

Abstract
Animal venoms can play an important role in drug discovery, as they are a rich source of evolutionarily tuned compounds that target a variety of ion channels and receptors. To date, there are six FDA-approved drugs derived from animal venoms, with recent work using high-throughput platforms providing a variety of new therapeutic candidates. However, high-throughput methods for screening animal venoms against purinoceptors, one of the oldest signaling receptor families, have not been reported. Here, we describe a variety of quantitative fluorescent-based high-throughput screening (HTS) cell-based assays for screening animal venoms against ligand-gated P2X receptors. A diverse selection of 180 venoms from arachnids, centipedes, hymenopterans, and cone snails were screened, analyzed, and validated, both analytically and pharmacologically. Using this approach, we performed screens against human P2X3, P2X4, and P2X7 using three different fluorescent-based dyes on stable cell lines and isolated the active venom components. Our HTS assays are performed in 96-well format and allow simultaneous screening of multiple venoms on multiple targets, improving testing characteristics while minimizing costs, specimen material, and testing time. Moreover, utilizing our assays and applying them to the other natural product libraries, rather than venoms, might yield other novel natural products that modulate P2X activity.
Natural products have a storied past as drug leads, with an estimated half of the top-selling drugs in the world originating from a natural product.1 Among them, animal venoms are no exception. Several research groups consider toxins from spiders,2,3 cone snails,4 snakes,5 sea anemones,6,7 jellyfishes,8 and scorpions9 as a reliable animal source to engage in therapeutic lead discovery. The underlying reason for this trend is that venoms offer a diversity of molecules that modulate a wide range of ion channels with high affinity and selectivity.10 However, the biochemical arsenal of these venomous creatures has barely been tapped due to biological, historical, technological, and even practical reasons.11
Fortunately, modern venom research is leveraging the recent revolution in high-throughput approaches in genomics, transcriptomics, proteomics, and metabolomics. The confluence of these technologies with advances in bioinformatics offers exciting new possibilities to exploit the remarkable chemical diversity of nature’s pharmacopeia in the quest for new drugs. While most of the six venom-derived drugs currently on the market have been developed from snakes,10 which yield large amounts of venom, the rapid progress in high-throughput screening (HTS) now enables efficient screening of venoms from animals that previously could not be studied because they yield only small amounts of venom.11 Still, many venoms have not been studied with respect to potential biological targets. In large part, this bottleneck is due to the fact that the development of robust HTS assays that could access this uncharted chemical space and determine the molecular targets of venom toxins is often a major challenge.12
Although ion channels are the third most common target of small-molecule drugs after kinases and G-protein-coupled receptors (GPCRs),13 they can be difficult targets to investigate using HTS approaches,14 in part due to the lack of high-throughput electrophysiological platforms for the characterization of compound activity. Although manual patch-clamp electrophysiological approaches are extremely information-rich, they are labor intensive, represent a challenge with regard to reproducibility of the cells being used, require highly skilled staff, and can only support the evaluation of small numbers of compounds. Consequently, HTS-based methodologies that use cell-based assays with membrane potential dyes, Ca2+-sensitive dyes, or ion-flux measurements have become integral components of ion-channel drug discovery programs.15 While it would have been irrational to expect HTS to directly deliver new molecular entities (NME) from a synthetic library, natural product libraries can be viewed as a population of structurally privileged NME selected by evolutionary pressures. By accessing their uncharted chemical space, HTS might inspire more rapid discoveries.16,17 Taken together with the unrealized potential of venoms, these natural products might be of renewed interest as a source of chemical diversity, HTS hit identification, and lead generation.16
Despite the current resurgence in the use of venoms as tools in biomedical research, purinergic receptors have been largely ignored in the quest for new toxins that modulate ligand-gated channels. The only study that explored whether venoms are capable of targeting purinergic receptors was from Grishin et al.,2 who reported a potent and selective peptidic modulator of human P2X3 from the venom of a wolf spider (family Lycosidae). In addition to the established role of the P2X3 receptor in chronic pain,18 the purinergic P2X-mediated system has been implicated in a wide range of disorders including hypertension,19 bladder incontinence,20 chronic cough,21 inflammatory and immune disorders,22 migraine,23 pain,24 irritable bowel syndrome,25 epilepsy,26 atherosclerosis,27 depression,28 diabetes,29 and cancer. However, we still continue to fall short in addressing the increasing need for novel, effective, safe, and well-tolerated treatments for these conditions, despite decades of innovation and effort in the purinergic field. To bridge this gap between the exciting progress that has been made in pursuit of P2X-targeted drugs for clinical development, we believe animal venoms might help populate unmet pharmacological space. Taken together with the need for high-quality HTS assays, we set out to develop a venom screen toward various P2X receptors that would be easily automated, fast, reliable, and robust and provide a quantitative output that correlates well with the validated data. Here, we report the design and development of three fluorescent-based high-throughput cell assays that can be used to screen animal venoms, or indeed other natural product sources, against the human purinergic receptors hP2X3, hP2X4, and hP2X7. These assays enable screening of multiple venoms against multiple targets, improving testing characteristics while minimizing costs, specimen material, and testing time. Moreover, application of our assay to other venom libraries or other natural products libraries might yield novel drug entities with P2X activity and thus promote the discovery of therapeutically beneficial agents for a variety of pathologies.
Results and Discussion
Assay Design
To develop heterologous expression systems for direct investigation of P2X modulation in adherent cell cultures, we chose 1321N1 and HEK293 cell lines. Previous screens designed to detect P2X activity against a background of endogenous, high-level promiscuous P2Y GPCR expression are often susceptible to artifacts or false positives derived presumably from P2Y cell-surface receptors hijacking P2X calcium signaling. Since the human astrocytoma cell line 1321N1 possesses no endogenous P2 receptors that might interfere with calcium signaling, we chose it as a suitable cell line for our studies. Additionally, we previously used stable HEK293 cell lines expressing hP2X4 or hP2X7 receptors to perform the preliminary fluorescent-based cell assays and successfully identified selected ginsenosides as novel allosteric modulators of the purinergic receptor family.30 A similar research effort was focused on other P2X receptors.31
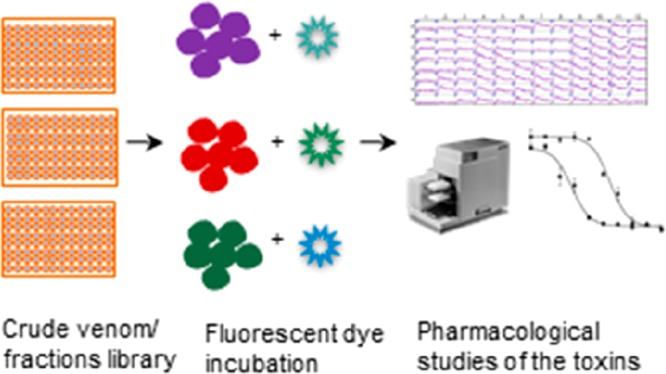
While these assays represent a good starting point to screen for potent P2X modulators, we still lack HTS that have been rigorously validated for analytical and biochemical relevance, especially when subjected to another class of natural products such as venoms. In order to streamline our HTS workflow and apply our assays to the growing field of venomics,11 we have developed high-quality HTS assays that would selectively detect toxin hits from different animal venoms toward stably expressed P2X channels. Figure 1 displays the screening and fractionation workflow that was developed to enable rapid interrogation of both crude venoms and semipure venom fractions. The general scheme involves the following steps: (A) fluorescent-based assays of crude venoms to identify “hits”; (B) fractionation of venoms and toxin hit identification; (C) toxin hit validation via a Flexstation 3 multimode plate reader to collect information about the calcium/dye flux in each well of a microplate simultaneously capturing the response kinetics of the P2X channel.32
Figure 1.

High-throughput screen of crude venoms against P2X receptors. (A) Crude venom (150 μL at a concentration of 1 g/L) is added to wells of a 96-well plate, then screened in triplicate using different fluorescent dyes (Fura-2-AM, YO-PRO-1, and FLIPR Calcium-6 assay) against 1321N1-hP2X4, HEK293-hP2X7, and HEK-hP2X3 stable cell lines. (B) Venoms identified as hits in the initial assay are fractionated using reverse-phase (RP) HPLC; then the fractions are screened in the HTS assays against the various P2X receptors to identify “hit” fractions with P2X activity. (C) Hit fractions identified are further fractionated using orthogonal chromatography techniques to identify the bioactive compound, which is then analyzed using mass spectrometry (MALDI-TOF, LC-MS, MS/MS). The toxin hit is then pharmacologically validated using two stable cell lines expressing the P2X receptor of interest.
Overall, this scheme proved to be robust and easy to implement. We measured fluorescence from the bottom of the well to reduce background fluorescence, although this requires that the cells are firmly adhered. If the cells detach or move during liquid addition, the signal is compromised.32 Thus, we developed stable adherent cell lines by transfecting 1321N1 and HEK293 cell lines with hP2X4 and hP2X7 plasmids: 1321N-hP2X4, HEK293-hP2X4, and HEK293-hP2X7.33 A plethora of assay formats have been enabled to support compound screening;34 however, we chose the 96-well plate format and evaluated hP2X4 and hP2X7 inhibition with Fura-2-AM and YOPRO-1 fluorescent dyes, respectively. By quantifying either agonist (ATP)-mediated increases in cytosolic Ca2+ concentrations (with Fura-2-AM) or dye uptake (with YOPRO-1), we monitored relative changes in the levels of intracellular Ca2+ or dye uptake in real time.
Assay Optimization
In developing the assays in a 96-well format, systematic variation in assay parameters led to the following optimal conditions that are outlined in the Experimental Section. Critically, the calcium-sensitive fluorescent dye Fura-2-AM and dye uptake probe YOPRO-1 utilize different incubation buffers. Whereas the Fura-2 assay on 1321N1-hP2X4 requires a medium containing calcium, the YOPRO uptake assay buffer is devoid of Mg2+ ions and contains a very low concentration of Ca2+ ions since these ions are known to inhibit hP2X7 pore formation.35 Since the real power of such in vitro assays lies in the possibility to perform high-throughput experiments, we decided to optimize our assay conditions for inhibition evaluation and identification studies. To determine whether our assays are pharmacologically predictive for P2X targets and capable of identifying inhibitors with the desired potency and mechanism of action, we systematically tested several commercially available small-molecule inhibitors of hP2X4 (BX430, 5-BDBD, PSB12062) and hP2X7 (AZ10606120). As a proof of principle, we screened each compound at various concentrations and generated concentration–response curves (Figure S1 in the Supporting Information). Either cells were preincubated with compounds for 10 min or antagonists or mock medium (buffer) was applied onto them via Flexstation 3 automated injection. The IC50 values we calculated for BX430, 5-BDBD, PSB12062, and AZ10606120 using this assay mostly corresponded well with reported potencies (Table 1);35−38 however, some IC50 values for 5-BDBD and PSB12062, in HEK293-hP2X4 and 1321N1-hP2X4, respectively, differed nearly 8-fold. This may be due to the assessment of IC50 values that were independently measured in different laboratories with different sets of assays.35−38 Ideally, IC50 values should be compared only under similar conditions since these values are often assay-specific.39
Table 1. IC50 Values of Known P2X Inhibitors Calculated Using Our HTS Assays against 1321N1-hP2X4, HEK293-hP2X4, and HEK293-hP2X7 Cell Lines.
| cell
line |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1321N1-hP2X4 |
HEK-hP2X4 |
HEK-hP2X7 |
|||||||
| inhibitor | IC50 [μM]a | IC50 [μM] | 95% Cl [μM] | IC50 [μM]a | IC50 [μM] | 95% Cl [μM] | IC50 [nM]a | IC50 [nM] | 95% Cl [nM] |
| BX430 | 1.640 | 0.55 | 0.34–0.87 | 0.5435 | 1.3 | 1.2–1.5 | N.A. | ||
| 5-BDBD | N.A. | 5.7 | 4.3–6.7 | 1.236 | 9.2 | 8.4–10.0 | N.A. | ||
| PSB12062 | 3.340 | 0.42 | 0.25–0.73 | 1.438 | 0.76 | 0.69–0.83 | N.A. | ||
| AZ10606120 | N.A. | N.A. | ∼10.037 | 92.0 | 81.0–103.0 | ||||
Literature values.
Based on these results, we chose BX430 and AZ10606120 as positive controls for the hP2X4 and hP2X7 assays, respectively, and utilized 10 μM concentrations throughout our studies. Critically, when these inhibitors were preincubated and the assay incubation time exceeded 60 min, these two inhibitors less effectively inhibited the P2Xs. Thus, screens were limited to <60 min. Moreover, cytotoxicity complications in assays that require long incubation periods are inevitable and in many cases can only be addressed by changing the assay configuration from preincubation.32 For this reason, the venoms (or inhibitors) were applied on top of the cells (or preincubated) after 30 s prior to injection of agonist (ATP) at 100 s. This incubation time of 70 s allowed inhibitors a sufficient time to inhibit either hP2X4 or hP2X7. We then monitored the fluorescent responses for a further 200 s per well. This result emphasizes the importance of assay optimization via pilot screens.
Screen of Animal Venoms against hP2X4
Once these conditions were defined, we proceeded into larger-size libraries, such as venoms, to ensure assay performance. For our typical crude venom screen, arranged in 96-well format, crude venoms were dissolved in water and diluted up to 25-fold from a stock solution of 1 g/L into the 96-well assay plate. In the HTS assays, outlined in Figure 1, toxins are not preincubated in discrete wells but are applied directly onto cells as previously discussed.
In total, 180 crude venoms (for details see Table S1 in the Supporting Information) from arachnids, centipedes, hymenopterans, and cone snails were arranged in standard 96-well drug plates and tested in duplicates. A subset of venoms were tested for dose-dependent effects in triplicates (10, 2, or 0.4 μg per well). Usually, chemical libraries are stored in organic solvents such as EtOH or DMSO41 and assays need to be configured so they are not sensitive to the concentration of these solvents. In contrast, the venoms (and later the fractionated toxins) were all dissolved in incubation buffer (see the Experimental Section), and thus the solvent effect was mitigated. After injection of crude venoms and agonist (10 μL each), fluorescent Fura2-AM Ca2+-based (Figure 2A,B) or YOPRO-1 (Figure 2C,D) dye uptake was measured as a function of time.
Figure 2.

Screen of crude spider venoms against 1321N1-hP2X4 and HEK293-hP2X4. (A) Example showing the effect of spider venom 1 (SV1) and 7 (SV7) and controls (buffer, ATP, and hP2X4-specific antagonist BX430) on 1321N1-hP2X4 and HEK-hP2X4 cells. While some spider venoms showed concentration-dependent inhibition (e.g., SV1), some venoms, such as SV7, had no effect (A). To examine whether SV1 and SV7 have an effect on their own on P2X4, they were applied via the Flexstation 3 automated injection system alone without later application of the P2X4 agonist ATP (this is denoted “Venom SV1//SV7 only”). (B) Kinetic responses for 1321N1-hP2X4. The inhibitory effect of one crude venom (e.g., SV1) was confirmed via dose-dependent inhibition in the HEK293-hP2X4 YOPRO-1 assay (C) and the kinetic responses shown in panel (D). Data points represent mean ± SD of three replicate experiments with triplicates on each plate except fraction injections. Significant differences between the control (10 μM ATP) and the venom are indicated by * (P < 0.05) using one-way ANOVA followed by Dunnett’s test.
Since venoms are complex mixtures of typically hundreds of components that differ in concentration, we could not predetermine the toxin concentrations used in the assays.
Thus, we performed our studies using a dilution series of toxin fractions, which helped us to identify venoms/toxins with higher or lower activity. Venom/toxin hits were defined as those venoms/fractions that gave concentration-dependent inhibition, at least 50% inhibition at the highest venom concentration (10 μg/well), and whose activities were confirmed upon retesting. The response for each crude venom was plotted as a function of time and is shown in Figure 2B and D. While venom SV7 did not show modulation of hP2X4, the representative trace for one hit venom, SV1, revealed a dose-dependent inhibition on 1321N1-hP2X4 with 10, 2, and 0.4 μg of crude venom yielding ∼69%, 27%, and 4% inhibition, respectively (Figure 2A). The inhibitory effect was validated and confirmed on HEK293-hP2X4 cells using the YOPRO-1 uptake assay (Figure 2C). Notably, 10 μg of SV1 venom yielded 69–80% inhibition, which is similar to the commercially available hP2X4 antagonist BX430 (75% inhibition at 10 μM).
Fractionation of Crude Venom Hits
Following identification of crude venom hits, we then sought to deconvolute the crude mixtures, as well as enhance the impact of minor components in the assay,32 through the creation of fractionated spider-venom product libraries. The crude venoms were fractionated using C18 RP-HPLC, which separates components on the basis of their relative hydrophobicity, with elution monitored via absorbance at 214 and 280 nm (Figure 3). Most active fractions from the first C18 RP-HPLC separation contained multiple components, and therefore an additional chromatography step was required to purify the hit compound. This was often as simple as an additional C18 RP-HPLC fractionation with a shallower gradient.
Figure 3.

Representative RP-HPLC chromatograms showing fractionation of crude venoms from various venomous animals. (A) Bahia scarlet tarantula (Lasiodora klugi); (B) Brazilian tarantula (Nhandu chromatus); (C) marine cone snail (Conus geographus); (D) German wasp (Vespula germanica); (E) European honeybee (Apis mellifera); (F) Asian hornet (Vespa velutina nigrithorax). Venoms were fractionated on an analytical C18 RP-HPLC column (Jupiter 5 μm; Phenomenex), and components eluted at a flow rate of 1 mL/min using a gradient of solvent B (90% MeCN, 0.05% trifluoroacetic acid (TFA) in H2O) in solvent A (0.05% TFA in H2O) as indicated by the dotted lines. Absorbance was monitored at 214, 254, and 280 nm, but only the 214 nm absorbance is plotted here.
Discrete fractions were automatically collected based on absorbance at 214 nm. The complexity of the crude of venoms from L. klugi (Figure 3A), V. germanica (Figure 3B), C. geographus (Figure 3C), and A. mellifera (Figure 3D), as judged by the complexity of the RP-HPLC chromatograms, is consistent with that reported previously for these species.4,41−43 A total of 25–49 fractions covering the entire elution profile were collected for each venom. Most fractions represented only a small percentage of the overall venom profile, although less than six fractions from L. klugi spider venom appeared to account for >75% of venom toxins.
A complicating fact in the HTS was that some venom components yielded nonspecific calcium responses prior to agonist application, and some wasp venoms, such as that from V. germanica, interfered with fluorescent signal generation or had cytotoxic pore-forming activity (Figure S2 in the Supporting Information).44 Interference with fluorescence signals represents a major challenge in assay development. We found that venoms containing highly colored components such as V. germanica (a number of yellow or red fractions were obtained upon fractionation) could potentially generate fluorescent signals. Fractions were subjected to a counter-screen without cells and were found to emit fluorescence at the tested wavelengths (340/380 excitation, 520 nm emission). Therefore, venoms with these characteristics could not be tested in these assays.
We believe some venom constituents to be concentrated biogenic polyamines (spermine, spermidine), cytotoxic peptides such as mellitin,45 or neurotransmitters such as histamine, acetylcholine, and serotonin. Venom fractions containing these compounds may modulate endogenously expressed receptors in these cell lines such as ionotropic glutamate receptors46 or GPCRs, and were thus considered nonspecific. Chelation of calcium by venom components would likely manifest as a reduction in extracellular calcium concentrations following application into the well. Modifying the extracellular calcium concentration 10-fold did not affect ATP-induced responses in the HEK-hP2X4 cells (not shown).
Assay Hit Validation
Here, the inhibitory behavior subsequently followed by agonist application was investigated. The purified fractions obtained were subjected to fluorescent-based bioassays on four stable cell lines: 1321N1-hP2X4, HEK293-hP2X4, HEK293-hP2X3, and HEK293-hP2X7. Forty-eight fractions from Nhandu chromatus venom, initially screened using 1321N1-hP2X4 (Ca2+ based Fura-2-AM assay, Figure 4A), were further evaluated on HEK293-hP2X4 (YOPRO-1 dye uptake assay, Figure 4B), HEK293-hP2X7 (Figure 4C), and HEK293-hP2X3 (Figure 4D) in order to both validate the fraction hits from the initial assay and test for target selectivity. P2X positive and negative controls (ATP, ivermectin,47 hP2X4-antagonist BX430;35 hP2X7-antagonists AZ1060612037 and JNJ47965567;48 α,β-methylene ATP;49 and the hP2X3 antagonist purotoxin-1 [PT12)]) were included as assay controls.
Figure 4.

Screening of N. chromatus venom fractions using the (A) 1321N1-hP2X4 cell line, (B) HEK293-hP2X4 cell line, (C) HEK293-hP2X7 cell line, and (D) HEK293-hP2X3 cell line. Fractions colored red selectively inhibited hP2X4. The dash represents 100% hP2X4 activity as followed by 10 μM ATP application. Data points represent mean ± SD of three replicate experiments, with triplicates on each plate except fraction injections. Significant differences between the positive control (ATP) and the fractions on either 1321N1-hP2X4 or HEK293-hP2X4 cell line are indicated by * (P < 0.05) using one-way ANOVA followed by Dunnett’s test.
A comparison of the P2X3, P2X4, and P2X7 assays provided some noteworthy inhibitory patterns. Ten of the 48 N. chromatus fractions inhibited 1321N1-hP2X4 by >75% (Figure 4A), and nine of them were validated on the HEK293- hP2X4 cell line (Figure 4B), which corresponds to a 90% validation rate. Fractions F39 and F42 from N. chromatus did not inhibit hP2X3 (Figure 4D) or hP2X7 (Figure 4C), whereas other fractions yielded inhibition of <20% (F10–F13, F40, F44, F45) or even slight potentiation of hP2X3 (F5, F44). This procedure further excluded several 1321N1-hP2X4 Fura-2-AM hit fractions that we could not validate in the YOPRO-1 HEK293-hP2X4 assay (“false positive hits”) and hits that had an inhibitory action on other P2X channels such as hP2X3 and hP2X7 (“nonspecific hits”). Since the entry point for any drug discovery program is generally the identification of modulators with adequate and specific activity against the target of interest, these initial hits from our screens provided a good starting point to rapidly trace pharmacologically relevant compounds.49 This establishes our fluorescent Fura-2-AM and YOPRO-1 assays as effective for measuring the inhibitory action of venom fractions on 1321N1-hP2X4.
Assay Specificity
After the initial screens of venom and venom fractions, the precision, reproducibility, specificity, and variability of the assays were evaluated. First, fractions F14, F28, and F47 from N. chromatus venom, which had no effect on any of the studied P2X receptors, were evaluated alongside fraction F5, which inhibited hP2X4. This F5 toxin fraction produced Ca2+ signals (Figure 5A) similar to YOPRO-1 dye uptake signals (Figure 5B) and when compared to the negative control (toxin F5 vs antagonist injection), which gave up to a 50-fold difference in signals, in both assays. However, when F5 was tested on HEK293-hP2X7, the difference between the control (300 μM ATP) and F5 application was not statistically significant (p > 0.05) (Figure 5C). These results confirm that the assay is highly specific for identifying toxin hits against hP2X4.
Figure 5.

Assay specificity. To assess assay specificity, commercially available compounds (BX430, PSB12062, AZ10606120, IVM) that are known to modulate hP2X4 and hP2X7 and inactive venom fractions (F14, F28, F47) were tested for a response in the (A) Fura-2 1321N1-hP2X4, (B) YOPRO-1 HEK293-hP2X4, and (C) YOPRO-1 HEK293-hP2X7 assays, together with a hit venom fraction (F5). In order to calculate the Z′ factor, data were collected over a period of one month with three experiments performed on different days and eight replicates per plate. Data points represent mean ± SD of three replicate experiments with triplicates on each plate except fraction injections. Significant differences between the control (ATP) and the venom are indicated by * (P < 0.05) using one-way ANOVA followed by Dunnett’s test.
Assay Reproducibility
Within a compound screening environment, it is a requirement that the assay is reproducible across assay plates, screen days, and the duration of the entire screening program.50 For that reason, we evaluated assay reproducibility using the Z′ factor statistical method.51 This is a common method for judging the quality of HTS assays, and it has become the standard method of measuring assay quality on a plate basis.15 The Z′ parameter considers not only the signal window in the assay but also the variance around both the high and low signals in the assay. Z′ ranges from 0 to 1; a value of >0.4 is considered appropriately robust for compound screening, although many industry groups prefer to work with a Z factor of >0.6.15 We calculated the mean and SD for positive [buffer + ATP] and negative [antagonist + ATP] wells and used them to determine the Z′ factor. The experiment was repeated once, and the averaged calculated Z′ factor for both experiments on 1321N1-hP2X4, HEK293-hP2X4, and HEK-hP2X7 cells was 0.57 ± 0.02 (CV 4.1%), 0.67 ± 0.032 (CV = 4.4%), and 0.56 ± 0.012 (CV = 2.2%), respectively. Our Z′ factors of >0.55 fall within the range expected for the robust and reproducible assays.51 This indicates that our assays are appropriate for HTS applications and that any plate or systematic errors potentially affecting the assay were not substantial.
Assay Variability
In addition to the Z factor, assay quality is also determined by monitoring intra- and interplate variability. Our data were compared to assess well-to-well (intraplate) variability as well as plate-to-plate (interplate) variability on six venom fractions and two controls (ATP, antagonist) in three different experiments throughout one month (see the text and Tables S1–3 in the Supporting Information).
For the 1321N1-hP2X4 assay (Table S1), interplate variability analysis yielded a mean CV of 9.98% (min: 6.43%, max: 13.82%, median: 8.83%). The calculated intraplate variability was 4.47% (min: 0.84%, max: 10.26%, median: 3.01%). For the HEK293-hP2X4 assay (Table S2), interplate variability analysis yielded a mean %CV of 13.59% (min: 11.68%, max: 14.97%, median: 14.13%). The calculated intraplate variability was 4.94% (min: 1.66%, max: 7.52%, median: 5.25%). For the HEK293-hP2X7 assay (Table S3), interplate variability analysis yielded a mean %CV of 14.88% (min: 12.88%, max: 17.49%, median: 14.82%). The calculated intraplate variability was 5.22% (min: 2.61%, max: 6.07%, median: 5.68%). Variability across the same plates was therefore low (<5%), and as many of the venom libraries to be tested would be measured on a single plate, this means that hit fractions can be identified with good accuracy.
Assay quality can also be monitored through the inclusion of pharmacological controls within each assay. Data for the controls (ATP and antagonist) fell within a predefined limit (CV = 1.9–5.3%), and thus the variability is deemed acceptable. Another quality control measure was the stability of the fractions used in these studies. The refrigerated samples used in these studies remained stable for the duration of the study (one month), as judged using RP-HPLC (data not shown). The results presented therefore indicate that our fluorescent-based assays provide a rapid and sensitive method for HTS screening of venoms and suggest that the assays can be adapted to other natural products screenings.
Summary
With recent advances in laboratory automation and HTS and MS methods, the use of venom and toxins as input into high-throughput assays has undergone a renaissance. Convergence of modern natural product isolation methods with chemical genomics and bioinformatics thus promises to further advance the rapid identification of potent natural products with novel mechanisms of action. Though a cell-based HTS may initially appear daunting, there are many targets for which a cell-based screen represents the fastest and cheapest path to lead generation.49 Furthermore, the two drivers for innovation in cell-based HTS methodologies are the need to miniaturize the assay volumes to 96-, 384-, and 1536-well format and the desire to capture temporal and spatial data on target activity.32 The HTS strategy we have developed for identifying P2X modulators from animal venoms provides a powerful tool in the hit generation process. Arranging venoms and venom fractions into 96-well plates allows for rapid screening of hundreds of samples, in principle, against multiple receptor targets.
In order to screen large chemical libraries (>104 compounds), these assays would need to be scaled-up to suit higher density formats such as 1536- or 3456-well plates. Higher density formats in combination with dedicated robotic workstations would make screening of large libraries highly feasible. Still, the P2X screens we developed provide a reliable, sensitive, and specific method for HTS assessment of venom fractions against hP2X3, hP2X4, and hP2X7. Using these assays, we first demonstrated that our HTS strategy allows screening of multiple targets, which provides significant cost and time savings. Second, the advantage of spending time to generate relatively pure natural products from a library is that it provides a more meaningful comparison between targets at early stages of the drug discovery process. Third, fractionation of the venoms allowed us to discriminate between fractions that are broadly cytolytic from those with a specific effect on a particular target. Finally, the majority of validated hits against hP2X4 were derived from spider venoms, further emphasizing the rich pharmacological diversity of this class of natural products.52−54 The availability of new and specific modulators from multiple chemical classes will be useful in understanding the biochemical, physiological, and clinical implications of venom toxins as well as providing functional insight into the P2X receptor family. In future work we aim to isolate and characterize new P2X inhibitors from these venomous animals in order to accelerate drug discovery in the purinergic field.
Experimental Section
Materials
Lyophilized hymenopteran venoms (species reported in Supporting Information, Table S4) were purchased from either Alphabiotoxine or Venomtech. Cone snail venoms were supplied by BioConus. Centipede venoms were provided by Dr. Eivind Undheim (The University of Queensland, Australia) and Dr. Ian Mellor (University of Nottingham, UK). All arachnid venoms were provided by Dr. Volker Herzig and Professor Glenn King (The University of Queensland) or were obtained from Alphabiotoxine. All other reagents were purchased from commercial sources and were of the highest purity commercially available.
Cell Cultures and Establishments of Stable Cell Lines
Human astrocytoma 1321N1 cells stably expressing hP2X4 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Bio-Whittaker) containing 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin (Fisher Scientific), and 400 μg/mL G418 (HelloBio). HEK293 cells stably expressing either hP2X3, hP2X4, or hP2X7 were maintained under the same condition in DMEM/F12 media (Gibco). The 1321N1-hP2X4 and HEK293-hP2X3 stable cell lines were generated by chemical transfection using Lipofectamine 2000 and plasmids encoding either hP2X3 or hP2X4. The hP2X3 plasmid was a kind gift from Dr. Lin-Hua Jiang (University of Leeds). Stable clones were selected using a positive selection marker (G418, 800 μg/mL). G418-resistant clones were further selected according to the strength of their ATP-induced increase in intracellular calcium ([Ca2+]I). All cells successfully expressing the receptor of interest were then expanded. All cells were maintained at 37 °C with 5% CO2 in a humidified incubator; P2X expression remained stable for at least 25–30 passages. All 96-well plates (Nunc catalogue number 167008, Fisher Scientific) were coated in-house with poly-d-lysine (Merck Millipore) at a concentration of 50 μg/mL.
[Ca2+]I Measurements (for Fura-2 AM Assay)
One day prior to measurements, 1321N1-hP2X4 cells were plated onto poly-d-lysine-coated 96-well plates at 2 × 104 cells/well. After 24 h, the cells were loaded for 1 h at 37 °C with 2 μM Fura-2 AM in Hank’s Balanced Salt Solution (HBSS, Gibco). The Fura-2 loading buffer dye was removed; then the cells were incubated in 80 μL of Etotal buffer, containing (in mM) 145 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 13 d-glucose, 10 HEPES; pH 7.33. Some of the compounds (IVM and/or BX430) were either pretreated with cells for 10 min or applied (crude venoms and fractions) before [Ca2+]I measurements on a Flexstation 3 (Molecular Devices) at 37 °C. The injection volume was 10 μL with 150 μL pipette height and rate of 4 (∼62 μL/s). The run time was 300 s with 3.5 s intervals and 3 reads/well. The change in [Ca2+]i concentration was calculated as the ratio of Fura-2 intensities at 520 emission from excitation at 340 and 380 nm (F ratio).
[Ca2+]I Measurements (for FLIPR Calcium 6 Assay)
One day prior to measurements, HEK293-hP2X3 cells were plated on poly-d-lysine-coated 96-well plates (Nunc catalogue number 167008, Fisher Scientific) at a concentration of 2 × 104 cells/well. After 24 h, the cells were loaded with the no-wash calcium-sensitive dye Calcium 6 using the FLIPR Calcium 6 assay kit (Molecular Devices) and incubated for 2 h prior to measurements on a Flexstation 3 (Molecular Devices) at 37 °C. The dye was diluted 1:3 in buffer containing (in mM) 145 NaCl, 5 KCl, 0.1 CaCl2, 13 d-glucose, and 10 HEPES; pH 7.35. The excitation and emission wavelengths were 485 and 525 nm, respectively, with 3 reads/well and a 1.3 s interval.
YOPRO-1 Dye Uptake Measurements
This method was adapted and further optimized from Patrice et al.50 One day prior to measurements, HEK293-hP2X4 and HEK293-hP2X7 cells were plated on poly-d-lysine-coated 96-well plates at 2 × 104 cells/well. After 24 h, the culture media was aspirated, and 80 μL of YOPRO-1 assay buffer (145 mM NaCl, 5 mM KCl, 0.1 mM CaCl2, 13 mM d-glucose, 10 mM HEPES; pH 7.35) with 2 μM YOPRO-1 was applied. Some of the compounds (IVM and/or BX430) were either preincubated with cells for 10 min or applied (crude venoms and fractions) before the measurements took place at 37 °C using a Flexstation 3 (Molecular Devices). The injection volume was 10 μL with a 150 μL pipet height and rate of 4 (∼62 μL/s). The run time was 300 s with a 3.9 s interval, 6 reads/well, and 77 reads in total. Measurement parameters were as follows: bottom reading, excitation wavelength (490 nm), emission wavelength (520 nm).
Isolation, Purification, and Mass Analysis of Venom Fractions
Venom (1 mg) was diluted with H2O, sterile filtered (0.22 μm; Merck Millipore), then loaded onto an analytical C18 RP-HPLC column (Jupiter 4.6 × 250 mm, 5 μm, 300 Å; Phenomenex) attached to an Agilent HPLC system. Components were eluted at 1 mL/min using isocratic elution at 5% solvent B (90% acetonitrile (MeCN), 0.05% trifluoroacetic acid (TFA) in H2O) for 5 min followed by a gradient of solvent B in solvent A (0.05% TFA in H2O): 5–20% over 5 min; 20–40% over 40 min; 40–80% over 5 min; 80–100% over 5 min. Absorbance was measured at 214, 254, and 280 nm using a UV detector (Shimadzu). Individual fractions were lyophilized, resuspended in 100 μL of H2O, and further purified using the same RP-HPLC system. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed on an Applied Biosystems 4700 Proteomics analyzer. The toxin fractions eluted from RP-HPLC were dissolved in 100–150 μL of H2O; then 2 μL was mixed with 2 μL of 10 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) matrix dissolved in 50% MeCN, 50% H2O, and 0.1% TFA. Toxins were then lyophilized in H2O and stored at −20 °C until further studies.
Assay Specificity
To assess assay specificity, we examined the response evoked by commercially available hP2X4 modulators (BX430, PSB12062, IVM), together with three fractions (F8, F28, F47) from N. chromatus venom that were not identified as hits in our initial assay. The positive control was a hit fraction (F5) from the same venom.
Assay Variability
Interplate and intraplate variability were evaluated using eight venom fractions in three different experiments. Venom fractions were prepared as described above and stored at 4 °C for the duration of the study. Each prepared fraction was tested on three different days, with eight replicates per plate. Eight replicates of positive controls (ATP), eight replicates of negative controls (buffer, antagonist), and eight replicates of a positive allosteric modulator (IVM) were included on each plate. Coefficients of variation were calculated using normalized results for each fraction by expressing the venom-fraction signal as a fraction of the averaged positive control signal from the same plate. For intraplate variability, unadjusted signal values were used to calculate variability between replicates for each fraction on a plate.
Assay Reproducibility
Assay reproducibility was assessed using the Z′ factor statistical method. This parameter assesses, in part, assay quality by calculating separation between positive and negative signals. Z′ values of 0.5–1.0 indicate a high level of reproducibility, whereas Z′ values of 0–0.5 indicate a less robust assay. The Z′ factor was calculated using the following formula:51
The Z′ experiment was performed twice with positive and negative controls (ATP and buffer, respectively) that were used throughout the assay development. In the first experiment, 60 positive controls (ATP) and 36 negative controls (hP2X4/hP2X7 antagonist) were tested. In the second experiment, 48 positive controls (ATP) and 48 negative controls (hP2X4/hP2X7 antagonist) were tested.
Data Analysis
GraphPad v. 8.0 was used to analyze data collected from the Flexstation 3 using SoftMax Pro v5.4 software; the baseline read delay was set to zero. Data are reported as mean ± SD, except where otherwise specified. For two groups, a paired t-test was performed. In the case of more than two groups, one-way ANOVA with multiple comparison (Dunnett’s post-test with ATP as the control sample) was used.
Acknowledgments
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (NRPDTP Grant 1794654) and the Royal Society (RG160939 to L.S.), and a Principal Research Fellowship to G.F.K. from the Australian Health & Medical Research Council. These funding bodies had no direct role in the design of the study, interpretation of the data, or content of the manuscript. We gratefully acknowledge members of the Deutsche Arachnologische Gesellschaft (DeArGe) for providing arachnids for venom extraction, in particular K. Fuchsgruber, M. Kesting, I. Wendt, and P. Zimmer. We thank Dr. E. Undheim (University of Queensland, Australia) and Dr. I. Mellor (University of Nottingham, UK) for providing centipede venoms. The hP2X3 plasmid was a kind gift from Dr. L.H. Jiang (University of Leeds, UK). We gratefully acknowledge the help of C. Smith in experiments with bee and wasp venoms.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.9b00410.
Additional information (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Paterson I.; Anderson E. A. Science 2005, 310, 451–453. 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- Grishin E. V.; Savchenko G. A.; Vassilevski A. A.; Korolkova Y. V.; Boychuk Y. A.; Viatchenko-Karpinski V. Y.; Nadezhdin K. D.; Arseniev A. S.; Pluzhnikov K. A.; Kulyk V. B.; Voitenko N. V.; Krishtal O. O. Ann. Neurol. 2009, 67, 680–683. 10.1002/ana.21949. [DOI] [PubMed] [Google Scholar]
- Klint J. K.; Smith J. J.; Vetter I.; Rupasinghe D. B.; Er S. Y.; Senff S.; Herzig V.; Mobli M.; Lewis R. J.; Bosmans F. J. Br. J. Pharmacol. 2015, 172, 2445–2458. 10.1111/bph.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera B. M.; Rivier J.; Clark C.; Ramilo C. A.; Corpuz G. P.; Abogadie F. C.; Mena E. E.; Woodward S. R.; Hillyard D. R.; Cruz L. J. Science 1990, 249, 257–263. 10.1126/science.2165278. [DOI] [PubMed] [Google Scholar]
- Diochot S.; Baron A.; Salinas M.; Douguet D.; Scarzello S.; Dabert-Gay A. S.; Debayle D.; Friend V.; Alloui A.; Lazdunski M.; Lingueglia E. Nature 2012, 490, 552. 10.1038/nature11494. [DOI] [PubMed] [Google Scholar]
- Diochot S.; Schweitz H.; Béress L.; Lazdunski M. J. J. Biol. Chem. 1998, 273, 6744–6749. 10.1074/jbc.273.12.6744. [DOI] [PubMed] [Google Scholar]
- Castañeda O.; Harvey A. L. Toxicon 2009, 54, 1119–1124. 10.1016/j.toxicon.2009.02.032. [DOI] [PubMed] [Google Scholar]
- Jouiaei M.; Casewell N.; Yanagihara A.; Nouwens A.; Cribb B.; Whitehead D.; Jackson T.; Ali S.; Wagstaff S.; Koludarov I.; Alewood P. Toxins 2015, 7, 936. 10.3390/toxins7030936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Gnanamani E.; Lynch S. R.; Zuñiga F. Z.; Jiménez-Vargas J. M.; Possani L. D.; Zare R. N. J. Nat. Prod. 2018, 81, 1899. 10.1021/acs.jnatprod.8b00527. [DOI] [PubMed] [Google Scholar]
- King G. F. Expert Opin. Biol. Ther. 2011, 11, 1469. 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- Holford M.; Daly M.; King G. F.; Norton R. S. Science 2018, 361, 842. 10.1126/science.aau7761. [DOI] [PubMed] [Google Scholar]
- Yu H.-b.; Li M.; Wang W.-p.; Wang X.-l. Acta Pharmacol. Sin. 2016, 37, 34. 10.1038/aps.2015.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; Overington J. P. Nat. Rev. Drug Discovery 2017, 16, 19. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J.; Bowlby M.; Peri R.; Vasilyev D.; Arias R. Nat. Rev. Drug Discovery 2008, 7, 358. 10.1038/nrd2552. [DOI] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Nat. Rev. Drug Discovery 2011, 10, 188. 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Keserü G. M.; Makara G. M. Nat. Rev. Drug Discovery 2009, 8, 203. 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- Koehn F. E.; Carter G. T. Nat. Rev. Drug Discovery 2005, 4, 206–220. 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- Bernier L. P.; Ase A. R.; Séguéla P. Br. J. Pharmacol. 2018, 175, 2219–2230. 10.1111/bph.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; Naito Y.; Hirokawa G.; Weng H.; Hiura Y.; Takahashi R.; Iwai N. Hypertens. Res. 2012, 35, 173. 10.1038/hr.2011.153. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Front. Pharmacol. 2017, 8, 661. 10.3389/fphar.2017.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford A.; Undem B. Front. Cell. Neurosci. 2013, 7, 267. 10.3389/fncel.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic Signalling 2016, 12, 59. 10.1007/s11302-015-9493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieślak M.; Czarnecka J.; Roszek K.; Komoszyński M. Purinergic Signalling 2015, 11, 307. 10.1007/s11302-015-9453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M. J. Neurosci. Res. 2017, 95, 1319. 10.1002/jnr.23816. [DOI] [PubMed] [Google Scholar]
- Neves A. R.; Castelo-Branco M. T.; Figliuolo V. R.; Bernardazzi C.; Buongusto F.; Yoshimoto A.; Nanini H. F.; Coutinho C. M.; Carneiro A. J. V.; Coutinho-Silva R. Inflamm Bowel Dis 2014, 20, 444. 10.1097/01.MIB.0000441201.10454.06. [DOI] [PubMed] [Google Scholar]
- Rassendren F.; Audinat E. J. Neurosci. Res. 2016, 94, 781. 10.1002/jnr.23770. [DOI] [PubMed] [Google Scholar]
- Ferrari D.; Vitiello L.; Idzko M.; la Sala A. Trends Mol. Med. 2015, 21, 184. 10.1016/j.molmed.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Liu J.; You X.; Kong P.; Song Y.; Cao L.; Yang S.; Wang W.; Fu Q.; Ma Z. Neurosci. Lett. 2016, 613, 60. 10.1016/j.neulet.2015.12.043. [DOI] [PubMed] [Google Scholar]
- Wu H.; Nie Y.; Xiong H.; Liu S.; Li G.; Huang A.; Guo L.; Wang S.; Xue Y.; Wu B. Inflammation 2015, 38, 2076. 10.1007/s10753-015-0189-y. [DOI] [PubMed] [Google Scholar]
- Helliwell R. M.; ShioukHuey C. O.; Dhuna K.; Molero J. C.; Ye J. M.; Xue C. C.; Stokes L. Br. J. Pharmacol. 2015, 172, 3326. 10.1111/bph.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai E.; Tsukimoto M.; Harada H.; Kojima S.. Purinergic Signalling 2014, 10, 487. 10.1007/s11302-014-9411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namovic M. T.; Jarvis M. F.; Donnelly-Roberts D.. Curr. Protoc Pharmacol 2012, 57, 10.1002/0471141755.ph0915s57. [DOI] [PubMed] [Google Scholar]
- Janzen W. P., Ed. High Throughput Screening: Methods and Protocols; Humana Press Scientific and Medical Publishers: Totowa, NJ, 2002. [Google Scholar]
- Hughes J. P.; Rees S.; Kalindjian S. B.; Philpott K. L. Br. J. Pharmacol. 2011, 162, 1239. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North R. A. Physiol. Rev. 2002, 82, 1013. 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Ase A. R.; Honson N. S.; Zaghdane H.; Pfeifer T. A.; Séguéla P. Mol. Pharmacol. 2015, 87, 606–616. 10.1124/mol.114.096222. [DOI] [PubMed] [Google Scholar]
- Balázs B.; Dankó T.; Kovács G.; Köles L.; Hediger M. A.; Zsembery Á. Cell. Physiol. Biochem. 2013, 32, 11. 10.1159/000350119. [DOI] [PubMed] [Google Scholar]
- Allsopp R. C.; Dayl S.; Schmid R.; Evans R. J. Sci. Rep. 2017, 7, 725. 10.1038/s41598-017-00732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Olmos V.; Abdelrahman A.; El-Tayeb A.; Freudendahl D.; Weinhausen S.; Müller C. E. J. Med. Chem. 2012, 55, 9576. 10.1021/jm300845v. [DOI] [PubMed] [Google Scholar]
- Kalliokoski T.; Kramer C.; Vulpetti A.; Gedeck P. PLoS One 2013, 8, e61007 10.1371/journal.pone.0061007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layhadi J. A.; Turner J.; Crossman D.; Fountain S. J. J. Immunol. 2018, 200, 1159. 10.4049/jimmunol.1700965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guette C.; Legros C.; Tournois G.; Goyffon M.; Celerier M. L. Toxicon 2006, 47, 640. 10.1016/j.toxicon.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Garenaux E.; Maes E.; Leveque S.; Brassart C.; Guerardel Y. Carbohydr. Res. 2011, 346 (9), 1093. 10.1016/j.carres.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Ferreira R. S. Jr.; Sciani J. M.; Marques-Porto R.; Junior A. L.; Orsi R. d. O.; Barraviera B.; Pimenta D. C. Toxicon 2010, 56, 355. 10.1016/j.toxicon.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Nakajima T. Venoms of the Hymenoptera 1986, 309. 10.1016/B978-0-12-554770-3.50010-3. [DOI] [Google Scholar]
- Lee M.-T.; Sun T.-L.; Hung W.-C.; Huang H. W. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 14243. 10.1073/pnas.1307010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. Neuron 1992, 8, 343. 10.1016/0896-6273(92)90300-3. [DOI] [PubMed] [Google Scholar]
- Priel A.; Silberberg S. D. J. Gen. Physiol. 2004, 123, 281. 10.1085/jgp.200308986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virginio C.; Robertson G.; Surprenant A.; North R. A. Mol. Pharmacol. 1998, 54, 53. 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- Bleicher K. H.; Böhm H.-J.; Müller K.; Alanine A. I. Nat. Rev. Drug Discovery 2003, 2, 369. 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- Patrice R.; Olivier E.; Tanter C.; Anaïs W.; Dutot M. J. Biol. Methods 2017, 4, e64. 10.14440/jbm.2017.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D.; Oldenburg K. R. J. Biomol. Screening 1999, 4, 67. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- King G. F.; Hardy M. C. Annu. Rev. Entomol. 2013, 58, 475. 10.1146/annurev-ento-120811-153650. [DOI] [PubMed] [Google Scholar]
- Smith J. J., Lau C. H. Y., Herzig V., Ikonomopoulou M. P., Rash L. D., King G. F.. Therapeutic applications of spider-venom peptides. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; Royal Society of Chemistry: Cambridge, United Kingdom, 2015; 221, 10.1039/9781849737876-00221. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.