

Abstract
(+)-Perseanol is an isoryanodane diterpene with potent antifeedant and insecticidal properties isolated from the tropical shrub Persea indica.1 It is structurally related to (+)-ryanodine, a high affinity ligand and modulator of ryanodine receptors (RyRs)—ligand-gated ion channels critical for intracellular Ca2+ signaling in vertebrates and invertebrates.2 Whereas ryanodine modulates RyR-dependent Ca2+ release across many organisms, including mammals, preliminary data indicate that ryanodane and isoryanodane congeners that lack the pyrrole-2-carboxylate ester, such as perseanol, may have selective activity in insects.3 Here we report the first chemical synthesis of (+)-perseanol, which proceeds in 16 steps from commercially available (R)-pulegone. The synthesis features a two-step annulation process that rapidly assembles the tetracyclic core from readily accessible cyclopentyl building blocks. This work demonstrates how convergent fragment coupling, when combined with strategic oxidation tactics, can enable the concise synthesis of complex and highly oxidized diterpene natural products.
The ryanodane and isoryanodane natural products are oxidized diterpenes with antifeedant and insecticidal activities against insects of the Hemiptera and Lepidoptera orders. Ryanodine (1, Figure 1a), isolated from Ryania speciosa Vahl, was the first of these natural products to be characterized, and powdered R. speciosa wood was marketed as a botanical insecticide with peak annual production reaching 200 metric tons.4 The insecticidal properties of 1 result from its modulation of Ca2+ release by RyR.2 In the early 2000s, renewed interest in the insect RyR as a biological target for pest control agents resulted in the discovery and development of the phthalic acid diamide and anthranilic diamide insecticides—which bind at an allosteric site in the transmembrane domain of the insect RyR—with sales of these products exceeding 1 billion USD.5–6
Figure 1. The ryanodane and isoryanodane diterpenes.
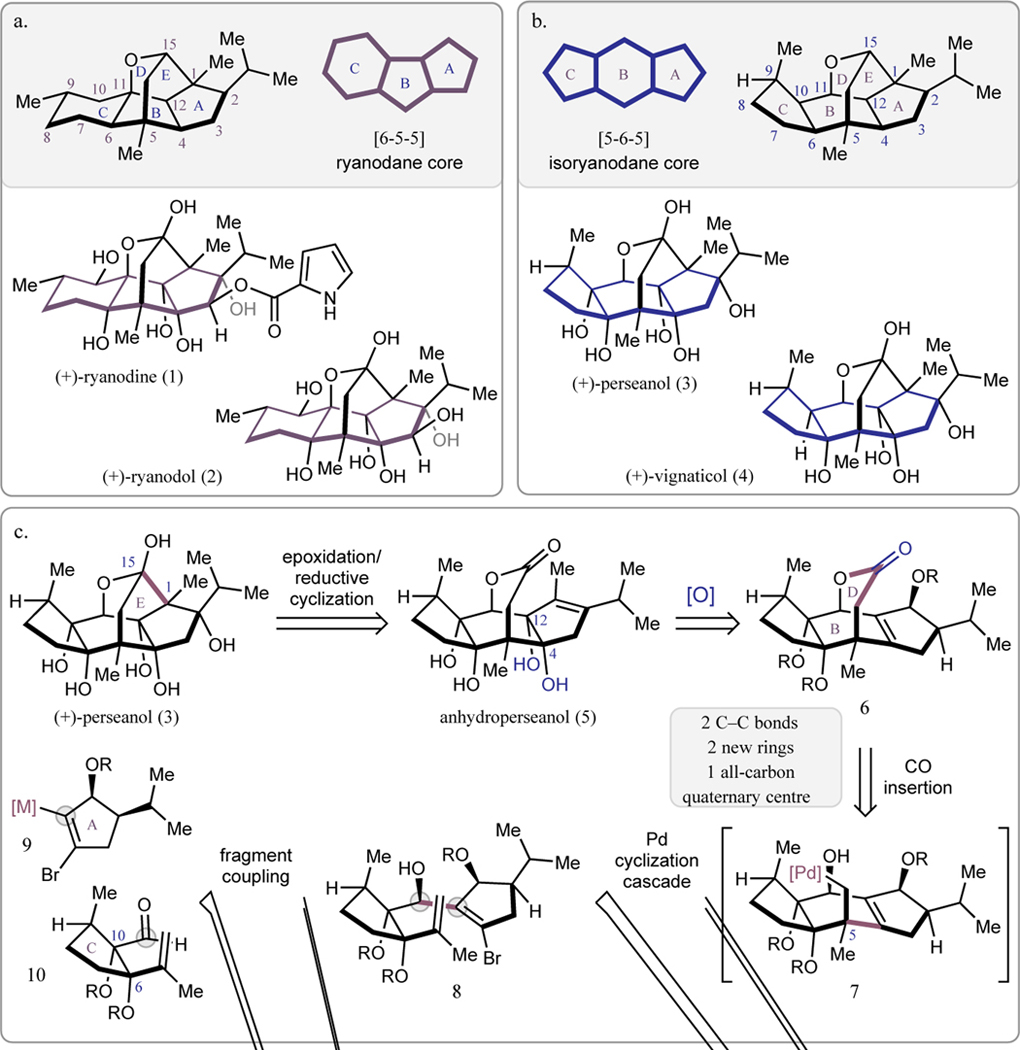
(a) Chemical structure, carbon numbering, and ring system letter assignment for the ryanodane diterpenes. (b) Chemical structure, carbon numbering, and ring system letter assignment for the isoryanodane diterpenes. (c) Retrosynthetic analysis of the isoryanodane diterpene (+)-perseanol.
Decades after the discovery of 1, Fraga and coworkers isolated the natural product (+)-perseanol (3, Figure 1b) and related congeners from the shrub Persea indica found in the Canarian Archipelago. Perseanol (3) features an isomeric carbon framework to 1 but bears a similar oxidation pattern and likely results from a shared biosynthetic pathway.1 A key difference between the structures of 3 and 1, in addition to their carbon skeletons, is that 3 lacks the pyrrole-2-carboxylate ester at C3, a functional group that is required for high affinity binding of 1 to mammalian isoforms of the RyR.2 Indeed, in preliminary assays, 3, 4, and related metabolites7–9 were found to exhibit potent antifeedant activity for lepidopteran pests with low toxicity toward mammalian cell lines (in contrast to 1), although the mode-of-action of 3 was not confirmed to be modulation of the insect RyR.10 Synthetic access to 3 could enable the elucidation of its mode-of-action and aid the identification of new approaches to target insect RyRs that have evolved resistance to the phthalic acid diamide and anthranilic diamide pesticides.11 Although Inoue and coworkers have reported an approach to the pentacyclic core of the isoryanodanes, there are no prior completed syntheses of this complex diterpene.12 Here we report the first chemical synthesis of (+)-perseanol (3), which proceeds in 16 steps from commercially available (R)-pulegone. The concise synthesis is enabled by a convergent fragment coupling approach that rapidly builds the anhydroperseanol tetracycle and uses strategic C–O bond constructions to minimize unnecessary functional group interconversions.
The structure of perseanol presents several synthetic challenges, including the central bridging 7-membered lactol and the two syn-diol motifs at the A–B and B–C ring fusions (Figure 1b). A critical aspect of our synthetic design was the strategic introduction of the six hydroxyl groups in order to minimize extraneous protecting group and oxidation state manipulations. With this in mind, we envisioned initially targeting the synthesis of anhydroperseanol (5), in which the C6–C10 diol would be introduced early in the synthetic sequence and the C4–C12 diol would be installed at a late stage (Figure 1c). Although the conversion of anhydroperseanol to perseanol had not previously been validated experimentally, this disconnection was guided by Deslongchamps’ synthesis of (+)-ryanodol (2),13 as well as our own synthesis of (+)-ryanodine.14–15 Having simplified our target to 5, we sought to identify a convergent fragment coupling that would rapidly assemble the tetracyclic lactone from two building blocks of similar size and complexity. Ultimately, lactone 6 was recognized as a strategic intermediate that could be accessed from simple cyclopentyl fragments by an annulation process involving two C–C bond forming steps: 1) the 1,2-addition of an organometallic species, such as 9, to aldehyde 10 to initially join the A and C rings, and 2) an intramolecular carbopalladation/carbonylation cascade reaction of 8 to close the B and D rings. In the key Pd-catalyzed cascade, it was envisioned that oxidative addition of alkenyl halide 8 to Pd0 followed by 6-exo-trig migratory insertion of the pendant 1,1-disubstituted alkene would give rise to σ-alkylpalladium species 7, which would be incapable of β-hydride elimination. Subsequent CO insertion of 7 and intramolecular capture by the C11 secondary alcohol would deliver 6, bearing the tetracyclic ring system of anhydroperseanol. In practice, this would require a bifunctional cyclopentene, 9, which we anticipated accessing via the selective lithiation of the corresponding iodide following precedent established by Vidari and coworkers.16 The second fragment, aldehyde 10, would be prepared from commercially available (R)-pulegone via the methyl pulegenate.17 The successful realization of this fragment coupling strategy would provide a modular route to 3 that we anticipated could ultimately give rise to additional designed and natural isoryanodanes.
Our investigations began with the preparation of C-ring aldehyde 10. Starting with (R)-(+)-pulegone (11), a known one-step oxidative ring contraction was performed to give methyl pulegenate (12) as an inconsequential mixture of diastereomers (Figure 2).18 Enolization of methyl ester 12 with potassium hexamethyldisilazide (KHMDS) followed by exposure to O2 then P(OMe)3 resulted in diastereoconvergent α-hydroxylation to furnish α-hydroxyester 13 (9:1 dr). Hydroxyl-directed epoxidation with meta-chloroperbenzoic acid (m-CPBA) provided epoxide 14 as a single diastereomer, and subsequent treatment of 14 with diethylaluminum 2,2,6,6-tetramethylpiperidide (Et2Al(TMP))19 induced epoxide isomerization to reveal syn-diol 15, bearing the requisite oxidation at C6 and C10 for elaboration to 3. Protection of the diol as the benzylidene acetal (16) followed by in situ diisobutylaluminum hydride (DIBAL) reduction of the ester provided alcohol 17 as a single diastereomer in 87% yield. Alcohol 17 was oxidized to aldehyde 18 via Stahl’s Cu-catalyzed aerobic conditions.20 This 6-step sequence provided gram scale access to a fully-elaborated C-ring precursor of (+)-perseanol (3).
Figure 2. Fragment preparation for the synthesis of (+)-perseanol.

Reagents and conditions as follows for C-ring fragment preparation: (1) Br2, NaHCO3, Et2O, –10 °C then NaOMe, MeOH, 55 °C, 78%. (2) KHMDS, THF then O2 (1 atm), P(OMe)3, −78 °C, 67%. (3) m-CPBA, NaHCO3, CH2Cl2, 0 °C, 92%. (4) Et2Al(TMP), PhMe, 0 °C, 68%. (5) benzaldehyde dimethyl acetal (PhCH(OMe)2), (±)-10-camphorsulfonic acid (CSA), 1,2-dichloroethane (DCE), 23 °C then DIBAL, 0 °C, 87%. (6) Cu(MeCN)4OTf, 4,4′-dimethoxy-2,3′-bipyridine (MeObpy), 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO), 1-methylimidazole (NMI), air, MeCN, 23 °C, 98%. Reagents and conditions as follows for A-ring fragment preparation: (1) 2-iodopropane (20), lithium diisopropylamide (LDA), diethylzinc (Et2Zn), hexamethylphosphoramide (HMPA), THF, −78 °C to 23 °C, 70%. (2) I2, CAN, MeCN, 0 °C to 23 °C, 73%. (3) 1.0 M NaOH (aq), 1,4-dioxane/MeOH (1:1), 23 °C. (4) oxalyl bromide ((COBr)2), DMF, CH2Cl2, 0 °C to 23 °C, 68%, 2 steps. (5) 25 (0.4 equiv), BH3•NEt2Ph (0.7 equiv), CH2Cl2, 23 °C, 44% (–)-27, 91% ee. (6) 28 (2.0 equiv), CSA, CH2Cl2, 23 °C, 81%.
Preparation of the A-ring fragment commenced with commercially available vinylogous ester 19 (Figure 2). Due to concerns about potential racemization under the conditions required to install the vicinal dihalide, we elected to prepare 24 first as a racemate, and then resolve the enantiomers in a subsequent asymmetric reduction step. To this end, the zinc enolate of 3-ethoxy-2-cyclopentenone (19) was alkylated under conditions reported by Overman and coworkers21 to generate rac-21. Iodination of the vinylogous ester with I2 and ammonium cerium(IV) nitrate (CAN) afforded iodide 22, which was hydrolyzed with aqueous sodium hydroxide. Diketone 23 was converted to rac-bromoiodocyclopentenone 24 upon treatment with a mixture of oxalyl bromide and N,N-dimethylformamide (DMF).22 The reaction proceeds with 5:1 regioselectivity, favoring bromination of the enol tautomer distal to the i-propyl group. Corey-Bakshi-Shibata (CBS) reduction of rac-24 using catalyst (R)-2523 resulted in a kinetic resolution to deliver alcohol (–)-(1S, 5R)-27 in 44% yield and 91% ee (S = 44, see Supplemental Information for details). The kinetic resolution is consistent with the stereochemical model developed by Corey (see 26),24 wherein the i-propyl substituent of (R)-24 projects away from the coordinated borane, resulting in reduction of (R)-24 at a faster rate than (S)-24. Unreacted enone (S)-24 could be recovered in 56% yield and 68% ee; resubjection of (R)-24 to (R)-25 allows it to be further enriched to 93% ee (79% recovery). Protection of alcohol 27 using Dudley’s conditions25 provided the C-ring fragment, para-methoxybenzyl (PMB) ether 29.
With the requisite fragments in hand, a two-step annulation to forge the anhydroperseanol tetracyclic ring system was investigated (Figure 3). First, the A and C ring fragments were joined by addition of aldehyde 18 to the alkenyllithium generated by selective lithium–iodide exchange of 29, which provided secondary alcohol 30 in 75% yield (3.2:1 dr, major diastereomer drawn). However, preliminary attempts to induce the subsequent carbopalladation/carbonylation cascade under canonical conditions, which involved exposure of the substrate to a Pd catalyst and base under a CO atmosphere, resulted in the clean recovery of alkenyl bromide 30 (Table 1, entry 1). A control experiment demonstrated that bromide 30 can undergo oxidative addition to bis(tri-ortho-tolylphosphine)palladium(0) (Pd(P(o-Tol)3)2) in the absence of CO, which led to the hypothesis that coordination of CO to Pd was inhibiting the rate of oxidative addition.26 To investigate the feasibility of the carbonylation step, bromide 30 was heated with stoichiometric Pd(P(o-Tol)3)2 to induce oxidative addition and alkene insertion, and upon consumption of starting material, CO was introduced. Gratifyingly, the desired tetracyclic lactone 31 was isolated in 52% yield under these stoichiometric conditions (entry 3). An extensive investigation of different Pd sources and ligands did not improve the yield further (entries 4 and 5, see Supplementary Information for further details). The major side product observed under these conditions was direct carbonylation of the bromide of 30 to give butyrolactone 32. Having validated that the cascade could be effected under stoichiometric conditions, we reasoned that in situ generation of CO, to maintain low concentrations of CO in solution,27–30 might enable the reaction to proceed with catalytic Pd. Ultimately, it was determined that the combination of 1.2 equiv N-formylsaccharin (36) and KF, in the presence of 50 mol % Pd(PPh3)4 and triethylamine (Et3N) provided the tetracyclic lactone 31 in 57% yield, as a single diastereomer at the newly formed quaternary carbon (entry 10). In contrast to the Manabe’s original report29 of Pd-catalyzed carbonylation with N-formylsaccharin, bisphosphine-ligated Pd complexes performed poorly (entries 12 and 13). This key transformation forges two C–C bonds, with perfect control over the C5 quaternary center, while forming the central 7-membered lactone of anhydroperseanol.
Figure 3. 16-step synthesis of (+)-perseanol.

Reagents and conditions as follows: (7) 29 (1.25 equiv), n-butyllithium (1.25 equiv), THF, −78 °C to −50 °C, 75%. (8) Pd(PPh3)4 (50 mol %), N-formylsaccharin (1.2 equiv), KF, Et3N, 1,4-dioxane, 100 °C, 57%. (9) DDQ, CH2Cl2/pH 7 buffer (5:1), 0 °C, 80%. (10) DMDO (3.0 equiv), Na2SO4, acetone, 23 °C. (11) MeMgCl (2.0 equiv), CeCl3•2LiCl (2.0 equiv), THF, 0 °C, 55%, 2 steps. (12) TFA, CH2Cl2, 0 °C, 90%. (13) SeO2, 1,4-dioxane, 100 °C, 78%. (14) VO(On-Pr)3, tert-butyl hydroperoxide (TBHP), PhMe, 60 °C, 68%. (15) 44 (4.5 equiv), PhH/THF (1:1), 10 °C, 25% (43% BRSM). (16) Pd(OH)2/C, H2 (1 atm), MeOH, 90%.
Table 1.
Evaluation of conditions for a Pd-catalyzed carbopalladation/carbonylation cascade.
| entrya | [Pd] | mol % | CO source | additive | N (min) | 30 (%)b | 31 (%)b | 32 (%)b |
|---|---|---|---|---|---|---|---|---|
| 1 | Pd(P(o-Tol)3)2 | 50 | CO (1 atm) | 0 | 92 | 1 | 5 | |
| 2 | Pd(P(o-Tol)3)2 | 50 | CO (1 atm) | 20 | 67 | 11 | 15 | |
| 3 | Pd(P(o-Tol)3)2 | 120 | CO (1 atm) | 90 | 0 | 52 | 0 | |
| 4 | Pd(P(o-Tol)3)2 | 120 | CO (10 atm) | 90 | 0 | 53 | 0 | |
| 5 | Pd(PPh3)4 | 120 | CO (1 atm) | 90 | 23 | 48 | 13 | |
| 6 | Pd(PPh3)4 | 50 | 33 | DBU | 85 | 0 | 8 | |
| 7 | Pd(PPh3)4 | 50 | 34 | 90 | 0 | 0 | ||
| 8 | Pd(PPh3)4 | 50 | 35 | KF | 14 | 7 | 4 | |
| 9 | Pd(PPh3)4 | 50 | 36 | KF | 22 | 31 | 10 | |
| 10 | Pd(PPh3)4 | 50 | 36 | KF | 1 | 57 | 14 | |
| 11 | Pd(P(o-Tol)3)2 | 50 | 36 | KF | 60 | 0 | 0 | |
| 12 | PdCl2(dppf) | 50 | 36 | KF | 80 | 0 | 0 | |
| 13 | PdCl2(Xantphos) | 50 | 36 | KF | 55 | 1 | 4 | |
Reactions performed on 0.01 mmol scale at 100 °C.
Yields determined by 1H NMR versus pyrazine as an added internal standard.
DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; dppf, 1,1′-bis(diphenylphosphino)ferrocene; Xantphos, 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
With the tetracyclic framework of anhydroperseanol (5) in place, our focus transitioned to the final adjustments of the A-ring oxidation pattern (Figure 3). To this end, PMB ether 31 was first subjected to 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to reveal C1 secondary alcohol 37, which was oxidized with dimethyldioxirane (DMDO) to the corresponding enone. In the presence of excess DMDO, the benzylidene acetal was unexpectedly oxidized to deliver hydroxybenzoate 38 (3:1 rr, major isomer drawn). Treatment of 38 with MeMgCl in the presence of CeCl3•2LiCl31 effected 1,2-addition to generate diol 39 (55% isolated yield of a single isomer, over two steps), an intermediate that now harbors all of the carbons present in the isoryanodane framework. Serendipitously, it was discovered that exposure of allylic alcohol 39 to trifluoroacetic acid (TFA) at 0 °C gives rise to orthobenzoate 41 in excellent yield. This 1,3-allylic transposition presumably proceeded by solvolysis under anchimeric assistance to generate dioxolenium ion 40, which is followed by intramolecular trapping with the C10 alcohol. Thus, over the course of these four steps, the benzylidene acetal protecting group was transiently repurposed as a directing group to guide the installation of the C4 tertiary alcohol and then reinstated as an orthobenzoate protecting group to mask the resulting triol for the rest of the synthesis.
With this fortuitous discovery, we were left to reconsider the final sequence of steps to prepare perseanol. Although we had initially targeted the preparation of anhydroperseanol (see Figure 1), the ability to prepare 41 led us to consider whether epoxide 43—potentially accessible from 41 by allylic C–H oxidation and hydroxyl-directed epoxidation—could undergo reductive cyclization. It was recognized that this cyclization might be challenging, given that formation of the C1–C15 bond via epoxylactone isomer 43 would require a Baldwin disfavored32 5-endo-tet epoxide ring opening, when viewed from the formation of the THF ring. Successful endo-ring openings of epoxides have been reported in the literature, but they generally rely on directing groups to stabilize the epoxonium intermediate under Brønsted or Lewis acidic conditions; related endo-cyclizations of epoxides under neutral or basic conditions are less common. Nevertheless, given the strategic advantage of this approach, we elected to investigate it.
To this end, exposure of 41 to SeO2 in 1,4-dioxane at 100 °C resulted in site-selective and stereospecific oxidation at C2 to give tertiary allylic alcohol 42 in 78% yield (Figure 3). Vanadium-mediated hydroxyl-directed epoxidation of 42 then provided epoxyalcohol 43 as a single diastereomer. The use of vanadium(V) oxytripropoxide (VO(On-Pr)3) proved essential to obtain full conversion of alkene 42; the more routinely used vanadium(III) acetylacetonate (VO(acac)2) gave only 5–10% conversion under otherwise identical conditions. Treatment of epoxylactone 43 with lithium 4,4′-di-tert-butylbiphenylide (LiDBB), the optimal conditions from our (+)-ryanodine synthesis,15 did produce small quantities of the desired pentacycle 46; however, significant decomposition was observed. Analysis of the side products revealed that reduction of the orthobenzoate was a competing process, prompting a screen of different reductants in order to prevent this undesired reactivity. Use of lithium naphthalenide (LiNap) provided the desired pentacycle in 17% isolated yield. Weaker reductants, like lithium anthracenide (LiAnth), gave rise to epoxide isomerization products instead of reductive cyclization. A further screen of modified naphthalenes revealed that use of lithium 2-phenylnaphthalenide (LiPhNap) effects cyclization to give the desired pentacycle 46 in 25% yield (43% yield based on recovered starting material). The use of PhH as a co-solvent, which had previously been reported by Carreira and coworkers to improve ketyl anion chemistry, was critical for the improved yield.33 We note that a similar substrate, lacking the C2 i-propyl substituent, undergoes the reductive cyclization mediated by LiDBB in 50% yield, demonstrating that the position of the epoxide itself is not chiefly responsible for the reduced efficiency in the cyclization. Deprotection of 46 with Pd(OH)2/C under an atmosphere of H2 afforded (+)-perseanol (3) in 90% yield. This approach provides (+)-perseanol (3) in 16 steps (longest linear sequence) from (R)-pulegone (11), and is the first total synthesis of an isoryanodane diterpene. The concision of the synthesis derives from the convergent union of two cyclopentyl fragments of comparable complexity, followed by a carbopalladation/carbonylation cascade to form two C–C bonds and rapidly constructs the tetracyclic lactone framework of anhydroperseanol. Strategic late-stage introduction of the A-ring oxidation pattern minimized lateral redox and protecting group manipulations. This synthetic framework should provide a versatile platform for the preparation of designed isoryanodanes and further studies of their mode-of-action.
Methods.
Unless otherwise stated, reactions were performed under an inert atmosphere (dry Ar) with freshly dried solvents utilizing standard Schlenk techniques. Tetrahydrofuran (THF), methylene chloride (CH2Cl2), acetonitrile (MeCN), diethyl ether (Et2O), 1,4-dioxane, toluene (PhMe), and benzene (PhH) were dried by passing through activated alumina columns. Yields refer to chromatographically and spectroscopically (1H and 13C) homogeneous materials, unless otherwise stated. Reagents were purchased at the highest commercial quality and used without purification, unless otherwise stated. Reactions were magnetically stirred and monitored by thin-layer chromatography. For full experimental details–including procedures for all reactions and characterization of all compounds (1H NMR, 13C NMR, mass spectrometry, infrared spectroscopy, retention factors)–see the Supplementary Information.
Supplementary Material
Acknowledgments.
Dr. Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis are gratefully acknowledged for access to analytical equipment. We thank Mr. Larry Henling (Caltech) and Dr. Julie Hofstra (Caltech) for X-ray data collection and data refinement, respectively, for the structure of 32 and S21. Prof. Yonghui Zhang (Huanzhong University of Science and Technology) is acknowledged for providing original spectral data of perseanol. Dr. Kangway Chuang (Caltech) is gratefully acknowledged for insightful contributions to the synthetic design. Fellowship support was provided by the NIH (A.H., Nos. 5T32GM007616-37 and 1F31GM120821). S.E.R. is a Heritage Medical Research Investigator. Financial support from the NIH (Nos. NIGMS RGM097582-01 and R35GM118191-01), Eli Lilly, and Novartis is gratefully acknowledged.
Footnotes
The authors declare no competing interests.
Data Availability. Characterization data for all compounds produced in this study are available in the Supplementary Information or on request from the corresponding author. Metrical parameters for the structure of 32 and S21 are available free of charge from the Cambridge Crystallographic Data Centre (CCDC) under reference number 1909375 and 1914686, respectively.
Supplementary Information is available in the online version of the paper.
References
- 1.Gonzalez-Coloma A; Terrero D; Perales A; Escoubas P; Fraga BM Insect antifeedant ryanodane diterpenes from Persea indica. J. Agric. Food. Chem 1996, 44, 296–300. [Google Scholar]
- 2.Sutko JL; Airey JA; Welch W; Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol. Rev 1997, 49, 53–98. [PubMed] [Google Scholar]
- 3.Gonzalez-Coloma A; Reina M; Gutierrez C; Fraga BM Natural insecticides: structure diversity, effects and structure-activity relationships. A case study. In Studies in Natural Products Chemistry, 2002; Vol. 26, pp 849–879. [Google Scholar]
- 4.United States Environmental Protection Agency R.E.D. Facts Ryanodine. https://archive.epa.gov/pesticides/reregistration/web/pdf/2595fact.pdf (accessed Oct 2018).
- 5.Lahm GP et al. RynaxypyrTM: a new anthranilic diamide insecticide acting at the Ryanodine receptor In Pesticide Chemistry: Crop Protection, Public Health, Environmental Safety, Ohkawa H; Miyagawa H; Lee PW, Eds. Wiley-VCH: Weinheim, 2007; pp 111–120. [Google Scholar]
- 6.Cordova D. et al. Elucidation of the mode of action of RynaxypyrTM, a selective Ryanodine receptor activator In Pesticide Chemistry: Crop Protection, Public Health, Environmental Safety, Ohkawa H; Miyagawa H; Lee PW, Eds. Wiley-VCH: Weinheim, 2007; pp 121–126. [Google Scholar]
- 7.Nohara T. et al. Cinncassiol D1 and its glucoside, novel pentacyclic diterpenes from Cinnamomi cortex. Tetrahedron Lett. 1980, 21, 2647–2648. [Google Scholar]
- 8.Chai XY et al. Six insecticidal isoryanodane diterpenoids from the bark and twigs of Itoa orientalis. Tetrahedron 2008, 64, 5743–5747. [Google Scholar]
- 9.Zeng JF et al. Diterpenoids with immunosuppressive activities from Cinnamomum cassia. J. Nat. Prod 2014, 77, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 10.Ling SQ; Xu YN; Gu YP; Liu SY; Tang WW Toxicity and biochemical effects of itol A on the brown planthopper, Nilaparvata lugens (Stål) (Hemiptera: Delphacidae). Pestic. Biochem. Physiol 2018, 152, 90–97. [DOI] [PubMed] [Google Scholar]
- 11.Sattelle DB; Cordova D; Cheek TR Insect ryanodine receptors: molecular targets for novel pest control chemicals. Invert. Neurosi 2008, 8, 107–19. [DOI] [PubMed] [Google Scholar]
- 12.Koshimizu M; Nagatomo M; Inoue M. Construction of a pentacyclic ring system of isoryanodane diterpenoids by SmI2-mediated transannular cyclization. Tetrahedron 2018, 74, 3384–3390. [Google Scholar]
- 13.Deslongchamps P. et al. Total synthesis of ryanodol. Can. J. Chem 1979, 57, 3348–3354. [Google Scholar]
- 14.Chuang KV; Xu C; Reisman SEA 15-step synthesis of (+)-ryanodol. Science 2016, 353, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu C; Han A; Virgil SC; Reisman SE Chemical synthesis of (+)-ryanodine and (+)-20-deoxyspiganthine. ACS Cent. Sci 2017, 3, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luparia M; Vadala A; Zanoni G; Vidari G. 1,2-Bisanionic coupling approach to 2,3-disubstituted cyclopentenols and cyclopentenones. Org. Lett 2006, 8, 2147–2150. [DOI] [PubMed] [Google Scholar]
- 17.Marx JN; Norman LR Synthesis of (–)-acorone and related spirocyclic sesquiterpenes. J. Org. Chem 1975, 40, 1602–1606. [Google Scholar]
- 18.Shen Y. et al. Protecting-group-free total synthesis of (–)-jiadifenolide: development of a [4+1] annulation toward multisubstituted tetrahydrofurans. Org. Lett 2015, 17, 5480–5483. [DOI] [PubMed] [Google Scholar]
- 19.Yasuda A; Yamamoto H; Nozaki H. Highly Stereospecific isomerization of oxiranes into allylic alcohols by means of organoaluminum amides. Bull. Chem. Soc. Jpn 1979, 52, 1705–1708. [Google Scholar]
- 20.Steves JE; Stahl SS Copper(I)/ABNO-catalyzed aerobic alcohol oxidation: alleviating steric and electronic constraints of Cu/TEMPO catalyst systems. J. Am. Chem. Soc 2013, 135, 15742–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iimura S; Overman LE; Paulini R; Zakarian A. Enantioselective total synthesis of guanacastepene N using an uncommon 7-endo Heck cyclization as a pivotal step. J. Am. Chem. Soc 2006, 128, 13095–13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mewshaw RE Vilsmeier reagents: preparation of β-halo-α,β-unsaturated ketones. Tetrahedron Lett. 1989, 30, 3753–3756. [Google Scholar]
- 23.Helal CJ; Meyer MP The Corey-Bakshi-Shibata reduction: mechanistic and synthetic considerations – bifuntional Lewis base catalysis and dual activation In Lewis Base Catalysis in Organic Synthesis, 1st ed.; Vedejs E; Denmark SE, Eds. Wiley-VCH: Weinheim, Germany, 2016; pp 387–455. [Google Scholar]
- 24.Corey EJ; Link JO A new chiral catalyst for the enantioselective synthesis of secondary alcohols and deuterated primary alcohols by carbonyl reduction. Tetrahedron Lett. 1989, 30, 6275–6278. [Google Scholar]
- 25.Nwoye EO; Dudley GB Synthesis of para-methoxybenzyl (PMB) ethers under neutral conditions. Chem. Commun 2007, 1436–1437. [DOI] [PubMed] [Google Scholar]
- 26.Wu LP et al. Palladium-catalyzed carbonylative transformation of C(sp3)-X bonds. ACS Catal. 2014, 4, 2977–2989. [Google Scholar]
- 27.Odell LR; Russo F; Larhed M. Molybdenum hexacarbonyl mediated CO gas-free carbonylative reactions. Synlett 2012, 685–698. [Google Scholar]
- 28.Ueda T; Konishi H; Manabe K. Palladium-catalyzed carbonylation of aryl, alkenyl, and allyl halides with phenyl formate. Org. Lett 2012, 14, 3100–3103. [DOI] [PubMed] [Google Scholar]
- 29.Ueda T; Konishi H; Manabe K. Palladium-catalyzed fluorocarbonylation using N-formylsaccharin as CO source: general access to carboxylic acid derivatives. Org. Lett 2013, 15, 5370–5373. [DOI] [PubMed] [Google Scholar]
- 30.Jiang X. et al. Palladium-catalyzed formylation of aryl halides with tert-butyl isocyanide. Org. Lett 2014, 16, 3492–3495. [DOI] [PubMed] [Google Scholar]
- 31.Krasovskiy A; Kopp F; Knochel P. Soluble lanthanide salts LnCl3•2LiCl for the improved addition of organomagnesium reagents to carbonyl compounds. Angew. Chem. Int. Ed 2006, 45, 497–500. [DOI] [PubMed] [Google Scholar]
- 32.Baldwin JE; Cutting J; Dupont W; Kruse L; Silberman L; Thomas RC 5-endo-trigonal reactions: disfavored ring-closure. J. Chem. Soc. Chem. Commun 1976, 736–738. [Google Scholar]
- 33.Wolleb H; Carreira EM Total synthesis of (+)-dendrowardol C. Angew. Chem. Int. Ed 2017, 56, 10890–10893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.