

Abstract
Background and Purpose:
We evaluated the association between two types of predictors of delayed cerebral ischemia (DCI) after nontraumatic subarachnoid hemorrhage (SAH), including biomarkers of the innate immune response and neurophysiologic changes on continuous EEG (cEEG).
Methods:
We studied SAH patients that had at least 72 hours of cEEG and blood samples collected within the first 5 days of symptom onset. We measured inflammatory biomarkers previously associated with DCI and functional outcome, including soluble ST2 (sST2), interleukin-6 (IL-6) and C-reactive protein (CRP). Serial plasma samples and cerebrospinal fluid (CSF) sST2 levels were available in a subgroup of patients. Neurophysiologic changes were categorized into new or worsening epileptiform abnormalities (EAs) or new background deterioration. The association of biomarkers with neurophysiologic changes were evaluated using the Wilcoxon Rank sum test. Plasma and CSF sST2 were further examined longitudinally using repeated measures mixed-effects models.
Results:
46 patients met inclusion criteria. 17 (37%) developed new or worsening EAs, 21 (46%) developed new background deterioration, and 8 (17%) developed neither. Early (day 0–5) plasma sST2 levels were higher among patients with new or worsening EAs (median 115ng/ml [IQR 73.8–197]) versus those without (74.7ng/ml [IQR 44.8–102], p=0.024). Plasma sST2 levels were similar between patients with or without new background deterioration. Repeated measures mixed-effects modeling that adjusted for admission risk factors showed that the association with new or worsening EAs remained independent for both plasma sST2 (β=0.41; 95%CI 0.09–0.73; p=0.01) and CSF sST2 (β=0.97; 95%CI 0.14–1.8; p=0.021). IL-6 and CRP were not associated with new background deterioration or with new or worsening EAs.
Conclusions:
In patients admitted with SAH, sST2 level was associated with new or worsening EAs but not new background deterioration. This association may identify a link between a specific innate immune response pathway and cEEG abnormalities in the pathogenesis of secondary brain injury after SAH.
Indexing terms: Subarachnoid Hemorrhage, Electroencephalography, Inflammation, Biomarkers
Introduction
Patients suffering delayed cerebral ischemia (DCI) after aneurysmal subarachnoid hemorrhage (SAH) have worse functional outcomes.1 The pathophysiologic mechanisms that contribute to DCI are uncertain, although cortical electrographic depolarizing events2 as well as inflammatory reaction have been implicated as having a role.3,4 Investigations of peripheral biomarkers linked to systemic inflammatory responses are associated with functional outcome, and this link appears to be mediated in part by nonconvulsive seizures.5,6 While nonconvulsive seizures occur in only 11–17%7 of patients with SAH, a wide spectrum of continuous electroencephalography (cEEG) abnormalities have been associated with subsequent DCI and poor outcome: new or worsening epileptiform abnormalities (EAs),8,9 deterioration of normal cEEG background frequencies and variability,10–15 and other depolarizing events.2,16–18
It is unclear whether the link between inflammation and seizures is isolated or extends to a broad range of cerebral dysfunction represented by other cEEG abnormalities. We sought to investigate whether two cEEG abnormalities associated with DCI, new or worsening EAs or cEEG background deterioration, were associated with several DCI-related pro-inflammatory biomarkers: C-reactive protein (CRP),19,20 interleukin-6 (IL-6),19,21,22 and soluble ST2 (sST2).5 We selected IL-6 and CRP because they have been studied extensively in the SAH literature19–22 and are believed to hold prognostic value. In a recent study, soluble ST2 (the protein product of the IL1RL1 gene) has demonstrated independent prognostic value when controlling for either IL-6 or CRP,5 indicating it may play an important role in secondary brain injury after SAH.
Methods
Patient Cohort
All data supporting the findings of this study are available from the corresponding authors upon reasonable request. We performed a post-hoc analysis examining cEEG abnormalities in a prospective single-center cohort study enrolling patients in a study examining inflammatory biomarkers and functional outcome following nontraumatic SAH between May 2013 and October 2015. The study was conducted under an Institutional Review Board-approved protocol; informed consent was obtained at the time of enrollment. Patient selection criteria were SAH from nontraumatic etiology and age ≥ 18 years. Patients were eligible if there was cEEG monitoring for at least 3 days and biomarker sampling within the first 5 days after SAH onset. At our institution, patients with either a high clinical grade (Hunt-Hess 3–5) and/or a high radiologic grade injury (modified Fisher score ≥3) undergo cEEG monitoring as standard of care. We recorded clinical, demographic and radiographic findings including age, gender, Hunt Hess (HH) and modified Fisher scores.
Electroencephalography
All cEEG recordings were acquired using 21 electrodes applied according to the standard International 10–20 system. Two clinical neurophysiologists, at least one board-certified, reviewed the raw EEG data prospectively in real time as part of clinical practice and generated twice-daily reports. Per American Clinical Neurophysiology Society (ACNS) nomenclature, new EAs were defined as worsening in frequency, prevalence, spatial extent, or epileptiform morphology of sporadic epileptiform discharges, lateralized rhythmic delta activity (LRDA), lateralized periodic discharges (LPD), or generalized periodic discharges (GPD).14,23 New background deterioration was defined as decreasing Alpha Delta Ratio (ADR), Relative Alpha Variability (RAV), or worsening focal slowing.14,23 As per our institutional guideline, patients are treated with anti-seizure prophylaxis until the culpable aneurysm was secured unless the clinical team opted to continue treatment for a separate clinical indication.
Biomarker assays
Biomarker assays were performed on venous plasma and CSF samples. Venous plasma samples were collected in ethylenediaminetetraacetic acid (EDTA)-containing tubes, and CSF was obtained from patients who had external ventricular drains (EVD) placed for clinical indications. Both plasma and CSF samples were centrifuged within 30 min of collection and stored at −80°C until analysis.5 The Presage ST2 Assay Kit (Critical Diagnostics, San Diego, CA) was utilized to measure sST2 from the stored plasma and CSF samples. CRP and IL-6 were measured using commercial ELISA kits (ThermoFisher, and R&D Systems, respectively) in accordance with the manufacturers’ instructions.
Statistical analysis
We pre-specified a primary hypothesis that plasma sST2, a biomarker of DCI and poor outcome, would be elevated in patients with new EAs or in patients with new background deterioration. We examined this association using the Wilcoxon rank sum test evaluating 95% confidence intervals and two-tailed significance testing. We pre-specified a secondary hypothesis that an association with new abnormalities in cEEG would be unique for sST2 compared with other inflammatory biomarkers. In an exploratory hypothesis, we examined serial biomarker measurements at early and late sampling time using a repeated measures linear mixed-effects model. CSF and plasma sST2 values were log-transformed prior to analysis. In all models, we adjusted for age, gender, and Hunt Hess grade. To adjust for the time between SAH onset and sample draw, we developed additional models that included time to sample draw as a covariate. To offset the potential for overfitting due to sample-size constraints, the mixed-effects models were repeated using a previously validated composite risk score14 that consolidated individual clinical and demographic risk factors into a numerical value.
Results
Patient Characteristics
Of 224 nontraumatic SAH patients admitted during the study period, 124 patients underwent informed consent and had at least one biomarker collection within 5 days of SAH onset. Of these, 46 patients underwent at least 72 hours of cEEG (median duration of cEEG monitoring: 8 days) (Figure 1). The median age of participants was 58.5 years (IQR 48.8–68.3) and 33 (72%) were female (Table 1). Deterioration in cEEG occurred in 29 (63%) patients: 17 (37%) developed new EAs, 21 (46%) developed new background deterioration, 9 (20%) developed both findings, and 17 (37%) developed neither. Of these patients, 3 (7%) had electrographic seizures. A subset of patients (n = 44) had IL-6 and CRP measurements collected at the initial timepoint (3.5 ± 0.9 days). Serial sST2 measurements were obtained at two additional timepoints: 18 patients had measurements acquired at timepoint 2, between days 5 and 10 after initial hemorrhage, and 16 patients had measurements sampled at timepoint 3, between days 10 and 15. Mean timepoint 2 collection occurred at 7.7 ± 1.2 days and timepoint 3 at 12.6 ± 1.6 days. Furthermore, CSF was collected in a subset of patients (n = 18) at timepoint 1 (n = 10), timepoint 2 (n = 9) and timepoint 3 (n = 9).
Figure 1.
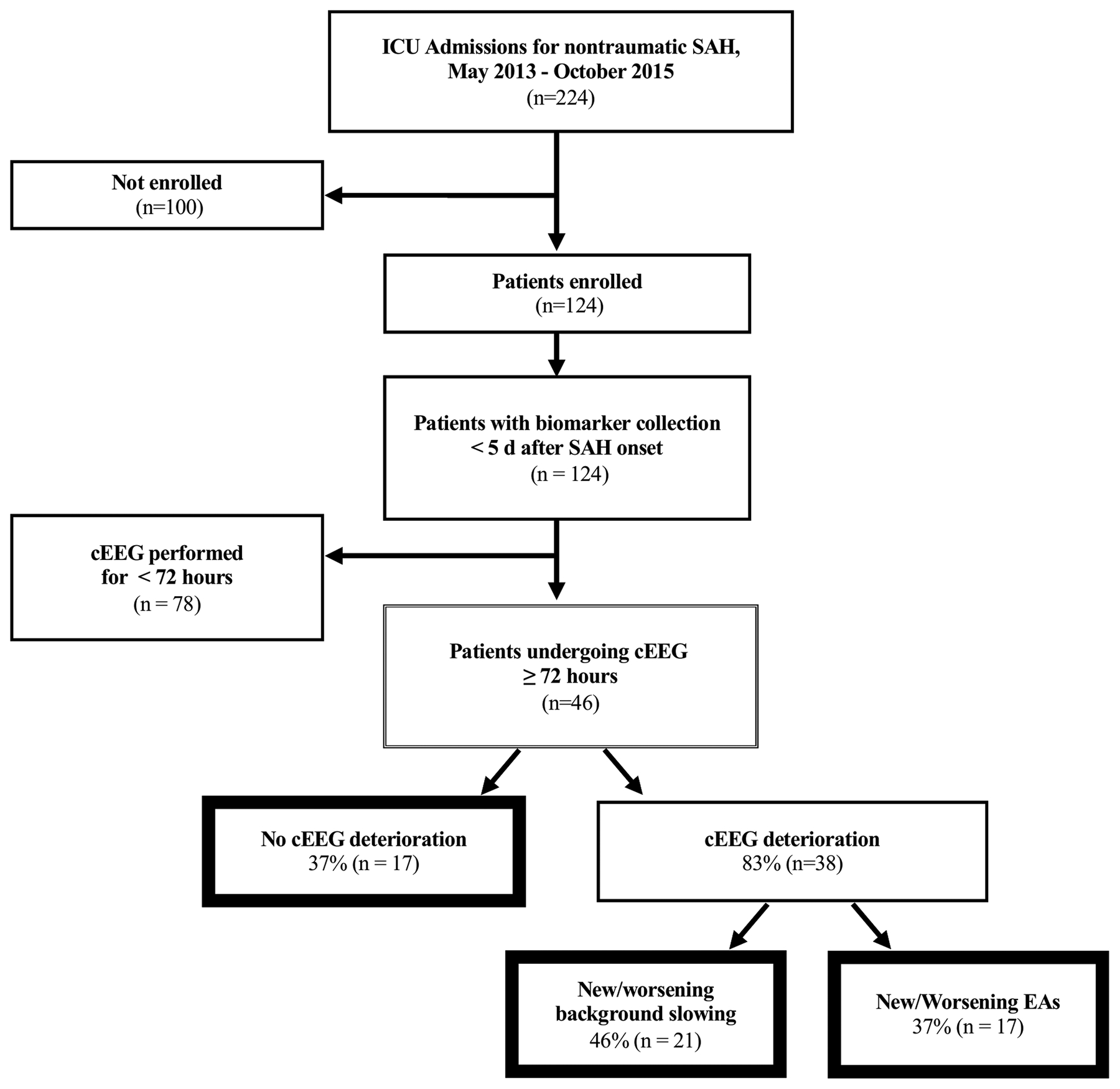
CONSORT Diagram
Table 1.
Patient Characteristics
| Enrolled Population (N=46) | |
|---|---|
| Demographics | |
| Age, median (IQR) | 58.5 (48.8–68.3) |
| Female gender, n (%) | 33 (72%) |
| Admission clinical grade, n (%) | |
| HH 1 | 10 (22%) |
| HH 2 | 6 (13%) |
| HH 3 | 10 (22%) |
| HH 4 | 12 (26%) |
| HH 5 | 8 (17%) |
| Admission radiologic grade, n (%) | |
| mFS 1 | 2 (4%) |
| mFS 2 | 1 (2%) |
| mFS 3 | 12 (26%) |
| mFS 4 | 31 (67%) |
| Electrographic findings, n (%) | |
| New/worsening EAs | 17 (37%) |
| New background deterioration | 21 (46%) |
| New background deterioration and new EAs | 9 (20%) |
| No new cEEG deterioration | 17 (37%) |
| Biomarker collection times, mean days ± SD | |
| Early | 3.5 ± 0.9 |
| Middle | 7.7 ± 1.2 |
| Late | 12.6 ± 1.6 |
EAs, epileptiform abnormalities; HH, Hunt and Hess; mFS, modified Fisher Grade
Early Biomarker Analysis
Median plasma sST2 in patients developing new EAs was higher (115 ng/ml [IQR 73.8–197]) than in patients who did not develop new EAs (74.7 ng/ml [IQR 44.8–102], p = 0.024) (Figure 2a). When patients with new-onset nonconvulsive seizures (n = 3) were excluded, the association persisted: median plasma sST2 was 125.7 ng/ml [IQR 80.1–202] for patients developing new EAs and 74.6 ng/ml [IQR 44.5–202] in those without (p = 0.0062). There was no difference between the median plasma sST2 level in patients who developed new background deterioration (75.7 ng/ml [IQR 44.5–120]) compared to those who did not (80.1 ng/ml [IQR 65.5–175], p = 0.56). There was no difference in sST2 levels for patients with high grade HH (4–5) at admission (94.8 ng/ml [IQR 65.3–179.0]) compared to those with lower grade HH (1–3) (75.4 ng/ml [IQR 46.5–122.1]) (p = 0.28) (Table 2).
Figure 2.
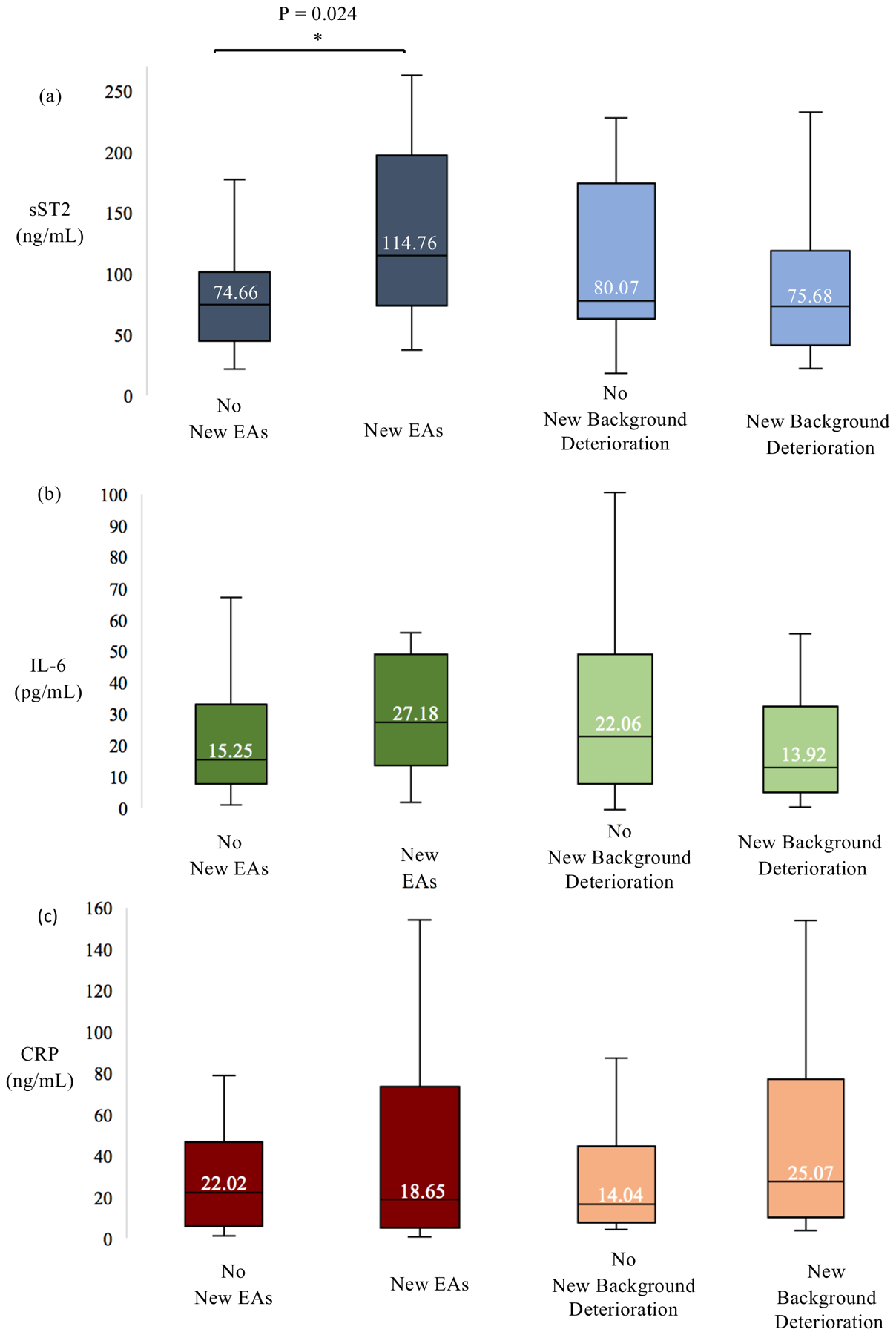
(a) Median sST2 sampled < 5 days from initial hemorrhage in patients developing new/worsening EAs was higher than in patients who did not develop new/worsening EAs (p = 0.024). There was no difference in median sST2 levels between patients who developed new background deterioration compared to those who did not. (b) IL-6 and (c) CRP were not significantly associated with new EAs or new background deterioration.
Table 2.
Biomarker Characteristics by SAH clinical severity.
| Hunt and Hess Grade 1–3 (n=28) | Hunt and Hess Grade 4–5 (n=18) | P-value | |
|---|---|---|---|
| sST2 level, median ng/mL (IQR) | 75.4 (46.5–122.1) | 94.8 (65.3–179.0) | 0.28 |
| IL-6 level, median pg/mL (IQR)* | 14.2 (8.1–27.2) | 36.1 (12.2–56.0) | 0.01 |
| CRP level, median ng/mL (IQR)* | 10.79 (3.6–38.1) | 41.04 (19.7–68.8) | 0.04 |
CRP, c-reactive protein; IL-6, interleukin 6; sST2, soluble ST2
n=43
Median IL-6 for patients developing new EAs (27.2 pg/mL [IQR 13.5–48.9]; n = 15) did not differ from those without (15.3 pg/mL [IQR 7.7 – 32.9], p = 0.28; n = 28; Figure 2b). Patients developing new background deterioration (13.9 pg/mL [IQR 6.5–32.9]; n = 20) had similar median IL-6 levels compared with those who did not (22.1 pg/mL [IQR 9.1–48.9], p = 0.13; n = 23). Patients with high grade HH at admission (14.2 [8.1–27.2]) had significantly elevated IL-6 levels compared to those without high grade HH (14.2 [8.1–27.2]) (p = 0.01) (Table 2).
CRP levels were not associated with cEEG abnormalities (Figure 2c). Patients developing new EAs (n = 15), (18.65 ng/mL [IQR 5.03–73.31]) did not differ from those not developing new EAs (n = 28), (22.02 ng/mL [IQR 5.77–46.68], p = 0.75). The median CRP levels for patients developing new background deterioration (n = 20) (25.07 ng/mL [IQR 5.95 −75.64]) and those not developing new background deterioration (n = 23), (17.29 ng/mL [IQR 5.03–38.12], p=0.41) were not different. CRP levels were significantly different for patients with high grade HH (41.04 [19.72–68.77]) versus those without high grade HH (10.79 [3.6–38.12]) at admission (p = 0.04) (Table 2).
Late Biomarker Analysis
The association between elevated plasma sST2 levels and new EAs persisted at timepoint 2. Patients developing new EAs had elevated median plasma sST2 levels at timepoint 2 (61.2 [IQR 44.9–106]), compared to those without new EAs (43.6 ng/mL [IQR 31.1–50.4], p=0.049). At timepoint 3, median plasma sST2 for patients with (67.3 ng/mL [IQR 37.0–96.1]) and without new EAs (38.7 ng/mL [IQR 32.9–74.3]) did not differ significantly (p = 0.37).
The occurrence of new background deterioration was not significantly associated with elevation in plasma sST2 levels at any of the late timepoints sampled. Median plasma sST2 for patients with new background deterioration versus those without new background deterioration, respectively, was 44.9 ng/mL [IQR 38.3–132] versus 47.9 ng/mL [IQR 33.6–61.2] at timepoint 2 (p = 0.6) and 38.5 ng/mL [IQR 35.1–56.6] versus 54.7 ng/mL [IQR 32.9–81.6] at timepoint 3 (p = 0.7).
Longitudinal Analysis
To account for repeated measures, we employed a linear mixed-effects model and found that new EAs were associated with elevated plasma sST2 over time (β=0.49; 95%CI 0.20 – 0.77; p = 0.001) (Figure 3a). This association persisted when adjusting for age, HH grade, and gender (β= 0.41; 95%CI 0.09–0.73; p = 0.01). No significant interactions between the covariates were identified. We also examined the effect of time to sample draw, and the association between sST2 level and new EAs remained independent (β=0.44; 95%CI 0.14–0.73; p = 0.003). In contrast, there was no association between worsening background deterioration and plasma sST2 level over time (β=−0.17; 95%CI −0.47–0.13; p = 0.26) (Figure 3b).
Figure 3.
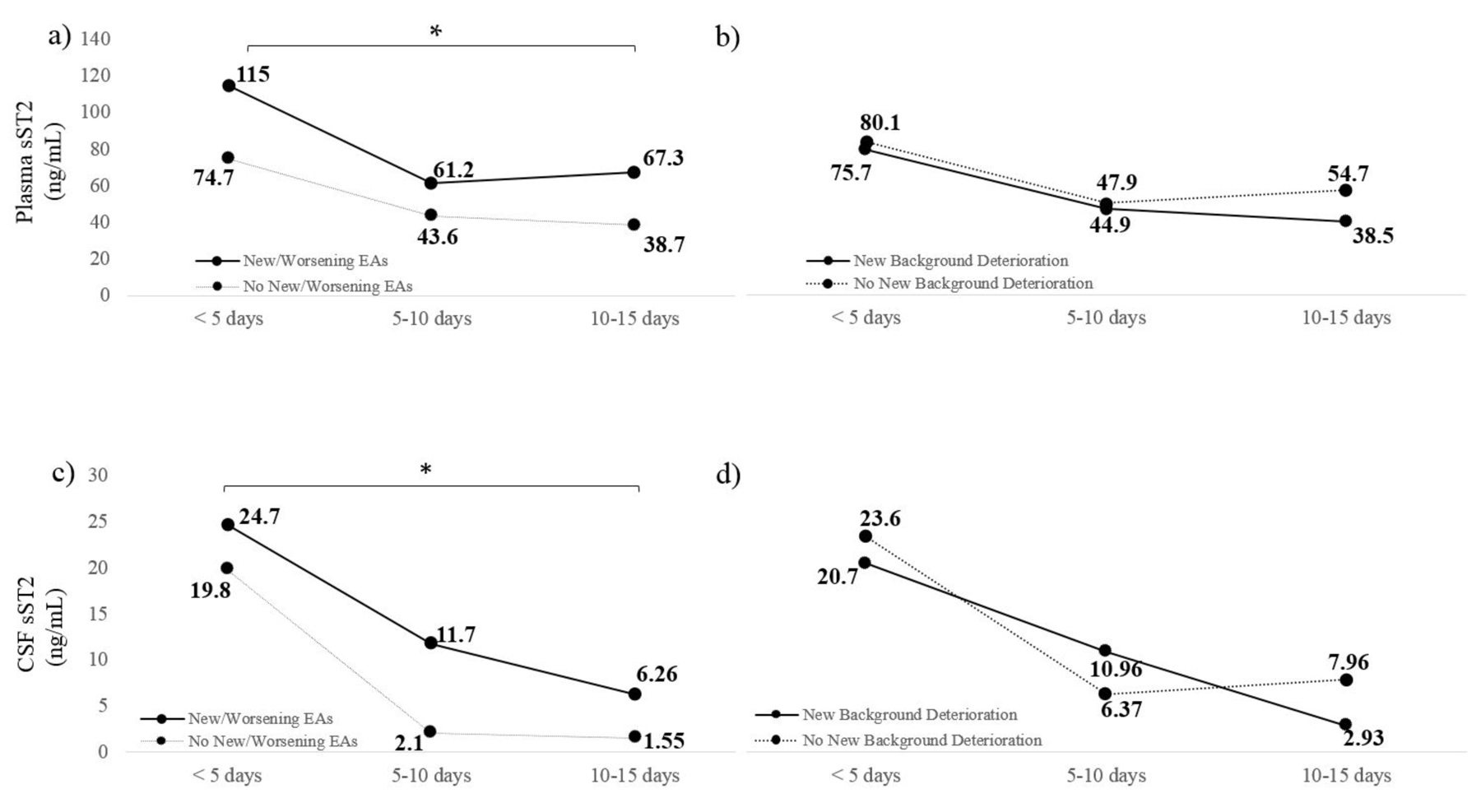
(a) Elevated plasma sST2 was significantly elevated over time in subjects who developed new or worsening EAs on EEG (p = 0.001). (b) There was no association between new or worsening background deterioration and plasma sST2 level over time (p = 0.56). (c) Elevated CSF sST2 was significantly associated with new or worsening EAs over time (0.003). (d) There was no association between elevated CSF sST2 and worsening background deterioration over time (p = 0.71).
At individual timepoints, CSF sST2 showed a trend towards an association with new EAs but not background decline. In a linear mixed-effects model, the association between new or worsening EAs and elevated CSF sST2 was significant (β=0.89; 95%CI 0.15–1.62; p = 0.018) (Figure 3c). When controlling for age, HH, and gender, the association between new or worsening EAs and elevated CSF sST2 persisted (β=0.97; 95%CI 0.14–1.8; p = 0.021). Using a mixed-effects model that incorporated a composite risk score rather than separate clinical and demographic variables revealed a similar association (β=0.89; 95%CI 0.14–1.6; p = 0.02). There was no association between CSF sST2 and background deterioration over time (β= −0.25; 95%CI −1.1–0.54; p = 0.53) (Figure 3d).
Discussion
Following nontraumatic subarachnoid hemorrhage, elevation of inflammatory plasma biomarkers were greatest in patients who develop new or worsening epileptiform activity. The relationship between systemic inflammation and cEEG changes did not extend to patients with non-epileptiform deterioration in cEEG background frequencies. Prior work has demonstrated a relationship between systemic inflammatory response syndrome and nonconvulsive seizures after SAH,6 and this study extends this association with systemic inflammation beyond seizure activity, to the occurrence of new or worsening EAs on the ictal-interictal continuum.24 Our primary finding demonstrates that sST2, a specific biomarker of inflammation, is uniquely associated with new or worsening EAs, whereas IL-6 and CRP are not.
sST2 is a decoy receptor for IL-33. When bound, it activates a pro-inflammatory response through the actions of toll-like receptor 4 (TLR-4), leading to immune cell population shifts.5,25 Like sST2, TLR-4 expression level on the cell surface of peripheral blood mononuclear cells (PBMC) is a risk factor predicting DCI and poor outcome at 3-months following SAH5,26 IL-6 is an interleukin secreted by T cells and macrophages that largely acts as a pro-inflammatory cytokine. CRP is an inflammatory protein synthesized downstream of IL-6.27 Both biomarkers have been studied widely in SAH and are believed to hold prognostic significance; however, there are a variety of conflicting studies calling this into question.3,19–22,28–32 While some evidence suggests sST2 may be a more reliable inflammatory marker in predicting DCI following SAH, additional studies are required to elucidate the principle inflammatory mediators.5 In predicting poor 90-day outcome, sST2 outperforms IL-6 or CRP.5
While new or worsening EAs and new cEEG background deterioration are also independently associated with developing DCI,8,13 their differential association with inflammation underscores that multiple pathogenic mechanisms lead to DCI.4 We speculate that the association of new or worsening EAs with inflammation may represent a pathophysiologic pathway to DCI distinct from that underlying the progression from non-epileptiform cEEG background deterioration. Inflammation-mediated changes to blood brain barrier permeability have been linked to seizures in experimental models,33,34 and septic patients without brain injury also have a higher likelihood of seizures.35 In this cohort, elevated sST2 in both plasma and CSF was associated with new or worsening EAs, even when patients with electrographic seizures were excluded. These results suggest that inflammation, like cerebral metabolism, may be associated with a range of epileptiform activity on the ictal-interictal continuum.36–39 It is not clear which is the initiating factor, the pro-inflammatory response or excitatory neurophysiological phenomena, or whether they are commonly related to another inciting factor. We accordingly focused on new or worsening EAs, once a 24-hour baseline period was established23 and examined associations adjusting for baseline admission severity.
Limitations of our study include sampling a subpopulation of all SAH patients undergoing cEEG that were enriched for greater severity, as well as those at a single institution so future studies are necessary to generalize these results to patients at other institutions. While the sample size is moderately sized, we did not conduct formal sample size or power calculations due to the retrospective nature of this cohort. While a larger cohort could reveal additional associations between worsening cEEG background deterioration in addition to new EAs, or between new EAs and additional biomarkers, the effect size for these nonsignificant relationships was small in our cohort, suggesting that the association of new EAs with sST2 is likely to be strongest in larger validation cohorts. Our dataset is unique in having carefully curated cEEG data as well as serial plasma and CSF samples; however, we were unable to obtain a plasma and CSF sample at each timepoint for every patient. Because sST2 early sampling occurred at mean 3.5±0.9 days, we were unable to determine whether elevation in sST2 preceded the development of new or worsening EAs, or vice versa. While we followed a strict protocol for handling and storing samples, our results may not generalize if these protocols are not followed. Future directions may include examining which factors contribute to inflammation following SAH; in prior studies, cEEG abnormalities14 and sST25 were associated with DCI and functional outcome independent of surgical treatment modality.
Our results demonstrate that elevation of the inflammatory marker sST2 in plasma and CSF is uniquely associated with new or worsening EAs following SAH. This association between inflammation and neurophysiologic markers of epileptiform brain dysfunction may represent an underlying mechanism contributing to DCI progression. Further studies are required to validate these findings and determine whether the acute inflammatory response after SAH acts as an upstream mechanism driving metabolically demanding cortical states.
Sources of Funding
NIH/NINDS K23 NS105950
R01 NS099209-03
AHA 17CSA 33440004
Heitman Neurovascular Research Foundation
Disclosures
S.F.Z has received grant support from Sage Therapeutics. A.B.P has received consulting fees from Penumbra, Microvention and Medtronic. W.T.K has received grant support and consulting fees from Biogen, Inc and from NControl Therapeutics, Inc. W.T.K. serves as a scientific advisory board member for Biogen, Inc. and NControl Therapeutics, Inc. W.T.K. has a US patent pending with NControl Therapeutics, Inc. E.S.R. receives grant support from the Department of Defense through a subcontract from Moberg ICU Solutions, Inc., and received financial compensation as a member of the scientific advisory board for UCB Pharmaceuticals and Ceribell Inc.
References
- 1.Eagles ME, Tso MK, Macdonald RL. Cognitive Impairment, Functional Outcome, and Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. World Neurosurg 2019;e558–62. [DOI] [PubMed] [Google Scholar]
- 2.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011;17:439–47. [DOI] [PubMed] [Google Scholar]
- 3.Chamling B, Gross S, Stoffel-Wagner B, Schubert GA, Clusmann H, Coburn M, et al. Early Diagnosis of Delayed Cerebral Ischemia: Possible Relevance for Inflammatory Biomarkers in Routine Clinical Practice? World Neurosurg 2017;104:152–7. [DOI] [PubMed] [Google Scholar]
- 4.Foreman B The pathophysiology of delayed cerebral ischemia. J. Clin. Neurophysiol 2016;33:174–82. [DOI] [PubMed] [Google Scholar]
- 5.Bevers MB, Wolcott Z, Bache S, Hansen C, Sastre C, Mylvaganam R, et al. Soluble ST2 links inflammation to outcome after subarachnoid hemorrhage. Ann Neurol 2019;86:384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claassen J, Albers D, Schmidt JM, De Marchis GM, Pugin D, Falo CM, et al. Nonconvulsive seizures in subarachnoid hemorrhage link inflammation and outcome. Ann Neurol 2014;75:771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Connor KL, Westover MB, Phillips MT, Iftimia NA, Buckley DA, Ogilvy CS, et al. High Risk for Seizures Following Subarachnoid Hemorrhage Regardless of Referral Bias. Neurocrit Care 2014;21:476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JA, Rosenthal ES, Biswal S, Zafar S, Shenoy AV, O’Connor KL, et al. Epileptiform abnormalities predict delayed cerebral ischemia in subarachnoid hemorrhage. Clin Neurophysiol 2017;128:1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zafar SF, Postma EN, Biswal S, Boyle EJ, Bechek S, O’Connor K, et al. Effect of epileptiform abnormality burden on neurologic outcome and antiepileptic drug management after subarachnoid hemorrhage. Clin Neurophysiol 2018;129:2219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labar DR, Fisch BJ, Pedley TA, Fink ME, Solomon RA. Quantitative EEG monitoring for patients with subarachnoid hemorrhage. Electroencephalogr Clin Neurophysiol 1991;78:325–32. [DOI] [PubMed] [Google Scholar]
- 11.Rots ML, van Putten MJAM, Hoedemaekers CWE, Horn J Continuous EEG Monitoring for Early Detection of Delayed Cerebral Ischemia in Subarachnoid Hemorrhage: A Pilot Study. Neurocrit Care 2016;24:207–16. [DOI] [PubMed] [Google Scholar]
- 12.Vespa PM, Nuwer MR, Juhász C, Alexander M, Nenov V, Martin N, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997;2699–710. [DOI] [PubMed] [Google Scholar]
- 13.Claassen J, Hirsch LJ, Kreiter KT, Du EY, Connolly ES, Emerson RG, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004;115:2699–710. [DOI] [PubMed] [Google Scholar]
- 14.Rosenthal ES, Biswal S, Zafar SF, O’Connor KL, Bechek S, Shenoy AV, et al. Continuous electroencephalography predicts delayed cerebral ischemia after subarachnoid hemorrhage: A prospective study of diagnostic accuracy. Ann Neurol 2018;83:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Wen D, Zheng J, Guo R, Li H, You C, et al. Predictive Accuracy of Alpha-Delta Ratio on Quantitative Electroencephalography for Delayed Cerebral Ischemia in Patients with Aneurysmal Subarachnoid Hemorrhage: Meta-Analysis. World Neurosurg 2019;126:e510–6. [DOI] [PubMed] [Google Scholar]
- 16.Chung DY, Oka F, Ayata C. Spreading depolarizations: A therapeutic target against delayed cerebral ischemia after subarachnoid hemorrhage. J. Clin. Neurophysiol 2016;33:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sánchez-Porras R, Zheng Z, Santos E, Schöll M, Unterberg AW, Sakowitz OW. The role of spreading depolarization in subarachnoid hemorrhage. Eur. J. Neurol 2013;20:1121–7. [DOI] [PubMed] [Google Scholar]
- 18.Hamming AM, Wermer MJH, Umesh Rudrapatna S, Lanier C, van Os HJA, van den Bergh WM, et al. Spreading depolarizations increase delayed brain injury in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab 2016;36:1224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muroi C, Hugelshofer M, Seule M, Tastan I, Fujioka M, Mishima K, et al. Correlation among systemic inflammatory parameter, occurrence of delayed neurological deficits, and outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery 2013;72:367–75. [DOI] [PubMed] [Google Scholar]
- 20.Fountas KN, Tasiou A, Kapsalaki EZ, Paterakis KN, Grigorian AA, Lee GP, et al. Serum and cerebrospinal fluid C-reactive protein levels as predictors of vasospasm in aneurysmal subarachnoid hemorrhage. Clinical article. Neurosurg Focus 2009;26:1–9. [DOI] [PubMed] [Google Scholar]
- 21.Kao HW, Lee KW, Kuo CL, Huang CS, Tseng WM, Liu CS, et al. Interleukin-6 as a prognostic biomarker in ruptured intracranial aneurysms. PLoS One 2015;10:e0132115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2010;13:339–46. [DOI] [PubMed] [Google Scholar]
- 23.Muniz C, Shenoy A, O’Connor KL, Bechek SC, Boyle EJ, Guanci MM, et al. Clinical development and implementation of an institutional guideline for prospective EEG monitoring and reporting of delayed cerebral ischemia CF. J Clin Neurophysiol 2017;33:217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirsch LJ, Laroche SM, Gaspard N, Gerard E, Svoronos A, Herman ST, et al. American clinical neurophysiology society’s standardized critical care EEG terminology: 2012 version. J. Clin. Neurophysiol 2013;30:1–27. [DOI] [PubMed] [Google Scholar]
- 25.Kakkar R, Lee RT. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov 2008;7:827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma C, Zhou W, Yan Z, Qu M, Bu X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J Neurol Sci 2015;359:67–71. [DOI] [PubMed] [Google Scholar]
- 27.Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front. Immunol 2018;9:754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopkins SJ, McMahon CJ, Singh N, Galea J, Hoadley M, Scarth S, et al. Cerebrospinal fluid and plasma cytokines after subarachnoid haemorrhage: CSF interleukin-6 may be an early marker of infection. J Neuroinflammation 2012;9:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMahon CJ, Hopkins S, Vail A, King AT, Smith D, Illingworth KJ, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J Neurointerv Surg 2013;5:512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen R, Bache S, Stavngaard T, Møller K. Plasma Levels of IL-6, IL-8, IL-10, ICAM-1, VCAM-1, IFNγ, and TNFα are not Associated with Delayed Cerebral Ischemia, Cerebral Vasospasm, or Clinical Outcome in Patients with Subarachnoid Hemorrhage. World Neurosurg 2019;128:e1131–6. [DOI] [PubMed] [Google Scholar]
- 31.Jeon YT, Lee JH, Lee H, Lee HK, Hwang JW, Lim YJ, et al. The postoperative C-reactive protein level can be a useful prognostic factor for poor outcome and symptomatic vasospasm in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg Anesthesiol 2012; 24:317–24. [DOI] [PubMed] [Google Scholar]
- 32.Romero FR, Cataneo DC, Cataneo AJM. C-reactive protein and vasospasm after aneurysmal subarachnoid hemorrhage. Acta Cir Bras 2014;29:340–5. [DOI] [PubMed] [Google Scholar]
- 33.Librizzi L, Noè F, Vezzani A, De Curtis M, Ravizza T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann Neurol 2012;72:82–90. [DOI] [PubMed] [Google Scholar]
- 34.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96(Part A): 70–82. [DOI] [PubMed] [Google Scholar]
- 35.Oddo M, Carrera E, Claassen J, Mayer SA, Hirsch LJ. Continuous electroencephalography in the medical intensive care unit. Crit Care Med 2009;37:2051–6. [DOI] [PubMed] [Google Scholar]
- 36.Struck AF, Westover MB, Hall LT, Deck GM, Cole AJ, Rosenthal ES. Metabolic Correlates of the Ictal-Interictal Continuum: FDG-PET During Continuous EEG. Neurocrit Care 2016;24:324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramaniam T, Jain A, Hall LT, Cole AJ, Westover MB, Rosenthal ES, et al. Lateralized periodic discharges frequency correlates with glucose metabolism. Neurology 2019;92:e670–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witsch J, Frey HP, Schmidt JM, Velazquez A, Falo CM, Reznik M, et al. Electroencephalographic periodic discharges and frequency-dependent brain tissue hypoxia in acute brain injury. JAMA Neurol 2017;74:301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vespa P, Tubi M, Claassen J, Buitrago-Blanco M, McArthur D, Velazquez AG, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol 2016;79:579–90. [DOI] [PubMed] [Google Scholar]