

Abstract
A robust biomarker screening and validation is crucial for overcoming the current limits in the clinical management of infectious diseases. In this chapter, a general workflow for metabolomics is summarized. Subsequently, an overview of the major contributions of this omics science to the field of biomarkers of infectious diseases is discussed. Different approaches using a variety of analytical platforms can be distinguished to unveil the key metabolites for the diagnosis, prognosis, response to treatment and susceptibility for infectious diseases. To allow the implementation of such biomarkers into the clinics, the performance of large-scale studies employing solid validation criteria becomes essential. Focusing on the etiological agents and after an extensive review of the field, we present a comprehensive revision of the main metabolic biomarkers of viral, bacterial, fungal, and parasitic diseases. Finally, we discussed several articles which show the strongest validation criteria. Following these research avenues, precious clinical resources will be revealed, allowing for reduced misdiagnosis, more efficient therapies, and affordable costs, ultimately leading to a better patient management.
Keywords: Metabolomics, Infectious diseases, Diagnostics, Biomarkers, Biomarker discovery
Introduction
The new challenge in the study of the biological processes is to understand the interactions among all biological agents (genes, transcripts, proteins, and metabolites). The metabolome, defined as the set of all metabolites (Oliver et al. 1998), represents the closest level to the phenotype which can be directly correlated with the status of the organism. In other words, changes in endogenous and environmental factors are likely to impact on the metabolome, which acts as a major chemical bridge connecting the environment with the different levels of a biological system (Fig. 7.1).
Fig. 7.1.
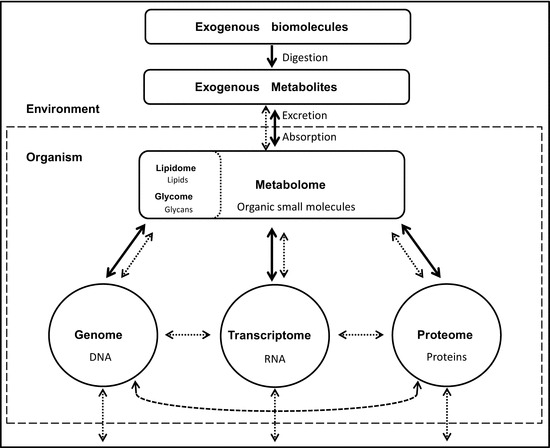
General scheme showing the major mass fluxes (normal arrows) and molecular interactions (dashed arrows) between the different systems of an organism and its environment
The interactions among organisms are uncountable. However, only a few of them produce a dysregulation of homeostatic processes, eventually leading to disease. Infectious diseases are one of the leading causes of mortality (WHO 2017a), and a better management of patients is undoubtedly necessary. Unfortunately, some diagnostic methods can be slow and tedious and lack adequate specificity and sensitivity (Brand et al. 2010; Jain 2010). In this context, metabolomics analyses can be performed to screen for metabolic biomarkers, which display novel potential advantageous features such as more accurate diagnosis, dynamic disease evaluation, non-invasive sampling, or personalized treatment assessment (Mayeux 2004). A biomarker can be defined as the biological molecule which is able to create a predictive model that can be used to classify a new sample or person into a specific group (e.g., healthy vs. diseased) with enough sensitivity and specificity (Xia et al. 2013). This task is technically challenging (Xia et al. 2013). Note that a PubMed search from 1990 to 2009 for the term “biomarker” revealed a total of 441,510 published articles, an enormous academic investment not translated into the expected improvements for patient care (Ptolemy and Rifai 2010).
Herein, we present a robust analytical workflow for biomarker discovery and validation with special attention to experimental design, sample treatment, analytical platforms, and data analysis. Subsequently, we will focus on the contributions of metabolomics to the identification of biomarkers in the context of infectious diseases. Table 7.1 depicts the strenghts, weaknesses and typical applications in metabolomics of the main analytical techniques.
Table 7.1.
Overview of the metabolome coverage of the main analytical techniques
| Analytical technique | Metabolome coverage | Compound requirements | Polarity | Examples |
|---|---|---|---|---|
| GC-MS | Low | Thermostable compounds with acidic H or volatiles | High–mid | Amino acids, small organic acids, carbohydrates, fatty acids, terpenes, cholesterol derivatives |
| LC-MS | High | Soluble compounds | Depending on column | Amino acids, oligopeptides, small organic acids, carbohydrates, phosphate compounds nucleosides, lipids, nucleotides, carnitines |
| CE-MS | Medium | Charged compounds | Only polar | Amino acids, oligopeptides, small organic acids, phosphate compounds nucleosides, nucleotides, carnitines |
| 1H-NMR | Low | Abundant compounds with non-exchangeable protons | – | Small organic acids, carbohydrates, amino acids |
| 31P-NMR | Low | Abundant phosphorus-containing compounds | – | Nucleotides, nucleosides, polyphosphate, phosphate enzyme cofactors, sugar phosphates |
Metabolomics Workflow for Biomarker Discovery
Sensu stricto, metabolomics corresponds with a non-target analysis—also named as global profiling or fingerprinting (Fiehn 2002)—in which the total metabolome is measured and the data are treated following differential analysis methodologies. Metabolomics is ideal for biomarker discovery, since no previous hypotheses are needed. Nevertheless, before they can be routinely used in clinics, biomarkers need to be validated which, from the analytical point of view, requires the performance of a classic target analysis.
The fingerprinting workflow for biomarkers screening (Fig. 7.2) encompasses several steps such as experimental design, sample treatment, analytical measurement and data processing, which will be below described in detail. Note that each step represents a potential source of error which may be accumulated. Special attention should be paid to the biospecimen selection and data meta-analysis, the two major bottlenecks in biomarker screening (Voskuil 2015).
Fig. 7.2.
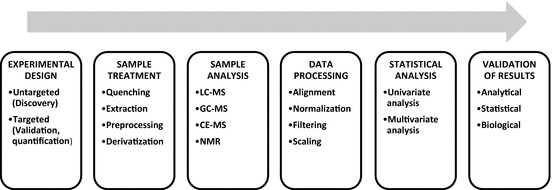
General workflow in a metabolomics experiment for biomarker discovery and validation
Experimental Design
A certain amount of variability is inherent to biology. However, ideally, the characteristic under study should be the only source of differences among the sample groups. Therefore, group selection becomes critical for a successful differential analysis. In order to minimize this problem, the size of the study cohort can be increased to mathematically dilute the interindividual variability. Nevertheless, a long analytical sequence can cause a problem in the signal stability. Although there are several normalization strategies (Godzien et al. 2013), it is highly recommended to periodically analyze quality controls (QC) through the analytical batch to monitor any potential bias. Ideally QCs should be a pool of aliquots obtained from each biological sample. However, when this is not possible, other alternatives are acceptable (Dunn et al. 2012; Godzien et al. 2015).
Sample Treatment
All the samples should be obtained following a strict protocol. What is more, their transport and storage are crucial to avoid metabolite degradation and subsequent potential bias between batches. Specific standard operating procedures (SOP) should be adapted to each type of matrix, although samples should be generally preserved at −80 °C unless otherwise recommended. Another turning point for sample stability is a fast and efficient metabolic quenching. Enzymes can be inactivated by the addition of organic solvents, or abrupt temperature, or pH variations (Mushtaq et al. 2014). Typical metabolic quenching strategies comprise the use of liquid nitrogen or cold methanol/water mixtures (Dettmer et al. 2007). However, caution must be taken as aggressive quenching methods can damage plasma membranes, causing potential metabolite leakage (Winder et al. 2008). To overcome this, a softer quenching methodology based on a fast vacuum filtration of cells and subsequent addition of a quenching solution has been proposed (da Luz et al. 2014), although its relatively longer quenching time can alter the metabolome.
The next step along sample preparation is metabolite extraction. If the sample is a solid (e.g., tissue), matrix disaggregation is previously needed. In a fingerprinting approach, the selected extraction solvent should solubilize as many metabolites as possible. Double extraction with both polar and non-polar solvents is always recommended to enhance the metabolite coverage (note that independent series of extractions with such solvents are always more efficient than a single extraction with the mix). On the other hand, in a target analysis, the solvent of interest is the one that maximizes the analyte recovery. Liquid–liquid and solid-phase extractions are commonly used methods for liquid samples such as biofluids and cell culture supernatants. Furthermore, samples can be derivatized to modify the chemical properties of analytes. Derivatization is usually required for gas chromatography (GC) analysis which needs thermally stable and volatile metabolites.
Analytical Measurement
In metabolomics fingerprinting, the main analytical platforms are nuclear magnetic resonance (NMR) and mass spectrometry (MS). NMR spectroscopy is a nondestructive technique which allows in vivo evaluation of the metabolism. However, the low sensitivity limits its applicability. On the other hand, MS is usually coupled to a separation technique as far as it is not possible to perform the direct analysis of the metabolome due to its chemical complexity. A mass spectrometer is composed by a source (where the molecules are ionized), an analyzer (in which the metabolites are separated by their m/z ratio), and a detector (which register the number of counts per hit). Different types of analyzers are available for metabolomics, being the time of flight (TOF) one of the most used, considering that it renders mass accuracy with only several part per million (ppm) of error. The quadrupole time of flight (QTOF) allows ion separation and subsequent collision-induced dissociation for MS/MS identification analysis. Finally, the triple quadrupole (QqQ) is ideal for target quantification by multiple reaction monitoring (MRM) mode.
GC separation of metabolites is based on their different vapor pressure and their affinity for the stationary phase of the column. Generally, low bleeding capillary columns are used in GC-MS, being the wall-coated open-tubular columns the most popular option (Mirnaghi and Caudy 2014). In metabolomics, the most frequently used stationary phases are DB-5 and DB-50 ((5%-phenyl)-methylpolysiloxane and (50%-phenyl)-methylpolysiloxane, respectively) (Rojo 2014). The main types of ion sources in GC-MS are electron ionization (EI) and chemical ionization (CI). GC-EI-MS renders well-separated efficient peaks, with high reproducibility in the MS spectra. These advantageous features have promoted the development of GC-MS spectral libraries (e.g., FiehnLib, NIST 14), which are extremely useful for metabolite identification. GC-MS metabolite coverage is limited to thermally stable and volatile compounds such as esters, terpenes, certain alcohols, aldehydes, thiols, or aromatic compounds (Rowan 2011). To overcome this limitation, chemical derivatization procedures are routinely applied (e.g., silylation with N,O-Bis(trimethylsilyl)trifluoroacetamide and trimethylchlorosilane for sugars, amino acids, amines, amides, fatty acids, etc.).
In liquid chromatography (LC), MS coupled systems samples are injected into a packed column containing a stationary phase. Metabolites are separated due to their different partition coefficients between the stationary phase and the eluotropic mobile phase (either isocratic or in a gradient). Based on the stationary phase, it can be distinguished among reverse-phase LC (e.g., C18, C8), hydrophilic interaction liquid chromatography (HILIC), and normal-phase LC. Reverse-phase LC is the gold standard technique for the analysis of hydrophobic and medium polarity compounds (e.g., lipids), whereas HILIC is preferred for polar metabolites although its reproducibility is challenging. Normal phase is not commonly used in metabolomics since highly hydrophobic mobile phases are required (e.g., hexane). The subsequent ionization of the metabolites is generally driven by an electrospray source either in positive or negative mode. The use of a QTOF analyzer enables metabolite annotation by comparing their accurate masses with public databases (e.g., HMDB, METLIN, and LIPID MAPS) and eventually their structural elucidation by MS/MS analysis.
Capillary electrophoresis (CE) comprises a wide variety of electrokinetic separation methods, from which capillary zone electrophoresis is the most commonly used in metabolomics (Zhang et al. 2017). Such technique is based on the application of high voltages between two electrodes, which are connected by a buffer solution that flows through a capillary. Net charges are produced in the capillary inner wall, which interacts with the oppositely charged ions contained in the buffer solution. When voltages are applied, ions migrate toward the oppositely charged electrode, carrying solvent molecules. Consequently, a plug-like flow defined as electroosmotic flow is produced. Electrophoretic mobility is an additional force that appears only in charged compounds and produces their displacement toward the electrode with opposite charge. CE-MS instruments can operate in two different modes depending on the charge of metabolites and MS detector polarity. Cation separation is mainly performed in bare fused silica capillaries by application of high voltage with positive MS ionization. It is ideal for metabolites such as nucleotides, nucleosides, amino acids, acylcarnitines, and other amines and amides. On the contrary, electrophoresis with reversed polarity required electroosmotic flow suppression or inversion by neutral or positively coated (e.g., polybrene-dextran sulfate-polybrene) capillaries (Ramautar et al. 2011). Although it is highly interesting for the analysis of short-chain organic acids or sugar phosphates, reversed polarity CE-MS capabilities have been limited by certain technical difficulties that occur whenever positive potential is applied to the stainless steel needle (e.g., a special ESI platinum needle is required under ESI in negative ionization mode).
In NMR, radio-frequency pulses are applied to the liquid samples which are placed in a magnetic field. Such pulses are capable of altering nuclei with non-zero magnetic momentum as 1H or 13C. Since the specific chemical environment of a nucleus determines its spin momentum dynamic evolution and coupling with other nuclei, precise acquisition information about the molecular structure information is acquired. Hence, accurate metabolite identification and quantification can be performed. Advantages of NMR are highly reproducible spectra, fast speed acquisition rates, minimum sample preparation, and relatively low cost of analysis. NMR can also be used for the analysis of phosphorus-containing metabolites, rendering relatively simplified spectra. These metabolites are either indicative of various cellular processes, energy metabolism (e.g., ATP, GMP, NADPH), central carbon metabolism processes (e.g., sugar phosphates, phosphoenol-3-pyruvate), or structural and signaling functions (e.g., phospholipids). To increase the sensitivity and metabolome coverage of NMR, chemical derivatization with 13C and 15N isotopic tags has been described (Gowda et al. 2012).
Data Processing
Data processing workflow is technique-dependent. Generally speaking, the first task once the analysis is finished is to check whether all profiles with the corresponding internal standard peaks and QC samples show a good repeatability and if the pressure or current curves are adequate. To deliver high-quality results, the reproducibility and suitability of every sample must be verified. Traceability of the analytical batch through an internal standard or based on QCs are the two main strategies to ensure a stable equipment performance.
A MS chromatogram/electropherogram contains three main pieces of information: MS spectrum, time, and abundance (ion current). Based on them, deconvolution algorithms reprocess the signal, and, according to the isotopic pattern distribution across the time, they create the metabolic features which ideally correspond to a chromatographic peak characterized by a defined area, elution time, and m/z. A particular case of deconvolution is done in GC-MS, in which the identification of the metabolic features is done by comparing the retention times and spectra from empirical data to reference libraries. Subsequently, features are aligned and a matrix is created. Alignment should be performed with a certain tolerance in retention time and mass windows to correct small fluctuations.
In some cases, normalization may be required to minimize alterations in the stability of analytical platforms. Data filtering is necessary to eliminate spurious signals produced after deconvolution (Godzien et al. 2015). First, a filtration by presence in QCs is performed. Secondly, filtration by presence in at least a minimum number of samples of one of the groups is also performed. As only stable signals pass the filter, random artifacts are reduced. As a result, only the metabolites that are strongly represented in the sample set are considered for statistical analysis. Data scaling is desirable to adjust the importance assigned to the elements of the data in fitting the model. By scaling, the weight of each variable is adjusted with a scaling factor estimated by either a dispersion criterion or a size measure.
Differential analysis statistical methods are used in fingerprinting analysis for biomarker discovery, considering each metabolite as a variable. Both univariate and multivariate statistical tests can be applied. Univariate models such as student’s t-test contemplate each metabolite as an independent variable, whereas multivariate can measure the influence of each metabolite on others. Regarding to multivariate tests, both unsupervised and supervised analysis can be performed. Principal component analysis (PCA) is the most common unsupervised model based on which any potential sample outlier can be detected. What is more, an adequate reproducibility of the analyses can be easily assessed by observing tight clustering of QCs in PCA plots. On the contrary, supervised analyses aim to generate models with a priori assigned groups. The most common supervised multivariate models are partial least squares regression discriminant analysis (PLS-DA) and orthogonal PLS-DA (OPLS-DA) analyses. To overcome the statistical bias induced by model overfitting and finding spurious correlations, different statistical methodologies such as permutation tests, cross-validation, or jackknifing have been used. Note that a detailed explanation of these algorithms is out of the scope of this chapter.
Metabolomics Workflow for Biomarker Validation
In a broad sense, validation is the assessment of an action, instrument, or technique to establish that it is delivering the intended outcome. Considering the multidisciplinary nature of the metabolomics experiment, different validation steps must be performed across the whole workflow. From the analytical point of view, validity is assessed as reporting several parameters for a certain method, such as the accuracy, precision, linearity, and limit of quantification. With respect to a mathematical model, validation ensures that overfitting is absent, which can be calculated by applying several tests mentioned above. Regarding the biological interpretation, validations aim to confirm the initial hypothesis using several strategies like detecting the same group of metabolites with a different technique in an independent set of samples, integrating multi-omics information, or developing ad hoc experiments.
Focusing on biomarker discovery, there is a relatively little consistency in the way of validating candidate biomarkers (Xia et al. 2013). Besides their identification by structural elucidation or standard spiking, to validate a biomarker means to (1) demonstrate that it is still significant in a second sample cohort, bigger than the previous one and with a broader interindividual variability, and (2) to assess on it the discriminant capability of the candidate by reporting its sensitivity and specificity which should be calculated based on accurate concentration values. In other words, a strong biomarker with a potential use in clinics’ daily routine should have a small ratio of false negatives (misdiagnosed diseased patients) and false positives (misdiagnosed healthy patients). A way to visualize both is performing the calculation of the receiver operator characteristic (ROC) curve. In this type of diagrams, specificity is plotted against selectivity for a given set of concentration values in order to determine the threshold between disease and healthy status. The area under the curve (AUC) of the ROC curve defines the strength of the biomarker. Generally, a strong biomarker displays AUC values bigger than 0.8, whereas fair biomarkers account for values between 0.8 and 0.6. An AUC below 0.6 does not satisfy the quality criteria for a good biomarker.
Any analytical platform is potentially suitable to measure accurate concentration values by using the classical quantification methods of standard curve, standard addition, or internal standard. Nevertheless, due to its superior capabilities in terms of sensitivity, the gold standard technique for quantification is a triple quadrupole (QqQ-MS), coupled to LC or GC and operating in MRM which reaches limits of detection of part per billion (μg/L). In brief, MRM mode is based on a selective fragmentation of the molecular ion of the MS spectra of the candidate, respectively, using its two most abundant fragments as quantification and qualification transitions. Thanks to MRM it is possible to avoid background MS noises.
Biomarkers for Infectious Diseases: Current Landscape
The analysis of biofluids or tissues from infected patients represents the most accurate methodology to describe the truly metabolic changes associated with disease. However, since sample availability is generally compromised in acute, low prevalent, or hazardous diseases, other experimental designs have been conceived, being most of these studies performed in human disease-mimicking models (Fig. 7.3). Although substantial differences between the metabolic profiles of disease models and human patients have been reported (Himmelreich et al. 2005; Salek et al. 2007), models show several advantages such as an enhanced evaluation of the disease prognosis. Model-based experiments comprise studies wherein animals are challenged with human or humanlike pathogens and in vitro studies wherein infection is performed under host cell lines. Other studies, while not containing truly present host–pathogen interactions, are capable of elucidating considerable aspects of the infectious process; these include, among others, experiments wherein a host–pathogen interaction has been previously established (e.g., analysis of cultures from clinical isolates) and studies wherein host–pathogen interactions are mimicked by means of challenging either host or pathogen cells with chemical conditions characteristics of disease (e.g., hypoxia (Eoh and Rhee 2013), addition of bacterial lipopolysaccharide (Laiakis et al. 2012)). Although a clear distinction can be observed between biomarker identification and unveiling disease’s mechanism, it should be highlighted that metabolomics studies which aim to understand the pathogenesis of infectious diseases can also reveal potential biomarkers. Due to the large number of the studies in this area, we focused on selected series of examples which illustrate an overview of the state of the art in the field.
Fig. 7.3.
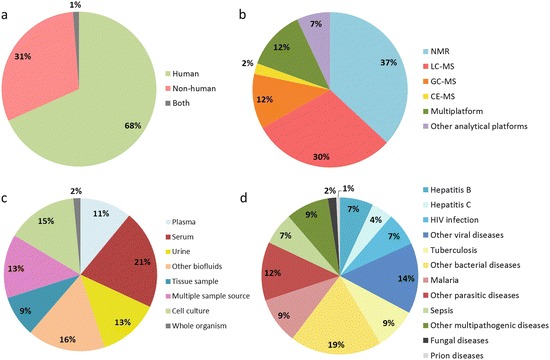
Classification of metabolomics experiments in infectious diseases attending to (a) host nature, (b) analytical technique, (c) sample type, and (d) disease under study. Items in pie charts are displayed clockwise
Biomarker Discovery
Metabolomics of Viral Diseases
The area of metabolomics of viral diseases has been subject of extensive study and review (Sanchez and Lagunoff 2015). In this book chapter, we will describe the major applications of metabolomics in three relevant viral diseases [human immunodeficiency virus infection (HIV), hepatitis C, and dengue fever]. Since the beginning of AIDS epidemics, HIV has infected more than 70 million people, from which 36.7 million live with HIV/AIDS worldwide (WHO 2017d). Although diverse therapeutic treatments have been developed, more than 1 million deaths per year are still caused by AIDS-related diseases (WHO 2017d). The first metabolomics studies regarding to HIV aimed to find metabolic biomarkers for HIV infection/response to antiretroviral (ART) treatment. In this context, Hewer et al. (2006) studied the serum metabolic profiles from ART-treated/ART-naïve HIV+ patients and HIV—controls by 1H-NMR. Changes in the levels of glucose, lipids, amino acids, glycerol, choline, and lipoproteins were observed, suggesting potentially diagnostic capabilities of this technique, alternative to the current gold standard (Western blot) (Syed et al. 2005). Other studies have been conducted to find a fast and non-invasive HIV diagnostic test; Ghannoum et al. (2013) compared the GC-MS and LC-MS metabolic profiles of oral lavage from healthy subjects and ART-naïve and ART-experienced patients. Interestingly, an increased phenylalanine/tyrosine ratio was found in ART-naïve patients, opening doors for new oral biomarkers. In contrast, a notable number of studies regarding the study of HIV-related conditions have been reported; since secondary lung infections are common among HIV+ patients, Cribbs et al. (2014) used LC-MS to study the differences between metabolic profiles of bronchoalveolar lavage fluid (BALF) of HIV+ patients and controls. Groups notably clustered in OPLS-DA models, and important altered levels of phospholipids, tripeptides, and pyochelin (a siderophore related with the opportunistic pathogen Pseudomonas aeruginosa) were observed. Additionally, HIV-related neurocognitive impairment has also been an object of study by Cassol et al. (2014). They performed a multiplatform, combined GC-MS and LC-MS analysis of the cerebrospinal fluid (CSF) of HIV+ individuals and controls, finding correlations between the levels of ketone bodies (1,2-propanediol, β-hydroxybutyrate), neurotransmitters (glutamate, N-acetylaspartate), and myoinositol with neurocognitive worsening.
Hepatitis C is a highly prevalent disease, affecting more than 71 million people worldwide (WHO 2017b). This liver disease is caused by the hepatitis C virus (HCV), a flavivirus comprehending six different genotypes. HCV infection usually displays mild symptoms in the acute phase of the disease and considerably high rates of chronification (85% of infected patients) (Pawlotsky 2004). Chronic HCV infection generates a progressive fibrosis of the liver, which is classified in four stages by the METAVIR system (F0 for absence of fibrosis, F1–2 for mild fibrosis, F3 for advanced fibrosis, and F4 for fully developed cirrhosis) (Bedossa and Poynard 1996). Such deterioration can ultimately lead to severe conditions, such as cirrhosis and hepatocellular carcinoma (HCC) (Pawlotsky 2004). Due to the considerable prevalence and such potentially fatal outcomes, HCV infection has been considerably studied by metabolomics (du Preez and Sithebe 2013). Additionally, given the invasiveness of the current gold standard for assessing the progression of HCV-related liver fibrosis (liver biopsy), many studies have focused on the development of alternative methods to assess disease progression. In this context, Zhang et al. (2006) conducted a targeted method using LC to quantify amino acids in plasma samples of HCV+ patients, generating models based on amino acid ratios from which a combined model ((Phe)/(Val) + (Thr + Met + Orn)/(Pro + Gly)) was capable of discriminating F0–2 from F3–F4 patients (ROC AUC = 0.92, 95% CI: 0.84–1.00) and F0–F3 from F4 patients (ROC AUC = 0.99, 95% CI: 0.96–1.01). Additionally, Cheng et al. (2012) performed a multiplatform LC-MS and GC-MS analysis of plasma metabolites comparing advanced fibrotic patients with hepatitis C patients not showing this condition. Notable alterations in primary bile acids (glycocholate, taurocholate), long-chain fatty acids (stearidonate, palmitoleate), and cysteine were observed. In another study conducted by Embade et al. (2016), a 1H-NMR analysis on serum samples from fibrotic and non-fibrotic patients was performed, finding significant alterations in amino acids and lipoproteins, among others. From such alterations, a multivariate model based on choline, acetoacetate, and LDL1 levels which effectively discriminated between F0 and F4 patients (ROC AUC = 0.922, 95% CI: 0.85–0.97) was generated. On the other hand, potential biomarkers to predict the treatment outcome of hepatitis C have also been studied. Before direct-acting antivirals (DAAs)—the current first-line treatments—were developed, predictive models of the outdated treatment comprising alpha-interferon plus ribavirin were generated (Saito et al. 2013). Following a similar line, Ceccotti et al. (2016) performed a longitudinal study wherein 1H-NMR was applied to serum samples from HCV+ patients before and after successful DAA therapy, finding differences in the levels of formate, acetate, and methionine between baseline and 12th week post-treatment samples from patients who achieved a sustained virological response.
Dengue fever (DF) is a widespread disease with an incidence of approximately 390 million people per year worldwide (Bhatt et al. 2013). DF is caused by dengue virus (DENV), from which four different serotypes have been described—DENV-1 to DENV-4. This mosquito-borne disease is usually asymptomatic (80% of cases) or manifested through mild symptoms (e.g., fever, rash, joint pain) (Martina et al. 2009). However, approximately 5% of DF cases progress to more severe forms, namely, dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), which seriously affect the cardiovascular system of patients, potentially leading to death (Martina et al. 2009). Given the heterogeneity of possible disease outcomes and the lack of an effective biomarker for prediction of a severe prognosis (John et al. 2015), different metabolomics studies have been performed to elucidate the mechanism of this disease and to find possible diagnostic and prognostic biomarkers. Preliminary studies of DENV-infected cell cultures have been described, such as the performance of a combined 1H-NMR and direct infusion MS analysis of DENV-infected endothelial cell lines (EA.hy926) by Birungi et al. (2010). These analyses revealed metabolic alterations in the levels of amino acids, fatty acids, and metabolites of the tricarboxylic acid cycle, showing a full separation among DENV serotypes in PCA plots, suggesting differences in the disease mechanism for each serotype. In another study from Fontaine et al. (2015), a combined LC-MS and GC-MS analysis of infected human foreskin fibroblasts unveiled alterations in the host central carbon metabolism (particularly in glycolytic intermediates). Different studies analyzing the serum, plasma, and urine of DF patients have been performed. For example, Shahfiza et al. (2015) identified by 1H-NMR a series of relevant gender-dependent alterations of urinary metabolites during DF (e.g., acetaminophen, glycine, betaine, creatinine, creatine phosphate, N-methylhydantoin, and β-hydroxybutyrate), while El-Bacha et al. (2016), using the same analytical technique, found plasma evidences for a DF-induced liver dysfunction (e.g., branched-chain amino acids, LDL, and VLDL). LC-MS and subsequent MS/MS ex vivo analyses have also revealed important contributions to DENV diagnostics. In this context, Voge et al. (2016) identified a differential metabolic profile between patients infected with DENV-1 and DENV-2 (octadecatrienal, octadecadienal, octadecadienoic acid, octadecenamide, sphingosine, 25-hydroxy-hexadehydrovitamin D3, 1,25-dihydroxycholesterol, and 3-deoxy-25-hydroxyvitamin D3, among others). Another LC-MS study, performed by Cui et al. (2016), suggested candidate biomarkers for the prediction of the progression of mild DF to DHF/DSS (e.g., kynurenine, serotonin, bilirubin, oleamide, 13-E-docosenamide, deoxyinosine, and several glycerophospholipids) from which particularly serotonin, in combination with γ-interferon, was able to build a robust predictive model (ROC AUC = 0.92).
Metabolomics of Bacterial Diseases
Prokaryotic cells encompass the biggest set of living organisms, comprising thousands of eubacterial and archaeal species. Interestingly, from the human health point of view, some of these organisms can potentially establish a symbiotic or a pathogenic relationship. In some cases, such relationship can be tracked trough different specific non-eukaryotic metabolites which unfortunately are not always easy to measure. We overview this vast area focusing on selected types of bacterial diseases.
Urinary tract infection (UTI) is a common disease, which is potentially capable of leading to severe excretory system complications. Although the etiology of UTI can be diverse, the major causative pathogen is uropathogenic Escherichia coli (UPEC) (up to 80% of cases) (Ronald 2003). Conventional bacterial identification tests display several limitations, suggesting the necessity of a better diagnostic tool (Pearle 2004). Candidate biomarkers for the potential diagnosis of UPEC-produced UTI were suggested by Lv et al. (2011), which compared the LC-MS urinary metabolic profile between both female UTI patients and healthy individuals. PLS-DA analysis notably discriminated these two sample groups, from which the metabolites most contributing to the group separation were identified as diphosphomevalonate, N-acetylneuraminic acid, tetrahydroaldosterone-3-glucuronide, 3-carboxy-1-hydroxypropylthiamine diphosphate, 6-ketoprostaglandin F1α, cortolone-3-glucuronide and 21-hydroxypregnenolone, desmosine, and pregnanediol-3-glucuronide. Interestingly, some UTI diagnostic biomarkers have been validated (see Sect. 7.4.2).
Clostridium difficile is a gram-positive, frequently nosocomial pathogen, which affects approximately 0.5–3 million people per year (Rojo et al. 2015). Clostridium difficile infection (CDI) affects the gastrointestinal tract and constitutes a potentially fatal disease which shows high rates of recurrence (20% after an initial infection, 40% after a second episode, and 60–80% in subsequent episodes) (Kao et al. 2016). Given the shortcomings derived from the current tests for the differential diagnosis or prediction of recurrent CDI (Burnham and Carroll 2013), several metabolomics studies have been focused on the identification of such diagnostic and prognostic biomarkers. For example, Kao et al. (2016) performed a combined non-targeted and targeted 1H-NMR analysis of the urine of patients with symptomatic CDI and healthy controls. Patients were grouped attending to the recurrence of CDI (one, two, three, or more than three episodes). When comparing patients and controls, choline was identified as the metabolite most contributing to the discrimination of both groups, while other relevant metabolites (e.g., hippurate, 3-methylhistidine, dimethylamine, trimethylamine, ethanolamine, leucine, valine, pyroglutamate, asparagine, threonine, and glutamine) were also identified. Differences between patients showing primary CDI and recurrent CDI infections were also clearly observed in PCA plots: recurrent patients showing more than two CDI episodes were effectively discriminated from the rest of CDI patients. Metabolites most contributing to this separation were identified as trans-aconitic acid and histidine, although other metabolites such as creatine, sucrose, O-acetylcarnitine, ethanol, tartrate, acetate, asparagine, 3-hydroxyvaleric acid, and carnitine were also found to be altered. In the same framework, Allegretti et al. (2016) performed a target LC-MS analysis directed toward the evaluation of bile salts on stool samples from patients with primary CDI and recurrent CDI and healthy controls. Statistically significant differences in the bile acid profile were found across the different groups under study; significantly decreased levels of litocholate and deoxycholate were found in all CDI patients when compared with controls. Similarly, the levels of deoxycholate were also found to be increased in primary CDI when compared to recurrent CDI patients. Recurrent CDI patients also showed higher abundance of primary bile salts and lower abundance of secondary bile salts than other groups, as well as higher levels of cholate and chenodeoxycholate compared with controls. Interestingly, the ratio of deoxycholate to glycoursodeoxycholate plus deoxycholate was identified as a potential biomarker combination model for the differential diagnosis between first and recurrent CDI. Regarding the search for potential biomarkers of CDI susceptibility, Dann et al. (2015) performed a longitudinal study wherein untargeted metabolomics were applied to stool samples of either patients at risk of developing CDI or patients being tested for CDI, finding increased concentrations of γ-aminobutyric acid in patients who developed CDI.
Enteric fever is a disease caused by the gram-negative bacteria Salmonella enterica, serotype typhi (typhoid fever), and Salmonella enterica, serotype paratyphi (paratyphoid fever). Enteric fever worldwide causes more than 25 million new infections per year (Buckle et al. 2012). Unfortunately, the current standard diagnostic tests for enteric fever lack of sensitivity and specificity, thus requesting the development of a potentially useful diagnostic biomarker (Parry et al. 2011). Furthermore, as both pathogens may display similar clinical symptoms and at the same time different antimicrobial resistance (Zaki and Karande 2011), biomarkers for the differential diagnosis may be potentially useful for a correct clinical practice. Näsström et al. (2014) compared the plasma metabolic profile of Salmonella typhi-/paratyphi-infected patients and healthy volunteers through GC-MS analysis. Although partial overlap in PCA scores was observed when comparing all groups, OPLS-DA analyses revealed correct group clustering, suggesting the potential discriminatory capacity of the analyzed metabolites. Subsequent selection of the most statistically significant metabolites identified a combination of six metabolites (gluconic acid, phenylalanine, ethanolamine, pipecolic acid, and two carbohydrates), which could discriminate between typhi and paratyphi patients (ROC AUC = 0.796), as well as between these patients and controls (ROC AUC = 0.923 and 0.948 for typhi and paratyphi patients, respectively).
The potential role of metabolomics in the diagnosis of lethal bacterial diseases can be exemplified by the studies of melioidosis. This disease, caused by the gram-negative bacteria Burkholderia pseudomallei, can have high mortality rates (up to 19%) (Currie et al. 2000). Melioidosis symptoms range from local abscesses to pneumonia or sepsis. Given the sensitivity lack of the current gold standard for the diagnosis of this disease (observation in cell cultures) (Limmathurotsakul et al. 2010), Lau et al. (2016) performed an untargeted LC-MS analysis of the plasma from patients with either melioidosis or bacteremia (distinct from B. pseudomallei) and healthy volunteers. PCA scores plot revealed notable clustering among the disease groups. About melioidosis patients, the most significant metabolites were found to be acylcarnitines, sphingomyelins, lysophosphatidylethanolamines, and phosphatidylcholines when compared to other disease groups/controls. Interestingly, sphingomyelin (d18:2/16:0) was found to be the best discriminator of melioidosis when compared with controls or other bacteremic patients.
Tuberculosis (TB) is another potentially lethal disease, which is primarily caused by Mycobacterium tuberculosis. Despite being treatable and curable, this widespread mycobacterial disease constitutes the first mortality cause among all infectious diseases, with estimated incidence and mortality rates of 10.4 and 1.4 million people per year worldwide, respectively (WHO 2017d). Given that infection with M. tuberculosis is usually acquired through bacilli inhalation, TB is most likely primarily developed in the lungs, causing pulmonary disease (active pulmonary TB). However, most of the infections remain subclinical, causing a latent infection (latent TB) that can progress to active pulmonary TB (10% of latent TB cases) (Fatima et al. 2017). In addition, M. tuberculosis can affect a wide range of extrapulmonary organs and tissues, such as the CNS, the genitourinary tract, and the lymphatic system (lymph node TB or LNTB) (Fatima et al. 2017). For instance, CNS invasion by M. tuberculosis can cause a varied range of serious conditions associated with high mortality and disability rates, such as tubercular meningitis (TBM), which is more common in children. In contrast, genital tuberculosis (GTB) specially constitutes a notable factor for women infertility. Active TB must be treated for at least 6 months, being the first-line treatment a combination of isoniazid, rifampicin, ethambutol, and pyrazinamide. Unfortunately, multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) constitute a major concern for public health, mainly in developing countries (Günther 2014). Given that current standard diagnostic methods (such as identification of the bacteria in cell cultures) show important drawbacks (time-consuming, low specificity, and sensitivity), most studies have focused on the development of biomarkers for the diagnosis of different forms of TB. To find such diagnostic biomarkers for active TB diagnosis, different experiments have compared the metabolic profile of active TB patients with controls. For instance, du Preez and Loots (2013) performed a non-targeted GC-MS analysis of sputum samples from patients suspected of having TB. After outlier removal, clustering of active TB patients and healthy controls was observed in PCA score plots. Altered levels of carbohydrates (glucosamine, N-acetylglucosamine, 2-deoxy-erythro-pentitol, glucopyranose, mannopyranose, gluconic acid δ-lactone), fatty acids (nonadecanoic acid, oleic acid, sebacic acid, C17:1ω7c), and other compounds (citramalic acid, glutaric acid, ethane, butanal, γ-aminobutyric acid, 3,4-dihydroxybutanoic acid, and normetanephrine) were found in TB-positive patients. In the same line, Frediani et al. (2014a) performed a LC-MS profiling of plasma. Interestingly, increased metabolites were found in the TB patient group from this study, such as lipids, choline derivatives, and glutamate. A similar experiment was designed by Zhong et al. (2016), who performed a non-targeted LC-MS analysis of serum from TB patients. The correspondent PCA scores plot revealed discrimination of TB-positive patients and controls. Interestingly, 99 significant metabolites were found to be statistically significant between patients and controls (e.g., fatty acids, phosphatidylcholines, and phosphatidylethanolamines). In the same framework, Luier and Loots (2016) performed a GC-MS analysis of urine samples from active TB and healthy controls, revealing significantly altered metabolites in the urine of TB patients (e.g., 5-hydroxyhexanoate, phenylacetate, 2-octenoate, 2-C-methylglycerol, 5-hydroxyhydantoin, oxalic acid, rhamnulose, quinolinic acid, ribitol, indole-3-carboxylate, kynurenate, and glycerol monostearate).
Other studies have also explored diagnostic biomarkers for latent TB and the differential diagnosis between active and latent TB. The typical experimental design of these studies comprises the grouping of individuals in active TB patients, latent TB patients, and controls. An example of this is the study published by Weiner et al. (2012), which evaluated the serum profiles of TB patients by means of a combined GC-MS and LC-MS analysis. Metabolites capable of discriminating between active TB patients and the rest of groups under study were identified as kynurenine, pyroglutamine, phenylalanine, histidine, cysteine, glutamine, citrulline, creatine, and tryptophan, among others. In contrast, 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid, N-acetylneuraminate, xanthine, hypoxanthine, and inosine levels were reported as statistically significant between active and latent TB patients. Even more interestingly, some compounds differed between latent TB and healthy controls (e.g., glycylvaline, 5-oxoproline, and inosine), suggesting the presence of potential biomarkers for latent TB diagnosis. Another potential application of metabolomics regarding the TB study is the suggestion of biomarkers that differentiate TB from other diseases that may show similar clinical features. These studies generally include groups of patients suffering from other diseases in their analysis cohort. For instance, Feng et al. (2015) compared the serum profiles obtained by LC-MS analysis of patients showing active TB, lung cancer, pneumonia, bronchiectasis, and chronic obstructive pulmonary disease. OPLS-DA models showed a full discrimination of TB patients when compared with other diseases, except for the pneumonia group. Interestingly, a combination of potential biomarkers (behenic acid, lysophosphatidylcholine (18:0), threoninyl-γ-glutamate, and presqualene diphosphate) displayed remarkable discriminatory capacity between TB patients and controls (ROC AUC = 0.991, 95% CI: 0.982–1.000). Only lysophosphatidylcholine (16:0) was able to statistically discriminate between TB patients and the rest of disease patients. Another approximation for the differential diagnosis of TB was conducted by Das et al. (2015) who compared the GC-MS urinary profile of symptomatic individuals suspected of TB infection. Urine analysis revealed notable clustering in PLS-DA analyses from which 44 statistically significant metabolites (e.g., lactic acid, norepinephrine, malic acid, hydroquinone, glucose, and 4-hydroxybenzoic acid) were used to build a model capable of differentiating TB patients (ROC AUC = 0.855, 95% CI: 0.72–0.96). Another potential capacity of metabolomics regarding TB is to unravel biomarkers capable of predicting early treatment response. In this context, Mahapatra et al. (2014) compared the LC-MS profiles of the urine from pulmonary TB at different times of treatment (baseline, month 1, month 2, and month 6). A comparison of metabolic profiles was further performed, ultimately leading to the selection of six molecular features that showed the highest statistical significance between baseline and early (1 month) treatment samples, from which four features were tentatively annotated as p-aminobenzoic acid, formimino-L-glutamic acid, Leu-Ala, and L-alpha-aspartyl-L-hydroxyproline. From another point of view, in the line of correct treatment management, few preliminary studies have been documented to evaluate possible biomarkers for distinguishing drug susceptible TB (DS-TB) from drug-resistant TB. Based on the results of an untargeted LC-MS analysis performed in MDR and DS-TB patients, Frediani et al. (2014a, b) suggested a relationship between the profile of D-resolvins, phosphatidylinositol, and trehalose-6-mycolate. In this context, the same group performed another LC-MS analysis comparing both MDR-TB and DS-TB patients. Statistical analysis revealed 66 significant features (e.g., mannose, isopentenyl diphosphate, etc.) (Frediani et al. 2015).
The aforementioned studies principally evaluated either latent or active pulmonary tuberculosis. Apart from this, other complications of TB have also been studied. With regard to the lymphatic system, Singh et al. (2014) performed a high-resolution magic-angle spinning (NMR technique used to perform experiments in solid state) analysis of biopsies from lymph node tissue of tuberculous cervical lymphadenitis and non-specific cervical lymphadenitis patients in order to find potential biomarkers for differential diagnosis. Successful separation between both patient groups was observed in PCA score plots. Statistically significant metabolites comprised monounsaturated and polyunsaturated fatty acids, amino acids (e.g., Ala, Lys, Gly, and Tyr), glucose, phosphocholine, and lactate, among others. Diagnostic biomarkers have also been suggested for tubercular meningitis in children. By 1H-NMR, Mason et al. (2016) compared the urinary profiles among TBM patients, non-TBM patients manifesting neurological disease, and healthy controls. In PCA analysis, the TBM group clustered and separated totally from controls, although partially from non-TBM patients. Further statistical analysis revealed a series of metabolites that widely contributed to the separation of groups. These included mostly small organic acids (4-hydroxyhippuric acid, quinolinic acid, 2-ketoglutaric acid, 3-hydroxyisovaleric acid, 2-hydroxybutyric acid, 3-hydroxybutyric acid, succinic acid, methylcitric acid) and amino acids (tryptophan, glutamic acid). Further evaluation of these metabolites revealed a combined model of four metabolites (4-hydroxyhippuric, 2-ketoglutaric, quinolinic, and methylcitric acids), which could differentiate TBM patients at early onset of disease (ROC AUC = 0.974). In order to find biomarkers for genital tuberculosis, Subramani et al. (2016) performed a 1H-NMR analysis of the serum from women with dormant GTB and controls (unexplained infertile women, women undergoing recurrent spontaneous miscarriage (RSM), and healthy proven fertile women undergoing voluntary sterilization). OPLS-DA revealed several metabolites that separate GTB women from the rest of the groups. These included mostly small organic acids (3-hydroxybutyrate, citrate, acetate, succinate) and amino acids (glutamine, glutamic acid, threonine), suggesting potential biomarkers for the differential diagnosis of this disease.
Metabolomics of Parasitic Diseases
Parasites are defined as a broad biological group encompassing different types of organisms ranging from protozoa to helminths. In this context, certain apicomplexan protozoa cause relevant human diseases, such as malaria (Plasmodium spp.), babesiosis (Babesia spp.), toxoplasmosis (Toxoplasma gondii), and cryptosporidiosis (Cryptosporidium spp.). There is a wide variety of metabolomics experiments of Plasmodium spp., which have been extensively reviewed (Lakshmanan et al. 2011; Olszewski and Llinás 2011; Sonawat and Sharma 2012). In contrast, few studies regarding Toxoplasma gondii and Cryptosporidium parvum have been described (Bisanz et al. 2006; Ng et al. 2012; Ramakrishnan et al. 2012). For these reasons, we focus on the major contributions of metabolomics to the study of malaria in truly infected patients.
With more than 212 million infected individuals and 429,000 deaths per year worldwide reported (2015) (WHO 2017d), malaria is a mosquito-borne disease shows high incidence and considerable mortality rates. Etiological agents of malaria comprehend different obligate intracellular parasites of the genus Plasmodium (P. falciparum, P. malariae, P. vivax, P. ovale, and P. knowlesi). These species parasitize erythrocytes and hepatocytes, causing human malaria with different disease severity and persistence of infection profiles. Although symptoms of mild malaria are often unspecific, severe malaria, usually caused by P. falciparum, can cause severe problems such as respiratory distress, renal failure, coagulopathy, shock, encephalopathy, and multiple organ dysfunction (Autino et al. 2012). The gold standard for malaria diagnosis implies direct observation of the parasites in blood smears. However, shortcomings are derived from this method (Hänscheid 2003), which could be potentially overcome by other diagnostic biomarkers. In this context, Lakshmanan et al. (2012) performed a LC-MS analysis of plasma samples from both P. falciparum-infected and P. falciparum-non-infected individuals, finding excellent group clustering in PLS-DA analyses. Traumatin, a plantlike metabolite was found to be considerably altered between both groups, as well as other statistically significant metabolites such as amino acids, amino acid derived compounds, lipids, and metabolites of the central carbon metabolism. Other studies, apart from suggesting potential diagnostic biomarkers, have been focused on the identification of biomarkers for the prognosis and differential diagnosis of malaria. Severe cases of malaria with CNS affectation—cerebral malaria (CM) or cerebral malaria with multiple organ dysfunction (CMMOD)—are an important source of mortality. With regard to this, Surowiec et al. (2015) performed a GC-MS analysis to compare the plasma profile of pediatric patients suffering from mild and severe malaria and healthy controls. Different metabolites were found to be altered between binary comparisons of disease groups that contributed to the generation of OPLS-DA models with calculated ROC curves. Mild malaria and severe malaria patients could be distinguished from the control groups, as well as mild malaria from severe malaria patients (ROC AUC values of 0.8442, 0.9165, and 0.7637, respectively). The most significantly altered metabolites found in this study included urea, glucuronic acid, histidine, β-hydroxybutyric acid, cysteine, tryptophan, palmitoleic acid, and octadecatrienoic acid. In the same line, Sengupta et al. (2016) performed a 1H-NMR experiment which compared the plasma profiles of differently affected (mild malaria, severe non-cerebral malaria, CM, and CMMOD) P. falciparum-infected patients, as well as patients presenting sepsis and viral encephalitis. Partial segregation between all patient groups (all forms of malaria, sepsis, viral encephalitis, and controls) was observed in PCA and OPLS-DA score plots. Important metabolites differentiating malaria patients and controls were lipoproteins (LDL/VLDL), lactate, and glycoproteins. Metabolites discriminating between P. falciparum-infected patients and patients suffering from sepsis/encephalitis included LDL/VLDL, lactic acid, and isoleucine. It is worthwhile to comment that a decrease in the glycoprotein levels was observed in CM and CMMOD patients when compared to other malaria patients, suggesting their potential applicability as CM diagnostic biomarkers. Pappa et al. (2015) studied the association between brain volume (a strong predictor of the outcome of cerebral malaria) and the plasma metabolic profile of children with CM. Based on a GC-MS and LC-MS multiplatform analysis, different metabolites were found to be correlated with brain volume. These included, among others, metabolites from the phospholipase A2 pathway (Arachidonic acid, 1-eicosatrienoylglycerophosphoethanolamine, 1-oleoylglycerophosphoetanolamine), mannitol, urobilin, fatty acids (pentadecanoic acid) and fatty acid-related metabolites (5-hydroxyhexanoic acid). Diagnostic biomarkers of vivax malaria have been proposed by Sengupta et al. (2011), who compared the 1H-NMR urinary profiles of P. vivax-infected patients, non-malarial fever patients, and healthy controls. Increased (valerylglycine, N-acetylornithine, salicylurate, phenylpyruvic acid, pipecolic acid, biopterin-3-hydroxybutyrate) and decreased (tyrosine, glucose, guanidinoacetate, alanine, creatine/phosphocreatine, N-acetylglutamate) metabolites were suggested as potential biomarkers for differentiating between malaria patients and controls. In contrast increased (glucose, glutamine, alanine, ornithine, hippurate, phenylalanine) and decreased (N-butyrate, acetate) metabolites were suggested as biomarkers for the differential diagnosis of fever due to either malaria or viral CNS affectation. Lastly, a 1H-NMR longitudinal study performed by the same group searched for diagnostic biomarkers and biomarkers for response to treatment in urine samples from P. vivax-infected patients and patients suffering from viral encephalitis (Sengupta et al. 2015). Urine samples were collected from day 1 (admission day) to day 5 and at day 30 after treatment, being this last sample set used as a self-negative control group. Day 30 samples were compared by PCA analyses with samples from day 1 to day 5, revealing progressive loss of observable clustering, which completely disappeared at day 5. Altered metabolites comprised, among others, alanine, glycine, 3-methylhistidine, hippuric acid, taurine, guanidinoacetate, and citric acid. Predictive models were generated, which were able to identify P. vivax malaria patients with up to a value of 85% success rate in a second sample cohort. Discriminatory metabolites were identified when comparing the urine of viral fever patients and P. vivax-infected patients from day 1 (particularly taurine and citrate, with ROC AUC values of 0.66 and 0.69, respectively).
From the clinical point of view, kinetoplastids are another group of relevant protozoal organisms in which various etiological agents of important human diseases such as human African trypanosomiasis (HAT) and Chagas disease are included. More than 65 million people are at risk of developing HAT, which is caused by Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense (WHO 2017c). Two stages of HAT can be clearly distinguished. In the first stage, parasites live and replicate extracellularly in the circulatory and lymphatic systems, whereas in the second stage (sleeping sickness) CNS invasion occurs, causing neurological disorders and, ultimately, coma and death if untreated (Kennedy 2004). Since important toxicity is derived from the current therapies for HAT advanced stages, a sensitive and specific test for disease staging is required (Kennedy 2004). However, the current surrogate standard for HAT staging lacks sensitivity and is invasive (requiring a lumbar puncture), since it implies the direct observation of either the parasite or an increase in the white blood cells count in the CSF (Vincent et al. 2016). To potentially overcome these limitations and study the global metabolic alterations of HAT, Vincent et al. (2016) performed a LC-MS analysis which compared the plasma, urine, and CSF metabolic profiles of T. brucei-infected patients of first and second HAT stages with controls. In CSF analysis, neopterin could discriminate between disease stages with 100% sensitivity and specificity. Furthermore, the levels of 5-hydroxytryptophan, a metabolite associated with somnolence, and other relevant metabolites, such as kynurenine, O-acetylcarnitine, N-acetylhistidine, tryptophan, N-acetyl-aspartylglutamate, linoleamide, and oleamide, were also found to be altered. Altered plasma metabolites were used to construct a model with two unidentified features that effectively discriminated both disease stages, (ROC AUC = 0.92) suggesting a potentially useful, less invasive method for the staging of this potentially fatal disease.
Chagas disease, caused by Trypanosoma cruzi, affects 8 million people worldwide, causing 15,000 deaths per year (Gironès et al. 2014). In contrast with T. brucei life cycle, T. cruzi requires to invade host cells to replicate as amastigotes, ultimately causing cellular lysis. Given that cardiac muscle cells are particularly invaded by this parasite, serious cardiovascular alterations are generally presented in T. cruzi-infected patients (Teixeira et al. 2011). Chagas disease can be manifested in an acute or chronic form and is usually asymptomatic (Teixeira et al. 2011). Current treatment efficacy is poorly assessed, due to the absence of a surrogate indicator of therapeutic success (Gironès et al. 2014). A preliminary study performed by Gironès et al. (2014) aimed to study the general metabolic alterations found in heart tissue and plasma of T. brucei-infected mice by untargeted GC-MS and LC-MS analysis. Metabolites such as p-cresol sulfate (a toxic metabolite from microbial origin), kynurenine, and allantoin were suggested as potential biomarkers of this disease, due to their considerable concentration changes found in serum.
Focusing on helminthiasis, several illustrative examples of how metabolomics has helped to identify biomarkers can be found in the study of onchocerciasis. This disease, commonly known as river blindness, affects more than 37 million people worldwide (Globisch et al. 2013). Onchocerciasis is a blackfly-borne (Simulium spp.) disease caused by filariae (Onchocerca volvulus), which typically affects the eyes and skin, causing inflammation and blindness (Globisch et al. 2013). Denery et al. (2010) performed a combined non-targeted and targeted analysis to evaluate the metabolic profile from serum and plasma of O. volvulus positive and negative individuals from different regions (African countries, Guatemala, USA, India) and compared such profile with other possible comorbidities (Chagas disease, leishmaniasis, HAT). After data filtering, 14 features were selected for LC-MS/MS and Fourier transform ion cyclotron resonance mass spectrometry analysis. These included hexacosenoic and pentacosenoic acids, a phosphorylated sphingolipid, a phosphorylated sphingolipid, sterol lipids, and fatty acids. Considering such candidate biomarkers, PCA analysis revealed discrimination of O. volvulus positive patients for the African samples, although group overlapping was observed (mainly in Guatemalan samples and samples from patients affected by either Chagas disease or leishmaniasis). Subsequent models considering these 14 features were generated by machine learning algorithms. The highest discriminatory capacity for the entire sample set was achieved using a random forests algorithm (88.6% of sensitivity and 88.3% of specificity), whereas for the African sample set, a functional trees algorithm achieved a 100% sensitivity and specificity. In another study of the same group, Globisch et al. (2013) performed a non-targeted LC-MS analysis in urine samples, identifying a highly enriched feature in O. volvulus positive African patients. Further validation of this compound will be discussed (see Sect. 7.4.2).
Metabolomics of Fungal Diseases
To the best of our knowledge, few metabolomics studies of fungal infections have been performed, being these particularly focused on the study of invasive fungal infections (IFIs) (de Francisco et al. 2015; Dessì et al. 2014; Koo et al. 2014; Liew et al. 2016). IFIs are generally caused by fungi from the genera Aspergillus (invasive aspergillosis or IA), Cryptococcus (cryptococcosis), and Candida (invasive candidiasis). Because of the inhalation of conidia, initial infection by Aspergillus spp. and Cryptococcus spp. is usually localized in the lungs. However, as in infections caused by Candida spp., fungi can spread to other regions such as the blood or CNS, developing potentially fatal diseases (Badiee and Hashemizadeh 2014). For this reason, IFIs constitute a notable source of mortality among immunocompromised patients (Enoch et al. 2017). The gold standard for IFI diagnosis is cell culture, which is usually slow (>72 h) and lacks sensitivity (Enoch et al. 2017). Koo et al. (2014) evaluated the volatile organic compounds (VOCs) from the breath of pulmonary IA suspected patients by GC-MS, finding a metabolic signature in IA patients (increased levels of β-trans-bergamotene, α-trans-bergamotene, β-vatirenene, and trans-geranylacetone). Focusing on cryptococcosis, a study published by Liew et al. (2016) simulated a primary C. neoformans infection using lung epithelial cell cultures. Two initial doses of yeast cells were used for the study, which consisted in the incubation and analysis of yeast positive and negative controls at 6, 12, and 18 h. GC-MS analysis revealed altered levels of metabolites in the supernatants of C. neoformans-infected cultures. Independently of the inoculation dose, cysteine was identified as a discriminative metabolite at 6 h of incubation. In contrast lactic acid, tyrosine, fructose, fumaric acid, and pantothenic acid were identified at 12 and 18 h of incubation in low inoculated cell cultures, whereas malic acid, 3-hydroxyvaleric acid, 3-phenyllactic acid, and adonitol were significantly altered in highly inoculated cell cultures. Another form of IFI, invasive candidiasis of the bloodstream, displays high mortality rates in newborns (up to 30%) (Dessì et al. 2014). A preliminary study performed by Dessì et al. (2014) compared the GC-MS urinary metabolic profiles between healthy controls and a newborn presenting fungal sepsis, obtained at different time points. Groups clustered separately in PCA analysis and the most relevant metabolites altered in the fungal septic patient were identified as amino acids (glycine, serine, threonine), carbohydrates (glucose, maltose), and both organic and fatty acids (citrate, hexadecanoate, octadecanoate), suggesting potential biomarkers for the diagnosis of this potentially fatal disease.
Metabolomics of Diseases with Diverse Etiological Agents
In clinics, one of the main potential goals of biomarkers is to discriminate between different etiological agents that can cause a disease with the same symptoms, contributing to a better patient management and a more sensitive use of pharmacological resources. In this line, Slupsky et al. (2009) performed a study which aimed to identify potential biomarkers for community-acquired pneumonia (CAP). CAP is a serious condition, generally caused by Streptococcus pneumoniae, wherein patients show a high mortality rate (up to 25%) (MMWR Recommendations and reports 1996). Currently, the standard diagnosis for CAP is based on pathogen isolation and culture from biofluids. This process has serious disadvantages: S. pneumoniae culture is time-consuming (typically more than 36 h) (Marston et al. 1997) and displays notable false positive rates (up to 65% in children) (Faden et al. 2002), since this microorganism can be found in the commensal human microbiota. In order to perform a preliminary approach for adequate biomarker identification, 1H-NMR analysis was performed in urine samples, wherein PCA analysis revealed notable discrimination of S. pneumoniae-infected and control groups. To further assess selectivity of these biomarkers, the S. pneumoniae-infected group was compared with other patients with lung infections that shared common clinical features with CAP (caused by Legionella pneumophila, Mycobacterium tuberculosis, Staphylococcus aureus, Coxiella burnetii, Haemophilus influenzae, Mycoplasma pneumoniae, Escherichia coli, Enterococcus faecalis, Moraxella catarrhalis, Streptococcus viridans, Streptococcus anginosus, influenza A virus, picornavirus, respiratory syncytial virus, parainfluenza viruses, coronaviruses, human metapneumovirus, or hantavirus). PCA analysis revealed notable discrimination between pneumococcal pneumonia and other bacterial and viral pulmonary infections except for L. pneumophila, evidencing the notable differential diagnostic capacity achieved by this study.
Other studies are specifically focused on the identification of biomarkers for differential diagnosis, such as the one performed by Li et al. (2017), who explored the possibility of discriminating tubercular meningitis from viral meningitis. Meningitis is a serious condition wherein inflammation of the meninges is produced, caused by a multitude of virus, bacteria (including mycobacteria), fungi, or parasites. Clinical symptoms (e.g., fever, neck stiffness, headache, photophobia, and vomiting) are common among diverse etiological agent, and therefore a fast patient management becomes crucial in order to reduce meningitis mortality. For this reason, given the wide variety of agents causing meningitis, pathogen-specific diagnostic methods have been developed (Bahr and Boulware 2014). In the same framework, this study reported a 1H-NMR analysis of the CSF from TBM and viral meningitis patients. While PCA analysis showed partial clustering of sample groups, OPLS-DA analysis allowed their complete discrimination. Metabolites most contributing to such discrimination were amino acids (glycine, tyrosine, glutamine, threonine, valine, alanine, aspartic acid, asparagine), carbohydrates (glucose, sucrose, fructose), small organic acids (acetate, citrate, malonic acid, pyruvic acid, malonate), lipoproteins, and others (betaine, glycerine, putrescine, choline), thus confirming a different metabolic signature among TBM and viral meningitis.
Another differential analysis example was proposed by Soga et al. (2011). In this study, the capacity of a CE-MS analysis of serum to discriminate among different forms of liver disease was evaluated. Patients suffering from infectious diseases at different disease stages, such as asymptomatic hepatitis B virus infection (AHB), chronic hepatitis B (CHB), chronic hepatitis C (CHC) hepatitis C with persistently normal alanine transaminase (CNALT), cirrhosis type C (CIR), hepatocellular carcinoma, non-alcoholic steatohepatitis (NASH), simple steatosis (SS), and drug-induced liver injury (DI), were included. Analysis revealed, among others, 49 statistically significant metabolites, mainly corresponding to γ-glutamyl dipeptides which were generally found to be increased in liver disease. The discriminatory capacity of models comprising the levels of γ-glutamyl dipeptides alone or in combination with biochemical parameters (aspartate aminotransferase (AST) and alanine aminotransferase (ALT), methionine sulfoxide) was tested in a first training and a second validation cohort. ROC AUC for both cohorts were able to effectively each liver disease with the rest of liver diseases and healthy controls (values of the ROC AUCs from the validation cohorts were 0.895, 0.707, 0.993, 0.803, 0.967, 0.849, 0.763, and 0.762 for CNALT (γ-Glu-Taurine, ALT), CHC (γ-Glu-Lys), CIR (γ-Glu-Ala, γ-Glu-Leu, γ-Glu-Ser, γ-Glu-Taurine), HCC (γ-Glu-Ala, γ-Glu-Citrulline, γ-Glu-Thr, γ-Glu-Phe), C (γ-Glu-Phe), DI (γ-Glu-Citrulline, ALT), AHB (Methionine sulfoxide, AST), and CHB (γ-Glu-Thr), respectively), brilliantly evidencing the potential of metabolomics in differential diagnosis.
Independently of the disease-causing pathogen, metabolic biomarkers for the diagnosis of multipathogenic diseases can be potentially identified. In this line, Su et al. (2014) identified several diagnostic and prognostic biomarkers of sepsis by LC-MS/MS analysis. Sepsis is a serious condition caused by multiple possible pathogens that spread from a local infection focus through the blood, generating a systemic infection, ultimately leading to septic shock and multiple organ failure. Even with the correct treatment, patients with sepsis show considerably high mortality rates (Martin 2012). The study was performed on controls, septic patients, and patients showing non-septic systemic inflammatory response syndrome (SIRS group), a sepsis-like condition. Further subdivision of the sepsis patients’ group was performed attending to different clinical criteria, classifying these in sepsis, severe sepsis, septic shock, and death patients (death occurred within 48 h after the analysis). Serum samples were obtained within 24 h after emergency room admission except for the death group, from which samples were collected within 48 h before death. Several metabolites were found to be altered between the different disease groups. Different statistical analyses were performed; PCA analysis revealed differentiation of controls from the rest of group patients, while OPLS-DA allowed the observation of discrimination between SIRS and sepsis groups. Further PLS-DA analysis differentiated mild sepsis from severe sepsis and septic shock. Additionally, PLS-DA analysis also differentiated patients of the death subgroup from the rest of septic patients. Eventually, the ROC curves for 12 selected significant metabolites among the different groups were calculated. Regarding sepsis diagnosis, lactitol dehydrate, N-nonanoyl-glycine, S-phenyl-cysteine, and S-(3-methylbutanoyl)-dihydrolipoamide-E were identified as potential biomarkers, from which this last obtained the best ROC values (AUC = 0.900; 95% CI: 0.799–1.001). Severity of sepsis could be determined by N,N-dimethyllysine, glycerylphosphorylethanolamine, cysteine, and 2-phenylacetamide, accounting this last metabolite for the highest ROC value between these potential biomarkers (AUC = 0.804; 95% CI: 0.647–0.962). Predictive biomarkers of death were identified as phosphatidylglycerol (22:2/0:0), S-(3-methylbutanoyl)-dihydrolipoamide-E, S-succinyl glutathione, and glycerophosphocholine), being phosphatidylglycerol (22:2/0:0) the best predictor of death within these groups (ROC AUC = 0.96; 95% CI: 0.863–1.057). In conclusion, this study was able to clearly assess disease diagnosis, differential diagnosis, and prognosis of sepsis between the different patients.
Biomarker Validation
To our knowledge, most of infectious disease metabolomics experiments have been focused on a discovery phase combining, in some cases, non-targeted and targeted analysis on the same samples. Fully validation following the criteria mentioned above is not so common.
One of the most successful examples is the validation of a diagnostic method for urinary tract infection (see Sect. 7.4.1.2) by Gupta et al. (2012). In this target experiment, the selection of the analytes was done based on the findings of previous fingerprinting studies (Gupta et al. 2009; Lenz et al. 2003, 2004; Sweatman et al. 1993), which suggested an increase of certain end products of bacterial metabolism in contaminated urine samples (lactate, acetate, succinate, formate, ethanol, citrate, trimethylamine, urea, glycine, hippurate, creatinine, trimethylamine, trimethylamine-N-oxide). For the biomarker validation, different experiments were designed: first, an ex vivo study was performed over a cohort of 682 suspected urine patients and 50 healthy controls to test the diagnostic capabilities of the method; secondly, an in vitro study was carried out in pathogen inoculated culture media in which samples were generated by urine sterilization supplementation and uropathogen inoculation. The results showed significant statistical variations in the levels of the measured from which acetate, formate, lactate, and succinate urine concentrations were selected to generate a model that was able to remarkably discriminate UTI patients from healthy controls (ROC AUC = 0.996; 95% CI: 0.992–1.001). Moreover, the levels of these metabolites were able to differentiate urine samples infected by either gram-positive cocci or gram-negative bacilli (ROC AUC = 0.975; 95% CI: 0.957–0.993), indicating the potential differential diagnostic capabilities of this method; finally, on the third place, a longitudinal study was performed, wherein the urine 10 UTI patients were sampled before and after 7, 14, and 21 days of antibiotic treatment. Further validation of the method was confirmed by the observation of a control-like urine profile in successfully antibiotic treatment patients, indicating eradication of the infection. Therefore, the remarkable ability of this large-scale study to identify and discriminate the causal agent of UTI was stated.
In relation with onchocerciasis, as mentioned above (see Sect. 7.4.1.3), Globisch et al. (2013) identified a highly increased feature in African O. volvulus positive urine samples (m/z 356.1340). In order to decipher the molecular structure of such feature, the fragmentation patterns of a subsequent LC-MS/MS analysis revealed losses of glucuronic acid, an aliphatic N-acetyl moiety, water, and ethylene. As a result, N-acetyltyramine-O,β-glucuronide (NATOG) was proposed as candidate structure. Structure assignment was confirmed by LC purification of NATOG from samples and subsequent comparative analysis of the m/z and retention times of both purified and de novo synthesized NATOG. However, when analysis of NATOG was performed in Guatemalan samples, lower levels of NATOG were detected, suggesting phylogenetic differences between African and American O. volvulus. NATOG capacities as prognostic biomarker were further tested in a comparative analysis wherein NATOG levels were measured in urine samples from O. volvulus positive patients receiving either doxycycline or placebo at 4–6 weeks posttreatment. Diminished levels of NATOG were observed in successfully treated patients, thus proving its potential use in disease progression evaluation.
Conclusions and Future Trends
The examples discussed in previous sections just illustrate selected representative possibilities of metabolomics which are far from being fully summarized. We have observed few biomarkers that have been integrated in clinics. Important obstacles to this are the lack of a consensus on validation criteria and standardization of metabolomics experiments. There is a need for a refinement of both analytical (e.g., pathogen isolation methods) and computational techniques (e.g., refinement of computational models of host–pathogen interactions) to ease the conclusions inferred from analysis. Given the current landscape of the actual development of metabolic biomarkers in this area, it can be concluded that it is time to take advantage of the evidence supported by previous knowledge found in preliminary studies. This would ultimately permit wider integration of metabolomics in the context of a more translational research, allowing this yet relatively recent omics science to reach one of its paramount objectives: to become a standard, well-stablished tool for biomarker identification which eventually will need to be competitive in cost/benefit with the classical clinical test.
Funding Statement
The co-authors would like to acknowledge funding from the Spanish Ministry of Economy and Competitiveness (CTQ2014-55279-R). Author M. F.-G. acknowledges Fundación Universitaria San Pablo CEU for his PhD fellowship.
Contributor Information
Ricardo Silvestre, Email: ricardosilvestre@med.uminho.pt.
Egídio Torrado, Email: egidiotorrado@ecsaude.uminho.pt.
Miguel Fernández-García, Email: mig.fernandez.ce@ceindo.ceu.es.
References
- Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, Gerber GK, Bry L, Clish CB, Alm E, Korzenik JR. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment Pharmacol Ther. 2016;43(11):1142–1153. doi: 10.1111/apt.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autino B, Corbett Y, Castelli F, Taramelli D (2012) Pathogenesis of malaria in tissues and blood. Mediterr J Hematol Infect Dis. 10.4084/mjhid.2012.061 [DOI] [PMC free article] [PubMed]
- Badiee P, Hashemizadeh Z. Opportunistic invasive fungal infections: diagnosis & clinical management. Indian J Med Res. 2014;139(2):195–204. [PMC free article] [PubMed] [Google Scholar]
- Bahr NC, Boulware DR. Methods of rapid diagnosis for the etiology of meningitis in adults. Biomark Med. 2014;8(9):1085–1103. doi: 10.2217/bmm.14.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. Hepatology. 1996;24(2):289–293. doi: 10.1002/hep.510240201. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GRW, Simmons CP, Scott TW, Farrar JJ, Hay SI. The global distribution and burden of dengue. Nature. 2013;496(7446):504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birungi G, Chen SM, Loy BP, Ng ML, Li SFY. Metabolomics approach for investigation of effects of dengue virus infection using the EA hy926 cell line. J Proteome Res. 2010;9(12):6523–6534. doi: 10.1021/pr100727m. [DOI] [PubMed] [Google Scholar]
- Bisanz C, Bastien O, Grando D, Jouhet J, Maréchal E, Cesbron-Delauw MF. Toxoplasma gondii acyl-lipid metabolism: de novo synthesis from apicoplast-generated fatty acids versus scavenging of host cell precursors. Biochem J. 2006;394:197–205. doi: 10.1042/BJ20050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand HK, Hermans PW, de Groot R. Host biomarkers and paediatric infectious diseases: from molecular profiles to clinical application. In: Finn A, Curtis N, Pollard AJ, editors. Hot topics in infection and immunity in children VI. 1. New York: Springer-Verlag; 2010. pp. 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle GC, Walker CL, Black RE. Typhoid fever and paratyphoid fever: systematic review to estimate global morbidity and mortality for 2010. J Glob Health. 2012;2(1):010401. doi: 10.7189/jogh.01.010401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham CA, Carroll KC. Diagnosis of Clostridium difficile infection: an ongoing conundrum for clinicians and for clinical laboratories. Clin Microbiol Rev. 2013;26(3):604–630. doi: 10.1128/CMR.00016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassol E, Misra V, Dutta A, Morgello S, Gabuzda D. Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS. 2014;28(11):1579–1591. doi: 10.1097/QAD.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccotti G, Meoni G, Tenori L, Gragnani L, Fognani E, Gianni E, Luchinat C, Zignego AL. A serum metabolomic analysis of HCV-infected patients successfully treated with IFN-free DAA regimens. Hepatology. 2016;64:378A–378A. [Google Scholar]
- Cheng J, Joyce A, Sterling RK, Sanyal AJ. Metabolomic profiling identifies fibrosis-associated differences in hepatitis C virus infection. Gastroenterology. 2012;142(5):S968. doi: 10.1016/S0016-5085(12)63750-0. [DOI] [Google Scholar]
- Cribbs SK, Park Y, Guidot DM, Martin GS, Brown LA, Lennox J, Jones DP. Metabolomics of bronchoalveolar lavage differentiate healthy HIV-1-infected subjects from controls. AIDS Res Hum Retroviruses. 2014;30(6):579–585. doi: 10.1089/aid.2013.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Lee YH, Thein TL, Fang J, Pang J, Ooi EE, Leo YS, Ong CN, Tannenbaum SR (2016) Serum metabolomics reveals serotonin as a predictor of severe dengue in the early phase of dengue fever. Plos Negl Trop Dis. 10.1371/journal.pntd.0004607 [DOI] [PMC free article] [PubMed]
- Currie BJ, Fisher DA, Howard DM, Burrow JN, Lo D, Selva-Nayagam S, Anstey NM, Huffam SE, Snelling PL, Marks PJ, Stephens DP, Lum GD, Jacups SP, Krause VL. Endemic melioidosis in tropical northern Australia: a 10-year prospective study and review of the literature. Clin Infect Dis. 2000;31(4):981–986. doi: 10.1086/318116. [DOI] [PubMed] [Google Scholar]
- da Luz JA, Hans E, Zeng A-P. Automated fast filtration and on-filter quenching improve the intracellular metabolite analysis of microorganisms. Eng Life Sci. 2014;14(2):135–142. doi: 10.1002/elsc.201300099. [DOI] [Google Scholar]
- Dann S, Aitken S, Ross C, Tessier MEM, Loeffelholz M, Koussoulas K, Bornstein JC, Feng H, Versalovic J, Pothoulakis C, Garey KW, Savidge T. Zolpidem confers disease susceptibility to Clostridium difficile infection. Gastroenterology. 2015;148(4):S727–S728. doi: 10.1016/S0016-5085(15)32484-7. [DOI] [Google Scholar]
- Das MK, Bishwal SC, Das A, Dabral D, Badireddy VK, Pandit B, Varghese GM, Nanda RK. Deregulated tyrosine-phenylalanine metabolism in pulmonary tuberculosis patients. J Proteome Res. 2015;14(4):1947–1956. doi: 10.1021/acs.jproteome.5b00016. [DOI] [PubMed] [Google Scholar]
- de Francisco TM, Zaramella IF, Gasparetto JC, Cerqueira LB, Piantavini MS, Pontarolo R, Campos FR. Rapid detection of aspergillosis in immunocompromised patients using DIMS and chemometric analysis. Anal Methods. 2015;7(15):6346–6351. doi: 10.1039/C5AY01146A. [DOI] [Google Scholar]
- Denery JR, Nunes AAK, Hixon MS, Dickerson TJ, Janda KD (2010) Metabolomics-based discovery of diagnostic biomarkers for onchocerciasis. PloS Negl Trop Dis. 10.1371/journal.pntd.0000834 [DOI] [PMC free article] [PubMed]
- Dessì A, Liori B, Caboni P, Corsello G, Giuffrè M, Noto A, Serraino F, Stronati M, Zaffanello M, Fanos V. Monitoring neonatal fungal infection with metabolomics. J Matern Fetal Neonatal Med. 2014;27:34–38. doi: 10.3109/14767058.2014.954787. [DOI] [PubMed] [Google Scholar]
- Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26(1):51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Preez I, Loots DT. New sputum metabolite markers implicating adaptations of the host to Mycobacterium tuberculosis, and vice versa. Tuberculosis (Edinb) 2013;93(3):330–337. doi: 10.1016/j.tube.2013.02.008. [DOI] [PubMed] [Google Scholar]
- du Preez I, Sithebe NP. The use of metabolomics as a tool to investigate hepatitis C. Metabolomics. 2013;9(2):497–505. doi: 10.1007/s11306-012-0467-8. [DOI] [Google Scholar]
- Dunn WB, Wilson ID, Nicholls AW, Broadhurst D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis. 2012;4(18):2249–2264. doi: 10.4155/bio.12.204. [DOI] [PubMed] [Google Scholar]
- El-Bacha T, Struchiner CJ, Cordeiro MT, Almeida FCL, Marques ET, Jr, Da Poian AT. 1H nuclear magnetic resonance metabolomics of plasma unveils liver dysfunction in dengue patients. J Virol. 2016;90(16):7429–7443. doi: 10.1128/JVI.00187-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embade N, Marino Z, Diercks T, Cano A, Lens S, Cabrera D, Navasa M, Falcón-Pérez JM, Caballería J, Castro A, Bosch J, Mato JM, Millet O (2016) Metabolic characterization of advanced liver fibrosis in HCV patients as studied by serum 1H-NMR spectroscopy. PLoS One 11(5). 10.1371/journal.pone.0155094 [DOI] [PMC free article] [PubMed]
- Enoch DA, Yang H, Aliyu SH, Micallef C. The changing epidemiology of invasive fungal infections. Methods Mol Biol. 2017;1508:17–65. doi: 10.1007/978-1-4939-6515-1_2. [DOI] [PubMed] [Google Scholar]
- Eoh H, Rhee KY. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2013;110(16):6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden H, Heimerl M, Varma C, Goodman G, Winkelstein P. Urinary excretion of pneumococcal cell wall polysaccharide in children. Pediatr Infect Dis J. 2002;21(8):791–793. doi: 10.1097/00006454-200208000-00020. [DOI] [PubMed] [Google Scholar]
- Fatima Z, Hameed S, Saibabu V, Sharma S, Hans S. Tuberculosis: propagation beyond lungs. In: Bhargava S, editor. Diagnosis & management of tuberculosis. 1. Wilmington: Open Access eBooks; 2017. pp. 1–21. [Google Scholar]
- Feng S, Du Y-Q, Zhang L, Zhang L, Feng R-R, Liu S-Y. Analysis of serum metabolic profile by ultra-performance liquid chromatography-mass spectrometry for biomarkers discovery: application in a pilot study to discriminate patients with tuberculosis. Chin Med J (Engl) 2015;128(2):159–168. doi: 10.4103/0366-6999.149188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics – the link between genotypes and phenotypes. Plant Mol Biol. 2002;48(1–2):155–171. doi: 10.1023/A:1013713905833. [DOI] [PubMed] [Google Scholar]
- Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue virus induces and requires glycolysis for optimal replication. J Virol. 2015;89(4):2358–2366. doi: 10.1128/JVI.02309-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frediani JK, Jones D, Tukvadze N, Sanikidze E, Kipiani M, Uppal K, Kurani S, Hebbar G, Colas R, Dalli J, Serhan C, Tangpricha V, Blumberg H, Ziegler T (2014a) Plasma high-resolution metabolomic profiling reveals upregulation of specific resolvins in patients with pulmonary tuberculosis. FASEB J 28(1)
- Frediani JK, Jones DP, Tukvadze N, Uppal K, Sanikidze E, Kipiani M, Tran VT, Hebbar G, Walker DI, Kempker RR, Kurani SS, Colas RA, Dalli J, Tangpricha V, Serhan CN, Blumberg HM, Ziegler TR (2014b) Plasma metabolomics in human pulmonary tuberculosis disease: a pilot study. PLoS ONE 9. 10.1371/journal.pone.0108854 [DOI] [PMC free article] [PubMed]
- Frediani JK, Chong E, Jones D, Yu T, Zhang L, Tukvadze N, Sanikidze E, Kipiani M, Hebbar G, Tran V, Kempker R, Tangpricha V, Blumberg H, Ziegler T (2015) Comparative plasma high-resolution metabolomic profiling in patients with drug-susceptible and multi-drug resistant pulmonary tuberculosis. FASEB J 29
- Ghannoum MA, Mukherjee PK, Jurevic RJ, Retuerto M, Brown RE, Sikaroodi M, Webster-Cyriaque J, Gillevet PM. Metabolomics reveals differential levels of oral metabolites in HIV-infected patients: toward novel diagnostic targets. OMICS. 2013;17(1):5–15. doi: 10.1089/omi.2011.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironès N, Carbajosa S, Guerrero NA, Poveda C, Chillón-Marinas C, Fresno M (2014) Global metabolomic profiling of acute myocarditis caused by Trypanosoma cruzi infection. PloS Negl Trop Dis. 10.1371/journal.pntd.0003337 [DOI] [PMC free article] [PubMed]
- Globisch D, Moreno AY, Hixon MS, Nunes AAK, Denery JR, Specht S, Hoerauf A, Janda KD. Onchocerca volvulus-neurotransmitter tyramine is a biomarker for river blindness. Proc Natl Acad Sci U S A. 2013;110(11):4218–4223. doi: 10.1073/pnas.1221969110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzien J, Ciborowski M, Angulo S, Barbas C. From numbers to a biological sense: how the strategy chosen for metabolomics data treatment may affect final results. A practical example based on urine fingerprints obtained by LC-MS. Electrophoresis. 2013;34(19):2812–2826. doi: 10.1002/elps.201300053. [DOI] [PubMed] [Google Scholar]
- Godzien J, Alonso-Herranz V, Barbas C, Armitage EG. Controlling the quality of metabolomics data: new strategies to get the best out of the QC sample. Metabolomics. 2015;11(3):518–528. doi: 10.1007/s11306-014-0712-4. [DOI] [Google Scholar]
- Gowda GA, Shanaiah N, Raftery D. Isotope enhanced approaches in metabolomics. Adv Exp Med Biol. 2012;992:147–164. doi: 10.1007/978-94-007-4954-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther G. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med (Lond) 2014;14(3):279–285. doi: 10.7861/clinmedicine.14-3-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Dwivedi M, Mahdi AA, Gowda GA, Khetrapal CL, Bhandari M. 1H-nuclear magnetic resonance spectroscopy for identifying and quantifying common uropathogens: a metabolic approach to the urinary tract infection. BJU Int. 2009;104(2):236–244. doi: 10.1111/j.1464-410X.2009.08448.x. [DOI] [PubMed] [Google Scholar]
- Gupta A, Dwivedi M, Mahdi AA, Khetrapal CL, Bhandari M. Broad identification of bacterial type in urinary tract infection using 1H NMR spectroscopy. J Proteome Res. 2012;11(3):1844–1854. doi: 10.1021/pr2010692. [DOI] [PubMed] [Google Scholar]
- Hänscheid T. Current strategies to avoid misdiagnosis of malaria. Clin Microbiol Infect. 2003;9(6):497–504. doi: 10.1046/j.1469-0691.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Hewer R, Vorster J, Steffens FE, Meyer D. Applying biofluid 1H NMR-based metabonomic techniques to distinguish between HIV-1 positive/AIDS patients on antiretroviral treatment and HIV-1 negative individuals. J Pharm Biomed Anal. 2006;41(4):1442–1446. doi: 10.1016/j.jpba.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Himmelreich U, Accurso R, Malik R, Dolenko B, Somorjai PR, Gupta RK, Gomes L, Mountford CE. Identification of Staphylococcus aureus brain abscesses: rat and human studies with 1H MR spectroscopy. Radiology. 2005;236(1):261–270. doi: 10.1148/radiol.2361040869. [DOI] [PubMed] [Google Scholar]
- Jain KK (2010) Role of Biomarkers in Health Care. In: Jain KK (ed) The Handbook of Biomarkers, 1st edn. Humana Press, New York, pp 134–149
- John DV, Lin Y-S, Perng GC. Biomarkers of severe dengue disease – a review. J Biomed Sci. 2015;22:83. doi: 10.1186/s12929-015-0191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao D, Ismond KP, Tso V, Millan B, Hotte N, Fedorak RN (2016) Urine-based metabolomic analysis of patients with Clostridium difficile infection: a pilot study. Metabolomics 12(8). 10.1007/s11306-016-1080-z
- Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113(4):496–504. doi: 10.1172/JCI200421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo S, Thomas HR, Daniels SD, Lynch RC, Fortier SM, Shea MM, Rearden P, Comolli JC, Baden LR, Marty FM. A breath fungal secondary metabolite signature to diagnose invasive aspergillosis. Clin Infect Dis. 2014;59(12):1733–1740. doi: 10.1093/cid/ciu725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiakis EC, Hyduke DR, Fornace AJ., Jr Comparison of mouse urinary metabolic profiles after exposure to the inflammatory stressors gamma radiation and lipopolysaccharide. Radiat Res. 2012;177(2):187–199. doi: 10.1667/RR2771.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan V, Rhee KY, Daily JP. Metabolomics and malaria biology. Mol Biochem Parasitol. 2011;175(2):104–111. doi: 10.1016/j.molbiopara.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan V, Rhee KY, Wang W, Yu Y, Khafizov K, Fiser A, Wu P, Ndir O, Mboup S, Ndiaye D, Daily JP. Metabolomic analysis of patient plasma yields evidence of plant-like alpha-linolenic acid metabolism in Plasmodium falciparum. J Infect Dis. 2012;206(2):238–248. doi: 10.1093/infdis/jis339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SK, Lee KC, Lo GC, Ding VS, Chow WN, Ke TY, Curreem SO, To KK, Ho DT, Sridhar S, Wong SC, Chan JF, Hung IF, Sze KH, Lam CW, Yuen KY, Woo PC (2016) Metabolomic profiling of plasma from melioidosis patients using UHPLC-QTOF MS reveals novel biomarkers for diagnosis. Int J Mol Sci 17(3). 10.3390/ijms17030307 [DOI] [PMC free article] [PubMed]
- Lenz EM, Bright J, Wilson ID, Morgan SR, Nash AF. A 1H NMR-based metabonomic study of urine and plasma samples obtained from healthy human subjects. J Pharm Biomed Anal. 2003;33(5):1103–1115. doi: 10.1016/S0731-7085(03)00410-2. [DOI] [PubMed] [Google Scholar]
- Lenz EM, Bright J, Wilson ID, Hughes A, Morrisson J, Lindberg H, Lockton A. Metabonomics, dietary influences and cultural differences: a 1H NMR-based study of urine samples obtained from healthy British and Swedish subjects. J Pharm Biomed Anal. 2004;36(4):841–849. doi: 10.1016/j.jpba.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Li Z, Du B, Li J, Zhang J, Zheng X, Jia H, Xing A, Sun Q, Liu F, Zhang Z. Cerebrospinal fluid metabolomic profiling in tuberculous and viral meningitis: screening potential markers for differential diagnosis. Clin Chim Acta. 2017;466:38–45. doi: 10.1016/j.cca.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Liew KL, Jee JM, Yap I, Yong PVC (2016) In vitro analysis of metabolites secreted during infection of lung epithelial cells by Cryptococcus neoformans. PLoS ONE. 10.1371/journal.pone.0153356 [DOI] [PMC free article] [PubMed]
- Limmathurotsakul D, Jamsen K, Arayawichanont A, Simpson JA, White LJ, Lee SJ, Wuthiekanun V, Chantratita N, Cheng A, Day NPJ, Verzilli C, Peacock SJ (2010) Defining the true sensitivity of culture for the diagnosis of melioidosis using Bayesian latent class models. PLoS One 5(8). 10.1371/journal.pone.0012485 [DOI] [PMC free article] [PubMed]
- Luier L, Loots DT (2016) Tuberculosis metabolomics reveals adaptations of man and microbe in order to outcompete and survive. Metabolomics 12(3). 10.1007/s11306-016-0969-x
- Lv H, Hung CS, Chaturvedi KS, Hooton TM, Henderson JP. Development of an integrated metabolomic profiling approach for infectious diseases research. Analyst. 2011;136(22):4752–4763. doi: 10.1039/c1an15590c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra S, Hess AM, Johnson JL, Eisenach KD, DeGroote MA, Gitta P, Joloba ML, Kaplan G, Walzl G, Boom WH, Belisle JT. A metabolic biosignature of early response to anti-tuberculosis treatment. BMC Infect Dis. 2014;14:53. doi: 10.1186/1471-2334-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston BJ, Plouffe JF, File TM, Hackman BA, Salstrom SJ, Lipman HB, Kolczak MS, Breiman RF. Incidence of community-acquired pneumonia requiring hospitalization – results of a population-based active surveillance study in Ohio. Arch Intern Med. 1997;157(15):1709–1718. doi: 10.1001/archinte.1997.00440360129015. [DOI] [PubMed] [Google Scholar]
- Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10(6):701–706. doi: 10.1586/eri.12.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina BE, Koraka P, Osterhaus AD. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009;22(4):564–581. doi: 10.1128/CMR.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason S, van Furth AMT, Solomons R, Wevers RA, van Reenen M, Reinecke CJ. A putative urinary biosignature for diagnosis and follow-up of tuberculous meningitis in children: outcome of a metabolomics study disclosing host-pathogen responses. Metabolomics. 2016;12(7):110. doi: 10.1007/s11306-016-1053-2. [DOI] [Google Scholar]
- Mayeux R. Biomarkers: potential uses and limitations. NeuroRx. 2004;1(2):182–188. doi: 10.1602/neurorx.1.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnaghi FS, Caudy AA. Challenges of analyzing different classes of metabolites by a single analytical method. Bioanalysis. 2014;6(24):3393–3416. doi: 10.4155/bio.14.236. [DOI] [PubMed] [Google Scholar]
- MMWR Recommendations and reports (1996) Defining the public health impact of drug-resistant Streptococcus pneumoniae: report of a working group. https://www.cdc.gov/mmwr/preview/mmwrhtml/00040449.htm. Accessed 16 May 2017 [PubMed]
- Mushtaq MY, Choi YH, Verpoorte R, Wilson EG. Extraction for metabolomics: access to the metabolome. Phytochem Anal. 2014;25(4):291–306. doi: 10.1002/pca.2505. [DOI] [PubMed] [Google Scholar]
- Näsström E, Thieu NT, Dongol S, Karkey A, Voong Vinh P, Ha Thanh T, Johansson A, Amit A, Thwaites G, Dolecek C, Basnyat B, Baker S, Antti H (2014) Salmonella typhi and Salmonella paratyphi A elaborate distinct systemic metabolite signatures during enteric fever. eLife. 10.7554/eLife.03100 [DOI] [PMC free article] [PubMed]
- Ng JS, Ryan U, Trengove RD, Maker GL. Development of an untargeted metabolomics method for the analysis of human faecal samples using Cryptosporidium-infected samples. Mol Biochem Parasitol. 2012;185(2):145–150. doi: 10.1016/j.molbiopara.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16(9):373–378. doi: 10.1016/S0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- Olszewski KL, Llinás M. Central carbon metabolism of Plasmodium parasites. Mol Biochem Parasitol. 2011;175(2):95–103. doi: 10.1016/j.molbiopara.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappa V, Seydel K, Gupta S, Feintuch CM, Potchen MJ, Kampondeni S, Goldman-Yassen A, Veenstra M, Lopez L, Kim RS, Berman JW, Taylor T, Daily JP. Lipid metabolites of the phospholipase A2 pathway and inflammatory cytokines are associated with brain volume in paediatric cerebral malaria. Malar J. 2015;14:513. doi: 10.1186/s12936-015-1036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry CM, Wijedoru L, Arjyal A, Baker S. The utility of diagnostic tests for enteric fever in endemic locations. Expert Rev Anti Infect Ther. 2011;9(6):711–725. doi: 10.1586/eri.11.47. [DOI] [PubMed] [Google Scholar]
- Pawlotsky JM. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004;12(2):96–102. doi: 10.1016/j.tim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Pearle MS. Does the midstream urine culture and sensitivity test reliably predict infected urine near a ureteral obstruction? Nat Clin Pract Urol. 2004;1(1):16–17. doi: 10.1038/ncpuro0004. [DOI] [Google Scholar]
- Ptolemy AS, Rifai N. What is a biomarker? Research investments and lack of clinical integration necessitate a review of biomarker terminology and validation schema. Scand J Clin Lab Invest Suppl. 2010;242:6–14. doi: 10.3109/00365513.2010.493354. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan S, Docampo MD, MacRae JI, Pujol FM, Brooks CF, van Dooren GG, Hiltunen JK, Kastaniotis AJ, McConville MJ, Striepen B. Apicoplast and endoplasmic reticulum cooperate in fatty acid biosynthesis in apicomplexan parasite Toxoplasma gondii. J Biol Chem. 2012;287(7):4957–4971. doi: 10.1074/jbc.M111.310144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramautar R, Nevedomskaya E, Mayboroda OA, Deelder AM, Wilson ID, Gika HG, Theodoridis GA, Somsen GW, de Jong GJ. Metabolic profiling of human urine by CE-MS using a positively charged capillary coating and comparison with UPLC-MS. Mol Biosyst. 2011;7(1):194–199. doi: 10.1039/C0MB00032A. [DOI] [PubMed] [Google Scholar]
- Rojo D (2014) Metabolómica e integración multiómica en organismos unicelulares. Hacia la comprensión de sistemas biológicos. Dissertation, CEU San Pablo University
- Rojo D, Gosalbes MJ, Ferrari R, Pérez-Cobas AE, Hernández E, Oltra R, Buesa J, Latorre A, Barbas C, Ferrer M, Moya A. Clostridium difficile heterogeneously impacts intestinal community architecture but drives stable metabolome responses. ISME J. 2015;9(10):2206–2220. doi: 10.1038/ismej.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald A. The etiology of urinary tract infection: traditional and emerging pathogens. Dis Mon. 2003;49(2):71–82. doi: 10.1067/mda.2003.8. [DOI] [PubMed] [Google Scholar]
- Rowan DD. Volatile metabolites. Metabolites. 2011;1(1):41–63. doi: 10.3390/metabo1010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Sugimoto M, Igarashi K, Saito K, Shao L, Katsumi T, Tomita K, Sato C, Okumoto K, Nishise Y, Watanabe H, Tomita M, Ueno Y, Soga T. Dynamics of serum metabolites in patients with chronic hepatitis C receiving pegylated interferon plus ribavirin: a metabolomics analysis. Metabolism. 2013;62(11):1577–1586. doi: 10.1016/j.metabol.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, Nunez D, Sweatman BC, Haselden JN, Cox RD, Connor SC, Griffin JL. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genomics. 2007;29(2):99–108. doi: 10.1152/physiolgenomics.00194.2006. [DOI] [PubMed] [Google Scholar]
- Sanchez EL, Lagunoff M. Viral activation of cellular metabolism. Virology. 2015;479–480:609–618. doi: 10.1016/j.virol.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Ghosh S, Basant A, Malusare S, Johri P, Pathak S, Sharma S, Sonawat HM. Global host metabolic response to Plasmodium vivax infection: a 1H NMR based urinary metabonomic study. Malar J. 2011;10:384. doi: 10.1186/1475-2875-10-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Ghosh S, Pathak S, Gogtay N, Thatte U, Doshi M, Sharma S, Sonawat HM. Metabolomic analysis of urine samples of vivax malaria in-patients for biomarker identification. Metabolomics. 2015;11(5):1351–1362. doi: 10.1007/s11306-015-0791-x. [DOI] [Google Scholar]
- Sengupta A, Ghosh S, Das BK, Panda A, Tripathy R, Pied S, Ravindran B, Pathak S, Sharma S, Sonawat HM. Host metabolic responses to Plasmodium falciparum infections evaluated by 1H NMR metabolomics. Mol Biosyst. 2016;12(11):3324–3332. doi: 10.1039/C6MB00362A. [DOI] [PubMed] [Google Scholar]
- Shahfiza N, Osman H, Hock TT, Shaari K, Abdel-Hamid AH. Metabolomics for characterization of gender differences in patients infected with dengue virus. Asian Pac J Trop Med. 2015;8(6):452–457. doi: 10.1016/j.apjtm.2015.05.012. [DOI] [PubMed] [Google Scholar]
- Singh S, Srivastava S, Roy R, Gaurav K, Kumar S, Sonkar AA, Goel MM, Garg R. Metabolic profiling of cervical tubercular lymphadenitis tissues by proton HR-MAS NMR spectroscopy. Metabolomics. 2014;10(5):975–985. doi: 10.1007/s11306-014-0626-1. [DOI] [Google Scholar]
- Slupsky CM, Rankin KN, Fu H, Chang D, Rowe BH, Charles PG, McGeer A, Low D, Long R, Kunimoto D, Sawyer MB, Fedorak RN, Adamko DJ, Saude EJ, Shah SL, Marrie TJ. Pneumococcal pneumonia: potential for diagnosis through a urinary metabolic profile. J Proteome Res. 2009;8(12):5550–5558. doi: 10.1021/pr9006427. [DOI] [PubMed] [Google Scholar]
- Soga T, Sugimoto M, Honma M, Mori M, Igarashi K, Kashikura K, Ikeda S, Hirayama A, Yamamoto T, Yoshida H, Otsuka M, Tsuji S, Yatomi Y, Sakuragawa T, Watanabe H, Nihei K, Saito T, Kawata S, Suzuki H, Tomita M, Suematsu M. Serum metabolomics reveals gamma-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J Hepatol. 2011;55(4):896–905. doi: 10.1016/j.jhep.2011.01.031. [DOI] [PubMed] [Google Scholar]
- Sonawat HM, Sharma S. Host responses in malaria disease evaluated through nuclear magnetic resonance-based metabonomics. Clin Lab Med. 2012;32(2):129–142. doi: 10.1016/j.cll.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Su L, Huang Y, Zhu Y, Xia L, Wang R, Xiao K, Wang H, Yan P, Wen B, Cao L, Meng N, Luan H, Liu C, Li X, Xie L (2014) Discrimination of sepsis stage metabolic profiles with an LC/MS-MS-based metabolomics approach. BMJ Open Respir Res. 10.1136/bmjresp-2014-000056 [DOI] [PMC free article] [PubMed]
- Subramani E, Dutta M, Jothiramajayam M, Joshi M, Srivastava S, Mukherjee A, Chakravarty B, Chaudhury K. Identification of serum metabolic markers for diagnosis of women with dormant genital tuberculosis. Metabolomics. 2016;12(6):99. doi: 10.1007/s11306-016-1042-5. [DOI] [Google Scholar]
- Surowiec I, Orikiiriza J, Karlsson E, Nelson M, Bonde M, Kyamanwa P, Karenzi B, Bergström S, Trygg J, Normark J. Metabolic signature profiling as a diagnostic and prognostic tool in pediatric Plasmodium falciparum malaria. Open Forum Infect Dis. 2015;2(2):ofv062. doi: 10.1093/ofid/ofv062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatman BC, Farrant RD, Lindon JC. NMR of biofluids: detection of 2H-acetate and 2H-formate in urine as an indicator of microbiological contamination. J Pharm Biomed Anal. 1993;11(2):169–172. doi: 10.1016/0731-7085(93)80138-Q. [DOI] [PubMed] [Google Scholar]
- Syed IH, Balakrishnan P, Solomon SS, Murugavel KG, Kumarasamy N, Vidya S, Martin SP, Thyagarajan SP, Mayer KH, Solomon S. HIV-1 western blot assay: what determines an indeterminate status? Indian J Med Sci. 2005;59(10):443–450. doi: 10.4103/0019-5359.17051. [DOI] [PubMed] [Google Scholar]
- Teixeira AR, Hecht MM, Guimaro MC, Sousa AO, Nitz N. Pathogenesis of Chagas’ disease: parasite persistence and autoimmunity. Clin Microbiol Rev. 2011;24(3):592–630. doi: 10.1128/CMR.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent IM, Daly R, Courtioux B, Cattanach AM, Bieler S, Ndung’u JM, Bisser S, Barrett MP (2016) Metabolomics identifies multiple candidate biomarkers to diagnose and stage human African trypanosomiasis. PloS Negl Trop Dis. 10.1371/journal.pntd.0005140 [DOI] [PMC free article] [PubMed]
- Voge NV, Perera R, Mahapatra S, Gresh L, Balmaseda A, Loroño-Pino MA, Hopf-Jannasch AS, Belisle JT, Harris E, Blair CD, Beaty BJ (2016) Metabolomics-based discovery of small molecule biomarkers in serum associated with dengue virus infections and disease outcomes. PLoS Negl Trop Dis. 10.1371/journal.pntd.0004449 [DOI] [PMC free article] [PubMed]
- Voskuil J. How difficult is the validation of clinical biomarkers? F1000Res. 2015;4:101. doi: 10.12688/f1000research.6395.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner J 3rd, Parida SK, Maertzdorf J, Black GF, Repsilber D, Telaar A, Mohney RP, Arndt-Sullivan C, Ganoza CA, Faé KC, Walzl G, Kaufmann SH (2012) Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS ONE 7(7). 10.1371/journal.pone.0040221 [DOI] [PMC free article] [PubMed]
- WHO (2017a) The top 10 causes of death. http://www.who.int/mediacentre/factsheets/fs310/en/. Accessed 29 May 2017
- WHO . Global hepatitis report 2017. Geneva: World Health Organization; 2017. [Google Scholar]
- WHO (2017c) Trypanosomiasis, human African (sleeping sickness). http://www.who.int/mediacentre/factsheets/fs259/en/. Accessed 28 May 2017
- WHO . World health statistics 2017: monitoring health for the SDGs, sustainable development goals. Geneva: World Health Organization; 2017. [Google Scholar]
- Winder CL, Dunn WB, Schuler S, Broadhurst D, Jarvis R, Stephens GM, Goodacre R. Global metabolic profiling of Escherichia coli cultures: an evaluation of methods for quenching and extraction of intracellular metabolites. Anal Chem. 2008;80(8):2939–2948. doi: 10.1021/ac7023409. [DOI] [PubMed] [Google Scholar]
- Xia J, Broadhurst DI, Wilson M, Wishart DS. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics. 2013;9(2):280–299. doi: 10.1007/s11306-012-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki SA, Karande S. Multidrug-resistant typhoid fever: a review. J Infect Dev Ctries. 2011;5(5):324–337. doi: 10.3855/jidc.1405. [DOI] [PubMed] [Google Scholar]
- Zhang QW, Takahashi M, Noguchi Y, Sugimoto T, Kimura T, Okumura A, Ishikawa T, Kakumu S. Plasma amino acid profiles applied for diagnosis of advanced liver fibrosis in patients with chronic hepatitis C infection. Hepatol Res. 2006;34(3):170–177. doi: 10.1016/j.hepres.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang W, Hankemeier T, Ramautar R. Next-generation capillary electrophoresis-mass spectrometry approaches in metabolomics. Curr Opin Biotechnol. 2017;43:1–7. doi: 10.1016/j.copbio.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Zhong L, Zhou J, Chen X, Yin Y. Serum metabolomic study for the detection of candidate biomarkers of tuberculosis. Int J of Clin Exp Pathol. 2016;9(3):3256–3266. [Google Scholar]