

Abstract
Colorectal cancer (CRC) is a major cause of mortality worldwide. Chemotherapy and radiation remain standard treatment for locally advanced disease, with current immune-targeting therapies applying to only a small subset of patients. Expression of the immuno-oncology target indoleamine 2,3 dioxygenase 1 (IDO1) is associated with poor CRC clinical outcomes but is understudied as a potential treatment target. In this study, we examined the interaction between the IDO1 pathway and radiation therapy in CRC. We used human and mouse CRC cell lines, organoids, mouse syngeneic CRC tumor graft models, and CRC tissues from patients who received radiation therapy. IDO1 activity was blocked using the clinical IDO1 inhibitor epacadostat and by genetic disruption. We found that radiation-induced IDO1 overexpression in CRC through Type I and II interferon signaling. IDO1 enzymatic activity directly influenced CRC radiation sensitivity. IDO1 inhibition sensitized CRC to radiation-induced cell death, whereas the IDO1 metabolite kynurenine promoted radioprotection. IDO1 inhibition also potentiated Th1 cytokines and myeloid cell–modulating factors in the tumor microenvironment and promoted an abscopal effect on tumors outside the radiation field. Conversely, IDO1 blockade protected the normal small intestinal epithelium from radiation toxicity and accelerated recovery from radiation-induced weight loss, indicating a role in limiting side-effects. These data demonstrated that IDO1 inhibition potentiates radiation therapy effectiveness in colorectal cancer. The findings also provide rationale and mechanistic insight for the study of IDO1 inhibitors as adjuvant therapy to radiation in patients with locally advanced sporadic and colitis-associated colorectal cancer.
Keywords: Immunotherapy, metabolism, TDO, IDO2, Tryptophan
INTRODUCTION
Colorectal cancer (CRC) remains a major cause of morbidity and mortality in the US and globally. With conventional chemotherapy and radiation, 5-year survival rates range from 49–80% for locally advanced CRC and ~10% for metastatic CRC (www.Cancer.org). These numbers are likely to improve as immunotherapy drugs targeting the programmed cell death pathway 1 (PD-1) are effective in patients with metastatic CRC bearing genetic features that prevent cells from repairing damaged DNA (mismatch repair deficiency [dMMR] or high microsatellite instability [MSI-H])(1,2). However, limitations to PD-1 therapies exist, as dMMR/MSI-H tumors represent a minority (10–20%) of all CRC (3). Gastrointestinal (GI) toxicities are common with immune checkpoint therapies and may further complicate treatment for those with a GI tract that is already compromised (4). Therefore, there remains an unmet need for new therapeutic approaches that apply across all CRC to improve response rates without enhancing toxicity.
Targeting the immunometabolic enzyme indoleamine 2,3 dioxygenase 1 (IDO1) represents one potential approach to address this unmet need. IDO1 is an enzyme that mediates the metabolism of the essential amino acid l-tryptophan to l-kynurenine. IDO1 is overexpressed across different malignancy types, including CRC, and is associated with poor prognosis independent of microsatellite stability (5–8). Mechanisms attributed to the pro-tumorigenic activity of IDO1 and kynurenine metabolites include shaping a tumor-favorable immune microenvironment and activation of the aryl hydrocarbon receptor (AHR) (9–13). IDO1 and the kynurenine pathways is also increased in human inflammatory bowel disease, a pre-neoplastic condition for colitis-associated cancer (CAC) (14,15). In preclinical models of CAC, we previously demonstrated that IDO1 activity directly promotes tumor growth through activation of PI3K/AKT signaling and nuclear localization of β-catenin (16,17). This finding is intriguing, as other studies indicate that IDO1 blockade can boost the effect of select DNA-damaging therapies in models of lung, melanoma, breast, and brain cancer (18–22).
In this study, we examined the interaction between the IDO1-tryptophan metabolism pathway and radiation therapy in CRC. Radiation is a primary or adjunctive therapy for several solid tumors, including locally advanced sporadic and colitis-associated rectal cancer. The PI3K/AKT pathway is known to promote tumor radio-resistance and reduce apoptosis in CRC (23,24). Thus, based on findings on IDO1 in CRC and its known immunomodulatory properties, we hypothesized that IDO1 inhibition would enhance the antitumor effects of radiation and efficacy of radiation therapy. To test this hypothesis, we examine several in vivo and in vitro models relevant to both CRC radiation therapy and radiation-relevant gastrointestinal toxicity. Our findings demonstrated a beneficial effect of the combination and provide mechanistic rationale for the design of a clinical trial examining the addition of IDO1 inhibition to radiation in patients with advanced rectal cancer.
MATERIALS AND METHODS
Human samples
All aspects of this study involving human tissues or specimens were approved by the Institutional Review Board of Washington University School of Medicine according to the U.S. Common Rule, and informed written consent was obtained. Human CRC samples of eight patients undergoing short-course radiation therapy (SCRT) for locally advanced (stage II or III) rectal cancer were identified from published cohort of patients who received five fractions of 5 Gy radiation followed by surgical resection within one week (Supplementary Table S1)(25). Sections from formalin-fixed, paraffin-embedded histology blocks of tissues were obtained from the Department of Pathology at Washington University. The reported histologic grades were from the official pathology report.
Cell lines and in vitro reagents
Human CRC cell lines, HT29 (HTB-38), HCT116 (CCL-247), DLD1 (CCL-221), and mouse CRC cell line CT26 (CRL-2638) were purchased from ATCC in 2016. Mouse CRC cell line MC38 (ENH204-FP) was purchased from Kerafast Inc. (Boston, MA) in 2017. All the CRCs were cultured and expanded in either RMPI 1640 or DMEM (Gibco) medium supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), streptomycin (100 μg/mL), and l-glutamine (2 mmol/L) according to supplier protocols and aliquoted at low passage rates (<10) for utilization across experiments. Cells were not reauthenticated. Mycoplasma testing was performed by the Tissue Culture Support Center at Washington University School of Medicine. Epacadostat was obtained under Material Transfer Agreement from Incyte, Inc (Wilmington, DE), stored at 4 °C and was reconstituted in DMSO at 50 mM and added to cell cultures at the experimentally described concentrations. L-kynurenine (Sigma, K8625–100mg) was reconstituted in H2O at 50 mM. IFNγ treatment (2 ng/mL) was used as described experimentally. Reagents and vendors are detailed in Supplementary Table S2.
Mouse CRC tumor models and irradiation
Six week old female wild-type (C57Bl/6 or BALB/c) or Rag1–/– mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animal procedures and maintenance were carried out in accordance to IACUC protocols approved by the Washington University School of Medicine Animal Studies Committee. As indicated in the figure legend, radiation was carried out as previously described using Gammacell 40 137Cs irradiator (Atomic Energy of Canada) at 78.8 cGy/min (whole body) or using RS-2000 irradiator (Rad Source Technologies, Suwanee, Georgia, USA) at 100 cGy/min with 160kVp X-rays using a 0.3 mm copper filter (focal fractionated IR)(26). Mice were anesthetized with 2% isoflurane using E-Z small animal anesthesia machine (E-Z systems, PA) prior to radiation. For irradiating the tumors, the rest of the body was shielded using a 5 mm-thick lead block (see Supplementary Fig. S1). Irradiators are calibrated yearly by Washington University Department of Radiation Safety. Experimentally, we confirmed the dose with Radcal 2186 for each experiment to the irradiated tumor and shielded areas. There was a minimal dose (0.000997 Gy) slightly above background to the organs and tumors shielded by lead.
Colon cancer heterotopic implantation models (CT26 in BALB/c, MC38 in C57Bl/6, HT29 or HCT116 in Rag1–/– mice) were established by injecting 1.0×106 CRC cells subcutaneously into the hind legs of the mice as previously described (26). Mouse body weight and tumor growth were assessed three times weekly until visible, then daily. The length, width, and depth of both right and left tumors were measured using an external digital caliper daily from when the tumors were palpable. The tumor volume was calculated using the formula V = (L × W × D)/2. After tumors were palpable (~250 mm3), mice were serpentine-sorted by the volume of index tumor (intended to treat) into experimental groups as described in the corresponding figure legends. Treatments by gavage included vehicle control (0.5% methylcellulose, Sigma) and IDO1 inhibitor epacadostat (Incyte, 300 mg/kg/day, suspended in 0.5% methylcellulose) as previously published (27). Lead shielding was used in focal irradiation experiments exposing only the “index” tumor-bearing limb and shielding the “contralateral” tumor, body, and spleen with a 5 mm lead plate as shown in Supplementary Fig. S1. In most experiments, treatment groups were euthanized on the same day to enable consistency in tumor tissue analyses. Detailed treatment strategies are described in corresponding figure legends.
Apoptosis and crypt survival in mouse intestine
The effect of radiation with 1-mT or epacadostat to the normal intestine was assessed as previously described (26). 6–8 week old female C57Bl/6 mice (without tumor) received total body irradiation (TBI) at 12 G. 1-DL-methyl tryptophan (cat# SX-999, 200 mg/pellet, releasing rate: 0.9 mg/hour, Innovative Research of America) were placed subcutaneously in the dorsum as previously described (28). Epacadostat (300 mg/kg) was gavaged two hours before irradiation (n=5–8 mice/group). For apoptosis analysis, the mice were gavaged once with epacadostat and sacrificed six hours after TBI. For crypt survival, the mice were gavaged once daily for three consecutive days and sacrificed 84 hours after TBI. The small intestine was harvested and fixed in 10% formalin for TUNEL and immunohistochemistry staining of BrdU. Briefly, six 5-mm fixed intestinal segments were taken from each mice and paraffin embedded for analysis. Apoptosis was scored on a cell-positional basis of 100 half-crypt sections per mouse by TUNEL staining using light microscopy. All crypts chosen were at least 20 cells in height, with cell position 1 located at the crypt base. For crypt survival analysis, each mouse received 120 mg/kg bromodeoxyuridine (BrdU) (Sigma) and 12 mg/kg fluorodeoxyuridine (Sigma) 90 minutes prior to sacrifice to label S-phase cells identified by immunohistochemistry using rat monoclonal anti-BrdU (1:1000, Accurate chemical & Scientific Corp). The viability of each surviving crypt was confirmed by incorporation of BrdU into 5 or more epithelial cells within each regenerative crypt. A minimum of 6 complete cross sections were scored for each mouse and the average was used to evaluate across experimental groups.
For mouse survival and weight loss experiments, 20 female C57Bl/6 female mice were randomized to receive gavage with epacadostat (300 mg/kg) or vehicle control (0.5% methylcellulose) two hours prior to focal abdominal irradiation (FAI) and daily until the end point. FAI was carried out in the RS-2000 irradiator where mice were anaesthetized with 2% isoflurane, and the mouse head, forelimbs, hind limbs, and thorax were shielded with lead as already described. Mice received 4 Gy FAI on 7 consecutive days as indicated.
CRC cell viability and clonogenic assay
All in vitro radiation experiments were performed using the Gammacell 40 137Cs irradiator (Atomic Energy of Canada) at 78.8 cGy/min. Epacadostat was obtained under Material Transfer Agreement from Incyte, Inc (Wilmington, DE). Epacadostat and L-kynurenine were prepared for culture experiments as previously described (29). For cell viability/proliferation assay, both wild type and IDO1–/– CRC cells were plated in 96 well plates. The cells were plated in 10% FBS containing complete medium overnight to allow the cells to attach and then replenished the cells with serum-free medium for six to eight hours to allow the cells synchronize. Then the cells were replenished with epacadostat (10 uM) or kynurenine (25 uM for CT26; 100 uM for HCT116 and HT29) containing 1–2% FBS medium for two hours before exposure to irradiation. 48 hours post irradiation, the cells were analyzed for the viability using Cell counting Kit-8 kit colorimetric assay (Dojindo Molecular Technologies) according to the manufacturer’s instruction using Synergy 2 microplate reader (Biotek, Winooski, VT).
Clonogenic assays were performed as previously described (26). Briefly, 6-well plates were used to plate a density of 200 cells/well for sham irradiated and 1000 cells/well irradiated cells. Cells were allowed to grow for 24 hours post plating, then new media containing epacadostat (10 uM) or vehicle control (DMSO: medium, 1:1000 by volume) was provided for 2 hours prior to irradiation at the described dosage (0, 2, 4, 6, 8 Gy). After 10~14 days of colony formation, plates were fixed in formalin for 20 minutes, washed in PBS, and stained with 0.5% crystal violet for 10 minutes. The plates were rinsed in water to remove background stains and air dried. The plates were photographed and colonies comprising of 50 cells or more were counted visually. The survival fractions were calculated after normalizing to plating efficiency and presented as surviving fraction relative to control.
Organoids and treatment
Mouse epithelial colonoids and enteroids were established from C57Bl/6 mice and those with azoxymethane (AOM, Sigma) and dextran sodium sulfate (DSS, Affymetrix) induced tumors (tumoroids) as previously described by our group (17,30). Spheroids were passaged and grown in 50% quality tested L-WRN CM prepared from our lab for ~18 hours then switched to 5% L-WRN CM and treated with vehicle control or 100 μM kynurenine for two hours prior to 6 Gy radiation. After 48 hours, viability was assessed using Cell Counting Kit-8 colorimetric assay. Absorbance was normalized by dividing by average absorbance of unirradiated, untreated controls for each genotype.
Generation of IDO1 knockout CRC cells using CRISPR
Alt-R CRISPR-Cas9 2-part guide RNA (crRNA+ tracrRNA, Integrated DNA Technologies [IDT], Coralville, IA) was used to generate IDO1 knockout cells according to manufacturer provided protocols. Briefly, two different crRNAs (type a and type b as shown in the Supplementary Table S3) targeting different exons of the IDO1 gene (human: exon 3 and 11; mouse: exon 3 and 7) were designed and synthesized in IDT. Both tracrRNA-ATTO 550 (Cat # 1075927) and Alt-R S.P. Cas9 nuclease (Cat# 1081058) were from IDT. Duplexes of crRNA: tracrRNA were incubated with Cas9 enzyme to make the ribonucleoprotein (RNP) complex. CT26 and HT29 CRC cells in Opti-MEM (Gibco) were transfected with equal amount of type a-RNP and type b-RNP using RNAiMAX (Cat# 13778100, Thermo Fisher Scientific). The cells were replenished with regular medium after overnight incubation. Transfected cells representing the brightest 3–5% ATTO550 signal were sorted using FACS (BD FACSAria II) and plated as single cells in each well of a 96 well plate. Individual clones were grown in regular culture medium for 1–2 weeks. DNA from cloned cells was isolated using QuickExtract DNA extraction solution (Cat# QE09050, Lucigen) and screened for edited IDO1 locus by PCR. Three different pairs of screening primers were used to screen the positive clones (31). Functional verification of IDO1 depletion was performed by comparing the IDO1 knockout cells and non-transfected controls after treatment with IFNγ (10 ng/mL) for 48 hours. Only both genomically and functionally verified IDO1–/– cells were used in the current study.
Inhibition of Type I and II interferon using antibodies both in vitro and in vivo
The recombinant murine IFNα4 was a gift by Kathleen Sheehan (Robert Schreiber Lab, Washington University Medical School), and the recombinant human IFNα2a was from Sigma-Aldrich (SRP4594–100 μg). Both human and murine IFNγ were obtained from PeproTech (Cat # 300–02; 315–05). The following monoclonal antibodies (mAbs) were produced by Leinco Technologies, Inc. (St. Louis, MO): MAR1–5A3 (neutralizing anti-murine IFNAR1), GIR-208 (neutralizing anti-human IFNAR1), H22 (neutralizing anti-murine IFNγ), and PIP (Armenian hamster IgG isotype control). The human IFNα/β R2 antibody was from R&D Systems (MAB4015–100). For in vitro experiments, The CRC cells were plated in 6 well plate, pretreated with the specific antibody or isotype control IgG (10 ug/mL) for 2 hours before exposure to the IFN cytokines with concentrations indicated in the legends, or irradiation (6 Gy). 48 hours post irradiation or IFN cytokine treatment, the cells were harvested for RNA using Trizol reagent for gene analysis.
For in vivo experiments, six-week-old female BALB/c mice were injected with 1×106 wild type CT26 cells in both hind legs. After tumors measured ~200 mm3, mice were randomized to treatment groups (n=3~4 mice/group or 6~8 tumors/group). For IFNα/β receptor blockade, mice were injected i.p. with a single 2 mg dose of MAR1–5A3 mAb or GIR-208 isotype control mAb as described previously (32,33). For IFNγ neutralization, 250 μg of IFNγ–specific H22 mAb(34) or IgG isotype control mAb (PIP clone) was injected i.p. All the antibodies or isotype IgG control antibodies were injected 20 hours before the tumors were irradiated. Tumors were harvested for RNA or histology 48 hours post irradiation.
Flow cytometry
Mouse spleens were excised and ground on 70μm nylon mesh before the filtrate was collected and centrifuged at 1.5 × 103 rpm for 5 minutes. The splenic cells were resuspended in phosphate buffered saline (PBS), counted, and stained for flow cytometric analysis. Tumor-infiltrating lymphocytes (TILs) were isolated by discontinuous Percoll gradient as described previously, with the following minor modifications (35). Briefly, the tumor tissues were minced, enzymatically digested with 0.1% collagenase (Sigma, Cat# C5138), hyaluronidase (0.1 mg/mL, Sigma, Cat# H6254), and DNase IV (20 U/mL, Sigma, Cat# D5025) containing HBSS solution, and ground on 70μm nylon mesh. The filtrate was collected, and the lymphocytes were separated from the tumor cells by centrifugation of the cell suspension on an 80%:40% two-layer Percoll gradient. Cell surface and intracellular marker staining was performed using 1–2 × 106 single-cell suspensions of TILs and splenocytes. The cells were first incubated with Zombie NIR Dye (BioLegend) to distinguish live and dead cells and then incubated with the following conjugated monoclonal antibodies (obtained from BioLegend unless otherwise stated) for 30 minutes at 4°C. Surface antibodies were diluted with staining buffer (2% FBS, 1 mM EDTA, and 0.02% NaN3 in PBS) into cocktails containing 10% 2.4G2 conditioned medium. Antibodies included: PerCP Cy-5–conjugated anti-CD3; APC-conjugated anti-NK1.1; FITC-conjugated anti-CD4; and PE-conjugated anti-CD8. The stained cells were permeabilized and fixed using True-Nuclear Transcription Factor Staining kit (BioLegend) and then processed for intracellular Foxp3 staining using Brilliant Violet 421-conjugated anti-Foxp3 for 30 minutes at 4°C. All data were collected using a BD Canto flow cytometer equipped with BD FACSDiva software. Data were analyzed using FlowJo v10.
Cytokine Analysis
Cytokine analysis on mouse plasma via cheek bleeding and tumor protein lysates was performed using Milliplex mouse cytokine panel 1 (Cat# MCYTMAG-70K-PX32) according to the instructions provided by the manufacture (Millipore) by the Washington University Center for Human Immunology and Immunotherapy Programs (www.chiips.wustl.edu). Data was analyzed on Milliplex Analyst 5.0 using the linear interpolation log scale curve fitting program. Multiple t test was used to compare the cytokine changes.
Determination of kynurenine and tryptophan in mouse plasma by mass spectrometry
The change of the ratio of tryptophan to kynurenine was used to reflect the IDO1 enzymatic activity. The concentrations of tryptophan and kynurenine in plasma obtained via cheek bleeding from mice that underwent various treatment were measured by using mass spectrometry at Incyte according to published protocols (36). The API 3000 mass spectrometer was operated in positive ion mode and utilized multiple reaction monitoring (SCIEX, Framingham, MA). The source voltage and temperature were 4000 V and 500 °C. The declustering, focusing, and exiting potentials were 16, 70, and 10. The quadropoles were set to unit resolution. For kynurenine, two transitions were monitored - 209.1/94.2 (collision energy, CE 19) and an alternate 209.1/191.8 (CE 13). For tryptophan, the C13 isotope parent ion was selected. The transitions were 205.9/188.8 (CE 15) and an alternate 206.9/147.2 (CE 25). For the deuterated internal standards, L-kynurenine 4, 5, 6, 8 -D4 and tryptophan 2, 4, 5, 6, 7 –D5 the transitions were 213.1/195.8 (CE 13) and 209.9/191.8 (CE 15), respectively. The internal standards kynurenine and tryptophan were sourced from Buchem BV (Apeldoorn, Netherlands) and CDN Isotopes (Quebec, Canada).
Western blotting
Western blotting was performed on tumor tissues or cultured CRC cells that were lysed in radioimmunoprecipitation buffer (1% Nonidet P-40, 0.5% deoxycholate, and 0.1% SDS in PBS) containing a protease inhibitor cocktail and phosphatase inhibitors (Sigma). Twenty micrograms of protein were separated in SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore) using iBlot unit (Thermo Fisher). The membrane was blocked in 5% nonfat dry milk in TBST (TBS with 0.05% Tween 20) for 1 hour and then incubated overnight at 4°C with one of the primary antibodies (Supplementary Table S2) in 5% nonfat dry milk in TBST. The blot was then incubated at room temperature for 2 hours in horseradish peroxidase-conjugated donkey anti-mouse, donkey anti-rabbit, or donkey anti-goat IgG secondary antibodies (Santa Cruz), as appropriate. The blots were washed and processed for luminescence with SuperSignal West Dura Extended Duration Substrate kit (Thermo Scientific). Densitometry of bands on films was assessed with ImageJ and normalized to actin expression.
Real time quantitative RT-PCR
Total RNA was extracted from CRC cells, organoids, or tissues using Trizol (Invitrogen) and quantitative RT-PCR was performed as previously described (28). 2 μL of 1:3 diluted cDNA template was used in a 20 μL PCR reaction. For each individual cDNA template, at least two replicates were carried out in each reaction. The gene expression is determined by 2–ΔΔCT method. Crossing threshold values for individual genes were normalized to actin or GAPDH gene expression. The forward and reverse primers were purchased from Integrated DNA Technologies as defined in Supplementary Table S4.
Immunofluorescence and immunohistochemistry
For the syngeneic mouse tumors, cryosection of tumor tissues were prepared by freezing tissues in TissueTek OCT Compound (Sakura Finetek USA, Inc). 5 micron frozen sections were cut and fixed in a 1:1 mixture of cold methanol and acetone for 20 min. For the human CRC tissues, paraffin-embedded tumor tissues were cut to 5 micron sections and deparaffinized and rehydrated before the following procedures. For immunohistochemistry staining, slides were rinsed in PBS, blocked with 2% normal goat serum for 1 hour, and then incubated with either anti-mouse IDO1 or anti-human IDO1 at the dilution of 1:200~400 overnight at 4°C. Biotin-conjugated secondary antibodies, streptavidin-horseradish peroxidase, and substrate were applied according to the manufacturer’s instructions (Vector Labs). The antibodies are listed in Supplementary Table S2. Each human CRC tissue section was semi-quantitatively scored according to the percentage of IDO1 positive cells and the staining intensity per protocol previously described (37). Briefly, we assigned the following proportion scores: 0 if 0–5 % of the tumor cells showed positive staining, 1 if 6–25 % of cells were stained, 2 if 26–50 % were stained, 3 if 51–75 % were stained, and 4 if over 75 % of the cells were stained. We rated the intensity of staining on a scale of 0 to 3: 0, negative; 1, weak; 2, moderate; and 3, strong. We then combined the proportion and intensity scores to obtain a total positive score (range, 0–12): score 0 was negative, a score of 1 to 6 was weakly positive, and a score of 7 to 12 was strongly positive. For immunofluorescence, the slides were blocked in PBS/5% BSA for 1 hour at room temperature, and then incubated for 2 hours at room temperature with monoclonal rabbit anti-human IDO1 monoclonal (Novus) or rabbit monoclonal anti-Ki67 (Abcam). After being washed with PBS, slides were incubated for 2 hours at room temperature with Alexa Fluor-labelled secondary antibody (Invitrogen). Vectashield with DAPI was used for nuclear counterstaining, and sections were viewed with a Zeiss Axiovert 200 with an AxioCam MRm camera. The percent of cells with positive staining was scored in five to ten random fields, containing over 1000 to 2000 nuclei each, with the observer blinded to the treatment condition.
Statistical analysis
All data presented as mean ± SEM unless described otherwise. All cell-based experiments were performed with ≥ 4 experimental replicates and repeated at least three times. Animal experiments had 4–7 mice/group and repeated ≥2 times/experiment. Unpaired and two-sided Student’s t-test, nonparametric t-test, or one-way or two-way ANOVA were performed in Graphpad Prism 5. Rate of change in tumor volumes and/or weight change over time was compared between the groups using general linear mixed models with random intercepts/slopes to allow estimation and comparison of within-subject rate of change in volume (38). All general linear mixed models in the longitudinal analyses assumed a subject level random effect and were fitted using the maximum likelihood method with unstructured covariance matrix. Statistical tests were based on the approximate F or t- tests with denominator degrees of freedom approximated by the Satterthwaite methods (39). General linear mixed model statistical analyses were performed with SAS version 9.4 (SAS Institute Inc.), and statistical significance was defined as P<0.05.
RESULTS
Radiation increases IDO1 expression in human and mouse CRC
To explore the relationship between IDO1 and radiation, we first examined if radiation impacted CRC expression of IDO1 and other tryptophan-metabolizing enzymes (TDO and IDO2). Cultured human and mouse CRC cell lines uniformly demonstrated upregulation of IDO1 mRNA expression after a single dose of gamma irradiation (Fig. 1A–B). Xeno- or syngeneic engraftment of CRC cell lines into the murine host increased Ido1 expression compared to in vitro culture (Supplementary Fig. S2). Focal radiation further augmented tumor Ido1 mRNA and protein expression (Fig. 1C–G, Supplementary Fig. S3A) in CRC tumor grafts in mice. In the tumor xenografts, human but not mouse IDO1 expression was increased, indicating that the implanted neoplastic cells were the primary source of gene upregulation, although IDO1+ tumor-infiltrating myeloid-derived cells were also present (Supplementary Fig. S3A–B and S4). Comparatively, the baseline and radiation-induced changes in the expression of TDO2 and IDO2 were inconsistent across cell lines (Fig. 1A–D).
Figure 1: Radiation increases IDO1 expression in colorectal cancer.
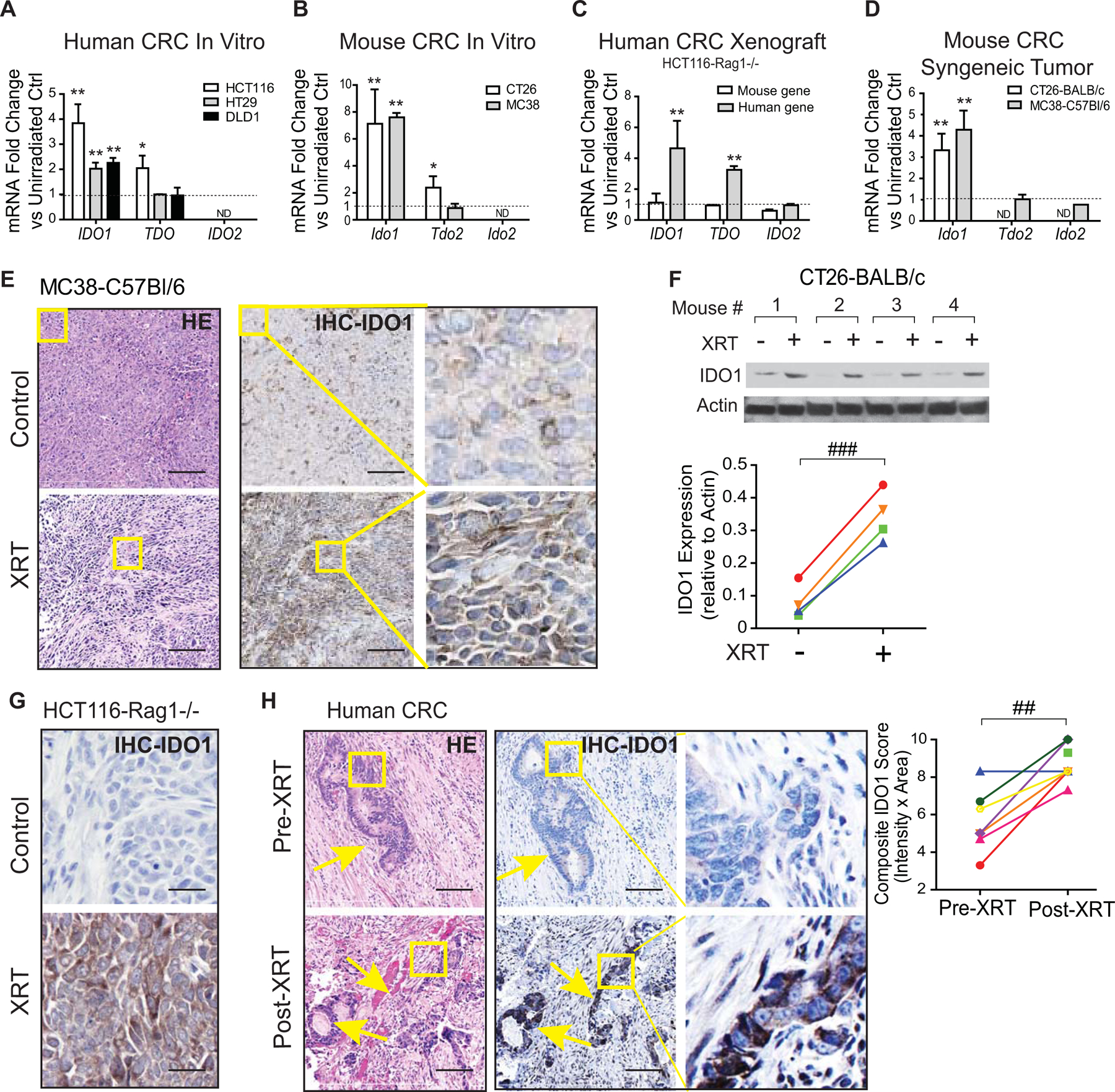
Human and mouse colorectal cancer (CRC) cell lines and tumor graft models were examined for change in IDO1 expression before and after irradiation. For in vitro experiments, mRNA and protein were extracted 48 hours after 6 Gy irradiation (IR). For in vivo models, 1×106 CRC cells were injected into the hind limbs of strain appropriate mice and allowed to grow to until palpable (~12 days). Tumors were then subjected to 6 Gy focal radiation therapy (XRT), and tissues were harvested 48 hours later. A-D, mRNA expression of tryptophan-metabolizing enzymes IDO1, TDO, and IDO2 (unirradiated controls equal to 1 and represented as a dashed line) in (A) human and (B) mouse CRC cell lines and (C) human xenograft and (D) syngeneic mouse models. E-G, IDO1 protein expression from in vivo tumor graft models (yellow box represents the zoomed areas for the images to the far right) using (E,G) IHC and (F) Western blotting (top) with protein quantification (bottom). Shown are 4 pairs of CT26 tumors. H, Human rectal cancer tumor samples from patients receiving 25 Gy XRT were examined for IDO1 expression. Representative images (left) and semi-quantitative IHC scoring (right) is presented for 7 pairs of pre- and post-RT and one additional post-RT sample from 8 total patients. Yellow arrow indicates malignant area. Yellow box represents the zoomed areas for the images to the far right. In vitro experiments were repeated ≥3 times with ≥3 experimental replicates. For in vivo animal experiments, n=4–7. Mean+SEM.*p<0.05, **p<0.01, ***p<0.001 by unpaired Student’s t-test or ##P<0.01, ###P<0.001 by paired Student’s t-test. Bars in micrograph=100 μm.
We next determined if CRC IDO1 expression was also increased in humans treated with radiation therapy (XRT). We examined tissue from a cohort of patients who underwent short-course radiation therapy of their rectal cancer followed by surgery within a week (25). In this cohort, XRT significantly increased tumor IDO1 expression (Fig. 1H, Supplementary Table S1). Together, these data indicated that IDO1 upregulation in the neoplastic epithelium was a common feature of CRC in response to ionizing radiation.
Type I and Type II interferon signaling contribute to radiation-induced IDO1
Interferons are potent cytokine inducers of IDO1, are present in the colon cancer tumor microenvironment (TME), and may be potentiated by radiation therapy (40–42). In cultured CRC cell lines, we found radiation increased expression of Type I (IFNA and IFNB) but not Type II (IFNG) interferons (Fig. 2A). Correspondingly, blockade of the common Type I IFN-α/β receptor (IFNAR-1) abrogated radiation-induced Ido1 expression, whereas anti-IFNγ had no effect (Fig. 2B). IFNAR-1 blockade consistently attenuated IDO1 expression across CRC cell lines and in tumor grafts (Fig. 2C–D). In irradiated mouse CRC tumor grafts, IFNγ blockade also significantly reduced Ido1 expression (Fig. 2D). These data suggested that Type I IFN signaling is necessary for cell-autonomous IDO1 expression, whereas both Type I and II interferons contribute to IDO1 expression in the TME.
Figure 2: Type I and Type II interferon signaling contribute to radiation-induced IDO1 in CRCs.
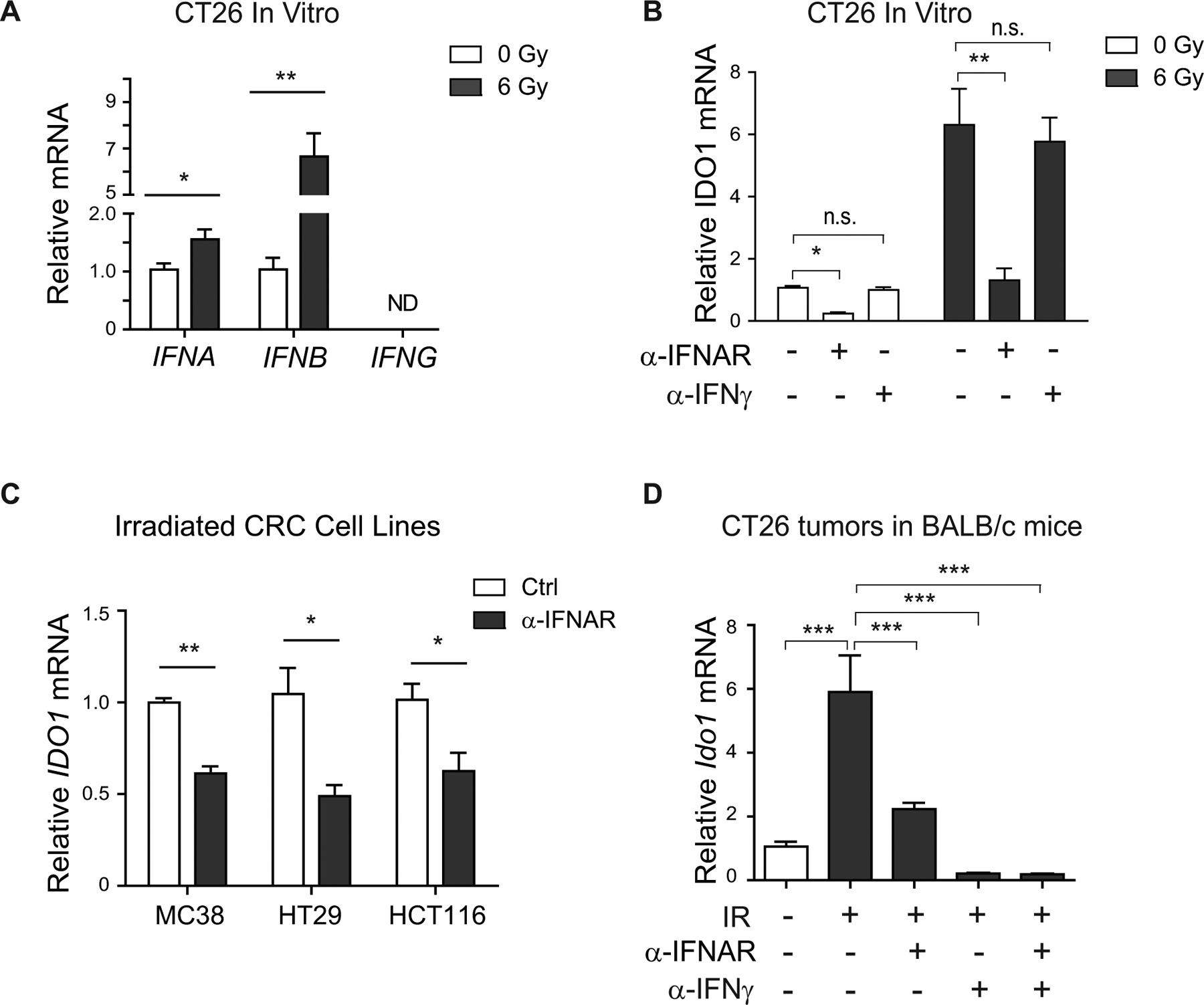
A, Expression of IFNα, IFNβ, and IFNγ in CT26 cells 48 hours after radiation. B, Effects of blocking the Type I IFN receptor (IFNAR) with a monoclonal antibody (10 μg/mL) on IDO1 expression in CT26 with or without irradiation (6 Gy at 48 hours). C, Effects of anti-IFNAR on IDO1 expression in irradiated (6 Gy at 48 hours) MC38, HT29, and HCT116 cells. D, Balb/C mice bearing CT26 tumors received isotype control, anti-IFNAR (2 mg), and/or anti-IFNγ (250 μg) I.P. 20 hours before 6 Gy focal irradiation. Tumor tissue was harvested for mRNA at 48 hours post-XRT. (A-C) n≥3 experimental replicates; (D) n=4 mice/group. Mean+SEM. *p<0.05, **p<0.01, ***p<0.001 by Student’s t-test versus control.
IDO1 inhibition enhances CRC radiation sensitivity in vitro
We next sought to determine if IDO1 activity is radioprotective to CRC. To address this, we used pharmacologic inhibition with a selective and potent IDO1 inhibitor, epacadostat (43). In dose-finding experiments, we found that 10 μM epacadostat consistently decreased proliferation across CRC cell lines with intact IDO1 but had no effect on CRISPR-edited IDO1–/– CRC cells (Fig. 3A, Supplementary Fig. S5–S6). Based on specificity of effect, 10 μM was used in subsequent experiments. Combining radiation and IDO1 inhibition further reduced CRC cell viability and colony forming capacity, while also enhancing cell death (Fig. 3B–D, Supplementary Fig. S6). CRC cells with CRISPR depletion of IDO1 also demonstrated enhanced radio-sensitivity (Fig. 3A). Together these findings demonstrated that IDO1 activity was radioprotective to CRC cells and that IDO1 inhibition enhanced the cytotoxic and anti-proliferative effects of radiation.
Figure 3: IDO1 inhibition enhances CRC radiation sensitivity in vitro.
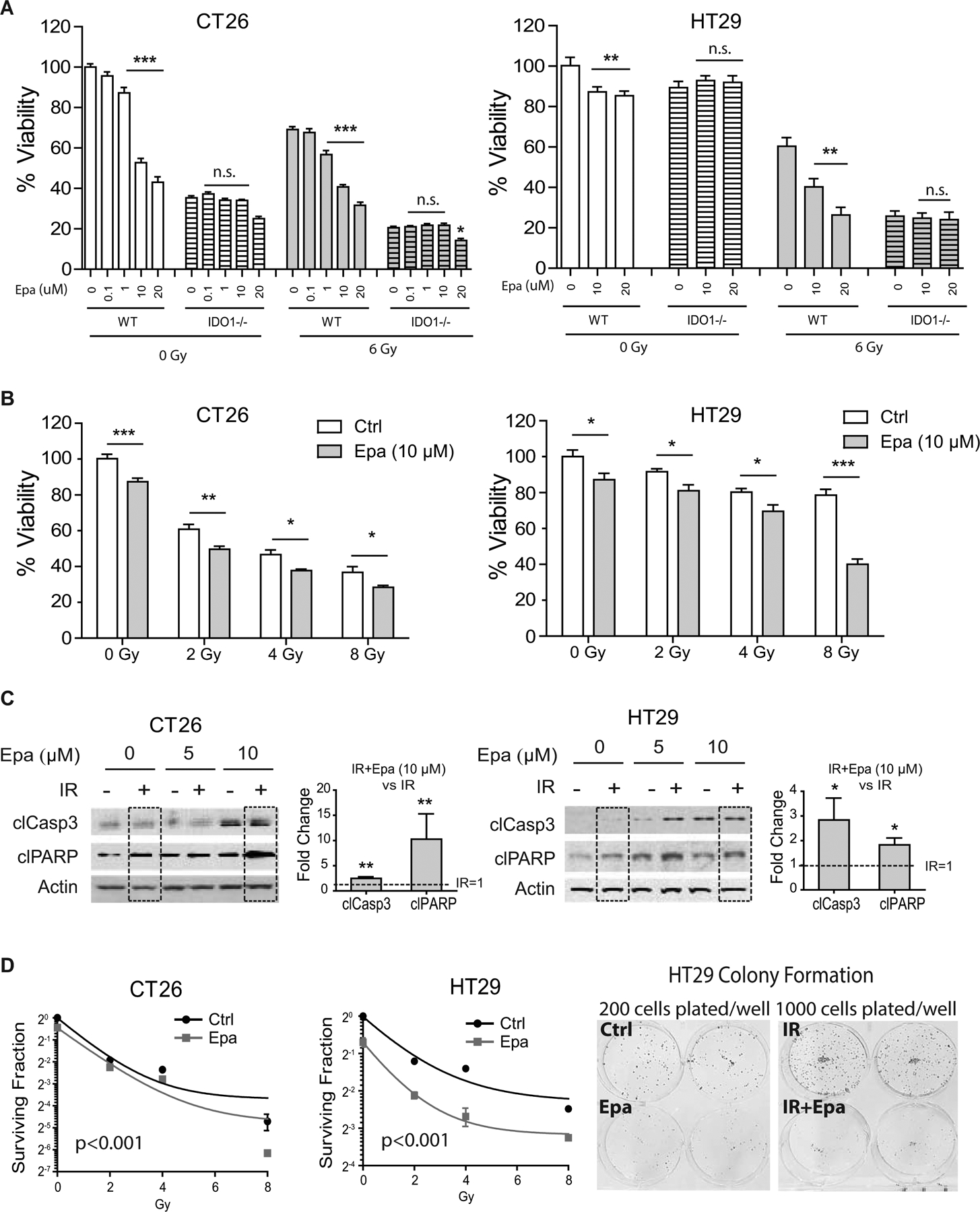
A, Cell viability assay of non-edited parental CT26 (left) and HT29 (right) cells (WT) vs. CRISPR-edited IDO1-null (IDO1–/–) cells. Where indicated, cells were analyzed 48 hours after irradiation. Epacadostat (Epa) titrations were used for IDO1 inhibition with or without 6 Gy irradiation (IR). B, Cell viability in the indicated CRC cells with increasing radiation doses and treatment with Epa (10 μM). C, Western blots of apoptotic markers cleaved caspase-3 (clCasp3) and cleaved PARP (clPARP) in CT26 (left) and HT29 (right) cells treated with 6 Gy in combination with Epa at 5 μM and 10 μM. Densitometry of clCasp3 and clPARP was performed comparing IR+Epa 10 μM and IR alone (IR=1, dashed lines/boxes). D, Clonogenic assay of CT26 and HT29 with or without 10 μM Epa after irradiation for 10~14 days. Representative images of colony formation are shown for HT29. Curves were constructed using an exponential decay nonlinear regression model. For cell viability experiments n=8–16; for colony formation experiments, n=3. Each experiment was repeated ≥3 times. Mean+SEM. *p<0.05, **p<0.01, ***p<0.01 by one-way ANOVA with Bonferroni posttest or Student’s t-test as appropriate.
IDO1 inhibition enhances CRC radio-sensitivity and shapes the tumor microenvironment
Having shown that IDO1 inhibition enhanced CRC radiation sensitivity in vitro, we next examined the impact of orally administered epacadostat in mice bearing CT26 tumors (an MSI-low CRC cell line) and receiving focal XRT (Fig. 4A, Supplementary Fig. S1). Once the tumors were palpable (~250 mm3), mice were serpentine sorted into groups with equivalent average tumor size to receive placebo, XRT (6 Gy × 2), epacadostat (300 mg/QD), or combined XRT and epacadostat. Tumor growth was delayed in the XRT and epacadostat monotherapy groups, whereas tumor growth was negligible in the combination treatment group (Fig. 4B). The epacadostat-treated groups exhibited lower circulating kynurenine concentrations and kynurenine to tryptophan (K/T) ratio (Fig. 4C–D), indicating efficacy of IDO1 enzymatic inhibition.
Figure 4: IDO1 inhibition enhances CRC radiation sensitivity in vivo.
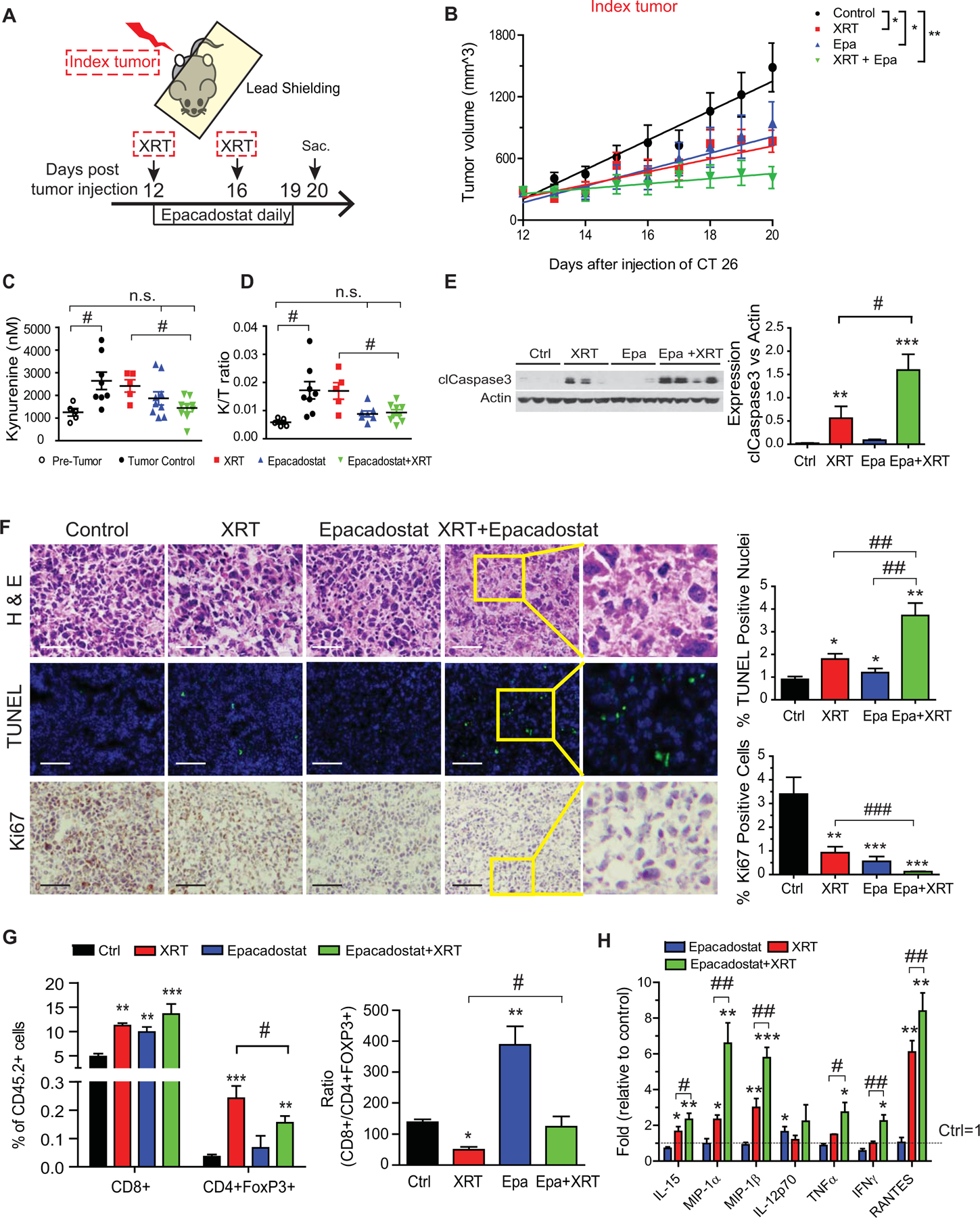
CRC was modeled using a syngeneic tumor graft of CT26 cells (1×106) injected into bilateral hind legs of BALB/c mice. Treatments included no therapy (control), radiation therapy alone (XRT, 6 Gy × 2), epacadostat (Epa) alone, and XRT plus Epa. n=5/group. A, Experimental setup. Mice were serpentine sorted into groups based on the size of the index tumors. Epa (6 mg) or vehicle control was gavaged daily 2 hours before XRT. Index tumor indicates the tumor that received XRT. Sac: sacrificed B, Changes in tumor size over time illustrated with average volume and best-fit linear representation based on mixed effects model. *p<0.05, **p<0.01. C, Plasma kynurenine and D, kynurenine to tryptophan (K/T) ratio of treatment groups and pre-tumor control mice (n=5–10 mice/group). E, Apoptosis as measured by cleaved caspase-3 (clCasp3) in tumor protein lysates harvested on day 20 post-implantation. Representative Western blot images with each lane reflecting an individual mouse tumor (left) and summarized quantification relative to actin (right). n=3–4. F, Representative images of tumors from treatment groups stained as indicated. TUNEL (apoptosis) and Ki67 (proliferation) quantitation provided at right. Bars in micrograph=40 μm. G, Tumor-infiltrating cytotoxic (CD8+) and regulatory (CD4+FoxP3+) T-cell populations expressed as a percentage (left) and ratio (right) of all tumor-infiltrating CD45.2+ lymphocytes (TILs). Results from 1 of 2 independent experiments. H, Cytokine analysis of index tumor protein lysates using Multiplex Cytokine Array. The values from mice receiving no treatment was set as 1. n=5–6/group. Mean+SEM. *P<0.05, **P<0.01, ***P<0.001 by Student’s t test compared to control. #P<0.05, ##P<0.01 by Student’s t test compared as indicated by the bracket.
To determine the antitumor mechanisms of combined XRT and IDO1 inhibition, we examined tumor cell viability and tumor-infiltrating lymphocytes (TIL). Tumor cells in mice receiving combined treatment exhibited higher apoptosis (cleaved caspase 3 and TUNEL) and lower proliferation (Ki67) than controls or either monotherapy (Fig. 4E–F). As shown in Figure 4G, TIL analysis revealed that XRT monotherapy created an unfavorable and lower ratio of cytotoxic (CD8+) to regulatory T cells (FoxP3+). IDO1 inhibition promoted a favorably higher CD8+/Treg ratio as a monotherapy and countered the adverse effect observed with XRT monotherapy.
Cytokine multiplex array revealed intriguing changes in the TME (Fig. 4H, Supplementary Table S5). Within this time frame, IDO1 inhibition promoted a modest increase in the active IL12 heterodimer (p70) but minimal other changes. XRT alone induced a mixed profile of inflammatory (macrophage inflammatory proteins [MIP] and IL15), as well as immunosuppressive (IL10), cytokines. By comparison, dual XRT and epacadostat–treated mice had tumors with the highest IL15, TNFα, IFNγ, MIPs, and the leukocyte chemotactic cytokine RANTES (CCL5). These data indicated a TME with enhanced Th1 cytokine activity and myeloid cell–modulating factors.
CT26 is an MSI-low CRC cell line. We therefore sought to confirm the effect in an MSI-high tumor graft model using MC38 cells. Epacadostat monotherapy was minimally effective at reducing MC38 tumor growth compared to CT26 tumor growth (Supplementary Fig. S7A). However, epacadostat+XRT slowed tumor growth more significantly than XRT alone (P=0.01 vs. P<0.001, Supplementary Fig. S7B). Overall, these data illustrated combining XRT and IDO1 inhibition is beneficial irrespective of MSI status.
IDO1 inhibition enhances the abscopal effect of radiation in distant CRC tumors
We next examined how IDO1 inhibition and XRT might affect cancer distant from the primary site, reflecting the phenomena of metastatic disease in humans. To do this, we first implanted CT26 cells bilaterally into mice and then, during the treatment phase, delivered XRT unilaterally to the index tumor only while shielding the rest of the body, including the contralateral tumor and spleen from radiation using a 5 mm lead plate (Fig. 4A, Supplementary Fig. S1). Growth of the contralateral (shielded) tumor (Fig. 5A) was assessed over time. Epacadostat monotherapy significantly slowed tumor growth similar to the index tumor, whereas XRT monotherapy did not (Fig 5B; P=0.04 and 0.08, respectively). The combination approach further slowed tumor growth (P=0.006) and led to regression in 40% of tumors. These findings suggested that IDO1 inhibition potentiates an abscopal effect of XRT, a phenomenon whereby local tumor therapy leads to regression of metastatic cancer at distant sites.
Figure 5: IDO1 inhibition enhances the abscopal effect of radiation in distant CRC tumors.
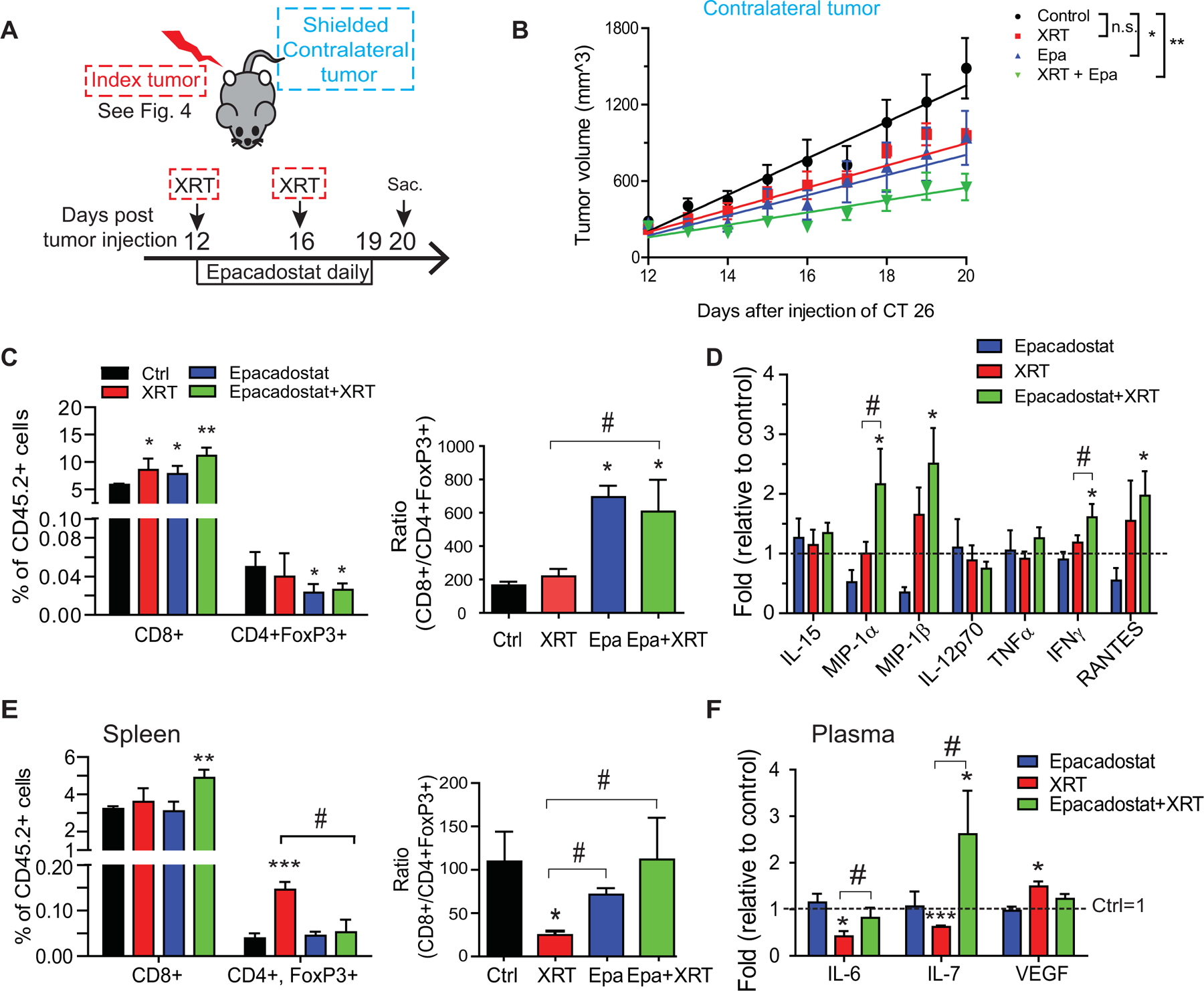
A, Experimental setup is as described in Figure 4. Data in this figure represents the contralateral (shielded) tumors. B, Changes in tumor volume over time. Best-fit linear curve and comparative statistics were assessed by mixed-effects modeling. Mean+SEM. *p<0.05, **p<0.01. n=5/group. C, Tumor-infiltrating cytotoxic (CD8+) and regulatory (CD4+FoxP3+) T-cell populations expressed as a percentage (left) and ratio (right) of all tumor-infiltrating CD45.2+ lymphocytes (TILs). Results from 1 of 2 independent experiments. D, Cytokine profile of contralateral tumor lysates. The values from mice receiving no treatment was set as 1. E, Changes in splenic lymphocyte populations. Left, percent of total CD45.2+ cells; right, the ratio of the CD8+ T cells to Tregs. F, Altered plasma cytokines. The values from mice receiving no treatment was set as 1. n=5–6/group. Mean+SEM. *P<0.05, **P<0.01, ***P<0.001 by Student’s t test compared to control. #P<0.05, ##P<0.01 by Student’s t-test compared as indicated by the bracket.
The abscopal effect is believed to be an immune-mediated phenomenon and may be potentiated by immune-stimulating therapies (44,45). Recognizing this, we evaluated for immune changes in the TME. In the distant tumors, XRT and IDO1 inhibition increased CD8+ T cells. However, only with IDO1 inhibition was Treg infiltration decreased and the CD8+/Treg ratio increased (Fig. 5C). Cytokine analysis provided further insight into the immune-mediated antitumor effects of combined XRT and IDO1 inhibition. XRT to the index tumor led to minimal changes in the contralateral tumor. However, significant pro-inflammatory signals were evident in the contralateral tumor in the combined treatment group, including elevated MIPs, IFNγ, and RANTES (Fig. 5D, Supplementary Table S6). This is consistent with a functional enhancement of cytotoxic T-cell activity and myeloid cell-modulating factors.
Systemic immune changes were also observed. In the spleen, tumor XRT promoted Treg expansion, leading to a lower CD8+/Treg ratio. This immunosuppressive effect was overcome by the addition of IDO1 inhibition (Fig. 5E). Tumor XRT suppressed plasma IL6 and IL7, while elevating circulating vascular endothelial growth factor (VEGF). IDO1 inhibition reversed each of these pro-tumorigenic changes. (Fig. 5F, Supplementary Table S7). The effect on VEGF raises the possibility that IDO1 inhibition is also affecting tumor neovascularization, an effect observed in other tumor models (46).
IDO1 inhibition slows secondary CRC growth after radiation to primary tumors
We next examined if pretreatment of IDO1 inhibition plus XRT prevented relapse by re-challenging mice with secondary tumors that previously received the treatment for their primary tumor (Fig. 6A). The primary tumor then received an additional XRT at 20 Gy, a dose shown to potentiate immunotherapy in MC38 tumors (47). In this model, we found that IDO1 inhibition+XRT slowed overall growth of the secondary tumors more effectively than XRT alone (P=0.04; Fig. 6B), with secondary tumor growth completely absent in 3 of 7 mice receiving both XRT and IDO1 inhibitor (Fig. 6C).
Figure 6: IDO1 inhibition slows secondary CRC growth.
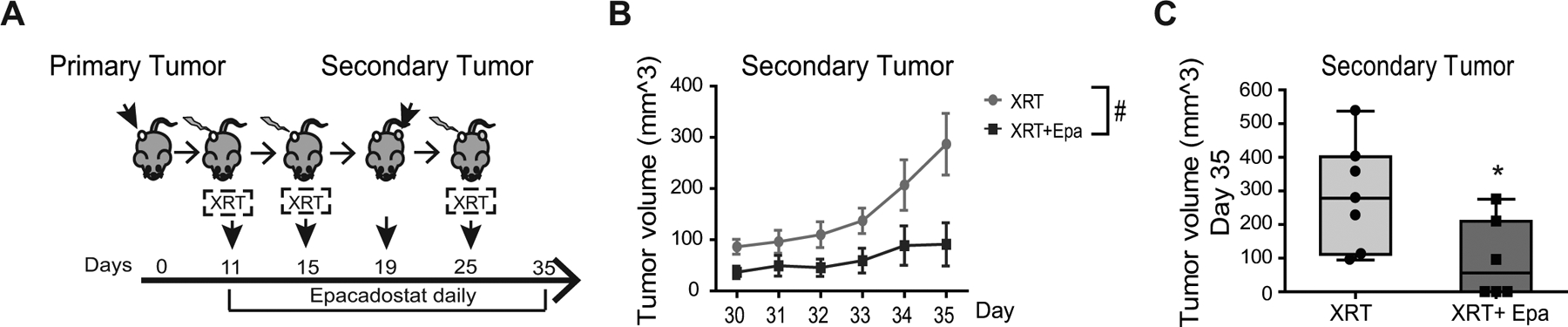
A, Experimental setup. C57Bl/6 mice received 1×106 MC38 cells (primary tumor) on Day 0 in the right hind leg. Two doses of focal XRT (6 Gy) was delivered as indicated and epacadostat (Epa, 6 mg) was administered by gavage daily. A secondary MC38 injection was placed in the contralateral leg on day 19. The mice then received a high dose focal RT (20 Gy) to the primary tumor. Both tumors were measured until 35 days post primary tumor implantation. B, Change in tumor volume over time in secondary tumors. Mean+SEM. #p<0.05 of secondary tumor growth by mixed-effect model accounting for interaction with the primary tumors. C, Comparison of endpoint secondary tumor volume at day 35. Mean+SEM. *P<0.05 by Student’s t-test. n=6–7/group.
IDO1 inhibition protects the normal intestinal epithelium from radiation injury
Combining anti-cancer therapies has the potential to augment normal tissue toxicity and side effects. Small bowel radiation injury is the principle toxicity of pelvic XRT used in rectal cancer (48). Diarrhea from small bowel damage can be severe and delay or require cessation of treatment. Given the clinical importance of this toxicity termed radiation enteritis, we next sought to evaluate the impact of IDO1 inhibition on radiotoxicity in the normal small intestine. Epithelial damage underlies the pathophysiology of radiation enteritis. Thus, we examined in vitro and in vivo models of XRT damage to the normal epithelium in the presence and absence of IDO1 activity. IDO1 blockade was protective of the normal small intestinal epithelium crypts in standard single-dose radiation assays of crypt survival and apoptosis (Fig. 7A–B). IDO1 inhibition hastened weight rebound and did not worsen survival in mice treated with fractionated abdominal radiation, a technique more reflective of radiotherapy for rectal cancer (Fig. 7C–D).
Figure 7: IDO1 inhibition protects the normal intestinal epithelium from radiation injury.
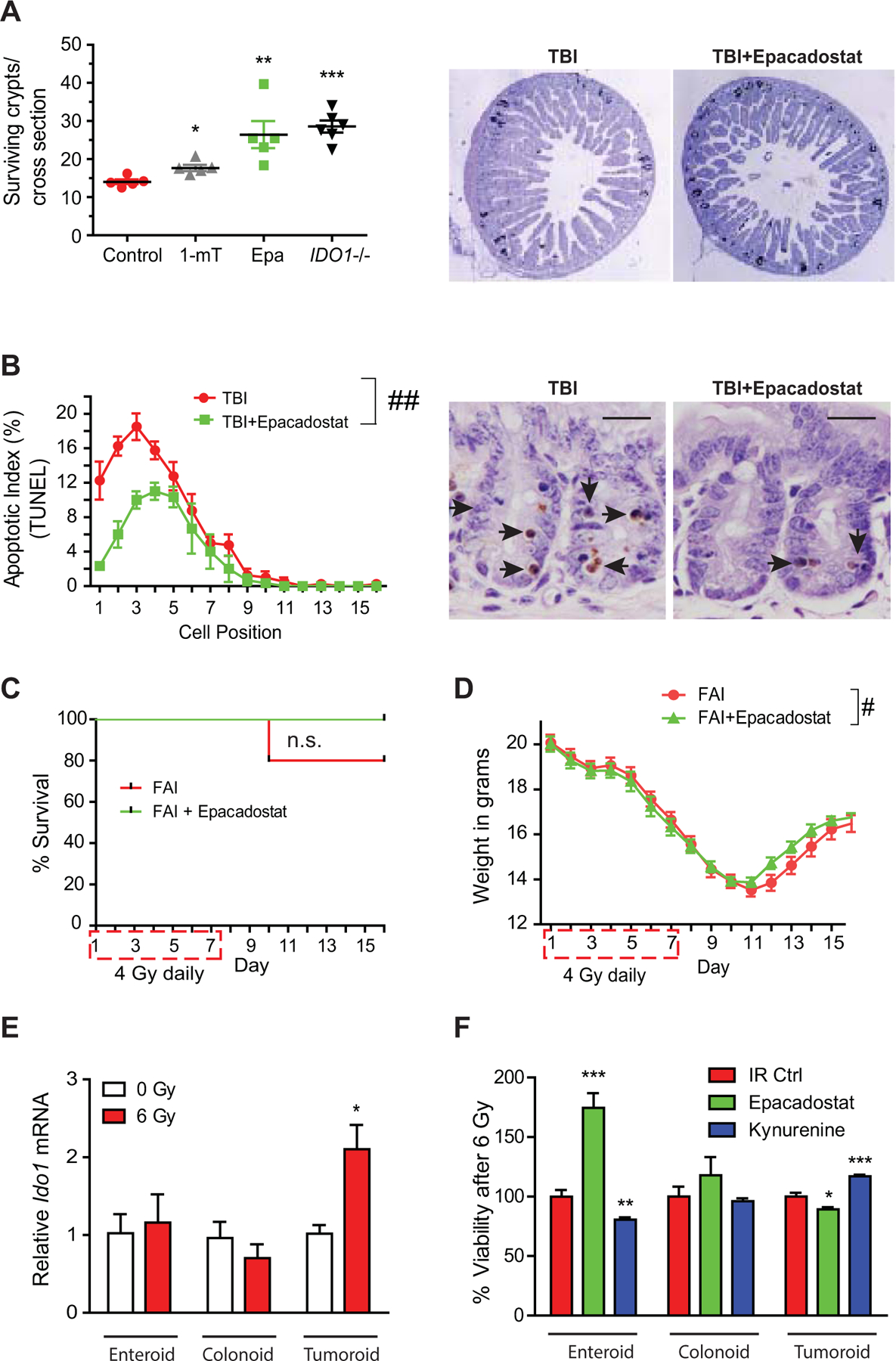
A, C57Bl/6 WT or IDO1–/– mice were treated with vehicle control, 1-mT, or epacadostat (Epa, 6 mg daily) and exposed to 12 Gy total body irradiation (TBI). Surviving epithelial crypts per jejunal cross section were assayed 84 hours after TBI. n=5–8/group. Results from 1 of 2 independent experiments. Right, representative images of BrdU stained jejunal cross sections. B, Apoptosis of jejunal epithelial cells by TUNEL 6 hours after 12 Gy TBI expressed by position within epithelial crypt. Right, representative TUNEL images. n=5. Bars in micrograph=20 μm. C, Survival and D, weight change over time for WT mice receiving fractionated abdominal irradiation (FAI, 4 Gy × 7 days) and either vehicle control or Epa by gavage. n=10/group. E, Relative IDO1 mRNA expression in mouse epithelium–derived enteroids, colonoids, and tumoroids following irradiation (6 Gy). n=3. F, Effect of Epa (10 μM) and kynurenine (100 μM) on cell viability of normal enteroids, colonoids, AOM/DSS tumoroids following irradiation (6 Gy). n=8. Mean+SEM. n.s., not significant; *p<0.05, **p<0.01, ***p<0.001 by Student’s t-test; ##P<0.01 by two-way ANOVA; #P<0.05 by mixed effects model.
We next addressed the possibility that IDO1 activity had an epithelial cell–intrinsic effect on radio-sensitivity using organoids derived from normal colon and small intestine, as well as colon tumors. Radiation did not alter Ido1 expression in normal colon- or ileum-derived organoids in vitro as it did with colon-derived tumoroids (Fig. 7E). Reflective of the in vivo findings, epacadostat improved viability of enteroids and reduced tumoroid viability with or without radiation (Fig. 7F, Supplementary Fig. S8A). Conversely, the IDO1 metabolite kynurenine reduced viability of irradiated enteroids and protected colon tumoroids. Kynurenine-mediated radioprotection was also observed in CRC cell lines (Supplementary Fig. S8B–C). Normal colon–derived epithelial cells (colonoids) were not significantly affected by either treatment. Together, these data demonstrated that IDO1 activity differentially affected radiation sensitivity based on tissue of origin (small intestine verses colon) and neoplastic state.
DISCUSSION
In this study, we demonstrated that radiation induced IDO1 overexpression in CRC and that IDO1 blockade enhanced the therapeutic effect of radiation. These observations were consistent across CRC cell lines, mouse models, and human tissues. The benefit of combining therapies was observed across models of primary, distant, and secondary CRC. Mechanistically, IDO1 inhibition augmented radiation’s cytotoxic and anti-proliferative effects while also promoting an antitumor immune microenvironment. Conversely, IDO1 inhibition reduced radiation damage to the normal small intestine, a primary tissue for XRT toxicity. Together, this study provides data to inform on the rational design of a clinical trial combining IDO1 inhibitors with radiation therapy for patients with advanced rectal cancer and perhaps other solid tumors.
Radiation increased IDO1 expression universally across human CRC samples, animal models, and cell lines regardless of MSI status. These findings have important conceptual implications, as restricted or undefined parameters for patient selection is a common challenge with immuno-oncology therapeutics (49). For example, anti–PD-1 therapies have shown efficacy only in patients with MSI-high or MMR-deficient CRC (1). However, this CRC subtype accounts for only a minority (10–20%) of all CRC and even fewer of rectal cancers (3). Heterogeneity of IDO1 expression has also been noted and is considered a potential limitation to this therapeutic approach. However, after radiation, IDO1 upregulation was nearly homogeneous. These findings suggest that baseline IDO1 expression may not be an obligatory inclusion criterion, as IDO1 overexpression will be common if not universal after radiation.
IDO1 inhibition enhanced the antitumor effects of radiation through two mechanisms. First, blockade of IDO1 augmented the cell-intrinsic antitumor effects of radiation in cultured CRC cells by further reducing proliferation and enhancing apoptosis. These results are consistent with the observation that IDO1 expression and kynurenine metabolites activate β-catenin signaling, as well as PI3K/AKT signaling, in colitis-associated and sporadic cancer (17). The AKT pathway promotes CRC proliferation and is intimately involved in preventing radiation-induced CRC apoptosis (24). The IDO1-kynurenine pathway also serves as a source for de novo generation of NAD+ (50). NAD+ is an important cofactor for the DNA repair enzymes activated during radiation therapy and is independently considered a target to increase radiation sensitivity (50,51). As a second mechanism, IDO1 inhibition also shaped a distinct immune response in irradiated tumors. Expression of IDO1 in tumor-infiltrating myeloid cells, as well as tumor cells, is well-recognized to promote growth by limiting antitumoral immunity through the suppression of tumor-reactive T cells (9,52). Consistent with this and the changes in T-cell populations, we found that TNFα and IFNγ were higher in irradiated tumors when mice received the IDO1 inhibitor. However, these tumors also demonstrated changes in myeloid cell–modulating factors including macrophage inflammatory proteins MIP1α and MIP1β (CCL3 and CCL4). This suggested activation of Th1 immune response as well as an environment favoring activation of granulocytes, both of which may result in the increased tumor killing. The addition of the IDO1 inhibitor produced a similar response in the contralateral (non-irradiated) tumor in mice bearing bilateral tumors where the index tumor was irradiated. Future studies should address the specificity and relative contributions of T-cell and myeloid cell-dependent tumor killing in this model.
This elevated immune response may explain how this combination abrogated secondary CRC growth and promoted an abscopal effect. The abscopal effect is an immune-mediated phenomenon whereby locally administered radiation can induce regression in tumor metastases at distant sites out of the radiation field (44). The abscopal effect depends on tumor type and concurrent therapies. However, immunotherapies have been observed to enhance this desired outcome (53). Anti–PD-L1 therapy is shown to facilitate the abscopal effect in mice, a finding that has spawned several clinical trials (47,54). We demonstrated here that IDO1 inhibition enhanced the abscopal effect in animal models of colon cancer. Further studies will be needed to confirm this effect across other CRC cell types and ideally using orthotopic and metastatic models of CRC.
Finally, our findings showed that IDO1 inhibition not only enhanced the antitumor effect of radiation, but limited radiotoxicity in the normal intestine. Intestinal radiation enteropathy is the primary dose-limiting toxicity of pelvic radiation therapy used for rectal cancer and is caused by epithelial stem cell death and crypt dropout (48). We demonstrated that IDO1 inhibition was radioprotective for normal intestinal epithelial stem cells and reduced crypt dropout. As a result, mice receiving IDO1 inhibition exhibited earlier weight rebound after fractionated radiation therapy. Kynurenine, the primary tryptophan metabolite from IDO1, had the opposite effect on radiation sensitivity in the normal small intestine compared to CRC. CRC has higher IDO1 expression and is more sensitive to kynurenine-mediated AKT activation than the normal epithelium (16,17). This provides explanation as to why IDO1 inhibition was radio-sensitizing to CRC but radioprotective of the normal epithelium. Additional studies are needed to fully dissect apart the mechanisms by which IDO1 inhibition protects the normal epithelium, and intestinal toxicity should be carefully examined in clinical trials combining IDO1 inhibitors with cytotoxic therapies.
Limitations exist in this study. This study included in vitro and heterotopic CRC models that do not fully recapitulate CRC biology, particularly as relates to immunity. Also, our study does not completely dissect apart the relative contribution towards radiation resistance that epithelial IDO1 expression plays verses IDO1 expression in other cell types (e.g. T cells, myeloid cells, etc). Although both likely contribute, the in vitro and in vivo experiments indicated that epithelial IDO1 was important in tumor radiation resistance. Further studies will be needed to fully dissect the mechanisms by which XRT-induced inflammation (cytokines and other damage-associated pathways) mediate induction of IDO1, TDO, and IDO2.
The current study can inform clinical trial development for IDO1 inhibitors. Although several of these drugs are being investigated for use in humans, the optimal strategy for applying IDO1 inhibitors to clinical cancer care remains unresolved (7,21,55). Epacadostat, the first IDO1 inhibitor to complete a phase I dose escalation trial, did not slow human tumor growth as monotherapy (56). A subsequent phase III trial failed to demonstrate a benefit for adding epacadostat to anti–PD-1 therapy in patients with metastatic melanoma (57). It was suggested that this trial may have failed due to inadequacies of patient selection and potentially insufficient dosing, but also that the preclinical evidence for combining IDO1 inhibition with anti–PD-1 is less compelling than for using adjunctively with DNA-damaging modalities (21). Indeed, 1-dl-methyl tryptophan (1-mT), a non-clinically tested compound with IDO1 inhibiting capacity, boosts the effect of select chemotherapeutics and/or radiation in models of melanoma, breast, and brain cancer (18–20). Also aligning with the current findings, the D isoform of 1-mT, in clinical trials as indoximod, acts downstream of IDO1 to stimulate mTORC1 and demonstrates a pattern of cooperation with DNA-damaging modalities (58). Another study found that IDO1 activity, as measured by serum K/T ratios before and after chemoradiotherapy, may predict survival in lung cancer patients (59). Together, this literature supports our findings that clinical grade IDO1 inhibition augmented the antitumor effects of radiation in CRC.
In summary, our data identified a therapeutic synergy of IDO1 inhibition with radiation in CRC, likely through a dual mechanism of enhancing direct tumor killing and breaking tumor immune tolerance. This strategy did not enhance XRT cytotoxicity to the normal small intestine epithelium. These results serve as the foundation for a Phase I study examining combined IDO1 inhibition and XRT in human rectal cancer ( NCT03516708). If demonstrated effective and safe, these findings may apply more widely to all forms of CRC as well as other solid tumors.
Supplementary Material
ACKNOWLEDGEMENTS
M.A. Ciorba has support from a Crohn’s and Colitis Foundation Daniel H Present Senior Research Award, Ref. 370763, NIH grants (DK109384, DK100737 and AI095776), philanthropic support from the Givin’ it all for Guts Foundation (https://givinitallforguts.org/), and the Emerson Collective. D.M. Alvarado was supported by DK077653 and The Lawrence C. Pakula MD IBD Innovation Fellowship. Core support from The Washington University Digestive Diseases Research Core Center (P30 DK052574), Siteman Cancer Center (P30 CA91842) and the Andrew M. and Jane M. Bursky Center for Human Immunology and Immunotherapy Programs. We thank Robert Schreiber Ph.D. and Kathleen Sheehan Ph.D. for guidance and resources on interferon experiments. We thank Peggy Scherle Ph.D. and Holly Koblish Ph.D at Incyte Corporation for critical comments and technical guidance on the use of epacadostat. We also thank Kevin Bowman and Michael Hansbury at Incyte for technical assistance with the K/T assessments. We also thank S. Santhanam and Caihong Wang for technical assistance and Lena McCue, Ph.D. in the Division of Biostatistics at Washington University for statistical support.
Footnotes
COI: MAC has received investigator-initiated research support and obtained epacadostat by an approved Material Transfer Agreement from Incyte, Corp (Wilmington, DE). All other authors declare no potential conflicts of interest.
REFERENCES
- 1.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372(26):2509–20 doi 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018;36(8):773–9 doi 10.1200/jco.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 3.Boland CR, Goel A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010;138(6):2073–87.e3 doi 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA oncology 2018;4(12):1721–8 doi 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003;9(10):1269–74 doi 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 6.Ferdinande L, Decaestecker C, Verset L, Mathieu A, Moles Lopez X, Negulescu AM, et al. Clinicopathological significance of indoleamine 2,3-dioxygenase 1 expression in colorectal cancer. Br J Cancer 2012;106(1):141–7 doi 10.1038/bjc.2011.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santhanam S, Alvarado DM, Ciorba MA. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl Res 2016;167(1):67–79 doi 10.1016/j.trsl.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meireson A, Chevolet I, Hulstaert E, Ferdinande L, Ost P, Geboes K, et al. Peritumoral endothelial indoleamine 2, 3-dioxygenase expression is an early independent marker of disease relapse in colorectal cancer and is influenced by DNA mismatch repair profile. Oncotarget 2018;9(38):25216–24 doi 10.18632/oncotarget.25393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother 2014;63(7):721–35 doi 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell reports 2015;13(2):412–24 doi 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest 2004;114(2):280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue P, Fu J, Zhou Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Frontiers in immunology 2018;9(286) doi 10.3389/fimmu.2018.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011;478(7368):197–203 doi 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 14.Sofia MA, Ciorba MA, Meckel K, Lim CK, Guillemin GJ, Weber CR, et al. Tryptophan Metabolism through the Kynurenine Pathway is Associated with Endoscopic Inflammation in Ulcerative Colitis. Inflamm Bowel Dis 2018;24(7):1471–80 doi 10.1093/ibd/izy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta NK, Thaker AI, Kanuri N, Riehl TE, Rowley CW, Stenson WF, et al. Serum analysis of tryptophan catabolism pathway: correlation with Crohn’s disease activity. Inflamm Bowel Dis 2012;18(7):1214–20 doi 10.1002/ibd.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thaker AI, Rao MS, Bishnupuri KS, Kerr TA, Foster L, Marinshaw JM, et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 2013;145(2):416–25 e1–4 doi 10.1053/j.gastro.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishnupuri KS, Alvarado DM, Khouri AN, Shabsovich M, Chen B, Dieckgraefe BK, et al. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res 2019;79(6):1138–50 doi 10.1158/0008-5472.CAN-18-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca S-B, Bolduc AK, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. Journal for immunotherapy of cancer 2014;2(1):21 doi 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monjazeb AM, Kent MS, Grossenbacher SK, Mall C, Zamora AE, Mirsoian A, et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clinical Cancer Research 2016;22(17):4328–40 doi 10.1158/1078-0432.ccr-15-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med 2005;11(3):312–9 doi 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 21.Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol 2019;41(1):41–8 doi 10.1007/s00281-018-0702-0. [DOI] [PubMed] [Google Scholar]
- 22.Li A, Barsoumian HB, Schoenhals JE, Caetano MS, Wang X, Menon H, et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int J Radiat Oncol Biol Phys 2019;104(4):903–12 doi 10.1016/j.ijrobp.2019.03.022. [DOI] [PubMed] [Google Scholar]
- 23.Brognard J, Clark AS, Ni Y, Dennis PA. Akt/Protein Kinase B Is Constitutively Active in Non-Small Cell Lung Cancer Cells and Promotes Cellular Survival and Resistance to Chemotherapy and Radiation. Cancer Research 2001;61(10):3986–97. [PubMed] [Google Scholar]
- 24.Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer 2002;94(12):3127–34 doi 10.1002/cncr.10591. [DOI] [PubMed] [Google Scholar]
- 25.Roy A, Mahasittiwat P, Weiner AA, Hunt SR, Mutch MG, Birnbaum EH, et al. Preoperative short-course radiation therapy for rectal cancer provides excellent disease control and toxicity: Results from a single US institution. Pract Radiat Oncol 2017;7(1):e51–e8 doi 10.1016/j.prro.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Riehl TE, Alvarado D, Ee X, Zuckerman A, Foster L, Kapoor V, et al. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut 2019;68(6):1003–13 doi 10.1136/gutjnl-2018-316226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. Journal for immunotherapy of cancer 2014;2(1):3 doi 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, et al. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. Journal of immunology 2010;184(7):3907–16 doi 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarado DM, Chen B, Iticovici M, Thaker AI, Dai N, VanDussen KL, et al. Epithelial Indoleamine 2,3-Dioxygenase 1 Modulates Aryl Hydrocarbon Receptor and Notch Signaling to Increase Differentiation of Secretory Cells and Alter Mucus-Associated Microbiota. Gastroenterology 2019;157(4):1093–108 e11 doi 10.1053/j.gastro.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 2015;64(6):911–20 doi 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauer DE, Canver MC, Orkin SH. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J Vis Exp 2015(95):e52118 doi 10.3791/52118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 2006;26(11):804–19 doi 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 33.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005;6(7):722–9 doi 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber RD, Hicks LJ, Celada A, Buchmeier NA, Gray PW. Monoclonal antibodies to murine gamma-interferon which differentially modulate macrophage activation and antiviral activity. J Immunol 1985;134(3):1609–18. [PubMed] [Google Scholar]
- 35.Liu Y, Chen K, Wang C, Gong W, Yoshimura T, Wang JM, et al. Isolation of Mouse Tumor-Infiltrating Leukocytes by Percoll Gradient Centrifugation. Bio-protocol 2013;3(17):e892 doi 10.21769/BioProtoc.892. [DOI] [Google Scholar]
- 36.Koblish HK, Hansbury MJ, Bowman KJ, Yang G, Neilan CL, Haley PJ, et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther 2010;9(2):489–98 doi 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- 37.Soliman H, Rawal B, Fulp J, Lee J-H, Lopez A, Bui MM, et al. Analysis of indoleamine 2–3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer immunology, immunotherapy : CII 2013;62(5):829–37 doi 10.1007/s00262-013-1393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laird NM, Ware JH. Random-Effects Models for Longitudinal Data. Biometrics 1982;38(4):963–74 doi 10.2307/2529876. [DOI] [PubMed] [Google Scholar]
- 39.Littell RC, Milliken GA, Stroup WW, Wolfinger RD, Oliver S. SAS for mixed models. SAS publishing; 2006. [Google Scholar]
- 40.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother 2014. doi 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014;41(5):843–52 doi 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Franco S, Turdo A, Todaro M, Stassi G. Role of Type I and II Interferons in Colorectal Cancer and Melanoma. Frontiers in immunology 2017;8:878- doi 10.3389/fimmu.2017.00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Medicinal Chemistry Letters 2017;8(5):486–91 doi 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012;366(10):925–31 doi 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 2018;18(5):313–22 doi 10.1038/nrc.2018.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mondal A, Smith C, DuHadaway JB, Sutanto-Ward E, Prendergast GC, Bravo-Nuevo A, et al. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine 2016;14:74–82 doi 10.1016/j.ebiom.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124(2):687–95 doi 10.1172/jci67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hauer-Jensen M, Denham JW, Andreyev HJ. Radiation enteropathy--pathogenesis, treatment and prevention. Nat Rev Gastroenterol Hepatol 2014;11(8):470–9 doi 10.1038/nrgastro.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salama AK, Moschos SJ. Next steps in immuno-oncology: enhancing antitumor effects through appropriate patient selection and rationally designed combination strategies. Ann Oncol 2017;28(1):57–74 doi 10.1093/annonc/mdw534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahm F, Oezen I, Opitz CA, Radlwimmer B, von Deimling A, Ahrendt T, et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res 2013;73(11):3225–34 doi 10.1158/0008-5472.CAN-12-3831. [DOI] [PubMed] [Google Scholar]
- 51.Lewis JE, Singh N, Holmila RJ, Sumer BD, Williams NS, Furdui CM, et al. Targeting NAD(+) Metabolism to Enhance Radiation Therapy Responses. Seminars in radiation oncology 2019;29(1):6–15 doi 10.1016/j.semradonc.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin Cancer Res 2015;21(24):5427–33 doi 10.1158/1078-0432.ccr-15-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynders K, Illidge T, Siva S, Chang JY, De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer treatment reviews 2015;41(6):503–10 doi 10.1016/j.ctrv.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spiotto M, Fu YX, Weichselbaum RR. The intersection of radiotherapy and immunotherapy: mechanisms and clinical implications. Science immunology 2016;1(3) doi 10.1126/sciimmunol.aag1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature reviews Drug discovery 2019. doi 10.1038/s41573-019-0016-5. [DOI] [PubMed] [Google Scholar]
- 56.Beatty GL, O’Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin Cancer Res 2017;23(13):3269–76 doi 10.1158/1078-0432.CCR-16-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long GV, Dummer R, Hamid O, Gajewski T, Caglevic C, Dalle S, et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J Clin Oncol 36, 2018 (suppl; abstr 108). [Google Scholar]
- 58.Fox E, Oliver T, Rowe M, Thomas S, Zakharia Y, Gilman PB, et al. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Frontiers in oncology 2018;8:370 doi 10.3389/fonc.2018.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Huang L, Jin JY, Jolly S, Zang Y, Wu H, et al. IDO Immune Status after Chemoradiation May Predict Survival in Lung Cancer Patients. Cancer Res 2018;78(3):809–16 doi 10.1158/0008-5472.can-17-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.