

Abstract
An efficient deuteration process of β-amino C─H bonds in various N-alkylamine-based pharmaceutical compounds has been developed. Catalytic reactions begin with the action of Lewis acidic B(C6F5)3 and Brønsted basic N-alkylamine, converting a drug molecule into the corresponding enamine. The acid/base catalysts also promote the dedeuteration of acetone-d6 to afford a deuterated ammonium ion. Ensuing deuteration of the enamine then leads to the formation of β-deuterated bioactive amines with up to 99% deuterium incorporation.
Graphical Abstract
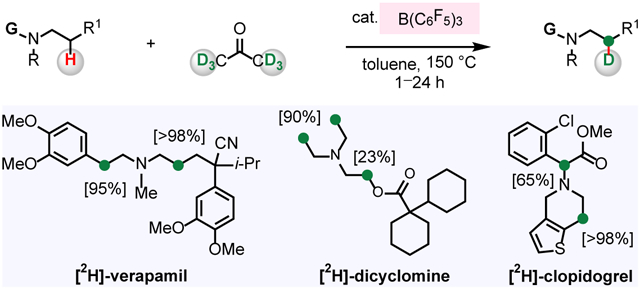
Deuterium-labeled pharmaceuticals are pivotal diagnostic tools in research aimed at determination of the corresponding biological outcomes and metabolites.1-6 Drugs containing C─D bonds have been prepared through multistep synthesis involving the reduction of unsaturated or halogenated intermediates.3 However, innovations in organometal-catalyzed C─H activation have enabled direct hydrogen isotope exchange (HIE) at C─H bonds for deuterium.4-6 In particular, HIE reaction targeting C(sp3)─H bonds of pharmaceuticals that contain an N-alkylamine unit is in high demand because these entities constitute over 50% of the top-selling commercial drugs.7 The state-of-the-art processes include Beller’s α- and β-amino C─H deuteration of metoclopramide (A1) and two other structurally related drug molecules promoted by Ru-based Shvo catalyst (Figure 1A).5 MacMillan’s photoredox-mediated α-deuteration and α-tritiation represents a notable strategy for isotopic labelling of a range of N-alkylamine-based drugs (Figure 1B).6 Still, development of methods for regioselective deuteration of poorly reactive β-amino C(sp3)─H bonds of drugs containing Lewis acid- and base-sensitive functional groups with an inexpensive deuterium source and promoted by non-precious metal-based catalysts is a significant challenge.8,9 Regioselective deuteration of metabolically stable β-amino C─H bonds (vs more labile α-amino C─H bonds) is particularly attractive as it minimizes the loss of the label due to exchange.1-3
Figure 1.


Amino C─H deuteration of biologically active molecules.
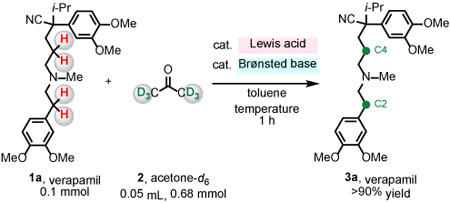
We began by contemplating a possible way to design a method for deuteration of biologically active compounds that contain an N-alkylamine unit (1) with readily available acetone-d6 2 as a source of deuterium (Figure 1C). We considered utilizing a combination of Lewis acid and Brønsted base catalysts that would function cooperatively.10-12 We envisioned that B(C6F5)3 could receive a hydride from an amine (1), generating a borohydride and an iminium ion (I).13-19 Subsequently, a Brønsted basic amine catalyst would deprotonate I, furnishing enamine II.13-16 Concurrently, the N-alkylamine could dedeuterate B(C6F5)3-activated acetone-d6 2, generating an enolate and a deuterated ammonium ion (III).20 Ensuing deuteration (IV) of the enamine II by III gives an iminium ion; subsequent borohydride reduction would afford β-deuterated product 3. Here, we report the development of a catalyst system for β-amino C─H deuteration of bioactive amines.
We first set out to identify a desirable combination of catalysts. We probed the ability of B(C6F5)3 and various Brønsted bases to catalyze the reaction between verapamil 1a and acetone-d6 2 (6.8 equivalent), generating 3a (Table 1). Treatment of 1a and 2 with 5.0 mol% B(C6F5)3 and 10 mol% NEt3, NBn3, or 1,2,2, 6,6-pentamethylpiperidine (PMP) afforded 3a in >90% yield (toluene, 125 °C, 1 h); 16-34% of β-amino C─H bonds were converted to C─D bonds (entries 1–3). With more Brønsted basic 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), no labeling was observed (entry 4). When the transformation was performed without a Brønsted base co-catalyst, 3a was generated with 35% and 21% deuterium incorporation (entry 5), suggesting that N-alkylamines 1a and/or 3a can promote deprotonation of the iminium ion (I, Figure 1C; NR3 = 1a and/or 3a). Deuterium incorporation diminished to <10% with 5.0 mol% of B(C6F5)3 and reaction temperature of 100 °C (entry 6), but when the reaction mixture was heated at 150 °C, 3a was obtained with >80% labeling (entry 7). With 10 mol% B(C6F5)3 there was only minor improvement (entry 8, 88% and 92%). However, by reacting 1a with two batches of 5.0 mol% of B(C6F5)3 and 2 (6.8 equivalent), we were able to obtain 3a with >95% deuterium incorporation (entry 9). There was no labeling without B(C6F5)3 or when the less hindered BF3 or the less Lewis acidic BPh3 were used (entries 10–12). These findings support the notion that strongly acidic B(C6F5)3 together with sterically demanding and electron-rich N-alkylamine constitute the most effective combination.10
Table 1.
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid | Brønsted base | temperature | d-incorporation (%) | |
| (mol%) | (mol%) | (°C) | [C2] | [C4] | |
| 1 | B(C6F5)3 (5.0) | NEt3 (10) | 125 | 17 | 26 |
| 2 | B(C6F5)3 (5.0) | NBn3 (10) | 125 | 20 | 34 |
| 3 | B(C6F5)3 (5.0) | PMP (10) | 125 | 16 | 26 |
| 4 | B(C6F5)3 (5.0) | DBU (10) | 125 | 0 | 0 |
| 5 | B(C6F5)3 (5.0) | none | 125 | 21 | 35 |
| 6 | B(C6F5)3 (5.0) | none | 100 | <5 | 7 |
| 7 | B(C6F5)3 (5.0) | none | 150 | 80 | 85 |
| 8 | B(C6F5)3 (10) | none | 150 | 88 | 92 |
| 9c | B(C6F5)3 (5.0 x 2) | none | 150 | 95 | >98 |
| 10 | none | none | 150 | 0 | 0 |
| 11 | BF3•OEt2 (5.0) | none | 150 | 0 | 0 |
| 12 | BPh3 (5.0) | none | 150 | 0 | 0 |
Conditions: verapamil (1a, 0.1 mmol), acetone-d6 (2, 0.68 mmol), organoborane, Brønsted base, toluene (0.4 mL), under N2, 1 h.
Yield and deuterium incorporation level was determined by 1H NMR analysis of unpurified reaction mixtures with mesitylene as the internal standard.
Conditions: verapamil (1a, 0.2 mmol), acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), toluene (0.8 mL), under N2, 150 °C, 1 h. Isolated and purified 3a was reacted with acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), toluene (0.8 mL), under N2, 150 °C, 1 h.
Green label indicates sites that are beta to N.
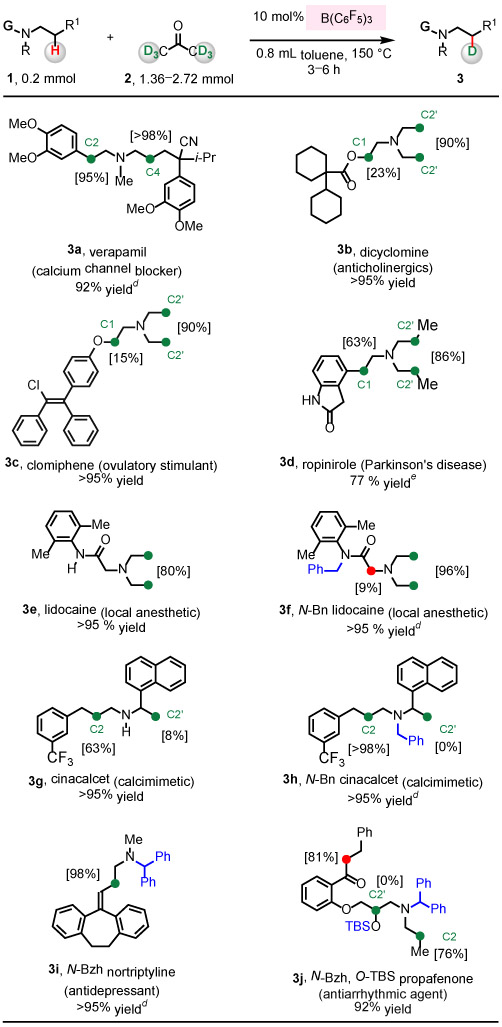
Acyclic β-amino C─H bonds in a number of pharmaceuticals (1a–1j) underwent efficient deuteration (Table 2). This protocol was found to be compatible with compounds that contain an array of Lewis acid-sensitive functional groups. In addition to the N-alkylamine units of 1a–1j, cyano (1a), ester (1b), amide (1d, 1e, 1f) and ketone (1j) were tolerated to give the deuteration products 3a–3j in 77 to >95% yield after purification by silica gel chromatography. Labeling took place with high regioselectivity for β-amino C─H bonds. In addition, drug molecules that possess acidic α-carbonyl C─H bonds also underwent efficient deuteration (3d, 3j) based on analysis of 1H NMR spectra of unpurified mixtures. Nonetheless, in case of 3d, α-carbonyl C─D bonds of indolin-2-one underwent H─D exchange during purification.
Table 2.
 |
Conditions: N-alkylamine (1, 0.2 mmol), acetone-d6 (2, 1.36 mmol), B(C6F5)3 (10 mol%), toluene (0.8 mL), under N2, 150 °C, 3 h.
Yield of isolated and purified product. Deuterium incorporation level was determined by 1H NMR analysis of the isolated and purified product.
Green label indicates sites that are beta to N. Red label is used for any other sites that undergo deuteration.
Conditions: N-alkylamine (1, 0.2 mmol), acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), toluene (0.8 mL), under N2, 150 °C, 3 h. After the filtration of the crude reaction mixture through a pad of silica gel and removal of volatiles, acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), and toluene (1.0 mL) were added under N2, and then heated at 150 °C, 3 h.
The reaction was carried out in two batches, using 10 mol% of B(C6F5)3 in the first batch, and 5.0 mol% in the second. For details, see the SI.
For substrates that possess electronically and sterically disparate β-amino C─H bonds (1a, 1b, 1c, 1d, 1g, 1j), deuterium labeling occurred at varying levels. With verapamil 1a, benzylic C2─H and non-benzylic C4─H bonds were converted to C─D bonds in >95%, but deuteration of non-benzylic C4─H bonds was more efficient (Table 1, entries 5-9). With dicyclomine 1b, while 90% of C2’─H bonds of N-ethyl groups was deuterated, only 23% of C1─H bonds adjacent to an ester group were converted to C─D bonds. Similar reactivity was observed with clomiphene 1c: 90% of C2’─H bonds were converted into C2’─D bonds, but 15% of α-aryloxy C1─H bonds were deuterated. Ropinirole 1d was labeled at benzylic C1─H bonds (63%), and C2’─H bonds of N-propyl group (86%).
Although the catalytic protocol tolerates an array of functional groups, isotopic labeling was more efficient with substrates bearing a protecting group. For instance, 80% of β-C─H bonds of lidocaine 1e, which possesses acidic amide N─H bonds, was converted to C─D bonds to give 3e. However, deuteration of N-benzyl-protected lidocaine 1f proceeded more efficiently to afford 3f with 96% d-incorporation; in addition, deuteration of α-carbonyl C─H bonds was also observed (9%). Cinacalcet 1g containing a secondary amine moiety was a compatible substrate to provide 3g (63% [C2] and 8% [C2’]), and N-benzyl-protected cinacalcet 3h was obtained with >98% of β-amino C2─H bonds selectively converted into C─D bonds. With less sterically hindered secondary amines nortriptyline 1i and propafenone 1j, their reaction with acetone-d6 may inhibit labeling. However, with an N-benzhydryl group installed, 3i and 3j could be readily generated. Silyl protection of the secondary alcohol proved to be effective in the case of propafenone 1j, giving 3j (76% [C2] and 0% at more sterically hindered [C2’]).
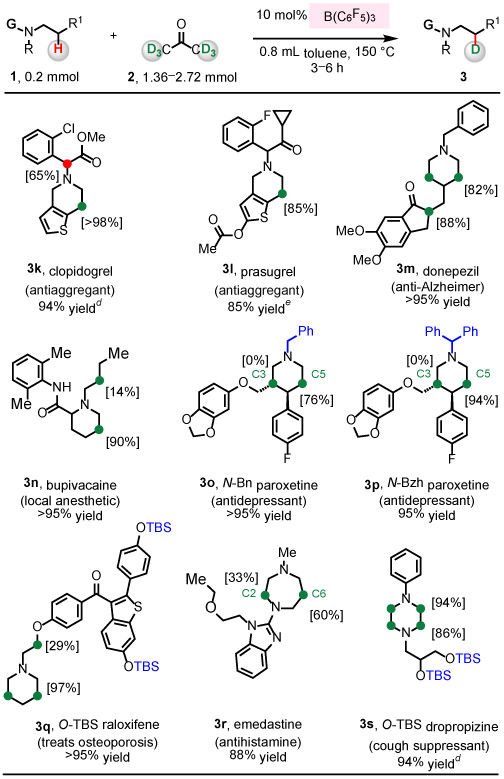
Next, we investigated possible labeling of various pharmaceuticals that contain cyclic β-amino C─H bonds (Table 3; 1k–1s). A variety of Lewis acid-sensitive heterocycles such as piperidine (1k–1q), 1,4-diazepane (1r), piperazine (1s), thiophene (1k, 1l), indanone (1m), benzodioxole (1o, 1p), benzothiophene (1q), as well as benzoimidazole (1r) were tolerated to give the corresponding deuteration products in 85 to >95% yield. With clopidogrel 1k, prasugrel 1l, and donepezil 1m, both β-amino C─H bonds and enolizable α-carbonyl C─H bonds underwent efficient deuteration to give 3k–3m, but acidic α-keto C─D bond of 3l was converted to C─H bond during purification. Less acidic α-amide C─H bond of bupivacaine 1n was not deuterated. With bupivacaine 1n and raloxifene 1q that contain acyclic and cyclic β-amino C─H bonds, labeling of the cyclic C─H bond was more efficient (>90% vs ≤29% for the acyclic C─H). N-Benzyl (1o) and N-benzhydryl (1p)-protected paroxetine gave 3o and 3p, respectively. The level of labeling for the more hindered 3p (94%) was superior to 3o (76%). Furthermore, deuteration of the C5─H bond occurred selectively, while the tertiary C3─H bond remained intact. Deuteration of emedastine 1r was found to take place at C2─H (33%) and C6─H (60%) bonds. All eight C─H bonds of piperazine ring of O-TBS-protected dropropizine 1s underwent deuteration to afford 3s (>86%). Using this protocol, α-amino C─H deuteration occurred only when these bonds were also alpha to a carbonyl group (3f) or beta to a N atom (3s).
Table 3.
 |
Conditions: N-alkylamine (1, 0.2 mmol), acetone-d6 (2, 1.36 mmol), B(C6F5)3 (10 mol%), toluene (0.8 mL), under N2, 150 °C, 3 h.
Yield of isolated and purified product. Deuterium incorporation level was determined by 1H NMR analysis of the isolated and purified product.
Green label indicates sites that are beta to N. Red label is used for any other sites that undergo deuteration.
Conditions: N-alkylamine (1, 0.2 mmol), acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), toluene (0.8 mL), under N2, 150 °C, 3 h. After the filtration of the crude reaction mixture through a pad of silica gel and removal of volatiles, acetone-d6 (2, 1.4 mmol), B(C6F5)3 (5.0 mol%), and toluene (1.0 mL) were added under N2, and then heated at 150 °C, 3 h.
The reaction was carried out in two batches, using 10 mol% of B(C6F5)3 in the first batch, and 5.0 mol% in the second. For details, see the SI.
The method is scalable. Treatment of 1.4 g (3.0 mmol) of verapamil 1a with 5.0 mol% B(C6F5)3, 20 mmol of acetone-d6 (toluene, 12 h, 150 °C), followed by filtration through a pad of silica gel and repeating the aforementioned procedure afforded 3a in 95% yield (2.9 mmol, 1.3 g) and >93% deuterium incorporation (Scheme 1).
Scheme 1.

Scale-up experiment
To summarize, we have designed an efficient and regioselective deuterium labeling of β-amino C─H bonds in various bioactive molecules, provided that sufficient steric congestion is present around the reacting amine. By implementing the cooperative action of B(C6F5)3 and N-alkylamine catalyst system, we show that it is possible to convert an N-alkylamine-based pharmaceutical compound to the corresponding enamine, and that the same catalyst system can generate a labeling agent from acetone-d6. The principles outlined herein, entailing conversion of amine containing drugs into enamines and its reaction with in situ generated electrophilic partner, provide a new rational framework for late-stage modification of a drug candidate. Studies along these lines are in progress.
Supplementary Material
Acknowledgements.
Financial support was provided by the NIH (GM-128695), the Sloan Foundation, and Boston College. We thank Professor Amir H. Hoveyda (Boston College) for helpful discussions.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) For selected reviews on hydrogen isotope exchange, see: Atzrodt J; Derdau V; Fey T; Zimmermann J The Renaissance of H/D Exchange. Angew. Chem., Int. Ed 2007, 46, 7744–7765. [DOI] [PubMed] [Google Scholar]; (b) Harbeson SL; Tung RD Deuterium Medicinal Chemistry: A New Approach to Drug Discovery and Development. MedChem News 2014, 2, 8–22. [Google Scholar]; (c) Atzrodt J; Derdau V; Kerr WJ; Reid M Deuterium- and Tritium-Labelled Compounds: Applications in the Life Sciences. Angew. Chem., Int. Ed 2018, 57, 1758–1784. [DOI] [PubMed] [Google Scholar]; (d) Atzrodt J; Derdau V; Kerr WJ; Reid M C─H Functionalisation for Hydrogen Isotope Exchange. Angew. Chem., Int. Ed 2018, 57, 3022–3047. [DOI] [PubMed] [Google Scholar]; (e) Pirali T; Serafini M; Cargnin S; Genazzani AA Applications of Deuterium in Medicinal Chemistry. J. Med. Chem 2019, 62, 5276–5297. [DOI] [PubMed] [Google Scholar]
- (2) (a).Penner N; Klunk LJ; Prakash C Human Radiolabeled Mass Balance Studies: Objectives, Utilities and Limitations. Biopharm. Drug Dispos 2009, 30, 185–203. [DOI] [PubMed] [Google Scholar]; (b) Miyoshi S; Mitsuoka K; Nishimura SA in Radioisotopes—Applications in Bio-Medical Science, Singh N, Ed.; InTech–Open Access Publisher, 2011; Chapter 5. [Google Scholar]
- (3).(a) For the multistep synthesis of hydrogen isotope-labeled drugs, see: Maltais F; Jung YC; Chen M; Tanoury J; Perni RB; Mani N; Laitinen L; Huang H; Liao S; Gao H; Tsao H; Block E; Ma C; Shawgo RS; Town C; Brummel CL; Howe D; Pazhanisamy S; Raybuck S; Namchuk M; Bennani YL In Vitro and In Vivo Isotope Effect with Hepatitis C Protease Inhibitors: Enhanced Plasma Exposure of Deuterated Telaprevir versus Telaprevir in Rats. J. Med. Chem 2009, 52, 7993–8001. [DOI] [PubMed] [Google Scholar]; (b) Allen PH; Hickey MJ; Kingston LP; Wilkinson DJ Metal-catalysed isotopic exchange labelling: 30 years of experience in pharmaceutical R&D. J. Labelled Comp. Radiopharm 2010, 53, 731–738. [Google Scholar]; (c) Lockey WJS; McEwen A; Cooke R Tritium: a coming of age for drug discovery and development ADME Studies. J. Labelled Comp. Radiopharm 2012, 55, 235–257. [Google Scholar]; (d) Elmore CS; Bragg RA Isotope chemistry; a useful tool in the drug discovery arsenal. Bioorg. Med. Chem. Lett 2015, 25, 167–171. [DOI] [PubMed] [Google Scholar]
- (4).(a) For organometal-catalyzed hydrogen isotope exchange reactions, see: Pieters G; Taglang C; Bonnefille E; Gutmann T; Puente C; Berthet J-C; Dugave C; Chaudret B; Rousseau B Regioselective and Stereospecific Deuteration of Bioactive Aza Compounds by the Use of Ruthenium Nanoparticles. Angew. Chem., Int. Ed 2014, 53, 230–234. [DOI] [PubMed] [Google Scholar]; (b) Taglang C; Martinez-Prieto LM; del Rosal I; Maron L; Poteau R; Philippot K; Chaudret B; Perato S; Lone AS; Puente C; Dugave C; Rousseau B; Pieters G Enantiospecific C─H Activation Using Ruthenium Nanocatalyst. Angew. Chem., Int. Ed 2015, 54, 10474–10477. [DOI] [PubMed] [Google Scholar]; (c) Yu RP; Hesk D; Rivera N; Pelczer I; Chirik PJ Iron-catalyzed tritiation of pharmaceuticals. Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]; (d) Hale LVA; Szymczak NK Stereoretentive Deuteration of α-Chiral Amines with D2O. J. Am. Chem. Soc 2016, 138, 13489–13492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chatterjee B; Krishnakumar V; Gunanathan C Selective α-Deuteration of Amines and Amino Acids using D2O. Org. Lett 2016, 18, 5892–5895. [DOI] [PubMed] [Google Scholar]; (f) Sawama Y; Nakano A; Matsuda T; Kawajiri T; Yamada T; Sajiki H H─D Exchange Deuteration of Arenes at Room Temperature. Organic Process Research & Development, 2019, 23, 648–653. KERR, Derdau, Kerr. [Google Scholar]
- (5).Neubert L; Michalik D; Bahn S; Imm S; Neumann H; Atzrodt J; Derdau V; Holla W; Beller M Ruthenium-Catalyzed Selective α,β-Deuteration of Bioactive Amines. J. Am. Chem. Soc 2012, 134, 12239–12244. [DOI] [PubMed] [Google Scholar]
- (6).Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC Photoredox-catalyzed deuteration and tritiation of pharmaceutical compounds. Science 2017, 358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ 2010, 53, 1348–1349. [Google Scholar]
- (8).(a) For reviews involving transformations of C(sp3)–H bonds, see: Dick AR; Sanford MS Transition metal catalyzed oxidative functionalization of carbon-hydrogen bonds. Tetrahedron 2006, 62, 2439–2463. [Google Scholar]; (b) Daugulis O; Roane J; Tran LD Bidentate, monoanionic auxiliary-directed functionalization of carbon-hydrogen bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-catalyzed transformations of alkyl C─H bonds. Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chu JCK; Rovis T Complementary strategies for directed C(sp3)–H functionalization: A comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbine/nitrene transfer. Angew. Chem., Int. Ed 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) He C; Whitehurst WG; Gaunt MJ Palladium-Catalyzed C(sp3)─H Bond Functionalization of Aliphatic Amines. Chem 2019, 5, 1031–1058. [Google Scholar]
- (9) (a).Yu P; Zheng S-C; Yang N-Y; Tan B; Liu X-Y Phosphine-catalyzed remote β-C─H functionalization of amines triggered by trifluoromethylation of alkenes: One-pot synthesis of bistrifluoromethylated enamides and oxazoles. Angew. Chem., Int. Ed 2015, 54, 4041–4045. [DOI] [PubMed] [Google Scholar]; (b) Ma L; Paul A; Breugst M; Seidel D Redox-neutral aromatization of cyclic amines: Mechanistic insights and harnessing of reactive intermediates for amine α- and β-C─H functionalization. Chem. Eur. J 2016, 22, 18179–18189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) For reviews of frustrated Lewis pair chemistry, see: Frustrated Lewis Pairs I: Uncovering and Understanding; Stephan DW; Erker G Eds.; Springer: Berlin, 2013; Vol. 332. [Google Scholar]; (b) Frustrated Lewis Pairs II: Expanding the Scope; Erker G; Stephan DW Eds.; Springer: Berlin, 2013; Vol. 334. [Google Scholar]; (c) Ashley AE; O'Hare D FLP-mediated activations and reductions of CO2 and CO. Top. Curr. Chem 2013, 334, 191–218. [DOI] [PubMed] [Google Scholar]; (d) Feng X; Du H Metal-free asymmetric hydrogenation and hydrosilylation catalyzed by frustrated Lewis pairs. Tetrahedron Lett. 2014, 55, 6959–6964. [Google Scholar]; (e) Stephan DW; Erker G Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem., Int. Ed 2015, 54, 6400–6441. [DOI] [PubMed] [Google Scholar]; (f) Stephan DW Frustrated Lewis pairs. J. Am. Chem. Soc 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]; (g) Oestreich M; Hermeke J; Mohr J A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev 2015, 44, 2202–2220. [DOI] [PubMed] [Google Scholar]; (h) Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- (11).(a) For selected reviews on cooperative catalysis, see: Tian S-K; Chen Y; Hang J; Tang L; McDaid P; Deng Li. Asymmetric Organic Catalysis with Modified Cinchona Alkaloids. Acc. Chem. Res 2004, 37, 621–631. [DOI] [PubMed] [Google Scholar]; (b) Shibasaki M; Kumagai N in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Peters R, Eds.; Wiley-VCH: New York, 2015; Chapter 1. [Google Scholar]
- (12).(a) For selected reviews on enantioselective non-covalent catalysis, see: Phipps RJ; Hamilton GL; Toste FD The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]; (b) Brak K; Jacobsen EN Asymmetric Ion-Pairing Catalysis. Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13) (a).Millot N; Santini CC; Fenet B; Basset JM Formation and characterization of zwitterionic stereoisomers from the reaction of B(C6F5)3 and NEt2Ph: (E)- and (Z)-[EtPhN+=CHCH2–B−(C6F5)3]. Eur. J. Inorg. Chem 2002, 2002, 3328–3335. [Google Scholar]; (b) Dureen MA; Brown CC; Stephan DW Addition of enamines or pyrroles and B(C6F5)3 “Frustrated Lewis Pairs” to alkynes. Organometallics 2010, 29, 6422–6432. [Google Scholar]; (c) Farrell JM; Heiden ZM; Stephan DW Metal-Free Transfer Hydrogenation Catalysis by B(C6F5)3. Organometallics 2011, 30, 4497–4500. [Google Scholar]
- (14).Focante F; Mercandelli P; Sironi A; Resconi L Complexes of tris(pentafluorophenyl)boron with nitrogen-containing compounds: Synthesis, reactivity and metallocene activation. Coord. Chem. Rev 2006, 250, 170–188. [Google Scholar]
- (15) (a).Zhang J; Park S; Chang S Catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C─Si bond formation. J. Am. Chem. Soc 2018, 140, 13209–13213. [DOI] [PubMed] [Google Scholar]; (b) Li R; Chen Y; Jiang K; Wang F; Lu C; Nie J; Chen Z; Yang G; Chen Y-C; Zhao Y; Ma C B(C6F5)3-catalyzed redox-neutral β-alkylation of tertiary amines using p-quinone methides via borrowing hydrogen. Chem. Commun 2019, 55, 1217–1220. [DOI] [PubMed] [Google Scholar]
- (16) (a).Maier AFG; Tussing S; Schneider T; Flörke U; Qu Z-W; Grimme S; Paradies J Frustrated Lewis pair catalyzed dehydrogenative oxidation of indolines and other heterocycles. Angew. Chem., Int. Ed 2016, 55, 12219–12223. [DOI] [PubMed] [Google Scholar]; (b) Kojima M; Kanai M Tris(pentafluorophenyl)borane-catalyzed acceptorless dehydrogenation of N-heterocycles. Angew. Chem., Int. Ed 2016, 55, 12224–12227. [DOI] [PubMed] [Google Scholar]
- (17) (a).Schwendemann S; Fröhlich R; Kehr G; Erker G Intramolecular frustrated N/B Lewis pairs by enamine hydroboration. Chem. Sci 2011, 2, 1842–1849. [Google Scholar]; (b) Chen G-Q; Kehr G; Daniliuc CG; Bursch M; Grimme S; Erker G Intermolecular Redox-Neutral Amine C─H Functionalization Induced by the Strong Boron Lewis Acid B(C6F5)3 in the Frustrated Lewis Pair Regime. Chem. Eur. J 2017, 23, 4723–4729. [DOI] [PubMed] [Google Scholar]
- (18).Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa M C─H Functionalization of amines via alkene-derived nucleophiles through cooperative action of chiral and achiral Lewis acid catalysts: Applications in enantioselective synthesis. J. Am. Chem. Soc 2018, 140, 10593–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tian J-J; Zeng N-N; Liu N; Tu X-S; Wang X-C Intramolecular cyclizations of vinyl-substituted N,N-dialkyl arylamines enabled by borane-assisted hydride transfer. ACS Catalysis 2019, 9, 295–300. [Google Scholar]
- (20).(a) For B(C6F5)3 and N-alkylamine-catalyzed deprotonation of carbonyl compounds, see: Shang M; Wang X; Koo SM; Youn J; Chan JZ; Yao W; Hastings BT; Wasa M Frustrated Lewis acid/Brønsted base catalysts for direct enantioselective α-amination of carbonyl compounds. J. Am. Chem. Soc 2017, 139, 95–98. [DOI] [PubMed] [Google Scholar]; (b) Cao M; Yesilcimen A; Wasa M Enantioselective Conia-Ene-Type Cyclizations of Alkynyl Ketones through Cooperative Action of B(C6F5)3, N-Alkylamine and a Zn-Based Catalyst. J. Am. Chem. Soc 2019, 141, 4199–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.