

Abstract
The development of high-throughput molecular technologies and associated bioinformatics has dramatically changed the capacities of scientists to produce, handle, and analyze large amounts of genomic, transcriptomic, and proteomic data. A clear example of this step-change is represented by the amount of DNA sequence data that can be now produced using next-generation sequencing (NGS) platforms. Similarly, recent improvements in protein and peptide separation efficiencies and highly accurate mass spectrometry have promoted the identification and quantification of proteins in a given sample. These advancements in biotechnology have increasingly been applied to the study of animal infectious diseases and are beginning to revolutionize the way that biological and evolutionary processes can be studied at the molecular level. Studies have demonstrated the value of NGS technologies for molecular characterization, ranging from metagenomic characterization of unknown pathogens or microbial communities to molecular epidemiology and evolution of viral quasispecies. Moreover, high-throughput technologies now allow detailed studies of host-pathogen interactions at the level of their genomes (genomics), transcriptomes (transcriptomics), or proteomes (proteomics). Ultimately, the interaction between pathogen and host biological networks can be questioned by analytically integrating these levels (integrative OMICS and systems biology). The application of high-throughput biotechnology platforms in these fields and their typical low-cost per information content has revolutionized the resolution with which these processes can now be studied.
The aim of this chapter is to provide a current and prospective view on the opportunities and challenges associated with the application of massive parallel sequencing technologies to veterinary medicine, with particular focus on applications that have a potential impact on disease control and management.
Key words: Next-generation sequencing, Animal infectious disease management, OMICS
Introduction
Genetic characterization of infectious agents plays a central role in the diagnosis, monitoring, and control of infectious diseases. The development of rapid DNA sequencing methods based on the selective incorporation of chain-terminating dideoxynucleotides ([1]; later termed “first-generation sequencing technologies”) and the polymerase chain reaction (PCR) DNA amplification technologies ([2]; reviewed in [3]) has paved the way for the study of biological and evolutionary processes at the molecular level. Such technologies have been extensively applied to the diagnosis and molecular epidemiology of infectious diseases of livestock and become important tools for targeted research on host-pathogen interactions. The most recent versions of these first-generation sequencing technologies are widely accessible and provide high-quality data. However, their application to projects such as whole genome sequencing is expensive and time-consuming, often requiring prior knowledge of the target genome for specific template amplification. These limitations have been particularly problematic for large sequencing projects and have motivated the development of alternative, post-Sanger sequencing technologies (“next-generation sequencing” or NGS).
Next-generation sequencing platforms provide unprecedented throughput, generating hundreds of gigabases of data in a single experiment. Although the initial capital investment and cost per experiment remain high, the price per information unit (nucleotide) has been dramatically reduced in comparison with first-generation sequencing. Moreover, these technologies allow unbiased sequencing without prior knowledge of the complete DNA content in a sample while retaining the flexibility to allow for targeted sequencing.
This paradigm shift in the scale of DNA sequence data has revolutionized the way biological and evolutionary processes can be studied at the molecular level, enabling genome projects previously restricted to high profile model organisms and human pathogens to target pathogens of lesser economic and medical significance.
Such advancements are now being increasingly applied to veterinary medicine. As a result, the increasing availability of these technologies combined with the rapid development of applied tools and protocols has provided a diverse array of applications for use in genomics and transcriptomics and even routine diagnostics.
In this chapter, we review recent advances in NGS technologies that are becoming commonplace in many laboratories, with an emphasis on the applications that have the potential to significantly impact on diagnosis, prevention, and control of infectious diseases in animals.
Massive Parallel Sequencing
Technologies
A number of different NGS platforms are currently available, with each utilizing different sequencing chemistries and detection strategies. This has led to individual systems having their own strengths and limitations (reviewed in [4–6]). Second-generation sequencing platforms vary in technology and chemistry used but have the following properties in common:
A DNA library is made from the sample. This library is either representing all DNA in sample without prior knowledge or a targeted library using PCR amplification or alternative enrichment methods. Adapter sequences are joined to the DNA molecules (by ligation or amplification) and can include a barcode sequence that allows multiplexing of several samples in an experiment.
Individual DNA molecules in each library are clonally amplified.
Clonal DNAs are sequenced by massive parallel sequencing.
Hundreds of thousands of DNA sequence reads result and need to be processed.
The second-generation sequencing platforms first emerged on the market with an emphasis on extreme high-throughput sequencing applications and initially were restricted to genome sequencing centers or core facilities. These technologies use different detection principles including pyrosequencing (454 Life Sciences, acquired by Roche, available since 2005, but planned to be discontinued by mid-2016), Illumina’s sequencing by synthesis (previously Solexa, available since 2007), SOLiD ligation-based sequencing (Life Technologies, available since 2006), and, more recently, the Ion Torrent semiconductor sequencing technology. Over the last 5–7 years, all of the major platforms have made significant improvements, with notable advancements made in terms of protocol complexity, overall performance (including read length, fidelity, lower input DNA), and cost efficiency. More recently, smaller benchtop sequencers [7] have become available, making the technology more accessible for use in routine microbiology laboratories, while academic core facilities and commercial service providers focused increasingly upon providing users with access to a wider diversity of the sequencing technologies available. These developments will bring NGS technologies within the reach of many more research groups and diagnostic laboratories where NGS analysis of a single isolate will generate significant quantities of data, many orders of magnitude greater than that generated by other typing methods.
In addition to the continuous improvement of existing platforms, newer methodologies are being developed. Third-generation sequencing technologies are defined as single-molecule sequencers (reviewed in [6, 8]). These approaches promise additional advantages such as scalability, simplicity, long read length, and low operational costs and do not require clonal amplification of template DNA molecules, thereby removing potential errors associated with clonal amplification. A single third-generation platform is currently available on the market since 2011 (PacBio, Pacific Biosciences) that sequences long single DNA molecules in real time, known as SMRT sequencing. Other technologies are still under development (e.g., [4]) such as DNA sequencing in nanopores that offer the potential of simple, inexpensive, single-molecule sequencing in miniaturized or highly scalable devices [9]. Although substantial validation data is still required, these technologies have the potential to make NGS even more widely available in diagnostic labs.
Challenges
While the advantages of NGS are numerous (unprecedented scale of genomic information, scalability, low-cost per information content, and high throughput), several challenges remain to be addressed.
Several processes in the NGS workflow, from sample selection to data interpretation, are potentially vulnerable to bias and/or error introduction (Fig. 1). This includes the error rates of the sequencing chemistry and library construction, as well as point mutations and insertions/deletions that may arise during reverse transcription and PCR amplification. The amplification of DNA by PCR to obtain clonal template sequences is subject to error introduction [8] and may result in an amplification bias impacting the relative frequency of sequence variants present in the sample. Sampling bias can be introduced when a relatively small number of samples are analyzed per epidemiological unit (e.g., single animal or herd) due to financial constraints restricting the thorough use of NGS. When only a small proportion of the nucleic acids in a single sample are subjected to sequence analysis, technical sampling bias occurs. While NGS data provides a high resolution of an individual sample, the resolution of higher epidemiological scales (Fig. 2) may thus be compromised due to insufficient sampling. Moreover, as a minimum amount of genetic material is needed as input for NGS workflows, this can result in bias towards samples with the highest pathogen titers. Errors and bias may also be introduced by methods to increase the sensitivity of the workflow, such as targeted pathogen genome amplification [10–12] or enrichment (e.g., [13]). Furthermore, reagent contamination has been reported to interfere with metagenomic analyses [14, 15]. Errors can also be made during the sequencing process itself via base miscalls by NGS machines. For example, the loss of synchronicity (dephasing) in a percentage of the clonally amplified DNA template [16] results in increased noise and sequencing errors [17, 8]. Each of the different NGS platforms available has its own distinct characteristics in terms of read and error profiles, with the Illumina platform often regarded as having the lowest error rate, while other platforms can produce longer reads [7, 18, 19]. In addition, certain biases and errors can be introduced during the analysis of NGS datasets, due to the limitations of the algorithms or reference data used [20, 21]. For example, genomic repeat regions are a well-known problem for sequence assembly algorithms [22]. However, software advancements combined with platform and chemistry developments are expected to further reduce operational costs, error rates, and input DNA quantities, allowing more careful sampling strategies and reducing experimentally introduced error and bias in the future.
Fig. 1.
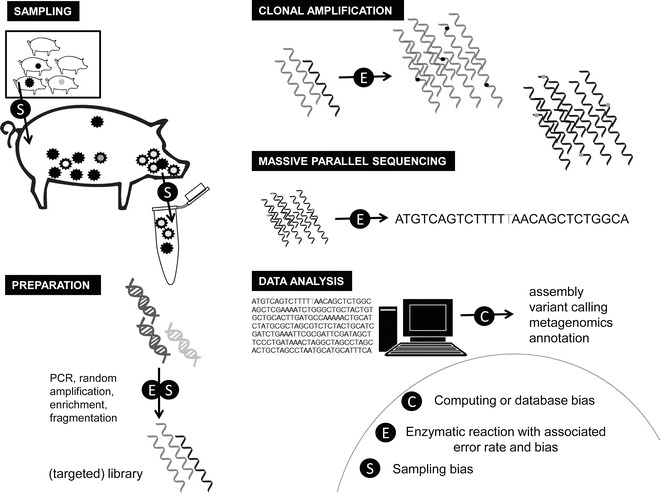
Steps in next-generation sequencing and data analysis workflows where error and bias introduction may occur
Fig. 2.
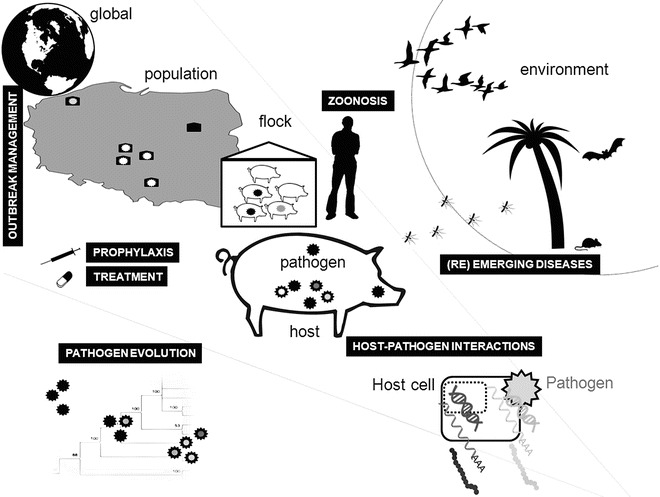
High-throughput technologies can be applied to numerous aspects of animal infectious diseases
As a result of errors and bias introductions, NGS data needs to be “cleaned”. This includes sequence filtering (removing low-quality sequences) and alignment followed by variant calling and error correction. Discriminating true biological variants from those due to experimental noise is an important issue when trying to identify low-frequency variants in a population, for example, in viral quasispecies or metagenomic analyses, and there are currently a number of bioinformatics tools to aid in this (e.g., [23–26, 19]). Currently, a multitude of software has been developed to address different aspects of NGS analyses [27, 28]. However, the available algorithms for both genome assembly and amplicon analysis can present some limitations [29], meaning that custom-made scripting and in-house resolution of bioinformatic problems are often needed to investigate novel datasets and specific hypotheses. In this context, researchers are frequently faced with the need to acquire computer skills and bioinformatics expertise. To evaluate the potential of NGS for a wider group of scientists and diagnosticians, there is a real need to develop flexible and practical bioinformatics workflows that can provide user-friendly tools for the analysis of massive datasets and that become publicly available. Although some software with a menu-driven approach is available (e.g., Geneious, CLC Workbench, Galaxy), most applications are optimized on UNIX-based operating systems and require some bioinformatics expertise. Although less user-friendly, UNIX-based pipelines are typically freely available to the NGS user community and are equipped with algorithms that track the high pace of innovation in the NGS field.
A further issue is the scale of genetic data produced by NGS technologies which presents a physical constraint in terms of data storage and analysis. Although limited datasets (e.g., resulting from the desktop-range 2nd-generation sequencers) can be managed using modest computing resources, like high-end desktop computers running virtual Linux machines [4], larger datasets typically require high-performance computational clusters, which present a considerable investment and require sufficient information technology (IT) support. Cloud computational resources (i.e., renting time from commercial high-performance computational clusters) may be a solution [30] although further developments are needed, given the data transfer issues resulting from huge file sizes [4]. Another issue for diagnostic laboratories is protection of data from unauthorized access, which cannot be guaranteed in the cloud, as data from diagnostic examinations need to be kept confidential. For labs frequently producing NGS data, data storage and backup costs can be substantial. Ideally, these huge genetic datasets should be made publicly available to the scientific community as they provide a source of information applicable to better understanding disease, design of targeted assays, systems biology, and integrated OMICS analysis approaches. To this end, online repositories such as the Sequence Read Archive (SRA; [31]) have been created to store both raw NGS and intermediate analysis files.
It will also be important to consider how results from complex and massive NGS datasets will be communicated to policy groups and the public and become a decision-supporting tool. To this end, it is necessary that scientists and diagnosticians develop and agree on data formats for the communication of NGS results for analyses that go beyond simple genome sequences, for instance, for reporting quasispecies compositions.
Application of NGS to Animal Infectious Disease
NGS technology is now being increasingly applied to study the etiology, genomics, evolution, and epidemiology of animal infectious diseases as well as host-pathogen interactions (Fig. 2). These applications have provided novel insights and illustrate the potential of this new technology to directly impact on our understanding and control strategies for animal infectious disease.
NGS platforms have been instrumental in the completion of large animal genomes and the documentation of genomic variation (reviewed in [32]). Available livestock genomes now include bovine, pig, sheep, equine, and avian [32] which provide an important source of knowledge for understanding food production and animal interaction with infectious pathogens. Additional livestock genome sequencing efforts have documented genomic variation providing information for the development of genetic markers applicable to animal breeding genetics [33–35], including traits related to pathogen resistance and interaction with microbial communities in poultry [36]. Others have used novel sequencing technologies for the targeted study of specific gene families occupying key roles in host immunology (e.g., Toll-like receptor (TLR) gene family [37]).
The high variability and large size of the mitochondrial genome (mtDNA) of eukaryotic parasites have been recently explored using NGS (reviewed by [38]). mtDNA sequences proved very informative in epidemiological studies [38] but also include comparative mtDNA sequencing of parasites with low and high zoonotic potential [39]. Targeting specific polymorphic genes in the Cryptosporidium parvum genome using NGS, extensive intra-host genetic diversity was documented [40]. Studies of the transcriptome (all mRNA transcripts in an organism, tissue, or cell; also called RNA-Seq) of different parasite species and/or developmental stages provide insights into aspects of gene expression, regulation, and function, which are major steps to understanding their biology (reviewed in [41]). Examples include the characterization of the transcriptome from Eimeria sp. from chicken [42] and Taenia sp. from sheep [43]. In addition, RNA-Seq data have been used to predict potential drug targets [44] and to identify key genes involved in anthelmintic resistance [45].
Over the last five years, NGS has been used as an extremely important tool in the tracing of transmission, genome characterization, and outbreak management of both viral and bacterial diseases. The sequencing of these two pathogen types poses very different sets of challenges and issues, where the large data output expressed typically in the Mb (megabase) to Gb (gigabase) range [4] is particularly suited for the sequencing of larger bacterial genomes. The high plasticity of some microbial genomes, with large mobile elements, gene-coding plasmids, chromosomal genes, and regions of extensive genetic variability, can frequently complicate genome assembly [46]. While most viral genomes are significantly smaller than their bacterial counterparts, the viral replication biology (particularly that of RNA viruses) poses its own unique problems. These involve the inherent variability of many viral genomes due to replication machinery lacking efficient proofreading mechanisms. This, combined with a short generation time and high replication rate, results in a complex mix of differing genomes (a “swarm” of closely related viruses) within a single host that are often termed as “quasispecies,” reviewed in [47]. In addition, recombination and reassortment of segmented viral genomes frequently occur. NGS techniques offer an unprecedented “step-change” increase in the amount of sequence data that can be generated from both types of these samples.
Figure 3 (different scales of sequence analysis) highlights where genetic analyses can target different biological scales and whether these are within an individual host, or between hosts, resulting in either host variation or inter-herd diversity/outbreak transmission.
Fig. 3.
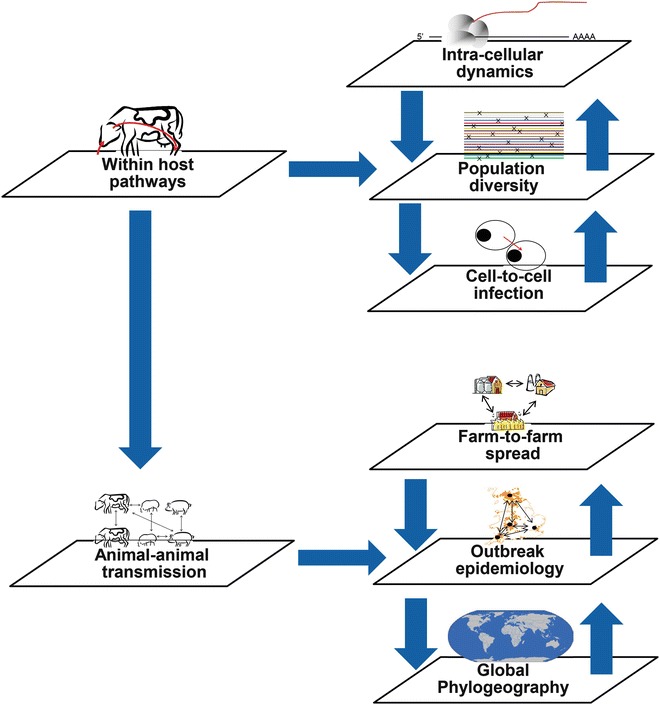
The differing levels of intra- and inter-host variation that can be explored using NGS technologies range from intracellular dynamics to epidemiological applications
At the level of the quasispecies, NGS technologies can now determine complete viral genomes to a fine-point resolution, allowing the quantification of viral diversity within samples [48] and making the sequencing of large numbers of samples economically feasible. The technology will allow the comparison of genetically diverse populations from different replication sites within a host [49, 50]. Wright and colleagues investigated the genetic diversity and resulting quasispecies population after inoculation of foot-and-mouth disease virus (FMDV) into a single animal and identified genetically distinct populations originating from different lesions [50]. Morelli and colleagues [51] studied the evolution of FMDV intra-sample sequence diversity during serial transmission in bovine hosts, providing novel insights into the fine-scale evolution of an RNA virus. NGS can also provide insights on microevolutionary processes of viruses at different scales, including the fine-point resolution molecular epidemiology analysis of outbreaks [52].
Recent studies on influenza A viruses have demonstrated that minority variants present in the donor population can be successfully transmitted to the recipient host and become prevalent with unpredictable impact on the virus biological properties [53, 54]. These findings suggest that the use of NGS approaches in RNA virus surveillance will be strategic to promptly detect biologically relevant viral quasispecies and will help in expanding our understanding of viral dynamics and emergence and the possible implications of mutation emergence for studies done using isolated viruses [55, 56].
The study of the viral swarm within individual hosts also has implications for understanding the evolutionary dynamics of viral populations under selection pressures, e.g., antiviral drugs. This has been a particularly active field in human medicine, e.g., with regard to human immunodeficiency virus (HIV) antiviral drugs response, drug resistance, and viral tropism (reviewed in [57–59]) and human influenza A (e.g., [60]) studies. The technologies’ application to personalize antiviral treatment as a function of genetic marker makeup in human medicine is just around the corner [61]. Although at present only an emerging field in veterinary science, the development of antiviral drugs has the potential to translate into efficient animal infectious disease control strategies (e.g., [62, 63]).
The majority of the papers using NGS to investigate animal infectious disease focus specifically upon the level of animal-to-animal transmission and the characterization of pathogens within a single host, as this yields the most useful data in terms of outbreak management and identifying mechanisms/sources of disease transmission. For example, Lefébure and colleagues [64] used NGS to study genome complexity and horizontal gene transfer in foodborne Campylobacter spp. Biek et al. [65] studied local transmission patterns of Mycobacterium bovis in cattle and wildlife reservoirs using whole genome sequences from 31 samples originating on five farms. These demonstrated enough diversity between individual outbreaks to determine evolutionary variation down to herd level. The identification of novel antimicrobial resistance genes in the foodborne pathogen Campylobacter coli [66] was possible using NGS, and the application of NGS technologies during a recent crisis involving foodborne enterohemorrhagic Escherichia coli O104:H4 allowed a swift genomic identification [67] that was key to the management of this crisis. Finally, the technology also proved very informative to study the molecular epidemiology and evolutionary history of extremely monomorphic Mycoplasma mycoides subsp. mycoides SC [68] in addition to studies tracing medically significant pathogens [69–71]. Samples from the US 2006–2007 West Nile virus (WNV) outbreak in birds were characterized using Illumina sequencing, resulting in the identification of a new genetic variant containing a 13-nucleotide indel [72]. A survey of Chinese domestic fowl using RNA-Seq on an Ion Torrent identified a novel Coronavirus, providing insights into the diversity and distribution of avian coronaviruses [73]. A further study has also investigated the role of Usutu virus in causing epizootic infection in blackbirds in Germany [74].
The advent of NGS has also led to the cost-efficient sequencing of complete viral genomes including avian influenza virus [75–78], classical swine fever virus [79], and bluetongue viruses [80]. An optimized method incorporating 454 sequencing for universal nonspecific RNA viral genomes from brain and cell culture material was applied to Lyssaviruses [81]. Other groups have reported the characterization of Louping ill virus in lambs [82], porcine reproductive and respiratory syndrome virus [83], and herpesviruses from Asian elephants [84]. Furthermore, studies using random amplification techniques have identified mixed infections of paramyxoviruses and avian influenza in bird populations [77, 85, 78]. Efficient influenza A-specific resequencing strategies [86, 87] have allowed the study of quasispecies-scale genetic variability with implications for immune response [88, 89], host cell line adaptation [90, 91], antiviral drug resistance [92], and pathogenicity [53]. Likewise, efficient targeted CSFV genome sequencing using NGS has led to insights in classical swine fever virus (CSFV) epidemiology based on isolates from an outbreak in wild boar from Germany [79] and in the role of quasispecies diversity in CSFV pathogenicity [93].
NGS technology has also allowed the characterization of complete microbial communities without prior knowledge. For instance, the unbiased characterization of conserved bacterial ribosomal RNA-encoding sequences (rRNA profiling) has been applicable to whole microbial community characterization (e.g., [94, 95]) and to molecular characterization of (uncultured) bacteria [96]. Metagenomics is the determination of the sequence content of a complete microbial community (reviewed in [97]). The analysis of the resulting data can be taxonomy oriented (identification and quantification of species diversity; [98]) or function based (identification of coding gene diversity, e.g., [99]). The latter has significant potential, e.g., in the screening for virulence-associated, antibiotic resistance genes, and vitamin production-associated genes in microbial communities [100]. NGS also offers the potential of unbiased sequencing of the nucleic acid content of a sample and has been applied to the characterization of the viral metagenome in samples [101] or the identification of unknown or unexpected viruses in diseased animals or insect vectors. Furthermore, metagenomic NGS workflows allow the study of the interaction of treatment with an animal’s microbiome [102]. In the microbiology lab, NGS has the potential for greater diagnostic resolution than any other typing method, and clinical microbiology labs are currently investigating its potential for routine diagnosis [103, 104].
Using NGS-based metagenomic approaches, multiple potential disease agents have been identified in a wide range of both domestic and wild animals (reviewed in [105–109]). Although the common goal is to identify potential pathogens, the studies can roughly be divided into three categories: (1) investigations of outbreaks of unknown etiology, (2) investigations of well-known disorders presumed to be of multifactorial etiology, and (3) metagenomic studies of reservoir species and vectors. Examples of the first category include the identification of a novel Orthobunyavirus affecting cattle (described in more detail below), an astrovirus in the brain of farmed minks suffering from encephalomyelitis [110], and a novel picornavirus as candidate etiologic agent for turkey viral hepatitis [111], among others. The second category encompasses investigations aimed at finding contributing infectious agents to complex diseases, such as colony collapse disorder of honey bees [112, 113] and postweaning multisystemic wasting syndrome in pigs [114]. Studies in the third category have been performed on diverse animal species suspected to be important reservoirs, such as bats [115, 116], African bush pigs [117], and red fox [118], as well as typical vector organisms, such as ticks [119].
Although it is an important first step, the identification and genetic characterization of candidate pathogens are not enough to establish causal relationships or understand how they may be associated with disease. It is therefore necessary to use a synergistic approach combining molecular diagnostic tools, such as NGS-based metagenomics and follow-up PCR-based assays targeting detected pathogen sequences, with more conventional diagnostic methods, including isolation and characterization. This is crucially important in situations where metagenomic data indicate the potential presence of multiple pathogens. While PCR-based prevalence studies in matching disease cases and healthy controls can provide further evidence for disease association, isolation of candidate pathogens is required to assign causality by addressing Koch’s postulates [120]. The assembled data from such a multidisciplinary (pathology, epidemiology, metagenomic data, PCR prevalence studies, isolation, characterization, etc.) should be used to identify the most likely candidate etiologic agent and to make informed intervention decisions. The synergetic and parallel use of molecular and classical methods not only results in detection of infectious agents and development of targeted diagnostic tests but also has the potential to make isolates or strains available shortly after the occurrence of outbreaks. The availability of isolates or strains is of special importance to allow the design of effective vaccines or antimicrobial drugs.
The power of NGS to boost the veterinary laboratory community’s responsiveness to emerging diseases was demonstrated through the discovery of a novel Orthobunyavirus in 2011 associated with fever, decreased milk production, and diarrhea in dairy cattle. Metagenomics, using 454 technology, allowed the identification of a novel virus, subsequently named Schmallenberg virus (SBV), in an epidemiological cluster of diseased cattle in Germany [121]. These viral sequences were used to rapidly design targeted molecular tests that were used to confirm a clear association between the presence of the virus and affected animals [110]. International adoption of these molecular tests identified a widespread occurrence of SBV in European countries (http://www.efsa.europa.eu/en/supporting/pub/429e.htm) and its detection in stillborn and malformed lambs [122, 123], as well in insect vectors [124, 125]. The molecular tests were also helpful in targeting samples for isolation of the virus, which ultimately led to the development of a prototype vaccine currently under evaluation [126].
Metagenomic NGS workflows also have the potential use for quality control of biological products [127] and vaccines [128–132] and provide a powerful approach for the identification and characterization of unexpected of highly divergent pathogen variants [133, 85] that may remain undetected using targeted diagnostic tests.
The technological possibility to study both the host and the pathogen with high resolution on the level of their genome, transcriptome, or proteome opens opportunities to study host/pathogen interactions at several levels ((genomics, transcriptomics, microRNAs (miRNA)) and ultimately to analytically integrate these levels (integrative omics or systems biology) aiming to study the interaction of pathogen, microbiome, and host biological networks with many examples in veterinary science. Nordentoft and colleagues [134] used NGS metagenomics to study the influence of livestock management parameters and infection with Salmonella enteritidis on the microbial community in the chicken intestinal tract. Another study [135] documented the effect of Campylobacter jejuni infection on the chicken fecal microbiome. The application of metagenomic techniques in poultry production could lead to the development of novel alternatives to antibiotic growth promoters and better understanding of the colonization of food production animals by foodborne pathogens such as Salmonella enterica and Campylobacter spp. [36]. Other studies investigated the host response to pathogen infection. Glass and colleagues [136] used NGS transcriptomics to document bovine resistance and tolerance traits to parasitic infection. The technology was also used to study the ferret transcriptome response to influenza infection [137], the chicken transcriptome response to Marek’s disease [138], the swine response to porcine reproductive and respiratory syndrome virus infection [139], and the changes in the mouse transcriptome after Brucella sp. infection [140].
microRNAs are considered to be a key mechanism of gene regulation in both parasites and viruses. Their characterization contributes to better understanding the complex biology of pathogens. Wang and coworkers [141] characterized microRNA sequences from Orientobilharzia turkestanicum, a fluke with zoonotic potential infecting sheep, and identified key target miRNAs for parasite energy metabolism, transcription initiation factors, signal transduction, and growth factor receptors. Virus-encoded microRNAs (vmiRNA) regulating viral or cellular transcripts can be targeted for virus discovery [142, 143]. miRNAs also play important roles in regulating host-pathogen interactions. NGS has been applied to investigate whether infection can modulate miRNA biogenesis and has also been used to identify miRNAs that influence pathogen replication, tropism, and pathogenic potential [144–149]. In particular, cellular miRNAs have been shown to interact with the viral genomic RNA or mRNA, facilitating or inhibiting the virus life cycle. These molecules have demonstrated immense potential as a source of antiviral therapeutics effective against a number of viruses (adenovirus, rabies, Venezuelan equine encephalitis, porcine reproductive and syndrome virus [150–153]) or for the design of live-attenuated virus vaccine based on miRNA-mediated gene silencing [154, 155, 147].
Conclusions
Next-generation sequencing technologies have the potential to revolutionize our understanding of the complex dimensions of animal infectious disease and infection biology (Fig. 2), ranging from the intracellular interactions to disease epidemiology. The application of high-throughput biotechnology platforms in these fields and their typical low-cost per information content has increased the resolution with which these processes can now be studied.
We now have high-resolution tools that provide veterinary diagnostic laboratories with the ability to undertake swift and flexible responses to emerging infectious diseases and unexpected pathogen variants. Moreover, these tools provide an increased resolution for the characterization of pathogens and provide important assets to improve our understanding. Fundamental research on pathogen evolution, adaptation, and virulence determinants can now be studied on a scale allowing within and between host dissections of genetic variability. Moreover, high-throughput tools open new perspectives to study the complex interaction between pathogen, host, and microbiome with very high resolution and to deepen our understanding of the key biological processes leading to protective immunity.
Not only will our increased understanding of pathogens and their interaction with livestock impact on future disease prevention, control, and management strategies, but the technologies may themselves become part of the intervention strategies, providing high-resolution data for molecular epidemiology to rapidly trace the origin and spread of outbreaks, for molecular typing, for predicting, and for optimizing the outcome of targeted treatment with antibiotics, antivirals, and anthelmintic.
The ready availability of high-resolution genomic and transcriptomic data will impact upon the targeted development of novel vaccines and drugs [156, 157], while NGS has the potential to become a powerful tool for the control of vaccines and other biological products.
As with any new technology, challenges remain. In the case of NGS, these include the requirement for expertise in both the laboratory and in the analysis of huge datasets and the current need for high investment in laboratory and data analysis hardware. As the technology is ever evolving towards lower cost, user-friendliness, and accessibility for smaller research and diagnostic labs, efforts are needed to make the data analysis more accessible to nonexpert users. This includes proper modeling of the sources of error introduction, solutions for public data storage, development of user-friendly but high standard analysis pipelines for routine applications, etc. Both the industry and the NGS user community can play a role in this evolution.
Similarly, recent improvements in protein and peptide separation efficiencies and highly accurate mass spectrometry have promoted the identification and quantification of proteins in a given sample [158]. Directly targeting peptide and protein content in a sample, proteomic approaches provide important additional information taking known issues, such as the quantitative discrepancy between mRNA transcript levels and final protein levels and posttranslational modification, into account [159].
Novel proteomic approaches have been applied to animal infectious disease research, including the study of E. coli response to chicken sera [160], proteomic profiling of porcine sera after FMDV infection [161], host-pathogen interaction during bovine mastitis [159], and metaproteomic studies characterizing the collective proteome of microbial communities [162].
This section contains excellent contributions exploring the application of high-throughput technologies to animal infectious diseases, including functional genomics of tick vectors infected with eukaryotic parasites, metagenomic approaches to detect bee viral pathogens, proteomics of vector-host-pathogen interactions, and NGS applications exploring parasites and intervention strategies.
Acknowledgments
The collaboration between the authors was supported by Epi-SEQ: a research project supported under the 2nd joint call for transnational research projects by EMIDA ERA-NET (FP7 project nr 219235). Additional support for this work in the United Kingdom was obtained from the Department of Environment, Food and Rural Affairs (Defra project SE2940) and BBSRC (BB/I014314/1).
Footnotes
The Epi-SEQ consortium (www.epi-seq.eu)
Contributor Information
Mónica V. Cunha, Email: monica.cunha@iniav.pt
João Inácio, Phone: +351 21 711 5360, Email: j.inacio@brighton.ac.uk.
Steven Van Borm, Email: stevenvanborm@coda-cerva.be.
References
- 1.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullis K, Faloona F, Scharf S, et al. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51:263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JM, Stirling D. A short history of the polymerase chain reaction. Methods Mol Biol. 2003;226:3–6. doi: 10.1385/1-59259-384-4:3. [DOI] [PubMed] [Google Scholar]
- 4.Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 5.Radford AD, Chapman D, Dixon L, et al. Application of next-generation sequencing technologies in virology. J Gen Virol. 2012;93:1853–1868. doi: 10.1099/vir.0.043182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52:413–435. doi: 10.1007/s13353-011-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loman NJ, Misra RV, Dallman TJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 8.Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19(R2):R227–240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 9.Eisenstein M. Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotechnol. 2012;30:295–296. doi: 10.1038/nbt0412-295. [DOI] [PubMed] [Google Scholar]
- 10.Brodin J, Mild M, Hedskog C, et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS One. 2013;8:e70388. doi: 10.1371/journal.pone.0070388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim KH, Bae JW. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl Environ Microbiol. 2011;77:7663–7668. doi: 10.1128/AEM.00289-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosseel T, Van Borm S, Vandenbussche F, et al. The origin of biased sequence depth in sequence-independent nucleic Acid amplification and optimization for efficient massive parallel sequencing. PLoS One. 2013;8:e76144. doi: 10.1371/journal.pone.0076144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pettengill JB, McAvoy E, White JR, et al. Using metagenomic analyses to estimate the consequences of enrichment bias for pathogen detection. BMC Res Notes. 2012;5:378. doi: 10.1186/1756-0500-5-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lysholm F, Wetterbom A, Lindau C, et al. Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS One. 2012;7:e30875. doi: 10.1371/journal.pone.0030875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naccache SN, Greninger AL, Lee D, et al. The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J Virol. 2013;87:11966–11977. doi: 10.1128/JVI.02323-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiteford N, Skelly T, Curtis C, et al. Swift: primary data analysis for the Illumina Solexa sequencing platform. Bioinformatics. 2009;25:2194–2199. doi: 10.1093/bioinformatics/btp383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 18.Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zagordi O, Klein R, Daumer M, Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archer J, Rambaut A, Taillon BE, et al. The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time—an ultra-deep approach. PLoS Comput Biol. 2010;6:e1001022. doi: 10.1371/journal.pcbi.1001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degner JF, Marioni JC, Pai AA, et al. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Archer J, Baillie G, Watson SJ, et al. Analysis of high-depth sequence data for studying viral diversity: a comparison of next generation sequencing platforms using Segminator II. BMC Bioinformatics. 2012;13:47. doi: 10.1186/1471-2105-13-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macalalad AR, Zody MC, Charlebois P, et al. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol. 2012;8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X, Chockalingam SP, Aluru S. A survey of error-correction methods for next-generation sequencing. Brief Bioinform. 2013;14:56–66. doi: 10.1093/bib/bbs015. [DOI] [PubMed] [Google Scholar]
- 27.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat Methods. 2009;6(11 Suppl):S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 28.Horner DS, Pavesi G, Castrignano T, et al. Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform. 2010;11:181–197. doi: 10.1093/bib/bbp046. [DOI] [PubMed] [Google Scholar]
- 29.Finotello F, Lavezzo E, Fontana P, et al. Comparative analysis of algorithms for whole-genome assembly of pyrosequencing data. Brief Bioinform. 2012;13:269–280. doi: 10.1093/bib/bbr063. [DOI] [PubMed] [Google Scholar]
- 30.Angiuoli SV, White JR, Matalka M, White O, Fricke WF. Resources and costs for microbial sequence analysis evaluated using virtual machines and cloud computing. PLoS One. 2011;6:e26624. doi: 10.1371/journal.pone.0026624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39(Database issue):D19–21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y, Sartor M, Cavalcoli J. Current status and future perspectives for sequencing livestock genomes. J Anim Sci Biotechnol. 2012;3:8. doi: 10.1186/2049-1891-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aslam ML, Bastiaansen JW, Elferink MG, et al. Whole genome SNP discovery and analysis of genetic diversity in Turkey (Meleagris gallopavo) BMC Genomics. 2012;13:391. doi: 10.1186/1471-2164-13-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos AM, Crooijmans RP, Affara NA, et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One. 2009;4:e6524. doi: 10.1371/journal.pone.0006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubin CJ, Zody MC, Eriksson J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 36.Diaz-Sanchez S, Hanning I, Pendleton S, D’Souza D. Next-generation sequencing: the future of molecular genetics in poultry production and food safety. Poult Sci. 2013;92:562–572. doi: 10.3382/ps.2012-02741. [DOI] [PubMed] [Google Scholar]
- 37.Fisher CA, Bhattarai EK, Osterstock JB, et al. Evolution of the bovine TLR gene family and member associations with Mycobacterium avium subspecies paratuberculosis infection. PLoS One. 2011;6:e27744. doi: 10.1371/journal.pone.0027744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jex AR, Littlewood DT, Gasser RB. Toward next-generation sequencing of mitochondrial genomes-focus on parasitic worms of animals and biotechnological implications. Biotechnol Adv. 2010;28:151–159. doi: 10.1016/j.biotechadv.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Webb KM, Rosenthal BM. Next-generation sequencing of the Trichinella murrelli mitochondrial genome allows comprehensive comparison of its divergence from the principal agent of human trichinellosis, Trichinella spiralis. Infect Genet Evol. 2011;11:116–123. doi: 10.1016/j.meegid.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Grinberg A, Biggs PJ, Dukkipati VS, George TT. Extensive intra-host genetic diversity uncovered in Cryptosporidium parvum using Next Generation Sequencing. Infect Genet Evol. 2013;15:18–24. doi: 10.1016/j.meegid.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Cantacessi C, Campbell BE, Gasser RB. Key strongylid nematodes of animals—Impact of next-generation transcriptomics on systems biology and biotechnology. Biotechnol Adv. 2012;30:469–488. doi: 10.1016/j.biotechadv.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Matsubayashi M, Hatta T, Miyoshi T, et al. High-throughput RNA sequencing profiles and transcriptional evidence of aerobic respiratory enzymes in sporulating oocysts and sporozoites of Eimeria tenella. Infect Genet Evol. 2013;18:269–276. doi: 10.1016/j.meegid.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Wu X, Fu Y, Yang D, et al. Detailed transcriptome description of the neglected cestode Taenia multiceps. PLoS One. 2012;7:e45830. doi: 10.1371/journal.pone.0045830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romine NM, Martin RJ, Beetham JK. Computational cloning of drug target genes of a parasitic nematode, Oesophagostomum dentatum. BMC Genet. 2013;14:55. doi: 10.1186/1471-2156-14-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cwiklinski K, Merga JY, Lake SL, et al. Transcriptome analysis of a parasitic clade V nematode: comparative analysis of potential molecular anthelmintic targets in Cylicostephanus goldi. Int J Parasitol. 2013;43:917–927. doi: 10.1016/j.ijpara.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 46.Tritt A, Eisen JA, Facciotti MT, Darling AE. An integrated pipeline for de novo assembly of microbial genomes. PLoS One. 2012;7:e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watson SJ, Welkers MR, Depledge DP, et al. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120205. doi: 10.1098/rstb.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stack JC, Murcia PR, Grenfell BT, Wood JL, Holmes EC. Inferring the inter-host transmission of influenza A virus using patterns of intra-host genetic variation. Proc Biol Sci. 2013;280:20122173. doi: 10.1098/rspb.2012.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright CF, Morelli MJ, Thebaud G, et al. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol. 2011;85:2266–2275. doi: 10.1128/JVI.01396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morelli MJ, Wright CF, Knowles NJ, et al. Evolution of foot-and-mouth disease virus intra-sample sequence diversity during serial transmission in bovine hosts. Vet Res. 2013;44:12. doi: 10.1186/1297-9716-44-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orton RJ, Wright CF, Morelli MJ, et al. Observing micro-evolutionary processes of viral populations at multiple scales. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120203. doi: 10.1098/rstb.2012.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fusaro A, Monne I, Hughes J, et al. (2013) Evolution of H7 viruses in poultry: convergent and divergent mutations. Paper presented at the Options for the Control of Influenza VIII, Cape Town, South Africa
- 54.Wilker PR, Dinis JM, Starrett G, et al. Selection on haemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat Commun. 2013;4:2636. doi: 10.1038/ncomms3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wellehan JF, Jr, Yu F, Venn-Watson SK, et al. Characterization of San Miguel sea lion virus populations using pyrosequencing-based methods. Infect Genet Evol. 2010;10:254–260. doi: 10.1016/j.meegid.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vrancken B, Lequime S, Theys K, Lemey P. Covering all bases in HIV research: unveiling a hidden world of viral evolution. AIDS Rev. 2010;12:89–102. [PubMed] [Google Scholar]
- 58.Swenson LC, Daumer M, Paredes R. Next-generation sequencing to assess HIV tropism. Curr Opin HIV AIDS. 2012;7:478–485. doi: 10.1097/COH.0b013e328356e9da. [DOI] [PubMed] [Google Scholar]
- 59.Swenson LC, Mo T, Dong WW, et al. Deep sequencing to infer HIV-1 co-receptor usage: application to three clinical trials of maraviroc in treatment-experienced patients. J Infect Dis. 2011;203:237–245. doi: 10.1093/infdis/jiq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghedin E, Laplante J, DePasse J, et al. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis. 2011;203:168–174. doi: 10.1093/infdis/jiq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Capobianchi MR, Giombini E, Rozera G. Next-generation sequencing technology in clinical virology. Clin Microbiol Infect. 2013;19:15–22. doi: 10.1111/1469-0691.12056. [DOI] [PubMed] [Google Scholar]
- 62.Backer JA, Vrancken R, Neyts J, Goris N. The potential of antiviral agents to control classical swine fever: a modelling study. Antiviral Res. 2013;99:245–250. doi: 10.1016/j.antiviral.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 63.Lefebvre DJ, De Vleeschauwer AR, Goris N, et al. Proof of Concept for the Inhibition of Foot-and-Mouth Disease Virus Replication by the Anti-Viral Drug 2′-C-Methylcytidine in Severe Combined Immunodeficient Mice. Transbound Emerg Dis. 2013 doi: 10.1111/tbed.12069. [DOI] [PubMed] [Google Scholar]
- 64.Lefebure T, Bitar PD, Suzuki H, Stanhope MJ. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol. 2010;2:646–655. doi: 10.1093/gbe/evq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Biek R, O’Hare A, Wright D, et al. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 2012;8:e1003008. doi: 10.1371/journal.ppat.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, Mukherjee S, Hoffmann M, et al. Whole-Genome Sequencing of Gentamicin-Resistant Campylobacter coli Isolated from U.S. Retail Meats Reveals Novel Plasmid-Mediated Aminoglycoside Resistance Genes. Antimicrob Agents Chemother. 2013;57:5398–5405. doi: 10.1128/AAC.00669-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mellmann A, Harmsen D, Cummings CA, et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011;6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dupuy V, Manso-Silvan L, Barbe V, et al. Evolutionary history of contagious bovine pleuropneumonia using next generation sequencing of Mycoplasma mycoides subsp. mycoides "Small Colony". PLoS One. 2012;7:e46821. doi: 10.1371/journal.pone.0046821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Das S, Roychowdhury T, Kumar P, et al. Genetic heterogeneity revealed by sequence analysis of Mycobacterium tuberculosis isolates from extra-pulmonary tuberculosis patients. BMC Genomics. 2013;14:404. doi: 10.1186/1471-2164-14-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eyre DW, Golubchik T, Gordon NC, et al. (2012) A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2 (doi: 10.1136/bmjopen-2012-001124) [DOI] [PMC free article] [PubMed]
- 71.Koser CU, Holden MT, Ellington MJ, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grinev A, Chancey C, Anez G, et al. Genetic analysis of West Nile virus isolates from an outbreak in Idaho, United States, 2006-2007. Int J Environ Res Public Health. 2013;10:4486–4506. doi: 10.3390/ijerph10094486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen GQ, Zhuang QY, Wang KC, et al. Identification and survey of a novel avian coronavirus in ducks. PLoS One. 2013;8:e72918. doi: 10.1371/journal.pone.0072918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Becker N, Jost H, Ziegler U, et al. Epizootic emergence of Usutu virus in wild and captive birds in Germany. PLoS One. 2012;7:e32604. doi: 10.1371/journal.pone.0032604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Croville G, Soubies SM, Barbieri J, et al. Field monitoring of avian influenza viruses: whole-genome sequencing and tracking of neuraminidase evolution using 454 pyrosequencing. J Clin Microbiol. 2012;50:2881–2887. doi: 10.1128/JCM.01142-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dugan VG, Dunham EJ, Jin G, et al. Phylogenetic analysis of low pathogenicity H5N1 and H7N3 influenza A virus isolates recovered from sentinel, free flying, wild mallards at one study site during 2006. Virology. 2011;417:98–105. doi: 10.1016/j.virol.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramakrishnan MA, Tu ZJ, Singh S, et al. The feasibility of using high resolution genome sequencing of influenza A viruses to detect mixed infections and quasispecies. PLoS One. 2009;4:e7105. doi: 10.1371/journal.pone.0007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Borm S, Rosseel T, Vangeluwe D, Vandenbussche F, van den Berg T, Lambrecht B. Phylogeographic analysis of avian influenza viruses isolated from Charadriiformes in Belgium confirms intercontinental reassortment in gulls. Arch Virol. 2012;157:1509–1522. doi: 10.1007/s00705-012-1323-x. [DOI] [PubMed] [Google Scholar]
- 79.Leifer I, Ruggli N, Blome S. Approaches to define the viral genetic basis of classical swine fever virus virulence. Virology. 2013;438:51–55. doi: 10.1016/j.virol.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 80.Rao PP, Reddy YN, Ganesh K, Nair SG, Niranjan V, Hegde NR. Deep sequencing as a method of typing bluetongue virus isolates. J Virol Methods. 2013;193:314–319. doi: 10.1016/j.jviromet.2013.06.033. [DOI] [PubMed] [Google Scholar]
- 81.Marston DA, McElhinney LM, Ellis RJ, et al. Next generation sequencing of viral RNA genomes. BMC Genomics. 2013;14:444. doi: 10.1186/1471-2164-14-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marston DA, Mansfield KL, Mearns R, Ellis RJ, Fooks AR, Johnson N (2013) Louping ill virus genome sequence derived from the spinal cord of an infected lamb. Genome Announc 1 (doi:10.1128/genomeA.00454-13) [DOI] [PMC free article] [PubMed]
- 83.Kvisgaard LK, Hjulsager CK, Fahnoe U, Breum SO, Ait-Ali T, Larsen LE. A fast and robust method for full genome sequencing of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 and Type 2. J Virol Methods. 2013;193:697–705. doi: 10.1016/j.jviromet.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 84.Wilkie GS, Davison AJ, Watson M, et al. Complete genome sequences of elephant endotheliotropic herpesviruses 1A and 1B determined directly from fatal cases. J Virol. 2013;87:6700–6712. doi: 10.1128/JVI.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosseel T, Lambrecht B, Vandenbussche F, van den Berg T, Van Borm S. Identification and complete genome sequencing of paramyxoviruses in mallard ducks (Anas platyrhynchos) using random access amplification and next generation sequencing technologies. Virol J. 2011;8:463. doi: 10.1186/1743-422X-8-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hoper D, Hoffmann B, Beer M. A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS One. 2011;6:e19075. doi: 10.1371/journal.pone.0019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kampmann ML, Fordyce SL, Avila-Arcos MC, et al. A simple method for the parallel deep sequencing of full influenza A genomes. J Virol Methods. 2011;178:243–248. doi: 10.1016/j.jviromet.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 88.Hoper D, Kalthoff D, Hoffmann B, Beer M. Highly pathogenic avian influenza virus subtype H5N1 escaping neutralization: more than HA variation. J Virol. 2012;86:1394–1404. doi: 10.1128/JVI.00797-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hutter J, Rodig JV, Hoper D, et al. Toward animal cell culture-based influenza vaccine design: viral hemagglutinin N-glycosylation markedly impacts immunogenicity. J Immunol. 2013;190:220–230. doi: 10.4049/jimmunol.1201060. [DOI] [PubMed] [Google Scholar]
- 90.Bourret V, Croville G, Mariette J, et al. Whole-genome, deep pyrosequencing analysis of a duck influenza A virus evolution in swine cells. Infect Genet Evol. 2013;18:31–41. doi: 10.1016/j.meegid.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 91.Roedig JV, Rapp E, Hoper D, Genzel Y, Reichl U. Impact of host cell line adaptation on quasispecies composition and glycosylation of influenza A virus hemagglutinin. PLoS One. 2011;6:e27989. doi: 10.1371/journal.pone.0027989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu NC, Young AP, Dandekar SW, et al. Systematic identification of H274Y compensatory mutations in influenza A virus neuraminidase by high-throughput screening. J Virol. 2013;87:1193–1199. doi: 10.1128/JVI.01658-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Topfer A, Hoper D, Blome S, et al. Sequencing approach to analyze the role of quasispecies for classical swine fever. Virology. 2013;438:14–19. doi: 10.1016/j.virol.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 94.Fouts DE, Szpakowski S, Purushe J, et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS One. 2012;7:e48289. doi: 10.1371/journal.pone.0048289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oikonomou G, Machado VS, Santisteban C, Schukken YH, Bicalho RC. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16 s rDNA. PLoS One. 2012;7:e47671. doi: 10.1371/journal.pone.0047671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kolbert CP, Rys PN, Hopkins M, et al. et al. 16S ribosomal DNA sequence analysis for identification of bacteria in a clinical microbiology laboratory. In: Persing DH, Tenover FD, Versalovic J, et al.et al., editors. Molecular microbiology: diagnostic principles and practice. Washington, DC: ASM Press; 2004. pp. 361–377. [Google Scholar]
- 97.Thomas T, Gilbert J, Meyer F. Metagenomics—a guide from sampling to data analysis. Microb Inform Exp. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferrer M, Ghazi A, Beloqui AV, et al. Functional metagenomics unveils a multifunctional glycosyl hydrolase from the family 43 catalysing the breakdown of plant polymers in the calf rumen. PLoS One. 2012;7:e38134. doi: 10.1371/journal.pone.0038134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Singh KM, Jakhesara SJ, Koringa PG, Rank DN, Joshi CG. Metagenomic analysis of virulence-associated and antibiotic resistance genes of microbes in rumen of Indian buffalo (Bubalus bubalis) Gene. 2012;507:146–151. doi: 10.1016/j.gene.2012.07.037. [DOI] [PubMed] [Google Scholar]
- 101.Day JM, Ballard LL, Duke MV, Scheffler BE, Zsak L. Metagenomic analysis of the turkey gut RNA virus community. Virol J. 2010;7:313. doi: 10.1186/1743-422X-7-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One. 2011;6:e27949. doi: 10.1371/journal.pone.0027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Long SW, Williams D, Valson C, et al. A genomic day in the life of a clinical microbiology laboratory. J Clin Microbiol. 2013;51:1272–1277. doi: 10.1128/JCM.03237-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sherry NL, Porter JL, Seemann T, et al. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J Clin Microbiol. 2013;51:1396–1401. doi: 10.1128/JCM.03332-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barzon L, Lavezzo E, Militello V, Toppo S, Palu G. Applications of next-generation sequencing technologies to diagnostic virology. Int J Mol Sci. 2011;12:7861–7884. doi: 10.3390/ijms12117861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Belak S, Karlsson OE, Blomstrom AL, Berg M, Granberg F. New viruses in veterinary medicine, detected by metagenomic approaches. Vet Microbiol. 2013;165:95–101. doi: 10.1016/j.vetmic.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 107.Bexfield N, Kellam P. Metagenomics and the molecular identification of novel viruses. Vet J. 2011;190:191–198. doi: 10.1016/j.tvjl.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bishop-Lilly KA, Turell MJ, Willner KM, et al. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl Trop Dis. 2010;4:e878. doi: 10.1371/journal.pntd.0000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blomstrom AL. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet Q. 2011;31:107–114. doi: 10.1080/01652176.2011.604971. [DOI] [PubMed] [Google Scholar]
- 110.Blomstrom AL, Widen F, Hammer AS, Belak S, Berg M. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol. 2010;48:4392–4396. doi: 10.1128/JCM.01040-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Honkavuori KS, Shivaprasad HL, Briese T, et al. Novel picornavirus in Turkey poults with hepatitis, California, USA. Emerg Infect Dis. 2011;17:480–487. doi: 10.3201/eid1703.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cox-Foster DL, Conlan S, Holmes EC, et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 113.Granberg F, Vicente-Rubiano M, Rubio-Guerri C, et al. Metagenomic detection of viral pathogens in Spanish honeybees: co-infection by Aphid Lethal Paralysis, Israel Acute Paralysis and Lake Sinai Viruses. PLoS One. 2013;8:e57459. doi: 10.1371/journal.pone.0057459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blomstrom AL, Belak S, Fossum C, et al. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 2009;146:125–129. doi: 10.1016/j.virusres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 115.Ge X, Li Y, Yang X, et al. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol. 2012;86:4620–4630. doi: 10.1128/JVI.06671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li L, Victoria JG, Wang C, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. 2010;84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Blomstrom AL, Stahl K, Masembe C, et al. Viral metagenomic analysis of bushpigs (Potamochoerus larvatus) in Uganda identifies novel variants of Porcine parvovirus 4 and Torque teno sus virus 1 and 2. Virol J. 2012;9:192. doi: 10.1186/1743-422X-9-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bodewes R, van der Giessen J, Haagmans BL, Osterhaus AD, Smits SL. Identification of multiple novel viruses, including a parvovirus and a hepevirus, in feces of red foxes. J Virol. 2013;87:7758–7764. doi: 10.1128/JVI.00568-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carpi G, Cagnacci F, Wittekindt NE, et al. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS One. 2011;6:e25604. doi: 10.1371/journal.pone.0025604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fredericks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hoffmann B, Scheuch M, Hoper D, et al. Novel orthobunyavirus in Cattle, Europe, 2011. Emerg Infect Dis. 2012;18:469–472. doi: 10.3201/eid1803.111905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Regge N, van den Berg T, Georges L, Cay B. Diagnosis of Schmallenberg virus infection in malformed lambs and calves and first indications for virus clearance in the fetus. Vet Microbiol. 2013;162:595–600. doi: 10.1016/j.vetmic.2012.11.029. [DOI] [PubMed] [Google Scholar]
- 123.van den Brom R, Luttikholt SJ, Lievaart-Peterson K, et al. Epizootic of ovine congenital malformations associated with Schmallenberg virus infection. Tijdschr Diergeneeskd. 2012;137:106–111. [PubMed] [Google Scholar]
- 124.De Regge N, Deblauwe I, De Deken R, et al. Detection of Schmallenberg virus in different Culicoides spp. by real-time RT-PCR. Transbound Emerg Dis. 2012;59:471–475. doi: 10.1111/tbed.12000. [DOI] [PubMed] [Google Scholar]
- 125.Elbers AR, Meiswinkel R, van Weezep E, van Oldruitenborgh-Oosterbaan MM, Kooi EA. Schmallenberg virus in Culicoides spp. biting midges, the Netherlands, 2011. Emerg Infect Dis. 2013;19:106–109. doi: 10.3201/eid1901.121054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wernike K, Nikolin VM, Hechinger S, Hoffmann B, Beer M. Inactivated Schmallenberg virus prototype vaccines. Vaccine. 2013;31:3558–3563. doi: 10.1016/j.vaccine.2013.05.062. [DOI] [PubMed] [Google Scholar]
- 127.Van Borm S, Rosseel T, Steensels M, van den Berg T, Lambrecht B. What’s in a strain? Viral metagenomics identifies genetic variation and contaminating circoviruses in laboratory isolates of pigeon paramyxovirus type 1. Virus Res. 2013;171:186–193. doi: 10.1016/j.virusres.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 128.Baylis SA, Finsterbusch T, Bannert N, Blumel J, Mankertz A. Analysis of porcine circovirus type 1 detected in Rotarix vaccine. Vaccine. 2011;29:690–697. doi: 10.1016/j.vaccine.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 129.Farsang A, Kulcsar G. Extraneous agent detection in vaccines-a review of technical aspects. Biologicals. 2012;40:225–230. doi: 10.1016/j.biologicals.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Neverov A, Chumakov K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc Natl Acad Sci U S A. 2010;107(46):20063–8. doi: 10.1073/pnas.1012537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Onions D, Kolman J. Massively parallel sequencing, a new method for detecting adventitious agents. Biologicals. 2010;38:377–380. doi: 10.1016/j.biologicals.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 132.Victoria JG, Wang C, Jones MS, et al. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J Virol. 2010;84:6033–6040. doi: 10.1128/JVI.02690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Miller PJ, Afonso CL, Spackman E, et al. Evidence for a new avian paramyxovirus serotype 10 detected in rockhopper penguins from the Falkland Islands. J Virol. 2010;84:11496–11504. doi: 10.1128/JVI.00822-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nordentoft S, Molbak L, Bjerrum L, et al. The influence of the cage system and colonisation of Salmonella Enteritidis on the microbial gut flora of laying hens studied by T-RFLP and 454 pyrosequencing. BMC Microbiol. 2011;11:187. doi: 10.1186/1471-2180-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qu A, Brulc JM, Wilson MK, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One. 2008;3:e2945. doi: 10.1371/journal.pone.0002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Glass EJ, Crutchley S, Jensen K. Living with the enemy or uninvited guests: functional genomics approaches to investigating host resistance or tolerance traits to a protozoan parasite, Theileria annulata, in cattle. Vet Immunol Immunopathol. 2012;148:178–189. doi: 10.1016/j.vetimm.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Leon AJ, Banner D, Xu L, et al. Sequencing, annotation, and characterization of the influenza ferret infectome. J Virol. 2013;87:1957–1966. doi: 10.1128/JVI.02476-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maceachern S, Muir WM, Crosby S, Cheng HH. Genome-wide identification of allele-specific expression (ASE) in response to Marek’s disease virus infection using next generation sequencing. BMC Proc. 2011;5(Suppl 4):S14. doi: 10.1186/1753-6561-5-S4-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miller LC, Fleming D, Arbogast A, et al. Analysis of the swine tracheobronchial lymph node transcriptomic response to infection with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. BMC Vet Res. 2012;8:208. doi: 10.1186/1746-6148-8-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang F, Hu S, Liu W, et al. Deep-sequencing analysis of the mouse transcriptome response to infection with Brucella melitensis strains of differing virulence. PLoS One. 2011;6:e28485. doi: 10.1371/journal.pone.0028485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang CR, Xu MJ, Fu JH, et al. Characterization of microRNAs from Orientobilharzia turkestanicum, a neglected blood fluke of human and animal health significance. PLoS One. 2012;7:e47001. doi: 10.1371/journal.pone.0047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kong BW. Identification of virus encoding microRNAs using 454 FLX sequencing platform. Methods Mol Biol. 2011;733:81–91. doi: 10.1007/978-1-61779-089-8_6. [DOI] [PubMed] [Google Scholar]
- 143.Ma M, Huang Y, Gong Z, et al. Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS One. 2011;6:e24758. doi: 10.1371/journal.pone.0024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cullen BR. Viruses and microRNAs. Nat Genet. 2006;38(Suppl):S25–30. doi: 10.1038/ng1793. [DOI] [PubMed] [Google Scholar]
- 145.Eulalio A, Schulte L, Vogel J. The mammalian microRNA response to bacterial infections. RNA Biol. 2012;9:742–750. doi: 10.4161/rna.20018. [DOI] [PubMed] [Google Scholar]
- 146.Gottwein E, Cullen BR. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. 2008;3:375–387. doi: 10.1016/j.chom.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Perez JT, Pham AM, Lorini MH, et al. MicroRNA-mediated species-specific attenuation of influenza A virus. Nat Biotechnol. 2009;27:572–576. doi: 10.1038/nbt.1542. [DOI] [PubMed] [Google Scholar]
- 148.Wang Y, Brahmakshatriya V, Lupiani B, et al. Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers. BMC Genomics. 2012;13:278. doi: 10.1186/1471-2164-13-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang Y, Brahmakshatriya V, Zhu H, et al. Identification of differentially expressed miRNAs in chicken lung and trachea with avian influenza virus infection by a deep sequencing approach. BMC Genomics. 2009;10:512. doi: 10.1186/1471-2164-10-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bhomia M, Sharma A, Gayen M, Gupta P, Maheshwari RK. Artificial microRNAs can effectively inhibit replication of Venezuelan equine encephalitis virus. Antiviral Res. 2013;100:429–434. doi: 10.1016/j.antiviral.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ibrisimovic M, Kneidinger D, Lion T, Klein R. An adenoviral vector-based expression and delivery system for the inhibition of wild-type adenovirus replication by artificial microRNAs. Antiviral Res. 2013;97:10–23. doi: 10.1016/j.antiviral.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Israsena N, Supavonwong P, Ratanasetyuth N, Khawplod P, Hemachudha T. Inhibition of rabies virus replication by multiple artificial microRNAs. Antiviral Res. 2009;84:76–83. doi: 10.1016/j.antiviral.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 153.Xia B, Song H, Chen Y, et al. Efficient inhibition of porcine reproductive and respiratory syndrome virus replication by artificial microRNAs targeting the untranslated regions. Arch Virol. 2013;158:55–61. doi: 10.1007/s00705-012-1455-z. [DOI] [PubMed] [Google Scholar]
- 154.Barnes D, Kunitomi M, Vignuzzi M, Saksela K, Andino R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe. 2008;4:239–248. doi: 10.1016/j.chom.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kelly EJ, Hadac EM, Greiner S, Russell SJ. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat Med. 2008;14:1278–1283. doi: 10.1038/nm.1776. [DOI] [PubMed] [Google Scholar]
- 156.Luciani F, Bull RA, Lloyd AR. Next generation deep sequencing and vaccine design: today and tomorrow. Trends Biotechnol. 2012;30:443–452. doi: 10.1016/j.tibtech.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Woollard PM, Mehta NA, Vamathevan JJ, et al. The application of next-generation sequencing technologies to drug discovery and development. Drug Discov Today. 2011;16:512–519. doi: 10.1016/j.drudis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 158.Mann M, Hendrickson R, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 159.Boehmer JL. Proteomic analyses of host and pathogen responses during bovine mastitis. J Mammary Gland Biol Neoplasia. 2011;16:323–338. doi: 10.1007/s10911-011-9229-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li G, Cai W, Hussein A, et al. Proteome response of an extraintestinal pathogenic Escherichia coli strain with zoonotic potential to human and chicken sera. J Proteomics. 2012;75:4853–4862. doi: 10.1016/j.jprot.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 161.Liu Y, Zhang K, Zheng H, et al. Proteomics analysis of porcine serum proteins by LC-MS/MS after foot-and-mouth disease virus (FMDV) infection. J Vet Med Sci. 2011;73:1569–1572. doi: 10.1292/jvms.11-0019. [DOI] [PubMed] [Google Scholar]
- 162.Kolmeder CA, de Vos WM. Metaproteomics of our microbiome—Developing insight in function and activity in man and model systems. J Proteomics. 2013;97:3–16. doi: 10.1016/j.jprot.2013.05.018. [DOI] [PubMed] [Google Scholar]