

Abstract
Nucleic acid sequencing is now commonplace in most research and diagnostic virology laboratories. The data generated can be used to compare novel strains with other viruses and allow the genetic basis of important phenotypic characteristics, such as antigenic determinants, to be elucidated. Furthermore, virus sequence data can also be used to address more fundamental questions relating to the evolution of viruses. Recent advances in laboratory methodologies allow rapid sequencing of virus genomes. For the first time, this opens up the potential for using genome sequencing to reconstruct virus transmission trees with extremely high resolution and to quickly reveal and identify the origin of unresolved transmission events within discrete infection clusters. Using foot-and-mouth disease virus as an example, this chapter describes strategies that can be successfully used to amplify and sequence the full genomes of RNA viruses. Practical considerations for protocol design and optimization are discussed, with particular emphasis on the software programs used to assemble large contigs and analyze the sequence data for high-resolution epidemiology.
Key words: Complete genome, foot-and-mouth disease virus, nucleotide sequence, virus
Introduction
During the past 15 years, a number of incremental improvements have been made to methods used to generate nucleotide sequence data. The principle underpinning the mostly widely used sequencing approaches is based on the dideoxynucleotide chain-termination method initially devised by Fred Sanger in the 1970s (1). The throughput and robustness of these methods have been improved by the use of fluorescent dyes and capillary separation technologies, such that the routine assembly of large fragments of genomic DNA (>10 kb) is now achievable by many modestly equipped laboratories. For the large part, protocols developed to sequence large fragments of nucleic acid can also be adapted to characterize the genomes of RNA viruses, which typically are 15 kb or less. Full-genome sequences of viruses can be used to address fundamental questions relating to evolution, identification of critical antigenic determinants, and viral molecular epidemiology. Although sequencing small numbers of some viral genomes can be straightforward, specific protocols and work flows are required to effectively manage projects that aim to characterize the molecular epidemiology of viral transmission.
Using foot-and-mouth disease virus (FMDV) as an example, this chapter describes strategies that can be successfully used to amplify and sequence the complete genomes of RNA viruses. Foot-and-mouth disease (FMD) is a highly contagious disease affecting cloven-hoofed livestock (cattle, sheep, pigs, goats, and water buffalo). The causative agent is a virus belonging to the genus Aphthovirus (family: Picornaviridae) that exists as seven antigenically distinct serotypes, each comprising numerous and constantly evolving variants (2). The genome of FMDV is approximately 8,300 nucleotides in length. It comprises a polyadenylated positive-sense RNA that encodes a single polyprotein, which is posttranslationally cleaved into constituent capsid proteins and nonstructural proteins involved in viral replication.
In common with most other RNA viruses, the enzyme (RNA-dependent RNA polymerase) responsible for replication of the FMDV genome has poor fidelity, such that changes to the nucleotide sequence frequently occur and are inherited to progeny viruses. This rapid evolution rate of FMDV allows virus transmission trees to be reconstructed with extremely high resolution, opening up the possibility of using these data to retrospectively reveal and identify the origin of unresolved transmission events (3,4). In addition to forensic molecular epidemiology, full-genome sequence data have also recently contributed to our understanding of a number of aspects of FMDV evolution, including (i) evolutionary rates (5); (ii) sites and importance of recombination (6,7); (iii) identification of ordered RNA structures (8); and (iv) contribution and significance of the quasi-species phenomenon to evolution (9). Sequence data from a wide variety of FMDV isolates also play an important role in the reiterative design of oligonucleotide primers used for molecular assays for routine diagnostic use in reference laboratories (for pan-reactive and serotype-specific detection and strain characterization).
Amplification Strategies: Design and Targeting of Polymerase Chain Reaction Primers
The extent of the run length obtained by capillary sequencers places a limit on the maximum distance between oligonucleotide primers (either in the polymerase chain reaction [PCR] amplification or cycle sequencing setup stages). In contrast to DNA targets, which are relatively stable, researchers who study RNA viruses, such as picornaviruses, are familiar with the plasticity of viral genomes. This high variability poses particular challenges for the design of pan-reactive oligonucleotide primers to reliably amplify complete viral complementary DNAs (cDNAs). For viruses such as FMDV, the existence of multiple serotypes (whose nucleotide sequences may vary by as much as 50% in some genome regions) can further complicate the identification of suitable target sequences.
As a consequence, the two extremes of the sequencing strategies used for FMDV are illustrated in Fig. 1 (see Fig. 1a,b) and shown by representative agarose gels in Fig. 2. In both of these approaches (adopted for the characterization of FMD outbreaks in the United Kingdom in 2001 and 2007), a large number of specific primers were required. Furthermore, these oligonucleotides are specific for defined lineages of FMDV, limiting their use for study of other genotypes of FMDV. Other FMD laboratories have used similar approaches also requiring large numbers of primers (11–15). Additional protocols, such as rapid amplification of cDNA ends (RACE), can be used to generate sequence data for the terminal ends of the genome and regions close to the poly (C) tract of FMDV. To reduce the complexity of the cycle-sequencing reactions, recognition sequences for universal sequencing primers (such as M13) can be incorporated into the 5′ ends of the primers used for PCR (see Fig. 1a).
Fig. 1.
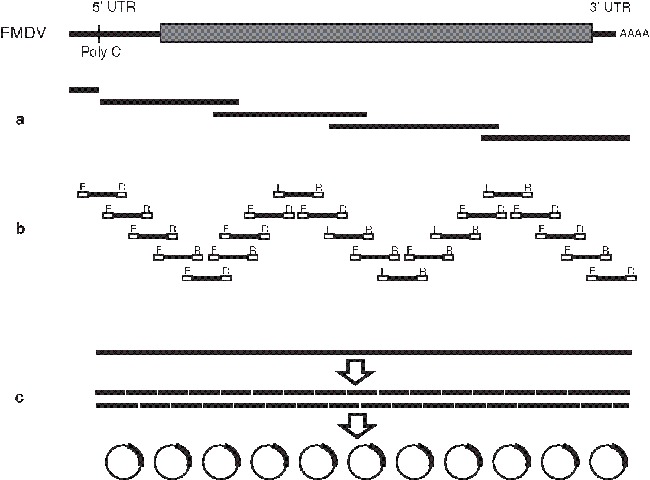
Outline of RT-PCR strategies that have been used to amplify the complete genome sequences of FMDV. (a) Long overlapping products (∼3 kb) are generated by PCR using full-length cDNA as a template. Sequences are obtained using a panel of specific sequencing primers (see ref. 3). (b) Short products (∼700 bp) are generated using FMDV-specific primers, which also incorporate regions (labelled F and R) targeted by the sequencing primers (10). (c) Long-range RT-PCR is used to amplify a product comprising the complete L fragment of FMDV. This may either be sequenced using many specific primers (11) or can be fragmented by restriction digest and cloned into a bacterial plasmid vector (pilot studies using this method have been undertaken by IAH in collaboration with the Wellcome Trust Sanger Institute, Cambridge).
Fig. 2.
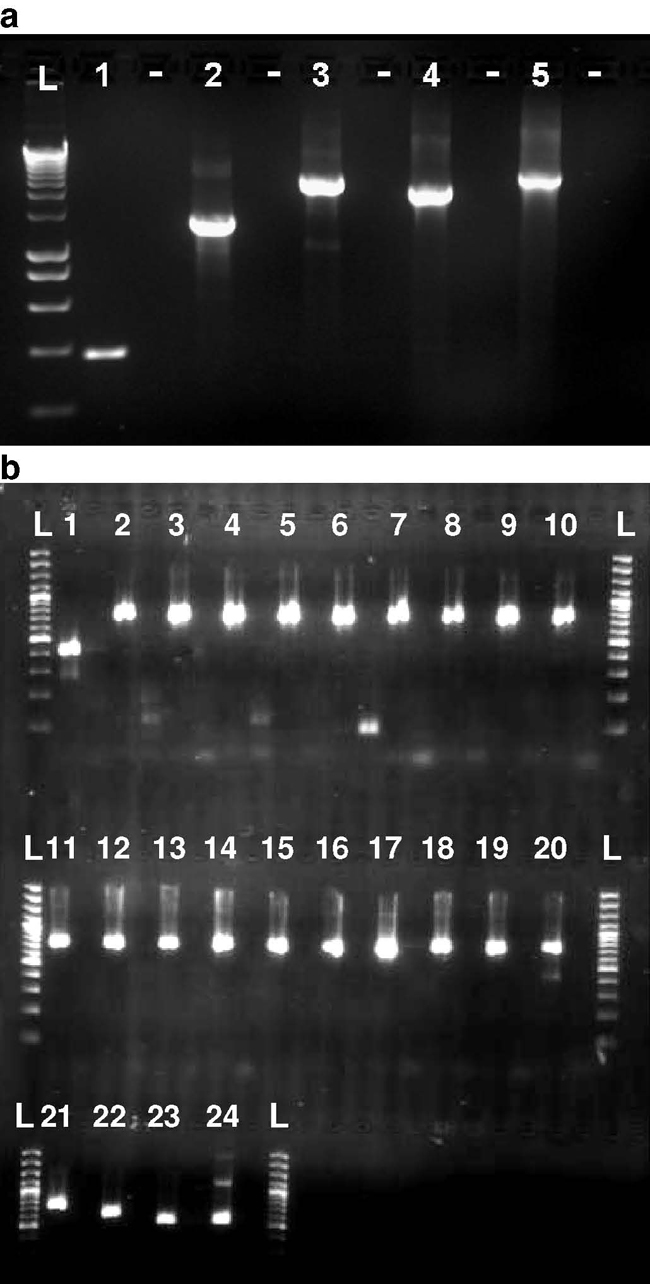
Amplification of full-FMDV genomes by RT-PCR. Agarose gel electrophoresis shows RT-PCR fragments representing amplification of the entire FMDV genome. (a) amplification of O/UKG/2001 genome using 5 RT-PCR fragments and (b) amplification of O/UKG/2007 genomes using 24 RT-PCR fragments.
Alternative approaches, such as shotgun cloning (for example, Fig. 1c) are also being considered for full-genome sequencing. Initially, these use long-range PCR to amplify large fragments of the virus genome (possibly even encompassing entire genomic sequences). These PCR products are subsequently fragmented and cloned into plasmid vectors prior to sequencing and reconstruction of the viral sequence. Since this approach uses only two viral-specific primers (which can be targeted to highly conserved regions) and is not reliant on internal virus-specific primers, this method may provide a more suitable approach that has a broader sensitivity to different viral variants. However, these methods need to balance the advantages in diagnostic sensitivity that are gained from using a smaller number of primers with the drawback of lower analytical sensitivity that may arise from amplifying large PCR products (in comparison to shorter fragments).
Overview of Approaches Used for FMDV Sequencing
In this chapter, a guide protocol that has been successfully used to sequence FMDV is presented. Although some of the finer details are specific to FMDV, the general approaches described are broadly applicable to other RNA viruses. Indeed, similar methods have been described recently to characterize the genomes of other viruses that infect humans, livestock, and plants (16–23).
Materials
RNA Extraction
0.04M phosphate-buffered saline: 35 mM Na2HPO4, 5.7 mM KH2PO4, pH 7.6. Store at room temperature.
Sterile sand (Fine Sifted, BDH). Small aliquots (∼3 g) are prepared and autoclaved prior to use. Store at room temperature.
Sterile pestle and mortar (Fisher); autoclave prior to use.
TRIzol Reagent (Invitrogen). Store at +2 to 8°C. This solution contains phenol and guanidine isothiocyanate; care should be taken to minimize skin contact and inhalation.
Chloroform (AnalaR Grade, BDH) (toxic and probable carcinogen; care should be taken to minimize inhalation and ingestion).
0.2M glycogen (Roche).
Isopropanol (propan-2-ol) (AnalaR Grade, BDH).
Ethanol (AnalaR Grade, BDH). Store at +2 to 8°C.
Nuclease-free water (deoxyribonuclease [DNase] and ribonuclease [RNase] free) (Invitrogen). Store at room temperature.
Reverse Transcription and PCR Amplification
Random hexamers (Promega). Store at −20°C.
Deoxynucleotide 5′-triphosphate (dNTP) mixture (Promega). The dNTP mix is a premixed solution containing sodium salts of dATP (deoxyadenosine 5′-triphosphate), dCTP (deoxycytosine 5′-triphosphate), dGTP (deoxyguanosine 5′-triphosphate), and dTTP (deoxythymidine 5′-triphosphate), each at 10 mM in water. Store at −20°C.
Oligonucleotide primers (Sigma-Aldrich).Complete list of primers used for PCR amplification of FMDV are described elsewhere (2,3,10).
Reverse transcription kit: SuperScript™ III RT (Invitrogen). Enzyme is supplied with a vial of 5X first-strand buffer (250 mM Tris-HCl, pH 8.3, 375 mM KCl, 15 mM MgCl2) and a vial of 100 mM dithiothreitol (DTT). Store at −20°C.
RNaseOUT (Invitrogen); store at −20°C.
GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare).
PCR Cleanup Prior to Setting Up Sequencing Reactions
Agarose (UltraPure™, Invitrogen). Store at room temperature.
Tris-borate-ethylenediaminetetraacetic acid (EDTA) buffer (National Diagnostics). 10X solution: When diluted, the 1X solution contains 89 mM Tris-base, 89 mM boric acid (pH 8.3), and 2 mM Na2EDTA. Store at room temperature.
Ethidium bromide (UltraPure). Of a 10 mg/mL stock solution, added 2 μL to 100 mL gels to visualize PCR bands. Ethidium bromide is a potent mutagen. Therefore, care should be taken to minimize exposure and to ensure correct disposal of material (solutions and gels) containing ethidium bromide. Store at room temperature.
6X loading buffer for samples to be tested by agarose gel electrophoresis (Invitrogen).
DNA standards, if required (Invitrogen).
Methods
RNA Extraction (see Notes 1 and 2)
Using sterile sand and a pestle and mortar, prepare a 10% (w/v) suspension of the tissue sample in phosphate-buffered saline. Liquid samples (such as serum) can be processed straight to step 3. Depending on application and nature of the sample to be tested (see Note 3), alternative RNA extraction protocols can also be used (such as commercially available silica-based spin columns).
Centrifuge at 300g for 10 min.
Add 200 μL of the sample supernatant to 1 mL of TRIzol reagent in a microfuge tube (see Note 4 ).
Add 240 μL of chloroform directly to the tube.
Mix the tube by inversion and centrifuge for 15 min at 10,000g at 2–8°C.
Transfer the top phase to a fresh microfuge tube and add 1μL of 0.2M glycogen.
Add an equivalent volume of isopropanol.
Mix the tube by inversion and centrifuge for 15 min at 10,000g at 2–8 °C.
Carefully wash the pellet (containing the RNA) with ice-cold 70% ethanol and recentrifuge for 15 min (10,000g at 2–8 °C).
Air-dry the pellet and resuspend RNA in nuclease-free water.
Reverse Transcription and PCR Amplification
Prepare primer mix (9 µL) containing 30 pmol reverse primer [5′-GGC GGC CGC TTT TTT TTT TTT TTT-3′], 50 ng random hexamers, and 30 nmol of each dNTP (3 µL of a 10 mM solution).
Add to 12 µL prepared RNA.
Denature the RNA by incubating the RNA/primer mix at 70°C for 3 min and place on ice for 3 min.
Add 17 µL of reverse-transcription (RT) mix containing 8 µL first-strand buffer, 2 µL 0.1 mM DTT, 2 µL RNaseOUT, 5 µL nuclease-free water.
Add 2 µL Superscript III Reverse Transcriptase.
Incubate at 42°C for 1–4 h followed by 85°C for 5 min. A specific PCR amplifying the 5′ end of the genome can be used to test that complete first-strand cDNAs have been generated.
Cleanup cDNA using GFX PCR DNA and Gel Band Purification kit according to manufacturers’ instructions and elute in 50 µL. This step removes unincorporated primers and dNTPs from the RT reaction.
Set up a PCR master mix in a clean room using the primer sets required for amplification of the genomic fragments.
Add 2.5 μL cDNA to each reaction in a separate area away from the PCR clean room (see Note 5).
Run thermocycling program (as described in refs. 2, 3, 10; see Note 6 ).
PCR Cleanup Prior to Setting Up Sequencing Reactions
Run 2 μL of PCR product on 1.2% (w/v) agarose gel at 105 V for 30 min to check reaction has worked.
Clean up cDNA using GFX PCR DNA and Gel Band Purification kit according to manufacturers’ instructions.
Quantify DNA concentration in purified PCR product. This can be done using a spectrophotometer (e.g., Nanodrop, Thermo Fisher Scientific) or by agarose gel electrophoresis using DNA standards (see Subheading 2.3 .).
Dilute products to give appropriate concentrations for sequencing.
Prepare sequencing reaction using diluted PCR product.
Analysis of Sequence Data
Sequencing viral genomes can quickly accumulate a large amount of data (see Note 7). Software programs (such as Lasergene, http://www.dnastar.com/) can be used to simplify the alignment of individual sequences and to rapidly assemble large contigs. The minimum criterion for acceptance of a final sequence is that each nucleotide position should be determined by sequencing reactions in either direction (forward and reverse).
Currently, the genetic evolution and relationships of viruses are studied by analyzing their genetic sequence data by phylogenetic methods. Phylogenetic trees are constructed and used to deduce the genetic relatedness of the viruses. There are different methods for constructing phylogenetic trees; the first approach developed was the maximum parsimony methodology, but more recently maximum likelihood (24) and Bayesian methods (25) are the preferred techniques for tree construction. Other methods based on distance matrixes, such as neighbor-joining (26) or unweighted pair-group method with arithmetic mean (UPGMA) (27), which calculate genetic distance from multiple sequence alignments, are simpler to implement but do not invoke an evolutionary model.
Maximum parsimony determines the most parsimonious tree requiring the least evolutionary steps. This method is simple and as such makes very few assumptions about the evolutionary process. However, certain features of genetic evolution of organisms present problems when using this method of tree construction. First, inaccuracies can occur as a result of the existence of homoplasy. Homoplasy describes processes, such as convergent evolution, by which a single mutation can occur twice on independent branches of a tree. Hence, it implies that two sequences sharing a mutation were not necessarily derived from a common ancestor that also contained this mutation. Another hurdle to overcome is back-mutation, by which a mutation reverts to its original genotype. This can cause the specific sequence to appear more ancestral than is necessarily the case. A further drawback to the method of maximum parsimony is that it takes no account of the rate at which mutations arise and the varying probabilities of different mutations occurring (i.e., transversions vs. transitions).
For these reasons, the parametric method of maximum likelihood is usually preferred as it provides the most probable tree that suits a specific determined evolutionary model. Providing that the model employed is a reasonable approximation of the evolutionary processes that gave rise to the observed genetic data, this analysis is potentially more powerful than other methods. The evolutionary model may include a large number of parameters accounting for differences in the probabilities of various character states, differences in the occurrence of particular substitutions/mutations, and differences in the probabilities of change among characters. With the sophisticated models such as the Hasegawa-Kishino-Yano (HKY) model (28) and the general time reversible (GTR) model (29), an improved idea of phylogeny is achieved, although fitting an incorrect model can give incorrect results. The suitability of models can be tested using a program such as model test (30).
Maximum likelihood estimation of tree phylogeny is generally preferable to maximum parsimony because it is statistically consistent with a better statistical foundation, and it allows complex modeling of evolutionary processes. However, the maximum likelihood method has a computing limitation for large numbers of sequences. To infer statistical confidence in either maximum parsimony or maximum likelihood, constructed phylogenies bootstrap analyses (31) are performed. A further method to infer phylogenies is that of Bayesian inference, which generates a posterior distribution for a parameter based on the prior for that parameter and the likelihood of the data (represented by the sequence alignment). In other words, whereas maximum likelihood analysis investigates the probability of the observed data given a specific evolutionary model, Bayesian inference looks at the probability that a model is correct given the observed data set. With the availability of Markov chain Monte Carlo methods (32), Bayesian inference can be a preferred choice for tree estimation because it can be faster than maximum likelihood, and no bootstrapping is required as the posterior probabilities determine the statistical confidence in the tree.
Although in the majority of incidences maximum likelihood or Bayesian inference is preferable for tree construction, in certain situations maximum parsimony can be a viable alternative. When studying closely related sequences over a short time period the likelihood of back-mutation is relatively low, and hence maximum parsimony tree construction is likely to give an accurate estimation of tree phylogeny. Phylogenetic analysis of virus sequences is often performed with the aim of tracing specific virus history, and in these cases the method of statistical parsimony can be used. The distances depicted by parsimony trees represent the actual number of differences between sequences, whereas for a maximum likelihood tree the probability of change is shown (Fig. 3 ).
Often when studying viruses, closely related sequences are being investigated, with a focus on the accumulation of changes, and in this case a simpler representation of the raw data as depicted by parsimony is desirable. The TCS statistical parsimony program (34) can position sequences internally on a branch, which assists in depicting directly ancestral sequences (see Fig. 3b). Although the statistical parsimony trees drawn by TCS are not bootstrapped, if the data comprise the complete genome sequences of the sampled viruses, then the tree is as accurate and as representative as it can be: It is not sensitive to the choice of a single arbitrary locus because there are no further genetic data retrievable. A useful Web site that lists available phylogenetic programs for analyzing sequence data is http://evolution.genetics.washington.edu/phylip/software.html.
Fig. 3.
Phylogenetic analysis of FMDV genomes from the 2001 outbreak in the United Kingdom. (a) Maximum likelihood phylogenetic tree representing 14 FMDV complete genomes rooted to sequence “1,” constructed using PhyloWIN95 (33), incorporating the HKY model of nucleotide substitution with gamma distributed rate heterogeneity. Bootstrap values from 1,000 replicates are shown. (b) Statistical parsimony representation of the same 14 complete FMDV sequences, constructed using TCS (Version 1.21; 34). Each line represents a nucleotide substitution and each dot a putative ancestor virus.
Future Technologies
Newer technologies are currently being developed that offer the potential to eliminate the use of capillary electrophoresis and even greater throughput. Resequencing microarrays have been developed and used to determine the sequence of the severe acute respiratory syndrome (SARS) coronavirus (35,36). However, development of specific arrays is heavily resource dependent and currently likely to be deployed only in niche markets. Of the newer technologies, sequential ligation systems (SOLiD), solid-phase primer amplification (Solexa), and bead-and-well-based pyrosequencing methods (such as the 454 platform) have the capacity to generate reads of 4–20 Mb in a single run. Although this might be considered excessive for characterization of individual viral genomes, these approaches may allow infrequent mutations within a viral population to be detected. Thus, these methods may be ideal for dissecting the genetic variability within viral populations.
Notes
In addition to ensuring that all solutions used for RNA extraction are RNase free, pipets and work surfaces should be cleaned using 10% bleach followed by DNAzap (Ambion) prior to and between each sample processed.
A logical work flow for processing the samples for sequencing projects is highly recommended (see Fig. 4 for an example). This is particularly important for high-resolution molecular epidemiological studies since the discrimination of samples may be dependent on the accurate determination of only a few nucleotide differences in the complete genome length (3). Therefore, it is important that care is taken to minimize cross-contamination between samples (particularly post-PCR products). If possible, samples should be processed independently (including suitable negative control material), and the study should be organized to attempt to maximize the differences between successive samples tested.
A variety of sample types (including blood, tissues, esophageal-pharyngeal fluid, and cell culture supernatant) can be tested; however, it is usually preferable to test primary material (such as clinical samples) since it is possible that cell culture passage or molecular cloning of viruses can introduce nucleotide changes that can influence the interpretation of results.
Once placed in TRIzol reagent, samples can be stored for extended periods (at a wide range of temperatures, −70 to +4°C).
The requirement to perform a high number of downstream sequencing reactions may necessitate that a relatively large volume of PCR product is generated requiring pooling of RNA, cDNA, or post-PCR products. An additional practical consideration is the fidelity of the DNA polymerase used for the PCR amplification step; if possible proofreading enzymes that are widely available should be used.
In common with other long PCR methods, the parameters of the protocol used for amplification of viral genomes should be optimized prior to routine use. Steps to be considered include the components of the RT or PCR mixes and the cycling times used for amplification. In initial experiments, a PCR targeting a fragment of the 5′ end of the genome can be used to confirm that full-length cDNA has been produced in the RT reaction.
In general, these methods provide an accurate estimation of the viral consensus sequence. However, it is important to recognize that this sequence will be a composite of the component variability that, to a greater or lesser extent, may be present. In spite of concerns that it is theoretically possible that the sequence generated will not represent an actual virus species present in the sample, studies with FMDV indicated that the majority of molecular clones have identical sequences to the consensus ( 37 ). Testing of duplicate samples can generate identical results ( 4), demonstrating that these methods are accurate, and as long as the viral concentrations are relatively high, consensus sequences obtained will mask any individual proofreading errors that might arise due to low fidelity of reverse transcriptase and polymerase enzymes. These aspects relating to accurate determination of the sequences of specific viral genomes (rather than consensus sequences) will be of particular concern in studies that aim to characterize the genetic population structure within samples (i.e., the quasi-species nature of a virus). New technologies and approaches (see Subheading 3.5.) may be utilized to address these important questions that underpin our understanding of viral evolution.
Fig. 4.
Physical separation of the laboratory activities: outline of the separate steps required for the amplification and sequencing of FMDV genomes.
Acknowledgments
This work was funded by Defra research project SE2936. We acknowledge the assistance of colleagues Guido König, Sasmita Upadhyaya, Nigel Ferris, and Geoff Hutchings and Michael Quail from the Wellcome Trust Sanger Institute, Cambridge, for collaboration with the shotgun sequencing approach.
References
- 1.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carrillo C, Tulman E R, Delhon G, Lu Z, Carreno A, Vagnozzi A, et al. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005;79:6487–6504. doi: 10.1128/JVI.79.10.6487-6504.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cottam E M, Haydon D T, Paton D J, Gloster J, Wilesmith J. W, Ferris N. P, et al. Molecular epidemiology of the foot-and-mouth disease virus outbreak in the United Kingdom in 2001. J. Virol. 2006;80:11274–11282. doi: 10.1128/JVI.01236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cottam E M, Thébaud G, Wadsworth J, Gloster J, Mansley L, Paton D. J, et al. Integrating genetic and epidemiological data to determine transmission pathways of foot-and-mouth disease virus. Proc. Biol. Sci. 2008;275:887–895. doi: 10.1098/rspb.2007.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrillo C, Lu Z, Borca M V, Vagnozzi A, Kutish G F, Rock D L. Genetic and phenotypic variation of foot-and-mouth disease virus during serial passages in a natural host. J. Virol. 2007;81:11341–11351. doi: 10.1128/JVI.00930-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heath L, van der Walt E, Varsani A, Martin D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006;80:11827–11832. doi: 10.1128/JVI.01100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson A L, O’Neill H, Maree F, Blignaut B, Carrillo C, Rodriguez L, et al. Mosaic structure of foot-and-mouth disease virus genomes. J. Gen. Virol. 2007;88:487–492. doi: 10.1099/vir.0.82555-0. [DOI] [PubMed] [Google Scholar]
- 8.Simmonds P, Tuplin A, Evans D J. Detection of genome-scale ordered RNA structure (GORS). in genomes of positive-stranded RNA viruses: implications for virus evolution and host persistence. RNA. 2004;10:1337–1351. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domingo E, Escarmis C, Lazaro E, Manrubia S C. Quasispecies dynamics and RNA extinction. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Cottam E M, Wadsworth J, Shaw A E, Rowlands R J, Goatley L, Maan S, et al. Transmission pathways of foot-and-mouth disease virus in the United Kingdom in 2007. PLoS Pathog. 2008;4:e1000050. doi: 10.1371/journal.ppat.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason P W, Pacheco J M, Zhao Q Z, Knowles N J. Comparisons of the complete genomes of Asian, African and European isolates of a recent foot-and-mouth disease virus type O pandemic strain (PanAsia) J. Gen. Virol. 2003;84:1583–1593. doi: 10.1099/vir.0.18669-0. [DOI] [PubMed] [Google Scholar]
- 12.Carrillo C, Tulman E R, Delhon G, Lu Z, Carreno A, Vagnozzi A, et al. High throughput sequencing and comparative genomics of foot-and-mouth disease virus. Dev. Biol. (Basel) 2006;126:23–30. doi: 10.1128/jvi.79.10.6487. [DOI] [PubMed] [Google Scholar]
- 13.Oem J K, Lee K N, Cho I S, Kye S J, Park J H, Joo Y S. Comparison and analysis of the complete nucleotide sequence of foot-and-mouth disease viruses from animals in Korea and other PanAsia strains. Virus Genes. 2004;29:63–71. doi: 10.1023/B:VIRU.0000032789.31134.eb. [DOI] [PubMed] [Google Scholar]
- 14.Nobiron I, Remond M, Kaiser C, Lebreton F, Zientara S, Delmas B. The nucleotide sequence of foot-and-mouth disease virus O/FRA/1/2001 and comparison with its British parental strain O/UKG/35/2001. Virus Res. 2205;108:225–229. doi: 10.1016/j.virusres.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 15.Du J, Chang H, Cong G, Shao J, Lin T, Shang Y, et al. Complete nucleotide sequence of a Chinese serotype Asia1 vaccine strain of foot-and-mouth disease virus. Virus Genes. 2007;35:635–642. doi: 10.1007/s11262-007-0126-8. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Xu Z, He Y, Yao Q, Zhang K, Jin M, et al. Genome comparison of a novel classical swine fever virus isolated in China in 2004 with other CSFV strains. Virus Genes. 2006;33:133–142. doi: 10.1007/s11262-005-0048-2. [DOI] [PubMed] [Google Scholar]
- 17.Herring B L, Bernardin F, Caglioti S, Stramer S, Tobler L, Andrews W, et al. Phylogenetic analysis of WNV in North American blood donors during the 2003–2004 epidemic seasons. Virology. 2007;363:220–228. doi: 10.1016/j.virol.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 18.Marston D A, McElhinney L M, Johnson N, Müller T, Conzelmann K K, Tordo N, et al. Comparative analysis of the full genome sequence of European bat lyssavirus type 1 and type 2 with other lyssaviruses and evidence for a conserved transcription termination and polyadenylation motif in the G-L 3′ non-translated region. J. Gen. Virol. 2007;88:1302–1314. doi: 10.1099/vir.0.82692-0. [DOI] [PubMed] [Google Scholar]
- 19.Lu L, Li C, Fu Y, Gao F, Pybus O G, Abe K, et al. Complete genomes of hepatitis C virus (HCV). subtypes 6c, 6l, 6o, 6p and 6q: completion of a full panel of genomes for HCV genotype 6. J. Gen. Virol. 2007;88:1519–1525. doi: 10.1099/vir.0.82820-0. [DOI] [PubMed] [Google Scholar]
- 20.Bialasiewicz S, Whiley D M, Lambert S B, Wang D, Nissen M D, Sloots T P. A newly reported human polyomavirus, KI virus, is present in the respiratory tract of Australian children. J. Clin. Virol. 2007;40:15–18. doi: 10.1016/j.jcv.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobs G B, Loxton A G, Laten A, Engelbrecht S. Complete genome sequencing of a non-syncytium-inducing HIV type 1 subtype D strain from Cape Town, South Africa. AIDS Res. Hum. Retroviruses. 2007;23:1575–1578. doi: 10.1089/aid.2007.0167. [DOI] [PubMed] [Google Scholar]
- 22.Borges M B, Caride E, Jabor A V, Malachias J M, Freire M S, Homma A, et al. Study of the genetic stability of measles virus CAM-70 vaccine strain after serial passages in chicken embryo fibroblasts primary cultures. Virus Genes. 2008;36:35–44. doi: 10.1007/s11262-007-0173-1. [DOI] [PubMed] [Google Scholar]
- 23.Xi D, Li J, Han C, Li D, Yu J, Zhou X. Complete nucleotide sequence of a new strain of Tobacco necrosis virus A infecting soybean in China and infectivity of its full-length cDNA clone. Virus Genes. 2008;36:259–266. doi: 10.1007/s11262-007-0185-x. [DOI] [PubMed] [Google Scholar]
- 24.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 25.Huelsenbeck J P, Ronquist F, Nielsen R, Bollback J P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- 26.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 27.Michener C D, Sokal R R. A quantitative approach to a problem in classification. Evolution. 1957;11:130–162. doi: 10.2307/2406046. [DOI] [Google Scholar]
- 28.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 29.Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 1984;20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 30.Posada D, Crandall K A. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 32.Yang Z, Rannala B. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo Method. Mol. Biol. Evol. 1997;14:717–724. doi: 10.1093/oxfordjournals.molbev.a025811. [DOI] [PubMed] [Google Scholar]
- 33.Galtier N, Gouy M, Gautier C. SeaView and Phylo_win, two graphic tools for sequence alignment and molecular phylogeny. Comput. Applic. Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 34.Clement M, Posada D, Crandall K A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 35.Sulaiman I M, Liu X, Frace M, Sulaiman N, Olsen-Rasmussen M, Neuhaus E, et al. Evaluation of affymetrix severe acute respiratory syndrome resequencing GeneChips in characterization of the genomes of two strains of coronavirus infecting humans. Appl. Environ. Microbiol. 2006;72:207–211. doi: 10.1128/AEM.72.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong C W, Albert T J, Vega V B, Norton J. E, Cutler D. J, Richmond T. A, et al. Tracking the evolution of the SARS coronavirus using high-throughput, high-density resequencing arrays. Genome Res. 2004;14:398–405. doi: 10.1101/gr.2141004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cottam, E. M., King, D. P., Wilson, A., Paton, D. J., and Haydon, D. T. (2009) Analysis of Foot-and-mouth disease virus nucleotide sequence variation within naturally infected epithelium. Virus Res. In press [DOI] [PubMed]