemble viral encephalitis.
Although rare, rabies should be considered in atypical cases of encephalitis or in cases where results of all investigations are negative. Because affected patients often do not have a history of having been bitten by an animal, a history of close contact with potentially infected animals, including bats, should be sought. Infection is almost always from inoculation but occasionally by inhalation. Rabies has been transmitted by corneal and solid organ allografts [96, 97]. The virus is transmitted to the CNS via nerve trunks. Proliferation in nerve cells in the brain and peripheral ganglia leads to an invariably fatal meningoencephalitis. The incubation period varies anywhere from 2 weeks more than year. Proximal bites with a relatively large inoculum of virus tend to be associated with shorter incubation periods. The onset is rapid with fever, anxiety, insomnia, headache, malaise, myalgia, fatigue, anorexia, nausea, vomiting, sore throat, and cough. The patient may complain of symptoms suggestive of paresthesia or fasciculation at the site of the original animal bite due to viral replication at the site of inoculation or in the dorsal ganglia of the sensory nerve supplying that area. The disease quickly progresses to an encephalitic phase consisting of agitation, excitation, and excessive motor activity. Patients may experience hallucinations, become combative, and develop muscle spasms with opisthotonus and involvement of the respiratory muscles. Painful spasms of the throat muscles, precipitated by attempts to swallow, may follow—this explains why patients tend to avoid drinking or swallowing. Spasms may be precipitated by mere air blowing onto the face; seizures are frequent. Periods of hallucinations and aberrant mentation may alternate with lucid periods that get progressively shorter as the disease progresses. Hyperesthesias with excessive reactivity to normal stimulation of light, sound, and touch are very common; autonomic nervous system changes such as dilated pupils, increased salivation, lacrimation, perspiration, and postural hypotension occur. Ultimately, brain stem function is affected with cranial nerve palsies, optic neuritis, and the characteristic hydrophobia due to the painful, violent involuntary contractions of the muscles of respiration and those in the pharynx and the larynx, initiated by attempts to swallow. Eventually the disease progresses to cardiorespiratory depression, coma, and death. Occasionally, rabies may present as an ascending paralysis clinically similar to the Guillain-Barré syndrome; corneal transplants from two patients presumed to have died from Guillain-Barré actually transmitted clinical rabies, resulting in the death of the recipients [98, 99]. The laboratory diagnosis of rabies requires viral isolation, positive serology (assuming the patient has not been immunized), or demonstration of the characteristic Negri bodies in brain tissue. Laboratory diagnostic evaluation for rabies includes serological testing plus demonstration of viral antigen by IFA in infected tissue, including corneal scrapings, skin biopsies or brain biopsies, and analysis of CSF or saliva specimens for rabies virus antigen or RNA [100]. Survival of patients with rabies is rare; most of the few who have survived received the rabies vaccine prior to the onset of illness.
In patients who have traveled overseas, encephalitis may be caused by various other infectious diseases, including Japanese B encephalitis, Murray Valley encephalitis, Omsk hemorrhagic fever, Kyasanur forest disease complex, Powassan virus, louping ill, Russian spring-summer encephalitis, Rift Valley fever, yellow fever, dengue, chikungunya, Hantaan virus, Puumala virus (a species of hantavirus), and the highly fatal hemorrhagic fevers caused by the Marburg, Ebola, and Lassa fever viruses.
B virus infection is caused by Macacine herpesvirus 1 (formerly Cercopithecine herpesvirus 1 (CHV-1)), an alphaherpesvirus closely related to herpes simplex virus. B virus is also commonly referred to as herpes B, monkey B virus, herpesvirus simiae, and herpesvirus B. The virus is commonly found among macaque monkeys, including rhesus macaques, pig-tailed macaques, and cynomolgus monkeys (also called crab-eating or long-tailed macaques), any of which can harbor latent B virus infection and appear to be natural hosts for the virus. Monkeys infected with B virus usually have no or only mild symptoms. In addition, rabbits, guinea pigs, and mice can be experimentally infected with B virus. The virus is related to human HSV but humans have little native ability to contain it in contrast to its natural host in whom it produces “cold sores.” Human infection usually results from bites or scratches from macaques or mucocutaneous exposure to monkey saliva; laboratory personnel who work with ostensibly healthy monkeys or their tissues are particularly at risk. Indirect contact transmission, such as a needlestick injury with a contaminated needle has been documented. B virus is transmitted to humans from saliva in monkeys and reaches the brain via nerves at the site of the monkey bite. Patients acquire an ascending myelitis and fulminant meningoencephalitis, which leads to death. Infection is diagnosed by a rise in antibody titer or by isolating the virus from the CNS. There are seven different exposures for which postexposure prophylaxis is recommended: if postexposure prophylaxis is administered, it should be started soon (within hours) after the exposure. Neurologic tests should include lumbar puncture and MRI of the brain; electroencephalography (EEG) should also be considered. CSF samples should be sent for culture, PCR detection of viral DNA, and serologic testing. While use of intravenous acyclovir and ganciclovir therapy for patients with the early stages of B virus disease, including patients with early signs of CNS disease has been associated with increased survival for some patients, antiviral therapy has not been effective in patients with advanced encephalomyelitis [101].
Treatment
No specific drug or serologic therapy is currently available for enterovirus or arbovirus infections. In general, viral meningitis due to enteroviruses is clinically mild, and most patients can be treated without admission to the hospital unless bacterial meningitis is a possibility in the differential diagnosis and needs to be ruled out. Patients with enteroviral meningitis usually recover within 7–10 days without antiviral therapy.
Intravenous acyclovir is indicated for patients with HSV or VZV meningitis. In patients with symptoms suggestive of encephalitis or brain parenchymal involvement, and in whom appropriate radiologic imaging studies have ruled out other pathology, such as brain abscess or subdural empyema, initiation of empiric intravenous acyclovir intravenously in doses appropriate for HSV encephalitis is indicated. Complications from acyclovir are relatively uncommon; timely diagnosis is of paramount importance since a successful outcome is largely associated with early institution of acyclovir therapy. Brain biopsy is not justified to prove the presence of herpes encephalitis prior to therapy. Before antiviral agents became available for the treatment of HSV encephalitis, the disease was fatal in approximately 70 % of patients, with an additional 20–25 % surviving with severe disabilities. The dose of acyclovir is 10 mg/kg IV every 8 h for 14–21 days.
Brain Abscess
Brain abscesses have been recognized since the days of Hippocrates in 460 BC. By definition, a brain abscess is a localized suppurative infection of the brain parenchyma [102]. The incidence in the general population has been estimated at 1.3–100,000 person-years, with the rates slightly higher in children between 5 and 9 years of age and after the age of 60 years. Most series document a male preponderance of between 2:1 and 3:1, and the age distribution is somewhat dependent on the associated underlying etiologies [103, 104]. While the etiology and distribution of associated diseases has remained essentially unchanged over the years for pyogenic brain abscesses, the AIDS epidemic has led to the occurrence of a large group of patients with brain abscess due to toxoplasmosis. Highly active antiretroviral therapy (HAART) has resulted in a reduction of morbidity and mortality in HIV-associated cerebral opportunistic infections.
Pathogenesis
Brain abscesses develop as localized areas of cerebritis (i.e., poorly demarcated areas of encephalitis), initially consisting of bacteria in the brain parenchyma together with inflammation and edema. Over the ensuing days this area of cerebritis becomes more localized with the development of necrosis in the middle and a ring-enhancing capsule. Ultimately, host defenses lead to the development of a well-formed fibrous capsule. The most common predisposing conditions for the development of a brain abscess are infections in the middle ear, paranasal sinuses, mastoids, and teeth (dental abscess). It is believed that bacteria reach the brain through valveless emissary veins, which traverse the cranium into the venous drainage system of the brain, or retrograde spread through the venous system. Alternatively, direct extension through an area of osteitis or osteomyelitis adjacent to the sinus or middle ear infection provides access to the CNS; chronic otitis media is a common predisposing factor with the abscesses most frequently forming in the temporal lobe or cerebellum. The other major mechanism by which the brain parenchyma becomes seeded is via metastatic transmission through the cerebral arteries from an extracranial focus of infection. Approximately 20 % of brain abscesses arise from a contiguous focus; 25 % are associated with hematogenous spread from a distant focus, such as a pyogenic lung abscess or bronchiectasis, and 25 % occur following trauma [105–107].
Hematogenous brain abscesses generally tend to be multiple and be located at the gray white matter junction; they also tend to follow a vascular distribution within the brain. Hematogenous dissemination from a contiguous focus of infection has also been described. Other distant foci that have been associated with brain abscess include wound infections, osteomyelitis, pelvic infection, cholecystitis, and other intra-abdominal foci. In fact, any procedure that results in a transient bacteremia can on occasion be associated with the subsequent development of a brain abscess. Despite its chronicity and relatively high frequency of bacteremia, and involvement of the brain in 20–40 % of cases, endocarditis accounts for only 1–5 % of cases of brain abscess [108, 109]. Patients with cyanotic congenital heart disease are at increased risk of acquiring brain abscesses. A significant number of brain abscesses are associated with penetrating trauma such as gunshot wounds, depressed skull fractures with retained with bone fragments, cranial penetration from objects such as pencils, animal bites, or even as a complication of cervical traction associated with pin-site infection. In patients with HIV infection, reactivation of toxoplasmosis can lead to brain abscesses. In approximately 25 % of cases no underlying etiology can be established.
Microbiology
The bacterial etiology of brain abscess is to a great extent dependent on the location of the abscess and the predisposing factors. Thus, aerobic, anaerobic, and microaerophilic streptococci are the most frequently isolated bacterial species. Staphylococcus aureus is the underlying cause of 25 % of brain abscesses and often associated with trauma, endocarditis, or following a neurosurgical procedure. In addition to streptococci, brain abscess associated with paranasal sinus or chronic otitis media infection may be caused by Haemophilus species, Bacteroides species, other anaerobes, and Pseudomonas aeruginosa in the case of chronic otitis media. If the source of bacteremia is intra-abdominal, Enterobacteriaceae, enterococci, and anaerobes are likely to be implicated; a urinary tract source is more likely associated with Pseudomonas spp. or Enterobacteriaceae, but not anaerobes. Brain abscess caused by anaerobes, including actinomyces, may be associated with spread from a lung abscess [110].
Although S. aureus is the most common organism complicating penetrating trauma, Clostridium species and Enterobacteriaceae are also commonly implicated. The nature of the precipitating trauma is important: for example, if trauma occurs in a water environment, Pseudomonas spp. and Aeromonas spp. would have to be among the organisms considered. Microorganisms associated with postoperative infections include S. aureus, Staphylococcus epidermidis and Enterobacteriaceae, and Pseudomonas spp.
Nocardia species are uncommon but important causes of brain abscesses (Fig. 22.15a, b), especially in immunocompromised populations [111–113]. The clinical presentation of Nocardia infections in the CNS is the same in normal and compromised hosts, although more frequent in compromised hosts. In a series of 11 cases and review of 120 cases of nocardial brain abscess in the literature, concomitant pulmonary disease was present in 34 %. Most of the brain abscesses were single; about one-third were multiple, and, overall, 38 % of the cases occurred in patients who were immunocompromised by virtue of HIV or other causes [113]. Rarely, Mycobacterium tuberculosis may produce a space-occupying lesion (tuberculoma), and while uncommon in the United States, tuberculomas are relatively common causes of brain abscesses in some less-developed countries [114, 115].
Fig. 22.15.
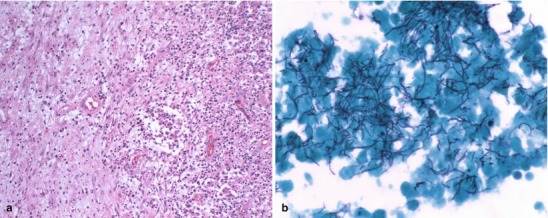
(a, b) A 52-year-old female with a left frontal lobe intraparenchymal abscess caused by Nocardia sp. Figure (a) shows the thickened collagenous abscess wall with neovascularization adjacent to acute inflammatory infiltrate. H&E, 10×. In figure (b), Gomori methenamine silver (GMS) stain highlights numerous branching, filamentous bacteria. GMS, 60× (Both courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Yeasts and fungi are important causes of brain abscess. Candida albicans almost never causes isolated brain abscesses but may cause microabscesses in association with disseminated candidiasis. Although cryptococcal infection of the CNS typically affects patients with HIV infection, cryptococcomas are rarely observed [116]. Agents of phaeohyphomycosis, such as Cladosporium, Bipolaris, Curvularia, and Wangiella, as well as the agents of chromoblastomycosis have all been reported as causes of brain abscesses.
Aspergillus spp. are well-recognized causes of brain abscess but are almost always limited to immunocompromised patient populations, transplant patients in particular. Aspergillus fumigatus is the most common species affecting humans; maxillary sinusitis of dental origin or the lungs are the most common sites of primary Aspergillus spp. infection [117]. Infection reaches the brain directly from the nasal sinuses via vascular channels or is blood-borne from the lungs and gastrointestinal tract [117]. Zygomycoses such as Mucor Rhizopus and Rhizomucor produce brain infection by direct extension from the paranasal sinuses in poorly controlled diabetics. Cerebral mucormycosis without rhino-orbital or systemic involvement is an extremely rare condition mostly associated with parenteral drug abuse [118].
Protozoa and other parasites are important causes of brain abscess. The incidence of protozoal and helminthic infestations of the CNS is less than 1 %, but these infestations tend to follow a fatal course and are more common among children, the elderly, and immunocompromised individuals. Toxoplasma gondii is probably the most frequent protozoal cause of brain abscess in the United States and is almost entirely associated with HIV infection. Strongyloides, entamoeba, echinococcus, paragonimus, trichinosis, sparganosis, and angiostrongylus have all been reported, particularly in less-developed nations. Rarely, brain abscess due to naegleria and acanthamoeba occur in the United States.
Clinical Manifestations
Presentation, generally, is that of an intracranial mass lesion: subacute onset, headache, and focal neurological deficits [106, 109]. However, the classic triad of focal neurological signs, fever, and headache is present in less than 50 % of cases; nuchal rigidity is not prominent in patients with brain abscess. Similarly, patients with subdural empyema seem to present with a more localized headache and focal neurologic symptoms and altered level of consciousness. Other common signs and symptoms include fever, chills, seizures, nausea, vomiting, and altered sensorium [119]. Depending on the location of the lesion, fever and stiff neck may be present. Headache of varying degree of severity is the most consistent symptom among patients with brain abscess. The headache is generally not well localized and may be mild and difficult to differentiate from ordinary headaches. Fever is present in 40–50 % of cases, and focal neurologic symptoms and signs, such as hemiparesis, aphasia, ataxia, and sensory deficits, may be present in one-third to one-half of cases. Papilledema as a reflection of increased ICP is present in only a minority of cases [112, 119]. Likewise, seizures are observed in approximately 25–45 % of patients by the time they present. The seizures are most often generalized and associated with frontal lobe lesions. Other frequent CNS findings include altered mental states—confusion, aberrant behavior, and somnolence. To some extent, the presenting signs and symptoms are dependent on the location of the abscess. For example, cerebellar abscesses often present with nystagmus, ataxia in the ipsilateral extremities, vomiting, and dysmetria [120, 121]. Frontal lobe abscesses generally present with headache, drowsiness, and deterioration of mental status or aphasia together with hemiparesis and unilateral motor signs. Temporal lobe abscesses may present with or without aphasia or dysphasia, depending on whether the abscess is in the dominant hemisphere. Patients with occipital lobe abscesses may present with homonymous hemianopia, while typical features in parietal lobe abscesses include hemianesthesia, homonymous hemianopia, neglect of one-half of the body, alexia, or impaired spatial perception. Pituitary abscesses may simulate a tumor and can present with visual field defects and endocrine abnormalities. Brain stem abscesses typically exhibit facial weakness, fever, headache, hemiparesis, dysphasia, and vomiting.
The differential diagnosis includes a wide range of other CNS infections, such as meningitis, subdural empyema, epidural abscess, viral encephalitis, and noninfectious causes, such as migraine, intracerebral and subarachnoid hemorrhage, venous sinus thrombosis, or malignancy.
Diagnosis
The diagnosis of a brain abscess is best established by neuroimaging [122, 123]. MRI is more sensitive than CT scans and provides information on the size, location, and stage of the abscess together with the extent of surrounding edema and presence or absence of mass effect, such as midline shift, hydrocephalus, and impending herniation. The time line for bacterial brain abscess evolution is reasonably predictable and is divided into early cerebritis, late cerebritis, early abscess, mature abscess, and hopefully resolution (Fig. 22.8). Each of these stages takes place in roughly 4 days, hence the rule of 4 s for intra-axial brain abscess. This evolution of brain abscess is predicated on a normal immune system and does not apply in the immunocompromised host. Success of treatment is measured by progressively diminishing cross-sectional diameter of the abscess cavity. Radiologic changes in the abscess wall (i.e., thickness and contrast enhancement), surrounding edema, or status of internal contents are not reliable predictors of treatment success. Characteristic findings of a brain abscess are a ring-enhancing lesion in the contrast CT or MRI studies with a hypodense center reflecting the necrotic center of the abscess surrounded by a variable zone of edema [124] (Fig. 22.16a, b).
Fig. 22.16.
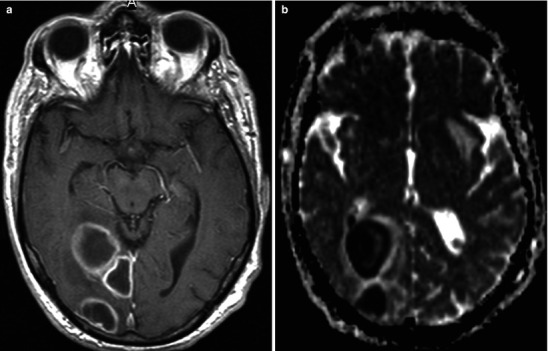
(a, b) Occipital abscess with daughter abscesses; images include two MRI images in the low convexity region of brain. Figure (a) is a post-contrast T1-w sequence and the left image is an ADC map (apparent diffusion coefficient) sequence, which provides information as to the diffusibility of the free water in the area of abnormality. The post-contrast TD1-w sequence (b) demonstrates contiguous ring-enhancing lesions in the right occipital brain. These findings do not distinguish between tumors versus tumefactive lesions, especially abscess. The diffusion sensitive imaging demonstrates free motion restriction within the central portions of the lesion. This in tandem with enhanced images is typical of brain abscess. The purulent material within the abscess contains increased water content (hyper intense of T2-w sequences), but it does not allow the water molecule to diffuse as well as fluid with the ventricles for instance. Hence, this ADC map shows the central water-restriction, often seen in pyogenic brain abscess
CT imaging studies are especially useful in defining contiguous head and neck pathology, which can be the source of infection. High detail CT imaging can detect the bone lysis of aggressive sinusitis and otomastoiditis better than MRI. CT is also better at defining bone dehiscence associated with prior trauma, which can also be the source for intracranial infection. MRI and CT are comparable in defining retropharyngeal abscess and chronic fungal sinus and skull base infections. CT is less sensitive than MRI for characterizing the stages of cerebritis and abscess formation and for excluding other causes of intra axial necrotic masses in the brain [125, 126]. MRI especially with diffusion brain imaging sequences maintains a high degree of sensitivity and specificity; see following discussion. However, some abscess remain in a chronic sequestered state and are difficult to differentiate from tumor; these are included in the category of tumefactive lesions along with others such as sequestered infarcts, thrombosed giant aneurysms, and Balo variant of multiple sclerosis, to name three.
The major difficulty with neuroimaging studies, especially CT imaging, is their sensitivity in differentiating an abscess from a tumor, including neuroblastomas and metastatic lesions. In one study, 8 out of 26 patients with a brain abscess were initially diagnosed as having a tumor [125]. Another study noted that in 18 % of CT scans from 100 patients with confirmed brain abscesses, the initial findings could not be radiographically distinguished from those typical of a malignancy [126].
MRI scanning provides soft tissue resolution and detail that is superior to that achieved with CT scanning [127]. In addition, there is no exposure to ionizing radiation, although the cost is substantially higher. On T1-weighted MRI scans, brain abscesses appear hypointense and show ring enhancement following administration of the contrast agent gadolinium. On T2-weighted sequences the central area of necrosis appears hyperintense and is surrounded by a well-defined hypointense capsule and readily discernible surrounding area of low density, representing cerebral edema. Another major advantage of MRI imaging studies is their ability to detect the cerebritis stage before the formation of the abscess with a fully developed capsule. The most helpful MR sequence is the diffusion techniques. This technique, most applicable once central necrosis has occurred, demonstrates the restriction of water movement within the forming fibrous abscess capsule. Other methods, such as magnetic resonance spectroscopy (MRS), can detect products of bacterial metabolism, such as lactate, acetate, or pyruvate, and may improve our ability to differentiate brain abscesses from malignancy [128–130].
Radionuclide brain scans using Indium-111-labeled leukocytes do not provide any advantage over conventional neuroimaging techniques. During the past several years, positron emission tomography (PET) with 18F-fluorodeoxyglucose (FDG) imaging have been playing larger roles in the diagnosis and management of patients with suspected brain abscesses [124, 131–134].
Routine laboratory studies are not particularly useful in the diagnosis of brain abscess. The white count is normal in 40 % of patients; the erythrocyte sedimentation rate is elevated in about 60 % while C-reactive protein levels are usually, but not invariably elevated. If the sedimentation rate is elevated, it may be useful to follow this over time to document a therapeutic response. Lumbar puncture in a patient with a space-occupying lesion is absolutely contraindicated unless an MRI or CT scan has indicated that herniation is unlikely to occur following an LP and there is a clear clinical suspicion of meningitis or meningeal carcinomatosis to justify taking the risk. The CSF in patients with brain abscess generally shows findings similar to any other parameningeal focus of infection, i.e., a pleocytosis with mixed neutrophils and lymphocytes, elevated protein levels, normal glucose levels, and a negative gram stain. CSF cultures are generally sterile, unless there is some anatomic connection between the abscess and the spinal fluid as may occur in cases in which the brain abscess is secondary to trauma or to a postoperative complication; blood cultures are positive in 10–20 % of patients. While reasonable empiric therapy can be devised for most common brain abscesses, culture of the material and transport to the laboratory under strictly anaerobic conditions is essential for optimal identification of the causative microorganism(s). In addition, a biopsy can be obtained and sent for pathologic evaluation to rule out malignancy, fungal invasion, and for detection of unusual microorganisms using special stains.
Treatment
Therapy for brain abscesses requires a combined medical and surgical approach [135]. Stereotactic-guided needle aspiration of pus drains the collection and enables procurement of specimens for gram stain, culture, and histology. Open craniotomy may be considered in cases where response to antimicrobial therapy is poor or the organism isolated from culture is antimicrobial-resistant. Antimicrobial therapy alone may be considered for patients with numerous abscesses that are not amenable to surgical drainage or for patients with small abscesses who have stable neurologic function. The choice of antimicrobials is determined both by the spectrum of microbiological agents known to cause brain abscess and the degree to which individual antimicrobials penetrate the blood–brain barrier and enter into the abscess cavity itself. For brain abscesses that develop in contiguity with frontal sinus infection, mixed aerobic and anaerobic flora may be assumed to be present. In this situation, even if anaerobic bacteria are not recovered, treatment should be given with high-dose penicillin 10–20 million units per day together with metronidazole 7.5 mg/kg IV every 6 h or 15 mg/kg IV every 12 h. If there is any suspicion that the abscess may have arisen from a dental focus, anaerobic cultures should be held for 7–14 days to enable detection of Actinomyces spp. growth. However, actinomycosis should respond to standard therapy with penicillin. Brain abscesses that are related to chronic otitis media and mastoiditis should be treated with a combination of antimicrobials that will cover anaerobes, Enterobacteriaceae, and Pseudomonas spp. Thus, a combination of cefotaxime, ceftazidime, or ceftriaxone plus metronidazole would work well in this clinical scenario. Although bacterial cultures may not always yield growth of anaerobes, particularly if the organisms are fastidious, the absence of growth of Enterobacteriaceae or Pseudomonas spp. on culturing abscess material from a patient who has not received prior intravenous antimicrobial therapy can be relied upon to rule out these particular organisms as the cause of infection. Similarly, the absence of S. aureus from culture of abscess matter would also be very good evidence that this agent is not involved in the pathogenesis of the abscess. In general, S. aureus is much more likely to be implicated in endocarditis, metastatic infection to the CNS, or in the setting of trauma or postsurgical wound infection. If a brain abscess is associated with a neurosurgical procedure, vancomycin should be included in the empirical therapeutic regimen to cover both methicillin-resistant S. aureus (MRSA) and coagulase-negative Staphylococcus spp.
A 6- to 8-week course of parenteral antimicrobial therapy plus regular follow-up computed tomography scans or MRI for at least 3 months to evaluate the therapeutic response is recommended. The antimicrobial regimen can be modified once antimicrobial susceptibility testing results become available. The dosage of third-generation cephalosporins is 2 g IV every 4 h for ceftazidime and 2 g IV every 12 h for ceftriaxone. Therapy is required for a minimum of 6 weeks. Patients with a brain abscess caused by Nocardia spp. should be treated with higher doses of trimethoprim/sulfamethoxazole (15 mg/kg/day of the trimethoprim component) in three to five divided doses until the infection is controlled; thereafter, the dose can be reduced to one double-strength trimethoprim/sulfamethoxazole tablet orally twice daily for 3–6 months in non-immunocompromised patients and up to a year in the immunocompromised person. In severely immunocompromised patients due to advanced AIDS lifelong treatment for Nocardia CNS infections may be required. The method of surgical intervention depends on the patient’s clinical status and the neuroradiographic characteristics of the abscess.
Spinal Epidural Abscess
The epidural space is defined by an area posterolateral to the spinal cord between the dura and the vertebral column. It extends from the foramen magnum to the sacrum; the space is largest in the midthoracic and lumbar regions. The epidural space contains lymphatics, spinal nerve roots, loose fatty tissue, and small arteries. An epidural abscess is a collection of pus between the dura and the vertebral column. A spinal epidural abscess is a medical emergency because of the risk of spinal cord compression and potential progression to irreversible paraplegia or quadriplegia.
Spinal epidural abscess is more common in the elderly with a peak incidence during the 6th and 7th decades [136]. The disease is rare among children and typically affects patients whose comorbid conditions predispose them to immunocompromised states [136–140]. Other populations at risk include the aged, IV drug abusers, immunosuppression due to HIV infection, and spinal surgical procedures [141].
Pathophysiology
The formation of spinal epidural abscesses can be spontaneous or secondary to direct inoculation of a pathogen into the epidural space. The most common cause of the spontaneous infection in the epidural space is hematogenous spread from distant foci (50 %), such as infections of the skin and oral cavity, pneumonia and other respiratory tract infections, endocarditis, intra-abdominal and pelvic sepsis, and urinary tract infections [136]. Other causes of spontaneous abscess formation include direct extension (i.e., contiguous spread) from preexisting discitis or osteomyelitis in an adjacent vertebral body, extension of a decubitus ulcer or paraspinal abscess, blunt spinal trauma, or penetrating injuries. Secondary causes include postoperative infections (16 % of all spinal epidural abscesses) and infections associated with epidural anesthesia catheters [141–143]. An epidural hematoma resulting from trauma can also become secondarily infected and progress to an epidural abscess [136].
The location of an epidural abscess within the spinal canal is determined largely by the underlying cause of the epidural abscess [104, 136, 140, 141]. The majority of spontaneous spinal epidural abscesses that result from hematogenous spread of bacteria are usually located posteriorly within the spinal canal; epidural abscesses secondary to preexisting vertebral osteomyelitis are usually confined to the anterior spinal canal [140–145]. The segregation and isolation of abscesses to the anterior or posterior spinal canal is thought to be secondary to septations within the epidural fat [140]. These septations not only divide the epidural space into anterior and posterior compartments but also divide the space longitudinally. The longitudinal septations usually limit the extent of epidural abscess formation to up to four vertebral levels [140]. Postsurgical spinal epidural abscesses often involve multiple compartments, extending several levels and circumferentially around the spinal cord, secondary to disruption of the epidural septations [144]. In addition to causing compression of the spinal cord, pus and granulation tissue in the epidural space can cause ischemia of the spinal cord by compromising and reducing arterial blood flow. The neurologic sequelae of epidural abscess formation can be slowly progressive or dramatically acute in nature; in the latter scenario, complete paralysis could occur within a matter of hours [141]. The neurological injury is thought to be secondary to both compression of the neural elements vascular thrombosis and ischemia [140, 141, 146, 147].
Risk Factors
The majority of patients who develop a spinal epidural abscess have one or more identifiable risk factors [141, 143]. Implicated comorbid conditions include osteomyelitis, discitis, diabetes, degenerative joint disease of the spine, IV drug abuse, alcoholism, chronic renal failure, immunodeficiency states, and cancer [148]. The spectrum of risk factors is fairly consistent between reports of other large case series [136, 137, 140, 144]. Certain comorbid states such as diabetes and alcoholism result in an immunodeficient state that predisposes a patient to the development of a spinal abscess [144, 146]. Other risk factors have a more direct role in the development of spinal epidural abscesses. Discitis and the bacteremia associated with IV drug abuse, directly seed the epidural space with pathogens responsible for the epidural infection [141].
Microbiology of Spinal Epidural Abscess Formation
S. aureus is the microorganism most commonly isolated from spinal epidural abscesses followed by gram-negative organisms particularly E. coli and Pseudomonas spp. (18 %), other gram-positive organisms including Streptococcus spp. (10 %), or anaerobes (2 %); polymicrobial growth is observed in 5–10 % of cases. Mycobacterium tuberculosis has been implicated in various cases, especially in economically less-developed countries. Pathogens less frequently implicated as causes of spinal epidural abscesses include Brucella spp., Actinomyces, Cryptococcus spp., and Aspergillus spp. [136, 142, 143].
Clinical Features and Diagnostic Considerations
The classic clinical presentation of a patient with spinal epidural abscess is back pain and fever; nerve root pain often ensues—a result of nerve root compression caused by a ruptured intervertebral disc. Further progression to spinal cord compression may lead to motor weakness, bowel and bladder dysfunction, limb weakness, and paralysis [141]. In reality, the presentation is highly variable; other common clinical findings include fever (61 %), paresis (53 %), bowel or bladder dysfunction (36 %), sepsis (17 %), radiculopathy (12 %), and plegia (14 %) [149, 150]. Because of the general and nonspecificity of symptoms and signs, patients, not infrequently, may be misdiagnosed initially [148]. Point tenderness over the involved vertebral levels is present in about one-quarter of patients and is associated with underlying bony involvement [148]. The most common location of spinal epidural abscess formation is in the lumbar region, but thoracic and cervical involvement is not uncommon [136].
Time between symptom onset and presentation is highly variable and does not correlate well with intraoperative findings [151]. Neurological deficits are present on physical examination in the majority of patients at the time of presentation [136, 137, 139, 141, 143, 148]. Neurological decline can occur chronically over months or precipitously over a few hours [137, 139, 141, 143–146, 149].
The most consistent laboratory abnormality is an elevated erythrocyte sedimentation rate (ESR), which is almost always present; ESR and C-reactive protein have been found to be highly sensitive and moderately specific in identifying patients in the emergency room with spinal epidural abscess [152, 153]. Leukocytosis is found in roughly 75 % of patients [141, 148]. Results of CSF analysis are variable, ranging from normal to frank pus [144]. Blood cultures have been reported to be positive in up to 60 % of cases [140]. Contrast-enhanced MRI scans give superior resolution of soft tissue, the spinal cord, and epidural space when compared to CT myelography and are the diagnostic procedure of choice in many facilities. An epidural abscess appears as a low-intensity image on T1-weighted MRI scans (Fig. 22.17). Gadolinium-enhanced magnetic resonance imaging remains one of the most sensitive, specific, and accurate imaging methods for confirming and defining the presence of a spinal epidural abscess and determining its location [54, 137–139]. If MRI services are not available, myelography should be carried out. Lumbar puncture to determine CSF protein concentrations is not needed for diagnosis and may introduce bacteria into the subarachnoid space with consequent meningitis and, therefore, should not be performed [154].
Fig. 22.17.
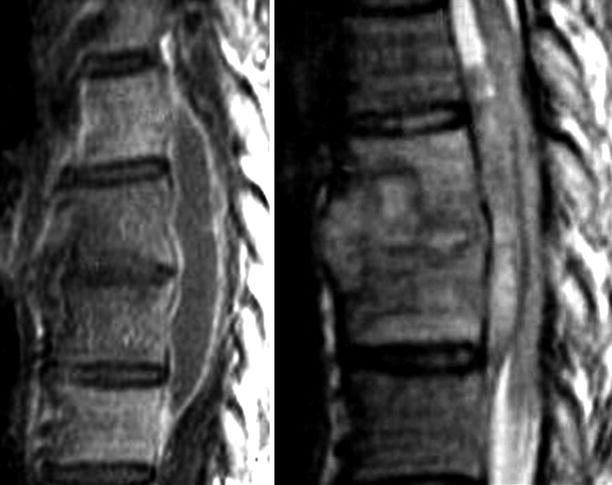
Acute discitis with secondary osteomyelitis and anterior epidural empyema; images include post-contrast T1-w (on left) and T2-w (on right) sequences. In this instance there is a central disc infection with adjacent osteomyelitis in vertebral bodies above and below the disc infection. This is the pattern usually associated with primary disc infection with secondary bone involvement and is usually of pyogenic causative agent. Additionally, there is an anterior epidural abscess that bridges the infected motion segment
Treatment
Surgical intervention for removal of pus and granulation tissue forms the basis of therapy, followed by a prolonged course of parenteral antimicrobials. If the patient has neurological signs at presentation, immediate surgical decompression of the spinal cord is absolutely essential. During the time that culture results are not available, and because S. aureus is a commonly implicated pathogen in spinal epidural abscesses, an antistaphylococcal penicillin, a first-generation cephalosporin, or vancomycin should be included in the treatment regimen together with an agent that is active against gram-negative organisms, such as a third-generation cephalosporin. If MRSA is suspected or implicated, vancomycin should be used. If the infection follows a neurosurgical procedure, an antistaphylococcal penicillin, a third-generation cephalosporin, and an aminoglycoside are prescribed in combination. Spinal epidural abscess carries with it a high mortality rate and significant long-term sequelae, including paralysis. The mortality rate is estimated to be 14 %, and 35–40 % of patients will have neurological sequelae, such as residual weakness or permanent paralysis [136, 138, 140, 148]. Prognosis depends upon the clinical and neurological condition of the patient at the time of presentation [136, 141, 146, 149]. Patients presenting with sepsis or plegia have higher mortality and long-term morbidity rates [137, 138, 142, 144, 145, 148, 150, 154].
Spinal epidural abscesses have been treated with medical therapy alone [147]. However, the majority of these patients fell into one of three categories: (1) panspinal infections not amendable to drainage; (2) operative candidates with a history of poor health and comorbid conditions, such as chronic cardiac or pulmonary disease; or (3) complete paralysis for greater than 24 h [155]. In order to treat patients nonoperatively the clinician must obtain the organism by another means, such as blood culture or needle aspirate, and be willing to perform serial neurological exams and monitor the response to therapy with serial MRI scans [155]. However, caution is warranted in treating spinal epidural abscesses nonoperatively [147].
Vertebral Osteomyelitis
Pyogenic spinal infections, including vertebral osteomyelitis, account for approximately 2–4 % of all cases of osteomyelitis, ranking third behind infections of the femur and tibia in adults [156–159]. Over the past two decades, the incidence of vertebral osteomyelitis appears to have been increasing; this increase is thought to be associated with a parallel increase in the number of immunocompromised patients [156, 157, 160–163]. With an incidence ranging between 0.3 cases per 100,000 among persons less than or equal to 20 years of age and 6.5 per 100,000 among persons greater than 70 years of age, vertebral osteomyelitis is the most common type of hematogenously acquired osteomyelitis [156, 157, 159, 161]. The condition occurs more frequently in men, especially those aged 50 years or older [157, 162, 164, 165]. Risk factors associated with spontaneous, acute vertebral osteomyelitis include diabetes mellitus, systemic steroid use, history of a genital or urinary infection, urinary tract procedures, bacteremia, protein calorie malnutrition, IV drug abuse, malignancy, various immunodeficient states, or advanced age [156, 157, 165–168].
Vertebral osteomyelitis in younger populations is frequently associated with IV drug abuse [166]. In fact, drug abusers account for over half the cases of pyogenic spinal osteomyelitis in some large series [157, 162, 169]. The overall incidence of diabetes in patients with vertebral osteomyelitis is between 18 % and 25 % [156]. However, in one series, 43 % of adult patients with hematogenous vertebral osteomyelitis at a tertiary care hospital had diabetes [158].
Prolonged steroid use is a risk factor for vertebral osteomyelitis caused by bacteria and atypical mycobacteria [166, 170]. The common denominator in each of these predisposing conditions appears to be a deficiency in some aspect of cellular or humoral immunity [166]. Postsurgical vertebral osteomyelitis accounts for approximately 2.5 % of all spinal osteomyelitis and occurs more frequently in malnourished patients, diabetics, and patients on steroid therapy [165, 167, 169, 171]. As the prevalence of AIDS increased, there has been resurgence in the incidence of spinal tuberculosis and the emergence of other fungal causes of vertebral osteomyelitis in this population [159, 163, 166, 168].
Pathogenesis
Spontaneous vertebral osteomyelitis usually results from hematogenous spread of organisms through the segmental spinal arteries to the subchondral plate region of the vertebral body adjacent to the disc space [167, 168]. In adults, the nidus of infection begins in the vertebral bodies at the level of the end arteriolar arcades and, after endplate destruction, spreads secondarily into the avascular disc space [156, 168]. In children, the disc space contains vascular channels that allow primary seeding of the intervertebral disc [156, 168, 169]. Segmental spinal arteries typically bifurcate to supply adjacent vertebral segments, this bifurcation is thought to account for the fact that vertebral osteomyelitis typically involves two adjacent vertebrae and the intervening disc space [168]. Postoperative vertebral osteomyelitis usually occurs due to direct inoculation of the microorganism at the time of surgery [156, 157, 161, 164, 167, 169]. The principal sources of wound contamination are surgery personnel or the patient’s own skin flora [167]. Unlike spontaneous vertebral osteomyelitis, the disc space is often the nidus of infection in patients who have undergone a surgical procedure [156, 167, 169]. Other sources or risk factors for vertebral osteomyelitis include inoculation via decubitus ulcers and trauma or IV drug abuse [165, 172–174].
Microbiology of Vertebral Osteomyelitis
Of the various microorganisms known to be associated with culture-positive, pyogenic vertebral osteomyelitis, gram-positive aerobic cocci are implicated 68 % of the time with S. aureus isolated in up to 60 % of cases, gram-negative aerobic bacilli in 29 % of the patients, and anaerobic bacteria in 3 % [156, 158, 163, 165–167, 175–181]. Radiological imaging is often unable to differentiate between vertebral osteomyelitis caused by mycobacteria and bacteria; thus, initial clinical assessment of a patient with spinal osteomyelitis should include tuberculosis in the differential diagnosis, especially in high-risk patients or in regions or countries with a high prevalence of mycobacterial infections or in countries where tuberculosis and HIV infection are endemic [163, 165, 166]. Coccidioides immitis, Blastomyces dermatitidis, Cryptococcus neoformans, Aspergillus species, and other less common fungi have all been implicated as causes of vertebral osteomyelitis [165, 182]. Pseudomonas aeruginosa and S. aureus are the most commonly implicated pathogens in vertebral osteomyelitis in IV drug abusers and occur at roughly equal rates [175, 183].
In elderly males with urinary tract infections or following invasive urological procedures, the most common causes of vertebral osteomyelitis are E. coli and Proteus species [165]. Postoperative spinal infections are usually caused by S. aureus [163, 165, 167, 168, 183]. Patients on long-term steroid treatment are susceptible to infections caused by atypical mycobacteria and Aspergillus spp. [117, 166, 170, 184–190]. Although vertebral osteomyelitis is generally caused by a single microorganism, contiguous sources of infection, such as a decubitus pressure ulcer or intra-abdominal abscess (e.g., psoas abscess), can lead to polymicrobial infection involving both aerobic and anaerobic microorganisms [165, 166].
Clinical Presentation and Diagnostic Considerations
Back pain is the most common (greater than 90 %) presentation of vertebral osteomyelitis [161, 166]. The pain is localized, continuous, and generally unrelated to movement or position [157]. Vertebral osteomyelitis acquired by the hematogenous mode tends to involve the lower dorsal and lumbar spine. Nearly all spine infections are associated with tenderness on palpation over the involved level: vertebral osteomyelitis tends to be localized to the lumbar and dorsal vertebrae in 58 and 30 % of cases, respectively [156, 157, 161, 166]. Involvement of the cervical vertebrae (11 %) has been noted among IV drug abusers and patients with pulmonary tuberculous [156]. Neurologic impairment, such as sensory loss, weakness, or radiculopathy, is reported in one-third of cases. The presence of radiculopathy, positive straight leg raise, and neurological deficit (4–16 %) is less reliable and often indicates the presence of epidural involvement [157, 166]. Fever is found in approximately half of patients with pyogenic spinal osteomyelitis, and constitutional symptoms, including malaise, night sweats, and anorexia, have been reported [157, 161, 167, 191].
Vertebral osteomyelitis has an insidious onset and may prove to be a diagnostic challenge because of the nonspecific nature of the symptoms and signs. For example, the differential diagnosis of back pain in a patient with fever includes various viral syndromes, aortitis, pyelonephritis, and pancreatitis [161]. In the absence of fever, other causes of back pain, such as an osteoporotic fracture, spondylarthritis, degenerative disc disease, or spinal stenosis, might have to be ruled out [161, 168]. In several large patient series, the delay between the onset of symptoms and eventual diagnosis has ranged from 3 weeks to 3 months [157, 162, 168, 192]. These delays in diagnosis can result in significant neurological morbidity [168]. Other complications arising from vertebral osteomyelitis include paraspinal abscesses, soft tissue extension, and spinal cord compression.
A leukocytosis is found in less than half of patients with vertebral osteomyelitis [157]. The most common laboratory abnormality is an elevated ESR. Blood cultures are critical in the workup of a patient with a putative diagnosis of vertebral osteomyelitis and, if positive (up to 78 % of blood cultures are positive), might obviate the need for an invasive diagnostic procedure to procure a biopsy specimen [157, 165, 178]. Plain X-ray findings include disc space narrowing and vertebral end plate changes that usually become apparent 2–4 weeks after symptom onset in approximately 80 % of patients [156, 157, 167, 191]. Technetium 99 m bone scanning combined with gallium scanning has a 90 % sensitivity and specificity for vertebral osteomyelitis [193]. MRI imaging is the most sensitive and specific imaging modality for vertebral osteomyelitis and has the added benefit of providing details regarding the presence of an epidural or paraspinal abscess and various other complications [156, 161, 194–196]. Characteristic MRI changes include decreased T1 signal and increased T2 signal of the involved vertebral endplates and disc space [156, 196] (Fig. 22.18). Positron emission tomographic (PET) scanning with 18F-fluorodeoxyglucose has a diagnostic accuracy similar to that of MRI and might be considered if the patient has metallic implants [195].
Fig. 22.18.
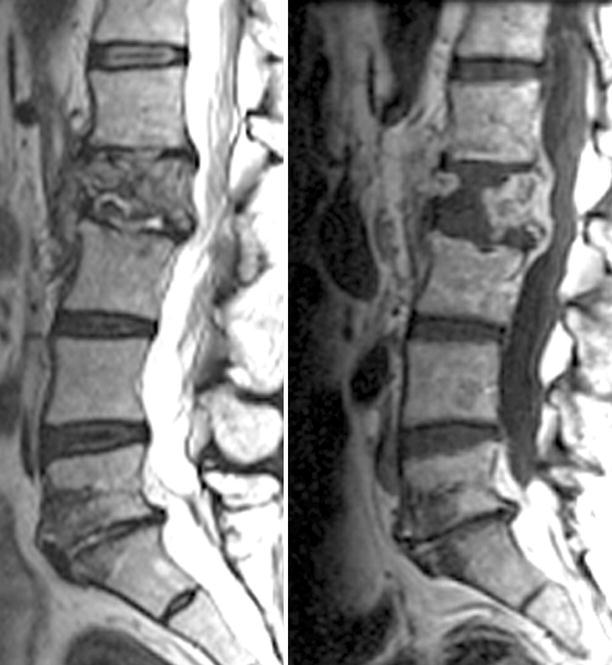
Spinal vertebral osteomyelitis with relative sparing of the disc space; images include T2-w sagittal image (left) and a post-contrast T1-w image (right). In this instance the spinal infection has virtually destroyed the L1 vertebral body. There is prominent residual contrast enhancement in the affected vertebral body. There is only minimal edema in the L1–2 disc space and relatively little enhancement. This complex would be consistent with hematogenous seeding the vascularized vertebral body rather than a primary disc infection with secondary osteomyelitis
Optimal pharmacologic treatment requires that the infecting microbe be identified and antimicrobial susceptibility profile be determined [157]. If imaging is suggestive of vertebral osteomyelitis but blood cultures are negative for bacterial growth, then a biopsy of the infected vertebra is mandatory [156, 157, 167, 191]. The procedure of choice would depend on the presence of underlying intrinsic patient risk factors with input from surgical and interventional radiology personnel regarding CT-guided versus open biopsy [161]. When blood cultures are negative, a CT-guided needle biopsy can be used to make a definitive microbiological diagnosis 60–90 % of the time [157]. An open biopsy should be considered when both blood cultures and CT-guided aspirates are negative [167]. A skin test for tuberculosis should be placed in all patients and CT aspirates or biopsy material should be sent for routine fungal stains and cultures [167, 192]. If polymicrobial osteomyelitis is suspected (e.g., a patient with concomitant intra-abdominal sepsis), a biopsy should be performed whether or not blood cultures are positive for bacterial growth [181].
Treatment
Targeted antimicrobial therapy, spinal immobilization, and, when necessary, surgical interventions are the mainstays of treatment of vertebral osteomyelitis [156, 157, 161, 165]. Seventy-five percent of patients with spinal osteomyelitis can be managed without surgical intervention [156]. Prolonged courses of parental antimicrobials directed by specific culture results and antimicrobial susceptibilities are the rule [161, 165, 191]. Although there are no data from controlled trials that suggest the optimal duration of therapy, 4–6 weeks of antimicrobial therapy is usually recommended [176, 197, 198]. One large review suggested that 4 weeks therapy with high-dose IV antimicrobials is sufficient to treat pyogenic spinal osteomyelitis as long as the following criteria are met: (1) there are no undrained abscesses, (2) the patient is clinically stable, and (3) the ESR has decreased to one-half its original value [165].
For methicillin-susceptible S. aureus (MSSA), high-dose oxacillin (2 g every 6 h or cefazolin, 1–2 g every 8 h) is recommended. Alternatively, levofloxacin 750 mg orally once daily plus rifampin 300 mg taken orally every 12 h may be considered. MRSA or coagulase-negative staphylococci can be treated with IV vancomycin (1 g every 12 h) or daptomycin (greater than or equal to 6 mg/kg of body weight once daily). Streptococcal species can be treated with IV penicillin G (5 million units every 6 h) or IV ceftriaxone (2 g once daily). Enterobacteriaceae can be treated with ciprofloxacin (750 mg taken orally every 12 h) or IV ceftriaxone (2 g once daily). Infections caused by quinolone-resistant Enterobacteriaceae (including extended-spectrum β-lactamase-producing E. coli), can be treated effectively with IV imipenem (500 mg every 6 h). For P. aeruginosa infections, cefepime or ceftazidime, or piperacillin/tazobactam every 6 h, followed by ciprofloxacin (750 mg taken orally every 12 h). Anaerobes can be treated with IV clindamycin (300–600 mg administered intravenously every 6–8 h), IV penicillin G (five million units every 6 h), or IV ceftriaxone (2 g once daily) with metronidazole (500 mg taken orally every 8 h.) Tuberculous vertebral osteomyelitis requires an average of 12 months treatment with a combination of isoniazid, rifampin, ethambutol, and pyrazinamide, depending on regional susceptibility [156, 199]. Treatment of other less common bacterial and fungal causes of vertebral osteomyelitis should be tailored to the individual pathogen and to regional antimicrobial susceptibility profiles. Therapy is guided by clinical response and the ESR as outlined previously. Continued elevation of the ESR may necessitate more prolonged therapy.
The indications for surgical intervention in spinal osteomyelitis are the presence of a neurological deficit, spinal instability, unresponsiveness to medical therapy, or a non-diagnostic CT-guided biopsy [161, 162]. In addition, surgery is recommended for the drainage of epidural or paraspinal abscesses that often accompany vertebral osteomyelitis [165] The goals of surgery are decompression of the neural elements, correction of spinal deformity, debridement of necrotic tissue, and the promotion of long-term stability [156, 162, 168, 192]. A variety of surgical approaches have been described in the literature, each with its own advantages and disadvantages [156, 162, 192]. The basic principles of surgical intervention include early instrumentation and fusion at the time of the initial operation in order to facilitate ambulation and avoid the complications associated with prolonged bed rest [156, 162, 192].
External Ventricular Drainage
Infectious Considerations
A ventriculostomy is an external ventricular drainage (EVD) device or ventricular catheter that is placed into the cerebral ventricles enabling drainage of CSF externally. It is typically connected by tubing to a CSF collection device that can be elevated or lowered at the bedside to vary the amount of CSF that is drained. EVD devices are an integral aspect of the intensive care management of neurosurgical patients [200–202]. Common indications for their use include management of hydrocephalus, elevated intracranial pressure, or intracranial hemorrhage and the administration of intrathecal medications [200–205]. EVD devices provide diagnostic information and also facilitate therapeutic CSF drainage [206–208]. The most common complication involving the use of EVDs is infection; rates of infection up to 27 % have been reported [203, 204, 209–215].
Risk factors for the development of EVD-associated ventriculomeningitis include EVD duration greater than 11 days, frequency of CSF sampling, intraventricular hemorrhage, surgical technique (subcutaneously tunneled EVD, Rickham reservoir with percutaneous CSF drainage), neurosurgical operative procedure, and irrigation or manipulation of the drainage system [204, 210–214]. Conflicting data exist regarding the association between duration of ventricular drainage and the rate of EVD-associated infections [205, 210, 216, 217]. Some clinical series have shown a linear relationship between infection and duration of ventricular drainage [218, 219]. Various authors have advocated the routine changing of ventricular catheters after day 5 in order to lower the risk of infection, while others have suggested that the duration of monitoring is not a risk factor associated with infection. In fact, later studies have shown that a constant daily rate of infection actually decreases after day 10 or 11 of drainage [217, 219].
Diagnostic clues for EVD-related ventriculomeningitis may stem from clinical signs, such as fever, meningism, reduced level of consciousness, photophobia, phonophobia, and abnormal laboratory parameters (e.g., reduced CSF glucose, increased CSF protein, CSF pleocytosis, positive CSF culture, or gram’s stain) [220]. Because the results of the CSF leukocyte count may be confounded by intraventricular hemorrhage, Pfausler and colleagues introduced the “cell index” as a new parameter for the diagnosis of EVD-related ventriculomeningitis [221]. They showed that a significant increase of this index is highly indicative of EVD-related ventriculitis in patients with hemorrhagic CSF and that calculation of the “cell index” on a daily basis allows the timely diagnosis and hence initiation of antimicrobial therapy of catheter-related ventriculomeningitis [221]. Coagulase-negative Staphylococcus (70 %), S. aureus (10 %), and gram-negative bacteria (15 %), including P. aeruginosa, Acinetobacter spp., Klebsiella spp., and E. coli, remain the most common microorganisms associated with ventriculomeningitis [220–224].
The role of perioperative and prophylactic antimicrobials in the prevention of EVD-associated infections has been extensively discussed and debated. Alleyne and colleagues reviewed over 300 patients who received either daily prophylactic antimicrobials or perioperative antimicrobials alone and found no significant difference in the infection rate between the two groups [225]. The benefit of systemic prophylactic antimicrobials for the first 24 h postoperatively to prevent shunt infection, regardless of the patient’s age or the type of internal shunt being used, has been demonstrated although the benefit of its use after this period remains uncertain [226–229].
Current evidence suggests that antibiotic-impregnated catheters reduce the incidence of shunt infection [226, 229–235]. Other factors thought to be important in preventing infection include perioperative antimicrobial administration prior to skin incision, tunneling of the ventricular catheter, proper surgical skin prep, strict adherence to aseptic technique, use of a closed ventricular drainage system, and meticulous sterile nursing care [210, 211, 218, 225, 236–243]. Antimicrobial impregnated catheters have been manufactured for a variety of applications and have been shown to be effective in preventing EVD-associated infections [225, 230, 231, 235–239, 244, 245]. Beer and colleagues suggest that the management of EVD-related ventriculitis should involve decisions related to catheter exchange, the type and route of administration (intravenous versus intrathecal) of antimicrobial therapy, based on the type of suspected organism and its resistance pattern, and the duration of antimicrobial therapy [220]. Once the diagnosis is made, broad-spectrum antimicrobials should be administered, followed by diagnostic testing, and targeted antibiotic therapy as soon as antimicrobial susceptibility testing results become available.
Mycobacterium Tuberculous Infections
Tuberculous Meningitis
Tuberculosis has been known to humankind since antiquity, having been demonstrated relatively recently by molecular methods in mummies from both the new and the old world dating to 1,000–1,500 years BC [246, 247]. Tuberculosis was recognized on clinical grounds in the eighteenth century, and with the isolation of the organism by Robert Koch in 1882, its ability to produce CNS disease was quickly recognized. Before the HIV pandemic, M. tuberculosis infection and disease was already endemic in many economically less-developed countries, though not at pandemic proportions. With the onset of the HIV pandemic, there has been a dramatic, parallel increase in rates of tuberculosis among HIV-infected patients in many of these countries, especially sub-Saharan Africa, Southeast Asia, and increasingly the Indian subcontinent. At the end of the first decade of the new millennium, tuberculosis remains the second leading cause of death from an infectious disease worldwide after HIV, despite the availability of highly efficacious treatment for decades. In 2010, there were an estimated 8.5–9.2 million cases and 1.2–1.5 million deaths attributable to tuberculosis [248]. Indeed, M. tuberculosis has now been established as the first or second most common cause of bloodstream infections in febrile adults who present to emergency rooms in sentinel hospitals in East Africa and Thailand [249–253].
Although comparative rates of M. tuberculosis infections are relatively lower among HIV-infected patients in North America and Western Europe, the World Health Organization generally considers M. tuberculosis infections a true pandemic that is unequivocally linked with immunosuppression resulting from HIV infection, though the problem has certainly been compounded by other factors associated with poverty, such as overcrowding, malnutrition, lack of access to healthcare, and poor sanitation, made particularly worse in refugee camps resulting from natural and man-made disasters and war [248]. The fact remains that at the end of the first decade of the twenty-first century, tuberculosis is the leading cause of death among HIV-infected patients worldwide, and tuberculosis meningitis remains the most rapidly progressive form of the disease.
Perhaps the most insidious occurrence resulting from the tuberculosis pandemic, with negative implications for effective therapy, is the emergence of multidrug-resistant (MDR) tuberculosis, defined as resistance to at least both isoniazid and rifampin, and extensively drug-resistant (XDR) strains of M. tuberculosis (i.e., strains resistant to practically all second-line agents) [248, 254–257]. Emerging resistance has made the management of pulmonary and extrapulmonary tuberculosis, including tuberculous meningitis, all the more difficult to manage.
Pathogenesis
Tuberculous meningitis can occur as a result of hematogenous seeding of the meninges; reactivation of metastatic M. tuberculosis foci in the meninges and brain parenchyma, which have been present asymptomatically for months to years following primary infection; or due to breakdown of an old tuberculous parameningeal granuloma with rupture into the subarachnoid space. In addition to HIV infection, other predisposing risk factors in adults include alcohol abuse, IV drug abuse, immunosuppression due to steroid and other immunosuppressive therapies, and chronic disorders, such as connective tissue diseases and chronic cardiopulmonary disease.
Pathologically, tuberculous meningitis leads to prodigious inflammation with production of a thick exudate at the base of the brain, particularly involving the optic nerves at the optic chiasm, the pons, and cerebellum. The histologic appearance depends on the stage of the disease. Initially it consists of polymorphonuclear leukocytes, macrophages, and lymphocytes. But later, after a phase of lymphocytic proliferation, granulomata with caseating centers become a prominent feature (Fig. 22.19). Another feature of tuberculous infection of the meninges is involvement of the blood vessels traversing the meninges: small- and medium-sized arteries are most often involved, although capillaries and veins may be similarly affected. The changes include granuloma formation and inflammation of the adventitia, which causes a reactive cellular proliferation of the intima, which in turn may lead to occlusion of the vessel and infarction of the areas supplied by the vessel. Clinically, this vasculitis is frequently found in the distribution of the middle cerebral artery due to its anatomic location at the base of the brain, where the inflammatory response is most intense. This profound inflammatory response with vasculitis causes compression of neural tissues and compromises cerebral blood flow leading to cerebral ischemia and infarction, obstruction of CSF free flow, resulting in hydrocephalus and cerebral edema.
Fig. 22.19.
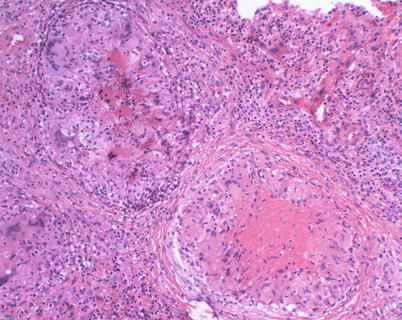
A 40-year-old female with chronic headaches, anosmia, and blurred vision. Biopsy revealed chronic granulomatous meningitis with giant cells and non-caseating and focal caseating (necrotizing) granulomas. Special stains for acid-fast bacilli demonstrated only rare acid-fast organisms (Courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Hydrocephalus is one of the most frequent complications of tuberculous meningitis, commonly accompanying symptomatic primary infection in children. Hydrocephalus occurs either by mechanical blockage of the spinal aqueduct or the foramina of Luschka due to the exudate at the base of the brain or to edema of the surrounding brain parenchyma. Hydrocephalus may also be caused by blockage of CSF reabsorption at the base of the brain due to the intense infiltrate. The former mechanism leads to noncommunicating hydrocephalus and the latter mechanism to communicating hydrocephalus.
Clinical Presentation
The classical clinical presentation of tuberculous meningitis in adults is that of fever and headache, together with meningismus that becomes progressively more severe over a period of 2–3weeks. However, the duration of prodromal symptoms can be quite variable and some patients have reported symptoms for several months before they actually sought medical attention. None of these symptoms or signs is universally present in all patients with tuberculous meningitis. Other frequently observed clinical features include lethargy and behavioral changes in 30–70 % of patients, seizures in 10–15 %, and cranial nerve palsies in up to 20–30 % of adults. Occasionally, abnormal movements such as chorea, hemiballismus, athetosis, myoclonus, and cerebellar signs and symptoms are observed. Localizing neurologic symptoms due to a tuberculoma depend on the size and location of the mass lesion. Strokes due to tuberculous vasculitis described previously usually involve the distribution of the middle cerebral artery and produce symptoms related to that vascular distribution. The most common cranial nerve abnormalities involve the sixth cranial nerve followed by the third, fourth, and the seventh but may involve the second, eighth, tenth, eleventh, and twelfth cranial nerves [258]. Coma is present or can develop in up to 30 % of adults and children. In some series as many as 50 % of children have a past history of tuberculosis whereas only 8–12 % of adults have such a history. Hydrocephalus is a serious and potentially devastating complication that may develop in up to 40 % and is associated with a variety of focal neurologic signs, including hemiparesis and blindness.
The differential diagnosis is quite wide and includes bacterial, viral, and fungal infections of the CNS as well as malignancies and noninfectious conditions such as CNS lupus (Table 22.5).
Diagnosis
Routine laboratory tests are not particularly helpful. The single most useful diagnostic procedure is CSF examination. The classic findings of elevated protein, depressed CSF glucose relative to serum, and pleocytosis with a lymphocytic predominance, together with a history of chronic illness over a matter of weeks as opposed to days as in acute bacterial meningitis, is strongly suggestive of a tuberculous or fungal etiology. Median CSF protein levels generally range between 100 and 400 mg/dL but may go as high 2 g/dL, although levels of that magnitude usually suggest mechanical blockage of CSF flow. The median white count generally runs between 100 and 200 WBC/mm3. The CSF glucose is less than 45 mg/dL in 70–80 % of patients. The glucose level tends to become progressively lower and the protein progressively higher as the duration of illness progresses without appropriate therapy; when treatment is successful, the glucose level tends to return towards normal. Although any one of these CSF parameters may be completely normal, it is extremely unusual for all three parameters to be completely normal in a patient with true tuberculous meningitis. If CSF analysis is completely normal and the laboratory reports M. tuberculosis, the report should be considered suspect until proven otherwise. Acid-fast smears of CSF are positive in less than 25 % of patients who ultimately have culture proven tuberculous meningitis, although CSF cultures may ultimately yield M. tuberculous growth in up to 70 % of patients.
The ESR varies considerably in series of patients with proven M. tuberculous meningitis, ranging from normal to more than 100 mm/h. Similarly, the syndrome of inappropriate antidiuretic hormone (SIADH), manifest by hyponatremia and hypochloremia, is not uncommon but is by no means diagnostic since the results are confounded by a variety of other events (e.g., vomiting and anorexia) that may accompany M. tuberculosis disease.
Chest radiographs are non-diagnostic although 25–50 % of adults may show radiographic evidence consistent with current or remote tuberculous infection [259]. In children who develop tuberculous meningitis on the heels of primary tuberculosis infection, chest radiographic evidence of M. tuberculosis infection has been observed in 50–80 % of cases. Miliary disease was commonly associated with tuberculous meningitis in the pre-antimicrobial era and remains common in HIV-endemic regions of the world; at the present time, though, it remains relatively uncommon in the United States.
Admission tuberculin skin testing as a diagnostic aid for the diagnosis of tuberculous meningitis is of limited utility: the performance of the test varies with age, BCG vaccination and nutritional status, HIV status (HIV-infected patients are often anergic to PPD skin testing), and technique of administration [260–262]. Thus, although skin testing may provide information regarding previous tuberculosis exposure or infection and might be useful in the diagnosis of meningitis in young children, it is not sufficiently sensitivity or specific for routine diagnosis of active M. tuberculosis meningitis [260, 263]. Similarly, interferon-gamma release assays (e.g., T-SPOT.TB), which are superior to tuberculin skin testing at diagnosing latent tuberculosis, are neither sufficiently sensitive nor specific for routine diagnosis of M. tuberculosis meningitis [264, 265].
Thus, in a patient with a nonspecific history that is suggestive of possible tuberculosis disease but with a CSF profile typical of aseptic meningitis, other causes of aseptic meningitis will have to be considered and ruled out if deemed necessary. Such infective causes include cryptococcal and other forms of fungal meningitis, early or partially treated bacterial meningitis, cerebral abscess and other parameningeal infections, Listeria spp. meningitis, leptospirosis, and syphilis. Noninfective causes that need to be considered in the differential diagnosis include carcinomatous meningitis, including lymphoma, connective tissue diseases, chronic subdural hematoma, and chemical- or drug-induced causes.
Modern radiographic techniques such as CT scan, MRI with gadolinium enhancement, and magnetic resonance angiography (MRA) are extremely sensitive in delineating CNS involvement, readily demonstrating meningeal inflammation and entrapment of cranial nerves in the basilar tuberculous exudate (Fig. 22.5c, d and 22.20) [266]. In addition, MRA can detect characteristic vascular narrowing which accompanies tuberculous meningitis but is less commonly seen with other pathologies. High-field MRA with contrast is more sensitive than conventional MRA in the detection of occlusion of smaller vessels, which are more commonly involved in the pathogenesis of tuberculous meningitis [267].
Fig. 22.20.
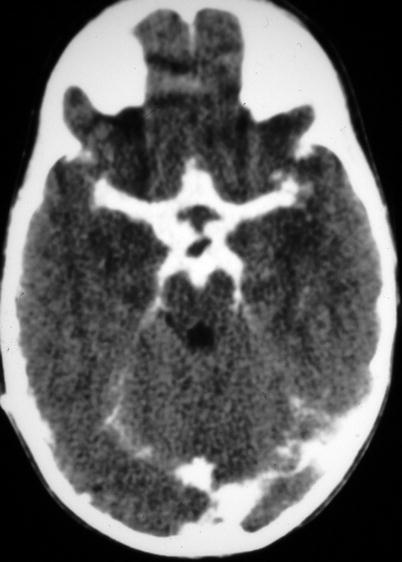
Basilar tuberculous exudate in a patient with tuberculous meningitis
Culture remains the gold standard for laboratory confirmation of tuberculosis and is required for isolating the organism, for drug-susceptibility testing and genotyping. Mycobacteria isolated from cultures are identified using standard biochemical analyses, nucleic acid probes, or 16S rRNA gene sequencing [268]. Real-time PCR assays that rapidly and specifically detect M. tuberculosis complex directly from acid-fast, smear-positive specimens and from broth cultures are now routinely conducted in various reference laboratories across the USA and now offer the potential to detect gene mutations responsible for drug resistance within hours of patient specimen collection compared with the average of 2 weeks required for traditional susceptibility testing methods. Real-time PCR assays used in the diagnosis of tuberculous meningitis have reported sensitivities ranging from 25 to 100 % and specificities of about 95 %. Thus, direct nucleic acid amplification tests should always be performed in conjunction with microscopy and culture, and each test result must be interpreted within the overall clinical setting in which it is used. To date, there is still no single diagnostic method that is both sufficiently rapid and sensitive for detecting M. tuberculosis infection [269].
Treatment
Treatment of tuberculous meningitis consists of at least three, and usually four, drugs until the susceptibilities are established: isoniazid (INH), rifampin, ethambutol, and pyrazinamide are the standard, together with pyridoxine at 25–50 mg daily to prevent depletion by INH; many authors recommend dexamethasone for the first month to improve outcome. Treatment must be continued for at least 12 months. INH is generally used at a dose of 300 mg/day in adults or 10 mg/kg/day in children. The most serious side effect is hepatitis, which ranges from asymptomatic enzyme elevations to fulminant hepatic necrosis. The true risk of INH side effects is approximately 0.1 % among those receiving INH alone for prophylaxis and greater than 1 % for those receiving INH as part of a treatment regimen for tuberculosis [270]. The incidence of hepatotoxicity is higher in persons over 35 years of age, as well as those with other conditions affecting liver function such as alcoholism and viral hepatitis. The dose of rifampin is 600 mg/day and is only infrequently associated with side effects such as a flu-like syndrome and a hypersensitivity reaction with renal, hepatic, and hematologic toxicity. Pyrazinamide is given in a dose of 25 mg/kg/day and has a relatively low incidence of side effects; there is little added toxicity when combined with INH and rifampin. INH, rifampin, ethambutol, and pyrazinamide all have good penetration of the CSF. Ethambutol is generally administered at a dose of 25 mg/kg for the first 1–2months of treatment with a reduction in dose to approximately 15 mg/kg/day because of the risk of optic neuritis, seen in approximately 25 % of patients. The first clue to the development of this complication is loss of red-green vision or diminished visual acuity; ophthalmologic consultation is suggested in these situations. Streptomycin was one of the first drugs found to be active against tuberculosis and is commonly administered in a dose of 20–40 mg/kg/day for children and 1 g/day for adults. Unfortunately, the irreversible ototoxicity is so frequent that it is not advisable to use this agent unless absolutely necessary. Second-line antituberculous drugs such as para-aminosalicylic (PAS), cycloserine, ethionamide, kanamycin, and amikacin should only be used based on treatment failure with primary agents and antimicrobial susceptibility studies. Of these agents, ethionamide and cycloserine penetrate well into the CNS.
Because one cannot rule out tuberculosis based on all of the immediately available diagnostic modalities, empiric therapy often has to be given while awaiting the results of cultures. A negative test for cryptococcal antigen, absence of encapsulated yeast forms on microscopy of an India ink smear, and no growth of the microorganism within 10–14 days of CSF culture essentially rules out cryptococcal meningitis. However, other fungal meningitides cannot be totally ruled out, and some patients may have to be placed on empirical therapy with both antituberculous medications and amphotericin B. Occasionally, CNS symptomatology in patients treated in this fashion turn out to have a noninfectious etiology; in these patients, further radiographic or invasive diagnostic studies may be indicated. In patients who show no signs of improvement within the first week or so, and particularly in the setting where the patient is known to have a malignancy, cytological analysis of the CSF should be requested to rule out meningeal carcinomatosis.
Case reports and various studies have demonstrated the immediate effects of steroids in terms of defervescence and clearing of the sensorium, even after a few doses. While there seems to be general agreement that survival from tuberculous meningitis is improved with the use of steroids, survivors often do so with severe sequelae [259]. However, most authorities currently recommend using steroids in all patients with M. tuberculosis meningitis, regardless of severity of disease at presentation, with a reducing course over 6–8 weeks [60, 260, 271]. The dose of prednisone is 60 mg/day or 1 mg/kg/day, or dexamethasone may also be used at a dose of 8–16 mg/day in divided doses. Steroids are given for 3–6 weeks and then tapered over the ensuing 2–4 weeks. If results of susceptibility testing implicate multidrug-resistant strains of M. tuberculosis, intrathecal therapy might have to be considered [272].
For HIV-infected patients, antiretroviral therapy and antituberculosis treatment should be initiated at the same time, regardless of CD4 cell counts. Of note, tuberculous meningitis may be a manifestation of paradoxical tuberculosis-associated immune reconstitution inflammatory syndrome. The mortality rate among HIV-infected patients with multidrug-resistant tuberculous meningitis is significantly higher than the comparable rate for meningitis caused by susceptible strains. In short, the best way to prevent HIV-associated tuberculous meningitis is to diagnose and isolate infectious cases of tuberculosis promptly and administer appropriate treatment promptly in all patients in whom the diagnosis of tuberculous meningitis is suspected or ascertained [273].
Prognosis and Sequelae
Prior to the availability of antituberculous therapy, survival from tuberculous meningitis was exceedingly rare; survival rates currently are 70–80 % in most recent series. Perhaps the most significant prognostic factor for survival is the degree to which the disease has advanced at the time of initial presentation. Other factors correlating with poor response to therapy include age and coexistent miliary disease. The earliest sign of response to therapy in most cases is reduction in peak daily temperatures within the first 1–2 weeks and subjective improvement in fatigue and malaise over the same period. However, early studies pointed out that it was not uncommon for some markers of disease, such as the presence of bacilli in a smear of the CSF or a rise in CSF protein, to occur shortly after the initiation of treatment. In general, the glucose level in the CSF rises with successful treatment while the protein returns to normal more slowly, a process that may take as long as 6 months.
Up to 50 % of survivors have a variety of neurologic deficits. As with survival itself, the more seriously ill the patient is upon presentation, the more likely complications or sequelae are to occur. Among children, the most common of these are seizure disorders, ataxia, incoordination, persistent cranial nerve abnormalities, and spastic hemiparesis. Adults are most frequently left with chronic organic brain syndrome, often with cranial nerve palsies, paraplegia, and hemiparesis. Optic atrophy can lead to varying degrees of visual impairment or blindness in both children and adults.
Tuberculoma
Tuberculomas are space-occupying mass lesions within the brain parenchyma ranging in size from less than 1 mm to greater than 10 cm. The pathogenesis of tuberculomas is similar to that of tuberculous meningitis in that a tuberculous granuloma, seeded during the phase of acquiring the primary infection, breaks down and, because of its location within the brain parenchyma, produces a mass lesion rather than meningitis. Patients may present with fever, headache, focal or generalized seizure, or change in mental status and may manifest signs of proptosis, papilledema, or transient neurologic deficits. Clinical features are related to the anatomic location of the tuberculoma. Generally, patients have a single lesion on presentation. However autopsy series and sophisticated radiologic studies have shown that in up to 70 % of patients, multiple lesions are present. The duration of symptoms prior to presentation is somewhat longer than in tuberculous meningitis, averaging weeks to months with occasional patients having symptoms for years prior to diagnosis. In fact, 30 % of patients with tuberculomas may remain asymptomatic throughout their life. Neuroimaging CT scan and MRI have a 100 % diagnostic sensitivity (Fig. 22.21); however, tissue confirmation is essential in order to rule out malignancies or other ring-enhancing, space-occupying lesions, such as pyogenic bacterial abscesses, neurocysticercosis, or toxoplasmosis [124, 260].
Fig. 22.21.
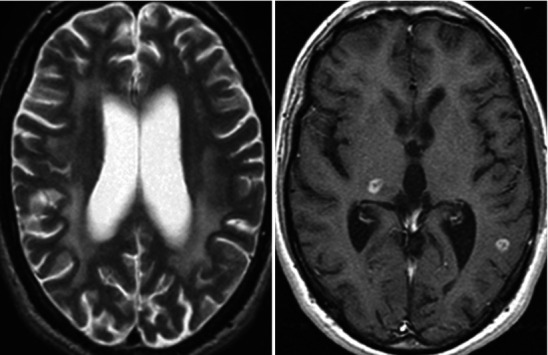
AIDS-related complex and tuberculomas; images include T2-w spine echo (left) and post-contrast T1-w sequence (right). The T2-w sequence demonstrates features of AIDS-related complex with global pronounced atrophy and diffuse bilateral loss of myelin within the centrum semiovale. The right image demonstrates multicentric brain nodules. Their distribution is consistent with a hematogenous-type of disease dissemination. These nodules were found to be tuberculomas, but evidence of small nodular enhancing lesion s has a wide differential diagnosis
Approximately 60 % of the specimens from tuberculomas stain positive for acid-fast bacilli on smear, and approximately the same number ultimately grow in culture. Caseating granulomata are invariably seen histologically. Treatment is essentially the same as for tuberculous meningitis, i.e., antituberculous therapy with adjunct corticosteroids if cerebral edema is present, rather than surgery. The utility of adjunct steroids for patients with intracranial tuberculomas without meningitis has not been fully characterized by randomized controlled trials However, current published data suggest that for this patient population, steroid therapy should be considered because it mitigates symptoms and reduces the frequency of seizure attacks, tuberculoma size, and perilesional edema [260, 274, 275]. The indications for surgical intervention include development of hydrocephalus or coalescence of multiple tuberculomas to form a tuberculous cerebral abscess [260, 276].
Spinal Tuberculosis
Spinal tuberculosis is a destructive form of tuberculosis and accounts for approximately half of all cases of musculoskeletal tuberculosis and is more common in children and young adults [277]. Tuberculous spinal meningitis may accompany tuberculous meningitis or may occur as an isolated entity. While tuberculous meningitis is essentially a disease of childhood, tuberculomas and spinal tuberculosis invariably are adult manifestations [275]. The pathogenesis and pathologic findings consist of characteristic exudate surrounding many parts of the spinal cord with symptoms due to compression and vasculitic changes in the arterial supply. Common clinical manifestations include constitutional symptoms (e.g., fever, malaise, weight loss), back pain, spinal tenderness, paraplegia, and spinal deformities [277]. Symptoms consistent with transverse myelitis (i.e., paraparesis, loss or disturbance of sensation with a sensory level on the trunk, bowel and bladder dysfunction) and spinal block, as well as nerve root pain, paresthesia, and motor weakness, may be seen [163].
The thoracic region of vertebral column is most frequently affected and formation of a “cold” abscess around the lesion is another characteristic feature [277]. The onset can be sudden or present as a slow ascending paralysis over several months to years. MRI is a more sensitive imaging technique than radiographs and more specific than computed tomography and is generally required for accurate diagnosis of spinal tuberculosis and to rule out intramedullary lesions [260, 277]. Neuroimaging-guided needle biopsy from the affected site in the center of the vertebral body remains the gold standard technique to establish a histopathologic diagnosis and underlying etiology [263, 277]. Antituberculous therapy remains the cornerstone of treatment and essentially is the same as for tuberculous meningitis. Indications for surgical intervention include development of compressive symptoms, large abscess formation, severe kyphosis, an evolving neurological deficit, or lack of response to medical treatment [163, 175, 277].
CNS Infections Caused by Rapidly Growing Mycobacteria
CNS infections associated with rapidly growing mycobacteria are rare. One of the few published reports has implicated Mycobacterium mucogenicum, which was isolated in pure culture and detected by PCR sequencing of CSF specimens from two immunocompetent patients who eventually died [278]. M. mucogenicum is frequently isolated from tap water or from respiratory specimens and is usually without clinical significance. Other case reports have described CNS infections caused by Mycobacterium fortuitum, another rapidly growing mycobacteria found in soil, dust, and water; these infections were all device-associated—a contaminated epidural catheter [279] and manipulation of ventriculoperitoneal shunt [280].
Patients with rapidly growing mycobacterial CNS infection usually present with subacute to chronic meningitis and neutrophilic pleocytosis and usually have a history of trauma or having undergone a neurosurgical procedure or manipulation of a device. CSF smears are often negative for acid-fast organisms but may show gram-positive rods [279–281]. Treatment requires a long course of two or more antimicrobial agents that have the ability to penetrate the blood–brain barrier, with adjunct immunomodulatory therapy with steroids, similar to that used in tuberculous meningitis [279–281].
Fungal CNS Infections
Fungal Meningitis
The most common causes of CNS mycoses include Cryptococcus spp. and the dimorphic fungi—Histoplasma spp., Coccidioides spp., and Blastomyces spp. In immunocompromised persons, Aspergillus spp., Candida spp., and the Mucorales are often implicated. Antifungal therapies that are useful in CNS infections include polyenes (e.g., amphotericin B and its lipid formulations) and azoles (e.g., fluconazole and itraconazole, voriconazole, and posaconazole). Antifungal agents that lack CNS penetration include echinocandins (e.g., caspofungin, micafungin, and anidulafungin). Amphotericin B and flucytosine are used to initiate treatment for CNS yeast infections caused by Candida and Cryptococcus neoformans. Amphotericin B particularly in lipid formulation and voriconazole are preferred for aspergillus infections.
Cryptococcus Meningitis
Cryptococcus spp. are perhaps the most common fungus implicated in CNS infections. C. neoformans has a worldwide distribution and is commonly found in soil that has been contaminated with bird droppings. There are two varieties of C. neoformans: C. neoformans var neoformans and C. neoformans var gattii. These two species differ in their ecology, distribution in nature, epidemiology, presentation, clinical course, and therapy. C. neoformans var neoformans is found worldwide and produces most of the infections in patients in the United States, while C. neoformans var gattii is a more commonly found in Southeast Asia, Africa, Australia, and parts of Southern California. The most important determinant of CNS infection caused by Cryptococcus spp. is the immune status of the host. This can be most easily demonstrated by the marked increase in the number of cases associated with HIV infection and the fact that as the CD4+ count decreases the incidence of cryptococcal infection increases markedly, particularly at CD4+ counts less than 200 cells/mm3. Although a variety of virulence factors have been described in Cryptococcus spp. infections, such as the production of the pigment melanin, its thick polysaccharide capsule is probably the most important virulence factor that protects the fungus from phagocytosis by the host.
The initial infection with Cryptococcus spp. is due to inhalation and the production of a pneumonitis, which is generally asymptomatic even in immunosuppressed patients. Symptomatic patients usually present with fever and cough and a variety of chest radiographic findings, such as nodular, pleural-based lesions and lobar infiltrates. The initial pneumonia generally clears without treatment, even in immunosuppressed patients. Dissemination from the pneumonitis results in seeding of various organs in the body; the CNS is particularly vulnerable (Fig. 22.22a–c). The organism may remain latent in the lung and at other sites indefinitely. Thus, most patients presenting with cryptococcal meningitis have no evidence of a concurrent pulmonary disease.
Fig. 22.22.
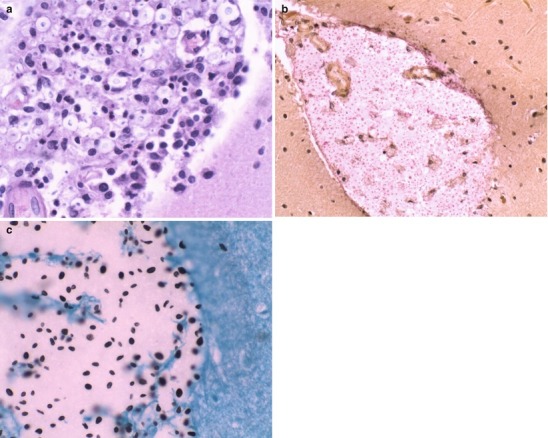
(a–c) CNS cryptococcosis in a patient with HIV infection. Figure (a) shows a distended cerebellar subarachnoid space containing numerous encapsulated Cryptococcus neoformans yeast forms with pericapsular clearing (H&E, 40×). Figure (b) shows how mucicarmine stains the yeast capsules pink to magenta (Mucicarmine, 20×). Figure (c) shows a Gomori methenamine silver (GMS) stain, highlights variably sized yeast forms, with a few yeasts exhibiting “teardrop” budding (GMS, 60×) (All courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
C. neoformans is the most common cause of fungal meningitis worldwide. Clinically, the presentation of cryptococcal meningitis is indistinguishable from that seen in chronic meningitis, associated with coccidioidomycosis, histoplasmosis, or tuberculous meningitis. Patients generally describe a history of 1–3 weeks of headache, fever, and stiff neck together with a variety of nonspecific symptoms, such as lethargy, confusion, nausea, vomiting, or rarely, symptoms suggestive of focal neurologic deficits. Patients may develop raised ICP with papilledema (33 %), cranial nerve palsies (20 %), and seizures; if left untreated, the condition progresses to obtundation and, ultimately, death. Occasionally, total visual loss develops secondary to fungal involvement of the optic tracts, as well as from adhesive arachnoiditis, chorioretinitis, or elevated ICP. Hydrocephalus is a frequent complication even in patients who responded to therapy. Neuroimaging can reveal the extent of CNS involvement: Figure 22.23 shows C. neoformans involvement of the basal ganglia.
Fig. 22.23.
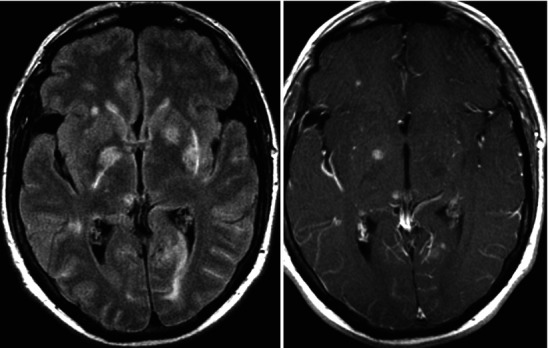
MRI showing Cryptococcus neoformans involvement of the basal ganglia
The diagnosis of cryptococcal meningitis is usually not difficult. The classical and time-honored method for diagnosis is demonstration of the yeast in spinal fluid by India ink stain. Microscopically the India ink particles serve to outline the very large clear polysaccharide capsule surrounding the yeast. The test is positive in over 90 % of HIV-infected patients, but in only 50 % of patients with normal immunity. Several latex agglutination tests have been developed that detect the excess polysaccharide capsule produced in the spinal fluid of patients with cryptococcal meningitis. This test is positive in over 95 % of patients with the condition. While the test is highly reliable, it is important to recognize that there can be both false-negative and false-positive reactions. For example, HIV-infected patients with overwhelming cryptococcal meningitis may have so much polysaccharide capsule in their CSF that a “prozone” effect occurs, resulting in the finding that undiluted CSF or CSF tested at a 1:2 dilution may appear negative. Generally, upon dilution of the spinal fluid to 1:10 or greater, a positive reaction will be observed. Laboratories must be aware of this prozone effect and physicians who suspect cryptococcal meningitis in HIV-infected patients should alert the laboratory to ensure that this possibility is not overlooked. In addition, cross-reactions with Trichosporon beigelii or Capnocytophaga may occasionally produce false-positive latex agglutination tests. The latex agglutination should be confirmed by growth of cryptococci in culture. If a positive cryptococcal antigen titer is found in the CSF from a patient for whom CSF culture did not yielded growth of Cryptococcus spp., the antigen test cannot be assumed to be false-positive—a large volume (10–20 mL) of CSF should be obtained for repeating the cryptococcal antigen test and reculturing for cryptococcus. The higher the cryptococcal antigen titer, the less likely it is to be a false-positive result. CSF changes in cryptococcal meningitis generally parallel those of chronic tuberculous meningitis and other chronic meningitides (Table 22.2). CSF analysis is typically abnormal as follows: glucose is generally less than 40 mg/dL, protein is elevated in the large majority of patients, and the white count is usually elevated with a lymphocytic predominance.
Treatment of cryptococcal meningitis depends in part on host susceptibility factors. In patients with no underlying chronic illness or history of HIV infection, IV amphotericin B at a dose of 0.5–0.8 mg/kg/day together with oral flucytosine 37.5 mg/kg every 8 h should be administered until the patient becomes afebrile and culture-negative; generally, this takes approximately 6 weeks. This should probably be followed by a course of fluconazole 400 mg/day for an additional 2–3 months. Flucytosine may cause severe leukopenia and thrombocytopenia, particularly in patients with impaired renal function, which, in turn, may develop as a result of amphotericin therapy. Therefore, patients need to be monitored carefully for these particular toxic side effects. Some authorities recommend measuring flucytosine levels and adjusting the dose to give a peak of 70–80 mcg/L and a trough of 30–40 mcg/L. However, services for routine measurement of these levels may not be readily available at many hospitals. Alternative therapeutic regimens include various lipid-soluble amphotericin B preparations, which may be used for patients who develop nephrotoxicity. If the patient is intolerant to flucytosine, therapy with amphotericin B at a higher dose of 0.7–1 mg/kg/day for 6–8 weeks may be considered. Fluconazole 400 mg/day orally for 8–10 weeks may be curative, particularly in non-immunocompromised patients who are less ill. However, it should be recognized that fluconazole alone is less effective than the combination of amphotericin B plus flucytosine: the length of time that CSF cultures remain positive is longer and rates of treatment failure tend to be higher for fluconazole-only therapeutic regimens.
Histoplasmosis
Histoplasma spp. are dimorphic fungi (i.e., they change their characteristic morphology depending on temperature.) Along with Coccidioides immitis and Blastomyces dermatitidis, H. capsulatum exists as a mold at room temperature (greater than 23 °C) and converts to a yeast form at body temperature of 37 °C. The organism is widely disseminated in nature in the soil and may reach high levels in areas where birds roost and in caves inhabited by large numbers of bats. In vitro, the mold phase is characterized by both macro- and microconidia. The microconidia are small, smooth oval bodies ranging in diameter from 2 to 5 μm and are believed to be the infective phase because their small size enables them to be readily carried down to the terminal bronchioles and alveoli. Although found worldwide, histoplasmosis has a very distinct geographical distribution in the United States. Most cases occur in the Ohio and Mississippi River valleys in the Central Midwestern part of the United States with extension as far eastward as Maryland, Delaware, and some parts of Georgia and Florida. The disease is rarely seen west of Texas, Oklahoma, and Kansas.
Once inhaled, the microconidia are ingested by alveolar macrophages and rapidly undergo conversion to the yeast phase. From the initial pulmonary foci, the yeast rapidly migrates to hilar lymph nodes from which they can disseminate to multiple foci in the body. As is the case with cryptococcus and tuberculosis, an asymptomatic pulmonary infection frequently develops approximately 2–3 weeks after exposure. The development of the cellular immune response limits the spread of the organism and generally clears the initial, early, pulmonary focus, leaving minimal to no calcifications in hilar lymph nodes and lung tissue. The disseminated lesions are most commonly manifest by widespread calcific lesions in the spleen and liver after they heal.
Although the vast majority of primary infections in immunologically normal hosts resolve spontaneously, leaving the patient with a positive histoplasma skin test, there are a number of unfavorable outcomes. Dormant infections may reactivate following severe immunosuppression—HIV infection, post transplantation, or after therapy with TNF blockers. Pulmonary disease may go on to produce chronic cavitary histoplasmosis that is radiographically identical to pulmonary tuberculosis. The initial pulmonary infection may result in an acute progressive disseminated infection that characteristically presents with fever, chills, weight loss, hepatosplenomegaly, and pancytopenia from bone marrow involvement. This form of disseminated histoplasmosis most often affects those who are highly immunosuppressed due to AIDS, lymphoma, or iatrogenic therapy.
CNS involvement in this syndrome includes encephalitis, acute meningitis, and encephalopathy. Occasionally, histoplasmomas or mass lesions in the CNS are observed [282–286]. A similar syndrome of disseminated histoplasmosis may also be observed in patients with normal immunity who present with more of a low-grade chronic illness, rather than the acute presentation of disseminated histoplasmosis typically seen in immunosuppressed patients. Symptoms of CNS involvement are secondary to intracranial mass lesions, isolated chronic meningitis with or without other manifestations of disseminated histoplasmosis or meningitis. Some patients may have CNS disease secondary to emboli from histoplasma endocarditis. Wheat and colleagues found that approximately 40 % of patients with histoplasma meningitis had chronic meningitis as part of their disseminated disease [284]; 25–30 % had isolated chronic meningitis, and the remainder presented with various forms of mass lesions and encephalitis. In general, the duration of symptoms of histoplasma meningitis prior to diagnosis tends to be somewhat longer than the duration of symptoms for cryptococcal or tuberculous meningitis. In Wheat’s series, approximately 30 % of patients had duration of symptoms less than 1 month; 44 % had symptoms for 2–6 months, and 27 % had symptoms lasting greater than 6 months before the diagnosis was made. Patients who present with localizing CNS signs and symptoms should have neuroimaging (CT scans or MRI) to rule out mass lesions. Those with systemic manifestations may have the diagnosis made by culture or biopsy of an enlarged lymph node, liver, or bone marrow. Diagnosis is best made by visualization of yeast in tissue or by culture. CSF findings are typical for chronic fungal and tuberculous meningitis, with 90 % of patients having a CSF pleocytosis with lymphocytic predominance (Table 22.2). At least 80 % of patients have an elevated CSF protein level and a glucose level less than 40 mg/dL. CSF white blood cells usually number between 50 and 500 cells/μl and are predominantly mononuclear.
Serologic testing for antibodies to H. capsulatum in blood is generally positive in 60–90 % of patients with CNS histoplasmosis. The standard assays are the complement fixation test that uses two separate antigens, yeast and mycelial (or histoplasmin), and the immunodiffusion assay [283]. Diagnosis is based on a fourfold rise in antibody titer; a single titer of ≥1:32 is suggestive but not diagnostic. However, in an endemic area, serological tests are often difficult to interpret because patients may have antibodies from prior exposure rather than active infection; likewise, skin testing in an endemic area is probably of no diagnostic value. In the series described by Wheat, CSF cultures were positive in only 26.7 % of patients [284].
Treatment
Histoplasmosis involving the CNS is difficult to treat. Amphotericin B given as a lipid formulation should be used for initial therapy in all patients at a dosage of 3–5 mg/kg daily for 3–4 months. Therapy can be assessed with serial weekly or biweekly lumbar punctures for CSF Histoplasma antigen levels, white blood cell counts, and complement fixation antibody titers. A rise in the CSF glucose and fall in the CSF cell count and protein levels would suggest successful response to treatment. Resolution of abnormal findings can help determine the duration of therapy [283]. Approximately 80 % of patients will respond to amphotericin, but at least half of those initial responders will relapse and approximately 20 % will die from the disease. Treatment with Amphotericin B should be followed by an oral azole agent for an undetermined period of time. Primary therapy with itraconazole or fluconazole is not useful in the treatment of CNS histoplasmosis—they do not cross the blood–brain barrier particularly well, and failure rates are remarkably high. Kauffman has categorically stated that primary azole therapy of CNS histoplasmosis should be discouraged [283]. Both azoles have been used successfully for secondary treatment following induction therapy with amphotericin B [283]. The dosage suggested for itraconazole is 200 mg twice or thrice daily, and that for fluconazole is 800 mg daily. In HIV-infected patients, suppressive treatment with itraconazole 200 mg orally daily should begin after the initial course of amphotericin.
Coccidioidal Meningitis
Infections with Coccidioides immitis have been recognized for over 100 years and were originally believed to be a nearly uniformly fatal. Like the other dimorphic fungi (Histoplasma capsulatum and Blastomyces dermatitidis), the natural habitat of C. immitis is soil. But unlike the other dimorphic fungi, C. immitis is not distributed worldwide but is limited to the lower Sonoran life zone, found primarily in the desert in the Southwest part of the United States, Mexico, and parts of South and Central America. The characteristics of this particular zone are an arid climate with a yearly rainfall of 5–20 in., hot summers, warm winters, and an alkaline soil. In the mold phase of the fungus, which is the form found in soil and other environmental sources, the hyphae fragment into specific structures known as arthrospores which are highly infectious and readily aerosolized when dust is produced. Thus, on occasion, dust storms have blown infectious spores of C. immitis as far north as Sacramento and beyond to produce outbreaks well outside the endemic area of the San Joaquin Valley. Transmission of the organism on fomites as far as the East Coast has been implicated in documented cases. The narrow environmental requirements of this fungus account for its endemic distribution within the United States. Epidemiologic studies of people who migrate into the Central Valley of California suggest that the annual risk of infection is approximately 15 %.
Pathogenesis
As is the case with the other dimorphic fungi, the initial route of infection is inhalation with an early focus of infection in lung tissue. The outcome of this infection is highly variable. Approximately one-half to two-thirds of patients demonstrated to be infected with this agent show little or absolutely no initial pulmonary infection. The majority of patients who become symptomatic develop a mild self-limited respiratory infection manifest by fever, cough, malaise, arthralgias, weight loss, and in some patients, a striking clinical syndrome of erythema nodosum and erythema multiforme. The vast majority of these symptomatic cases resolve within 2–4 weeks, occasionally taking up to several months, without treatment. Complications occur in no more than about 10 % of all patients with clinically symptomatic primary infections. Occasional patients will present with fulminant pneumonia and shock-like syndrome, possibly resulting from a particularly high inoculum or perhaps as a result of fungemia and miliary dissemination of the disease. Such a presentation is not uncommon among HIV patients with severe depression of the CD4+ count. Other manifestations include pulmonary nodules, cavities, and chronic lung disease indistinguishable from chronic tuberculosis. The most common site of disseminated lesions is the skin where maculopapular lesions may be progress to keratotic and verrucous ulcers with subcutaneous fluctuant abscesses.
The most serious form of disseminated disease following initial pulmonary infection is coccidioidal meningitis which is a result of lymphohematogenous spread from lungs to meninges [287]. Without treatment it is nearly uniformly fatal within 2 years of diagnosis [287–290]. Observational studies suggest about 80 % of patients who develop meningitis become symptomatic within 6 months of the initial infection [291]. The signs and symptoms of coccidioidal meningitis are very similar to those of other chronic fungal and tuberculous meningitides. Patients generally present with headache, fever, varying degrees of nuchal rigidity, nausea, vomiting, seizures, and altered mental status [287–291]. Factors that predispose to C. immitis dissemination and the development of meningitis include immunosuppression due to HIV infection, diabetes mellitus, alcohol abuse, pregnancy, steroids or other immunosuppressive drugs, and non-Caucasian race [289].
Diagnosis of coccidioidal meningitis is based on analysis of the CSF, which shows typical findings of elevated opening pressure (greater than 25 cm H2O), elevated leukocyte count with a predominance of lymphocytes, elevated protein, and depressed glucose (Table 22.2). Occasionally, patients may have a significant CSF eosinophilia [292]. Thus, in a patient who presents with a history suggestive of chronic meningitis together with typical CSF findings, a history of travel to or having lived in an endemic area must be carefully sought after. Complications, such as meningitis, may occur as late as 2 years after the initial exposure, which may be exceedingly brief, such as a mere drive through the California’s Central Valley [286, 288–291].
Although C. immitis grows well on typical fungal media, only about one-third of CSF cultures yield growth of the pathogen [289]. The most reliable method of diagnosis is the detection of complement fixing antibodies in the CSF. Although this testing may be negative in a few patients during the early phase of the disease, they eventually become positive over the ensuing months. In patients with pulmonary disease or extrapulmonary manifestations, biopsy with culture and histopathologic examination of involved tissue may give positive results. The demonstration of a spherule in tissue or a positive culture is diagnostic. MRI scans of the brain invariably are abnormal with evidence of basilar meningitis, hydrocephalus, or cerebral infarcts [289].
Treatment
Fluconazole at 400 mg/day is currently recommended as the treatment of choice for coccidioidal meningitis because the response rate of approximately 70 % is very close to that achieved with intrathecal amphotericin B, which was used in the past. In patients who do not respond to the 400 mg/day dose of fluconazole, higher doses may be used, otherwise options are limited. If there is no response to azole therapy, amphotericin B at a dose of 0.1–0.3 mg/kg/day may be given intrathecally, optimally through an Ommaya reservoir. Intravenous amphotericin B has poor CSF penetration and has a limited role in the management of coccidioidal meningitis [289]. Recent studies on rabbit models suggest that lipid preparations of amphotericin B tend to have better penetration of brain parenchyma and meninges and might be effective in treating coccidioidal meningitis. However, such studies have yet to be duplicated in humans [293, 294]. With the demonstration of rising CSF glucose and falling white cell count and protein, this schedule of amphotericin B administration may be decreased to three times a week after 2–3 weeks of daily treatment, and maintenance may be achieved with twice weekly or once weekly injections. Treatment with either oral fluconazole or intrathecal amphotericin B must be prolonged for at least 2 years after the spinal fluid becomes completely normal. In patients with HIV infection, therapy is continued for life. Patients who have disseminated extrapulmonary disease in addition to meningitis should also receive systemic amphotericin at 0.6–1.0 mg/kg/day for 7 days followed by 0.8 mg/kg every other day, to a total dose of 2.5–3 g. The amphotericin should then be followed by oral fluconazole 400 mg/day for up to a year after the course of amphotericin. It should be noted that the response rate is by no means 100 % in either meningitis or disseminated disease, and that relapses are not uncommon even in patients who respond initially.
As is the case with other fungi and tuberculosis, patients occasionally present with mass lesions in brain parenchyma caused by C. immitis. These lesions invariably require surgical drainage or excision. In addition, hydrocephalus is relatively common in patients with C. immitis meningitis, particularly in children, and must be managed with ventricular shunting. On occasion, C. immitis may actually grow in the shunt and cause obstruction. Patients who have a ventricular shunt in place cannot be administered intrathecal amphotericin B in an Ommaya reservoir. Rather, Amphotericin B must be administered intrathecally via intracisternal puncture or lateral neck injection under radiographic guidance. Although intrathecal therapy can be given via the lumbar route, this inevitably leads to varying degrees of potentially severe and debilitating arachnoiditis after several weeks.
Blastomycosis
Blastomycosis is a systemic pyogranulomatous disease caused by Blastomyces dermatitidis, another dimorphic fungus that grows as a yeast form at 37 º C and in a hyphal form at room temperature. B. dermatitidis exists in nature in the warm moist soils of wooded areas rich in organic debris, such as decaying vegetation. However, the reports of isolation of the organism in nature have been relatively few and somewhat inconsistent. B. dermatitidis occurrence follows the distribution of the Mississippi River and is most commonly reported in states such as Louisiana, Western Alabama, Central Arkansas, Missouri, Kentucky, Western Tennessee, and as far north as Minnesota. Outbreaks have occurred along the St. Lawrence River in Canada and in many parts of North and South Carolina [295].
Pathogenesis
Pulmonary infection occurs by the inhalation of conidia, which convert to the yeast form after deposition in the airways. Pulmonary manifestations vary from asymptomatic to overwhelming multilobar infection with involvement of hilar lymph nodes. From there, dissemination to skin and other organs may occur via lymphohematogenous spread. Normal host responses generate a characteristic pyogranulomatous reaction. Extrapulmonary involvement in descending order of frequency include skin (25 %), osteoarticular (25 %), genitourinary (17 %), and CNS (5 %) disease [295–299]. CNS disease is more common in immunocompromised patients, for example, 40 % of patients with AIDS who have blastomycosis also have CNS involvement as meningitis or mass lesions [295, 300, 301]. Less common metastatic sites include the liver, spleen, and naso-/oropharynx. Chronic pulmonary infection resembling tuberculosis or other chronic fungal disease may develop. Dissemination to the skin produces a variety of clinical manifestations, such as chronic verrucous lesions with crusting and purulent drainage, giant keratoacanthoma, and lesions mimicking pyoderma gangrenosum or squamous cell carcinoma. Suppurative granulomatous lesions of the skin and bone may develop and become chronic.
Pathologically, the histological response is that of neutrophils and non-caseating granulomata with epithelioid and giant cells. Because of a vigorous proliferative response, the pseudoepitheliomatous changes seen on skin and mucosal surfaces in response to B. dermatitidis may resemble squamous cell carcinoma.
Clinical Manifestations
Most acutely infected patients are asymptomatic or develop a self-limited respiratory illness. The vast majority of patients present with symptoms of pneumonia after an incubation period of 30–45 days. Symptoms are nonspecific, with fever, cough, myalgias, arthralgias, and pleuritic chest pain. Chest examination may reveal lobar or segmental consolidation, including mass lesions mimicking carcinoma with hilar lymphadenopathy. Involvement of the CNS is unusual, with cases generally presenting with meningitis, and rarely as intracranial mass lesion or abscesses that might be solitary or multiple [296, 300, 302].
Clinical involvement of the CNS accounts for 5–10 % of extrapulmonary blastomycosis in clinical reviews [299, 303, 304]. Although CNS blastomycosis may develop in patients without evidence of disseminated disease, most reported cases involving the CNS also have evidence of systemic infection. In addition to meningitis, patients with blastomycosis may present with blastomycomas or mass lesions in the CNS more frequently than is seen with other fungal CNS infections [296, 302]. While steroids and immunosuppressive treatment do not predispose to meningitis per se, they may predispose to dissemination of pulmonary blastomycosis and, hence, increase the probability of neurologic involvement.
Diagnosis
Because there is no highly specific serologic or diagnostic skin test material available, the diagnosis rests upon culture of the organism from pulmonary secretions, skin biopsy material, or direct visualization of round, multinucleated yeast forms that produce daughter cells from a single broad-based bud [305]. Histopathologic analysis of tissue specimens stained with methenamine silver or periodic acid-Schiff (PAS) reagents is the standard diagnostic method for extrapulmonary infection [295]. A urinary antigen is available to aid in diagnosis, but it is not particularly specific (79 %) and shows cross-reactivity by testing positive (96.3 %) in patients with histoplasmosis [306, 307]. Antibody assays by complement fixation or immunodiffusion remain nonspecific and insensitive, and the confirmatory diagnostic test at present remains growth of the organism in culture [308].
Treatment
Prior to the availability of amphotericin B and itraconazole, the disease was progressive and was associated with mortality rates greater than 60 %. Some patients with pulmonary disease have a self-limited infection, but it remains unclear whether the initial primary pulmonary infection should be treated. However, because of the inherent limited ability to predict which cases will disseminate, it certainly seems reasonable to treat most cases of pulmonary infection caused by Blastomyces spp. In mild to moderate cases of pulmonary disease, the treatment of choice is itraconazole 200 mg once or twice daily for 6–12 months. In moderately severe to severe disseminated blastomycosis, lipid amphotericin B 3–5 mg/kg/day or deoxycholate amphotericin B 0.3–1.0 mg/kg/day for 1–2 weeks should be used followed itraconazole, 200 mg twice daily for 12 months [295]. Mild to moderate disseminated CNS disease should be treated with high-dose lipid amphotericin B 5 mg/kg/day for 1–2 weeks followed itraconazole 200 mg twice daily for 12 months. If the patient is immunosuppressed or is HIV-infected, the recommended therapeutic regimen is lipid amphotericin B 3–5 mg/kg/day or deoxycholate amphotericin B 0.7–1.0 mg/kg/day for 1–2 weeks followed by itraconazole 200 mg twice daily for 12 months; lifelong suppressive therapy may be indicated in some patients, especially if immunosuppression cannot be reversed [295]. In patients with life-threatening disease, the total dose of amphotericin B may have to be increased.
Candida CNS Infections
Candida species are a part of the normal human enteric flora and rarely produce CNS disease. Meningitis is uncommon except as a complication of an operation or ventricular shunt. Neonates and premature infants seem to be at higher risk than adults. CNS infections caused by C. albicans account for a majority of the infections; infections caused by non-albicans species (e.g., C. parapsilosis, C. tropicalis, C. glabrata, C. pseudotropicalis, C. guilliermondii, C. krusei, or C. lusitaniae) are relatively less common. In general, CNS disease due to Candida species occurs as part of disseminated infection in hospitalized patients who have predisposing risk factors, such as prematurity, intravenous hyperalimentation, indwelling catheters, treatment with corticosteroids, neutropenia, diabetes, HIV infection, patients with leukemia, post neurosurgery, broad-spectrum antimicrobial use, or and indwelling bladder and intravenous catheters [309–313].
Four clinicopathologic disease groups of neurocandidiasis have been identified: (1) cerebral microabscesses that cause a diffuse encephalopathy. Patients with microabscesses are generally diagnosed with systemic candidiasis before death, and the neurologic involvement is usually only detected at autopsy [311, 313, 314]. (2) Candida meningitis that occurs together with disseminated microabscesses or alone as chronic meningitis with a dense exudate at the base of the brain and brain stem. Microscopic examination reveals changes consistent with granulomatous meningitis, sometimes with yeasts and hyphae; the onset is usually subacute with fever and headache [313, 315]. A cerebral CT scan might reveal hydrocephalus and CSF culture invariably yields the microorganism. (3) Cerebral macroabscesses are less frequent and may result in seizures and focal neurological signs; diagnosis is based on neuroimaging studies and biopsy [316]. And (4) vasculitis and mycotic aneurysms vascular complications lead to cerebral infarction and subarachnoid hemorrhage [317].
Zygomycetes and Zygomycosis
Zygomycosis is the term used to describe infection caused any of the zygomycetes—Absidia, Rhizopus, and Mucor. These saprophytic organisms are ubiquitous in the environment, especially soil and commonly found on bread. The genuses Absidia, Rhizopus, and Mucor can invade the CNS by direct extension or from hematogenous spread. Disseminated disease occurring via the hematogenous route may produce CNS mucormycosis resulting in infarction and abscess formation. The Zygomycetes tend to invade the paranasal sinuses or palate with subsequent progression through tissue, nerves, blood vessels, and fascial planes, followed by the vital structures at the base of the brain (Fig. 22.24a, b). In patients with diabetes, this disease typically presents as rhinocerebral mucor (Fig. 22.25). Patients are generally predisposed to this complication if their serum glucose has remained uncontrolled, and the patient has been acidotic for several weeks. Initial symptoms usually include sinus pain, headache, fever, and nasal stuffiness and discharge, which quickly progresses to facial cellulitis, swelling, proptosis, carotid artery occlusion, cavernous sinus thrombosis, CNS infarction due to fungal thrombosis, CNS hemorrhage, abscess, cerebritis, blindness, and airway obstruction caused by head and neck infections. Death ensues if the condition is not treated. Other at-risk populations include persons who have been neutropenic for more than 2–3 weeks, iron overload, burns, HIV infection and AIDS, blood dyscrasias, transplantation, immunosuppression, chemotherapy, IV drug abuse, and high-dose steroids. Patients with rhinocerebral mucormycosis may manifest hyperglycemia and a metabolic acidosis. Lumbar puncture and CSF analysis invariably reveal a raised opening pressure, a neutrophilic pleocytosis, normal or slightly elevated protein levels, and low glucose levels. In most cases, however, CSF study findings are normal [318]. Neuroimaging studies help to support the diagnosis of rhinocerebral mucormycosis and to precisely determine the extent of disease. CT scanning and MRI reveal soft tissue extent of infection, mucosal thickening, and bony destruction of the sinuses and orbit, presence of cavernous sinus thrombosis, cerebritis, and cerebral edema (Fig. 22.25). Tissue biopsy is necessary to demonstrate the invasive hyphae. The Zygomycetes all appear as ribbon-like nonseptate hyphae in tissue with right- or obtuse-angle branching (Fig. 22.24b). Treatment of the condition is based on three main principles: rapid reversal of underlying predisposing factors, antifungal therapy with amphotericin B, and timely surgical intervention. Treatment includes control of the underlying predisposing condition (e.g., control of diabetic ketoacidosis, hypoxia, and electrolyte abnormalities), IV amphotericin B in a dose of at least 1 mg/kg/day, together with vigorous surgical debridement of all involved tissue. Dexamethasone has been used to treat brain edema. Rhinocerebral mucormycosis carries a prognosis of high morbidity and mortality. Survival depends on the reversibility of underlying risk factors and early surgical intervention [319].
Fig. 22.24.
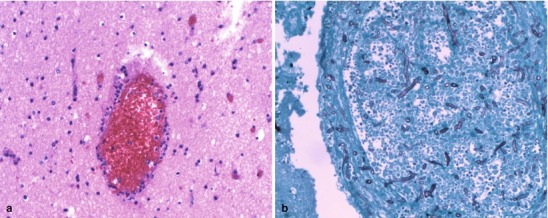
(a) A 63-year-old male with disseminated mucor infection. Shows CNS vasculitis with neutrophils invading CNS parenchymal vessels with early vessel wall destruction (H&E, 20×). (b) Same patient. Features a Gomori methenamine silver (GMS)-stained specimen demonstrating angioinvasion by fungi with broad aseptate hyphae and right-angle branching (GMS, 20×) (Both courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Fig. 22.25.
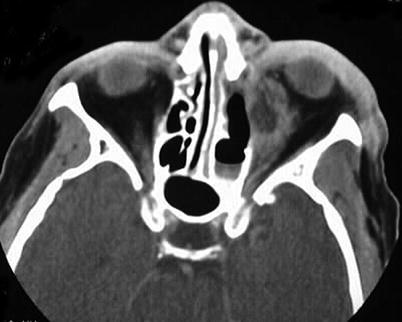
Orbital mucormycosis. Image is a post-contrast axial CT section through the ethmoidal bridge. This case illustrates changes of a left subperiosteal, mesial, orbital abscess. There is little evidence of ethmoid sinusitis. However, it is not uncommon, as in this case, that invasive fungal infections arising either from sinusitis or rhinitis may have only subtle mucosal thickening on imaging, yet still can permeate bone creating soft tissue abscesses in the skull base
CNS Infections Caused by Aspergillus Species
Aspergillus fumigatus and Aspergillus flavus can produce CNS disease very similar to that described earlier for mucormycosis. Invasive aspergillosis can occur in patients with preexisting bronchiectasis, chronic bronchitis, asthma, tuberculosis, or in persons who are immunosuppressed (e.g., solid organ transplant recipients). Patients with prolonged and profound neutropenia (less than 100 cells /μL) are at high risk for invasive aspergillosis. Colonization with Aspergillus spp. can lead to tissue invasion with branching septate hyphae that are best visualized in tissue with silver stains and histologically distinct from other fungi—i.e., Aspergillus spp. have frequent septae that branch at 45° angles. Lung invasion can involve blood vessels leading to hematogenous spread. The majority of invasive CNS infections occur in immunosuppressed transplant patients in the hospital who had pulmonary disease that progressed to brain involvement via hematogenous dissemination. Compared with persons with uncontrolled diabetes who are more susceptible to infections caused by mucor, patients with prolonged neutropenia appear to be more susceptible to invasive Aspergillus spp. sinus infection with direct extension from the maxillary and ethmoid sinuses with progression to cavernous sinus thrombosis. Voriconazole which has good CNS tissue penetration is usually first-line therapy with surgical debridement of all infected tissue. High-dose amphotericin B may be considered in treatment failure [320].
Opportunistic Infections Associated with HIV Infection
Despite the marked improvement in outcomes and outlook for HIV-infected patients with the introduction of highly active antiretroviral therapy (HAART) in 1995, there are still approximately 50,000 new cases of HIV infection per year in the United States [321]. HIV infection predisposes individuals to a variety of opportunistic infections of the brain, including infections caused by Cryptococcus spp., Toxoplasma gondii, CMV, or the polyomavirus (JC virus) that causes progressive multifocal leukoencephalopathy (PML). In addition, the virus itself is associated with a variety of neuropathologic manifestations.
Toxoplasma Gondii
Toxoplasmic encephalitis is caused by the protozoan T. gondii. Disease appears to occur almost exclusively because of reactivation of latent tissue cysts. Primary infection occasionally is associated with acute cerebral or disseminated disease. T. gondii causes latent infection in a significant proportion (10–90 %) of the world’s population but uncommonly causes clinically significant disease [322]. In addition, T. gondii infects numerous wild and domestic animals. Human infection generally occurs through the ingestion of raw or undercooked meat that contains cysts, or by the ingestion of food or water contaminated by the oocysts shed in the stool of infected animals. In the United States, the major animal reservoir for this infection is the domestic cat. The life cycle is complex. The sexual phase of the cycle takes place in the cat with formation of oocysts in the mucosal lining of the intestine for approximately 3 weeks after initial infection. During this time, as many as ten million oocysts may be shed daily. Oocysts require 1–5 days to become infectious after being shed by the cat, a process that depends on temperature and availability of oxygen. During intestinal infection, the tachyzoite form of the organism, a 2–4 μm wide by 4–8 μm long crescent-like structure, is produced and disseminates to many different areas in the host. Wide ranges of intermediate hosts ingest the oocysts, which form latest cysts in many tissues, including muscle. In humans, as in many domestic and wild animals, this dissemination is asymptomatic and results in the formation of cysts in brain parenchyma as well as muscle and numerous other organs. Transmission to humans is a result of exposure to excreta from cats, ingestion of undercooked infected meat, or from contaminated water supplies.
Serologic surveys in the United States show that approximately 15 % of the general population have been infected with toxoplasmosis at some time in the past [323]. In contrast, the prevalence is much higher in economically underdeveloped countries and in certain parts of Europe where rates may range as high as 75 %. Although reactivation of toxoplasmosis in the brain generally leads to local replication with the production of single or multiple abscesses, hematogenous dissemination and infection in the lung and in other parts of the body have been documented. The incidence and attributable mortality in Europe and the United States have decreased substantially since the introduction of antiretroviral therapy and the broad use of prophylaxis regimens active against T. gondii.
Clinical Manifestations
In humans, primary infection with toxoplasma may, on occasion, produce a mononucleosis-like illness characterized by lymphadenopathy, fever, malaise, liver function abnormalities, and, occasionally, myocarditis. However, in the large majority of cases, primary infection is asymptomatic and only becomes recognized under conditions of extreme immunosuppression, such as occurs when CD4+ counts in HIV-infected individuals fall to levels less than 100 cells/mm3, when the cysts break down and initiate symptomatic infection. In immunocompromised patients, reactivation of latent disease can cause life-threatening encephalitis. Patients with toxoplasmic encephalitis generally present with a syndrome of fever, headache and varying degrees of confusion, lethargy, obtundation, focal neurologic signs developing over a period of 1–3 weeks, lymphadenopathy, and splenomegaly. In some patients, the presentation can be abrupt, with seizures or cerebral hemorrhage. Hemiparesis and abnormalities of speech are quite common. Less commonly, brain stem involvement may result in cranial nerve deficits, while dyskinesias such as Parkinsonism, dystonia, tremor, and hemiballismus may also accompany the presenting syndromes. Patients occasionally present with endocrine abnormalities due to involvement of the pituitary axis as well as psychiatric manifestations, such as psychoses and dementia.
Diagnosis
The diagnosis is made by a combination of clinical, imaging, morphological, and serological investigations. In an HIV-infected patient, the finding of single or multiple ring-enhancing lesions by CT or MRI scanning strongly suggests toxoplasma encephalitis (Fig. 22.26a–c). If the patient is also known to be, or is found to be, serologically positive for antibodies to toxoplasma, the combination is virtually diagnostic. In patients with a single lesion, the major differential is lymphoma, and a brain biopsy may be required to make a definitive diagnosis. MRI has superior sensitivity when compared with CT scanning and should therefore be used as the initial diagnostic procedure when feasible. Toxoplasmic encephalitis lesions on MRI appear as high-signal abnormalities on T2-weighted studies and have a rim of enhancement surrounding the edema on T1-weighted, contrast-enhanced images. Other imaging techniques, such as PET, radionuclide (e.g., Thallium 201) scanning, and magnetic resonance techniques have been used to evaluate patients with AIDS who have focal CNS lesions and to specifically differentiate between toxoplasmosis and primary CNS lymphoma. Grossly, the acute lesions are hemorrhagic, necrotic, space-occupying lesions with surrounding edema. Histologically, there are cysts, free parasites, necrosis, and vasculitis (Fig. 22.27a, b). These lesions regress with treatment.
Fig. 22.26.
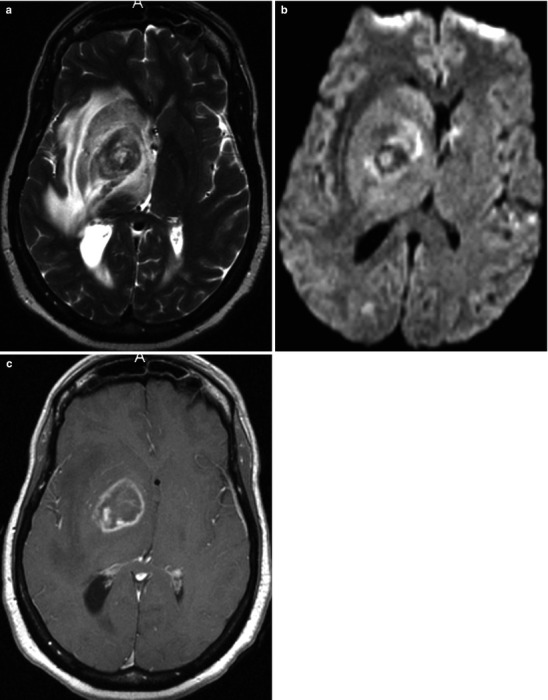
(a–c) Toxoplasmosis in the right basal ganglia with a T2-w sequence (a), a diffusion-weighted image (b) and a post gadolinium image (c). Toxoplasmosis has a propensity for basal ganglia involvement. It has features which are often very similar to glioblastoma and primary CNS lymphoma and often different from pyogenic abscess. Note in this case that despite the marginal enhancement (similar to pyogenic abscess) the central portion of the abscess forms neither a distinct suppurative cavity nor exhibits water in its center as usually occurs in a pyogenic abscess cavity. In the context of an immunocompromised host, toxoplasmosis should be included as a potential tumefactive entity in the differential of any central nuclear mass
Fig. 22.27.
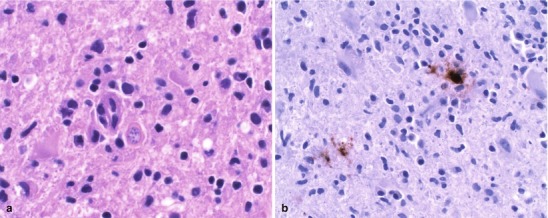
(a) CNS toxoplasmosis. This figure features cysts (bradyzoites) of Toxoplasma gondii in brain tissue (H&E, 60×). (b) Shows an anti-Toxoplasmagondii antibody immunohistochemical study that is immunoreactive for both bradyzoites and trophozoites (Both courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Laboratory studies, such as complete blood cell count (CBC), chemistries, and liver function tests, are typically normal. Affected patients may have a lymphocytosis. CSF analysis shows nonspecific abnormalities, such as elevated white count and protein levels. Attempts to improve the sensitivity by the detection of toxoplasma oligoclonal antibody bands have been reported but do not add any more diagnostic value than a positive serum antibody test. PCR for toxoplasmosis in CSF has been reported to have a sensitivity of 81 % in untreated patients with acute disease; the drawback is that many “in-house” PCR assays suffer from lack of standardization and variable performance according to the laboratory performing the assays [324].
Although a wide variety of antibody tests are available, including the original Sabin Feldman Dye Test, current commercial ELISA (enzyme-linked immunosorbent assay), IgG, and IgM tests, along with the indirect fluorescent antibody test, are readily available and highly specific. Because the disease in HIV patients is almost always due to reactivation of an old focus, the IgG test should be positive and the IgM test should be negative in these individuals. However, diagnosis based on classical serological testing is often inconclusive as immunodeficient individuals often fail to produce significant titers of specific antibodies [324].
Treatment
Treatment should be instituted empirically for HIV-infected patients with compatible clinical and imaging studies. The standard drug regimens include 6 weeks of therapy with pyrimethamine 200 mg as a loading dose followed by 75–100 mg daily together with either sulfadiazine 1–1.5 g every 6 h or clindamycin 600–1,200 mg IV every 6 h. Folinic acid (leucovorin), 10–20 mg/day, is given to reduce bone marrow toxicity. If the patient is intolerant of sulfadiazine or clindamycin, the following may be given in addition to pyrimethamine and folinic acid: clarithromycin 1 g orally every 12 h, atovaquone 700 mg orally every 6 h, azithromycin 1,200–1,500 mg/day orally, or dapsone 100 mg/day.
A systematic review of therapeutic regimens for the management of toxoplasmic encephalitis in HIV-infected adults concluded that pyrimethamine + sulfadiazine and pyrimethamine + clindamycin were equivalent in effectiveness in the treatment of acute toxoplasmic encephalitis in HIV-infected individuals [324, 325]. The review also found that trimethoprim/sulfamethoxazole, which is cheap and readily available in developing countries, may be suitable first-line therapy for acute toxoplasmic encephalitis in HIV-infected individuals and, in fact, was not found to be inferior compared with pyrimethamine + sulfadiazine [325]. More recent studies indicate that trimethoprim/sulfamethoxazole is indeed an alternative treatment for toxoplasmic encephalitis because it is inexpensive, well tolerated, and as effective as pyrimethamine-sulfadiazine [326]. Trimethoprim/sulfamethoxazole may be given 3–5 mg/kg of the trimethoprim component orally or IV every 12 h for 4–6 weeks. Other potential advantages of trimethoprim/sulfamethoxazole include less adverse events, ease of dosing, parenteral formulation, cost, and accessibility [326–328]. Pereira-Chioccola and colleagues recommend that complications, such as expansive brain lesions with a mass effect (e.g., deviation of the middle line structures or imminent risk of cerebral herniation) and cases with diffuse encephalitis should be administered adjunctive corticosteroid therapy. Seizures should be treated with appropriate anticonvulsant agents [327].
Signs of improvement are generally seen in the level of consciousness and in a decrease of fever within 5–7 days, with over 90 % generally responding by day 14 [329]. If there is no clinical or radiographic response within 10 days, alternative diagnoses and brain biopsy must be considered. Reactivation of latent infection in the CNS is a common HIV/AIDS-related complication. Thus, current guidelines recommend that patients with toxoplasmic encephalitis should be treated with the initial regimen for 4–6 weeks, depending upon the degree and rapidity of improvement, and then receive lifelong suppression with sulfadiazine 500–1,000 mg orally four times daily and with pyrimethamine 25–75 mg and folinic acid 10–20 mg both by mouth daily. Patients on long-term maintenance treatment for cerebral toxoplasmosis have a low risk for subsequently developing Pneumocystis carinii pneumonia. This decreased risk is thought to be the result of chronic suppressive treatment with pyrimethamine and sulfonamides [330]. More recent data suggest that discontinuation of maintenance therapy against toxoplasmic encephalitis for individuals infected with HIV/AIDS, who are receiving successful antiretroviral therapy, might be safe and that patients who remain clinically and radiologically free of relapse at 6 months after discontinuation are unlikely to experience a relapse of toxoplasmic encephalitis [331, 332]. However, the overall body of published data in support of a policy that recommends discontinuing secondary prophylaxis remains relatively limited at the present time.
Cytomegalovirus (CMV)
CMV is a DNA herpes virus. CMV is similar to HSV and VZV in its ability to establish latent infection. CMV tends to infect epithelial cells and leukocytes. In HIV-infected adults, serologic evidence of prior CMV infection is present in over 90 % of individuals. Serologic surveys in the general adult population show a seroprevalence rate ranging between 50 and 80 %, depending upon the socioeconomic status and the particular group studied; the seroprevalence of CMV rises with age. Approximately 10 % of all infants born in the United States either have CMV infection at birth or acquire it within the neonatal period. The vast majority of these infections is asymptomatic and result from exposure to reactivated virus that is transmitted transplacentally in seropositive mothers. Subsequently, both children and adults may become infected through exposure to infected urine often from infants in day care; the virus may also be acquired from sexual contact, generally during the late teens and twenties, or by respiratory droplet infection. In non-immunocompromised adults, primary CMV infection is clinically identical to that of infectious mononucleosis and runs a self-limited course of 2–3 weeks characterized by fever, fatigue, malaise, lymphadenopathy, sore throat, and elevated liver enzymes, together with atypical lymphocytes in the blood smear.
In HIV-infected individuals, as long as cell-mediated immunity remains intact, symptomatic reactivation of CMV is not generally seen. However, as the disease progresses and the CD4+ count falls to less than 50 cells/mm3, reactivation of CMV becomes more frequent. CMV retinitis is the most common form recognized clinically, and in the pre-HAART era, up to 30 % of HIV-infected patients showed clinical evidence of CMV retinitis. The diagnosis of CMV retinitis is a based on the puffy white retinal infiltrates seen together with retinal hemorrhage. It may or may not be associated with systemic or other manifestations of CMV. Either one or both eyes may be involved at a given time.
There are several forms of CNS involvement with CMV and the pathogenesis appears to follow two different routes: (1) via the ependymal cells in the ventricles and (2) via the blood through capillary endothelial cells. Infection via the ependymal cells and CSF is manifest as necrotizing encephalitis limited to the periventricular areas, with numerous cytomegalic inclusions in and around the lesions. In contrast, infection acquired via hematogenous dissemination results in microglial nodular encephalitis, which is manifest pathologically as glial nodules formed by rod cells, few lymphocytes, and macrophages. Very little tissue damage is associated with these lesions [333]. The microglial nodular disease may involve multiple parts of the brain including the periventricular areas. As with CMV retinitis, these CNS complications occur very late in HIV infection and almost always in patients with CD4+ counts less than 50 cells/mm3. In the series reported by Grassi and coworkers, the mean CD4+ count for patients with microglial nodular encephalitis was 21 cells/mm3, while the average CD4+ count of patients with ventriculoencephalitis was 11 cells/mm3 [333].
Although there is considerable overlap of symptomatology between these two pathologic conditions, microglial nodular encephalitis is characterized by the onset of acute confusion associated with delirium and psychomotor agitation, whereas the onset of ventriculoencephalitis is insidious and generally characterized by cognitive disturbances, memory deficits, and mental sluggishness. Patients with either type of encephalopathy may complain of headaches and have seizures. CSF examination in these conditions reveals a slightly elevated protein, which tends to be lower in microglial nodular encephalitis, averaging approximately 60 mg/dL as compared with 172 mg/dL in ventriculoencephalitis. Glucose levels are normal and cells are generally, although not always, absent. Although MRI is more sensitive than CT scan in the diagnosis of these conditions, the MRI may be normal or show nonspecific changes [334, 335]. In patients with ventriculoencephalitis, typical MRI findings include a hyperintense ventricular rim on T2-weighted imaging or enhancements with gadolinium. Almost all cases show nonspecific cerebral atrophy. Systemic involvement with CMV is more frequently associated with microglial nodular encephalitis, which may be documented with CMV antigenemia testing or viral culture. Patients with ventriculoencephalitis are more likely to have the clinical syndrome of CMV radiculopathy [333, 335–338]. CMV infection in HIV patients may also involve the spinal cord. A syndrome characterized by ascending weakness of the lower extremities associated with loss of deep tendon reflexes progressing to loss of bowel and bladder control has been described [339]. The syndrome may begin with low back pain with radiation down to the legs or into the groin or anal area, followed over 1–3 weeks by the development of progressive weakness.
Laboratory diagnosis is carried out in several ways: (1) seroconversion, (2) DNA detection in infected tissues using PCR, (3) antigen detection in tissues, (4) cytopathology, (5) isolation of virus from tissue or secretions, or (vi) CMV cytopathology. Pathologically, the CMV radiculopathy is characterized by mononuclear infiltration of the cauda equina and lumbar sacral nerve roots together with CMV inclusions seen in the Schwann’s cells and epithelial cells, leading to axonal destruction. Untreated, this condition generally progresses to irreversible paralysis. Analysis of CSF in patients with this syndrome characteristically shows elevation of neutrophils, sometimes as high as 5,000 cells/mm3. Although the CSF protein is only mildly elevated and the glucose generally normal, some patients may have marked hypoglycorrhachia with glucose levels as low as 5–10 mg/ dL.
Treatment
Ganciclovir 5 mg/kg IV every 12 h should be given for 14–21 days followed by maintenance with oral or IV doses that must be continued indefinitely to avoid or delay relapses, unless CD4+ recovery occurs. Valganciclovir, a prodrug of ganciclovir, possessing excellent oral bioavailability and antiviral activity is effective in both the induction phase and the maintenance phase of CMV retinitis therapy [340, 341]. Ganciclovir blocks CMV replication by inhibiting CMV DNA polymerase. Alternatively, foscarnet 90 mg/kg, adjusted for renal function, can be given IV twice daily for 2–3 weeks. Following induction with ganciclovir or foscarnet, HAART therapy can then be given in the hope of sustained improvement in CD4+ count and maintenance of a response to therapy. In the pre-HAART era, CMV encephalitis responded relatively poorly to ganciclovir, with survival generally in the 3–4 month range. The suppression of HIV replication and elevation in CD4 cells observed during HAART may allow AIDS patients to undergo immune reconstitution. Discontinuation of maintenance CMV therapy for these patients may be considered in these patients [332, 342, 343]. The drawback to withdrawing maintenance therapy is the risk of acquiring sight-threatening inflammatory conditions, including immune recovery uveitis [344].
HIV Encephalopathy and Dementia
HIV is a neurotropic virus that appears to enter the brain via infected macrophages early in the course of infection. HIV infection itself causes CNS complications, including encephalitis and dementia. Encephalopathy usually develops as part of the acute HIV syndrome during the seroconversion phase. Two pathogenic mechanisms are thought to underlie these CNS conditions. In the first mechanism, HIV and its fragments induce damage directly or indirectly through the accumulation of infected or activated macrophage and microglia cells that release neurotoxic mediators including both cellular activation products and viral proteins [345, 346]. The accumulation of these activated macrophage/microglia cells, some of which are infected, release a number of cytokines and small molecule mediators and viral proteins that act on bystander cells. These viral proteins and cellular products have neurotoxic properties and act directly and through induction of astrocyte dysfunction, leading to neuronal injury [346–348]. The second, less predominant, pathogenic mechanism is the ability of HIV to impair neurogenesis [349, 350]. Kaul suggests that both proposed pathogenic mechanisms occur side by side with other host-virus interactions [345].
Pathologically, the most frequent findings in persons with HIV encephalopathy are brain atrophy characterized by sulcal widening and ventricular dilatation together with varying degrees of meningeal fibrosis. The most distinctive histologic feature of this condition is white matter pallor, chiefly seen in a periventricular distribution together with microglial nodules, diffuse astrocytosis, and perivascular mononuclear inflammation. HIV can be readily demonstrated in these nodules by immunohistochemical techniques. HIV-associated dementia is a late phenomenon caused by cortical neuronal loss and abnormalities of the dendritic connections; cerebral atrophy in these patients invariably is confirmed on gross necropsy. Histologically, the brain of a person with HIV encephalitis is characterized by multiple nodules of infected microglia and microglial giant cells in the white matter (Fig. 22.10). These lesions also affect neural function by secreting toxic cytokines.
Clinically, HIV encephalopathy presents with altered mental status characterized by mental slowing frequently accompanied by clinical evidence of subcortical dementia, such as bradykinesia, postural instability, slow and clumsy gait, and altered muscle tone. Radiographically, the most common features are generalized cerebral atrophy together with widespread symmetric hyperintense white matter abnormalities seen on T2 imaging, which generally have a periventricular distribution. Examples are shown in Fig. 22.28. CSF studies are non-diagnostic: the CSF is commonly acellular and a mononuclear pleocytosis is seen in about 25 % of patients with cell counts generally less than 50 cells/mm3 [351, 352]. CSF protein levels are elevated (usually less than 200 mg/dL) in 60 % of patients, while CSF immunoglobulin G levels are raised in about 80 %. Glucose levels in the CSF of HIV-infected patients are usually within normal limits.
Fig. 22.28.
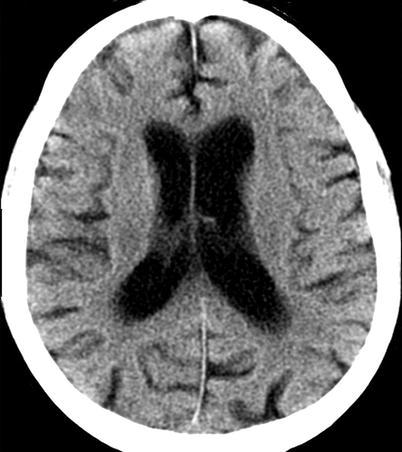
Primary HIV infection of the CNS. Ventriculomegaly and sulcal dilatation (indicating cortical brain atrophy). There is no transependymal fluid to suggest elevated CSF pressure. These findings are indicative of both central and cortical brain atrophy which, in this case, is out of proportion to chronological age. Generalized atrophy is one of the features of primary HIV involvement of brain (Reproduced with permission from Rand et al. [521])
HIV may be detected in CSF by a variety of techniques, including PCR, during all the clinical stages of infection, i.e., HIV is present in the CSF in the absence of neurologic abnormalities. Thus, the presence of HIV in the CSF is not diagnostic for HIV encephalopathy and HIV RNA levels in the CSF do not necessarily correlate with the corresponding plasma levels. Neuroimaging studies can support a diagnosis of AIDS dementia complex by revealing cortical atrophy and ventricular enlargement and hyperintense lesions in the periventricular white matter on MRI. HAART remains the main mode of medical treatment for HIV encephalopathy and other HIV-related cognitive disorders.
Parasitic Infections of the Central Nervous System
Cerebral Malaria
Despite its eradication from North America and Europe, it is estimated that malaria infects over 2.5 billion people worldwide and causes between one and three million deaths each year. The disease is caused by one of five different Plasmodium species: P. vivax, P. ovale, P. malariae, P. knowlesi, and P. falciparum. Most of the deaths and serious complications of malaria, especially CNS involvement, are caused by P. falciparum. Malaria sporozoites are transmitted from the female Anopheles mosquito to the patient at the time the mosquito bites a person for its blood meal. Sporozoites are carried rapidly to the liver where they multiply in approximately 1 week to become tissue schizonts or the dormant hypnozoites produced by P. vivax and P. ovale. Infected liver cells then burst, releasing thousands of merozoites, each of which in turn infects red blood cells in the blood stream. Continued asexual replication in the bloodstream through repeated cycles of maturation and rupture of red cells with release of merozoites eventually results in symptomatic infection. During this process, some of the parasites develop into sexual forms called gametocytes which produce no symptoms themselves but which may circulate for a prolonged period of time. It is the ingestion of these gametocytes that leads to the sexual reproduction cycle in the Anopheles mosquito resulting in the motile sporozoites which invade the mosquito salivary glands and can be transmitted back to humans at the time of the next feeding.
Malaria is widely distributed in economically less-developed countries, particularly sub-Saharan Africa, Central America and the Caribbean, South America, the Middle East, Far East, and Indonesia. The reader is strongly urged to access the CDC’s website for the most up-to-date availability on the distribution and drug resistance among Plasmodium spp. on a country-by-country basis.
Pathogenesis
P. falciparum is the cause of the most malignant form of malaria and is associated with almost all serious complications associated with the infection. Cerebral malaria, in particular, is the most prominent and serious of these complications. Attributable mortality remains relatively high (20 %) and is often associated with delays in diagnosis and treatment. As P. falciparum trophozoites mature in the red blood cells, they induce the formation of small knobs on the surface of the red cell. These knobs bind to adhesion molecules (also known as intercellular adhesion molecule-1) on the microvascular endothelial cells, leading to sequestration. Sequestration is the process whereby erythrocytes containing mature forms of P. falciparum adhere to microvascular endothelial cells resulting in marked reduction or disappearance of these cells from the circulation. Sequestration of erythrocytes in small blood vessels and consequent obstruction of microcirculatory flow is a specific property of P falciparum and an important mechanism causing coma and death in cerebral malaria. The second important factor in the pathogenesis of cerebral malaria is the increase in cytokine production. In an attempt to control the infection, the host immune system produces a potent proinflammatory response in which cells of the macrophage-monocyte series are induced to release various cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, and IL-8 [353]. However, this response may also induce complications, such as severe anemia, hypoglycemia, and cerebral malaria.
The adhesion molecules are upregulated in malaria as a result of cytokine productions, TNF-α in particular. Furthermore, parasitized erythrocytes tend to adhere to adjacent uninfected cells leading to rosetting. In addition, as the parasite matures inside the erythrocyte, the normally flexible cell becomes more spherical and rigid. Because of the rosetting and increased rigidity of parasitized erythrocytes, the erythrocytes become trapped in the capillaries. The end result of cytoadherence, rosetting, and rigidity is the enhancement of sequestration of P. falciparum-parasitized erythrocytes in the cerebral vasculature, stagnation of the cerebral blood flow, and secondary ischemia leading to tissue hypoxia, lactic acidosis, hypoglycemia, and prevention of delivery of nutrients to the tissues. High concentrations of TNF-α can precipitate cerebral malaria by increasing the sequestration of parasitized erythrocytes. Although all tissues potentially can become involved, the brain is the most profoundly affected [354–362].
In the CNS, this process results in delirium, impaired consciousness, convulsions, paralysis, coma, and, ultimately, rapid death if not treated. Systemic manifestations of severe falciparum malaria include anemia, lactic acidosis, hypoglycemia, pulmonary edema, adult respiratory distress syndrome, and disseminated intravascular coagulation. Of note, the pathophysiology of malaria does not include vasculitis or inflammatory cellular infiltration in or around the cerebral vasculature, and most patients have no evidence of cerebral edema. Raised intracranial pressure likely arises from an increase in the overall cerebral blood volume rather than brain swelling arising from cerebral edema and capillary leakage. Coma in malaria is generally not associated with raised intracranial pressure. Clinical features of P. falciparum malaria include fever and chills (83 %), altered sensorium (48 %), jaundice (27 %), anemia (75 %), cerebral involvement (45 %), thrombocytopenia (41 %), and renal failure (25 %).
The diagnosis should be considered in a person with altered consciousness, fever, and a relevant travel history, which is critically important to elicit, as is the history of whether the patient took or was compliant with prophylaxis for malaria. The location of the travel is particularly critical as P. falciparum is typically resistant to chloroquine. Resistance to trimethoprim/sulfamethoxazole, mefloquine, and other agents has been documented in many parts of the world, particularly Southeast Asia and sub-Saharan Africa. Because there is no latent form of P. falciparum in the liver, as there is for P. vivax and P. ovale, cases of P. falciparum malaria should become clinically evident within a month after leaving an endemic area.
The laboratory diagnosis of malaria is made from examination of the blood smear. Although delay is common because of the time needed for their preparation and reading, thick and thin blood smears remain the cornerstone of laboratory diagnosis of malaria in current practice. Despite the availability of rapid diagnostic testing for the detection of malaria based on lateral-flow immunochromatography in which clinicians can detect malaria parasite antigens from finger-prick blood specimens within 10–15 min, microscopic examination of blood smears remains the most cost-effective methodology for diagnosis of malaria, provided the results reach those who need to know in a timely manner.
Rapid diagnostic testing kits based on molecular platforms with high sensitivity and high negative predictive value for P. falciparum would be of particular use in acute care settings in regions of low malaria endemicity, where the diagnosis is suspected but lack of laboratory expertise precludes the diagnosis and reading of blood smears. Lastly, rapid diagnostic testing may benefit severely ill patients by confirming or excluding a malaria diagnosis rapidly and facilitating prompt intervention. Rapid test kits for malaria have limitations that preclude replacing microscopy of blood smears any time soon. These limitations include inability to ascertain parasitemia quantitatively or to differentiate between the four plasmodium species.
Treatment
Untreated, cerebral malaria is fatal. In cases of severe malaria with CNS involvement, the patient must be treated as though they have falciparum malaria regardless of the preliminary interpretation of the blood smear. According to CDC, if severe malaria is strongly suspected but a laboratory diagnosis cannot be made at that time, blood should be collected for diagnostic testing as soon as it is available and parenteral antimalarial drugs started empirically. Once the diagnosis is considered likely, parenteral quinidine gluconate should be started. The recommended therapeutic regimen includes a loading dose of 6.25 mg base/kg (=10 mg salt/kg) infused intravenously over 1–2 h followed by a continuous infusion of 0.0125 mg base/kg/min (=0.02 mg salt/kg/min). An alternative regimen is an intravenous loading dose of 15 mg base/kg (=24 mg salt/kg) of quinidine gluconate infused intravenously over 4 h, followed by 7.5 mg base/kg (=12 mg/kg salt) infused over 4 h every 8 h, starting 8 h after the loading dose. Quinidine gluconate therapy should be combined with doxycycline, tetracycline, or clindamycin. If the patient is unable to tolerate oral therapy, doxycycline (100 mg every 12 h) or clindamycin (5 mg base/kg every 8 h) may be given intravenously until the patient can be switched to oral therapy [354, 356, 359]. More recently, artemisinin derivatives, such as parenteral artesunate, have been shown to substantially reduce mortality in African children with severe malaria; the authors go on to suggest that parenteral artesunate should replace quinine as the treatment of choice for severe falciparum malaria worldwide [363].
Parenteral quinidine gluconate is cardiotoxic and can induce hyperinsulinemic hypoglycemia. Thus, a baseline EKG should be obtained before initiating therapy and glucose levels must be monitored closely. Critical care management includes continuous cardiac and blood pressure monitoring with appropriate supportive management of coexisting medical complications often associated with severe malaria: convulsions, renal failure, adult respiratory distress syndrome, disseminated intravascular coagulation, lactic acidosis, hypoglycemia, fluid and electrolyte abnormalities, circulatory collapse, acute renal failure, secondary bacterial infections, and severe anemia.
Intravenous corticosteroids are associated with poor outcomes and are absolutely contraindicated [361]. Brain swelling on CT scan is a common finding in adult patients with cerebral malaria but is not related to coma depth or survival. Mannitol therapy as adjunctive treatment for brain swelling in adult cerebral malaria prolongs coma duration and may be harmful [364]. Results of studies of antipyretics, anticonvulsants (phenobarbitone), anticytokine/anti-inflammatory agents (anti-TNF antibodies, pentoxifylline, dexamethasone), iron chelators, and hyperimmune sera have not proven beneficial in improving patient outcomes [359].
With treatment, almost all patients with CNS malaria recover completely if they survive the acute episode. However, globally, overall mortality in children and adults remains unacceptably high. Approximately 12 % of patients with cerebral malaria may have lasting neurologic sequelae, including cortical blindness, tremor, cranial nerve palsies, and sensory and motor deficits, although approximately 50 % of these sequelae resolve with time.
Amoebic Meningoencephalitis
A dramatic and almost uniformly fatal primary meningoencephalitis can be seen with infection caused by the free-living amoeba Naegleria fowleri and Acanthamoeba species. Free-living N. fowleri are found widespread in nature, particularly in the upper surface layers of lakes or shallow fresh water in warm climates. Acanthamoeba spp. are usually found in soil and in fresh and brackish water. Most cases in the United States, typically children or young adults, occur in the southeastern states. Clinically, patients present with sudden onset of high fever, photophobia, and headaches; progression to obtundation occurs relatively quickly. There is usually a classic history of swimming or waterskiing in warm, freshwater lakes. The amoebae invade the nasal cavity along the blood vessels associated with the olfactory nerves and traverse the cribriform plate to reach the frontal lobes and the surrounding meninges, where they rapidly produce a highly necrotizing, purulent, and destructive encephalitis. Because of the olfactory involvement, there may be alterations of smell or taste. Otherwise, nonspecific symptoms such as confusion, irritability, restlessness, and seizures with rapid progression to delirium, stupor, and coma ensue. The CSF is usually bloody, shows a leukocytosis with neutrophil predominance, low glucose levels, and elevated protein. If the diagnosis is strongly suspected, one can examine the unstained CSF with a slide warmer to look for the typical, mobile, amoeboid motion of the trophozoites; gram stain and culture are usually negative. A PCR assay for Naegleria, Acanthamoeba, and Balamuthia has been developed by CDC [365, 366].
Frontal lobe involvement is readily seen on MRI but the diagnosis is often delayed because of the rarity of the condition and paucity of immediately diagnostic signs. Only a small number of patients have been reported to have survived and all received amphotericin B to which the pathogens are susceptible in vitro [367–370]. The optimal treatment regimen is not known for this condition and some authors recommend maximal systemic doses of amphotericin B with intracisternal amphotericin B adjunctive rifampin and doxycycline [371]. Surgical intervention usually involves placement of a reservoir for intrathecal amphotericin B and shunting to alleviate hydrocephalus.
Neurocysticercosis
Neurocysticercosis is the most common helminthic infection of the nervous system, and a leading cause of acquired epilepsy worldwide [372–374]. Cysticercosis is widespread in areas such as Mexico, Central America, South America, Africa, Southeast Asia, India, the Philippines, and Southern Europe. More recent data indicated that it is an increasing problem in Europe [372]. Neurocysticercosis in adults results from infection of the brain with the larval cysts of the cestode, Taenia solium, the pork tapeworm. Human infection occurs by two different mechanisms: [1] ingestion of the eggs leading to embryonation of the eggs and penetration of the intestinal wall with hematogenous transport of cysticerci to many different tissues, primarily muscle and brain, where they encyst and remain potentially infectious for long period of time. Alternatively, ingestion of undercooked pork can result in ingestion of the cysticerci by humans. In the latter instance the cysticerci mature into the typical tapeworm which attaches to the intestine and may grow to lengths of up to 3 m and live for up to 25 years. During this time it produces egg-filled segments, called proglottids, which are excreted in the feces. Ingestion of the eggs from these proglottids leads to neurocysticercosis, occasionally even by autoinfection from the patient’s own intestinal tapeworm.
Symptomatic neurocysticercosis results from the enlargement of the cysticercal cysts in the brain parenchyma over a period of months to years. In the United States, patients may present with seizures due to neurocysticercosis that was acquired after a visit to an endemic part of the world 30 years previously. The clinical manifestations are nonspecific and varied depending on the number, size, and anatomic location of the cystic lesions. Seizures and headaches are common presenting symptoms. If the cysts block the flow of CSF, symptoms associated with raised ICP, such as headache, nausea, vomiting, changes in vision, dizziness, ataxia, and confusion, may result. When the cysts are located in the meninges, chronic meningitis may occur. An unusual form called “racemes” cysticercosis is caused by the proliferation of cysts at the base of the brain and can result in severe disease, including mental deterioration and death. Intraspinal cysts are common and may produce symptoms of cord compression; severity again depends on location and size of cysts.
The most frequent clinical presentations are seizures (64.8 %), symptoms associated with raised intracranial hypertension, and meningism [375]. Radiological studies of the skull show intracranial calcifications suggestive of cysticercosis in up to 50 % of patients [375]. Analysis of CSF invariably shows increased pressure, elevated lymphocyte and eosinophil counts, elevated protein levels, and reduced glucose levels. Neurocysticercosis is readily diagnosed by neuroimaging (CT scanning or MRI) studies in which multiple cysts of varying sizes and stages are demonstrated (Fig. 22.29a, b). MRI may show the parasites as well as the changes they induce in the nervous system [374, 376, 377]. Figure 22.29c, d shows the histology of the cyst wall with outer, middle, and inner layers. Serological testing is available with sensitivities as high as 94 % in patients with multiple cysts but significantly lower in those with single cysts [378]. Older cysts are often calcified. PCR assays have the highest sensitivity (95.9 %) but variable specificity (80 % or 100 %) depending on the controls used [378]. Serology is generally available through the CDC or at specific national commercial laboratories. However, the identification of specific antibodies and antigens is currently used only to support the diagnosis because of the limited specificity and sensitivity of current tests.
Fig. 22.29.
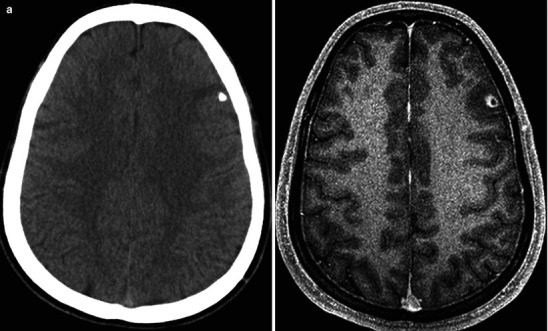
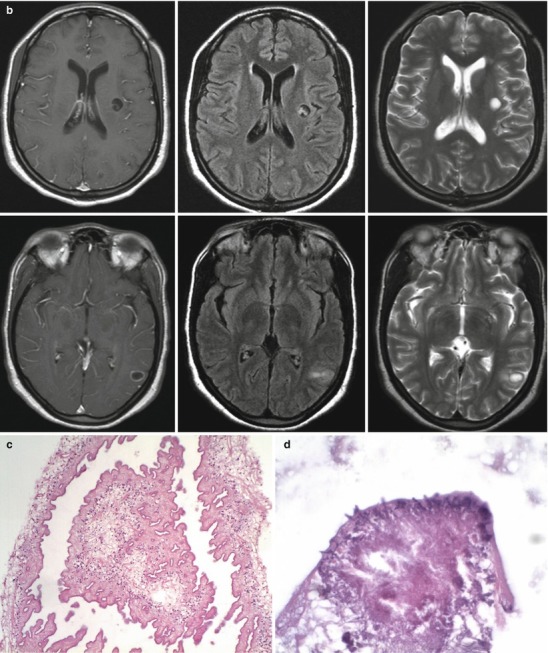
(a) Neurocysticercosis. Images include non-contrast CT (left) and post-gadolinium-enhanced T1-w MRI (right). These images demonstrate the features of chronic neurocysticercosis. The frontal lesion is calcified as evident on the CT. However, it also produces local inflammation evident on the enhanced MR image. This local inflammatory reaction frequently can produce seizure activity. Hence, late state neurocysticercosis can be a mimic for cortical oligodendroglioma. (b) Neurocysticercosis; images include a panorama of the MRI sequences in the mid-convexity level of the brain including pre and post-contrast DT1-w sequences and T2-w spin echo sequences which illustrate typical findings of neurocysticercosis. The findings in this case are those of cystic nodules scattered in brain and in the depths of the sulci. The margins of the lesion show reactive hyperemia. Within some of the lesions an organism scolex is evident confirming the basis for the nodules. (c) Neurocysticercosis. A 25-year-old male from Latin America with a lateral ventricular cyst. The figure shows the cyst wall with outer cuticular layer, middle cellular layer, and inner reticular layer (H&E 20×). (d) Features a high-power view of a portion of the scolex (H&E 60×) (d: Courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
Treatment
Based on neuroimaging studies, CNS lesions can be classified into active and inactive neurocysticercosis (Fig. 22.29a). Patients with inactive parenchymal neurocysticercosis generally have no evidence of viable or degenerating parasites; the utility of antiparasitic drugs in these patients remains limited. However, these patients are at increased risk of developing seizures, and standard anticonvulsive therapy with phenytoin, phenobarbital, or carbamazepine is indicated. Patients who develop hydrocephalus need to be treated with ventricular peritoneal shunting. Virtually all patients with active neurocysticercosis have seizures, which must be treated with anticonvulsants. Probably the majority of these patients can be treated symptomatically and followed by MRI because the cysticerci typically undergo complete degeneration over a 1- to 2-year period. This process results in either calcified inactive cysticerci that continue to induce seizures thereby requiring continued therapy with anticonvulsants or, in a majority, a normal MRI in which case anticonvulsants may be tapered as long as the patients remain seizure free.
Cysticidal drugs (albendazole and praziquantel) have improved the prognosis of this condition and can be given with praziquantel at doses of 50–100 mg/kg/day for 15–30 days or albendazole 10–15 mg/kg/day for 8 days. Albendazole has been superior to praziquantel in trials comparing the efficacy of these drugs [376, 377, 379]. Another advantage of albendazole is that it also destroys subarachnoid and ventricular cysts [380, 381]. In some of these cases, particularly in patients with large subarachnoid cysts, higher doses (up to 30 mg/kg/day) or more prolonged, or even repeated, courses of albendazole may be needed. Although these agents kill the cysticerci, some controlled trials have not shown any clinical benefit over symptomatic treatment alone [382–384]. The main adverse side effect of praziquantel is worsening neurologic function, i.e., headaches, dizziness, seizures, and increased ICP probably as a result of an increase in the host inflammatory response and cerebral edema due to the larval death. There is a strong consensus that there is no role for anti-cysticidal drugs in patients with only calcified lesions and those patients with single enhancing lesions will do well regardless of antiparasitic therapy [385]. Riley and White point out that while antiparasitic therapy is indicated in patients with multiple subarachnoid cysticerci or giant cysticerci they are contraindicated in patients with cerebral edema (cysticercal encephalitis) [385].
Surgery plays an important role in the management of some forms of the disease, particularly hydrocephalus and intraventricular cysts. Standard treatment of ventricular neurocysticercosis has been the surgical removal of cysts that block CSF flow; in patients with ventricular cysticerci, endoscopic removal remains the preferred therapy [385]. Recent studies have found fewer shunt failures when such patients are treated with antiparasitic drugs [386–388]. Cysticercosis involving the basilar cisterns is associated with a prominent inflammatory arachnoiditis and can be complicated by both vasculitis, resulting in lacunar infarctions, and invasion of the cysticerci into larger vessels, resulting in strokes. Thus, some authors have recommended the addition of corticosteroids in the treatment of patients with cisternal cysticercosis [387–390].
A consensus guideline panel headed by Garcia and coworkers underscored four major tenets of managing neurocysticercosis [391, 392]: (1) individualize therapeutic decisions, including whether to use antiparasitic drugs, based on the number, location, and viability of the parasites within the CNS; (2) actively manage growing cysticerci either with antiparasitic drugs or surgical excision; (3) prioritize the management of intracranial hypertension secondary to neurocysticercosis before considering any other form of therapy; and (4) manage seizures as done for seizures due to other causes of secondary seizures because they are due to an organic focus that has been present for a long time [391].
Echinococcus
Echinococcal disease is caused by tapeworms that commonly infect dogs, cats, wolves, and other carnivores. It is found worldwide but is particularly common in countries surrounding the Mediterranean, parts of East Africa, Russia and South America. There is very little echinococcal disease in the United States, but it is important to recognize that bears, foxes, and wolves in Canada and Alaska are commonly infected with this parasite. Disease is produced when an egg from an infected animal is ingested and the oncosphere within is activated, penetrates the gut wall, and travels via veins or lymphatics to various tissues in the body where they form hydatid cysts. Up to 80 % of cysts occur in the liver and about 10 % in the lungs. CNS involvement is uncommon and occurs in only about 2 % of cases. At all anatomic sites, cysts may be single or multiple.
Of the four species that infect humans, Echinococcus granulosus and E. multilocularis account for the vast majority of cases. CNS disease is commonly characterized by slowly enlarging solitary cysts in the brain. Depending on the size and location of the cyst, the patient may remain asymptomatic. However, as the CNS lesion enlarges in size, symptoms may arise from the local effects of the lesion itself or secondary to raised ICP, resulting in headache, nausea and vomiting, seizures, hemiparesis, dysarthria, and cranial nerve palsies. The diagnosis may be suspected from radiographic appearance of the cyst itself seen on CT scanning or MRI. Classic radiographic features include a sharp, spherical border lacking a rim of enhancement or surrounding edema, although a fine rim of peripheral enhancement with perilesional edema may be seen if active inflammation is present. CT is superior for the detection of extrahepatic disease; MRI does not appear to add any diagnostic benefit.
Immunodiagnostic testing is available from CDC; sensitivity of these tests varies from 60 to 90 % and they are highly sensitive for liver cysts although less so for cysts in the brain. Thus, a negative test does not absolutely rule out cerebral echinococcal disease. History of travel to or living in an endemic area especially with exposure to sheep also increases the likelihood of the diagnosis of echinococcus. Patients with intracranial hydatid cysts usually present with focal neurological deficit and features of raised intracranial pressure due to interference with CSF flow. The treatment of hydatid cyst is surgical and the aim of the surgery is to excise the cyst in toto without rupture to prevent recurrence and anaphylactic reaction. Albendazole therapy is given in a daily dose of 10 mg/kg, taken three times for 4 months. It is a broad-spectrum oral antihelminthic drug, which acts by blocking glucose uptake of the larva and adult worm. The glycogen stores are thus depleted thereby decreasing the ATP formation resulting in death of the parasite. Albendazole can decrease the size of large cysts and cause smaller ones to disappear.
Strongyloidiasis
Strongyloides stercoralis is a small nematode with free-living forms found in soil, while parasitic forms (i.e., the adult female) live within intestinal crypts in the duodenum, the jejunal mucosal villi, or in the submucosa; the male does not enter the intestinal mucosa but are passed in stool. Normally, the adult worms bore into the mucosa and produce eggs, which pass out with stool. The eggs deposited by the female may hatch the rhabditiform larvae that enter the lumen of the intestine to be passed out in stool. Eggs released from these organisms normally mature in the soil to produce more rhabditiform larvae. In the environment, these larvae transform to the filariform infective larvae that can directly penetrate intact skin of humans and other mammals. For reasons not fully understood, in some patients the transformation from rhabditiform to the filariform infective larvae can also occur while still in the lower bowel or perianal area, before being passed out in the stool. The filariform larvae burrow through the intestinal wall and perianal skin to reinfect the patient, a phenomenon known as autoinfection. After burrowing through the skin, the filariform larvae enter the lymphatics and, ultimately, the venous system where they are carried to the pulmonary capillaries. Here, they migrate out of the blood vessels into alveoli, up the airways and then down through the esophagus to reach the small bowel. Symptoms include ground itch, urticarial, and pulmonary symptoms. A worm burden in the intestines might lead to a malabsorption syndrome. Chronic strongyloidiasis and autoinfection have been observed in World War II veterans who were in POW camps on the Pacific front up to 30 years after their return to the United States and the United Kingdom.
The most serious consequence of S. stercoralis infection occurs as a result of massive autoinfection caused by immunosuppression (e.g., persons on steroid therapy, organ transplant, HTLV-1 coinfection, cancer, or malnutrition) or following treatment of lymphoma, leukemia, or leprosy with corticosteroids or cytotoxic drugs. In this condition, known as the hyperinfection syndrome, the autoinfection cycle escalates to generate massive infection with millions of parasites throughout the whole intestine and hematogenous dissemination of the invasive form of the filariform larvae to all organs, including the liver, lungs, and brain.
As part of this hyperinfection syndrome in immunocompromised patients, CNS involvement may be manifest by headache, altered mentation, meningismus, focal or generalized seizures, or motor weakness. Encephalopathy is common and pyogenic meningitis caused by strongyloides larvae in the meninges can occur. A unique aspect of the hyperinfection syndrome is the likelihood that meningitis and septic shock due to E. coli and other gram-negative enteric organisms can occur. These gram-negative infections are thought to be caused by the enteric organisms being carried either on the larvae or within the gut of the larvae as they migrate through the tissues, resulting in bacterial meningitis once the CNS is invaded. Although an eosinophilia is common in strongyloidiasis, it is almost never seen in patients with the hyperinfection syndrome because of existing immunosuppression (usually corticosteroids) and is an indication of poor prognosis—the lower the eosinophil count, the worse the prognosis. The diagnosis can be made by identifying the larvae in stool, duodenal aspirate, or sputum. For massive strongyloidiasis, treatment with thiabendazole 25 mg/kg twice daily for 10 days has been effective. Ivermectin therapy has also proven effective in the treatment of the hyperinfection syndrome.
Toxocariasis
Toxocara canis and Toxocara cati are nematodes that infect the intestines of dogs and cats, respectively. As a result of this infection in domestic animals, the eggs of these organisms are distributed widely in the soil to which humans may be exposed. Human infection occurs when eggs are ingested and hatch in the small intestine. Larvae then migrate through the intestinal wall and into various tissues of the body, and most often manifest as visceral larvae migrans (VLM). Symptoms include abdominal pain, hepatomegaly, anorexia, nausea, vomiting, lethargy, behavioral changes, pneumonia, cough, wheezing, lymphadenopathy, or fever. The hallmark of the disease is striking eosinophilia. Older children, adolescents, or young adults may develop unilateral loss of vision; ophthalmoscopy reveals a lesion not unlike a retinoblastoma. When eosinophilia is seen in small children between the ages of 2 and 4, it readily suggests a clinical diagnosis of VLM. Though very uncommon, CNS involvement can manifest as dementia, meningoencephalitis, myelitis, cerebral vasculitis, epilepsy, or optic neuritis [393–396]. CNS involvement in patients with VLM may also present as encephalopathy with seizures. Other manifestations include meningoencephalitis, transverse myelitis, and psychiatric disturbances.
Treatment is diethylcarbamazine 2 mg/kg orally three times daily for 10 days or albendazole 400 mg orally twice a day for 5 days. Steroids are indicated for ocular disease and may be necessary for severe lung, heart, or CNS involvement. The differential diagnosis of parasitic eosinophilic meningitis includes infections caused by Angiostrongylus cantonensis and Gnathostoma species; meningitis caused by these organisms is caused by their random migration into the CNS.
Syphilis
Syphilis is caused by the spirochete Treponema pallidum, belonging to the family Spirochaetaceae. The organisms are thin, tightly coiled bacteria that exhibit a characteristic undulating movement under direct dark-field observation. Syphilis is categorized as early (primary, secondary, and tertiary) or late (late latent, late benign, and late clinical infection). Before penicillin became available, the prevalence of primary and secondary syphilis was high but fell rapidly with the introduction of antimicrobials after the Second World War. Syphilis is transmitted primarily through sexual contact between infected and uninfected persons. Less common modes of transmission include nonsexual contact with infectious lesions, such as breaks in the skin or mucous membrane that come into contact with infectious lesions containing spirochetes, transplacentally from mother to fetus in utero, transfusion of contaminated blood products, or laboratory accidents.
Pathogenesis
The initial manifestation is the primary chancre, which begins at the site of inoculation as a painless papule and rapidly ulcerates into a relatively painless and indurated ulcer that is dark-field positive. The primary chancre is frequently associated with regional, nonsuppurative, nontender lymphadenopathy. The chancre generally heals spontaneously within 3–6 weeks. The incubation period from exposure to clinical disease can range anywhere from 3 weeks to 3 months. Spirochetemia occurs at the onset of the primary chancre. For a period of 2–8 weeks following the primary chancre, the treponemes migrate to the lymphatics and gain access to the circulation to cause a systemic illness (the secondary syphilis phase) in approximately 30 % of untreated patients. Systemic manifestations of secondary syphilis include fever, malaise, headache, pharyngitis, anorexia, weight loss, and arthralgias. The classic manifestations are maculopapular, papular, or pustular skin lesions, which are distributed over the entire body, including the palms and soles and patients may develop patchy alopecia. Genital ulcerations, such as condylomata lata and mucous patches, develop in about 20–35 % of those with clinically evident secondary syphilis.
During the secondary syphilis phase, between 8 and 40 % of affected persons develop some evidence of CNS involvement, including meningitis and cranial nerve involvement; patients may complain of headache, decreased vision, tinnitus, and vertigo. During the secondary stage, spirochetes can be found in the blood, CNS, and aqueous humor of the eye. However, direct dark-field examination of the CSF in these patients rarely reveals spirochetes. Using rabbit inoculation to test CSF for viable T. pallidum, Lukehart and coworkers found that 30 % of 40 patients with primary and secondary syphilis had viable treponemes in CSF and that CNS invasion by the spirochete is common in early syphilis and is apparently independent of HIV infection [397].
The clinical manifestations of secondary syphilis resolve spontaneously, without antimicrobial therapy after a period of weeks to several months to enter the latent stage. Patients, however, remain seroreactive. During the secondary syphilis phase, patients generally remain asymptomatic until manifestations of tertiary syphilis appear years later in one-third of untreated patients. Clinically, tertiary syphilis is divided into three general categories: neurosyphilis, cardiovascular syphilis, and gummatous syphilis.
Neurosyphilis
Neurosyphilis is due to damage produced by meningovasculitis and degenerative parenchymal changes in the entire CNS. Since spirochete invasion of the CNS occurs during the primary stage, a small population of patients has continuing CNS involvement. Syphilitic meningitis is an early manifestation, usually occurring within the first 2 years of the primary infection and resulting from small vessel arteritis in the meninges, which accounts for the typical symptoms of headache, nausea, and vomiting seen in approximately 90 % of patients. In addition, up to 45 % of patients with syphilitic meningitis may have cranial nerve palsies. Seizures have been reported in 17 % and fever occurs in less than 50 %. The CSF white count is almost invariably abnormal, but there is only a mild decrease in CSF glucose.
Although there is some overlap of symptomatology, meningovascular syphilis presents with findings of meningitis together with focal neurologic findings due to syphilitic arteritis. The peak incidence of this condition is approximately 7 years after acquisition of syphilis and accounts for approximately 12 % of patients with CNS involvement [398–401]. Patients with meningovascular syphilis generally present with a history of several weeks to months of prodromal symptoms and signs, such as headache, vertigo, personality changes, behavioral changes, insomnia or seizures, and stroke-like neurologic deficits, most frequently involving the distribution of vessels in the territory of the middle cerebral artery followed by that of the basilar artery. Thus, while the distribution of strokes in such patients may be similar to that of the patient with atherosclerotic disease, the occlusive symptoms develop gradually over a period of time in meningeal vascular syphilis as opposed to sudden onset in patients with atherosclerotic strokes [401]. In contrast to the findings from the preantibiotic era, neurosyphilis is now most often identified in young patients with HIV coinfection [402, 403].
The majority of neurologic manifestations of tertiary syphilis involves the CNS parenchyma and is classified as parenchymatous neurosyphilis. This category includes two classical syndromes: general paresis and tabes dorsalis. In contradistinction to the pathogenesis of syphilitic meningitis or meningovascular syphilis, these syndromes result from progressive neuronal destruction with fibrosis and atrophy rather than ischemic damage from vasculitis. General paresis, also known as general paralysis of the insane, is a chronic progressive meningoencephalitis with a peak incidence 10–20 years after acquisition of syphilis. Patients generally present with gradual deterioration of mental functioning characterized by difficulties in concentration, irritability, and deficits of higher cognitive function. As the condition progresses, these manifestations become more obvious and symptoms may mimic psychiatric disease, including delusions, paranoia, emotional lability, memory loss, or dementia. Difficulties with motor control then develop with a loss of facial muscle and extremity tone, loss of fine motor control, and the development of tremors and dysarthria. Subsequently, patients may have seizures, loss of bowel and bladder control, and paralysis; the pupils become unresponsive to light and painful stimuli and may be constricted and unequal in size (the Argyll Robertson pupil). Untreated, the disease follows a progressive or subacute course over 3–4 years. Pathologically there is diffuse cortical atrophy, dilatation of the ventricles, and neuronal dropout with accompanying gliosis. Spirochetes can be demonstrated in 25–40 % of patients with silver stain. The diagnosis is established by a combination of the clinical presentation, positive serology, and elevated CSF white count and protein.
Tabes dorsalis results from progressive neuronal degeneration, especially the dorsal roots and the posterior column of the spinal cord. It has a peak incidence that is generally later than that of paresis, approximately 15–20 years after infection, and the progression of this condition is somewhat slower than that of paresis. The classical early symptomatology is “lightning pains” in the distribution of nerve roots. These pains are described as lancinating, lasting for minutes to hours, and most often involve the lower extremities. Ten percent to 20 % of patients with tabes may also present with episodic attacks of abdominal pain. In addition to pain, some patients experience episodic paresthesias. Ultimately, patients experience progressive loss of vibration and proprioceptive sensation, particularly in the lower extremities. As a result, the patients exhibit a characteristic broad-based, shuffling gait and may develop Charcot joints. Muscular atrophy develops in approximately 20 % of patients. The Argyll Robertson pupil, in which one or both pupils constrict with accommodation but do not react directly to light, is a characteristic feature of both general paresis and tabes dorsalis. Pathologically there is atrophy of the posterior columns of the spinal cord with inflammatory infiltrates and loss of neurons. In contrast to paresis, it is unusual to be able to stain the spirochete in nerve tissue. The diagnosis is readily made from the characteristic neurologic findings together with positive serology. However, CSF leukocytosis is observed in only 50 % of patients, and protein elevation is seen in approximately 53 % of patients.
Syphilitic gummas are progressive granulomatous tumorlike lesions primarily involving skin, mucous membranes, and bone but which can develop in any organ in the body including the brain. Gummas may arise in almost any part of the CNS but are most often associated with the pia mater and consist of rubbery masses varying in size from several millimeters to centimeters [404, 405]. Localized findings range from small superficial nodules to large radiating lesions. CNS symptoms depend on the anatomic location of the lesions.
Serologic Testing
Laboratory diagnosis of syphilis depends on the stage of the disease and clinical manifestations. Patients with lesions on moist skin or mucous membranes during either primary or secondary syphilis can usually be diagnosed by the demonstration of treponemes on dark-field microscopy. While treponemes can be demonstrated in dry lesions or lymph nodes by biopsy or saline aspiration, the yield is considerably lower. Serologic tests for syphilis are generally divided into two different types: nontreponemal and treponemal tests. Nontreponemal tests measure IgG or IgM antibodies directed at cardiolipin, which is released when the treponeme damages cells during an infection. Thus, a positive nontreponemal test is indicative of an active infection, but a confirmatory test with a treponemal test is required to verify that it is indeed a syphilis infection that is causing elevated cardiolipin levels. Currently, a standardized mixture of cardiolipin, cholesterol, and lecithin which has fewer false-positive reactions is used and forms the basis of today’s standardized tests. The most common of these are the classic venereal disease research laboratory (VDRL), in which agglutination of the cardiolipin, cholesterol, and lecithin antigen is carried out on a slide using heated serum; the rapid plasma reagin (RPR) card test; the automated reagin test (ART); or the toluidine red unheated test (TRUST). Because of the stringency of the technical requirements for the test, the VDRL is generally performed only on CSF and serum screening tests are generally done with the RPR and its variants. Specific treponemal tests for syphilis include the T. pallidum hemagglutination (TPHA) test, the microhemagglutination test with T. pallidum antigen, the fluorescent treponemal antibody-absorption test (FTA-abs), the enzyme-linked immunosorbent assay (ELISA), and the hemagglutination treponemal test for syphilis (HATTS).
Nontreponemal tests (RPR and VDRL) rise during primary syphilis and reach their peak in secondary syphilis. They slowly decline with advancing age. With treatment, they revert to normal over a few weeks. It is important to note that the serologic response to syphilis increases gradually over the course of the primary infection, so that when the chancre is first observed no more than 10–20 % of patients may be seropositive by any method. However, this will increase with the duration of the chancre during primary infection to approximately 70 % for both the treponemal and nontreponemal tests by the time the chancre heals. During secondary syphilis, serologic tests, whether treponemal or nontreponemal, are positive in almost 100 % of patients.
Early treatment of the primary infection should render the patient seronegative within a year. Treatment of syphilis during the secondary and latent stages will generally result in a significant fall in the titer of the nontreponemal tests. These tests should be negative within 1 year in a patient treated for primary syphilis or within 2 years for a patient treated for secondary syphilis. Patients who remain seropositive by nontreponemal tests after treatment probably have either persistent infection or the so-called biologic false-positive sometimes seen in patients with HIV infection [406, 407].
In general, once the treponemal tests become positive, they remain so for life even if the patient has been successfully treated. Serologic tests can be used to diagnose neurosyphilis during the latent and late latent stages by testing CSF for VDRL antibodies. With the exception of rare false-positive results, possibly resulting from blood contamination, a reactive CSF-VDRL in the absence of substantial contamination of CSF with blood is diagnostic of neurosyphilis.
Although the specificity of the CSF-VDRL in diagnosing likely active neurosyphilis is 100 %, the sensitivity is only about 27 % and, in early syphilis, is of unknown prognostic significance [408, 409]. The insensitivity of the CSF-VDRL test limits its usefulness as a screening test for neurosyphilis. The CSF-FTA-abs test appears more sensitive for screening CSF but is less specific than the CSF-VDRL test in distinguishing currently active neurosyphilis from past syphilis. These findings imply that clinical judgment remains the sine qua non for establishing the diagnosis of active neurosyphilis. Most other tests are both insensitive and nonspecific and must be interpreted in relation to other test results and the clinical assessment. Furthermore, one cannot use the more sensitive treponemal antibody tests with CSF to diagnose neurosyphilis as these antibodies cross the blood–brain barrier and are therefore likely to be present in the CSF of all patients who have positive serum treponemal tests for syphilis. Thus, a positive CSF treponemal test does not provide any additional information to that obtained by testing serum alone. For example, in one study the CSF-FTA-abs test was positive in 48 patients of whom only 15 had clinical neurosyphilis [408].
While one can resort to sophisticated methods of CSF analysis, such as levels of CSF treponemal antibody compared with serum levels adjusted for changes in the blood–brain barrier by using serum/CSF albumin ratios, and demonstrating a significantly higher than expected CSF level of specific treponemal antibody, it is probably safer to treat patients who have a reactive serum testing if they have any clinical signs of neurosyphilis. If the patient has no clinical manifestations of neurosyphilis and preventive treatment for latent neurosyphilis is being considered, treatment should be based on the presence of an abnormal number of white blood cells in the CSF rather than trying to fine tune the serologic diagnosis. Various studies using PCR have shown that DNA from T. pallidum can be detected in the CSF of patients with neurosyphilis. However, it remains unclear whether a positive PCR assay means that the patient has to be treated for latent neurosyphilis or that a negative test excludes the diagnosis of neurosyphilis. Neuroimaging using CT scanning or MRI can be used to document CNS gummas or other complications of tertiary syphilis.
In a recent review, Ghanem underscored the point that there is no gold standard for the diagnosis of neurosyphilis [403]. He also made the point that CDC’s criteria for the diagnosis of neurosyphilis are based on surveillance definitions used mainly for epidemiologic purposes. He rendered two clinical categories of neurosyphilis: (1) “confirmed” neurosyphilis, which is defined as any stage of syphilis plus a reactive CSF-VDRL, and (2) presumptive neurosyphilis defined as any stage of syphilis, a nonreactive CSF-VDRL, a CSF pleocytosis, or elevated protein, with clinical signs or symptoms consistent with syphilis without an alternate diagnosis to account for these clinical features [403].
Asymptomatic neurosyphilis is defined by the presence of CSF abnormalities consistent with neurosyphilis in persons with serological evidence of syphilis and no neurological symptoms or signs [403]. Also, asymptomatic neurosyphilis can occur in both early and latent stages of syphilis. For example, among a large population with defined latent syphilis and no evidence of symptomatic neurological disease, 13.5 % were found to have asymptomatic neurosyphilis [410]. Moreover, patients with latent syphilis and asymptomatic neurosyphilis are more likely to have syphilitic involvement of the skin [410].
Treatment
The CSF is always abnormal in active disease, and only active disease responds to treatment. The preferred treatment for all manifestations of neurosyphilis is intravenous aqueous crystalline penicillin G 12–24 million units/day given in six divided doses for 10–14 days. Alternatively, 2.4 million units of procaine penicillin G can be given intramuscularly together with 500 mg/day of probenecid four times a day for 10–14 days. In penicillin-allergic patients, doxycycline 200 mg orally each day for 21 days or ceftriaxone 1 g IM or IV for 14 days has been recommended. However, treatment failures have been documented with ceftriaxone, especially in HIV-infected patients. Patients with syphilitic meningitis or meningovascular syphilis generally respond to treatment with the exception of cases in which the patient has focal cranial nerve abnormalities associated with syphilitic meningitis or larger ischemic defects caused by the arteritis associated with meningovascular syphilis.
For patients with tabes dorsalis or general paresis, improvement approaching cure is relatively uncommon and, in fact, for a majority of patients, disease progression continues despite “adequate” penicillin treatment [411]. Penicillin remains the drug of choice for all forms of neurosyphilis, but disease progression has been frequently reported following the use of penicillin G benzathine [398]. Thus, documentation of CSF resolution over the months following penicillin therapy is required to confirm curative treatment.
Patients with asymptomatic neurosyphilis appear to respond very well to treatment. In one study, 89 % of 454 patients who initially had ≥10 leukocytes/mm3 of CSF had normalized their cell counts at a 1-year follow-up, as had 69 % of those with abnormal protein prior to treatment [412]. Because patients with primary and secondary syphilis are curable with standard treatment of benzathine penicillin G 2.4 million units IM weekly for three weekly doses, the question of proper treatment always arises when an asymptomatic patient is found to have a positive VDRL, RPR, or other screening test for syphilis. The problem is that this regimen does not reliably provide CSF levels in excess of 0.018 μg/mL of CSF in all patients, which is believed to be necessary to kill spirochetes within the CNS [413–415]. The treatment of cerebral gummas is IV penicillin G with neuroimaging follow-up recommended for most patients [416]. Surgery should be reserved for those unresponsive to antibiotics or those with acutely elevated intracranial pressure [416]. All patients found to have serologic evidence of syphilis, assuming that false-positive tests such as those due to pregnancy and other intercurrent illness can be ruled out, should have serologic testing for HIV because the ability to eradicate syphilis is considerably lower in HIV-infected patients and re-treatment may be necessary.
Lyme Disease
Lyme disease is due to systemic infection with the microaerophilic spirochete Borrelia burgdorferi and the body’s immune response to the infection. Lyme disease was first recognized in the United States in the early 1970s when Dr. Allen Steere at Yale University investigated an outbreak of juvenile rheumatoid arthritis in the small towns of Lyme, Old Lyme, and East Haddam, Connecticut. In the initial report, they identified and ascertained 39 children and 12 adults who presented with a classic, characteristic, remitting, relapsing oligoarticular arthritis with onset in the summer or early fall. All of these patients lived in rural areas and half of the patients lived on two adjacent country roads. In addition, 13 of these patients had noted an unusual skin lesion an average of 4 months before the onset of the arthritis [417]. Subsequent prospective studies then defined neurologic abnormalities such as Bell’s palsy, sensory radiculoneuritis, lymphocytic meningitis, and cardiac conduction abnormalities also associated with Lyme disease. These studies showed that at least a quarter of the patients remembered a tick bite at the site of the initial skin lesion, and based on examination of the actual tick from one of these patients, the vector was identified as Ixodes scapularis [418–420]. The agent of Lyme disease was finally isolated by Dr. Willy Burgdorfer from the Rocky Mountain Laboratory in Hamilton, Montana, when he was searching for evidence of Rocky Mountain spotted fever in ticks isolated from New York State. No rickettsia were found; however, spirochetes were seen in stains of the insect’s digestive tract [421, 422].
Pathogenesis
The clinical manifestations fall broadly into three stages: early localized, early disseminated, and chronic disseminated. Approximately 50 % of untreated patients progress to disseminated disease. During the first stage, the earliest manifestations of Lyme disease occur at the site of the tick bite beginning as a red macule or papule that may expand to an area 10–15 cm with red outer borders and partial central clearing—the so-called “bull’s eye” skin rash. The lesion develops as early as 3 days and as late as 30 days following the initial bite and generally lasts 3–4 weeks. It is most commonly located on the thigh or groin and develops in approximately 80 % of patients [419, 423, 424]. Following the entry of the spirochete into the patient via the tick bite, dissemination of the spirochete occurs during the development of this initial lesion, and while some patients may develop multiple secondary annular lesions that are similar to the primary site lesion, others may clear the infection without developing symptoms. All patients largely remain seropositive. In the second stage, the organism produces symptoms by direct invasion. Systemic symptoms of fatigue, lethargy, and malaise along with generalized lymphadenopathy, meningismus, encephalopathy, migratory musculoskeletal pain, splenomegaly, sore throat, and cough may develop in varying degrees during this early disseminated phase [423, 424]. For the most part, the systemic manifestations as well the erythema chronicum migrans lesions themselves usually resolve without treatment in 3–4 weeks. Untreated, however, the Lyme disease spirochete becomes sequestered and persists in various tissues, particularly the CNS, joints, heart, and the skin.
In some patients with meningitis, particularly in Europe, where the disease is caused by a different species of spirochete, significant neurologic abnormalities develop, including cranial neuritis which can present as an isolated facial palsy (motor and sensory), radicular mononeuritis multiplex, or myelitis alone or in varying combinations. Although these patients may have some neck stiffness on extreme flexion, typical Kernig’s and Brudzinski’s signs are not present. Patients may complain of excruciating headache as well as severe musculoskeletal pain. Early on, examination of the CSF may be normal, but patients may develop a lymphocytic pleocytosis with a normal glucose level.
During this early dissemination stage, cardiac symptoms and signs manifest, usually atrioventricular block with varying degrees of other forms occasionally noted including complete heart block, which rarely persists for more than a week and generally does not require the insertion of a pacemaker [425, 426]. Occasional patients have been described with osteomyelitis, myositis, panniculitis, eosinophilic fasciitis, conjunctivitis, or even deeper involvement of the orbital structures including panophthalmitis and choroid retinitis with exudative retinal detachment or interstitial keratitis.
The third stage of Lyme disease is characterized by arthritis, which develops in about 60 % of untreated patients. Symptoms include intermittent attacks of pain particularly involving the large joints (e.g., the knee), in an asymmetric pattern. Attacks of acute arthritis generally last weeks to months followed by periods of remission. Joint fluid counts range from 500 to 100,000 cells/mm3 with a high percentage of polymorphonuclear leukocytes. Even untreated, this condition resolves gradually over a period of years.
The late manifestations of CNS involvement of Lyme disease generally develop a year or more after the onset of illness and generally do not improve spontaneously. In both North American and European forms of this disease, persistence of the B. burgdorferi spirochete has been demonstrated in CSF and in brain parenchyma up to 9 years after the onset of illness [425–429]. The most common neurologic symptoms are speech abnormalities, limb weakness, gait difficulties, ataxia, bladder dysfunction, visual changes, hearing loss, mood changes, sleep disorders, and deteriorating memory and concentration. Symptoms of late progressive Lyme encephalomyelitis may develop either acutely or gradually and then worsen progressively over the ensuing months to years [427, 429–438]. Progression may be gradual or stepwise, characterized by sudden deterioration and only partial improvement between episodes. Headaches, nausea, vomiting, and neck stiffness have been reported but occur less often, while mental deficits such as behavioral changes, poor memory, and concentration are common. More severe changes including confusion, disorientation, dementia, delirium, and somnolence can occur. Symptoms such as apraxia, myoclonus, hemiparesthesia, and visual field abnormalities have been reported. Spinal cord involvement is common and myelitis may present as a progressive paraparesis or quadriparesis that can become very severe. Approximately 45 % of patients have cranial nerve palsies; involvement of the optic, facial, oculomotor, and vestibulocochlear cranial nerves have all been reported [426, 431, 439]. Peripheral radiculoneuritis occurs in less than 10 % of patients.
The CSF is abnormal in almost all cases of CNS involvement in Lyme disease. Generally, there is CSF pleocytosis, predominantly monocytic in the range of 100–200 cells/mm3, although levels as high as 2,300 cells/mm3 have been reported. CSF protein concentrations are usually greater than 50 mg/dL, usually in the range of 100–200 mg/dL, although concentrations levels as high as 1,800 mg/dL have been reported; glucose is generally normal to low. Oligoclonal bands specific for B. burgdorferi may be present.
EEG and CT abnormalities, including infarcts in the internal capsule, thalamus, lentiform nucleus, hydrocephalus, and cerebral atrophy, have been reported [430, 432, 440–443]. MRI shows additional lesions such as multifocal white matter abnormalities, infarcts, periventricular and sub-insular cavities, as well as atrophy of the pons and medulla [432, 440, 442–445]. MRI imaging in patients with myelitis have shown diffuse or focal signal abnormalities in relevant parts of the spinal cord [432, 446–449].
In contrast to the dramatic and objectively documented neurologic abnormalities and syndromes seen in a small number of patients with Lyme encephalopathy, a certain number of North American patients have reported the development of a less dramatic but nonetheless disabling CNS symptom complex. These patients complain of overwhelming fatigue, accompanied by loss of memory and concentration, and almost always without physical neurologic abnormalities. Psychological testing shows abnormalities or dysfunction in memory, ability to learn or acquire new information, attention span, concentration, problem solving, perceptual motor performance, and verbal fluency. Although depression and irritability are frequently reported, general fatigue appears to be the overriding complaint. Many of these patients fit the definition of the chronic fatigue syndrome as defined by the CDC. Objective laboratory and radiographic signs of infection are generally absent. CSF pleocytosis is present in less than 5 % of the cases, and CSF protein is elevated in only a minority of patients. Oligoclonal bands for B. burgdorferi are absent and patients with these complaints may or may not have antibodies to Lyme disease in the serum. Some of these patients have been reported to have MRI abnormalities, such as focal areas of increased signal in deep cerebral white matter [441, 450–452]. In general, symptoms in these patients do not improve spontaneously. A variable number of persons with this condition apparently do respond to courses of antimicrobial therapy, sometimes for 6 months or more [441, 450–453].
A number of North American patients have developed a mild multifocal polyneuropathy distinct from the meningopolyneuritis of early disseminated Lyme disease as a manifestation of late Lyme disease. Intermittent tingling and paresthesia of the extremities are the most common symptoms, occurring in approximately 50 % of patients with this form of late Lyme disease. The onset is generally 8 months to several years after the initial infection. The symptoms are usually distal, may be symmetric or asymmetric, and can involve both arms and legs. About 25 % of patients present with carpal tunnel syndrome or develop it at some point. Radicular pain occurs in 25–50 % of those with this syndrome; this pain is intermittent, asymmetric, and multifocal, typically radiating from the spine into the limbs or trunk [441, 452, 454–457]. Sensory changes such as mild stocking and glove distal sensory loss, as well as distal asymmetrical or truncal sensory loss also occur [441, 452, 454, 455, 458, 459].
Objective evidence of organic disease is much more common in these patients than in those reporting symptoms of chronic fatigue, with up to 83 % of patients having electromyographic abnormalities demonstrable particularly among those with distal paresthesia. In addition, CSF abnormalities, mostly in the form of increased protein concentration and intrathecal antibody synthesis specific for B. burgdorferi are found in up to 70 % of patients with these symptoms [441, 458]. Treatment with antimicrobials may improve the paresthesia and electrophysiologic conduction abnormalities but may require prolonged regimens from 3 to 7 months [441, 450, 454–456, 458]. Improvement among patients with radicular pain is less frequent and is only seen in about 50 %.
The diagnosis of Lyme disease can be made with reasonable assurance by a well-documented history of a tick bite and together with clinical evidence of typical skin lesions (erythema chronicum migrans). However, during this early stage of the disease, only 30–40 % of patients will have a serologic test positive for Lyme disease in an acute serum specimen, and only 60–70 % of these patients will be positive in the convalescent sera 2–4 weeks later. CDC recommends a two-step approach for serological testing: the first step in the workup of patients with putative Lyme symptoms is to obtain an antibody titer using an ELISA test to screen for the presence of antibody; the second step is confirmation of positive titers with a Western blot assay; both IgG and IgM antibodies are formed. However, persistence of the IgM antibody alone in the absence of an IgG response after the first month of illness may signal a false-positive reaction. After the first 1–2 months of infection, over 90 % of patients will have a specific IgG antibody response to the spirochete. It has been noted that patients treated effectively early in the course of erythema chronicum migrans may never develop a humoral immune response, although cellular immunity may be demonstrated and persist for years.
Treatment
Treatment of early dissemination and localized erythema chronicum migrans consists of doxycycline 100 mg twice a day for 20–30 days or amoxicillin 500 mg three times daily for 20–30 days, with some experts recommending the addition of probenecid 500 mg three times daily to the amoxicillin regimen. Cefuroxime axetil 500 mg orally three times daily for 2–3 weeks has also been recommended as has erythromycin 400 kg/mg/day in four divided doses for 2–4 weeks in children. For patients with arthritis, treatment with doxycycline or amoxicillin is extended to 1–2 months, and intravenous ceftriaxone 2 g a day for 14–30 days is also recommended.
For patients with early or late neurologic abnormalities, ceftriaxone 2 g IV daily for 2–4 weeks is generally recommended. Alternatives include penicillin G 20 million units IV in four divided doses daily for up to 30 days, as well as doxycycline 100 mg orally three times per day for 14–30 days. Treatment failures have been reported for all of these regimens and treatment may need to be repeated.
Cardiac abnormalities are treated as for early infection in those patients with first-degree AV block; IV ceftriaxone or penicillin is used for higher degrees of AV block. In patients with neurologic manifestations of early disseminated Lyme disease, IV therapy with penicillin or ceftriaxone can lead to a mild Herxheimer-like reaction with worsening of pain and fever during the first 18–20 h [425, 460]. In general, meningismus, radicular pain, and systemic symptoms improve within days although residual fatigue, arthralgias, and muscular skeletal pain can persist for some time thereafter. Motor deficits improve more slowly, over 2–3 months, and sometimes never fully recover. CNS abnormalities usually stop progressing and begin to improve slowly, but residual deficits may remain [428, 460–464]. CSF cell counts respond over the course of treatment but may not return to normal for several months, and the protein concentration falls even more slowly and may remain elevated for up to 1 year in some patients. If the patient does not respond by the end of the second week, treatment should be extended for at least another 2 weeks.
The severe abnormalities of late Lyme disease generally respond well to high-dose penicillin, doxycycline, or ceftriaxone [430, 432, 439, 442, 443, 449, 451, 452, 462–467]. Altogether 80–90 % of patients improve with IV cephalosporins, but recovery is slow and often incomplete, with little change occurring during the treatment itself and only developing over the subsequent weeks after treatment has stopped. At this time, there are no published data that support the routine use of steroid therapy in the management of CNS complications of Lyme disease. Finally, although a controversial issue, there are no convincing data that show prolonged antimicrobial therapy is effective for patients in whom symptoms persist after completing the recommended antimicrobial therapy for acute Lyme disease [468].
Miscellaneous Infections with CNS Complications
Rickettsial Disease
Rickettsiae are small gram-negative, obligate intracellular coccobacilli that are transmitted by tick bites. Rickettsiae grow freely in the cytoplasm of eukaryotic host cells on which they are dependent for nucleotide cofactors and ATP; they do not grow well outside the host cell. They can only be grown in living host cells, such as cell cultures and embryonated eggs. Most rickettsiae have animal reservoirs and are spread by infected arthropod vectors. Infections generally follow the distribution of the main vectors: Dermacentor andersoni (the wood tick) is the primary vector in the Western United States; Dermacentor variabilis (dog tick) is the principal vector in the Eastern United States. Infections are limited to warmer months.
Following the tick bite, Rickettsiae infect the vascular endothelium lining the small blood vessels causing a vasculitis—the primary pathologic feature of rickettsial infection, particularly RMSF. A combination of endothelial proliferation, necrosis, and perivascular inflammation, predominantly with a mononuclear cell infiltrate, leads to thrombosis and leakage of erythrocytes into surrounding tissues resulting in petechial lesions. Should systemic infection ensue, these vascular lesions occur throughout the body. This is in contrast to the infiltrate of polymorphonuclear leukocytes seen in typical immunocomplex vasculitis.
Rickettsial disease is characterized by fever, headache, rash, myalgias, and myositis. The most important rickettsiosis in the United States is Rocky Mountain spotted fever (RMSF), which is an acute febrile illness caused by Rickettsia rickettsii. As the name implies, the disease is seen in the Rocky Mountain States, such as Colorado and Wyoming, but also occurs in the inland parts of North Carolina, Virginia, and other Southeastern States. For RMSF, the incubation period between tick bite and onset of illness is 2–6 days. Fever, headache, rash, mental confusion, and myalgia are the predominant clinical features [469–471]. The rash usually develops on the second or third day of illness, initially on the wrists and ankles followed by spread to the trunk within hours. In contrast with a viral exanthem, the RMSF rash can appear on the palms and soles. About 25 % of patients may have an encephalitis at presentation with lethargy, confusion, or delirium. This may deteriorate to seizures and coma. As a result of the variability of the vessels involved in the CNS, a wide range of clinical neurologic features are associated with RMSF, including seizures, deafness, facial diplegia, gaze palsies, nystagmus, ataxia, dysphasia, transverse myelitis, neurogenic bladder, hemiplegia, and paraplegia or quadriplegia.
The diagnosis of RMSF is fundamentally a clinical diagnosis and depends on the history of a tick bite together with a compatible systemic illness with or without a rash [471]. The diagnosis of RMSF must be considered in all febrile patients who have known or possible exposure to ticks, especially if they live in or have traveled to endemic regions during warmer months [471]. Because culture of rickettsiae is difficult, serological testing remains the mainstay of confirming the diagnosis. However, though serologic diagnosis is highly accurate, it is not readily available in most institutions or as a routine service in the acute setting, and therefore treatment must be started empirically; patients with RMSF who received antirickettsial therapy within 5 days of the onset of symptoms are significantly less likely to die than those in whom therapy, for whatever reasons, was delayed [472].
The basic tenet of RMSF management, then, is the commencement of appropriate antimicrobial therapy without delay in conjunction with supportive therapy where indicated, i.e., IV hydration for hypovolemia or circulatory collapse, mechanical ventilatory support especially if the patient has developed ARDS, blood transfusion, and management of DIC. In adults and children, the recommended dose is doxycycline 100 mg per dose administered twice daily (orally or intravenously) for adults or 2.2 mg/kg body weight per dose administered twice daily (orally or intravenously) for children weighing less than 100 lbs (45.4 kg) [470]. Previously, for children under the age of 8 years, chloramphenicol was recommended because of the tooth discoloration associated with tetracyclines. However, in 1997, the American Academy of Pediatrics Committee on Infectious Diseases revised its recommendations and identified doxycycline as the drug of choice for treating presumed or confirmed RMSF in children of any age, because of the effectiveness of this antimicrobial in reducing attributable morbidity and potential mortality. Sulfonamides may worsen the disease and are therefore contraindicated.
Most patients show improvement within 2 days but may require up 7–10 days in severe cases. Despite modern therapeutic modalities, the fatality rate of RMSF is still in the range of 5–10 % [470]. Even after recovery, CNS abnormalities may persist in a significant number of patients with RMSF, including intellectual defects, impaired fine motor skills, aphasia, and EEG changes. Long-term neurological sequelae includes paraparesis; hearing loss; peripheral neuropathy; bladder and bowel incontinence; cerebellar, vestibular, and motor dysfunction; and language disorders [473]. Sexton and colleague made the point that the mechanism of such abnormalities is rickettsia-induced vasculitis with subsequent infarction of neural tissue and that although early treatment may prevent many, but not all, such complications, other factors, such as age and race, might be playing a role in the pathogenesis of severe disease [474].
Human Ehrlichiosis
Ehrlichia species are tiny, obligate, leukocyte-associated, gram-negative bacteria that replicate in membrane-bound compartments inside the host white blood cells to cause human disease. Anaplasma phagocytophilum causes human granulocytotropic anaplasmosis (HGA), previously known as human granulocytotropic ehrlichiosis (HGE). A. phagocytophilum is transmitted by Ixodes scapularis, which also transmits the agents that cause Lyme disease and babesiosis. Ehrlichia chaffeensis, the cause of human monocytic ehrlichiosis (HME), is transmitted by the Lone Star tick found on the whitetail deer and prefers to invade mononuclear leukocytes (e.g., macrophages). Both diseases are transmitted by deer or dog tick bites—HME is relatively common in the South and Southeast United States, while most cases of human HGA so far have been recorded from the upper Midwest, Northeast, and some parts of the South. HGA is the predominant form of ehrlichiosis in the United States and runs second only to Lyme disease as a tick-borne transmitted infection in the United States. More than 2,900 cases of HGA have been reported to the CDC between 1994 and 2005, with the annual number of cases of HGA exceeding that of HME at an estimated annual incidence of 1.6 cases per million in the US [475]. Ehrlichiosis is a seasonal disease with highest rates of occurrence from April to September. DNA studies of the agent that causes HGA show that it is mostly related to Ehrlichia canis but probably represents a different subspecies.
Both HGA and HME have incubation periods of about 7 (range 5–14) days after the tick bite. Symptoms of ehrlichiosis include high fever, chills, rigors, malaise, severe headache, myalgias, nausea, and vomiting and may include confusion, disorientation, obtundation, and ataxia in some patients [475]. Presentation may be abrupt or subacute in nature. Compared with RMSF, rashes are relatively uncommon (36 % of HME and 2 % of HGA) and are characteristically maculopapular with a distribution on the upper and lower limbs, trunk, or face. The petechial rash of RMSF is absent in ehrlichiosis. Patients with HME can develop aseptic meningitis and meningoencephalitis or progress to respiratory and renal insufficiency. Patients may develop hepatosplenomegaly. Although respiratory insufficiency can also occur in HGA, meningoencephalitis is uncommon in this type of human ehrlichiosis.
CSF is frequently normal; other laboratory studies are non-diagnostic or nonspecific; thrombocytopenia is common in both types of ehrlichiosis. Wright stain of a peripheral blood and buffy coat smear might reveal the characteristic intracellular inclusions known as morulae; these inclusions are present in the cytoplasm of neutrophils and monocytes of HGA and HME, respectively. However, presence of morulae in smears is variable. Serologic confirmation of illness is the usual method of diagnosis and is highly accurate but available only from state and other reference laboratories; a fourfold rise of titers is considered diagnostic. The differential diagnosis includes RMSF, Lyme disease, and babesiosis. As with RMSF, treatment with doxycycline (100 mg twice daily) for 14 days or chloramphenicol must be given empirically [470, 475]. Untreated, mortality of all patients with human ehrlichiosis ranges remains significant, ranging from 2 to 10 %.
Human Bartonellosis (Cat-Scratch Disease)
Cat-scratch disease is caused by Bartonella henselae, a pleomorphic gram-negative bacillus that binds silver and can be identified by Warthin-Starry stains. The condition develops predominantly in persons under the age of 21 years after receiving a scratch from a kitten or feral cat; the organism is transmitted less commonly by cat fleas. The organism proliferates at the site of the injury and, following an incubation period of 3–21 days, one or more papules develop at the site of the bite or scratch. Regional lymphadenitis ensues with enlargement of epitrochlear, axillary, and cervical lymph nodes, along with fever and malaise. In the typical case of cat-scratch disease, a single enlarged, fluctuant lymph node is the most common occurrence; generalized lymphadenopathy is relatively uncommon. About one-third of patients will develop systemic symptoms, such as fever and malaise. Other clinical manifestations include endocarditis and granulomatous or suppurative hepatosplenic and osseous lesions.
B. henselae involvement of the CNS is not uncommon: patients may actually have a variety of CNS symptoms, including encephalopathy, retinitis, or Parinaud’s ocular glandular syndrome (i.e., ocular granuloma or conjunctivitis with preauricular lymphadenopathy). In a large series of 130 seropositive patients with cat-scratch disease, the occurrence of neuroretinitis and encephalopathy was approximately 22 and 15 %, respectively.
Bartonella grows slowly on solid and liquid media and, if suspected, the medical microbiology laboratory should be contacted to hold the cultures for an extended period. Histological features of lymph nodes generally are nonspecific, showing features of chronic granulomatous and acute inflammatory changes. Diagnosis can be made serologically using an indirect immunofluorescence assay or enzyme immunosorbent assay to detect B. henselae antibodies; PCR assays have proven are more sensitive and specific at detecting B. henselae. Although most cases of cat-scratch disease in the normal host are self-limited and resolve over a period of 3–8 weeks, treatment is recommended. A 5-day course of azithromycin is the treatment of choice. Alternatives are clarithromycin, doxycycline, or ciprofloxacin for 10–14 days [476]. In severe cases, including patients with encephalopathy, IV azithromycin or gentamicin plus rifampin is recommended. Although there are reports of the effectiveness of adjunct steroid therapy in the management of patients with cat-scratch encephalitis, no randomized controlled trials have been carried out to determine whether steroids actually improve patient outcomes [477, 478].
Whipple’s Disease
Whipple’s disease is a slowly progressive systemic disease caused by Tropheryma whippelii, a gram-positive rod-shaped bacterium. The disease primarily affects Caucasian men over the age of 40 years. Whipple’s disease is relatively uncommon with most reported cases from North America and Western Europe. The typical clinical manifestations are arthralgias (67 %), diarrhea (76 %), weight loss (92 %), abdominal pain (55 %), and other features of malabsorption [479]. Less commonly, patients may present with fever, chills, CNS abnormalities, seizures, cardiovascular symptoms, endocarditis, or darkening of the skin. CNS involvement is the most serious complication of Whipple’s disease with neurological signs documented in 10–40 % of affected patients [479, 480]; damage to the CNS may be irreversible. T. whippelii is an unusual bacterium capable of intracellular survival. Left untreated, Whipple’s disease is invariably fatal [481]. Although humans are the only known host for T. whippelii, a natural source has not been defined. The organism itself is ubiquitous in the environment and has been isolated from the stool of sewage workers suggesting that a possible mode of transmission might be fecal-oral [482]. It appears that the genetic predisposition of the host rather than the genotype of the bacterium is the key risk factor for infection [483, 484].
CNS involvement is one of the more frequent complications of this disease and can, on occasion, occur in the absence of recognized gastrointestinal or systemic involvement. The most frequent neurologic manifestations of Whipple’s disease are dementia, decreasing levels of consciousness, progressive supranuclear ophthalmoplegia, myoclonus, and hypothalamic dysfunction. The dementia progresses slowly and is characterized by memory impairment, confusion, personality change, paranoia, emotional instability, and depression [479, 480, 485–491]. An unusual syndrome of synchronized ocular movements and contractions of the jaw, known as oculomasticatory myorhythmia, is sometimes seen and is unique to Whipple’s disease [487–489].
The disease is most often diagnosed through upper gastrointestinal endoscopy with biopsy of the small bowel or a mesenteric lymph node. Macroscopically, the duodenal mucosa is pale yellow with dilated villi and ecstatic lymph vessels [479]. Periodic acid-Schiff (PAS) stains will reveal the presence of PAS positive material (e.g., mucin and carbohydrate) with occasional bacilliform organisms. Pathologic involvement of the brain is most often manifest by chalky, yellowish white, 1–2 mm nodules distributed diffusely throughout the cortical and subcortical gray matter of both the cerebrum and the cerebellum. The most frequently involved sites are the temporal, periventricular, and periaqueductal gray matter as well as the hippocampus, hypothalamus, and basal ganglia. Histologically, the nodules are made up of microglia that stain strongly positive with PAS. Although neuroimaging studies with CT or MRI may reveal cerebral atrophy, ring-enhancing lesions, hydrocephalus, or changes consistent with demyelination, these findings are nonspecific for Whipple’s disease.
Although PCR diagnosis of Whipple’s disease is possible, healthy carriers can have positive PCR even if they do not have Whipple’s disease. Thus, PCR is better used when there is clinical suspicion of Whipple’s disease rather than for screening [479]. Moreover, because of the wide range of conditions in the differential diagnosis of degenerative CNS diseases, tissue biopsy is generally required for diagnosis. For CNS disease, immunohistochemistry testing using specific antibodies is more sensitive than PAS staining by being able to identify T. whipplei in tissues, including those with negative PAS staining [479, 492]. CNS involvement, consistent with the characteristic clinical syndromes of Whipple’s disease, and occurring in the setting of neuroimaging studies, biopsy proven systemic Whipple’s disease, or PCR are also sufficiently diagnostic [493, 494].
Untreated, Whipple’s disease is associated with high mortality rates. And because of the risk of CNS relapse, even while the patient is on oral trimethoprim/sulfamethoxazole, a regimen that includes antimicrobial agents (e.g., a third-generation cephalosporin) that penetrate the CNS is indicated [495, 496]. The initial treatment of CNS involvement in Whipple’s disease is ceftriaxone 2 g IV once daily or procaine penicillin G 1–2 million units IM once daily, with streptomycin 1 g IM daily and trimethoprim/sulfamethoxazole one DS tablet three times daily for 14 days, followed by trimethoprim/sulfamethoxazole one DS tablet twice daily for at least 1 year [479, 484]. Some clinicians continue therapy for 2 years to life. For severe CNS disease with cerebral lesions, Schneider and colleagues recommend consideration of adjunct corticosteroid therapy as outlined in the guidelines for tuberculous meningitis [271, 479]. Unfortunately, the overall prognosis of CNS Whipple’s disease is not good. With antimicrobial therapy, disease progression can be halted, but significant clinical improvement is limited, relapses are frequent, and established neurological defects are difficult to reverse.
Subacute Sclerosing Panencephalitis (SSPE)
This rare, progressive encephalitis usually occurs 2–10 years after an uncomplicated bout of measles. It occurs most frequently between the ages of 4 and 20 years and has a prolonged clinical course. It is thought to be due to reactivation of latent measles virus with high levels of IgM and IgG in the blood and CSF. Most of the pathologic features of the disease are localized to the CNS and retina. Histological findings include those of a subacute meningoencephalitis. Neuronophagia (neuronal degeneration) is common and residual neurons may contain intranuclear or cytoplasmic inclusion bodies. Progressive neurologic disease is characterized by seizures, behavioral abnormalities, and by gradual deterioration in intellectual, motor, and autonomic nervous system function. Management of the disease includes medical control of seizures. Although some authors have advocated the use of antiviral and immunomodulatory therapies (e.g., interferon, ribavirin, and Isoprinosine) to slow the progression of the disease and improve life expectancy in patients, the results are conflicting—the condition continues to show relentless progression. Only 5 % of individuals with SSPE undergo spontaneous remission; the remaining 95 % invariably die within 5 years of diagnosis [497].
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an intractable, severe, usually fatal demyelinating disease that is caused by the JC virus, a small double-stranded, nonenveloped DNA virus, closely related to two other polyomavirus: BK virus and the simian virus SV 40 of monkeys. PML is primarily seen in highly immunosuppressed patients, especially those with AIDS and reticuloendothelial malignancies and in patients on immunosuppressive therapy [498]. In the latter case, the immunomodulatory agent most often implicated in the development of PML is the monoclonal antibody natalizumab [499]. It is believed that under conditions of immunosuppression, replication of JC virus in the CNS increases.
Polyomaviruses are not thought to cause disease in immunocompetent individuals. In the pre-AIDS era, it was recognized that the main risk factor for PML was reduced resistance in persons of any age. The typical clinical features in these patients were impaired vision, motor weakness, and change in mentation, including personality changes [500]. It is estimated that the seroprevalence of the JC virus is 50 % in the 20–29 years age group; this rises to about 68 % in older persons [501]. Even normal brain tissue from immunocompetent individuals with no evidence of PML or demyelinating lesions has been found to harbor JC virus DNA sequences by PCR methodology, suggesting that the JC virus remains latent in the brain before immunosuppression [502–506]. The JC virus has also been detected in the blood of up to 22 % of immunosuppressed patients in the absence of PML [505, 507].
During 1979–1987, deaths related to PML increased fourfold from 1.5/10,000,000 persons in 1979 to 6.1/10,000,000 persons in 1987; this increase was largely attributed to the parallel increase in the incidence of HIV infection in the United States during this period [508]. PML-associated death rates peaked in the mid-1990s and have been decreasing since. For example, death rates have decreased from 2.7 deaths per one million persons during 1992–1995 to 0.6 per one million during 2002–2005 [509]. This significant decrease has been attributed to HAART which became the standard of care in the United States in 1996 [509].
CNS symptoms associated with PML reflect the anatomic location of pathologic brain lesions. PML characteristically presents with progressive focal neurologic defects, primarily hemiparesis, visual field defects, and cognitive deterioration [504, 509–514]. As the disease progresses, aphasia, ataxia, and cranial nerve defects may occur, ultimately resulting in cortical blindness, quadriparesis, profound dementia, seizures, and coma. The average duration of survival is approximately 4 months, although a subset of 5–10 % of patients, including some persons with HIV infection, survives for over a year following diagnosis.
The diagnosis is made from a combination of clinical features with characteristic findings on CT or MRI (Fig. 22.30a, b). The combination of a characteristic clinical picture and typical imaging findings supports a confident presumptive diagnosis of PML [323]. Definitive diagnosis requires brain biopsy and histologic examination, which should demonstrate characteristic cytopathic changes in the oligodendrocytes (Fig. 22.31a, b). These cells typically contain a homogeneous basophilic nuclear inclusion, and virus can be demonstrated by in situ hybridization. Viral damage to the oligodendrocytes, which synthesize and maintain myelin, ultimately leads to widespread, but patchy, multifocal demyelination, surrounded by giant, bizarre astrocytes containing intranuclear inclusions. The pathologic changes generally correlate with the patient’s clinical status. Virus particles can be demonstrated in the inclusions by electron microscopy [515].
Fig. 22.30.
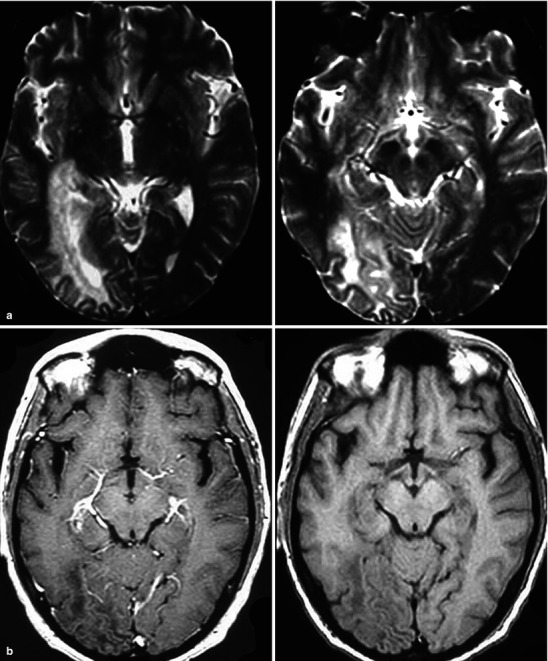
(a, b) Progressive multifocal leukoencephalopathy (JC virus). Figure (a) includes two adjacent T2-w sections. Figure (b) includes a post-contrast (left) and pre contrast (right) images of the same area. These images demonstrate the features of PML with a focal area of abnormality affecting mainly white matter with no appreciable internal contrast enhancement. These findings are nonspecific but in the context of an immune compromised host, PML is an important consideration. PML can cross the midline through the corpus callosum in which case it simulates both lymphoma and diffusely infiltrating astrocytoma
Fig. 22.31.
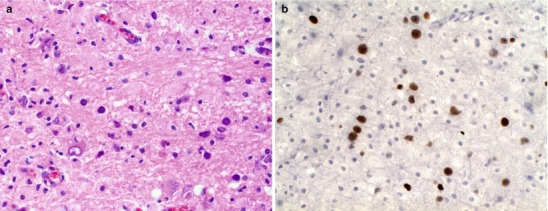
(a) Progressive multifocal leukoencephalopathy (JC virus). Numerous foamy macrophages infiltrating CNS white matter and bizarre reactive astrocytes. Scattered oligodendroglia exhibit smudged glassy nuclei (H&E 40×). (b) Progressive multifocal leukoencephalopathy (JC virus) Immunohistochemical study for JC virus antibody is immunoreactive in infected glial cells (Both courtesy of Anthony Yachnis, MD, and Kelly Devers, MD, University of Florida College of Medicine)
The CSF is invariably normal in the majority of patients with PML, although slight increases in protein levels and leukocyte counts have been documented for some. JC virus can be detected in the CSF of most patients with PML, whether immunosuppressed or not [503, 516, 517]. In general, however, patients with PML are highly likely to have JC virus DNA in their CSF as compared with normal or immunosuppressed patients without PML. Despite the sensitivity of PCR testing for JC virus, occasional cases of PML have been observed in which no JC virus can be found even in brain tissue at autopsy [518].
Immune reconstitution represents the mainstay of treatment for PML. Thus, for HIV-infected persons with PML, the basis of therapy remains the initiation of antiretroviral therapy for persons who are not on therapy and optimizing the antiretroviral regimen to achieve virologic suppression in patients who are already receiving antiretroviral therapy [323]. With rapid reversal of immunosuppression followed by immunologic recovery, patients may suffer a paradoxical clinical deterioration termed immune reconstitution inflammatory syndrome (IRIS) [499]. High-dose corticosteroids are often recommended if a clinical and imaging syndrome resembling IRIS develops after immune restoration. The JC virus DNA detection in CSF by nucleic acid amplification techniques and the CD4+ cell count are the most promising prognostic marker; higher levels of CD4+ cell counts are associated with improved survival [512].
Cidofovir, an agent used in the treatment of CMV infection, does have activity against polyomaviruses in vitro and in animal models. In a non-blinded, multicenter trial, the 1-year survival among patients with PML was 61 % in HIV-1-infected patients who received HAART with cidofovir compared with 29 % in those who received HAART without cidofovir [519]. Thus, although blinded studies are needed to confirm this observation and treatment guidelines do not recommend its use, cidofovir should be strongly considered in the empiric treatment of PML, keeping in mind the risk of renal toxicity associated with its use. Initial reports of success with cytosine arabinoside (ara-C) have not been supported by a formal clinical trial [520].
Acknowledgment
The first edition of this chapter on central nervous system (CNS) infections was authored by the following individuals: Dr. Kenneth H. Rand (Division of Infectious Diseases, Department of Medicine, University of Florida College of Medicine) and Drs. Arthur J. Ulm and David W. Pincus (Department of Neurological Surgery, University of Florida College of Medicine). Their chapter served as an inspiration and guideline for the organization and content of the second edition chapter on central nervous system (CNS) infections. For that, we are grateful.
Contributor Information
A Joseph Layon, Email: ajlayon@geisinger.edu.
Andrea Gabrielli, Email: agabrielli@anest.ufl.edu.
William A. Friedman, Email: friedman@neurosurgery.ufl.edu
Lennox K. Archibald, Email: lennox.archibald@medicine.ufl.edu.
Ronald G. Quisling, Email: quislr@radiology.ufl.edu.
References
- 1.Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med. 1997;337:970–6. doi: 10.1056/NEJM199710023371404. [DOI] [PubMed] [Google Scholar]
- 2.Kim KS. Acute bacterial meningitis in infants and children. Lancet Infect Dis. 2010;10:32–42. doi: 10.1016/S1473-3099(09)70306-8. [DOI] [PubMed] [Google Scholar]
- 3.Prevention CfDCa Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2005;54:1–21. [PubMed] [Google Scholar]
- 4.Hsu HE, Shutt KA, Moore MR, et al. Effect of pneumococcal conjugate vaccine on pneumococcal meningitis. N Engl J Med. 2009;360:244–56. doi: 10.1056/NEJMoa0800836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fothergill LD, Wright J. Influenzal meningitis: relation of age incidence to bacterial power of blood against causal organism. J Immunol. 1933;24:273–84. [Google Scholar]
- 6.Thigpen MC, Whitney CG, Messonnier NE, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011;364:2016–25. doi: 10.1056/NEJMoa1005384. [DOI] [PubMed] [Google Scholar]
- 7.Unhanand M, Mustafa MM, McCracken GH, Jr, Nelson JD. Gram-negative enteric bacillary meningitis: a twenty-one-year experience. J Pediatr. 1993;122:15–21. doi: 10.1016/S0022-3476(05)83480-8. [DOI] [PubMed] [Google Scholar]
- 8.Sarff LD, Platt LH, McCracken GH., Jr Cerebrospinal fluid evaluation in neonates: comparison of high-risk infants with and without meningitis. J Pediatr. 1976;88:473–7. doi: 10.1016/S0022-3476(76)80271-5. [DOI] [PubMed] [Google Scholar]
- 9.Sarff LD, McCracken GH, Schiffer MS, et al. Epidemiology of Escherichia coli K1 in healthy and diseased newborns. Lancet. 1975;1:1099–104. doi: 10.1016/S0140-6736(75)92496-4. [DOI] [PubMed] [Google Scholar]
- 10.Schiffer MS, Oliveira E, Glode MP, McCracken GH, Jr, Sarff LM, Robbins JB. A review: relation between invasiveness and the K1 capsular polysaccharide of Escherichia coli. Pediatr Res. 1976;10:82–7. doi: 10.1203/00006450-197602000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Nigrovic LE, Kuppermann N, Malley R. Children with bacterial meningitis presenting to the emergency department during the pneumococcal conjugate vaccine era. Acad Emerg Med. 2008;15:522–8. doi: 10.1111/j.1553-2712.2008.00117.x. [DOI] [PubMed] [Google Scholar]
- 12.Nigrovic LE, Kuppermann N, Macias CG, et al. Clinical prediction rule for identifying children with cerebrospinal fluid pleocytosis at very low risk of bacterial meningitis. JAMA. 2007;297:52–60. doi: 10.1001/jama.297.1.52. [DOI] [PubMed] [Google Scholar]
- 13.May M, Daley AJ, Donath S, Isaacs D. Early onset neonatal meningitis in Australia and New Zealand, 1992–2002. Arch Dis Child Fetal Neonatal Ed. 2005;90:F324–7. doi: 10.1136/adc.2004.066134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weisfelt M, van de Beek D, Spanjaard L, de Gans J. Nosocomial bacterial meningitis in adults: a prospective series of 50 cases. J Hosp Infect. 2007;66:71–8. doi: 10.1016/j.jhin.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Helbok R, Broessner G, Pfausler B, Schmutzhard E. Chronic meningitis. J Neurol. 2009;256:168–75. doi: 10.1007/s00415-009-0122-0. [DOI] [PubMed] [Google Scholar]
- 16.Kallstrom H, Blackmer Gill D, Albiger B, Liszewski MK, Atkinson JP, Jonsson AB. Attachment of Neisseria gonorrhoeae to the cellular pilus receptor CD46: identification of domains important for bacterial adherence. Cell Microbiol. 2001;3:133–43. doi: 10.1046/j.1462-5822.2001.00095.x. [DOI] [PubMed] [Google Scholar]
- 17.Kallstrom H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–47. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- 18.Toleman M, Aho E, Virji M. Expression of pathogen-like Opa adhesins in commensal Neisseria: genetic and functional analysis. Cell Microbiol. 2001;3:33–44. doi: 10.1046/j.1462-5822.2001.00089.x. [DOI] [PubMed] [Google Scholar]
- 19.Mulks MH, Plaut AG. IgA protease production as a characteristic distinguishing pathogenic from harmless neisseriaceae. N Engl J Med. 1978;299:973–6. doi: 10.1056/NEJM197811022991802. [DOI] [PubMed] [Google Scholar]
- 20.Hill DJ, Griffiths NJ, Borodina E, Virji M. Cellular and molecular biology of Neisseria meningitidis colonization and invasive disease. Clin Sci (Lond) 2010;118:547–64. doi: 10.1042/CS20090513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouphael NG, Stephens DS. Neisseria meningitidis: biology, microbiology, and epidemiology. Methods Mol Biol. 2012;799:1–20. doi: 10.1007/978-1-61779-346-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wurzner R, Orren A, Lachmann PJ. Inherited deficiencies of the terminal components of human complement. Immunodefic Rev. 1992;3:123–47. [PubMed] [Google Scholar]
- 23.Song JH, Dagan R, Klugman KP, Fritzell B. The relationship between pneumococcal serotypes and antibiotic resistance. Vaccine. 2012;30(17):2728–37. doi: 10.1016/j.vaccine.2012.01.091. [DOI] [PubMed] [Google Scholar]
- 24.Hausdorff WP, Feikin DR, Klugman KP. Epidemiological differences among pneumococcal serotypes. Lancet Infect Dis. 2005;5:83–93. doi: 10.1016/S1473-3099(05)01280-6. [DOI] [PubMed] [Google Scholar]
- 25.Pfister HW, Fontana A, Tauber MG, Tomasz A, Scheld WM. Mechanisms of brain injury in bacterial meningitis: workshop summary. Clin Infect Dis. 1994;19:463–79. doi: 10.1093/clinids/19.3.463. [DOI] [PubMed] [Google Scholar]
- 26.Pichichero ME, Loeb M, Anderson, Smith DH. Do pili play a role in pathogenicity of Haemophilus influenzae type B? Lancet. 1982;2:960–2. doi: 10.1016/S0140-6736(82)90161-1. [DOI] [PubMed] [Google Scholar]
- 27.Mason EO, Jr, Kaplan SL, Wiedermann BL, Norrod EP, Stenback WA. Frequency and properties of naturally occurring adherent piliated strains of Haemophilus influenzae type b. Infect Immun. 1985;49:98–103. doi: 10.1128/iai.49.1.98-103.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Filippidis A, Fountas KN. Nasal lymphatics as a novel invasion and dissemination route of bacterial meningitis. Med Hypotheses. 2009;72:694–7. doi: 10.1016/j.mehy.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 29.Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011;24:557–91. doi: 10.1128/CMR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Join-Lambert O, Morand PC, Carbonnelle E, et al. Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog Neurobiol. 2010;91:130–9. doi: 10.1016/j.pneurobio.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Flierl MA, Rittirsch D, Huber-Lang MS, Stahel PF. Pathophysiology of septic encephalopathy – an unsolved puzzle. Crit Care. 2010;14:165. doi: 10.1186/cc9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tunkel AR, Scheld WM. Pathogenesis and pathophysiology of bacterial meningitis. Clin Microbiol Rev. 1993;6:118–36. doi: 10.1128/cmr.6.2.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carpenter RR, Petersdorf RG. The clinical spectrum of bacterial meningitis. Am J Med. 1962;33:262–75. doi: 10.1016/0002-9343(62)90024-4. [DOI] [PubMed] [Google Scholar]
- 34.Durand ML, Calderwood SB, Weber DJ, et al. Acute bacterial meningitis in adults. A review of 493 episodes. N Engl J Med. 1993;328:21–8. doi: 10.1056/NEJM199301073280104. [DOI] [PubMed] [Google Scholar]
- 35.Gorse GJ, Thrupp LD, Nudleman KL, Wyle FA, Hawkins B, Cesario TC. Bacterial meningitis in the elderly. Arch Intern Med. 1984;144:1603–7. doi: 10.1001/archinte.1984.00350200107016. [DOI] [PubMed] [Google Scholar]
- 36.Verghese A, Gallemore G. Kernig’s and Brudzinski’s signs revisited. Rev Infect Dis. 1987;9:1187–92. doi: 10.1093/clinids/9.6.1187. [DOI] [PubMed] [Google Scholar]
- 37.Ward MA, Greenwood TM, Kumar DR, Mazza JJ, Yale SH. Josef Brudzinski and Vladimir Mikhailovich Kernig: signs for diagnosing meningitis. Clin Med Res. 2010;8:13–7. doi: 10.3121/cmr.2010.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joffe AR. Lumbar puncture and brain herniation in acute bacterial meningitis: a review. J Intensive Care Med. 2007;22:194–207. doi: 10.1177/0885066607299516. [DOI] [PubMed] [Google Scholar]
- 39.Joffe AR. Prognostic factors in adults with bacterial meningitis. N Engl J Med. 2005;352:512–5. doi: 10.1056/NEJM200502033520519. [DOI] [PubMed] [Google Scholar]
- 40.Oliver WJ, Shope TC, Kuhns LR. Fatal lumbar puncture: fact versus fiction – an approach to a clinical dilemma. Pediatrics. 2003;112:e174–6. doi: 10.1542/peds.112.3.e174. [DOI] [PubMed] [Google Scholar]
- 41.Tattevin P, Bruneel F, Regnier B. Cranial CT before lumbar puncture in suspected meningitis. N Engl J Med. 2002;346:1248–51. doi: 10.1056/NEJM200204183461615. [DOI] [PubMed] [Google Scholar]
- 42.van Crevel H, Hijdra A, de Gans J. Lumbar puncture and the risk of herniation: when should we first perform CT? J Neurol. 2002;249:129–37. doi: 10.1007/PL00007855. [DOI] [PubMed] [Google Scholar]
- 43.Fishman RA. Cerebrospinal fluid in diseases of the nervous system. Philadelphia: WB Saunders Co; 1992. [Google Scholar]
- 44.Skipper BJ, Davis LE. Ascertaining hypoglycorrhachia in an acute patient. Am J Emerg Med. 1997;15:378–80. doi: 10.1016/S0735-6757(97)90131-5. [DOI] [PubMed] [Google Scholar]
- 45.Negrini B, Kelleher KJ, Wald ER. Cerebrospinal fluid findings in aseptic versus bacterial meningitis. Pediatrics. 2000;105:316–9. doi: 10.1542/peds.105.2.316. [DOI] [PubMed] [Google Scholar]
- 46.Feigin RD, Shackelford PG. Value of repeat lumbar puncture in the differential diagnosis of meningitis. N Engl J Med. 1973;289:571–4. doi: 10.1056/NEJM197309132891108. [DOI] [PubMed] [Google Scholar]
- 47.Herndon RM, Brumback RA. The cerebrospinal spinal fluid. Boston: Kluwer Academic Publishers; 1989. [Google Scholar]
- 48.Neuman MI, Tolford S, Harper MB. Test characteristics and interpretation of cerebrospinal fluid gram stain in children. Pediatr Infect Dis J. 2008;27:309–13. doi: 10.1097/INF.0b013e31815f53ba. [DOI] [PubMed] [Google Scholar]
- 49.Hayden RT, Frenkel LD. More laboratory testing: greater cost but not necessarily better. Pediatr Infect Dis J. 2000;19:290–2. doi: 10.1097/00006454-200004000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Mein J, Lum G. CSF bacterial antigen detection tests offer no advantage over Gram’s stain in the diagnosis of bacterial meningitis. Pathology. 1999;31:67–9. doi: 10.1080/003130299105601. [DOI] [PubMed] [Google Scholar]
- 51.Gray BM, Simmons DR, Mason H, Barnum S, Volanakis JE. Quantitative levels of C-reactive protein in cerebrospinal fluid in patients with bacterial meningitis and other conditions. J Pediatr. 1986;108:665–70. doi: 10.1016/S0022-3476(86)81038-1. [DOI] [PubMed] [Google Scholar]
- 52.Skull SA, Leach AJ, Currie BJ. Streptococcus pneumoniae carriage and penicillin/ceftriaxone resistance in hospitalised children in Darwin. Aust N Z J Med. 1996;26:391–5. doi: 10.1111/j.1445-5994.1996.tb01928.x. [DOI] [PubMed] [Google Scholar]
- 53.Jones ME, Draghi DC, Karlowsky JA, Sahm DF, Bradley JS. Prevalence of antimicrobial resistance in bacteria isolated from central nervous system specimens as reported by U.S. hospital laboratories from 2000 to 2002. Ann Clin Microbiol Antimicrob. 2004;3:3. doi: 10.1186/1476-0711-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gouveia EL, Reis JN, Flannery B, et al. Clinical outcome of pneumococcal meningitis during the emergence of penicillin-resistant Streptococcus pneumoniae: an observational study. BMC Infect Dis. 2011;11:323. doi: 10.1186/1471-2334-11-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deghmane AE, Alonso JM, Taha MK. Emerging drugs for acute bacterial meningitis. Expert Opin Emerg Drugs. 2009;14:381–93. doi: 10.1517/14728210903120887. [DOI] [PubMed] [Google Scholar]
- 56.Ohga S, Okada K, Ueda K, et al. Cerebrospinal fluid cytokine levels and dexamethasone therapy in bacterial meningitis. J Infect. 1999;39:55–60. doi: 10.1016/S0163-4453(99)90103-2. [DOI] [PubMed] [Google Scholar]
- 57.Ahmed A, Jafri H, Lutsar I, et al. Pharmacodynamics of vancomycin for the treatment of experimental penicillin- and cephalosporin-resistant pneumococcal meningitis. Antimicrob Agents Chemother. 1999;43:876–81. doi: 10.1128/aac.43.4.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lutsar I, Friedland IR, Jafri HS, et al. Factors influencing the anti-inflammatory effect of dexamethasone therapy in experimental pneumococcal meningitis. J Antimicrob Chemother. 2003;52:651–5. doi: 10.1093/jac/dkg417. [DOI] [PubMed] [Google Scholar]
- 59.McIntyre PB, Berkey CS, King SM, et al. Dexamethasone as adjunctive therapy in bacterial meningitis. A meta-analysis of randomized clinical trials since 1988. JAMA. 1997;278:925–31. doi: 10.1001/jama.278.11.925. [DOI] [PubMed] [Google Scholar]
- 60.van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol. 2010;9:254–63. doi: 10.1016/S1474-4422(10)70023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roos KL, Scheld WM. The management of fulminant meningitis in the intensive care unit. Infect Dis Clin North Am. 1989;3:137–54. [PubMed] [Google Scholar]
- 62.Roos KL, van de Beek D. Bacterial meningitis. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. Elsevier: vol. 96. 2010. p. 51–63. [DOI] [PubMed]
- 63.Gwer S, Gatakaa H, Mwai L, Idro R, Newton CR. The role for osmotic agents in children with acute encephalopathies: a systematic review. BMC Pediatr. 2010;10:23. doi: 10.1186/1471-2431-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hinson HE, Stein D, Sheth KN. Hypertonic saline and mannitol therapy in critical care neurology. J Intensive Care Med. 2013;28(1):3–11. doi: 10.1177/0885066611400688. [DOI] [PubMed] [Google Scholar]
- 65.Liu S, Li L, Luo Z, et al. Superior effect of hypertonic saline over mannitol to attenuate cerebral edema in a rabbit bacterial meningitis model. Crit Care Med. 2011;39:1467–73. doi: 10.1097/CCM.0b013e3182120d13. [DOI] [PubMed] [Google Scholar]
- 66.Murthy JM. Management of intracranial pressure in tuberculous meningitis. Neurocrit Care. 2005;2:306–12. doi: 10.1385/NCC:2:3:306. [DOI] [PubMed] [Google Scholar]
- 67.Qureshi AI, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301–13. doi: 10.1097/00003246-200009000-00032. [DOI] [PubMed] [Google Scholar]
- 68.Singhi S, Singhi P, Baranwal AK. Bacterial meningitis in children: critical care needs. Indian J Pediatr. 2001;68:737–47. doi: 10.1007/BF03191900. [DOI] [PubMed] [Google Scholar]
- 69.Czosnyka M, Pickard JD. Monitoring and interpretation of intracranial pressure. J Neurol Neurosurg Psychiatry. 2004;75:813–21. doi: 10.1136/jnnp.2003.033126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar G, Kalita J, Misra UK. Raised intracranial pressure in acute viral encephalitis. Clin Neurol Neurosurg. 2009;111:399–406. doi: 10.1016/j.clineuro.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 71.Sala F, Abbruzzese C, Galli D, et al. Intracranial pressure monitoring in pediatric bacterial meningitis: a fancy or useful tool? A case report. Minerva Anestesiol. 2009;75:746–9. [PubMed] [Google Scholar]
- 72.Whitney CG, Farley MM, Hadler J, et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med. 2003;348:1737–46. doi: 10.1056/NEJMoa022823. [DOI] [PubMed] [Google Scholar]
- 73.Pilishvili T, Lexau C, Farley MM, et al. Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J Infect Dis. 2010;201:32–41. doi: 10.1086/648593. [DOI] [PubMed] [Google Scholar]
- 74.CDC Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2005;54:1–21. [PubMed] [Google Scholar]
- 75.CDC Updated recommendations for use of meningococcal conjugate vaccines – Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb Mortal Wkly Rep. 2011;60:72–6. [PubMed] [Google Scholar]
- 76.CDC Meningococcal conjugate vaccines policy update: booster dose recommendations. Pediatrics. 2011;128:1213–8. doi: 10.1542/peds.2011-2380. [DOI] [PubMed] [Google Scholar]
- 77.CDC Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23) MMWR Morb Mortal Wkly Rep. 2010;59:1102–6. [PubMed] [Google Scholar]
- 78.CDC Enterovirus surveillance – United States, 1970–2005. MMWR Surveill Summ. 2006;55:1–20. [PubMed] [Google Scholar]
- 79.Asnis DS, Conetta R, Teixeira AA, Waldman G, Sampson BA. The West Nile Virus outbreak of 1999 in New York: the Flushing Hospital experience. Clin Infect Dis. 2000;30:413–8. doi: 10.1086/313737. [DOI] [PubMed] [Google Scholar]
- 80.Rappole JH, Derrickson SR, Hubalek Z. Migratory birds and spread of West Nile virus in the Western Hemisphere. Emerg Infect Dis. 2000;6:319–28. doi: 10.3201/eid0604.000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rappole JH, Hubalek Z. Migratory birds and West Nile virus. J Appl Microbiol. 2003;94(Suppl):47S–58. doi: 10.1046/j.1365-2672.94.s1.6.x. [DOI] [PubMed] [Google Scholar]
- 82.CDC Human rabies prevention – United States, 2008: recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm Rep. 2008;57:1–28. [PubMed] [Google Scholar]
- 83.Blanton JD, Palmer D, Dyer J, Rupprecht CE. Rabies surveillance in the United States during 2010. J Am Vet Med Assoc. 2011;239:773–83. doi: 10.2460/javma.239.6.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharpe AH, Fields BN. Pathogenesis of viral infections. Basic concepts derived from the reovirus model. N Engl J Med. 1985;312:486–97. doi: 10.1056/NEJM198502213120806. [DOI] [PubMed] [Google Scholar]
- 85.Iwasaka T, Kidera Y, Tsugitomi H, Sugimori H. The cellular changes in primary and recurrent infection with herpes simplex virus type 2 in an in vitro model. Acta Cytol. 1987;31:935–40. [PubMed] [Google Scholar]
- 86.Esiri MM. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci. 1982;54:209–26. doi: 10.1016/0022-510X(82)90183-6. [DOI] [PubMed] [Google Scholar]
- 87.Pleasure SJ, Fischbein NJ. Correlation of clinical and neuroimaging findings in a case of rabies encephalitis. Arch Neurol. 2000;57:1765–9. doi: 10.1001/archneur.57.12.1765. [DOI] [PubMed] [Google Scholar]
- 88.Peters CJ. Arenaviruses. In: Richman DD, Whitley RJ, Hayden FG, editors. Clinical virology. Washington, D.C.: American Society of Microbiology Press; 2009. pp. 1009–29. [Google Scholar]
- 89.Jolles S, Sewell WA, Leighton C. Drug-induced aseptic meningitis: diagnosis and management. Drug Saf. 2000;22:215–26. doi: 10.2165/00002018-200022030-00005. [DOI] [PubMed] [Google Scholar]
- 90.Dupuis M, Hull R, Wang H, et al. Molecular detection of viral causes of encephalitis and meningitis in New York State. J Med Virol. 2011;83:2172–81. doi: 10.1002/jmv.22169. [DOI] [PubMed] [Google Scholar]
- 91.Murphy RF, Caliendo AM. Relative quantity of cerebrospinal fluid herpes simplex virus DNA in adult cases of encephalitis and meningitis. Am J Clin Pathol. 2009;132:687–90. doi: 10.1309/AJCP0KN1PCHEYSIK. [DOI] [PubMed] [Google Scholar]
- 92.Cinque P, Cleator GM, Weber T, Monteyne P, Sindic CJ, van Loon AM. The role of laboratory investigation in the diagnosis and management of patients with suspected herpes simplex encephalitis: a consensus report. The EU Concerted Action on Virus Meningitis and Encephalitis. J Neurol Neurosurg Psychiatry. 1996;61:339–45. doi: 10.1136/jnnp.61.4.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Espy MJ, Uhl JR, Mitchell PS, et al. Diagnosis of herpes simplex virus infections in the clinical laboratory by LightCycler PCR. J Clin Microbiol. 2000;38:795–9. doi: 10.1128/jcm.38.2.795-799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Whitley RJ, Alford CA, Hirsch MS, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314:144–9. doi: 10.1056/NEJM198601163140303. [DOI] [PubMed] [Google Scholar]
- 95.Whitley RJ, Cobbs CG, Alford CA, Jr, et al. Diseases that mimic herpes simplex encephalitis. Diagnosis, presentation, and outcome. NIAD Collaborative Antiviral Study Group. JAMA. 1989;262:234–9. doi: 10.1001/jama.1989.03430020076032. [DOI] [PubMed] [Google Scholar]
- 96.CDC Investigation of rabies infections in organ donor and transplant recipients – Alabama, Arkansas, Oklahoma, and Texas, 2004. MMWR Morb Mortal Wkly Rep. 2004;53:586–9. [PubMed] [Google Scholar]
- 97.CDC Human-to-human transmission of rabies via corneal transplant – Thailand. MMWR Morb Mortal Wkly Rep. 1981;30:473–4. [PubMed] [Google Scholar]
- 98.CDC Human-to-human transmission of rabies by a corneal transplant-Idaho. MMWR. 1979;28:109–11. [Google Scholar]
- 99.Houff SA, Burton RC, Wilson RW, et al. Human-to-human transmission of rabies virus by corneal transplant. N Engl J Med. 1979;300:603–4. doi: 10.1056/NEJM197903153001105. [DOI] [PubMed] [Google Scholar]
- 100.Jackson AC. Rabies in the critical care unit: diagnostic and therapeutic approaches. Can J Neurol Sci. 2011;38:689–95. doi: 10.1017/s0317167100054056. [DOI] [PubMed] [Google Scholar]
- 101.Cohen JI, Davenport DS, Stewart JA, Deitchman S, Hilliard JK, Chapman LE. Recommendations for prevention of and therapy for exposure to B virus (cercopithecine herpesvirus 1) Clin Infect Dis. 2002;35:1191–203. doi: 10.1086/344754. [DOI] [PubMed] [Google Scholar]
- 102.Canale DJ. William Macewen and the treatment of brain abscesses: revisited after one hundred years. J Neurosurg. 1996;84:133–42. doi: 10.3171/jns.1996.84.1.0133. [DOI] [PubMed] [Google Scholar]
- 103.Carpenter J, Stapleton S, Holliman R. Retrospective analysis of 49 cases of brain abscess and review of the literature. Eur J Clin Microbiol Infect Dis. 2007;26:1–11. doi: 10.1007/s10096-006-0236-6. [DOI] [PubMed] [Google Scholar]
- 104.Nicolosi A, Hauser WA, Musicco M, Kurland LT. Incidence and prognosis of brain abscess in a defined population: Olmsted County, Minnesota, 1935–1981. Neuroepidemiology. 1991;10:122–31. doi: 10.1159/000110257. [DOI] [PubMed] [Google Scholar]
- 105.Nielsen H, Harmsen A, Gyldensted C. Cerebral abscess. A long-term follow-up. Acta Neurol Scand. 1983;67:330–7. doi: 10.1111/j.1600-0404.1983.tb03150.x. [DOI] [PubMed] [Google Scholar]
- 106.Nielsen H, Gyldensted C, Harmsen A. Cerebral abscess. Aetiology and pathogenesis, symptoms, diagnosis and treatment. A review of 200 cases from 1935–1976. Acta Neurol Scand. 1982;65:609–22. doi: 10.1111/j.1600-0404.1982.tb03114.x. [DOI] [PubMed] [Google Scholar]
- 107.Honda H, Warren DK. Central nervous system infections: meningitis and brain abscess. Infect Dis Clin North Am. 2009;23:609–23. doi: 10.1016/j.idc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 108.Harris PS, Cobbs CG. Cardiac, cerebral, and vascular complications of infective endocarditis. Cardiol Clin. 1996;14:437–50. doi: 10.1016/S0733-8651(05)70294-0. [DOI] [PubMed] [Google Scholar]
- 109.Muzumdar D, Jhawar S, Goel A. Brain abscess: an overview. Int J Surg. 2011;9:136–44. doi: 10.1016/j.ijsu.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 110.Le Moal G, Landron C, Grollier G, et al. Characteristics of brain abscess with isolation of anaerobic bacteria. Scand J Infect Dis. 2003;35:318–21. doi: 10.1080/00365540310000265. [DOI] [PubMed] [Google Scholar]
- 111.Cunha BA. Central nervous system infections in the compromised host: a diagnostic approach. Infect Dis Clin North Am. 2001;15:567–90. doi: 10.1016/S0891-5520(05)70160-4. [DOI] [PubMed] [Google Scholar]
- 112.Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–79. doi: 10.1086/515541. [DOI] [PubMed] [Google Scholar]
- 113.Mamelak AN, Obana WG, Flaherty JF, Rosenblum ML. Nocardial brain abscess: treatment strategies and factors influencing outcome. Neurosurgery. 1994;35:622–31. doi: 10.1227/00006123-199410000-00007. [DOI] [PubMed] [Google Scholar]
- 114.Bartzatt R. Tuberculosis infections of the central nervous system. Cent Nerv Syst Agents Med Chem. 2011;11:321–7. doi: 10.2174/1871524911106040321. [DOI] [PubMed] [Google Scholar]
- 115.Bathla G, Khandelwal G, Maller VG, Gupta A. Manifestations of cerebral tuberculosis. Singapore Med J. 2011;52:124–30. [PubMed] [Google Scholar]
- 116.Jung A, Korsukewitz C, Kuhlmann T, et al. Intracerebral mass lesion diagnosed as cryptococcoma in a patient with sarcoidosis, a rare opportunistic manifestation induced by immunosuppression with corticosteroids. J Neurol. 2012;259(10):2147–50. doi: 10.1007/s00415-012-6473-y. [DOI] [PubMed] [Google Scholar]
- 117.Nadkarni T, Goel A. Aspergilloma of the brain: an overview. J Postgrad Med. 2005;51(Suppl 1):S37–41. [PubMed] [Google Scholar]
- 118.Metellus P, Laghmari M, Fuentes S, et al. Successful treatment of a giant isolated cerebral mucormycotic (zygomycotic) abscess using endoscopic debridement: case report and therapeutic considerations. Surg Neurol. 2008;69:510–5. doi: 10.1016/j.surneu.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 119.Chun CH, Johnson JD, Hofstetter M, Raff MJ. Brain abscess. A study of 45 consecutive cases. Medicine. 1986;65:415–31. doi: 10.1097/00005792-198611000-00006. [DOI] [PubMed] [Google Scholar]
- 120.Arseni C, Ciurea AV. Cerebellar abscesses. A report on 119 cases. Zentralbl Neurochir. 1982;43:359–70. [PubMed] [Google Scholar]
- 121.Turner RC, Dodson SC, Rosen CL. Medical management of cerebellar abscess: a case report and review of the literature. W V Med J. 2011;107:21–3. [PubMed] [Google Scholar]
- 122.Kastrup O, Wanke I, Maschke M. Neuroimaging of infections of the central nervous system. Semin Neurol. 2008;28:511–22. doi: 10.1055/s-0028-1083688. [DOI] [PubMed] [Google Scholar]
- 123.Nathoo N, Nadvi SS, Narotam PK, van Dellen JR. Brain abscess: management and outcome analysis of a computed tomography era experience with 973 patients. World Neurosurg. 2011;75:716–26. doi: 10.1016/j.wneu.2010.11.043. [DOI] [PubMed] [Google Scholar]
- 124.Garg RK, Sinha MK. Multiple ring-enhancing lesions of the brain. J Postgrad Med. 2010;56:307–16. doi: 10.4103/0022-3859.70939. [DOI] [PubMed] [Google Scholar]
- 125.Holtas S, Tornquist C, Cronqvist S. Diagnostic difficulties in computed tomography of brain abscesses. J Comput Assist Tomogr. 1982;6:683–8. doi: 10.1097/00004728-198208000-00004. [DOI] [PubMed] [Google Scholar]
- 126.Miller ES, Dias PS, Uttley D. CT scanning in the management of intracranial abscess: a review of 100 cases. Br J Neurosurg. 1988;2:439–46. doi: 10.3109/02688698809029597. [DOI] [PubMed] [Google Scholar]
- 127.Nguyen JB, Black BR, Leimkuehler MM, Halder V, Nguyen JV, Ahktar N. Intracranial pyogenic abscess: imaging diagnosis utilizing recent advances in computed tomography and magnetic resonance imaging. Crit Rev Comput Tomogr. 2004;45:181–224. [PubMed] [Google Scholar]
- 128.Lai PH, Hsu SS, Ding SW, et al. Proton magnetic resonance spectroscopy and diffusion-weighted imaging in intracranial cystic mass lesions. Surg Neurol. 2007;68(Suppl 1):S25–36. doi: 10.1016/j.surneu.2007.07.080. [DOI] [PubMed] [Google Scholar]
- 129.Lai PH, Hsu SS, Lo YK, Ding SW. Role of diffusion-weighted imaging and proton MR spectroscopy in distinguishing between pyogenic brain abscess and necrotic brain tumor. Acta Neurol Taiwan. 2004;13:107–13. [PubMed] [Google Scholar]
- 130.Desprechins B, Stadnik T, Koerts G, Shabana W, Breucq C, Osteaux M. Use of diffusion-weighted MR imaging in differential diagnosis between intracerebral necrotic tumors and cerebral abscesses. AJNR Am J Neuroradiol. 1999;20:1252–7. [PMC free article] [PubMed] [Google Scholar]
- 131.Omuro AM, Leite CC, Mokhtari K, Delattre JY. Pitfalls in the diagnosis of brain tumours. Lancet Neurol. 2006;5:937–48. doi: 10.1016/S1474-4422(06)70597-X. [DOI] [PubMed] [Google Scholar]
- 132.Kang K, Lim I, Roh JK. Positron emission tomographic findings in a tuberculous brain abscess. Ann Nucl Med. 2007;21:303–6. doi: 10.1007/s12149-007-0023-1. [DOI] [PubMed] [Google Scholar]
- 133.Kosterink JG. Positron emission tomography in the diagnosis and treatment management of tuberculosis. Curr Pharm Des. 2011;17:2875–80. doi: 10.2174/138161211797470183. [DOI] [PubMed] [Google Scholar]
- 134.Kumar R, Basu S, Torigian D, Anand V, Zhuang H, Alavi A. Role of modern imaging techniques for diagnosis of infection in the era of 18F-fluorodeoxyglucose positron emission tomography. Clin Microbiol Rev. 2008;21:209–24. doi: 10.1128/CMR.00025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu CH, Chang WN, Lui CC. Strategies for the management of bacterial brain abscess. J Clin Neurosci. 2006;13:979–85. doi: 10.1016/j.jocn.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 136.Mackenzie AR, Laing RB, Smith CC, Kaar GF, Smith FW. Spinal epidural abscess: the importance of early diagnosis and treatment. J Neurol Neurosurg Psychiatry. 1998;65:209–12. doi: 10.1136/jnnp.65.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bluman EM, Palumbo MA, Lucas PR. Spinal epidural abscess in adults. J Am Acad Orthop Surg. 2004;12:155–63. doi: 10.5435/00124635-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 138.Tompkins M, Panuncialman I, Lucas P, Palumbo M. Spinal epidural abscess. J Emerg Med. 2010;39:384–90. doi: 10.1016/j.jemermed.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 139.Sendi P, Bregenzer T, Zimmerli W. Spinal epidural abscess in clinical practice. QJM. 2008;101:1–12. doi: 10.1093/qjmed/hcm100. [DOI] [PubMed] [Google Scholar]
- 140.Martin RJ, Yuan HA. Neurosurgical care of spinal epidural, subdural, and intramedullary abscesses and arachnoiditis. Orthop Clin North Am. 1996;27:125–36. [PubMed] [Google Scholar]
- 141.Pradilla G, Ardila GP, Hsu W, Rigamonti D. Epidural abscesses of the CNS. Lancet Neurol. 2009;8:292–300. doi: 10.1016/S1474-4422(09)70044-4. [DOI] [PubMed] [Google Scholar]
- 142.Soehle M, Wallenfang T. Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51:79–85. doi: 10.1097/00006123-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 143.Curry WT, Jr, Hoh BL, Amin-Hanjani S, Eskandar EN. Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol. 2005;63:364–71. doi: 10.1016/j.surneu.2004.08.081. [DOI] [PubMed] [Google Scholar]
- 144.Hlavin ML, Kaminski HJ, Ross JS, Ganz E. Spinal epidural abscess: a ten-year perspective. Neurosurgery. 1990;27:177–84. doi: 10.1227/00006123-199008000-00001. [DOI] [PubMed] [Google Scholar]
- 145.Nussbaum ES, Rigamonti D, Standiford H, Numaguchi Y, Wolf AL, Robinson WL. Spinal epidural abscess: a report of 40 cases and review. Surg Neurol. 1992;38:225–31. doi: 10.1016/0090-3019(92)90173-K. [DOI] [PubMed] [Google Scholar]
- 146.Huang PY, Chen SF, Chang WN, et al. Spinal epidural abscess in adults caused by Staphylococcus aureus: clinical characteristics and prognostic factors. Clin Neurol Neurosurg. 2012;114(6):572–6. doi: 10.1016/j.clineuro.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 147.Wheeler D, Keiser P, Rigamonti D, Keay S. Medical management of spinal epidural abscesses: case report and review. Clin Infect Dis. 1992;15:22–7. doi: 10.1093/clinids/15.1.22. [DOI] [PubMed] [Google Scholar]
- 148.Maslen DR, Jones SR, Crislip MA, Bracis R, Dworkin RJ, Flemming JE. Spinal epidural abscess. Optimizing patient care. Arch Intern Med. 1993;153:1713–21. doi: 10.1001/archinte.1993.00410140107012. [DOI] [PubMed] [Google Scholar]
- 149.Khanna RK, Malik GM, Rock JP, Rosenblum ML. Spinal epidural abscess: evaluation of factors influencing outcome. Neurosurgery. 1996;39:958–64. doi: 10.1097/00006123-199611000-00016. [DOI] [PubMed] [Google Scholar]
- 150.Tang HJ, Lin HJ, Liu YC, Li CM. Spinal epidural abscess – experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45:76–81. doi: 10.1053/jinf.2002.1013. [DOI] [PubMed] [Google Scholar]
- 151.Del Curling O, Jr, Gower DJ, McWhorter JM. Changing concepts in spinal epidural abscess: a report of 29 cases. Neurosurgery. 1990;27:185–92. doi: 10.1227/00006123-199008000-00002. [DOI] [PubMed] [Google Scholar]
- 152.Davis DP, Salazar A, Chan TC, Vilke GM. Prospective evaluation of a clinical decision guideline to diagnose spinal epidural abscess in patients who present to the emergency department with spine pain. J Neurosurg Spine. 2011;14:765–70. doi: 10.3171/2011.1.SPINE1091. [DOI] [PubMed] [Google Scholar]
- 153.Davis DP, Wold RM, Patel RJ, et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26:285–91. doi: 10.1016/j.jemermed.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 154.Reihsaus E, Waldbaur H, Seeling W. Spinal epidural abscess: a meta-analysis of 915 patients. Neurosurg Rev. 2000;23:175–204. doi: 10.1007/PL00011954. [DOI] [PubMed] [Google Scholar]
- 155.Obrador GT, Levenson DJ. Spinal epidural abscess in hemodialysis patients: report of three cases and review of the literature. Am J Kidney Dis. 1996;27:75–83. doi: 10.1016/S0272-6386(96)90033-5. [DOI] [PubMed] [Google Scholar]
- 156.Khan IA, Vaccaro AR, Zlotolow DA. Management of vertebral diskitis and osteomyelitis. Orthopedics. 1999;22:758–65. doi: 10.3928/0147-7447-19990801-07. [DOI] [PubMed] [Google Scholar]
- 157.Strausbaugh LJ. Vertebral osteomyelitis. How to differentiate it from other causes of back and neck pain. Postgrad Med. 1995;97(147–8):51–4. [PubMed] [Google Scholar]
- 158.Bhavan KP, Marschall J, Olsen MA, Fraser VJ, Wright NM, Warren DK. The epidemiology of hematogenous vertebral osteomyelitis: a cohort study in a tertiary care hospital. BMC Infect Dis. 2010;10:158. doi: 10.1186/1471-2334-10-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Grammatico L, Baron S, Rusch E, et al. Epidemiology of vertebral osteomyelitis (VO) in France: analysis of hospital-discharge data 2002–2003. Epidemiol Infect. 2008;136:653–60. doi: 10.1017/S0950268807008850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Digby JM, Kersley JB. Pyogenic non-tuberculous spinal infection: an analysis of thirty cases. J Bone Joint Surg Br. 1979;61:47–55. doi: 10.1302/0301-620X.61B1.370121. [DOI] [PubMed] [Google Scholar]
- 161.Zimmerli W. Clinical practice. Vertebral osteomyelitis. N Engl J Med. 2010;362:1022–9. doi: 10.1056/NEJMcp0910753. [DOI] [PubMed] [Google Scholar]
- 162.Rezai AR, Woo HH, Errico TJ, Cooper PR. Contemporary management of spinal osteomyelitis. Neurosurgery. 1999;44:1018–25. doi: 10.1097/00006123-199905000-00047. [DOI] [PubMed] [Google Scholar]
- 163.Nussbaum ES, Rockswold GL, Bergman TA, Erickson DL, Seljeskog EL. Spinal tuberculosis: a diagnostic and management challenge. J Neurosurg. 1995;83:243–7. doi: 10.3171/jns.1995.83.2.0243. [DOI] [PubMed] [Google Scholar]
- 164.Priest DH, Peacock JE., Jr Hematogenous vertebral osteomyelitis due to Staphylococcus aureus in the adult: clinical features and therapeutic outcomes. South Med J. 2005;98:854–62. doi: 10.1097/01.smj.0000168666.98129.33. [DOI] [PubMed] [Google Scholar]
- 165.Sapico FL. Microbiology and antimicrobial therapy of spinal infections. Orthop Clin North Am. 1996;27:9–13. [PubMed] [Google Scholar]
- 166.Broner FA, Garland DE, Zigler JE. Spinal infections in the immunocompromised host. Orthop Clin North Am. 1996;27:37–46. [PubMed] [Google Scholar]
- 167.Ozuna RM, Delamarter RB. Pyogenic vertebral osteomyelitis and postsurgical disc space infections. Orthop Clin North Am. 1996;27:87–94. [PubMed] [Google Scholar]
- 168.Cahill DW, Love LC, Rechtine GR. Pyogenic osteomyelitis of the spine in the elderly. J Neurosurg. 1991;74:878–86. doi: 10.3171/jns.1991.74.6.0878. [DOI] [PubMed] [Google Scholar]
- 169.Acosta FL, Jr, Chin CT, Quinones-Hinojosa A, Ames CP, Weinstein PR, Chou D. Diagnosis and management of adult pyogenic osteomyelitis of the cervical spine. Neurosurg Focus. 2004;17:E2. doi: 10.3171/foc.2004.17.6.2. [DOI] [PubMed] [Google Scholar]
- 170.Sarria JC, Chutkan NB, Figueroa JE, Hull A. Atypical mycobacterial vertebral osteomyelitis: case report and review. Clin Infect Dis. 1998;26:503–5. doi: 10.1086/517096. [DOI] [PubMed] [Google Scholar]
- 171.Klein JD, Garfin SR. Nutritional status in the patient with spinal infection. Orthop Clin North Am. 1996;27:33–6. [PubMed] [Google Scholar]
- 172.Endress C, Guyot DR, Fata J, Salciccioli G. Cervical osteomyelitis due to i.v. heroin use: radiologic findings in 14 patients. AJR Am J Roentgenol. 1990;155:333–5. doi: 10.2214/ajr.155.2.2115262. [DOI] [PubMed] [Google Scholar]
- 173.Lafont A, Olive A, Gelman M, Roca-Burniols J, Cots R, Carbonell J. Candida albicans spondylodiscitis and vertebral osteomyelitis in patients with intravenous heroin drug addiction. Report of 3 new cases. J Rheumatol. 1994;21:953–6. [PubMed] [Google Scholar]
- 174.Rahman I, Bhatt H, Chillag S, Duffus W. Mycobacterium chelonae vertebral osteomyelitis. South Med J. 2009;102:1167–9. doi: 10.1097/SMJ.0b013e3181bae784. [DOI] [PubMed] [Google Scholar]
- 175.Colmenero JD, Jimenez-Mejias ME, Sanchez-Lora FJ, et al. Pyogenic, tuberculous, and brucellar vertebral osteomyelitis: a descriptive and comparative study of 219 cases. Ann Rheum Dis. 1997;56:709–15. doi: 10.1136/ard.56.12.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Livorsi DJ, Daver NG, Atmar RL, Shelburne SA, White AC, Jr, Musher DM. Outcomes of treatment for hematogenous Staphylococcus aureus vertebral osteomyelitis in the MRSA ERA. J Infect. 2008;57:128–31. doi: 10.1016/j.jinf.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 177.Lora-Tamayo J, Euba G, Narvaez JA, et al. Changing trends in the epidemiology of pyogenic vertebral osteomyelitis: the impact of cases with no microbiologic diagnosis. Semin Arthritis Rheum. 2011;41:247–55. doi: 10.1016/j.semarthrit.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 178.Mylona E, Samarkos M, Kakalou E, Fanourgiakis P, Skoutelis A. Pyogenic vertebral osteomyelitis: a systematic review of clinical characteristics. Semin Arthritis Rheum. 2009;39:10–7. doi: 10.1016/j.semarthrit.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 179.Nolla JM, Ariza J, Gomez-Vaquero C, et al. Spontaneous pyogenic vertebral osteomyelitis in nondrug users. Semin Arthritis Rheum. 2002;31:271–8. doi: 10.1053/sarh.2002.29492. [DOI] [PubMed] [Google Scholar]
- 180.Osenbach RK, Hitchon PW, Menezes AH. Diagnosis and management of pyogenic vertebral osteomyelitis in adults. Surg Neurol. 1990;33:266–75. doi: 10.1016/0090-3019(90)90047-S. [DOI] [PubMed] [Google Scholar]
- 181.Patzakis MJ, Rao S, Wilkins J, Moore TM, Harvey PJ. Analysis of 61 cases of vertebral osteomyelitis. Clin Orthop Relat Res. 1991;(264):178–83. [PubMed]
- 182.Mete B, Kurt C, Yilmaz MH, et al. Vertebral osteomyelitis: eight years’ experience of 100 cases. Rheumatol Int. 2012;32(11):3591–7. doi: 10.1007/s00296-011-2233-z. [DOI] [PubMed] [Google Scholar]
- 183.Barnes B, Alexander JT, Branch CL., Jr Cervical osteomyelitis: a brief review. Neurosurg Focus. 2004;17:E11. doi: 10.3171/foc.2004.17.6.11. [DOI] [PubMed] [Google Scholar]
- 184.Beluffi G, Bernardo ME, Meloni G, Spinazzola A, Locatelli F. Spinal osteomyelitis due to Aspergillus flavus in a child: a rare complication after haematopoietic stem cell transplantation. Pediatr Radiol. 2008;38:709–12. doi: 10.1007/s00247-008-0789-x. [DOI] [PubMed] [Google Scholar]
- 185.Chang HM, Yu HH, Yang YH, et al. Successful treatment of Aspergillus flavus spondylodiscitis with epidural abscess in a patient with chronic granulomatous disease. Pediatr Infect Dis J. 2012;31:100–1. doi: 10.1097/INF.0b013e3182309ec0. [DOI] [PubMed] [Google Scholar]
- 186.Ranjan R, Mishra S, Ranjan S. Aspergillus vertebral osteomyelitis in an immunocompetent person. Neurol India. 2010;58:806–8. doi: 10.4103/0028-3886.72196. [DOI] [PubMed] [Google Scholar]
- 187.Sethi S, Siraj F, Kalra K, Chopra P. Aspergillus vertebral osteomyelitis in immunocompetent patients. Ind J Orthop. 2012;46:246–50. doi: 10.4103/0019-5413.93693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Studemeister A, Stevens DA. Aspergillus vertebral osteomyelitis in immunocompetent hosts: role of triazole antifungal therapy. Clin Infect Dis. 2011;52:e1–6. doi: 10.1093/cid/ciq039. [DOI] [PubMed] [Google Scholar]
- 189.Tew CW, Han FC, Jureen R, Tey BH. Aspergillus vertebral osteomyelitis and epidural abscess. Singapore Med J. 2009;50:e151–4. [PubMed] [Google Scholar]
- 190.Zhu LP, Chen XS, Wu JQ, Yang FF, Weng XH. Aspergillus vertebral osteomyelitis and ureteral obstruction after liver transplantation. Transpl Infect Dis. 2011;13:192–9. doi: 10.1111/j.1399-3062.2011.00599.x. [DOI] [PubMed] [Google Scholar]
- 191.An HS, Seldomridge JA. Spinal infections: diagnostic tests and imaging studies. Clin Orthop Relat Res. 2006;444:27–33. doi: 10.1097/01.blo.0000203452.36522.97. [DOI] [PubMed] [Google Scholar]
- 192.Rath SA, Neff U, Schneider O, Richter HP. Neurosurgical management of thoracic and lumbar vertebral osteomyelitis and discitis in adults: a review of 43 consecutive surgically treated patients. Neurosurgery. 1996;38:926–33. doi: 10.1097/00006123-199605000-00013. [DOI] [PubMed] [Google Scholar]
- 193.Turpin S, Lambert R. Role of scintigraphy in musculoskeletal and spinal infections. Radiol Clin North Am. 2001;39:169–89. doi: 10.1016/S0033-8389(05)70271-2. [DOI] [PubMed] [Google Scholar]
- 194.Palestro CJ, Love C, Miller TT. Diagnostic imaging tests and microbial infections. Cell Microbiol. 2007;9:2323–33. doi: 10.1111/j.1462-5822.2007.01013.x. [DOI] [PubMed] [Google Scholar]
- 195.Palestro CJ, Love C, Miller TT. Infection and musculoskeletal conditions: Imaging of musculoskeletal infections. Best Pract Res Clin Rheumatol. 2006;20:1197–218. doi: 10.1016/j.berh.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 196.Vaccaro AR, Shah SH, Schweitzer ME, Rosenfeld JF, Cotler JM. MRI description of vertebral osteomyelitis, neoplasm, and compression fracture. Orthopedics. 1999;22:67–73. doi: 10.3928/0147-7447-19990101-09. [DOI] [PubMed] [Google Scholar]
- 197.Roblot F, Besnier JM, Juhel L, et al. Optimal duration of antibiotic therapy in vertebral osteomyelitis. Semin Arthritis Rheum. 2007;36:269–77. doi: 10.1016/j.semarthrit.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 198.Hadjipavlou AG, Mader JT, Necessary JT, Muffoletto AJ. Hematogenous pyogenic spinal infections and their surgical management. Spine. 2000;25:1668–79. doi: 10.1097/00007632-200007010-00010. [DOI] [PubMed] [Google Scholar]
- 199.Perronne C, Saba J, Behloul Z, et al. Pyogenic and tuberculous spondylodiskitis (vertebral osteomyelitis) in 80 adult patients. Clin Infect Dis. 1994;19:746–50. doi: 10.1093/clinids/19.4.746. [DOI] [PubMed] [Google Scholar]
- 200.Dey M, Jaffe J, Stadnik A, Awad IA. External ventricular drainage for intraventricular hemorrhage. Curr Neurol Neurosci Rep. 2012;12:24–33. doi: 10.1007/s11910-011-0231-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li LM, Timofeev I, Czosnyka M, Hutchinson PJ. Review article: the surgical approach to the management of increased intracranial pressure after traumatic brain injury. Anesth Analg. 2010;111:736–48. doi: 10.1213/ANE.0b013e3181e75cd1. [DOI] [PubMed] [Google Scholar]
- 202.Gigante P, Hwang BY, Appelboom G, Kellner CP, Kellner MA, Connolly ES. External ventricular drainage following aneurysmal subarachnoid haemorrhage. Br J Neurosurg. 2010;24:625–32. doi: 10.3109/02688697.2010.505989. [DOI] [PubMed] [Google Scholar]
- 203.The Brain Trauma Foundation. The American Association of Neurological Surgeons Recommendations for intracranial pressure monitoring technology. J Neurotrauma. 2000;17:497–506. doi: 10.1089/neu.2000.17.497. [DOI] [PubMed] [Google Scholar]
- 204.Gutierrez-Gonzalez R, Boto GR, Perez-Zamarron A. Cerebrospinal fluid diversion devices and infection. A comprehensive review. Eur J Clin Microbiol Infect Dis. 2012;31(6):889–97. doi: 10.1007/s10096-011-1420-x. [DOI] [PubMed] [Google Scholar]
- 205.Ngo QN, Ranger A, Singh RN, Kornecki A, Seabrook JA, Fraser DD. External ventricular drains in pediatric patients. Pediatr Crit Care Med. 2009;10:346–51. doi: 10.1097/PCC.0b013e3181a320cd. [DOI] [PubMed] [Google Scholar]
- 206.Speck V, Staykov D, Huttner HB, Sauer R, Schwab S, Bardutzky J. Lumbar catheter for monitoring of intracranial pressure in patients with post-hemorrhagic communicating hydrocephalus. Neurocrit Care. 2011;14:208–15. doi: 10.1007/s12028-010-9459-6. [DOI] [PubMed] [Google Scholar]
- 207.Zhong J, Dujovny M, Park HK, Perez E, Perlin AR, Diaz FG. Advances in ICP monitoring techniques. Neurol Res. 2003;25:339–50. doi: 10.1179/016164103101201661. [DOI] [PubMed] [Google Scholar]
- 208.Ghajar J. Intracranial pressure monitoring techniques. New Horiz. 1995;3:395–9. [PubMed] [Google Scholar]
- 209.Dasic D, Hanna SJ, Bojanic S, Kerr RS. External ventricular drain infection: the effect of a strict protocol on infection rates and a review of the literature. Br J Neurosurg. 2006;20:296–300. doi: 10.1080/02688690600999901. [DOI] [PubMed] [Google Scholar]
- 210.Hoefnagel D, Dammers R, Ter Laak-Poort MP, Avezaat CJ. Risk factors for infections related to external ventricular drainage. Acta Neurochir. 2008;150:209–14. doi: 10.1007/s00701-007-1458-9. [DOI] [PubMed] [Google Scholar]
- 211.Kitchen WJ, Singh N, Hulme S, Galea J, Patel HC, King AT. External ventricular drain infection: improved technique can reduce infection rates. Br J Neurosurg. 2011;25:632–5. doi: 10.3109/02688697.2011.578770. [DOI] [PubMed] [Google Scholar]
- 212.Kourbeti IS, Jacobs AV, Koslow M, Karabetsos D, Holzman RS. Risk factors associated with postcraniotomy meningitis. Neurosurgery. 2007;60:317–25. doi: 10.1227/01.NEU.0000249266.26322.25. [DOI] [PubMed] [Google Scholar]
- 213.O’Brien D, Stevens NT, Lim CH, et al. Candida infection of the central nervous system following neurosurgery: a 12-year review. Acta Neurochir. 2011;153:1347–50. doi: 10.1007/s00701-011-0990-9. [DOI] [PubMed] [Google Scholar]
- 214.Schade RP, Schinkel J, Visser LG, Van Dijk JM, Voormolen JH, Kuijper EJ. Bacterial meningitis caused by the use of ventricular or lumbar cerebrospinal fluid catheters. J Neurosurg. 2005;102:229–34. doi: 10.3171/jns.2005.102.2.0229. [DOI] [PubMed] [Google Scholar]
- 215.von der Brelie C, Simon A, Groner A, Molitor E, Simon M. Evaluation of an institutional guideline for the treatment of cerebrospinal fluid shunt-associated infections. Acta Neurochir (Wien) 2012;154(9):1691–7. doi: 10.1007/s00701-012-1329-x. [DOI] [PubMed] [Google Scholar]
- 216.Korinek AM, Reina M, Boch AL, Rivera AO, De Bels D, Puybasset L. Prevention of external ventricular drain – related ventriculitis. Acta Neurochir. 2005;147:39–45. doi: 10.1007/s00701-004-0416-z. [DOI] [PubMed] [Google Scholar]
- 217.Lo CH, Spelman D, Bailey M, Cooper DJ, Rosenfeld JV, Brecknell JE. External ventricular drain infections are independent of drain duration: an argument against elective revision. J Neurosurg. 2007;106:378–83. doi: 10.3171/jns.2007.106.3.378. [DOI] [PubMed] [Google Scholar]
- 218.Mayhall CG, Archer NH, Lamb VA, et al. Ventriculostomy-related infections. A prospective epidemiologic study. N Engl J Med. 1984;310:553–9. doi: 10.1056/NEJM198403013100903. [DOI] [PubMed] [Google Scholar]
- 219.Winfield JA, Rosenthal P, Kanter RK, Casella G. Duration of intracranial pressure monitoring does not predict daily risk of infectious complications. Neurosurgery. 1993;33:424–30. doi: 10.1227/00006123-199309000-00011. [DOI] [PubMed] [Google Scholar]
- 220.Beer R, Pfausler B, Schmutzhard E. Management of nosocomial external ventricular drain-related ventriculomeningitis. Neurocrit Care. 2009;10:363–7. doi: 10.1007/s12028-008-9155-y. [DOI] [PubMed] [Google Scholar]
- 221.Pfausler B, Beer R, Engelhardt K, Kemmler G, Mohsenipour I, Schmutzhard E. Cell index – a new parameter for the early diagnosis of ventriculostomy (external ventricular drainage)-related ventriculitis in patients with intraventricular hemorrhage? Acta Neurochir. 2004;146:477–81. doi: 10.1007/s00701-004-0258-8. [DOI] [PubMed] [Google Scholar]
- 222.Kim JH, Desai NS, Ricci J, et al. Factors contributing to ventriculostomy infection. World Neurosurg. 2012;77:135–40. doi: 10.1016/j.wneu.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 223.Chi H, Chang KY, Chang HC, Chiu NC, Huang FY. Infections associated with indwelling ventriculostomy catheters in a teaching hospital. Int J Infect Dis. 2010;14:e216–9. doi: 10.1016/j.ijid.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 224.Stenehjem E, Armstrong WS. Central nervous system device infections. Infect Dis Clin North Am. 2012;26:89–110. doi: 10.1016/j.idc.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 225.Alleyne CH, Jr, Hassan M, Zabramski JM. The efficacy and cost of prophylactic and perioprocedural antibiotics in patients with external ventricular drains. Neurosurgery. 2000;47:1124–7. doi: 10.1097/00006123-200011000-00020. [DOI] [PubMed] [Google Scholar]
- 226.Ratilal B, Costa J, Sampaio C. Antibiotic prophylaxis for surgical introduction of intracranial ventricular shunts. Cochrane Database Syst Rev. 2006;(3):CD005365. [DOI] [PMC free article] [PubMed]
- 227.Munch TN, Juhler M. Antibiotic prophylaxis in insertion of intracranial ventricular shunts. A survey of a Cochrane review. Ugeskr Laeger. 2008;170:131–5. [PubMed] [Google Scholar]
- 228.Ratilal B, Sampaio C. Prophylactic antibiotics and anticonvulsants in neurosurgery. Adv Tech Stand Neurosurg. 2011;36:139–85. doi: 10.1007/978-3-7091-0179-7_6. [DOI] [PubMed] [Google Scholar]
- 229.Ratilal B, Costa J, Sampaio C. Antibiotic prophylaxis for surgical introduction of intracranial ventricular shunts: a systematic review. J Neurosurg Pediatr. 2008;1:48–56. doi: 10.3171/PED-08/01/048. [DOI] [PubMed] [Google Scholar]
- 230.Muttaiyah S, Ritchie S, John S, Mee E, Roberts S. Efficacy of antibiotic-impregnated external ventricular drain catheters. J Clin Neurosci. 2010;17:296–8. doi: 10.1016/j.jocn.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 231.Abla AA, Zabramski JM, Jahnke HK, Fusco D, Nakaji P. Comparison of two antibiotic-impregnated ventricular catheters: a prospective sequential series trial. Neurosurgery. 2011;68:437–42. doi: 10.1227/NEU.0b013e3182039a14. [DOI] [PubMed] [Google Scholar]
- 232.Eymann R, Chehab S, Strowitzki M, Steudel WI, Kiefer M. Clinical and economic consequences of antibiotic-impregnated cerebrospinal fluid shunt catheters. J Neurosurg Pediatr. 2008;1:444–50. doi: 10.3171/PED/2008/1/6/444. [DOI] [PubMed] [Google Scholar]
- 233.Gutierrez-Gonzalez R, Boto GR. Do antibiotic-impregnated catheters prevent infection in CSF diversion procedures? Review of the literature. J Infect. 2010;61:9–20. doi: 10.1016/j.jinf.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 234.Lemcke J, Depner F, Meier U. The impact of silver nanoparticle-coated and antibiotic-impregnated external ventricular drainage catheters on the risk of infections: a clinical comparison of 95 patients. Acta Neurochir Suppl. 2012;114:347–50. doi: 10.1007/978-3-7091-0956-4_67. [DOI] [PubMed] [Google Scholar]
- 235.Soleman J, Marbacher S, Fandino J, Fathi AR. Is the use of antibiotic-impregnated external ventricular drainage beneficial in the management of iatrogenic ventriculitis? Acta Neurochir. 2012;154:161–4. doi: 10.1007/s00701-011-1156-5. [DOI] [PubMed] [Google Scholar]
- 236.Zingale A, Ippolito S, Pappalardo P, Chibbaro S, Amoroso R. Infections and re-infections in long-term external ventricular drainage. A variation upon a theme. J Neurosurg Sci. 1999;43:125–32. [PubMed] [Google Scholar]
- 237.Babu MA, Patel R, Marsh WR, Wijdicks EF. Strategies to decrease the risk of ventricular catheter infections: a review of the evidence. Neurocrit Care. 2012;16:194–202. doi: 10.1007/s12028-011-9647-z. [DOI] [PubMed] [Google Scholar]
- 238.Fichtner J, Guresir E, Seifert V, Raabe A. Efficacy of silver-bearing external ventricular drainage catheters: a retrospective analysis. J Neurosurg. 2010;112:840–6. doi: 10.3171/2009.8.JNS091297. [DOI] [PubMed] [Google Scholar]
- 239.McCarthy PJ, Patil S, Conrad SA, Scott LK. International and specialty trends in the use of prophylactic antibiotics to prevent infectious complications after insertion of external ventricular drainage devices. Neurocrit Care. 2010;12:220–4. doi: 10.1007/s12028-009-9284-y. [DOI] [PubMed] [Google Scholar]
- 240.Razmkon A, Bakhtazad A. Maintaining CSF drainage at external ventricular drains may help prevent catheter-related infections. Acta Neurochir. 2009;151:985. doi: 10.1007/s00701-009-0292-7. [DOI] [PubMed] [Google Scholar]
- 241.Rivero-Garvia M, Marquez-Rivas J, Jimenez-Mejias ME, Neth O, Rueda-Torres AB. Reduction in external ventricular drain infection rate. Impact of a minimal handling protocol and antibiotic-impregnated catheters. Acta Neurochir. 2011;153:647–51. doi: 10.1007/s00701-010-0905-1. [DOI] [PubMed] [Google Scholar]
- 242.Cummings R. Understanding external ventricular drainage. J Neurosci Nurs. 1992;24:84–7. doi: 10.1097/01376517-199204000-00006. [DOI] [PubMed] [Google Scholar]
- 243.Narayan RK, Kishore PR, Becker DP, et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56:650–9. doi: 10.3171/jns.1982.56.5.0650. [DOI] [PubMed] [Google Scholar]
- 244.Tamburrini G, Massimi L, Caldarelli M, Di Rocco C. Antibiotic impregnated external ventricular drainage and third ventriculostomy in the management of hydrocephalus associated with posterior cranial fossa tumours. Acta Neurochir. 2008;150:1049–55. doi: 10.1007/s00701-008-0022-6. [DOI] [PubMed] [Google Scholar]
- 245.Lopez-Alvarez B, Martin-Laez R, Farinas MC, Paternina-Vidal B, Garcia-Palomo JD, Vazquez-Barquero A. Multidrug-resistant Acinetobacter baumannii ventriculitis: successful treatment with intraventricular colistin. Acta Neurochir. 2009;151:1465–72. doi: 10.1007/s00701-009-0382-6. [DOI] [PubMed] [Google Scholar]
- 246.Nerlich AG, Haas CJ, Zink A, Szeimies U, Hagedorn HG. Molecular evidence for tuberculosis in an ancient Egyptian mummy. Lancet. 1997;350:1404. doi: 10.1016/S0140-6736(05)65185-9. [DOI] [PubMed] [Google Scholar]
- 247.Salo WL, Aufderheide AC, Buikstra J, Holcomb TA. Identification of Mycobacterium tuberculosis DNA in a pre-Columbian Peruvian mummy. Proc Natl Acad Sci U S A. 1994;91:2091–4. doi: 10.1073/pnas.91.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Organization WH. WHO Report 2011: Global Tuberculosis Control. Geneva: World Health Organization; 2011. [Google Scholar]
- 249.Archibald LK, den Dulk MO, Pallangyo KJ, Reller LB. Fatal Mycobacterium tuberculosis bloodstream infections in febrile hospitalized adults in Dar es Salaam, Tanzania. Clin Infect Dis. 1998;26:290–6. doi: 10.1086/516297. [DOI] [PubMed] [Google Scholar]
- 250.Archibald LK, McDonald LC, Nwanyanwu O, et al. A hospital-based prevalence survey of bloodstream infections in febrile patients in Malawi: implications for diagnosis and therapy. J Infect Dis. 2000;181:1414–20. doi: 10.1086/315367. [DOI] [PubMed] [Google Scholar]
- 251.Archibald LK, McDonald LC, Rheanpumikankit S, et al. Fever and human immunodeficiency virus infection as sentinels for emerging mycobacterial and fungal bloodstream infections in hospitalized patients greater than /=15 years old, Bangkok. J Infect Dis. 1999;180:87–92. doi: 10.1086/314836. [DOI] [PubMed] [Google Scholar]
- 252.Bell M, Archibald LK, Nwanyanwu O, et al. Seasonal variation in the etiology of bloodstream infections in a febrile inpatient population in a developing country. Int J Infect Dis. 2001;5:63–9. doi: 10.1016/S1201-9712(01)90027-X. [DOI] [PubMed] [Google Scholar]
- 253.McDonald LC, Archibald LK, Rheanpumikankit S, et al. Unrecognised Mycobacterium tuberculosis bacteraemia among hospital inpatients in less developed countries. Lancet. 1999;354:1159–63. doi: 10.1016/S0140-6736(98)12325-5. [DOI] [PubMed] [Google Scholar]
- 254.CDC Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs – worldwide, 2000–2004. MMWR Morb Mortal Wkly Rep. 2006;55:301–5. [PubMed] [Google Scholar]
- 255.CDC Two simultaneous outbreaks of multidrug-resistant tuberculosis – Federated States of Micronesia, 2007–2009. MMWR Morb Mortal Wkly Rep. 2009;58:253–6. [PubMed] [Google Scholar]
- 256.Cegielski JP. Extensively drug-resistant tuberculosis: “there must be some kind of way out of here”. Clin Infect Dis. 2010;50(Suppl 3):S195–200. doi: 10.1086/651491. [DOI] [PubMed] [Google Scholar]
- 257.Shah NS, Wright A, Bai GH, et al. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg Infect Dis. 2007;13:380–7. doi: 10.3201/eid1303.061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kent SJ, Crowe SM, Yung A, Lucas CR, Mijch AM. Tuberculous meningitis: a 30-year review. Clin Infect Dis. 1993;17:987–94. doi: 10.1093/clinids/17.6.987. [DOI] [PubMed] [Google Scholar]
- 259.Thwaites GE, Tran TH. Tuberculous meningitis: many questions, too few answers. Lancet Neurol. 2005;4:160–70. doi: 10.1016/S1474-4422(05)01013-6. [DOI] [PubMed] [Google Scholar]
- 260.Thwaites G, Fisher M, Hemingway C, Scott G, Solomon T, Innes J. British Infection Society guidelines for the diagnosis and treatment of tuberculosis of the central nervous system in adults and children. J Infect. 2009;59:167–87. doi: 10.1016/j.jinf.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 261.Joos TJ, Miller WC, Murdoch DM. Tuberculin reactivity in bacille Calmette-Guerin vaccinated populations: a compilation of international data. Int J Tuberc Lung Dis. 2006;10:883–91. [PubMed] [Google Scholar]
- 262.Kilpatrick ME, Girgis NI, Tribble D, Farid Z. The value of the tuberculin skin test in patients with tuberculous meningitis. J Egypt Public Health Assoc. 1996;71:1–8. [PubMed] [Google Scholar]
- 263.Thwaites GE, Schoeman JF. Update on tuberculosis of the central nervous system: pathogenesis, diagnosis, and treatment. Clin Chest Med. 2009;30:745–54. doi: 10.1016/j.ccm.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 264.Ferrara G, Losi M, D’Amico R, et al. Use in routine clinical practice of two commercial blood tests for diagnosis of infection with Mycobacterium tuberculosis: a prospective study. Lancet. 2006;367:1328–34. doi: 10.1016/S0140-6736(06)68579-6. [DOI] [PubMed] [Google Scholar]
- 265.Ferrara G, Losi M, Meacci M, et al. Routine hospital use of a new commercial whole blood interferon-gamma assay for the diagnosis of tuberculosis infection. Am J Respir Crit Care Med. 2005;172:631–5. doi: 10.1164/rccm.200502-196OC. [DOI] [PubMed] [Google Scholar]
- 266.Foerster BR, Thurnher MM, Malani PN, Petrou M, Carets-Zumelzu F, Sundgren PC. Intracranial infections: clinical and imaging characteristics. Acta Radiol. 2007;48:875–93. doi: 10.1080/02841850701477728. [DOI] [PubMed] [Google Scholar]
- 267.Trivedi R, Saksena S, Gupta RK. Magnetic resonance imaging in central nervous system tuberculosis. Indian J Radiol Imaging. 2009;19:256–65. doi: 10.4103/0971-3026.57205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Parsons LM, Somoskovi A, Gutierrez C, et al. Laboratory diagnosis of tuberculosis in resource-poor countries: challenges and opportunities. Clin Microbiol Rev. 2011;24:314–50. doi: 10.1128/CMR.00059-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Murthy JM. Tuberculous meningitis: the challenges. Neurol India. 2010;58:716–22. doi: 10.4103/0028-3886.72178. [DOI] [PubMed] [Google Scholar]
- 270.Nolan CM, Goldberg SV, Buskin SE. Hepatotoxicity associated with isoniazid preventive therapy: a 7-year survey from a public health tuberculosis clinic. JAMA. 1999;281:1014–8. doi: 10.1001/jama.281.11.1014. [DOI] [PubMed] [Google Scholar]
- 271.Thwaites GE, Nguyen DB, Nguyen HD, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741–51. doi: 10.1056/NEJMoa040573. [DOI] [PubMed] [Google Scholar]
- 272.Berning SE, Cherry TA, Iseman MD. Novel treatment of meningitis caused by multidrug-resistant Mycobacterium tuberculosis with intrathecal levofloxacin and amikacin: case report. Clin Infect Dis. 2001;32:643–6. doi: 10.1086/318698. [DOI] [PubMed] [Google Scholar]
- 273.Garg RK, Sinha MK. Tuberculous meningitis in patients infected with human immunodeficiency virus. J Neurol. 2011;258:3–13. doi: 10.1007/s00415-010-5744-8. [DOI] [PubMed] [Google Scholar]
- 274.Afghani B, Lieberman JM. Paradoxical enlargement or development of intracranial tuberculomas during therapy: case report and review. Clin Infect Dis. 1994;19:1092–9. doi: 10.1093/clinids/19.6.1092. [DOI] [PubMed] [Google Scholar]
- 275.Galimi R. Extrapulmonary tuberculosis: tuberculous meningitis new developments. Eur Rev Med Pharmacol Sci. 2011;15:365–86. [PubMed] [Google Scholar]
- 276.Hall WA, Truwit CL. The surgical management of infections involving the cerebrum. Neurosurgery. 2008;62(Suppl 2):519–30. doi: 10.1227/01.neu.0000316255.36726.5b. [DOI] [PubMed] [Google Scholar]
- 277.Garg RK, Somvanshi DS. Spinal tuberculosis: a review. J Spinal Cord Med. 2011;34:440–54. doi: 10.1179/2045772311Y.0000000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Adekambi T, Foucault C, La Scola B, Drancourt M. Report of two fatal cases of Mycobacterium mucogenicum central nervous system infection in immunocompetent patients. J Clin Microbiol. 2006;44:837–40. doi: 10.1128/JCM.44.3.837-840.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Madaras-Kelly KJ, DeMasters TA, Stevens DL. Mycobacterium fortuitum meningitis associated with an epidural catheter: case report and a review of the literature. Pharmacotherapy. 1999;19:661–6. doi: 10.1592/phco.19.8.661.31530. [DOI] [PubMed] [Google Scholar]
- 280.Midani S, Rathore MH. Mycobacterium fortuitum infection of ventriculoperitoneal shunt. South Med J. 1999;92:705–7. doi: 10.1097/00007611-199907000-00009. [DOI] [PubMed] [Google Scholar]
- 281.Talati NJ, Rouphael N, Kuppalli K, Franco-Paredes C. Spectrum of CNS disease caused by rapidly growing mycobacteria. Lancet Infect Dis. 2008;8:390–8. doi: 10.1016/S1473-3099(08)70127-0. [DOI] [PubMed] [Google Scholar]
- 282.Bradsher RW. Histoplasmosis and blastomycosis. Clin Infect Dis. 1996;22(Suppl 2):S102–11. doi: 10.1093/clinids/22.Supplement_2.S102. [DOI] [PubMed] [Google Scholar]
- 283.Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20:115–32. doi: 10.1128/CMR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system. A clinical review. Medicine. 1990;69:244–60. doi: 10.1097/00005792-199007000-00006. [DOI] [PubMed] [Google Scholar]
- 285.Gottfredsson M, Perfect JR. Fungal meningitis. Semin Neurol. 2000;20:307–22. doi: 10.1055/s-2000-9394. [DOI] [PubMed] [Google Scholar]
- 286.Chakrabarti A. Epidemiology of central nervous system mycoses. Neurol India. 2007;55:191–7. doi: 10.4103/0028-3886.35679. [DOI] [PubMed] [Google Scholar]
- 287.Williams PL. Coccidioidal meningitis. Ann N Y Acad Sci. 2007;1111:377–84. doi: 10.1196/annals.1406.037. [DOI] [PubMed] [Google Scholar]
- 288.Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42:103–7. doi: 10.1086/497596. [DOI] [PubMed] [Google Scholar]
- 289.Mathisen G, Shelub A, Truong J, Wigen C. Coccidioidal meningitis: clinical presentation and management in the fluconazole era. Medicine. 2010;89:251–84. doi: 10.1097/MD.0b013e3181f378a8. [DOI] [PubMed] [Google Scholar]
- 290.Bouza E, Dreyer JS, Hewitt WL, Meyer RD. Coccidioidal meningitis. An analysis of thirty-one cases and review of the literature. Medicine. 1981;60:139–72. doi: 10.1097/00005792-198105000-00001. [DOI] [PubMed] [Google Scholar]
- 291.Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis. 1993;16:247–54. doi: 10.1093/clind/16.2.247. [DOI] [PubMed] [Google Scholar]
- 292.Ragland AS, Arsura E, Ismail Y, Johnson R. Eosinophilic pleocytosis in coccidioidal meningitis: frequency and significance. Am J Med. 1993;95:254–7. doi: 10.1016/0002-9343(93)90276-U. [DOI] [PubMed] [Google Scholar]
- 293.Capilla J, Clemons KV, Sobel RA, Stevens DA. Efficacy of amphotericin B lipid complex in a rabbit model of coccidioidal meningitis. J Antimicrob Chemother. 2007;60:673–6. doi: 10.1093/jac/dkm264. [DOI] [PubMed] [Google Scholar]
- 294.Clemons KV, Capilla J, Sobel RA, Martinez M, Tong AJ, Stevens DA. Comparative efficacies of lipid-complexed amphotericin B and liposomal amphotericin B against coccidioidal meningitis in rabbits. Antimicrob Agents Chemother. 2009;53:1858–62. doi: 10.1128/AAC.01538-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801–12. doi: 10.1086/588300. [DOI] [PubMed] [Google Scholar]
- 296.Bariola JR, Perry P, Pappas PG, et al. Blastomycosis of the central nervous system: a multicenter review of diagnosis and treatment in the modern era. Clin Infect Dis. 2010;50:797–804. doi: 10.1086/650579. [DOI] [PubMed] [Google Scholar]
- 297.Borgia SM, Fuller JD, Sarabia A, El-Helou P. Cerebral blastomycosis: a case series incorporating voriconazole in the treatment regimen. Med Mycol. 2006;44:659–64. doi: 10.1080/13693780600803870. [DOI] [PubMed] [Google Scholar]
- 298.Chowfin A, Tight R, Mitchell S. Recurrent blastomycosis of the central nervous system: case report and review. Clin Infect Dis. 2000;30:969–71. doi: 10.1086/313828. [DOI] [PubMed] [Google Scholar]
- 299.Gonyea EF. The spectrum of primary blastomycotic meningitis: a review of central nervous system blastomycosis. Ann Neurol. 1978;3:26–39. doi: 10.1002/ana.410030106. [DOI] [PubMed] [Google Scholar]
- 300.Bakleh M, Aksamit AJ, Tleyjeh IM, Marshall WF. Successful treatment of cerebral blastomycosis with voriconazole. Clin Infect Dis. 2005;40:e69–71. doi: 10.1086/429319. [DOI] [PubMed] [Google Scholar]
- 301.Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847–53. doi: 10.7326/0003-4819-116-10-847. [DOI] [PubMed] [Google Scholar]
- 302.Chander B, Deb P, Sarkar C, Garg A, Mehta VS, Sharma MC. Cerebral blastomycosis: a case report. Indian J Pathol Microbiol. 2007;50:821–4. [PubMed] [Google Scholar]
- 303.Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21–40. doi: 10.1016/S0891-5520(02)00038-7. [DOI] [PubMed] [Google Scholar]
- 304.Pappas PG. Blastomycosis. Semin Respir Crit Care Med. 2004;25:113–21. doi: 10.1055/s-2004-824896. [DOI] [PubMed] [Google Scholar]
- 305.Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367–81. doi: 10.1128/CMR.00056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Durkin M, Witt J, Lemonte A, Wheat B, Connolly P. Antigen assay with the potential to aid in diagnosis of blastomycosis. J Clin Microbiol. 2004;42:4873–5. doi: 10.1128/JCM.42.10.4873-4875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Smith JA, Kauffman CA. Blastomycosis. Proc Am Thorac Soc. 2010;7:173–80. doi: 10.1513/pats.200906-040AL. [DOI] [PubMed] [Google Scholar]
- 308.Martynowicz MA, Prakash UB. Pulmonary blastomycosis: an appraisal of diagnostic techniques. Chest. 2002;121:768–73. doi: 10.1378/chest.121.3.768. [DOI] [PubMed] [Google Scholar]
- 309.Casado JL, Quereda C, Corral I. Candidal meningitis in HIV-infected patients. AIDS Patient Care STDS. 1998;12:681–6. doi: 10.1089/apc.1998.12.681. [DOI] [PubMed] [Google Scholar]
- 310.Goldani LZ, Santos RP. Candida tropicalis as an emerging pathogen in Candida meningitis: case report and review. Braz J Infect Dis. 2010;14:631–3. [PubMed] [Google Scholar]
- 311.Montero A, Romero J, Vargas JA, et al. Candida infection of cerebrospinal fluid shunt devices: report of two cases and review of the literature. Acta Neurochir. 2000;142:67–74. doi: 10.1007/s007010050009. [DOI] [PubMed] [Google Scholar]
- 312.Rodriguez-Arrondo F, Aguirrebengoa K, De Arce A, et al. Candidal meningitis in HIV-infected patients: treatment with fluconazole. Scand J Infect Dis. 1998;30:417–8. doi: 10.1080/00365549850160747. [DOI] [PubMed] [Google Scholar]
- 313.Sanchez-Portocarrero J, Perez-Cecilia E, Corral O, Romero-Vivas J, Picazo JJ. The central nervous system and infection by Candida species. Diagn Microbiol Infect Dis. 2000;37:169–79. doi: 10.1016/S0732-8893(00)00140-1. [DOI] [PubMed] [Google Scholar]
- 314.Parker JC, Jr, McCloskey JJ, Lee RS. Human cerebral candidosis – a postmortem evaluation of 19 patients. Hum Pathol. 1981;12:23–8. doi: 10.1016/S0046-8177(81)80238-9. [DOI] [PubMed] [Google Scholar]
- 315.Bayer AS, Edwards JE, Jr, Seidel JS, Guze LB. Candida meningitis. Report of seven cases and review of the english literature. Medicine. 1976;55:477–86. doi: 10.1097/00005792-197611000-00004. [DOI] [PubMed] [Google Scholar]
- 316.Black JT. Cerebral candidiasis: case report of brain abscess secondary to Candida albicans, and review of literature. J Neurol Neurosurg Psychiatry. 1970;33:864–70. doi: 10.1136/jnnp.33.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Lipton SA, Hickey WF, Morris JH, Loscalzo J. Candidal infection in the central nervous system. Am J Med. 1984;76:101–8. doi: 10.1016/0002-9343(84)90757-5. [DOI] [PubMed] [Google Scholar]
- 318.Bengel D, Susa M, Schreiber H, Ludolph AC, Tumani H. Early diagnosis of rhinocerebral mucormycosis by cerebrospinal fluid analysis and determination of 16s rRNA gene sequence. Eur J Neurol. 2007;14:1067–70. doi: 10.1111/j.1468-1331.2007.01878.x. [DOI] [PubMed] [Google Scholar]
- 319.Mallis A, Mastronikolis SN, Naxakis SS, Papadas AT. Rhinocerebral mucormycosis: an update. Eur Rev Med Pharmacol Sci. 2010;14:987–92. [PubMed] [Google Scholar]
- 320.Schwartz S, Thiel E. Cerebral aspergillosis: tissue penetration is the key. Medical mycology. 2009;47(Suppl 1):S387–93. doi: 10.1080/13693780802537953. [DOI] [PubMed] [Google Scholar]
- 321.Prejean J, Song R, Hernandez A, et al. Estimated HIV incidence in the United States, 2006–2009. PLoS One. 2011;6:e17502. doi: 10.1371/journal.pone.0017502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–76. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 323.Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58:1–207. [PubMed] [Google Scholar]
- 324.Contini C. Clinical and diagnostic management of toxoplasmosis in the immunocompromised patient. Parassitologia. 2008;50:45–50. [PubMed] [Google Scholar]
- 325.Dedicoat M, Livesley N. Management of toxoplasmic encephalitis in HIV-infected adults – a review. S Afr Med J. 2008;98:31–2. [PubMed] [Google Scholar]
- 326.Beraud G, Pierre-Francois S, Foltzer A, et al. Cotrimoxazole for treatment of cerebral toxoplasmosis: an observational cohort study during 1994–2006. Am J Trop Med Hyg. 2009;80:583–7. [PubMed] [Google Scholar]
- 327.Pereira-Chioccola VL, Vidal JE, Su C. Toxoplasma gondii infection and cerebral toxoplasmosis in HIV-infected patients. Future Microbiol. 2009;4:1363–79. doi: 10.2217/fmb.09.89. [DOI] [PubMed] [Google Scholar]
- 328.Portegies P, Solod L, Cinque P, et al. Guidelines for the diagnosis and management of neurological complications of HIV infection. Eur J Neurol. 2004;11:297–304. doi: 10.1111/j.1468-1331.2004.00856.x. [DOI] [PubMed] [Google Scholar]
- 329.Luft BJ, Hafner R, Korzun AH, et al. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. Members of the ACTG 077p/ANRS 009 Study Team. N Engl J Med. 1993;329:995–1000. doi: 10.1056/NEJM199309303291403. [DOI] [PubMed] [Google Scholar]
- 330.Heald A, Flepp M, Chave JP, et al. Treatment for cerebral toxoplasmosis protects against Pneumocystis carinii pneumonia in patients with AIDS. The Swiss HIV Cohort Study. Ann Intern Med. 1991;115:760–3. doi: 10.7326/0003-4819-115-10-760. [DOI] [PubMed] [Google Scholar]
- 331.Bertschy S, Opravil M, Cavassini M, et al. Discontinuation of maintenance therapy against toxoplasma encephalitis in AIDS patients with sustained response to anti-retroviral therapy. Clin Microbiol Infect. 2006;12:666–71. doi: 10.1111/j.1469-0691.2006.01459.x. [DOI] [PubMed] [Google Scholar]
- 332.Soriano V, Dona C, Rodriguez-Rosado R, Barreiro P, Gonzalez-Lahoz J. Discontinuation of secondary prophylaxis for opportunistic infections in HIV-infected patients receiving highly active antiretroviral therapy. AIDS. 2000;14:383–6. doi: 10.1097/00002030-200003100-00011. [DOI] [PubMed] [Google Scholar]
- 333.Grassi MP, Clerici F, Perin C, et al. Microglial nodular encephalitis and ventriculoencephalitis due to cytomegalovirus infection in patients with AIDS: two distinct clinical patterns. Clin Infect Dis. 1998;27:504–8. doi: 10.1086/514682. [DOI] [PubMed] [Google Scholar]
- 334.Clifford DB, Arribas JR, Storch GA, Tourtellote W, Wippold FJ. Magnetic resonance brain imaging lacks sensitivity for AIDS associated cytomegalovirus encephalitis. J Neurovirol. 1996;2:397–403. doi: 10.3109/13550289609146905. [DOI] [PubMed] [Google Scholar]
- 335.Arribas JR, Storch GA, Clifford DB, Tselis AC. Cytomegalovirus encephalitis. Ann Intern Med. 1996;125:577–87. doi: 10.7326/0003-4819-125-7-199610010-00008. [DOI] [PubMed] [Google Scholar]
- 336.Anders HJ, Goebel FD. Neurological manifestations of cytomegalovirus infection in the acquired immunodeficiency syndrome. Int J STD AIDS. 1999;10:151–9. doi: 10.1258/0956462991913817. [DOI] [PubMed] [Google Scholar]
- 337.Cinque P, Cleator GM, Weber T, et al. Diagnosis and clinical management of neurological disorders caused by cytomegalovirus in AIDS patients. European Union Concerted Action on Virus Meningitis and Encephalitis. J Neurovirol. 1998;4:120–32. doi: 10.3109/13550289809113490. [DOI] [PubMed] [Google Scholar]
- 338.Silva CA, Oliveira AC, Vilas-Boas L, Fink MC, Pannuti CS, Vidal JE. Neurologic cytomegalovirus complications in patients with AIDS: retrospective review of 13 cases and review of the literature. Rev Inst Med Trop São Paulo. 2010;52:305–10. doi: 10.1590/s0036-46652010000600004. [DOI] [PubMed] [Google Scholar]
- 339.Eidelberg D, Sotrel A, Vogel H, Walker P, Kleefield J, Crumpacker CS., 3rd Progressive polyradiculopathy in acquired immune deficiency syndrome. Neurology. 1986;36:912–6. doi: 10.1212/WNL.36.7.912. [DOI] [PubMed] [Google Scholar]
- 340.Kurup A, Torriani FJ. Therapy and prevention of cytomegalovirus retinitis. Curr Infect Dis Rep. 2001;3:371–7. doi: 10.1007/s11908-001-0078-4. [DOI] [PubMed] [Google Scholar]
- 341.Spector SA, McKinley GF, Lalezari JP, et al. Oral ganciclovir for the prevention of cytomegalovirus disease in persons with AIDS. Roche Cooperative Oral Ganciclovir Study Group. N Engl J Med. 1996;334:1491–7. doi: 10.1056/NEJM199606063342302. [DOI] [PubMed] [Google Scholar]
- 342.Waib LF, Bonon SH, Salles AC, et al. Withdrawal of maintenance therapy for cytomegalovirus retinitis in AIDS patients exhibiting immunological response to HAART. Rev Inst Med Trop São Paulo. 2007;49:215–9. doi: 10.1590/S0036-46652007000400004. [DOI] [PubMed] [Google Scholar]
- 343.Macdonald JC, Torriani FJ, Morse LS, Karavellas MP, Reed JB, Freeman WR. Lack of reactivation of cytomegalovirus (CMV) retinitis after stopping CMV maintenance therapy in AIDS patients with sustained elevations in CD4 T cells in response to highly active antiretroviral therapy. J Infect Dis. 1998;177:1182–7. doi: 10.1086/515281. [DOI] [PubMed] [Google Scholar]
- 344.Wohl DA, Kendall MA, Owens S, et al. The safety of discontinuation of maintenance therapy for cytomegalovirus (CMV) retinitis and incidence of immune recovery uveitis following potent antiretroviral therapy. HIV Clin Trials. 2005;6:136–46. doi: 10.1310/4J65-4YX1-4ET6-E5KR. [DOI] [PubMed] [Google Scholar]
- 345.Kaul M. HIV-1 associated dementia: update on pathological mechanisms and therapeutic approaches. Curr Opin Neurol. 2009;22:315–20. doi: 10.1097/WCO.0b013e328329cf3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Yadav A, Collman RG. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol. 2009;4:430–47. doi: 10.1007/s11481-009-9174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–6. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 348.Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci U S A. 1993;90:2769–73. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Krathwohl MD, Kaiser JL. HIV-1 promotes quiescence in human neural progenitor cells. J Infect Dis. 2004;190:216–26. doi: 10.1086/422008. [DOI] [PubMed] [Google Scholar]
- 350.Okamoto S, Kang YJ, Brechtel CW, et al. HIV/gp120 decreases adult neural progenitor cell proliferation via checkpoint kinase-mediated cell-cycle withdrawal and G1 arrest. Cell Stem Cell. 2007;1:230–6. doi: 10.1016/j.stem.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 351.Nogales-Gaete J, Syndulko K, Tourtellotte WW. Cerebrospinal fluid (CSF) analyses in HIV-1 primary neurological disease. Ital J Neurol Sci. 1992;13:667–83. doi: 10.1007/BF02334971. [DOI] [PubMed] [Google Scholar]
- 352.Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517–24. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 353.Freitas do Rosario AP, Langhorne J. T cell-derived IL-10 and its impact on the regulation of host responses during malaria. Int J Parasitol. 2012;42(6):549–55. doi: 10.1016/j.ijpara.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 354.Garg RK. Cerebral malaria. J Assoc Physicians India. 2000;48:1004–13. [PubMed] [Google Scholar]
- 355.Higgins SJ, Kain KC, Liles WC. Immunopathogenesis of falciparum malaria: implications for adjunctive therapy in the management of severe and cerebral malaria. Expert Rev Anti Infect Ther. 2011;9:803–19. doi: 10.1586/eri.11.96. [DOI] [PubMed] [Google Scholar]
- 356.Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20:597–603. doi: 10.1016/j.pt.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 357.Medana IM, Day NP, Sachanonta N, et al. Coma in fatal adult human malaria is not caused by cerebral oedema. Malar J. 2011;10:267. doi: 10.1186/1475-2875-10-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Ponsford MJ, Medana IM, Prapansilp P, et al. Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J Infect Dis. 2012;205:663–71. doi: 10.1093/infdis/jir812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Warrell DA. Management of severe malaria. Parassitologia. 1999;41:287–94. [PubMed] [Google Scholar]
- 360.Warrell DA, Looareesuwan S, Phillips RE, et al. Function of the blood-cerebrospinal fluid barrier in human cerebral malaria: rejection of the permeability hypothesis. Am J Trop Med Hyg. 1986;35:882–9. doi: 10.4269/ajtmh.1986.35.882. [DOI] [PubMed] [Google Scholar]
- 361.Warrell DA, Looareesuwan S, Warrell MJ, et al. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313–9. doi: 10.1056/NEJM198202113060601. [DOI] [PubMed] [Google Scholar]
- 362.Warrell DA, White NJ, Warrell MJ. Dexamethasone deleterious in cerebral malaria. Br Med J (Clin Res Ed) 1982;285:1652. doi: 10.1136/bmj.285.6355.1652-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Dondorp AM, Fanello CI, Hendriksen IC, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–57. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Mohanty S, Mishra SK, Patnaik R, et al. Brain swelling and mannitol therapy in adult cerebral malaria: a randomized trial. Clin Infect Dis. 2011;53:349–55. doi: 10.1093/cid/cir405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Qvarnstrom Y, James C, Xayavong M, et al. Comparison of real-time PCR protocols for differential laboratory diagnosis of amebiasis. J Clin Microbiol. 2005;43:5491–7. doi: 10.1128/JCM.43.11.5491-5497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Qvarnstrom Y, Visvesvara GS, Sriram R, da Silva AJ. Multiplex real-time PCR assay for simultaneous detection of Acanthamoeba spp., Balamuthia mandrillaris, and Naegleria fowleri. J Clin Microbiol. 2006;44:3589–95. doi: 10.1128/JCM.00875-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Seidel JS, Harmatz P, Visvesvara GS, Cohen A, Edwards J, Turner J. Successful treatment of primary amebic meningoencephalitis. N Engl J Med. 1982;306:346–8. doi: 10.1056/NEJM198202113060607. [DOI] [PubMed] [Google Scholar]
- 368.CDC Primary amebic meningoencephalitis – Arizona, Florida, and Texas, 2007. MMWR Morb Mortal Wkly Rep. 2008;57:573–7. [PubMed] [Google Scholar]
- 369.Lopez C, Budge P, Chen J, et al. Primary amebic meningoencephalitis: a case report and literature review. Pediatr Emerg Care. 2012;28:272–6. doi: 10.1097/PEC.0b013e3182495589. [DOI] [PubMed] [Google Scholar]
- 370.Soltow SM, Brenner GM. Synergistic activities of azithromycin and amphotericin B against Naegleria fowleri in vitro and in a mouse model of primary amebic meningoencephalitis. Antimicrob Agents Chemother. 2007;51:23–7. doi: 10.1128/AAC.00788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Visvesvara GS. Amebic meningoencephalitides and keratitis: challenges in diagnosis and treatment. Curr Opin Infect Dis. 2010;23:590–4. doi: 10.1097/QCO.0b013e32833ed78b. [DOI] [PubMed] [Google Scholar]
- 372.Del Brutto OH. Neurocysticercosis in Western Europe: a re-emerging disease? Acta Neurol Belg. 2012;112(4):335–43. doi: 10.1007/s13760-012-0068-3. [DOI] [PubMed] [Google Scholar]
- 373.Del Brutto OH. Neurocysticercosis among international travelers to disease-endemic areas. J Travel Med. 2012;19:112–7. doi: 10.1111/j.1708-8305.2011.00592.x. [DOI] [PubMed] [Google Scholar]
- 374.Del Brutto OH. Neurocysticercosis: a review. ScientificWorldJournal. 2012;2012:159821. doi: 10.1100/2012/159821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Takayanagui OM, Jardim E. Clinical aspects of neurocysticercosis: analysis of 500 cases. Arq Neuropsiquiatr. 1983;41:50–63. doi: 10.1590/S0004-282X1983000100004. [DOI] [PubMed] [Google Scholar]
- 376.Sotelo J, Del Brutto OH. Review of neurocysticercosis. Neurosurg Focus. 2002;12:e1. doi: 10.3171/foc.2002.12.6.2. [DOI] [PubMed] [Google Scholar]
- 377.Sotelo J, Del Brutto OH. Brain cysticercosis. Arch Med Res. 2000;31:3–14. doi: 10.1016/S0188-4409(99)00073-9. [DOI] [PubMed] [Google Scholar]
- 378.Michelet L, Fleury A, Sciutto E, et al. Human neurocysticercosis: comparison of different diagnostic tests using cerebrospinal fluid. J Clin Microbiol. 2011;49:195–200. doi: 10.1128/JCM.01554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Sotelo J, del Brutto OH, Penagos P, et al. Comparison of therapeutic regimen of anticysticercal drugs for parenchymal brain cysticercosis. J Neurol. 1990;237:69–72. doi: 10.1007/BF00314663. [DOI] [PubMed] [Google Scholar]
- 380.Del Brutto OH. Albendazole therapy for subarachnoid cysticerci: clinical and neuroimaging analysis of 17 patients. J Neurol Neurosurg Psychiatry. 1997;62:659–61. doi: 10.1136/jnnp.62.6.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Takayanagui OM, Jardim E. Therapy for neurocysticercosis. Comparison between albendazole and praziquantel. Arch Neurol. 1992;49:290–4. doi: 10.1001/archneur.1992.00530270106026. [DOI] [PubMed] [Google Scholar]
- 382.Padma MV, Behari M, Misra NK, Ahuja GK. Albendazole in neurocysticercosis. Natl Med J India. 1995;8:255–8. [PubMed] [Google Scholar]
- 383.Carpio A, Santillan F, Leon P, Flores C, Hauser WA. Is the course of neurocysticercosis modified by treatment with antihelminthic agents? Arch Intern Med. 1995;155:1982–8. doi: 10.1001/archinte.1995.00430180088010. [DOI] [PubMed] [Google Scholar]
- 384.Padma MV, Behari M, Misra NK, Ahuja GK. Albendazole in single CT ring lesions in epilepsy. Neurology. 1994;44:1344–6. doi: 10.1212/WNL.44.7.1344. [DOI] [PubMed] [Google Scholar]
- 385.Riley T, White AC., Jr Management of neurocysticercosis. CNS Drugs. 2003;17:577–91. doi: 10.2165/00023210-200317080-00003. [DOI] [PubMed] [Google Scholar]
- 386.Shandera WX, White AC, Jr, Chen JC, Diaz P, Armstrong R. Neurocysticercosis in Houston, Texas. A report of 112 cases. Medicine. 1994;73:37–52. doi: 10.1097/00005792-199401000-00004. [DOI] [PubMed] [Google Scholar]
- 387.White AC, Garcia HH. Recent developments in the epidemiology, diagnosis, treatment, and prevention of neurocysticercosis. Current infectious disease reports. 1999;1:434–40. doi: 10.1007/s11908-999-0055-x. [DOI] [PubMed] [Google Scholar]
- 388.White AC., Jr Neurocysticercosis: a major cause of neurological disease worldwide. Clin Infect Dis. 1997;24:101–13. doi: 10.1093/clinids/24.2.101. [DOI] [PubMed] [Google Scholar]
- 389.Jimenez-Vazquez OH, Nagore N. Cisternal neurocysticercosis. Br J Neurosurg. 2008;22:774–5. doi: 10.1080/02688690802379142. [DOI] [PubMed] [Google Scholar]
- 390.Sotelo J. Clinical manifestations, diagnosis, and treatment of neurocysticercosis. Curr Neurol Neurosci Rep. 2011;11:529–35. doi: 10.1007/s11910-011-0226-7. [DOI] [PubMed] [Google Scholar]
- 391.Garcia HH, Evans CA, Nash TE, et al. Current consensus guidelines for treatment of neurocysticercosis. Clin Microbiol Rev. 2002;15:747–56. doi: 10.1128/CMR.15.4.747-756.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Garcia HH, Del Brutto OH, Nash TE, White AC, Jr, Tsang VC, Gilman RH. New concepts in the diagnosis and management of neurocysticercosis (Taenia solium) Am J Trop Med Hyg. 2005;72:3–9. [PubMed] [Google Scholar]
- 393.Sommer C, Ringelstein EB, Biniek R, Glockner WM. Adult Toxocara canis encephalitis. J Neurol Neurosurg Psychiatry. 1994;57:229–31. doi: 10.1136/jnnp.57.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Moreira-Silva SF, Rodrigues MG, Pimenta JL, Gomes CP, Freire LH, Pereira FE. Toxocariasis of the central nervous system: with report of two cases. Rev Soc Bras Med Trop. 2004;37:169–74. doi: 10.1590/S0037-86822004000200011. [DOI] [PubMed] [Google Scholar]
- 395.Maiga Y, Wiertlewski S, Desal H, Marjolet M, Damier P. Presentation of cerebral toxocariasis with mental confusion in an adult: case report and review of the literature. Bull Soc Pathol Exot. 2007;100:101–4. [PubMed] [Google Scholar]
- 396.Finsterer J, Auer H. Neurotoxocarosis. Rev Inst Med Trop São Paulo. 2007;49:279–87. doi: 10.1590/S0036-46652007000500002. [DOI] [PubMed] [Google Scholar]
- 397.Lukehart SA, Hook EW, 3rd, Baker-Zander SA, Collier AC, Critchlow CW, Handsfield HH. Invasion of the central nervous system by Treponema pallidum: implications for diagnosis and treatment. Ann Intern Med. 1988;109:855–62. doi: 10.7326/0003-4819-109-11-855. [DOI] [PubMed] [Google Scholar]
- 398.Simon RP. Neurosyphilis. Arch Neurol. 1985;42:606–13. doi: 10.1001/archneur.1985.04060060112021. [DOI] [PubMed] [Google Scholar]
- 399.Hotson JR. Modern neurosyphilis: a partially treated chronic meningitis. West J Med. 1981;135:191–200. [PMC free article] [PubMed] [Google Scholar]
- 400.Hooshmand H, Escobar MR, Kopf SW. Neurosyphilis. A study of 241 patients. JAMA. 1972;219:726–9. doi: 10.1001/jama.1972.03190320032011. [DOI] [PubMed] [Google Scholar]
- 401.Holmes MD, Brant-Zawadzki MM, Simon RP. Clinical features of meningovascular syphilis. Neurology. 1984;34:553–6. doi: 10.1212/WNL.34.4.553. [DOI] [PubMed] [Google Scholar]
- 402.Flood JM, Weinstock HS, Guroy ME, Bayne L, Simon RP, Bolan G. Neurosyphilis during the AIDS epidemic, San Francisco, 1985–1992. J Infect Dis. 1998;177:931–40. doi: 10.1086/515245. [DOI] [PubMed] [Google Scholar]
- 403.Ghanem KG. REVIEW: neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010;16:e157–68. doi: 10.1111/j.1755-5949.2010.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Weinert LS, Scheffel RS, Zoratto G, et al. Cerebral syphilitic gumma in HIV-infected patients: case report and review. Int J STD AIDS. 2008;19:62–4. doi: 10.1258/ijsa.2007.007007. [DOI] [PubMed] [Google Scholar]
- 405.Horowitz HW, Valsamis MP, Wicher V, et al. Brief report: cerebral syphilitic gumma confirmed by the polymerase chain reaction in a man with human immunodeficiency virus infection. N Engl J Med. 1994;331:1488–91. doi: 10.1056/NEJM199412013312204. [DOI] [PubMed] [Google Scholar]
- 406.Fiumara NJ. Treatment of primary and secondary syphilis. Serological response. JAMA. 1980;243:2500–2. doi: 10.1001/jama.1980.03300500026022. [DOI] [PubMed] [Google Scholar]
- 407.Brown ST, Zaidi A, Larsen SA, Reynolds GH. Serological response to syphilis treatment. A new analysis of old data. JAMA. 1985;253:1296–9. doi: 10.1001/jama.1985.03350330094030. [DOI] [PubMed] [Google Scholar]
- 408.Davis LE, Schmitt JW. Clinical significance of cerebrospinal fluid tests for neurosyphilis. Ann Neurol. 1989;25:50–5. doi: 10.1002/ana.410250108. [DOI] [PubMed] [Google Scholar]
- 409.Workowski KA, Berman S. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1–110. [PubMed] [Google Scholar]
- 410.Moore JE. Asymptomatic neurosyphilis V: a comparison of early and late asymptomatic neurosyphilis. Arch Derm Syphilol. 1928;18:99–108. doi: 10.1001/archderm.1928.02380130102009. [DOI] [Google Scholar]
- 411.Wilner E, Brody JA. Prognosis of general paresis after treatment. Lancet. 1968;2:1370–1. doi: 10.1016/S0140-6736(68)92674-3. [DOI] [PubMed] [Google Scholar]
- 412.Curtis AC, Cutler JC, Gammon G, et al. Penicillin treatment of asymptomatic central nervous system syphilis. I. Probability of progression to symptomatic neurosyphilis. AMA Arch Dermatol. 1956;74:355–66. doi: 10.1001/archderm.1956.01550100023006. [DOI] [PubMed] [Google Scholar]
- 413.Dunlop EM, Al-Egaily SS, Houang ET. Penicillin levels in blood and CSF achieved by treatment of syphilis. JAMA. 1979;241:2538–40. doi: 10.1001/jama.1979.03290490044025. [DOI] [PubMed] [Google Scholar]
- 414.Mohr JA, Griffiths W, Jackson R, Saadah H, Bird P, Riddle J. Neurosyphilis and penicillin levels in cerebrospinal fluid. JAMA. 1976;236:2208–9. doi: 10.1001/jama.1976.03270200046031. [DOI] [PubMed] [Google Scholar]
- 415.Schoth PE, Wolters EC. Penicillin concentrations in serum and CSF during high-dose intravenous treatment for neurosyphilis. Neurology. 1987;37:1214–6. doi: 10.1212/WNL.37.7.1214. [DOI] [PubMed] [Google Scholar]
- 416.Fargen KM, Alvernia JE, Lin CS, Melgar M. Cerebral syphilitic gummata: a case presentation and analysis of 156 reported cases. Neurosurgery. 2009;64:568–75. doi: 10.1227/01.NEU.0000337079.12137.89. [DOI] [PubMed] [Google Scholar]
- 417.Steere AC, Malawista SE, Snydman DR, et al. Lyme arthritis: an epidemic of oligoarticular arthritis in children and adults in three connecticut communities. Arthritis Rheum. 1977;20:7–17. doi: 10.1002/art.1780200102. [DOI] [PubMed] [Google Scholar]
- 418.Steere AC, Malawista SE, Hardin JA, Ruddy S, Askenase W, Andiman WA. Erythema chronicum migrans and Lyme arthritis. The enlarging clinical spectrum. Ann Intern Med. 1977;86:685–98. doi: 10.7326/0003-4819-86-6-685. [DOI] [PubMed] [Google Scholar]
- 419.Steere AC, Broderick TF, Malawista SE. Erythema chronicum migrans and Lyme arthritis: epidemiologic evidence for a tick vector. Am J Epidemiol. 1978;108:312–21. doi: 10.1093/oxfordjournals.aje.a112625. [DOI] [PubMed] [Google Scholar]
- 420.Wallis RC, Brown SE, Kloter KO, Main AJ., Jr Erythema chronicum migrans and lyme arthritis: field study of ticks. Am J Epidemiol. 1978;108:322–7. doi: 10.1093/oxfordjournals.aje.a112626. [DOI] [PubMed] [Google Scholar]
- 421.Burgdorfer W. Discovery of the Lyme disease spirochete and its relation to tick vectors. Yale J Biol Med. 1984;57:515–20. [PMC free article] [PubMed] [Google Scholar]
- 422.Burgdorfer W. The enlarging spectrum of tick-borne spirochetoses: R. R. Parker Memorial Address. Rev Infect Dis. 1986;8:932–40. doi: 10.1093/clinids/8.6.932. [DOI] [PubMed] [Google Scholar]
- 423.Steere AC, Bartenhagen NH, Craft JE, et al. Clinical manifestations of Lyme disease. Zentralbl Bakteriol Mikrobiol Hyg A. 1986;263:201–5. doi: 10.1016/s0176-6724(86)80123-7. [DOI] [PubMed] [Google Scholar]
- 424.Steere AC, Bartenhagen NH, Craft JE, et al. The early clinical manifestations of Lyme disease. Ann Intern Med. 1983;99:76–82. doi: 10.7326/0003-4819-99-1-76. [DOI] [PubMed] [Google Scholar]
- 425.Steere AC. Lyme disease. N Engl J Med. 1989;321:586–96. doi: 10.1056/NEJM198908313210906. [DOI] [PubMed] [Google Scholar]
- 426.Yoshinari NH, Steere AC, Cossermelli W. A review of Lyme disease. Rev Assoc Med Bras. 1989;35:34–8. [PubMed] [Google Scholar]
- 427.Keller TL, Halperin JJ, Whitman M. PCR detection of Borrelia burgdorferi DNA in cerebrospinal fluid of Lyme neuroborreliosis patients. Neurology. 1992;42:32–42. doi: 10.1212/WNL.42.1.32. [DOI] [PubMed] [Google Scholar]
- 428.Preac-Mursic V, Weber K, Pfister HW, et al. Survival of Borrelia burgdorferi in antibiotically treated patients with Lyme borreliosis. Infection. 1989;17:355–9. doi: 10.1007/BF01645543. [DOI] [PubMed] [Google Scholar]
- 429.Shadick NA, Phillips CB, Logigian EL, et al. The long-term clinical outcomes of Lyme disease. A population-based retrospective cohort study. Ann Intern Med. 1994;121:560–7. doi: 10.7326/0003-4819-121-8-199410150-00002. [DOI] [PubMed] [Google Scholar]
- 430.Ackermann R, Rehse-Kupper B, Gollmer E, Schmidt R. Chronic neurologic manifestations of erythema migrans borreliosis. Ann N Y Acad Sci. 1988;539:16–23. doi: 10.1111/j.1749-6632.1988.tb31834.x. [DOI] [PubMed] [Google Scholar]
- 431.Hanny PE, Hauselmann HJ. Lyme disease from the neurologist’s viewpoint. Schweiz Med Wochenschr. 1987;117:901–15. [PubMed] [Google Scholar]
- 432.Kohler J, Kern U, Kasper J, Rhese-Kupper B, Thoden U. Chronic central nervous system involvement in Lyme borreliosis. Neurology. 1988;38:863–7. doi: 10.1212/WNL.38.6.863. [DOI] [PubMed] [Google Scholar]
- 433.Kuntzer T, Bogousslavsky J, Miklossy J, Steck AJ, Janzer R, Regli F. Borrelia rhombencephalomyelopathy. Arch Neurol. 1991;48:832–6. doi: 10.1001/archneur.1991.00530200072021. [DOI] [PubMed] [Google Scholar]
- 434.Meurers B, Kohlhepp W, Gold R, Rohrbach E, Mertens HG. Histopathological findings in the central and peripheral nervous systems in neuroborreliosis. A report of three cases. J Neurol. 1990;237:113–6. doi: 10.1007/BF00314674. [DOI] [PubMed] [Google Scholar]
- 435.Mokry M, Flaschka G, Kleinert G, Kleinert R, Fazekas F, Kopp W. Chronic Lyme disease with an expansive granulomatous lesion in the cerebellopontine angle. Neurosurgery. 1990;27:446–51. doi: 10.1227/00006123-199009000-00018. [DOI] [PubMed] [Google Scholar]
- 436.Pachner AR. Borrelia burgdorferi in the nervous system: the new “great imitator”. Ann N Y Acad Sci. 1988;539:56–64. doi: 10.1111/j.1749-6632.1988.tb31838.x. [DOI] [PubMed] [Google Scholar]
- 437.Rafto SE, Milton WJ, Galetta SL, Grossman RI. Biopsy-confirmed CNS Lyme disease: MR appearance at 1.5 T. AJNR Am J Neuroradiol. 1990;11:482–4. [PMC free article] [PubMed] [Google Scholar]
- 438.Crisp D, Ashby P. Lyme radiculoneuritis treated with intravenous immunoglobulin. Neurology. 1996;46:1174–5. doi: 10.1212/WNL.46.4.1174. [DOI] [PubMed] [Google Scholar]
- 439.O’Connell S. Lyme borreliosis: current issues in diagnosis and management. Curr Opin Infect Dis. 2010;23:231–5. doi: 10.1097/QCO.0b013e32833890e2. [DOI] [PubMed] [Google Scholar]
- 440.Mertens HG, Martin R, Kohlhepp W. Clinical and neuroimmunological findings in chronic Borrelia burgdorferi radiculomyelitis (Lyme disease) J Neuroimmunol. 1988;20:309–14. doi: 10.1016/0165-5728(88)90180-4. [DOI] [PubMed] [Google Scholar]
- 441.Logigian EL, Kaplan RF, Steere AC. Chronic neurologic manifestations of Lyme disease. N Engl J Med. 1990;323:1438–44. doi: 10.1056/NEJM199011223232102. [DOI] [PubMed] [Google Scholar]
- 442.Reik L., Jr Stroke due to Lyme disease. Neurology. 1993;43:2705–7. doi: 10.1212/WNL.43.12.2705. [DOI] [PubMed] [Google Scholar]
- 443.Weder B, Wiedersheim P, Matter L, Steck A, Otto F. Chronic progressive neurological involvement in Borrelia burgdorferi infection. J Neurol. 1987;234:40–3. doi: 10.1007/BF00314008. [DOI] [PubMed] [Google Scholar]
- 444.Agarwal R, Sze G. Neuro-lyme disease: MR imaging findings. Radiology. 2009;253:167–73. doi: 10.1148/radiol.2531081103. [DOI] [PubMed] [Google Scholar]
- 445.Hildenbrand P, Craven DE, Jones R, Nemeskal P. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am J Neuroradiol. 2009;30:1079–87. doi: 10.3174/ajnr.A1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Kohler J, Kasper J, Kern U, Thoden U, Rehse-Kupper B. Borrelia encephalomyelitis. Lancet. 1986;2:35. doi: 10.1016/S0140-6736(86)92574-2. [DOI] [PubMed] [Google Scholar]
- 447.Agosta F, Rocca MA, Benedetti B, Capra R, Cordioli C, Filippi M. MR imaging assessment of brain and cervical cord damage in patients with neuroborreliosis. AJNR Am J Neuroradiol. 2006;27:892–4. [PMC free article] [PubMed] [Google Scholar]
- 448.Bigi S, Aebi C, Nauer C, Bigler S, Steinlin M. Acute transverse myelitis in Lyme neuroborreliosis. Infection. 2010;38:413–6. doi: 10.1007/s15010-010-0028-x. [DOI] [PubMed] [Google Scholar]
- 449.Chang BL, Shih CM, Ro LS, et al. Acute neuroborreliosis with involvement of the central nervous system. J Neurol Sci. 2010;295:10–5. doi: 10.1016/j.jns.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 450.Halperin JJ, Luft BJ, Anand AK, et al. Lyme neuroborreliosis: central nervous system manifestations. Neurology. 1989;39:753–9. doi: 10.1212/WNL.39.6.753. [DOI] [PubMed] [Google Scholar]
- 451.Halperin JJ. Nervous system lyme disease: diagnosis and treatment. Rev Neurol Dis. 2009;6:4–12. [PubMed] [Google Scholar]
- 452.Halperin JJ. Nervous system Lyme disease. J R Coll Physicians Edinb. 2010;40:248–55. doi: 10.4997/JRCPE.2010.314. [DOI] [PubMed] [Google Scholar]
- 453.Halperin JJ. Central nervous system Lyme disease. Curr Neurol Neurosci Rep. 2005;5:446–52. doi: 10.1007/s11910-005-0032-1. [DOI] [PubMed] [Google Scholar]
- 454.Halperin JJ, Pass HL, Anand AK, Luft BJ, Volkman DJ, Dattwyler RJ. Nervous system abnormalities in Lyme disease. Ann N Y Acad Sci. 1988;539:24–34. doi: 10.1111/j.1749-6632.1988.tb31835.x. [DOI] [PubMed] [Google Scholar]
- 455.Halperin J, Luft BJ, Volkman DJ, Dattwyler RJ. Lyme neuroborreliosis. Peripheral nervous system manifestations. Brain. 1990;113(Pt 4):1207–21. doi: 10.1093/brain/113.4.1207. [DOI] [PubMed] [Google Scholar]
- 456.Halperin JJ. Neuroborreliosis: central nervous system involvement. Semin Neurol. 1997;17:19–24. doi: 10.1055/s-2008-1040908. [DOI] [PubMed] [Google Scholar]
- 457.Halperin JJ. Neuroborreliosis (nervous system Lyme disease) Curr Treat Options Neurol. 1999;1:139–46. doi: 10.1007/s11940-999-0013-9. [DOI] [PubMed] [Google Scholar]
- 458.Logigian EL, Steere AC. Clinical and electrophysiologic findings in chronic neuropathy of Lyme disease. Neurology. 1992;42:303–11. doi: 10.1212/WNL.42.2.303. [DOI] [PubMed] [Google Scholar]
- 459.Halperin JJ. North American Lyme neuroborreliosis. Scand J Infect Dis Suppl. 1991;77:74–80. [PubMed] [Google Scholar]
- 460.Steere AC, Pachner AR, Malawista SE. Neurologic abnormalities of Lyme disease: successful treatment with high-dose intravenous penicillin. Ann Intern Med. 1983;99:767–72. doi: 10.7326/0003-4819-99-6-767. [DOI] [PubMed] [Google Scholar]
- 461.Kalish RA, Kaplan RF, Taylor E, Jones-Woodward L, Workman K, Steere AC. Evaluation of study patients with Lyme disease, 10-20-year follow-up. J Infect Dis. 2001;183:453–60. doi: 10.1086/318082. [DOI] [PubMed] [Google Scholar]
- 462.Pfister HW, Preac-Mursic V, Wilske B, Einhaupl KM. Cefotaxime vs penicillin G for acute neurologic manifestations in Lyme borreliosis. A prospective randomized study. Arch Neurol. 1989;46:1190–4. doi: 10.1001/archneur.1989.00520470044025. [DOI] [PubMed] [Google Scholar]
- 463.Pfister HW, Preac-Mursic V, Wilske B, Schielke E, Sorgel F, Einhaupl KM. Randomized comparison of ceftriaxone and cefotaxime in Lyme neuroborreliosis. J Infect Dis. 1991;163:311–8. doi: 10.1093/infdis/163.2.311. [DOI] [PubMed] [Google Scholar]
- 464.Pfister HW, Rupprecht TA. Clinical aspects of neuroborreliosis and post-Lyme disease syndrome in adult patients. Int J Med Microbiol. 2006;296(Suppl 40):11–6. doi: 10.1016/j.ijmm.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 465.Halperin JJ. Diagnosis and treatment of the neuromuscular manifestations of lyme disease. Curr Treat Options Neurol. 2007;9:93–100. doi: 10.1007/s11940-007-0035-0. [DOI] [PubMed] [Google Scholar]
- 466.Logigian EL, Kaplan RF, Steere AC. Successful treatment of Lyme encephalopathy with intravenous ceftriaxone. J Infect Dis. 1999;180:377–83. doi: 10.1086/314860. [DOI] [PubMed] [Google Scholar]
- 467.Halperin JJ, Shapiro ED, Logigian E, et al. Practice parameter: treatment of nervous system Lyme disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2007;69:91–102. doi: 10.1212/01.wnl.0000265517.66976.28. [DOI] [PubMed] [Google Scholar]
- 468.Klempner MS, Hu LT, Evans J, et al. Two controlled trials of antibiotic treatment in patients with persistent symptoms and a history of Lyme disease. N Engl J Med. 2001;345:85–92. doi: 10.1056/NEJM200107123450202. [DOI] [PubMed] [Google Scholar]
- 469.Sexton DJ, Kaye KS. Rocky mountain spotted fever. Med Clin North Am. 2002;86:351–60. doi: 10.1016/S0025-7125(03)00091-9. [DOI] [PubMed] [Google Scholar]
- 470.Chapman AS, Bakken JS, Folk SM, et al. Diagnosis and management of tickborne rickettsial diseases: Rocky Mountain spotted fever, ehrlichioses, and anaplasmosis – United States: a practical guide for physicians and other health-care and public health professionals. MMWR Recomm Rep. 2006;55:1–27. [PubMed] [Google Scholar]
- 471.Chen LF, Sexton DJ. What’s new in Rocky Mountain spotted fever? Infect Dis Clin North Am. 2008;22:415–32. doi: 10.1016/j.idc.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 472.Kirkland KB, Wilkinson WE, Sexton DJ. Therapeutic delay and mortality in cases of Rocky Mountain spotted fever. Clin Infect Dis. 1995;20:1118–21. doi: 10.1093/clinids/20.5.1118. [DOI] [PubMed] [Google Scholar]
- 473.Archibald LK, Sexton DJ. Long-term sequelae of Rocky Mountain spotted fever. Clin Infect Dis. 1995;20:1122–5. doi: 10.1093/clinids/20.5.1122. [DOI] [PubMed] [Google Scholar]
- 474.Sexton DJ, Kirkland KB. Rickettsial infections and the central nervous system. Clin Infect Dis. 1998;26:247–8. doi: 10.1086/517043. [DOI] [PubMed] [Google Scholar]
- 475.Ismail N, Bloch KC, McBride JW. Human ehrlichiosis and anaplasmosis. Clin Lab Med. 2010;30:261–92. doi: 10.1016/j.cll.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Windsor JJ. Cat-scratch disease: epidemiology, aetiology and treatment. Br J Biomed Sci. 2001;58:101–10. [PubMed] [Google Scholar]
- 477.Weston KD, Tran T, Kimmel KN, Maria BL. Possible role of high-dose corticosteroids in the treatment of cat-scratch disease encephalopathy. J Child Neurol. 2001;16:762–3. doi: 10.1177/088307380101601010. [DOI] [PubMed] [Google Scholar]
- 478.Genizi J, Kasis I, Schif A, Shahar E. Effect of high-dose methyl-prednisolone on brainstem encephalopathy and basal ganglia impairment complicating cat scratch disease. Brain Dev. 2007;29:377–9. doi: 10.1016/j.braindev.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 479.Schneider T, Moos V, Loddenkemper C, Marth T, Fenollar F, Raoult D. Whipple’s disease: new aspects of pathogenesis and treatment. Lancet Infect Dis. 2008;8:179–90. doi: 10.1016/S1473-3099(08)70042-2. [DOI] [PubMed] [Google Scholar]
- 480.Slowik A, Szczudlik A. Whipple’s disease – a rare cause of neurological symptoms and disorders. Neurol Neurochir Pol. 2002;36:959–70. [PubMed] [Google Scholar]
- 481.Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med. 1992;327:293–301. doi: 10.1056/NEJM199207303270501. [DOI] [PubMed] [Google Scholar]
- 482.Schoniger-Hekele M, Petermann D, Weber B, Muller C. Tropheryma whipplei in the environment: survey of sewage plant influxes and sewage plant workers. Appl Environ Microbiol. 2007;73:2033–5. doi: 10.1128/AEM.02335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Moos V, Loddenkemper C, Schneider T. Tropheryma whipplei infection. Colonization, self-limiting infection and Whipple’s disease. Pathologe. 2011;32:362–70. doi: 10.1007/s00292-011-1446-y. [DOI] [PubMed] [Google Scholar]
- 484.Moos V, Schneider T. Changing paradigms in Whipple’s disease and infection with Tropheryma whipplei. Eur J Clin Microbiol Infect Dis. 2011;30:1151–8. doi: 10.1007/s10096-011-1209-y. [DOI] [PubMed] [Google Scholar]
- 485.Abreu P, Azevedo E, Lobo L, Moura CS, Pontes C. Whipple disease and central nervous system. Acta Med Port. 2005;18:199–208. [PubMed] [Google Scholar]
- 486.Bermejo P, Burgos A. Whipple’s disease and central nervous system. Med Clin (Barc) 2006;127:379–85. doi: 10.1157/13092439. [DOI] [PubMed] [Google Scholar]
- 487.Durand DV, Lecomte C, Cathebras P, Rousset H, Godeau P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Societe Nationale Francaise de Medecine Interne. Medicine. 1997;76:170–84. doi: 10.1097/00005792-199705000-00003. [DOI] [PubMed] [Google Scholar]
- 488.Louis ED. Whipple disease. Curr Neurol Neurosci Rep. 2003;3:470–5. doi: 10.1007/s11910-003-0049-2. [DOI] [PubMed] [Google Scholar]
- 489.Louis ED, Lynch T, Kaufmann P, Fahn S, Odel J. Diagnostic guidelines in central nervous system Whipple’s disease. Ann Neurol. 1996;40:561–8. doi: 10.1002/ana.410400404. [DOI] [PubMed] [Google Scholar]
- 490.Scheld WM. Whipple disease of the central nervous system. J Infect Dis. 2003;188:797–800. doi: 10.1086/378075. [DOI] [PubMed] [Google Scholar]
- 491.Vital Durand D, Gerard A, Rousset H. Neurological manifestations of Whipple disease. Rev Neurol. 2002;158:988–92. [PubMed] [Google Scholar]
- 492.Baisden BL, Lepidi H, Raoult D, Argani P, Yardley JH, Dumler JS. Diagnosis of Whipple disease by immunohistochemical analysis: a sensitive and specific method for the detection of Tropheryma whipplei (the Whipple bacillus) in paraffin-embedded tissue. Am J Clin Pathol. 2002;118:742–8. doi: 10.1309/8YGR-FE7L-39LL-L37C. [DOI] [PubMed] [Google Scholar]
- 493.Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine. 2002;81:443–57. doi: 10.1097/00005792-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 494.Panegyres PK, Edis R, Beaman M, Fallon M. Primary Whipple’s disease of the brain: characterization of the clinical syndrome and molecular diagnosis. QJM. 2006;99:609–23. doi: 10.1093/qjmed/hcl081. [DOI] [PubMed] [Google Scholar]
- 495.Schnider PJ, Reisinger EC, Berger T, Krejs GJ, Auff E. Treatment guidelines in central nervous system Whipple’s disease. Ann Neurol. 1997;41:561–2. doi: 10.1002/ana.410410425. [DOI] [PubMed] [Google Scholar]
- 496.Schnider PJ, Reisinger EC, Gerschlager W, et al. Long-term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8:899–903. [PubMed] [Google Scholar]
- 497.Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52:901–7. doi: 10.1111/j.1469-8749.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 498.Weissert R. Progressive multifocal leukoencephalopathy. J Neuroimmunol. 2011;231:73–7. doi: 10.1016/j.jneuroim.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 499.Fox R. Advances in the management of PML: focus on natalizumab. Cleve Clin J Med. 2011;78(Suppl 2):S33–7. doi: 10.3949/ccjm.78.s2.08. [DOI] [PubMed] [Google Scholar]
- 500.Brooks BR, Walker DL. Progressive multifocal leukoencephalopathy. Neurol Clin. 1984;2:299–313. [PubMed] [Google Scholar]
- 501.Egli A, Infanti L, Dumoulin A, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199:837–46. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 502.White FA, 3rd, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66:5726–34. doi: 10.1128/jvi.66.10.5726-5734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Vago L, Cinque P, Sala E, et al. JCV-DNA and BKV-DNA in the CNS tissue and CSF of AIDS patients and normal subjects. Study of 41 cases and review of the literature. J Acquir Immune Defic Syndr. 1996;12:139–46. doi: 10.1097/00042560-199606010-00006. [DOI] [PubMed] [Google Scholar]
- 504.Quinlivan EB, Norris M, Bouldin TW, et al. Subclinical central nervous system infection with JC virus in patients with AIDS. J Infect Dis. 1992;166:80–5. doi: 10.1093/infdis/166.1.80. [DOI] [PubMed] [Google Scholar]
- 505.Koralnik IJ, Boden D, Mai VX, Lord CI, Letvin NL. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253–60. doi: 10.1212/WNL.52.2.253. [DOI] [PubMed] [Google Scholar]
- 506.Ferrante P, Caldarelli-Stefano R, Omodeo-Zorini E, Vago L, Boldorini R, Costanzi G. PCR detection of JC virus DNA in brain tissue from patients with and without progressive multifocal leukoencephalopathy. J Med Virol. 1995;47:219–25. doi: 10.1002/jmv.1890470306. [DOI] [PubMed] [Google Scholar]
- 507.Dubois V, Dutronc H, Lafon ME, et al. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J Clin Microbiol. 1997;35:2288–92. doi: 10.1128/jcm.35.9.2288-2292.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Holman RC, Janssen RS, Buehler JW, Zelasky MT, Hooper WC. Epidemiology of progressive multifocal leukoencephalopathy in the United States: analysis of national mortality and AIDS surveillance data. Neurology. 1991;41:1733–6. doi: 10.1212/WNL.41.11.1733. [DOI] [PubMed] [Google Scholar]
- 509.Christensen KL, Holman RC, Hammett TA, Belay ED, Schonberger LB. Progressive multifocal leukoencephalopathy deaths in the USA, 1979–2005. Neuroepidemiology. 2010;35:178–84. doi: 10.1159/000311014. [DOI] [PubMed] [Google Scholar]
- 510.Amend KL, Turnbull B, Foskett N, Napalkov P, Kurth T, Seeger J. Incidence of progressive multifocal leukoencephalopathy in patients without HIV. Neurology. 2010;75:1326–32. doi: 10.1212/WNL.0b013e3181f73600. [DOI] [PubMed] [Google Scholar]
- 511.Focosi D, Marco T, Kast RE, Maggi F, Ceccherini-Nelli L, Petrini M. Progressive multifocal leukoencephalopathy: what’s new? Neuroscientist. 2010;16:308–23. doi: 10.1177/1073858409356594. [DOI] [PubMed] [Google Scholar]
- 512.Hernandez B, Dronda F, Moreno S. Treatment options for AIDS patients with progressive multifocal leukoencephalopathy. Expert Opin Pharmacother. 2009;10:403–16. doi: 10.1517/14656560802707994. [DOI] [PubMed] [Google Scholar]
- 513.Lima MA, Bernal-Cano F, Clifford DB, Gandhi RT, Koralnik IJ. Clinical outcome of long-term survivors of progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 2010;81:1288–91. doi: 10.1136/jnnp.2009.179002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Tavazzi E, White MK, Khalili K. Progressive multifocal leukoencephalopathy: clinical and molecular aspects. Rev Med Virol. 2012;22:18–32. doi: 10.1002/rmv.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Silverman L, Rubinstein LJ. Electron microscopic observations on a case of progressive multifocal leukoencephalopathy. Acta Neuropathol. 1965;5:215–24. doi: 10.1007/BF00686519. [DOI] [PubMed] [Google Scholar]
- 516.McGuire D, Barhite S, Hollander H, Miles M. JC virus DNA in cerebrospinal fluid of human immunodeficiency virus-infected patients: predictive value for progressive multifocal leukoencephalopathy. Ann Neurol. 1995;37:395–9. doi: 10.1002/ana.410370316. [DOI] [PubMed] [Google Scholar]
- 517.Hammarin AL, Bogdanovic G, Svedhem V, Pirskanen R, Morfeldt L, Grandien M. Analysis of PCR as a tool for detection of JC virus DNA in cerebrospinal fluid for diagnosis of progressive multifocal leukoencephalopathy. J Clin Microbiol. 1996;34:2929–32. doi: 10.1128/jcm.34.12.2929-2932.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Dorries K, Arendt G, Eggers C, Roggendorf W, Dorries R. Nucleic acid detection as a diagnostic tool in polyomavirus JC induced progressive multifocal leukoencephalopathy. J Med Virol. 1998;54:196–203. doi: 10.1002/(SICI)1096-9071(199803)54:3<196::AID-JMV10>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 519.De Luca A, Giancola ML, Ammassari A, et al. Potent anti-retroviral therapy with or without cidofovir for AIDS-associated progressive multifocal leukoencephalopathy: extended follow-up of an observational study. J Neurovirol. 2001;7:364–8. doi: 10.1080/13550280152537256. [DOI] [PubMed] [Google Scholar]
- 520.Hall CD, Dafni U, Simpson D, et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. AIDS Clinical Trials Group 243 Team. N Engl J Med. 1998;338:1345–51. doi: 10.1056/NEJM199805073381903. [DOI] [PubMed] [Google Scholar]
- 521.Rand KH, Ulm AJ, Pincus DW. Central nervous system infections. In: Layon AJ, Gabrielli A, Friedman WA, editors. Textbook of neurointensive care. Philadelphia: WB Saunders; 2004. [Google Scholar]