

Abstract
Spleen tyrosine kinase (Syk) binds ITAM-bearing receptors in a wide variety of cell types. One such example is the activation of mast cells, basophils and eosinophils via the stimulation of the FcεRI receptor by IgE/allergen complexes. The possible role of Syk in inflammatory signaling cascades has led to the development of pharmacological agents designed to block the Syk catalytic domain as potential novel therapeutics. Whilst the enzymatic activity of Syk lends towards the design of small-molecule inhibitors, other attention has focused on the possibility of targeting Syk expression using anti-sense oligonucleotides as an alternate means of anti-inflammatory therapy. In this study, we compared the ability of multiple optimized Syk siRNA sequences and small-molecule Syk inhibitors to block FcεRI-mediated signal transduction, degranulation and TNFα secretion in the basophilic cell line RBL-2H3. We also characterized the specificity of each siRNA sequence with regards to off-target induction of the interferon-inducible gene IFIT1. We identified a single siRNA sequence, which displayed a favorable profile of efficient Syk knockdown, blockage of FcεRI-mediated signal transduction, degranulation and TNFα secretion and a lack of IFIT1 induction. The effect of this siRNA was comparable to that of the Syk kinase domain inhibitors BAY61-3606 and R406. The identification of an active and specific Syk siRNA could be a basis for the development of therapeutic Syk siRNAs against inflammatory diseases.
Keywords: Allergy, Inflammation, Spleen tyrosine kinase (Syk), RBL-2H3, siRNA, IgE, FcεRI, R406, Piceatannol, BAY61-3606
1. Introduction
Spleen tyrosine kinase (Syk) is expressed widely in the immune system including in leukocytes, platelets and dendritic cells [1]. A wide variety of cell surface receptors on immune system cells and other cell types have been postulated to transduce signals via the binding and activation of Syk [2], [3], [4], [5]. For the most part these include receptors bearing the immunoreceptor tyrosine-based activation motif (ITAM) [2]. However, other non-ITAM-bearing receptors such as those for IL-2, IL-15, erythropoietin and lipopolysaccharide (endotoxin/LPS) have also been postulated to signal via the activation of Syk [6], [7], [8], [9].
The FcεRI receptor complex is expressed on mast cells, basophils and eosinophils and signal transduction following the binding of IgE/allergen leads to Syk binding and phosphorylation [3], [10]. Syk is then believed to mediate the direct phosphorylation several proteins within an inflammatory complex including linker for activator of T cells (LAT), SH2-domain-containing leukocyte protein-76 (SLP-76) and Vav [3]. Following the assembly and activation of this complex of proteins, activation of MAPKs, calcium-flux and PLCγ1 and PLCγ2 phosphorylation lead to degranulation, lipid mediator and cytokine synthesis. Syk has gathered a great deal of attention as a potentially novel target for the treatment of allergic and other inflammatory disorders [3], [10], [11]. In particular, several small-molecule inhibitors of the Syk kinase domain have shown efficacy both human and animal models of allergic disease [12], [13], [14], [15], [16].
Recent advances in the use of small interfering RNA (siRNA) technologies as a means of post-transcriptional gene silencing have provided a potential alternative to the use of small-molecule inhibitors for the inhibition of disease targets [17], [18], [19]. In this study, we designed a range of different Syk siRNA sequences and tested them for their ability to knockdown Syk in the rat basophilic RBL-2H3 cell line. Syk siRNAs were also tested for their specificity by investigating the induction of an interferon response, as analyzed by the expression of IFN-induced protein with tetratricopeptide repeats-1 (IFIT1/ISG56), which is a characteristic inflammatory response to viral dsRNA [20]. Selected siRNAs displaying a combination of efficient and specific knockdown of Syk were compared with the previously published Syk kinase inhibitors R406 [14], BAY61-3606 [15] and piceatannol [21] for their effect on FcεRI signal transduction and basophilic degranulation. The identification of specific and cellularly active Syk sequences in this study provides a basis for the development of Syk siRNAs in more complex in vivo models of inflammatory diseases.
2. Materials and methods
2.1. Reagents and siRNAs
Individual Duplex siRNAs were purchased from Thermo Fisher Scientific (Lafayette, CO) and Applied Biosystems/Ambion (Austin, TX). Antibodies to Syk (SYK-01), PLCγ1 (1F1), β-actin (C4) and LAT (FL-233) were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Antibodies to phospho-PLCγ1 (Tyr783), phospho-Zap70 (Tyr319)/Syk (Tyr352), Phospho-Erk (Thr202/Tyr204) and p44/42 MAP Kinase were obtained from Cell Signaling Technology (Danvers, MA). The SLP-76 (Tyr128), SLP-76, and high affinity IgE receptor (FcεRI) (BC4) antibodies were from BD Biosciences (San Jose, CA). The phospho-LAT (Tyr226) and phosphotyrosine (4G10) antibodies were purchased from Millipore (Billerica, MA). The mouse anti-DNP IgE antibody (clone TIB142) was a kind gift from Dr. Takeshi Kono (Nippon Boehringer Ingelheim Co., LTD, Tokyo). Bovine serum albumin-2,4-dinitrophenyl (DNP-BSA) was purchased from Invitrogen (Karlsruhe, Germany). Piceatannol was purchased from Merck (Darmstadt, Germany).
2.2. Culture and treatment of RBL-2H3 cells with pharmacological agents
RBL-2H3 cells (ATCC: CRL-2256) were routinely cultured in minimum essential medium (MEM) supplemented with 10% fetal calf serum (FCS). For the treatment of cells with pharmacological agents for later analysis of proteins by Western blotting, 5 × 105 cells were seeded into 24-well plates and cultured overnight. Cells were then washed once with phosphate buffered saline (PBS) and then incubated for 30 min in MEM without FBS plus either inhibitors or the vehicle dimethyl sulfoxide (DMSO). Cells were then activated for 10 min by the addition of the anti-FcεRI (BC4) antibody (0.02 μg/ml). Cells were then washed once with ice-cold PBS and lysed at 4 °C for 30 min in Lysis Buffer (150 mM NaCl, 20 mM Tris, pH 7.5, 1% NP40, 5 mM EDTA, 50 mM NaF, 20 μM Na3VO4) supplemented with Halt Protease Inhibitor Cocktail and Halt Phosphatase Inhibitor Cocktail (both from Thermo Fisher Scientific, Rockford, IL). Lysates were then cleared by centrifugation and prepared for Western blotting by the addition of NuPAGE® Sample Reducing Agent and LDS Sample Buffer (both from Invitrogen).
2.3. Transfection of RBL-2H3 and MEF cells with siRNAs
The Nucleofector L Kit (#VCA-1005) (Lonza Cologne AG, Germany) was used for electroporation of RBL-2H3 cells with siRNAs. 1 × 106 cells were electroporated in cuvettes in Nucleofector L Solution with 420 nM of each siRNA using a Nucleofector® Device (Lonza, #AAD-1001) with program L-029. Cells were then diluted in 6 well plates with MEM/10% FCS to give a final siRNA concentration of 20 nM. Cells were cultivated for a further 2 days prior to application in cellular assays or for the analysis of Syk mRNA levels by TaqMan PCR as described in the following section.
DharmaFECT 2 siRNA Transfection Reagent (#T-2002-01) (Thermo Fisher Scientific) was used for the transfection of mouse embryonic fibroblast (MEF) cells for the analysis of the induction of the interferon (IFN)-inducible gene IFIT1. MEF cells were routinely cultured in DMEM/10% FCS and seeded at a concentration of 4.2 × 105 in 6-well plates and grown overnight until they reached 90% confluence. Cells were then transfected with 20 nM of each siRNA mixed with DharmaFECT 2 siRNA Transfection Reagent. Following 24 h of culture, mRNA was analyzed by TaqMan PCR.
2.4. Gene expression analysis
For quantitative analysis of gene expression total RNA was isolated from cell culture lysates according to the RNeasy protocol (Qiagen, Hilden, Germany). The purified total RNA was stored at −20 °C. The gene expression levels were determined by TaqMan® analysis in a 7900HT Sequence Detection System (Applied Biosystems Inc., Foster City, CA) using the TaqMan® EZ RT-PCR reagent kit (Applied Biosystems) for reverse transcription and PCR amplification in ABI PRISM 384-well optical reaction plates (Applied Biosystems). For the detection of the respective transcripts, the following primers and probes were used: Syk_rat (Applied Biosystems, # Rn00562684_m1), Syk_mouse (Applied Biosystems, # Mm00441649_m1), RNA PolII_rat: forward primer: 5′-GCAGGCGAGAGCGTTGAG-3′, reverse primer: 5′-CATTGGTATAAT CAAAACGGAACTTC-3′, probe: 5′-FAM-CTGGCTACACTTAAGCC TTCTAATAAAGC-TAMRA-3′, RNA PolII_mouse: forward primer: 5′-GCCAAAGACTCCTTCACTCACT GT-3′, reverse primer: 5′-TTCCAAGCGGCAAAGAATGT-3′, probe: 5′-FAM-TGGCTCT TTCAGCATCTCGTGCAGATT-TAMRA-3′ and Ifit_mouse: forward primer: 5′-GGCAGG TTTCTGAGGAGTTCTG-3′, reverse primer: 5′-CATCAGCATTCTCTCCCATGGT-3′, probe: 5′-FAM-AAAACCCAGAGAACAGCTACCACCTTTACAGC-TAMRA-3′. Relative quantities of expression levels were determined by comparison of ct-values with a dilution series of a standard total RNA sample and normalized for the 18S rRNA quantity. For the presentation of the expression levels, normalized relative quantities of a transcript were divided by the mean value of the respective control group and displayed as%-control values.
2.5. Western blot analysis of proteins
Cell lysates were loaded onto NuPAGE® 4–12% Bis–Tris gels (Invitrogen) and transferred to Immobilon-P Transfer Membranes (Millipore, Billerica, MA) by wet blotting. After blocking with blocking buffer (5% non-fat milk in TBS), membranes were incubated under gentle agitation with the respective primary antibodies diluted in blocking buffer for 1 h at room temperature. Membranes were then washed four times for 10 min with TBS-T (TBS with 0.05% Tween 20) and then incubated with peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Europe Ltd., Suffolk, UK) at a dilution of 1/5000 in blocking buffer (Jackson ImmunoResearch Europe Ltd, Suffolk, UK). Membranes were then washed again in TBS-T as described above and then Western blots developed using the Enhanced Chemiluminescence (ECL) reagents (PerkinElmer, Waltham, MA) according to the manufacturer’s instructions. Membranes were in some cases stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) and then analyzed with additional antibodies.
2.6. β-Hexosaminidase assay
The release of β-hexosaminidase enzyme activity from RBL-2H3 cells was used as a quantifiable readout for degranulation [22]. One day following transfection of cells with siRNAs, media was replaced by MEM/10% FCS containing 1 μg/ml of the mouse anti-DNP IgE antibody (clone TIB142) and the cells then cultured for a further 24 h. The following day, cells were washed twice with PBS and then cultured for 30 min in sterile filtered Tyrode’s Buffer (135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.6 mM Glucose, 20 mM HEPES, 1 mg/ml BSA, pH 7.4). For testing of the effects of pharmacological agents on degranulation, non-transfected cells were treated for 30 min with Tyrode’s Buffer plus inhibitors. Cells were then activated by the addition of DNP-BSA (100 ng/ml) in Tyrode’s Buffer for 60 min. Supernatants were collected and cleared by centrifugation prior to analysis using the β-hexosaminidase assay as follows. 50 μl of supernatants were combined with 50 μl of substrate solution (4 mM 4-nitrophenyl N-acetyl-β-d-glucosaminide in McIlvaine’s Buffer, pH 4.5) in 96-well plates and incubated for 30 min at 37 °C. Reactions were then terminated by the addition of 150 μl of 0.2 M Glycine, pH 10.7. Absorbance was measured at 405 nm.
2.7. TNFα and histamine measurements
Following transfection with siRNAs, cells were incubated as described above with the mouse anti-DNP IgE antibody. The following day, cells were washed and then cultured for 6 h in MEM/10% FCS plus DNP-BSA (100 ng/ml). Supernatants were then cleared by centrifugation and analyzed for levels of rat TNFα using the rat TNFα Immunoassay (R&D Systems, Minneapolis, MN) and histamine using a specific enzyme immunoassay (EIA) (IMMUNOTECH, Czech Republic), according to the manufacture’s instructions.
3. Results
3.1. Design of siRNAs against Syk
Ten siRNA sequences (A–J) were designed against Syk with the aim of knocking down Syk in the rat RBL-2H3 cell line (Fig. 1 ). Sequences were based on the RefSeq information for human (NM_003177), mouse (NM_011518) and rat (NM_012758) Syk mRNAs. Sequences A–F had 100% homology with rat Syk alone, whereas sequences G–J were pan siRNAs with complete homology to the rat, mouse and human Syk transcripts.
Fig. 1.

Species specificity and sequence of each siRNA against Syk. The sequence of the Syk siRNAs A–J is shown along with the specific of location homology to the rat Syk mRNA (RefSeq_dna: NM_012758). The species specificity indicates that this siRNA has 100% homology with the given mRNA for either rat, mouse or human Syk. CDS; coding sequence, 3′UTR; 3′ untranslated region, bp; base pair number.
3.2. Identification of active siRNAs against rat Syk
RBL-2H3 cells were transfected with each siRNA as described in Section 2. Syk mRNA levels were determined using TaqMan PCR (Fig. 2 A). Syk levels were normalized to RNA Polymerase II in each sample. An siRNA against GAPDH, as well as cells electroporated in the absence of siRNAs were used as negative controls. These two controls led to a minor decrease in Syk mRNA levels compared to untreated cells. Sequence A was the most potent siRNA with regards to Syk mRNA knockdown. Meanwhile sequence D, was the least potent. Whole cell protein extracts were also analyzed for Syk expression by Western blotting with a specific antibody (Fig. 2B). As a control, levels of β-actin were also examined to ensure equal loading of cellular proteins for each sample. Syk protein levels were reduced in a manner consistent with the potency of each siRNA towards Syk mRNA levels, with sequence A displaying the most potent knockdown of Syk protein.
Fig. 2.
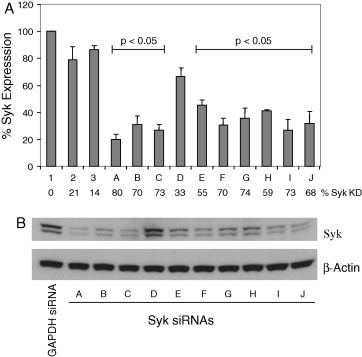
Identification of active siRNAs against rat Syk. (A) RBL-2H3 cells were transfected with Syk siRNAs A-J. In addition, three controls were used. (1) untreated cells, (2) cells transfected with GAPDH siRNA, (3) cells electroporated in the absence of siRNAs. Syk mRNA levels were measured 2 days after transfection by TaqMan PCR. Expression of Syk was normalized to levels of RNA polymerase II mRNA and then expressed as the percentage expression compared to that in untreated cells. Error bars represent the standard deviation of the mean for triplicate determinations from three separate experiments. A paired Student’s t-test was used to compare the statistical significance of the difference between data points. With the exception of Syk siRNA D, each Syk siRNA significantly reduced Syk mRNA expression relative to the GAPDH siRNA control with a p-value of less than 0.05. The percentage knockdown of Syk mRNA compared to untreated cells is also presented. (B) Total cell lysates were analyzed for levels of Syk protein by Western blot. Levels of β-actin were examined to control for protein loading. Note that two Syk proteins (Syk and the smaller SykB) are expressed in RBL-2H3 cells as a result of alternate mRNA splicing [48].
3.3. Induction of the interferon-responsive gene IFIT1 by active Syk siRNAs
Induction of interferon (IFN) expression by double stranded RNA (dsRNA) comprises a first line of cellular defense against viral infection [20]. Viral dsRNA activates a range of cellular pathways including the dsRNA recognition protein PKR [23], toll-like receptor 3 (TLR3) [24] and the 2′-5′-oligoadenylate-dependent ribonuclease L (RNase L) [25], [26] leading to inhibition of cellular translation and the expression of stress response genes including IFNs. Following IFN signaling, a wave of other genes are expressed including the IFN-stimulated genes (ISG), which function to block viral replication [20], [27]. Activation of the IFN response has also been demonstrated in siRNA transfected cells and this complicates the therapeutic potential of these molecules [28], [29]. In particular, Syk is considered a major target for inflammatory disorders and an induction of inflammatory mediators due to an anti-viral cellular response would be an unacceptable side-effect when designing Syk siRNA therapeutics.
The expression of IFN-induced protein with tetratricopeptide repeats-1 (IFIT1/ISG56) was analyzed in mouse embryonic fibroblast (MEF) cells transfected with a selection of active Syk siRNAs (A, B, C, G, H, I and J) in order to determine if these siRNAs activated anti-viral cellular pathways. MEF cells have previously been shown to express interferon-responsive genes in response to siRNAs and therefore represent a valid experimental system [28], [30]. Syk siRNAs C, H and I increased the expression of IFIT1 by twofold suggestive of an activation of the IFN response (Fig. 3 ). The GAPDH siRNA as well as Syk siRNAs A, B, G and J were without effect on IFIT1 expression.
Fig. 3.
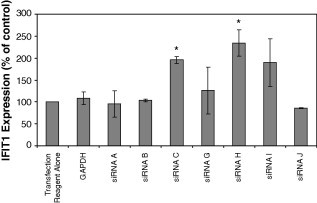
Expression of IFIT1 in response to siRNA transfection. MEF cells were transfected with Syk siRNAs A, B, C, G, H, I and J and the control siRNA against GAPDH and the expression of IFIT1 determined 2 days later using TaqMan PCR. IFIT1 expression was normalized to levels of RNA polymerase II mRNA and then expressed as the percentage expression compared to that in cell treated with the transfection reagent alone. Error bars represent the standard deviation of the mean for duplicate determinations from two separate experiments. A paired Student’s t-test was used to compare the statistical significance of the difference between data points. With the exception of Syk siRNAs C and H (marked with an asterisk), no significant increase in IFIT1 expression relative to the GAPDH siRNA control was observed with a p-value of less than 0.05.
3.4. Knockdown of Syk reduces FcεRI-mediated signal transduction in a manner similar to Syk kinase inhibitors
The ability of a selection of active Syk siRNAs (A, G and J) to block FcεRI-mediated signal transduction was examined in RBL-2H3 cells. Crosslinking of the FcεRI receptor in these cells led to a rapid phosphorylation Syk at tyrosine 346 (Fig. 4 A). In addition, the Syk substrates LAT (tyrosine 226) and SLP-76 (tyrosine 128) as well as the associated protein PLCγ1 (tyrosine 783) and downstream protein Erk (threonine 202/tyrosine 204) were also phosphorylated. A general activation of protein tyrosine phosphorylation with predominant signals at 100, 70–75 and 55 kDa was also observed in response to FcεRI receptor crosslinking. Consistent with results presented above (Fig. 2), Syk siRNA A was the most potent in reducing levels of Syk protein. Sequences J and H were respectively less potent than sequence A. The phosphorylation of LAT, SLP-76, PLCγ1 and Erk was decreased by Syk siRNAs in a manner that directly correlated with their effects on Syk expression. Interestingly, whilst the tyrosine phosphorylation of proteins at 100 and 70–75 kDa was reduced by Syk siRNAs, the phosphorylation of the 55 kDa protein was slightly enhanced. This suggests that the phosphorylation of this unidentified 55 kDa protein could be negatively regulated by Syk in response to FcεRI receptor crosslinking. The total levels of LAT, SLP-76, PLCγ1, Erk and β-actin were not affected by the Syk siRNAs indicating that the observed reduction in their respective phospho-forms was due to a decrease in their phosphorylation and not a decrease in their stability or expression.
Fig. 4.
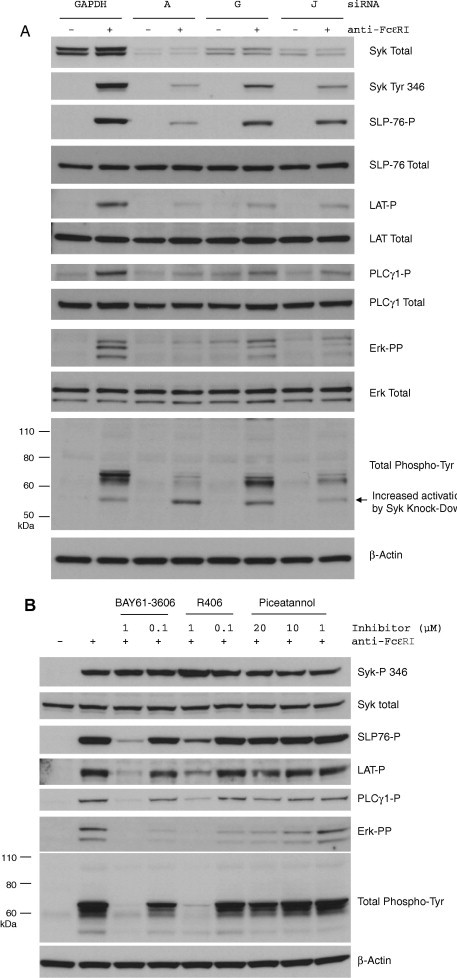
Knockdown of Syk reduces FcεRI-mediated signal transduction in a manner similar to Syk kinase inhibitors. (A) RBL-2H3 cells were transfected with active Syk siRNAs A, G and J or the control siRNA against GAPDH and then treated with or without the FcεRI-crosslinking antibody. Total cell lysates were analyzed by Western blot for expression and phosphorylation of Syk (tyrosine 346), SLP-76 (tyrosine 128), LAT (tyrosine 226), PLCγ1 (tyrosine 783), Erk (threonine 202/tyrosine 204) and total tyrosine-phosphorylated proteins. Total levels of each protein as well as β-actin were also analyzed. (B) Untransfected RBL-2H3 cells were treated with the Syk small molecule kinase inhibitors BAY61-3606 (1 and 0.1 μM), R406 (1 and 0.1 μM) and piceatannol (20, 10 and 1 μM) and then activated with the FcεRI-crosslinking antibody. The phosphorylation of the same proteins mentioned in part A of this figure was analyzed by Western blot. Total Syk and β-actin levels were analyzed to control for protein loading.
We used the small-molecule inhibitors of the Syk kinase domain BAY61-3606, R406 and piceatannol to investigate if a similar inhibition of FcεRI signaling could be observed with Syk inhibitors and Syk siRNAs. The FcεRI-induced phosphorylation of LAT, SLP-76, PLCγ1, Erk and proteins at 100, 70–75 and 55 kDa were dose-dependently inhibited by the BAY61-3606 and R406 compounds (Fig. 4B). Interestingly, in contrast to the effects of Syk siRNAs, the phosphorylation of the 55 kDa protein decreased with BAY61-3606 and R406. This may represent a discrepancy in the mechanism of action of these two inhibitory approaches. The phosphorylation of Syk at tyrosine 346 was unaffected by these inhibitors suggesting that although it is activated following FcεRI crosslinking, that it is not a substrate for Syk autocatalytic activity. When used at 20 μM, piceatannol led to a minor decrease in the phosphorylation of LAT, SLP-76, PLCγ1, Erk and the 100, 70–75 and 55 kDa proteins, yet the inhibitor was without effect at the lower concentrations of 10 and 1 μM.
3.5. Syk siRNAs and inhibitors block degranulation and TNFα expression
The activation of mast cells and basophils via IgE complexes leads to degranulation and secretion of cytokines. We tested the ability of Syk siRNAs A, G and J and the Syk inhibitors to block these cellular responses in RBL-2H3 cells. All three Syk siRNAs potently blocked the release of β-hexosaminidase from activated RBL-2H3 cells (Fig. 5 A). Sequence G was shown above to knockdown Syk protein to a lesser extent than siRNAs A and J. Consistent with this, the release of β-hexosaminidase was inhibited to a lesser extent. The BAY61-3606 and R406 Syk inhibitors also blocked β-hexosaminidase release (Fig. 5B). Interestingly, although 10 and 1 μM piceatannol was shown above to have no impact on FcεRI signal transduction, these concentrations effectively blocked β-hexosaminidase release. This indicates that although piceatannol has no impact on Syk-mediated signal transduction, it exerts unrelated anti-inflammatory effects in activated RBL-2H3 cells. Each Syk siRNA and inhibitor also blocked the release of TNFα and histamine by RBL-2H3 cells activated via FcεRI-crosslinking (Table 1 ).
Fig. 5.
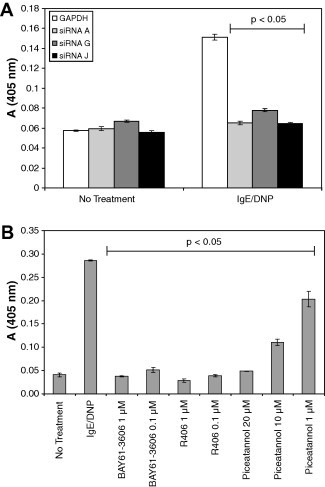
Syk siRNAs and inhibitors block FcεRI-mediated β-hexosaminidase release. RBL-2H3 cells were treated with Syk siRNAs (A) and kinase inhibitors (B) and then activated by overnight incubation in the mouse anti-DNP IgE antibody followed by addition of DNP-BSA. β-Hexosaminidase activity was measure in culture supernatants as described in Section 2. Error bars represent the standard deviation of the mean for quadruplicate determinations. A paired Student’s t-test was used to compare the statistical significance of the difference between data points. All treatments with Syk siRNAs and inhibitors led to a significantly reduced secretion of β-hexosaminidase compared to the control with a p-value of less than 0.05.
Table 1.
Syk Knockdown and kinase inhibitors block TNFα expression and degranulation of histamine. RBL-2H3 cells were treated with the small-molecule inhibitors and siRNAs prior to activation of the FcεRI receptor with IgE and DNP-albumin as described in Section 2. The percentage release of TNFα and histamine compared to IgE/DNP alone is presented. For reference, the IgE/DNP positive control resulted in the release of 30 pg/ml TNFα and 21.3 nM histamine. Results are typical of two identical experiments.
| TNFα (% control) | Histamine (% control) | |
|---|---|---|
| No Treatment | 0 | 0 |
| IgE/DNP | 100 | 100 |
| BAY61-3606 1 μM | 0 | 0 |
| BAY61-3606 0.1 μM | 0 | 0 |
| R406 1 μM | 0 | 0 |
| R406 0.1 μM | 9 | 0 |
| Piceatannol 20 μM | 23 | 13 |
| Piceatannol 1 μM | 29 | 79 |
| siRNA A | 0 | 0 |
| siRNA G | 28 | 12 |
| siRNA J | 34 | 21 |
4. Discussion
Syk is widely expressed in the immune system where it is implicated in the transmission of signals via Fc receptors and other cell surface proteins [1]. This has led to Syk gaining a great deal of interest as a potential therapeutic target for the treatment of inflammatory diseases such as asthma [3], rheumatoid arthritis (RA) [31] and lupus [32]. Meanwhile, recent reports have suggested that Syk may represent a novel therapeutic target for the treatment of B and T cell lymphomas [33], [34]. Current approaches towards Syk inhibition include the use of small molecule kinase domain inhibitors and in particular the R406 compound has shown efficacy in animal and human models of RA [16], [32] and immune thrombocytopenic purpura (ITP) [35].
A novel approach towards Syk inhibition could be realized in the blockage of Syk expression using techniques such as anti-sense oligonucleotides or RNAi. These approaches potentially enable the reduction of gene expression as a means of target modulation as an alternative to the action of small-molecule inhibitors for which there are only a limited number of ‘druggable’ targets. Aerolized Syk anti-sense oligonucleotides have been show to reduce Syk expression in alveolar macrophages and also blocked IgG/antigen-complex induced nitric oxide and TNFα release, pulmonary infiltration of inflammatory cells and allergic contraction of the trachea [36], [37], [38]. These studies highlight the potential efficacy of targeting Syk expression levels as a novel therapeutic principle in airway inflammatory diseases. Yet the potency of different siRNAa towards Syk knockdown and also the specificity of these sequences have not been directly investigated.
In this study, we designed a range of different Syk siRNA sequences and tested their ability to knockdown Syk in the rat basophilic RBL-2H3 cell line. Syk siRNAs A–C and E–J all knocked down Syk mRNA and protein expression and each tested siRNA was also able to block FcεRI-induced signal transduction, degranulation and TNFα release in a manner similar to the Syk inhibitors BAY61-3606 and R406. Interestingly, the commonly used and published Syk inhibitor piceatannol was shown to block RBL-2H3 cell degranulation and TNFα release at concentrations which did not affect Syk-mediated signal transduction. This highlights the Syk-independent anti-inflammatory properties of this compound that must be considered in future studies. The blockage of FcεRI signaling by each siRNA was directly proportional to their effect on Syk expression. These results not only suggest a fundamental role of Syk in FcεRI signaling, but also demonstrate the efficacy of the siRNA approach in the targeting of FcεRI-dependent allergic responses.
The clinical potential of siRNAs is somewhat limited by complications associated with activation of cellular anti-viral interferon-inducible genes and unanticipated off-target effects on other mRNAs. Syk siRNA A was a unique sequence in that it displayed the most potent knockdown of Syk expression and reduction of FcεRI-activated signaling and cellular responses, coupled with a lack of effect on the cellular IFN response. Furthermore, Affymetrics analysis of the effect of Syk siRNA A on the global expression of rat mRNAs in RBL-2H3 cells revealed no off-target regulatory effects (data not shown). The identification of this sequence and its desirable in vitro profile, raises the possibility of this sequence becoming a candidate for progress into Syk-dependent diseases models in vivo. Other limiting factors for potential siRNA therapeutics include complications regarding the efficient delivery to specific target cells and the maintenance of siRNA in vivo stability. Airway delivery of siRNAs via inhalation has the advantage of potentially limiting systemic distribution of the molecules, whilst locally targeting airway cells and their genes [39], [40], [41]. Airway siRNAs have already demonstrated efficient local knockdown of targets including heme oxygenase-1 [42], glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [43] as well as the blockage of the replication and pathogenicity influenza virus A [44], respiratory syncytial virus (RSV) [45], [46] and SARS coronavirus (SCV) [47]. Future studies are warranted to clarify the potential of inhaled Syk siRNAs in models of airway inflammatory cells in vivo.
Acknowledgments
We would like to thank the following people at Boehringer Ingelheim Pharma for their excellent technical assistance and support of this work; Andreas Grieser, Bernd Guilliard, Susanne Müller, Christine Pischzan, Margit Ried, Christine Strasser and Eva Wex. We would also like to thank the following academic staff members at Boehringer Ingelheim Pharma for their support and contributions; Drs. Thierry Bouyssou, Florian Gantner, Matthias Grauert, Tobias Hildebrandt, Silke Hobbie, Matthias Hoffmann, Takeshi Kono, Peter Seither, Eric Simon and Stefan-Lutz Wollin.
References
- 1.Turner M., Schweighoffer E., Colucci F., Di Santo J.P., Tybulewicz V.L. Immunol. Today. 2000;21:148–154. doi: 10.1016/s0167-5699(99)01574-1. [DOI] [PubMed] [Google Scholar]
- 2.Underhill D.M., Goodridge H.S. Trends Immunol. 2007;28:66–73. doi: 10.1016/j.it.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Wong B.R., Grossbard E.B., Payan D.G., Masuda E.S. Expert. Opin. Investig. Drugs. 2004;13:743–762. doi: 10.1517/13543784.13.7.743. [DOI] [PubMed] [Google Scholar]
- 4.Ohtsuka M., Arase H., Takeuchi A., Yamasaki S., Shiina R., Suenaga T., Sakurai D., Yokosuka T., Arase N., Iwashima M., Kitamura T., Moriya H., Saito T. Proc. Natl. Acad. Sci. USA. 2004;101:8126–8131. doi: 10.1073/pnas.0401119101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki-Inoue K., Fuller G.L., Garcia A., Eble J.A., Pohlmann S., Inoue O., Gartner T.K., Hughan S.C., Pearce A.C., Laing G.D., Theakston R.D., Schweighoffer E., Zitzmann N., Morita T., Tybulewicz V.L., Ozaki Y., Watson S.P. Blood. 2006;107:542–549. doi: 10.1182/blood-2005-05-1994. [DOI] [PubMed] [Google Scholar]
- 6.Ratthe C., Girard D. J. Leukoc. Biol. 2004;76:162–168. doi: 10.1189/jlb.0605298. [DOI] [PubMed] [Google Scholar]
- 7.Minami Y., Nakagawa Y., Kawahara A., Miyazaki T., Sada K., Yamamura H., Taniguchi T. Immunity. 1995;2:89–100. doi: 10.1016/1074-7613(95)90081-0. [DOI] [PubMed] [Google Scholar]
- 8.Duprez V., Blank U., Chretien S., Gisselbrecht S., Mayeux P. J. Biol. Chem. 1998;273:33985–33990. doi: 10.1074/jbc.273.51.33985. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhary A., Fresquez T.M., Naranjo M.J. Immunol. Cell Biol. 2007 doi: 10.1038/sj.icb7100030. [DOI] [PubMed] [Google Scholar]
- 10.Ulanova M., Duta F., Puttagunta L., Schreiber A.D., Befus A.D. Expert. Opin. Ther. Targets. 2005;9:901–921. doi: 10.1517/14728222.9.5.901. [DOI] [PubMed] [Google Scholar]
- 11.Bajpai M., Chopra P., Dastidar S.G., Ray A. Expert. Opin. Investig. Drugs. 2008;17:641–659. doi: 10.1517/13543784.17.5.641. [DOI] [PubMed] [Google Scholar]
- 12.Meltzer E.O., Berkowitz R.B., Grossbard E.B. J. Allergy Clin. Immunol. 2005;115:791–796. doi: 10.1016/j.jaci.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 13.Guyer B.J., Shimamoto S.R., Bradhurst A.L., Grossbard E.B., Dreskin S.C., Nelson H.S. Allergy Asthma Proc. 2006;27:208–213. doi: 10.2500/aap.2006.27.2861. [DOI] [PubMed] [Google Scholar]
- 14.Braselmann S., Taylor V., Zhao H., Wang S., Sylvain C., Baluom M., Qu K., Herlaar E., Lau A., Young C., Wong B.R., Lovell S., Sun T., Park G., Argade A., Jurcevic S., Pine P., Singh R., Grossbard E.B., Payan D.G., Masuda E.S. J. Pharmacol. Exp. Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto N., Takeshita K., Shichijo M., Kokubo T., Sato M., Nakashima K., Ishimori M., Nagai H., Li Y.F., Yura T., Bacon K.B. J. Pharmacol. Exp. Ther. 2003;306:1174–1181. doi: 10.1124/jpet.103.052316. [DOI] [PubMed] [Google Scholar]
- 16.Weinblatt M.E., Kavanaugh A., Burgos-Vargas R., Dikranian A.H., Medrano-Ramirez G., Morales-Torres J.L., Murphy F.T., Musser T.K., Straniero N., Vicente-Gonzales A.V., Grossbard E. Arthritis Rheum. 2008;58:3309–3318. doi: 10.1002/art.23992. [DOI] [PubMed] [Google Scholar]
- 17.Lu P.Y., Woodle M.C. Methods Mol. Biol. 2008;437:93–107. doi: 10.1007/978-1-59745-210-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood M., Yin H., McClorey G. PLoS Genet. 2007;3:e109. doi: 10.1371/journal.pgen.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C., Li M., Chen C., Yao Q. Expert Opin. Ther. Targets. 2008;12:637–645. doi: 10.1517/14728222.12.5.637. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar S.N., Sen G.C. Pharmacol. Ther. 2004;103:245–259. doi: 10.1016/j.pharmthera.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Oliver J.M., Burg D.L., Wilson B.S., McLaughlin J.L., Geahlen R.L. J. Biol. Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- 22.Silverman M.A., Shoag J., Wu J., Koretzky G.A. Mol. Cell. Biol. 2006;26:1826–1838. doi: 10.1128/MCB.26.5.1826-1838.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A., Haque J., Lacoste J., Hiscott J., Williams B.R. Proc. Natl. Acad. Sci. USA. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 25.Liang S.L., Quirk D., Zhou A. IUBMB Life. 2006;58:508–514. doi: 10.1080/15216540600838232. [DOI] [PubMed] [Google Scholar]
- 26.Zhou A., Paranjape J.M., Der S.D., Williams B.R., Silverman R.H. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- 27.Der S.D., Zhou A., Williams B.R., Silverman R.H. Proc. Natl. Acad. Sci. USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sledz C.A., Holko M., de Veer M.J., Silverman R.H., Williams B.R. Nat. Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 29.Siolas D., Lerner C., Burchard J., Ge W., Linsley P.S., Paddison P.J., Hannon G.J., Cleary M.A. Nat. Biotechnol. 2005;23:227–231. doi: 10.1038/nbt1052. [DOI] [PubMed] [Google Scholar]
- 30.Terenzi F., White C., Pal S., Williams B.R., Sen G.C. J. Virol. 2007;81:8656–8665. doi: 10.1128/JVI.00322-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pine P.R., Chang B., Schoettler N., Banquerigo M.L., Wang S., Lau A., Zhao F., Grossbard E.B., Payan D.G., Brahn E. Clin. Immunol. 2007;124:244–257. doi: 10.1016/j.clim.2007.03.543. [DOI] [PubMed] [Google Scholar]
- 32.Bahjat F.R., Pine P.R., Reitsma A., Cassafer G., Baluom M., Grillo S., Chang B., Zhao F.F., Payan D.G., Grossbard E.B., Daikh D.I. Arthritis Rheum. 2008;58:1433–1444. doi: 10.1002/art.23428. [DOI] [PubMed] [Google Scholar]
- 33.Young R.M., Hardy I.R., Clarke R.L., Lundy N., Pine P., Turner B.C., Potter T.A., Refaeli Y. Blood. 2009;113:2508–2516. doi: 10.1182/blood-2008-05-158618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldman A.L., Sun D.X., Law M.E., Novak A.J., Attygalle A.D., Thorland E.C., Fink S.R., Vrana J.A., Caron B.L., Morice W.G., Remstein E.D., Grogg K.L., Kurtin P.J., Macon W.R., Dogan A. Leukemia. 2008;22:1139–1143. doi: 10.1038/leu.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Podolanczuk A., Lazarus A.H., Crow A.R., Grossbard E., Bussel J.B. Blood. 2008 doi: 10.1182/blood-2008-07-166439. [DOI] [PubMed] [Google Scholar]
- 36.Stenton G.R., Ulanova M., Dery R.E., Merani S., Kim M.K., Gilchrist M., Puttagunta L., Musat-Marcu S., James D., Schreiber A.D., Befus A.D. J. Immunol. 2002;169:1028–1036. doi: 10.4049/jimmunol.169.2.1028. [DOI] [PubMed] [Google Scholar]
- 37.Stenton G.R., Kim M.K., Nohara O., Chen C.F., Hirji N., Wills F.L., Gilchrist M., Hwang P.H., Park J.G., Finlay W., Jones R.L., Befus A.D., Schreiber A.D. J. Immunol. 2000;164:3790–3797. doi: 10.4049/jimmunol.164.7.3790. [DOI] [PubMed] [Google Scholar]
- 38.Ulanova M., Puttagunta L., Kim M.K., Schreiber A.D., Befus A.D. Curr. Opin. Investig. Drugs. 2003;4:552–555. [PubMed] [Google Scholar]
- 39.Bitko V., Barik S. Methods Mol. Biol. 2008;442:75–82. doi: 10.1007/978-1-59745-191-8_6. [DOI] [PubMed] [Google Scholar]
- 40.Durcan N., Murphy C., Cryan S.A. Mol. Pharm. 2008;5:559–566. doi: 10.1021/mp070048k. [DOI] [PubMed] [Google Scholar]
- 41.Popescu F.D. J. Cell. Mol. Med. 2005;9:840–853. doi: 10.1111/j.1582-4934.2005.tb00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X., Shan P., Jiang D., Noble P.W., Abraham N.G., Kappas A., Lee P.J. J. Biol. Chem. 2004;279:10677–10684. doi: 10.1074/jbc.M312941200. [DOI] [PubMed] [Google Scholar]
- 43.Massaro D., Massaro G.D., Clerch L.B. Am J. Physiol. Lung Cell Mol. Physiol. 2004;287:L1066–L1070. doi: 10.1152/ajplung.00067.2004. [DOI] [PubMed] [Google Scholar]
- 44.Tompkins S.M., Lo C.Y., Tumpey T.M., Epstein S.L. Proc. Natl. Acad. Sci. USA. 2004;101:8682–8686. doi: 10.1073/pnas.0402630101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong X., Zhang W., Lockey R.F., Auais A., Piedimonte G., Mohapatra S.S. Genet.Vaccines Ther. 2007;5:4. doi: 10.1186/1479-0556-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bitko V., Musiyenko A., Shulyayeva O., Barik S. Nat. Med. 2005;11:50–55. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- 47.Li B.J., Tang Q., Cheng D., Qin C., Xie F.Y., Wei Q., Xu J., Liu Y., Zheng B.J., Woodle M.C., Zhong N., Lu P.Y. Nat. Med. 2005;11:944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Latour S., Zhang J., Siraganian R.P., Veillette A. EMBO J. 1998;17:2584–2595. doi: 10.1093/emboj/17.9.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]