

Abstract
Body-on-a-chip in vitro systems are a promising technology that aims to increase the predictive power of drug efficacy and toxicity in humans when compared to traditional animal models. Here, we developed a new heart-liver body-on-a-chip system with a skin surrogate to assess the toxicity of drugs that are topically administered. In order to test the utility of the system, diclofenac, ketoconazol, hydrocortisone and acetaminophen were applied topically through a synthetic skin surrogate (Strat-M membrane) and the toxicity results were compared to acute drug exposures from systemically applying the compounds. The heart-liver system was successful in predicting the effects for both cardiac and liver function changes due to the compounds. The difference in the concentrations of drugs applied topically compared to systemically indicate that the barrier properties of the skin surrogate was efficient. One important advantage of this heart-liver system was the capability of showing differential effects of acute and chronic drug exposure which is necessary as part of the International Conference in Harmonisation (ICH) tri-partate guidelines. In conclusion, this work indicates a promising heart-liver body-on-a-chip system that can be used for the assessment of potential drug toxicity from dermal absorption as well as evaluate transport dynamics through the skin in the same system.
Graphical Abstract
Introduction
One of the most challenging problems faced by pharmacology and biotechnology companies is the low success rate of compounds that make it to regulatory approval. Success rates of compounds range from 26% (for haematology compounds) to 5% (oncology drugs) with an overall success rate of 9.6% for all indications.1 This low percentage of approval is mostly due to the low accuracy of prediction of preclinical in vitro and in vivo models for drug efficacy and toxicity in humans.2 In the last few decades multi-organ microphysiological systems technology (also known as “body-on-a-chip”) has emerged as a possible solution to increase the predictive power for human response to drugs.3 To achieve this purpose there is a need to improve the complexity of the systems by adding different organ tissues to the same chip and to be able to test different drug delivery methods.
Drug toxicity can affect several tissues, however compounds that have been withdrawn from trials were mostly those that cause hepatoxicity and cardiotoxicity.4 Although most drugs have better bioavailability and consequently are more effective through oral administration, transdermal drug delivery is a successful method for administrating a compound for prolonged periods of time that can reduce this risk of toxicity.5 For that reason, it is relevant to develop a multi-organ system that efficiently evaluates the toxicity of topically administrated drugs to human heart and liver tissues.
Diclofenac sodium, ketoconazol, acetaminophen and hydrocortisone are well known drugs with topical and oral formulations clinically available. Diclofenac sodium is a phenylacetic acid derivative and a non-steroidal anti-inflammatory drug indicated for pain and inflammation management. Oral formulations of diclofenac sodium have been shown to induce a variety of gastrointestinal side effects such as perforations and ulcerations and in some cases hepatic and cardiovascular complications have been reported. To minimize the potential side effects from oral diclofenac formulations, topical formulations have been developed.6 Ketoconazole is a broad-spectrum antifungal derived from imidazole and is mostly indicated for superficial and systemic mycosis (ketoconazole hepatoxicity). Recently, the United States Food and Drug Administration (FDA) issued a drug safety communication involving the use of oral ketoconazole due to potential fatal drug induced liver injury.7 Ketoconazole is also administered topically, generally in the form of cream or suspensions.8 Hydrocortisone is a synthetic version of the hormone cortisol which is produced by the adrenal cortex. Hydrocortisone is a glucocorticoid and is often used as a standard therapy for patients with adrenal insufficiency.9 Finally, acetaminophen is a widely used non-narcotic analgesic and antipyretic agent used for pain relief and to treat fever.10 Acetaminophen is relatively safe when consumed in the recommended doses, however, ingestion of large doses can cause severe liver injury.11 Recent reports have suggested that acetaminophen could induce acute liver failure even at lower doses.12
Current in vitro models have a variety of limitations and there are few manuscripts detailing systems demonstrating inclusion of skin and other barrier tissues in a functional multi-organ system 13. Cell based skin-on-a-chip models have been more physiologically relevant alternative model compared to traditional monolayer cell culture, however the precise control of the skin microenviroment, the inability to recapitulate the heterogeneous nature of the skin which can affect the permeation rate of drugs are important challenges that need to be addressed 14. Furthermore, few skin-on-a-chip models integrate other organs such as heart and liver, and are not able to evaluate the toxicity and effect of drugs in these important tissues.
In this work, we have developed a new microphysiological heart-liver (HL) body-on-a-chip system with a skin surrogate for testing drug toxicity associated with transdermal drug delivery. We have evaluated diclofenac, ketoconazole, hydrocortisone and acetaminophen by topical and systemic administration to evaluate the toxicity prediction of the system. This platform efficiently predicted drug toxicity not only from acute drug exposure but also for chronic drug treatment, which is an important feature needed for in vitro technology for drug testing. Another advantage of the system is the real time assessment of cardiac organ function. This new technological finding ultimately could assist in the design of clinical trial protocols and reduce the cost, time and labour of drug development.
Experimental
Microfabrication
Microfluidic housings were designed and fabricated as previously described 15,16 using poly (methyl methacrylate) and polydimethylsiloxane (PDMS) elastomer sheets. The medium recirculates between 2 reservoirs through a microfluidic network formed by the PDMS gaskets, with flow driven by a rocking platform. Drug dosing was applied either A) at a construct containing a Strat-M membrane (Millipore) acting as a barrier between the air and the medium for topical application or B) at the reservoir closest to the liver chamber for systemic application. A cantilever bioMEMS device for measuring cardiac force contained an array of two rows of microscale cantilevers (4 μm thick, 100 μm wide, and 750 μm long), with 16 cantilevers per row. Custom patterned MEA chips, for cardiac electrical measurements, were fabricated using standard microfabrication procedures following a previously described protocol 17. The surface of each chip was coated with poly (ethylene glycol) (PEG)-containing silane, and this PEG layer was patterned by selective ablation using a 193 nm ArF excimer laser (Lambda Physik, Santa Clara, CA). The ablated PEG surfaces were coated with human plasma fibronectin (Millipore) diluted in 1X phosphate buffered saline (PBS) (Thermo Scientific), then incubated at 37 °C for 30 minutes followed by a PBS rinse.
CYP 1A1 and 3A4 enzymatic activity
1A1 and 3A4 cytochrome p450 enzymatic activity was measured with a commercial kit (Promega, Madison, WI, USA) at the end point of the experiment as described previously.15 CYP activities were calculated as pmol D-Luciferin produced per hour by 1 × 106 hepatocytes and subsequently normalized to control.
Cardiac Electrical Measurements
Electrical activity of the cardiomyocytes was measured using a commercially available MEA amplifier (MEA 1040, Multichannel Systems, Reutlingen, BW, Germany) adapted to the housing system. The cMEA contact pads were connected to the MEA amplifier through a Zebra® elastomeric electrical connector and a custom printed circuit board. The temperature inside the housing was controlled to 37 °C by a PID controller (Multichannel Systems, Reutlingen, BW, Germany). Electrical signals were recorded using a commercially available data acquisition software package (MC_Rack, Multichannel Systems, Reutlingen, BW, Germany), with beat frequency quantified from spontaneous recordings (20 s recording period). Cardiomyocytes were electrically stimulated using an STG 1002 stimulator (Multichannel Systems, Reutlingen, BW, Germany) with a 1 ms bipolar pulse (600–1000 mV) applied to a single electrode at increasing frequencies from 0.5 Hz to 4 Hz to control the origin of the conduction path to measure conduction velocity. The minimum interspike interval (mISI, a QT-interval analogue) was measured using the data obtained from the frequency sweep, as described in Stancescu et al. 19. Data analysis was performed in Clampfit 10.3 (Molecular Devices, San Jose, CA, USA), and to measure the electrical parameters of conduction velocity, QT-interval and spontaneous beat frequency. Changes over time are presented as values relative to measurements taken on day 4 (immediately before drug addition) and are compared to control conditions (no drug treatment).
Cardiac Force Measurements
Cardiac force data was gathered from cantilevers using a previously described optical detection system which measures the deflection of the cantilever in direct response to the contraction exerted by cardiomyocytes.16–17 This deflection in cantilever position can be converted to force output using a modified version of Stoney’s equation as thoroughly described in Pirozzi et al.20 Data analysis was performed using custom peak detection software written in Python to determine beat frequency and average peak amplitude.
Viability assay
Cell viability was measured at experiment end using a standard MTT assay (Thermo Fisher Scientific, V13154, Waltham, MA, USA). Cardiac cell viability was evaluated using 10% alamarBlue® solution in culture medium (Thermo Fisher Scientific, DAL1100, Waltham, MA, USA) on day 7 of the experiment with a 24 h incubation at 37 °C. Conversion of the alamarBlue® was measured with a Synergy HT plate reader and KC4 software (Excitation 530 nm; Emission 590 nm, Winooski, VT, USA). The viability results are presented relative to the control values.
Cell Culture
Cryopreserved human iPSc derived cardiomyocytes (Cellular Dynamic International, CDI, Madison, WI, USA) were plated onto fibronectin coated photolithographically patterned microelectrode arrays and silicon-based cantilevers to measure parameters of cardiac function (conduction velocity, beat frequency, minimum inter-spike interval, and contractile force) as described in detail previously. 19 Cryopreserved human primary hepatocytes (Massachusetts General Hospital (MGH, Boston, MA, USA, lot #Hw54) were plated (1700 cells/mm2) on collagen coated glass coverslips (60 μg/mL acidified collagen type I for 30 min at 37 °C) according to providers recommended protocol. Full medium changes were performed every other day for 7 days before assembly into multi-organ housings. These surfaces were incorporated into self-contained, multi-organ, microfluidic device systems as described in Oleaga et al.17 Briefly, PDMS gaskets were designed and fabricated to designate and hold in location MEAs and cantilevers, define channel flow and seal the poly methyl methacrylate outer housing. Systems were placed on a rocker platform inside a 5% CO2, 37°C, 100% relative humidity incubator to facilitate gravity driven medium flow. Daily medium changes of 30% of the volume (total volume 2 mL) were performed and collected for further analysis. The defined serum-free medium base composition used to maintain the heart-liver culture systems was previously described. 15
Albumin and LDH Measurements
Albumin production (BioAssay Systems (Bethyl Laboratory Inc., Montgomery, TX, USA,) and LDH release (Promega, Madison, WI, USA) were quantified from the supernatant removed from systems using commercial kits as described by the manufacturer. Assays were read with a Synergy HT plate reader and KC4 (Winooski, VT, USA). Albumin production was calculated as albumin produced daily normalized to 1 × 106 hepatocytes and LDH was calculated as Relative Fluorescence Units (RFU) and then each was further normalized to the production on the initial day and control.
Drug solutions and application
Drug permeation studies were performed with each compound applied topically in the form of a solution. Diclofenac sodium, ketoconazole, hydrocortisone and acetaminophen were resuspended in a 10% DMSO (v/v) in water solution to achieve the desired concentrations of 1.5% and 3% (w/w) for diclofenac (47200 μM and 94400 μM, respectively), 0.11% w/w for ketoconazole (2070 μM), 1% (w/w) for hydrocortisone (27.6 mM) and 1.5% (w/w) for acetaminophen (100 mM). A 50 μl dose of each of the prepared drug solutions was applied on the synthetic membrane through the donor chamber of the skin module in the HL+skin system with an area for drug absorption of 0.283 cm2. The total amounts of diclofenac sodium added topically from either the 1.5% or 3% solutions were 750 μg or 1500 μg, respectively. The total amounts of ketoconazole, hydrocortisone, and acetaminophen added topically were 55 μg, 500 μg, and 750 μg, respectively. The compound solutions were applied on the synthetic membrane for 24 hours and then removed. For acute systemic delivery, diclofenac, ketoconazole and hydrocortisone were resuspended with DMSO to prepare stock solutions at 200 times the systemic concentration used and were delivered as mentioned above. Acetaminophen was resuspended in cell culture medium at the appropriate concentration to yield 20 mM in the HL system.
HPLC MS/MS Quantitation of Drug Compounds
Quantitation was performed using an Agilent 6490 Triple Quadrupole LCMS system with an electrospray source interfaced to an Agilent 1200 HPLC with a binary pump. Data Analysis was performed using Agilent MassHunter software. The HPLC column used was an Agilent Zorbax Eclipse Plus C-18, 4.6 mm ID (internal diameter) X 100 mm (length), 3.6 μm particles. Separation was achieved using reverse phase gradient elution. For each separation, the aqueous mobile phase was 0.1% formic acid. Methanol was used as the organic modifier.
Samples were received for analysis frozen, in polypropylene centrifuge tubes. After thawing, the samples were vortex mixed and then 50 μl of sample and 50 μl of internal standard solution were combined and diluted to 400 μl with methanol in a 1.6 ml polypropylene centrifuge tube. The diluted sample was vortex mixed and then centrifuged at 1300 RPM for five minutes to precipitate proteins. The supernatant was further diluted 50 μl to 1050 μl and placed in an autosampler vial for analysis. Any further dilutions needed were made using either water or aqueous mobile phase as the solvent (depending on the literature reported solubility of the compound of interest).
Each compound was quantitated using Multiple Reaction Monitoring (MRM). Nitrogen was used as the fragmentation gas. Diclofenac was determined using negative ion mode, and positive ion mode was used for all other compounds. Daily calibrations were performed, and at least five calibration levels over approximately three decades of concentration were measured for each drug. Approximate detection limits, the identity of the internal standard used, and the instrument parameters, are reported in Table 1.
TABLE 1.
Detection Limits, Identity of Internal Standard and Instrumentation Parameters
| Compound | Internal Standard | transition (Daltons) | Collision Energy | Detection Limit (nM) |
|---|---|---|---|---|
| acetaminophen | antipyrine | 152.1→110.1 | 16 | 50 |
| diclofenac | antipyrine | 296.1→214.1 | 45 | 200 |
| ketoconazole | acetaminophen | 531.2→82.0 | 35 | 300 |
| hydrocortisone | acetaminophen | 363.2→120.0 | 36 | 90 |
Statistical analysis
All results are presented as mean, and error bars are standard error of the mean (SEM). Data were analysed with One-way Anova using SigmaPlot Systat Software (SigmaPlot Systat Software Inc. 12.5). Differences between groups were considered statistically significant when p < 0.05.
Results and discussion
Acute drug exposure toxicity evaluation
In order to determine the cardiotoxicity and hepatotoxicity of compounds and to compare the efficacy of the barrier function provided by the Strat-M membrane, acute exposures of the compounds were topically and systemically administrated in the heart-liver body-on-a-chip system.
Diclofenac.
Although diclofenac sodium permeation through the skin is relatively low, its topical application allows it to reach an effective concentration in the pain-affected area and surrounding tissues while maintaining a low systemic concentration.21 Topical application of diclofenac sodium is denoted as safe and is widely used as an alternative to reduce the side effects associated with high systemic concentrations from oral doses. The recommended dose for diclofenac from 1.5% solutions for knee pain due to osteoarthritis is approximately 5 ml per day applied on each knee.22 In this work, diclofenac sodium solution was applied topically to the heart-liver platform at a concentration of 1.5% and 3% (w/w). No significant changes were observed in cardiac or hepatic function (Figure 2) following the application of a 1.5% diclofenac solution. In the same manner, no significant changes were observed for cardiac or liver viability at the end of the culture period (Figure 2H), in accordance with LDH levels that indicated no significant changes compared to the control (Figure 2F). Whereas the 1.5% diclofenac sodium dosage showed no significant toxicity for the cardiomyocytes, acute topical administration of a 3% diclofenac sodium solution decreased conduction velocity (96% ± 4%), contractile force (44 ± 12%) and spontaneous frequency (90 ± 10%) after 24 hours of topical delivery (Figure 2A, B and C) with no recovery after 72 hours. For hepatic function, no significant changes were observed in CYP activity or albumin secretion (Figure 2E and G) after acute topical administration of a 3% diclofenac solution.
Figure 2: Cardiac and liver function and viability upon acute topical and systemic diclofenac administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 24 and 72 hours after the acute topical and systemic application of diclofenac sodium solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (72 hours after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (72 hours after drug addition). Diclofenac effects were plotted normalized to function before drug addition and to the topical vehicle control. * = p < 0.05; ** = p < 0.001
The concentrations used for topical application in the clinic translates into approximately 520 to 1040 μg/(day*cm2) (considering a knee area of 150 cm2).23 In the heart-liver system the applied dose of 1.5% solution is approximately 26,500 μg/(day*cm2), thus the acute topical exposure is 25-50 times the recommended dose per area which represents exposure of a very large body surface area to diclofenac. In addition, the system can be modified to either reduce or increase the skin area of exposure thus regulating the rate of drug permeation.
To compare the differences between topical and systemic application of diclofenac, the heart-liver system was also dosed with acute systemic diclofenac at different concentrations. Acute treatment affected the electrical activity and contractile force (Figure 2A, B, C and D). The contractile force decreased 39 ± 18% with recovery after exposure to a 300 μM concentration (Figure 2A). The electrical activity in the system was affected in a dose dependent manner to increasing acute concentrations. When the system was exposed to a 150 μM acute dose, it produced a significant reduction in the spontaneous beat frequency and conduction velocity (57 ± 10% and 36 ± 10%, respectively) during the first 24 hours with a recovery after 72 hours (Figure 2B and C). This difference between the systemic and topical results may be due to differences in drug exposure profile. A comparison of diclofenac concentrations in the heart-liver system indicated that diclofenac exposure to either 300 μM systemic or 3% topical yield similar levels of concentrations after 24 hours, however, diclofenac concentration in the subsequent days was higher in the 3% topical application (Figure 3). A significant decrease over time in albumin production was observed in the systems exposed to 150 μM and 300 μM diclofenac (Figure 2E). For liver CYP activity, the induction of 1A1, 3A4 and 2C9 was observed at 300 μM, (Figure 2G). Cell viability was not affected in the system exposed to any of the concentrations tested of diclofenac and correlates with no significant changes in LDH levels compared to the vehicle control (Figure 2F and H).
Figure 3: Diclofenac concentration over time from acute topical and systemic delivery in the heart-liver system.

Diclofenac concentration comparison from either topical application (1.5% or 3%) or systemic delivery (150 and 300 μM) over the period of 3 days.
Results obtained with a 1.5% diclofenac sodium solution demonstrate the safety of these drugs at the concentration applied. In contrast, a 3% diclofenac sodium solution produced significant changes in organ function, especially in cardiac tissue. The use of diclofenac has been associated with several adverse effects, especially hepatic and cardiovascular effects.24,25 In vitro studies have shown that diclofenac affects electrical parameters of cardiac tissue and may reduce cardiac excitability and contractility.25 Diclofenac sodium can inhibit voltage dependent Na+ channels and L-type Ca2+ channels in rat myoblasts.25,26 In our results, the acute systemic delivery of diclofenac increased CYP3A4 and CYP2C9 activities in hepatocytes and reduced albumin secretion. This result agrees with studies showing that the 5-hydroxylation of diclofenac is mainly catalysed by CYP3A4 and to a much less extent by CYP2C9.27 Interestingly, diclofenac also induced CYP1A1 activity at the highest concentration tested. Although there are no reports of diclofenac and increased CYP1A1 activity in primary hepatocytes, diclofenac is known to interact with aryl hydrocarbon receptor and induce CYP1A1 activity in aquatic fauna.28
Ketoconazole.
Ketoconazole solution was applied topically to the heart-liver platform at a concentration of 0.11% (w/w) on the synthetic membrane through the donor chamber for 24 h. No significant changes were observed compared to the topical vehicle control in cardiac or hepatic function following the application of the 0.11% ketoconazole solution (Figure 4). In addition, no significant changes were observed for cardiac or liver viability (Figure 4H) or in LDH levels compared to the control at the end of the culture period (Figure 4F).
Figure 4: Cardiac and liver function and viability upon acute topical and systemic ketoconazole administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 24 and 72 hours after the acute topical and systemic application of ketoconazole solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (72 hours after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (72 hours after drug addition). Ketoconazole effects were plotted normalized to function before drug addition and to the topical vehicle control. p > 0.05 in all data points.
The acute systemic dosing of ketoconazole in the heart-liver system displayed a significant reduction in the spontaneous beat frequency (20 ± 4% and 52 ± 21%, respectively) during the first 24 hours with a recovery after 72 hours at 10 μM and 60 μM (Figure 4C). The MISI interval increased only for the acute systemic exposure to 60 μM after 72 hours (Figure 4D). Azole antifungals such as ketoconazole, are associated with long QT syndrome and ventricular arrhythmias.29 In addition, in vitro studies have found that ketoconazole prolongs the QT interval by blocking rapidly rectifier potassium channels (IKr).30,31 No significant changes were observed in contractile force or conduction velocity (Figure 4A and B). A significant decrease over time in albumin production was observed in the systems exposed to 10 μM and 60 μM ketoconazole (Figure 4E). These results are in accordance with literature findings that ketoconazole and its metabolite, N-desacetyl-ketoconazole, are hepatotoxic due to covalent binding to hepatic proteins.32 In addition, Azole antifungal agents have been reported to cause both hepatocellular and cholestatic injury.33 For liver CYP activity, inhibition of 3A4 was observed at 10 μM and 60 μM (Figure 4G), which was expected as ketoconazole is a well known inhibitor of CYP3A isoforms and is often used in drug metabolism research and drug development processes.34 Furthermore, studies have shown that ketoconazole also has an affinity for CYP2C9, although to a lesser extent.35 Cell viability was not affected in the heart-liver system exposed to any of the concentrations tested which correlated with no significant changes in LDH levels compared to the vehicle control (Figure 4F and H). Ketoconazole concentrations after 24 hours in the system exposed to either 10 or 60 μM were 2.8 ± 0.1 and 18.3± 0.3 μM (Figure 5), respectively. In contrast, topical ketoconazole concentration after 24 hours was relatively low (0.19 ± 0.01 μM) (Figure 5).
Figure 5: Ketoconazole concentration over time from acute topical and systemic delivery in the heart-liver system.

Ketoconazole concentration comparison from either topical application (0.11%) or systemic delivery (10 and 60 μM) over the period of 3 days.
Drug delivery of compounds through the skin requires crossing the barrier generated by the multiple layers of skin. This barrier property makes the permeation of drugs through the skin difficult. Permeation of drug molecules and xenobiotics through the skin greatly decreases with increasing molecular weight (especially after 500 daltons) and high logP values.36 Ketoconazole has a molecular weight of 531 and a log P of 4.31 and although its topical application is widely used to treat localized superficial skin fungal infections, its penetration into the skin is very low.37 Actually, topical ketoconazole concentration in heart systems only reached a 0.19 μM concentration after 24 hours. In our experiments, without the skin barrier, the dose applied from the 0.11% ketoconazole solution would be equivalent to a 51 μM systemic exposure, thus relatively close to the 60 μM dose that affected cardiac activity. These results demonstrate the efficacy of the barrier properties imposed by the use of the Strat-M membrane in the heart-liver system.
Hydrocortisone.
A concentration of 1% (w/w) of topical hydrocortisone solution was applied to the heart-liver system. No significant changes were observed compared to the topical vehicle control in cardiac or hepatic function following the application of the 1% hydrocortisone solution (Figure 6). No significant changes were observed for cardiac or liver viability at the end of the culture period (Figure 6H) and this result agrees with no significant changes in LDH levels compared to the control (Figure 6F).
Figure 6: Cardiac and liver function and viability upon acute topical and systemic hydrocortisone administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 24 and 72 hours after the acute topical and systemic application of hydrocortisone solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (72 hours after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (72 hours after drug addition). Hydrocortisone effects were plotted normalized to function before drug addition and to the topical vehicle control. * = p < 0.05; ** = p < 0.001
However, the acute systemic delivery of hydrocortisone at high concentrations produced significant changes in both the cardiac and hepatic tissue. In the cardiac module, hydrocortisone decreased conduction velocity, beat frequency and contractile force. The heart-liver system exposed to 750 μM and 1500 μM produced a significant reduction in the conduction velocity (26 ± 7% and 91 ± 9%, respectively) (Figure 6B) and spontaneous beat frequency (86 ± 5% and 95 ± 3%, respectively) during the first 24 hours with a partial or full recovery after 72 hours (Figure 6C). The contractile force decreased for both concentrations tested in a dose dependent manner after 24 hours with no recovery (Figure 6A). No significant changes were observed in the MISI interval (Figure 6D). Although glucocorticoids are relatively safe when administered at therapeutic concentrations, the use of high doses of corticosteroids has been associated with adverse cardiovascular effects and atrial fibrillation.38,39 In addition, chronic use of corticosteroids is also associated with hypertension and heart failure, cardiac hypertrophy, and ventricular dysfunction.40,41 Interestingly, our results showed that the conduction velocity and beat frequency values recover after 72 hours while the contractile force did not. These findings suggest a specific hydrocortisone mechanism of action. Studies have shown that dexamethasone (another glucocorticoid) could have an important role in the Ca2+ handling in cardiomyocytes and may alter the expression of ion channel genes related to Ca2+ kinetics and contractile function.42 It has also been proposed that high doses of corticosteroids interfere with potassium efflux.43
In the liver module, a significant decrease over time in albumin production was observed in the systems exposed to 750 μM and 1500 μM hydrocortisone (Figure 6E). The decrease in albumin secretion indicated that high concentrations of hydrocortisone affects liver function. In vivo studies have shown that the use of chronic glucocorticoids is associated with several liver adverse effects including steatosis and steatohepatitis.44,45 Corticosteroids cause steatosis by inhibiting mitochondrial β-oxidation, decrease hepatic triglyceride secretion and produce lipid peroxidation.46,47 For liver CYP activity, the induction of 3A4 was observed at 750 μM and 1500 μM (Figure 6G) and induction of 2C9 was observed at 1500 μM. Induction of CYP3A4 was expected since glucocorticoids are well known 3A4 inducers.48 Cell viability was not affected in heart-liver systems exposed to any of the concentrations tested of hydrocortisone and correlates with no significant changes in LDH levels compared to the vehicle control (Figure 6F). Hydrocortisone concentration after 24 hours in a heart-Liver system exposed to either 750 or 1500 μM was 262 ± 4 and 506 ± 4 μM (Figure 7), respectively. In contrast, the hydrocortisone concentration after 24 hours in the system dosed topically was relatively low (0.27 ± 0.01 μM) (Figure 7).
Figure 7: Hydrocortisone concentration over time for acute topical and systemic delivery in the heart-liver system.

Hydrocortisone concentration comparison from either topical application (1%) or systemic delivery (750 and 1500 μM) over the period of 3 days.
Acetaminophen.
Acetaminophen is a phenol derivative nonsteroidal anti-inflammatory (NSAID) drug widely used for pain management. Although the main route of application for acetaminophen is oral, a few topical and transdermal applications have been developed.49 In general acetaminophen and other phenol derivative drugs have poor skin absorption.50
The topical acetaminophen solution was applied to the heart-liver platform at a concentration of 1.5% (w/w). No significant changes were observed compared to the topical vehicle control in cardiac or hepatic function following the application of a 1.5% acetaminophen solution (Figure 8). A small but significant decrease in hepatocyte viability (18 ± 4%) was observed at the end of the culture period (Figure 8H) however, no significant changes in LDH levels were observed compared to the control (Figure 8F).
Figure 8: Cardiac and liver function and viability upon acute topical and systemic acetaminophen administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 24 and 72 hours after the acute topical and systemic application of acetaminophen solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (72 hours after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (72 hours after drug addition). Acetaminophen effects were plotted normalized to function before drug addition and to the topical vehicle control. * = p < 0.05
The acute systemic delivery of acetaminophen at high concentrations affected the cardiac and hepatic tissue. Acetaminophen at 20 mM was systemically applied to the heart-liver system and drastically affected the electrical activity and contractile force. The spontaneous beat frequency was reduced (90 ± 10%) during the first 24 hours and ceased after 72 hours (Figure 8C). The conduction velocity and MISI were not detected either at 24 hour or 72 hours following drug treatment (Figure 8B and D). The contractile force decreased at 24 hours (78 ± 13%) with no recovery (Figure 8A). Cell viability was affected, especially for cardiomyocytes (Figure 8H). Acetaminophen concentration after 24 hours in the systems exposed to 20 mM was 12.7 ± 0.6 mM (Figure 9). In contrast, the acetaminophen concentration found after 24 hours in dosed topically was relatively low (1.59 μM) (Figure 9). These results suggest that although acetaminophen is not known to be a cardiotoxic drug, it drastically reduces cardiac electrical activity and contractile force at high concentrations. There are several reports that indicate that acetaminophen might cause toxic myocarditis and acute myocardial necrosis when ingested in large doses.51 Hypothesis for the direct toxic effect on the heart suggests that myocardial injury occurs due to a similar mechanism which causes hepatic damage, with acetaminophen partly converted to a toxic metabolite, N-acetyl-p-benzoquinonimine, which is normally inactivated by reduction with glutathione.51–53 High doses of acetaminophen might cause glutathione depletion and therefore N-acetyl-p-benzoquinonimine induced cardiac damage.51 Other studies have found that acetaminophen is not only hepatotoxic but also cardiotoxic in neonatal rat heart cells with an IC50 ratio (heart/hepatocyte) of approximately 1.54 Furthermore, the data suggest that acetaminophen decreased albumin production and inhibited CYP3A4 function (Figure 8E and G). Hepatotoxicity is well established for acetaminophen and is related to the conversion of a small proportion of the ingested dose to N-acetyl-p-benzoquinoneimine,55 in addition, acetaminophen is known to inhibit CYP3A4.56
Figure 9: Acetaminophen concentration over time for acute topical and systemic delivery in the heart-liver system.

Acetaminophen concentration comparison from either topical application (1.5%) or systemic delivery (20 mM) over the period of 3 days.
Chronic drug exposure toxicity evaluation
One of the challenges faced by in vitro models is the capability of maintaining viable cells for long periods of time. Thus, in order to determine if our system could be used to study chronic topical drug exposure, we applied diclofenac and acetaminophen daily for 10 days and measured cardiac and hepatic function.
Diclofenac chronic exposure.
The results obtained with the chronic topical applications of diclofenac at 1.5% showed that cardiac and hepatic functions were affected over time. In the system, chronic treatment with 1.5% diclofenac was enough to affect the electrical activity of cardiomyocytes, with increased MISI interval and decreased conduction velocity and spontaneous beat frequency (Figure 10B, C and D). In the liver module, chronic topical application of 1.5% diclofenac increased CYP 3A4 and 2C9 and decreased albumin secretion (Figure 10E and G). No significant changes in cell viability were observed. A small increase in LDH was detected at days 5 and 6 (Figure 10F).
Figure 10: Cardiac and liver function and viability upon chronic topical diclofenac administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 10 days after topical application of diclofenac solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (10 days after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (10 days after drug addition). Diclofenac effects were plotted normalized to function before drug addition and to the topical vehicle control. * = p < 0.05; ** = p < 0.001
The acute topical drug exposure results indicated that a 1.5% solution of diclofenac had no cardiotoxic and hepatotoxic effects, in contrast with our chronic results where it was enough to alter cardiac and hepatic functions with repeat doses. This observation can be explained by the differences in drug concentration over time in our system. Since the average concentration in systems exposed to 1.5% diclofenac topical solution after 24 hours was 59 μM, thus subsequent dosing would increase the concentration to similar levels of those observed from an acute exposure to 300 μM (data not shown).
Acetaminophen chronic exposure.
Chronic exposure of a topical 1.5% acetaminophen solution in the heart-liver system showed no significant changes in cardiac and hepatic function. It was observed that at day 7 the conduction velocity of cardiomyocytes was decreased but no other significant changes were observed (Figure 11). In the liver module there was a small decrease in albumin secretion detected at day 4 followed by a recovery (Figure 11E). No significant changes were observed in CYP activity (Figure 11G). No significant changes were observed in hepatocyte or cardiomyocyte viability (Figure 11H).
Figure 11: Cardiac and liver function and viability upon chronic topical acetaminophen administration on the heart-liver system.

The cardiac function was segregated into contractile force (A) and electrical activity (B, C, D) and was evaluated non-invasively for 10 days after topical application of acetaminophen solution. For liver function albumin (E) and LDH (F) were evaluated over time and cytochrome p450 activity (G) was evaluated at the endpoint (10 days after drug addition). Hepatocyte and cardiomyocyte viabilities (H) were evaluated at the end point (10 days after drug addition). Acetaminophen effects were plotted normalized to function before drug addition and to the topical vehicle control. p > 0.05 in all data points.
Conclusions
In this work, we have successfully demonstrated that our heart-liver body-on-a-chip system efficiently predicted drug toxicity not only from acute drug exposure but also for chronic drug treatment associated with transdermal drug delivery. Topical and systemic administration of diclofenac, ketoconazole, hydrocortisone and acetaminophen have shown that our systems have reliable toxicity prediction with the advantage of the real time assessment of cardiac organ function. This new in vitro technological finding ultimately could assist in the design of clinical trial protocols as well as reduce the cost, time and labour of drug development. In conclusion, this work indicates a promising in vitro platform that can be simultaneously used for the assessment of potential drug toxicity from dermal absorption and evaluate transport dynamics through the skin.
Supplementary Material
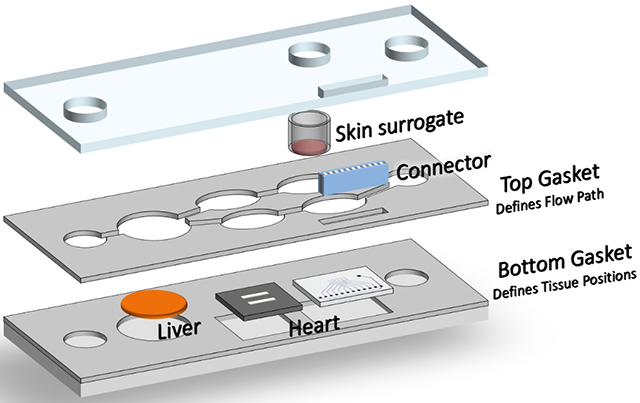
Figure 1: Schematic of the Heart-Liver body-on-a-chip system with skin module containing.

1) liver module on a coverslip, 2) cardiomyocytes on bioMEMS cantilevers, 3) cariomyocytes on a microelectrode array, and 4) skin module. An electrical connector was used to interface the microelectrode array to an external amplifier and stimulation system.
Acknowledgements
Funding for this research was provided by the National Institutes of Health, SBIR Phase II grant number R44TR001326 and the L’Oreal Corporation.
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x
Conflicts of interest
The authors confirm that competing financial interests exist but there has been no financial support for this research that could have influenced its outcome. JJH has a potential competing financial interest, in that a company has been formed to market services for types of cells like this in body-on-a-chip devices.
References
- 1.Mullard A, Nature Reviews Drug Discovery, 2016, 15, 447. [DOI] [PubMed] [Google Scholar]
- 2.Esh MB, Smith AS, Prot JM, Oleaga C, Hickman JJ, Shuler ML. Adv. Drug Deliv Rev 2014, 69-70, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang YI, Oleaga C, Long CJ, Esch MB, McAleer CW, Miller PG, Hickman JJ, Shuler ML. Exp. Biol. Med, 2017, 17, 1701–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daly AK, Genome Medicine, 2013, 5,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darwhekar G, Jain DK, Paditar VK. Asi. J. Pharmacy Life Sci, 2011, 1, 269–278. [Google Scholar]
- 6.Gan TJ. Current medical research and opinion, 2010, 26, 1715–1731. [DOI] [PubMed] [Google Scholar]
- 7.Khoza S, Moyo I, Ncube D. Journal of toxicology, 2017, 6746989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Symoens J, Cauwenbergh G. Jucker E (Ed.), Progress in Drug Research / Fortschritte der Arzneimittelforschung / Progrès des recherches pharmaceutiques, Birkhäuser Basel, Basel, 1983, 63–84. [DOI] [PubMed] [Google Scholar]
- 9.Mallappa A, Debono M. Endocr Dev 2016, 30, 42–53. [DOI] [PubMed] [Google Scholar]
- 10.Larson AM. Clin Liver Dis, 2007, 11, 525–548. [DOI] [PubMed] [Google Scholar]
- 11.Thomson JS, Prescott LF. Br Med J,1996, 2, 506–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. J Clin Transl Hepatol, 2016, 4, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Broek LJ, Bergers LIJC, Reijnders CMA, Gibbs S. Stem Cell Rev Rep., 2017, 13(3), 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Sito L, Mao M, He J, Zhang YS, Zhao X. Microphysiol Syst., 2018, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oleaga C, Bernabini C, Smith AS, Srinivasan B, Jackson M, McLamb W, Platt V, Bridges R, Cai Y, Santhanam N, Berry B, Najjar S, Akanda N, Guo X, Martin C, Ekman G, Esch MB, Langer J, Ouedraogo G, Cotovio J, Breton L, Shuler ML, and Hickman JJ. Sci Rep, 2016, 6: 20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McAleer CW, Pointon A, Long CJ, Brighton RL, Wilkin BD, Bridges LR, Narasimhan Sriram N, Fabre K, McDougall R, Muse VP, Mettetal JT, Srivastava A, Williams D, Schnepper MT, Roles JL, Shuler ML, Hickman JJ, Ewart L. Sci Rep., 2019, 9(1), 9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oleaga C, Legters G, Bridges LR, Kumanchik L, Martin C, Cai Y, Schnepper M, McAleer CW, Long CJ, Hickman JJ. Stem Cell-Derived Models in Toxicology (Springer New York: New York, NY: ), 2017. [Google Scholar]
- 18.Oleaga C, Riu A, Rothemund S, Lavado A, McAleer CW, Long CJ, Persaud K, Narasimhan NS, Tran M, Roles J, Carmona-Moran CA, Sasserath T, Elbrecht DH, Kumanchik L, Bridges LR, Martin C, Schnepper MT, Ekman G, Jackson M, Wang YI, Note R, Langer J, Teissier S, Hickman JJ. Biomaterials, 2018, 182: 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stancescu M, Molnar P, McAleer CW, McLamb W, Long CJ, Oleaga C, Prot JM, and Hickman JJ. Biomaterials, 2015, 60: 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pirozzi KL, Long CJ, McAleer CW, Smith AS, and Hickman JJ. Appl Phys Lett, 2013, 103: 83108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radermacher J, Jentsch D, Scholl MA, Lustinetz T, Frolich JC. British journal of clinical pharmacology, 1991, 31(5), 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth SH, Shainhouse JZ. Archives of internal medicine, 2004, 164(18), 2017–2023. [DOI] [PubMed] [Google Scholar]
- 23.Hohe J, Ateshian G, Reiser M, Englmeier KH, Eckstein F. Magnetic resonance in medicine, 2002, 47(3), 554–561. [DOI] [PubMed] [Google Scholar]
- 24.Bort R, Ponsoda X, Jover R, Gomez-Lechon MJ, Castell JV. The Journal of pharmacology and experimental therapeutics, 1999, 288(1), 65–72. [PubMed] [Google Scholar]
- 25.Yarishkin OV, Hwang EM, Kim D, Yoo JC, Kang SS, Kim DR, Shin J-H-J, Chung H-J, Jeong H-S, Kang D, Han J, Park J-Y, Hong S-G. The Korean Journal of Physiology & Pharmacology : Official Journal of the Korean Physiological Society and the Korean Society of Pharmacology, 2009, 13(6), 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fei XW, Liu LY, Xu JG, Zhang ZH, Mei YA. Biochemical and biophysical research communications, 2006, 346(4), 1275–1283. [DOI] [PubMed] [Google Scholar]
- 27.Shen S, Marchick MR, Davis MR, Doss GA, Pohl LR. Chem Res Toxicol, 1999, 12(2), 214–22. [DOI] [PubMed] [Google Scholar]
- 28.Prokkola JM, Nikinmaa M, Lubiana P, Kanerva M, McCairns RJS, Götting M. Aquatic Toxicology, 2015, 158, 116–124. [DOI] [PubMed] [Google Scholar]
- 29.Tonini M, De Ponti F, Di Nucci A, Crema F. Alimentary pharmacology & therapeutics, 1999, 13(12), 1585–1591. [DOI] [PubMed] [Google Scholar]
- 30.Yap YG, Camm AJ. Heart, 2003, 89(11), 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dumaine R, Roy M-L, Brown AM. Journal of Pharmacology and Experimental Therapeutics, 1998, 286(2), 727. [PubMed] [Google Scholar]
- 32.Rodriguez RJ, Buckholz CJ. Xenobiotica; the fate of foreign compounds in biological systems, 2003, 33(4), 429–441. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez RJ, Acosta D. Toxicology 1997, 117(2), 123–131. [DOI] [PubMed] [Google Scholar]
- 34.Greenblatt DJ. Clinical Pharmacology in Drug Development, 2014, 3(1), 1–3. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, E. M Drug metabolism and disposition: the biological fate of chemicals, 2002, 30(3), 314–8. [DOI] [PubMed] [Google Scholar]
- 36.Bos JD, Meinardi MM. Experimental dermatology, 2000, 9(3), 165–9. [DOI] [PubMed] [Google Scholar]
- 37.Jacobs GA, Gerber M, Malan MM, du Preez JL, Fox LT, du Plessis J. Drug delivery, 2016, 23(2), 631–41. [DOI] [PubMed] [Google Scholar]
- 38.Chiappini B, El Khoury G. Expert Opin Drug Saf, 2006, 5, 811–814. [DOI] [PubMed] [Google Scholar]
- 39.Pimenta E, Wolley M, Stowasser M. Endocrinology, 2012, 153, 5137–2142. [DOI] [PubMed] [Google Scholar]
- 40.Grunfeld JP, Eloy L. Hypertension, 1987, 10, 608–618. [DOI] [PubMed] [Google Scholar]
- 41.Oakley RH, Cidlowski JA. J Steroid Biochem Mol Biol, 2015, 153, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De P, Roy SG, Kar D, Bandyopadhyay A. J Endocrinol, 2011, 209, 105–114. [DOI] [PubMed] [Google Scholar]
- 43.Fujimoto S, Kondoh H, Yamamoto Y, Hisanaga S, Tanaka K. Am J Nephrol, 1990, 10, 231–236. [DOI] [PubMed] [Google Scholar]
- 44.Patel V, Sanyal AJ. Clin Liver Dis, 2013, 17, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Büttner R, Schölmerich J, Hellerbrand C. Cell Research, 2009, 19, 996. [DOI] [PubMed] [Google Scholar]
- 46.Kneeman JM, Misdraji J, Corey KE. Therapeutic Advances in Gastroenterology, 2012, 5, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons PS, Miles JM, Gerich JE, Haymond MW. J Clin Invest, 1984, 73, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hewitt NJ, Lechón MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG. Drug Metab Rev, 2007, 39, 159–234. [DOI] [PubMed] [Google Scholar]
- 49.Sintov Amnon C, Krymberk I, Gavrilov V, Gorodischer R. Journal of Pharmacy and Pharmacology, 2010, 55(7), 911–919. [DOI] [PubMed] [Google Scholar]
- 50.Thomas DJ, Majumdar S, Sloan BK. Molecules,2009, 14(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armour A, Slater SD. Postgrad Med J, 1993, 69, 52–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin SM, Park K. Environ Health Toxicol, 2012, 27, e2012011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ralapanawa U, Jayawickreme KP, Ekanayake EMM, Dissanayake AMSDM. BMC Pharmacol Toxicol, 2016, 17, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue T, Tanaka K, Mishima M, Watanabe K. AATEX, 2008, 14, 457–462. [Google Scholar]
- 55.Routledge P, Vale JA, Bateman DN, Johnston GD, Jones A, Judd A, Thomas S, Volans G, Prescott LF, Proudfoot A. BMJ, 1998, 317, 1609–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feierman DE, Melnikov Z, Zhang J. Arch Biochem Biophys, 2002, 398, 109–117. [DOI] [PubMed] [Google Scholar]
- 57.Alexander A, Dwivedi S, Ajazuddin, Giri TK, Saraf S, Saraf S, Tripathi DK. J Control Release, 2012, 164, 26–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.