

Abstract
Non-PCR-based target amplification technologies, including transcription-mediated amplification (TMA), nucleic acid sequence-based amplification (NASBA), and strand displacement amplification (SDA), are currently the basis for a broad range of clinical infectious-disease molecular diagnostics. These amplification technologies are very sensitive and specific and can be used in combination with traditional end-point or “real-time” detection formats. For several nucleic acid targets, TMA, NASBA, and SDA have certain advantages over PCR-based applications. This two-part article will review the molecular basis of each technology and how the technology has been applied to clinical diagnostic systems. The articles will describe the current testing platforms available, U.S. Food and Drug Administration (FDA)- and non-FDA-approved assays, and availability of analyte-specific reagents. In addition, an open-platform system is described that utilizes standardized reagents and methods and allows the user to develop in-house protocols. Finally, applications for the future are discussed.
Introduction
Over the past 10 years, nucleic acid amplification tests (NAAT) have moved from the research setting to routine clinical laboratory use. Molecular-based testing is now widely accepted and recognized as the new “gold standard” for the diagnosis of many infectious agents. NAAT are highly sensitive and specific and more rapid than traditional microbial culture. These molecular technologies have enabled laboratories to identify infectious agents that may not have been considered in the original diagnoses, and to confirm clinical diagnoses that previously would have been impossible or difficult to make (e.g., herpes simplex virus encephalitis) and have aided in the discovery of new infectious agents, such as the recently identified human metapneumovirus. These assays are particularly useful for infectious agents that cannot be cultured (e.g., hepatitis C virus), that are difficult to culture (e.g., Mycobacterium leprae), or that, for safety reasons, the laboratory would not want to culture (e.g., variola virus or the SARS-related coronavirus). Rapid diagnostics can greatly impact patient care, direct the use of appropriate antimicrobial therapies, and reduce hospitalization and overall medical costs.
The enhanced detection of infectious agents can be achieved either by amplifying the signal used to detect the target nucleic acid, or by directly amplifying the target nucleic acid, followed by probe-based detection. Examples of signal amplification methods used in infectious-disease diagnostics are branched DNA (bDNA, Bayer Diagnostics, Tarrytown, NY) (1), ligase chain reaction (LCR, Abbott Laboratories, Abbott Park, IL) (2), signal-mediated amplification of RNA technology (SMART, Cytocell Technologies, Ltd, Cambridge, UK) (3), ramification amp-lification (RAM) (4), and the Invader assays (Third Wave Technologies, Madison, WI) (5). Generally, the process of nucleic acid target amplification is associated with PCR, which is an excellent method and the most widely used amplification technology. However, other, non-PCR-based target amplification technologies, including nucleic acid sequence-based amplification (NASBA) (6, 7), transcription-mediated amplification (TMA) (8), and strand displacement amplification (SDA) (9, 10), have been used successfully in a variety of commercial assays for detecting infectious agents. Non-PCR target amplification assays generally offer sensitivity and specificity equal to PCR and, in some cases, have advantages for certain applications. As with PCR, these applications are adaptable to all nucleic acid targets (DNA, RNA, mRNA, and rRNA), have multiplex capabilities, and can be qualitative or quantitative. The products of the amplification reactions are single-stranded nucleic acids that do not require denaturation prior to hybridization with traditional probes for end-point detection or with the fluorescent probes used in real-time detection procedures. The amplification reactions are isothermal and can be performed in a heat block, water bath, or incubator/fluorometer, thus greatly reducing equipment costs compared to real-time multi-dye PCR thermocyclers.
This article will focus on NASBA, TMA, and SDA, the non-PCR-based target amplification technologies, and their current applications for use in clinical infectious disease diagnostics.
Nucleic Acid Sequence-Based Amplification (NASBA)
NASBA is an isothermal (41°C) amplification method achieved through the coordinated activities of three enzymes — avian myeloblastosis virus reverse transcriptase (AMV-RT), RNase H, and T7 RNA polymerase — and two DNA oligonucleotide primers that are specific for the target sequence of interest (Fig. 1) (6, 7). Amplification is based on primer extension and RNA transcription. In phase I, the target serves as a template for the extension of primer 1 (containing the T7 RNA polymerase promoter site) by AMV-RT to form an RNA-DNA duplex. Extension is followed by degradation of the template RNAs by RNase H, resulting in single-stranded DNA containing the T7 promoter sequence. The extension of primer 2 by AMV-RT results in the synthesis of the second strand of DNA and the formation of a double-stranded T7 RNA polymerase promoter. RNA synthesis by T7-RNA polymerase follows. With RNA transcription the system enters the isothermal cyclic phase II, resulting in the accumulation of large amounts of single-stranded RNA, which is antisense (complementary) to the original target RNA. The single-stranded RNA can be detected by using traditional methods, such as electrochemiluminescence (ECL), or in real time, using fluorescent probes, such as molecular beacons. The assay can also be adapted for DNA targets.
Figure 1.
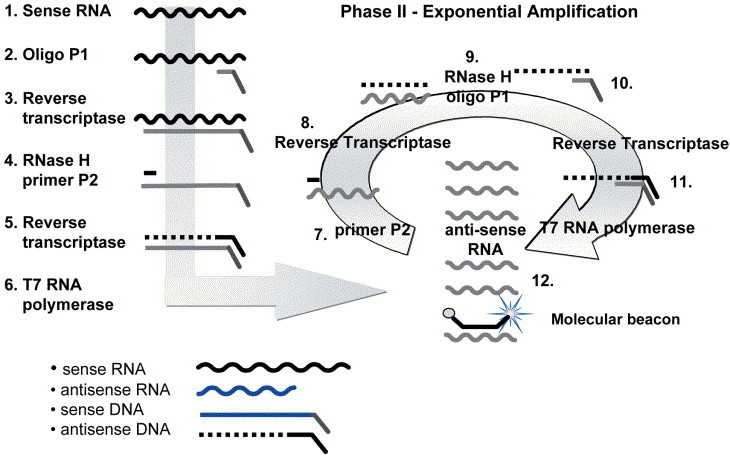
Nucleic acid sequenced based amplification (NASBA). An example of an RNA target is shown. Included in the amplification reaction are reverse transcriptase (RT), RNase H, T7 RNA polymerase, nucleotides, primers, and molecular beacons if performing real-time detection.
Phase I. Template amplification
1. Single-stranded sense RNA.
2. Oligonucleotide P1, containing the T7 promoter recognition sequence, binds to the complementary sequence on the target.
3. RT makes a DNA copy of the RNA template.
4. RNase H removes the RNA from the duplex, and P2 binds the antisense DNA strand.
5. RT copies the antisense DNA to form a double-stranded DNA complex.
6. T7 RNA polymerase recognizes the double-stranded T7 promoter sequence and initiates transcription, making hundreds of antisense RNA copies.
Phase II. Exponential amplification
7. P2 binds the complementary sequence on the antisense RNA strands.
8. RT makes a DNA copy of the RNA template.
9. RNase H removes the RNA from the duplex.
10. P2 binds the antisense DNA strand.
11. RT creates a double-stranded T7 RNA polymerase promoter.
12. Additional rounds of transcription occur, resulting in 108 to 1010 copies of antisense RNA that are templates both for more rounds of amplification and for detection by using probes and electrochemiluminescence or in real time with molecular beacons incorporated into the amplification reaction, as shown. (Figure reprinted with permission of bioMérieux, Inc., Durham, NC.)
NucliSens technology
NucliSens technology (bioMérieux, Durham, NC) is comprised of four major procedural components: nucleic acid release, nucleic acid extraction, NASBA amplification, and target detection. By following the procedure of Boom et al. (11) using the NucliSens isolation reagents, total nucleic acids can be purified from a variety of sample types, including whole blood, dried blood spots, blood film slides, plasma, serum, tissue, peripheral blood mononuclear cells, body fluids, stool, respiratory samples, and cerebrospinal fluid (CSF). Samples in volumes ranging from 2 to 10 ml are added to NucliSens lysis buffer containing guanidine thiocyanate and Triton X-100. Viral particles, bacterial particles, or cells are disintegrated, and the nucleic acids are released. Any RNases and DNases present in the sample are inactivated. Silicon dioxide particles are added to the lysed sample. Under high salt conditions, these particles act as a solid phase to capture the nucleic acids. The particles are washed several times to remove proteins and inhibitors. Finally, the highly purified concentrated nucleic acids are eluted from the silica particles in RNase/DNase-free water. Extractions can be performed manually or with automated or semi-automated extraction systems (see “Automation for NucliSens assays” below).
NASBA amplification is performed using primers specific for the target of interest. The assays can be multiplexed to include more than one target or an internal control to monitor the entire process from nucleic acid extraction through detection. Detection of the RNA products can be achieved by using two different formats, either end-point ECL detection or in real time using molecular-beacon technology (12). For ECL detection (Fig. 2), aliquots of diluted amplicons are added to hybridization solutions containing a streptavidin paramagnetic bead with a bound target-specific oligonucleotide that functions as a solid phase to capture the amplified targets. Also included in the hybridization reaction mixture is a ruthenium (Ru2+)-labeled detection probe. The Ru2+ probe can either be target specific (e.g., NucliSens HIV-1 QT assay) or generic (as supplied in the NucliSens Basic Kit detection module). If generic Ru2+ probes are used, the P2 amplification primer is designed to contain a sequence complementary to the sequence on the generic Ru2+ probe. The resulting amplicons are detected by using the target-specific bead-oligonucleotide and the Ru2+ detection probe (Fig. 2D). ECL hybridizations are performed at 41°C, followed by ECL detection using the NASBA QR reader (bioMérieux).
Figure 2.
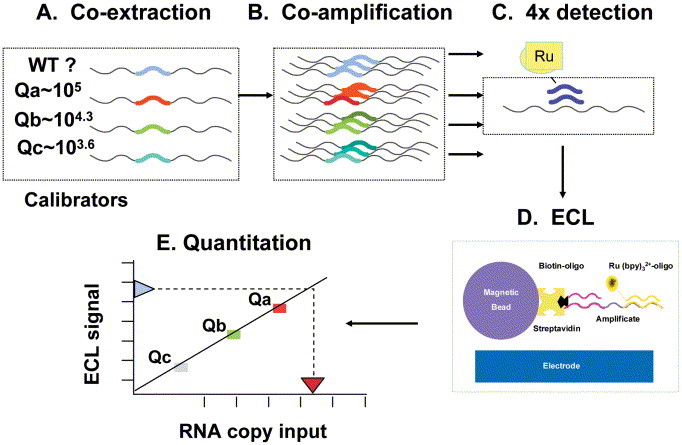
Schematic of HIV-1 QT assay. (A) Three internal calibrators, each with a unique altered 20-nucleotide internal sequence and of known RNA concentration, are added to lysis buffer with patient sample, and total nucleic acids are co-isolated. (B) Calibrators and wild-type patient HIV-1 RNA are co-amplified by NASBA in a single tube. (C) Four separate ECL detections are performed using specific ruthenium-labeled probes targeted to the altered internal calibrator sequences or the wild-type HIV-1 sequence. (D) Streptavidin paramagnetic beads with a bound HIV-1-specific biotinylated oligonucleotide capture the amplified targets. Voltage is applied, and ECL signals are read by the NucliSens Analyzer. (E) Wild-type HIV-1 RNA quantitation is based on unique calibration curves generated by the calibrators Qa, Qb, and Qc for each sample. (Figure reprinted with permission of bioMérieux, Inc., Durham, NC.)
Alternatively, the assays can be adapted to a real-time format by using molecular-beacon technology (12). Molecular beacons (Fig. 3) are hairpin structures that consist of a single-stranded loop sequence complementary to the target and a short 6- to 7-bp stem to which a fluorophore, e.g., rhodamine or fluorescein, is attached to the 5′ end and a quencher, such as dabcyl, is attached to the 3′ end of the beacon sequence. In the native state, the beacon is a closed structure, held together by the complementary stem sequences. Since the fluorophore and quencher are in close proximity, little or no fluorescent light is emitted. In the presence of complementary target sequences, the beacons bind to this target and undergo a structural change, thereby separating the fluorophore and quencher. Light is emitted and measured at specific UV wavelengths. Both the target and the internal-control molecular beacons, each with its specific fluorophore, are added at the start of the amplification reaction. Target amplification at 41°C and continuous monitoring of the emitted fluorescence of both molecular beacon probes are performed simultaneously using a NucliSens EasyQ Analyzer (bioMérieux, Boxtel, The Netherlands).
Figure 3.
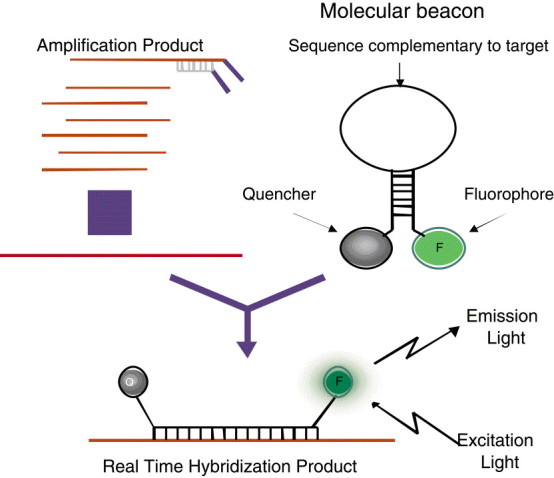
Real-time amplification and detection using molecular beacons. Added to the amplification reaction are molecular beacons specific for the target of interest. When the target is present, beacons open and bind to the complementary target sequence separating the fluorophore and the quencher. Upon excitation, light is emitted and measured. (Figure reprinted with permission of bioMérieux, Inc., Durham, NC.)
Commercially available FDA-approved complete kits that use ECL detection include the NucliSens HIV-1 QT assay and the NucliSens CMV pp67 assay. A new NucliSens EasyQ HIV-1 assay, which uses molecular-beacon technology, is currently available outside the United States. In addition to these assays, the NucliSens Basic Kit reagents can be used to develop in-house assays or used with analyte-specific reagents (ASR).
NucliSens HIV-1 viral-load assays
The NucliSens HIV-1 QT assay is a quantitative assay for measuring the HIV-1 viral load from plasma (13, 14). Total plasma nucleic acids and three internal calibrators (supplied with the kit) of known low, medium, and high HIV-1 RNA concentrations are co-extracted (Fig. 2A). Extractions can be performed manually or with the automated NucliSens Extractor. Co-amplification of wild-type patient HIV-1 RNA (sense RNA) and the internal calibrators (Fig. 2B) is performed in a single tube using two DNA oligonucleotides that are specific for the gag region (encoding core proteins) of the HIV-1 target sequence. The single-stranded RNA product can then be readily detected by ECL after hybridization of wild-type or calibrator-specific Ru2+-labeled oligonucleotide probes (Fig. 2C and D). The amount of light generated (ECL units) is proportional to the amount of the product amplificate (RNA copies/ml) (Fig. 2E). Calculation is based on the relative amounts of RNAs of the four amplificates (wild type patient sample and three calibrators) that determine the original amount of wild-type HIV-1 RNA in the sample. No additional external controls or standards are necessary for the quantitation. However, the manufacturer recommends that a high positive control, a low positive control, and a negative control be included with the first run of each kit lot and a negative control and a low positive control with each subsequent run of the same kit to verify product performance. An external control can also be used to monitor assay reproducibility from lot to lot, run to run, and technician to technician. A multicenter clinical evaluation of the HIV-1 QT assay determined that the linear range of the assay was 51 to 5,390,000 RNA copies/ml, with a cutoff of 25 RNA copies/ml (14). The 50% detection rate was 41 RNA copies/ml, and the 95% detection rate was 176 RNA copies/ml. The mean reproducibility of the assay over the dynamic range was 0.15 log standard deviation, and the specificity was 100%.
The NucliSens EasyQ HIV-1 version 1.1 assay (15) is a quantitative assay for determining the HIV-1 viral load in plasma samples and is based on NASBA amplification and molecular-beacon detection. The assay uses a single calibrator RNA, which is co-extracted, co-amplified, and co-detected in real time in the same tube as the HIV-1 wild-type RNA (Fig. 4). During the 1-h amplification and detection process, two fluorescent signal curves are generated, which are interpreted by the NucliSens Director software algorithm to calculate the HIV-1 viral load in the patient sample. An independent study showed a correlation coefficient r of 0.97 when the NucliSens EasyQ HIV-1 version 1.1 assay was compared to the NucliSens HIV-1 QT assay and an r of 0.93 when it was compared to the Roche Amplicor Monitor HIV-1 ultrasensitive assay (15). The assay detects all M group subtypes. NucliSens EasyQ HIV-1 version 1.1 has obtained CE marking in Europe. CE marking on a product is a manufacturer's declaration that the product complies with the essential requirements of the relevant European health, safety, and environmental protection legislation. The performance levels and technical specifications must be in accordance with those established by several European standards agencies.
Figure 4.
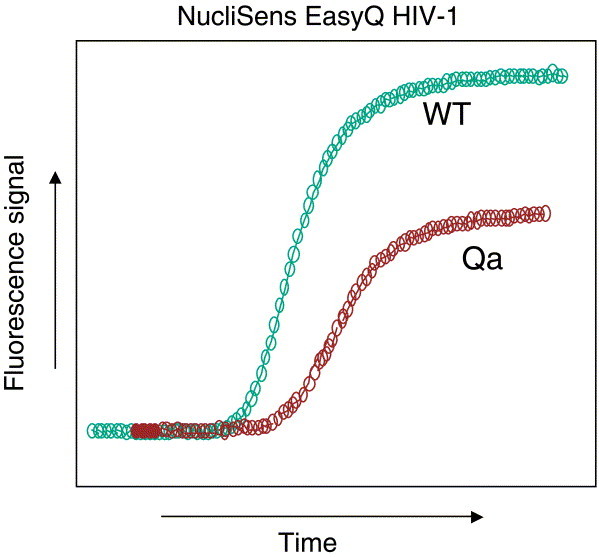
HIV-1 EasyQ quantitation using molecular beacons. Real-time HIV-1 quantitation is based on the presence of a single calibrator (Qa) of known RNA concentration, which is co-extracted and co-amplified with the patient sample. Algorithms based on multiple parameters (including internal calibrator values, kinetics of the assay, molecular-beacon binding, time to detection, and fluorescence units) determine the number of wild-type HIV-1 RNA copies in the patient sample. (Figure reprinted with permission of bioMérieux, Inc., Durham, NC.)
NucliSens cytomegalovirus pp67 assay
The NucliSens CMV pp67 assay is used to monitor immunosuppressed patients at risk for cytomegalovirus (CMV) disease, for the diagnosis of CMV disease, and for assessing response to anti-CMV therapy (16, 17, 18). The assay targets pp67 mRNA, which encodes a phosphorylated matrix tegument protein transcribed from the UL 65 gene. pp67 mRNA is one of the most abundant late gene transcripts and is detectable only during viral replication. The presence of pp67 transcripts, therefore, is indicative of active CMV infection and should not be detected during latent infection. Studies have demonstrated that CMV pp67 late gene transcripts are an alternative target for determining active CMV infection in renal allograft recipients and heart, lung, and bone marrow transplant recipients (17). Nucleic acids from plasma and an internal control are co-extracted and co-amplified by NASBA, followed by ECL detection. The lower limit of detection for the assay is less than 25 mRNA copies. A multicenter clinical trial study demonstrated an 86% concordance between the pp67 assay and current reference tests, including cell culture and a CMV p65 antigenemia assay, when active CMV disease was present (16). Although not FDA approved for testing CSF, studies in our laboratory have demonstrated that the detection of pp67 mRNA in CSF was a better predictor of active CMV-related central nervous system disease than the detection of CMV DNA by PCR (18).
NucliSens Basic Kit applications
The NucliSens Basic Kit reagents have been used by numerous investigators to develop their own assays. Currently, over 90 applications have been validated that target bacteria, viruses, mycobacteria, fungi, parasites, chemokines, cytokines, and genetic markers. The NucliSens Basic Kit permits standardization of all assays to one easy format. The NucliSens Kit components include four separate modules for each stage of analysis: sample lysis, nucleic acid isolation, amplification, and detection. Detection can be performed by using ECL and the generic Ru2+ probe supplied in the basic kit or in real time by adding target-specific molecular beacons to the amplification reaction. The user designs the primers and capture probes (ECL detection) or molecular beacons. Currently, one ASR that uses NASBA and molecular beacons is available for the detection of enterovirus from CSF (bioMérieux) (19). The assay targets the 5′ non-coding region of the enteroviral genome and has broad reactivity, detecting echoviruses, coxsackieviruses, numbered enteroviruses, and poliovirus, with no cross-reactivity to numerous other viruses tested (20). The assay is internally controlled for monitoring the efficiency of the entire process, from isolation to detection. Studies from our laboratory have shown that the rate of amplification inhibition is less than 0.5%, and all inhibited samples were grossly bloody. The sensitivity of the assay is approximately 10 to 100 RNA copies per amplification reaction (20). The enterovirus NASBA assay detected 23% more positive CSF samples than routine viral culture (21). The rapid diagnosis of enteroviral infections can significantly reduce the length of hospital stay and reduce overall medical costs (21).
By combining different detection formats with NASBA, an effective method is available for targeting sequences not always detected with molecular beacons because of AT-rich sequences. A NASBA assay developed in our laboratory for detecting malaria parasites combines the use of molecular beacons for a generic screening assay and ECL detection for species identification in positive samples (22). Whole-blood samples, dried blood spots, or scrapings from stained blood film slides are screened using primers and a molecular beacon generic for all four Plasmodium species. The assay can detect all four species with comparable sensitivities (0.0001 parasites/reaction) in as little as 12 min. Amplicons from positive samples are hybridized with species-specific capture probes and the generic Ru2+ probe, followed by ECL detection. The assay was 100% sensitive compared to traditional microscopy. The NASBA assay was more specific than microscopy, particularly for slides that were difficult to interpret or when very low levels of parasitemia were present. The assay was superior to microscopy for the detection of mixed infections.
Automation for NucliSens assays
Nucleic acid extractions from a variety of sample types can be performed manually or with the automated NucliSens Extractor. The reagents used in the Extractor system are identical to those used in the manual NucliSens (11) isolation procedure. Lysis buffer containing patient sample, calibrators or internal controls, and silica are added to extraction cartridges and placed on the instrument. The fully automated washing cycles and nucleic acid elution take place in the closed, disposable filter cartridges, guaranteeing complete contamination control during the nucleic acid extraction process. The extractor processes 10 samples in approximately 60 min. Routine molecular diagnostic laboratories and blood bank testing laboratories have demonstrated excellent performance isolating nucleic acids from plasma and serum for use in the NucliSens HIV-1 RNA assays and in assays for detecting other bloodborne viruses, such as hepatitis viruses. Alternatively, extractions can be performed using magnetic silica particles and the miniMag, a semi-automated extraction system (bioMérieux). A fully automated version, the Mag-Extractor, is currently in development.
The NucliSens EasyQ system consists of an incubator, an analyzer, and a computer with Director software (15, 19, 22). The compact incubator performs the short pre-amplification step. The analyzer reads fluorescence from the 8-well reaction strips in a temperature-controlled environment. Up to 48 samples can be run in less than 2 h. The assay-specific software enables real-time monitoring of individual sample amplification, automated data analysis, and reporting of quantitative or qualitative results.
Transcription Mediated Amplification (TMA)
TMA is an autocatalytic, isothermal (42°C) amplification technology (Fig. 5) that utilizes two enzymes, RT and T7 RNA polymerase, and two primers that are complementary to the target sequence (8). One primer contains the promoter sequence for T7 RNA polymerase. This promoter primer binds to the target sequence, and RT creates a DNA copy of the target strand by extension from the 5′ end of the promoter primer, resulting in an RNA-DNA hybrid. The RNase H activity of the RT degrades the RNA in the RNA-DNA hybrid, resulting in single-stranded DNA that contains the T7 RNA polymerase promoter sequence. The second primer binds the DNA copy, and a new DNA strand is synthesized by the RT, resulting in a double-stranded DNA copy that contains the T7 polymerase promoter sequence. T7 polymerase binds to the promoter sequence and initiates transcription off the DNA template, resulting in many copies of RNA (100 to 1,000 per DNA template). Each of the newly synthesized RNA copies enters into the amplification cycle and serves as a template for the TMA process. There is an exponential expansion of the RNA, resulting in the production of up to 10 billion amplicons within 15 to 30 min. RNA amplicons can then be detected by using a variety of technologies that will be described in the next issue (CMN vol. 26, no. 17).
Figure 5.
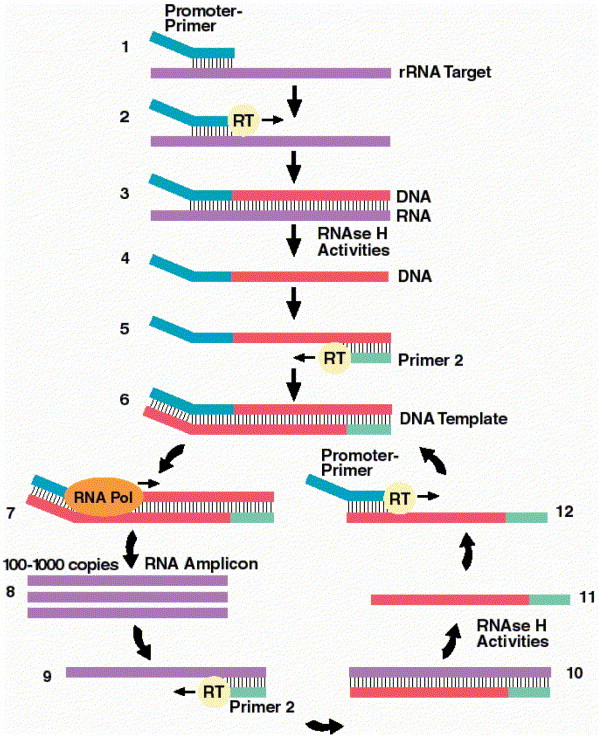
Transcription-mediated amplification. (Step 1) Promoter-primer binds to rRNA target. (Step 2) Reverse transcriptase (RT) creates DNA copy of rRNA target. (Step 3) RNA-DNA duplex. (Step 4) RNase H activities of RT degrades the rRNA. (Step 5) Primer 2 binds to the DNA, and RT creates a new DNA copy. (Step 6) Double-stranded DNA template with a promoter sequence. (Step 7) RNA polymerase (RNA Pol) initiates transcription of RNA from DNA template. (Step 8) 100 to 1,000 copies of RNA amplicon are produced. (Step 9) Primer 2 binds to each RNA amplicon and RT creates a DNA copy. (Step 10) RNA-DNA duplex. (Step 11) RNase H activities of RT degrade the rRNA. (Step 12) Promoter-primer binds to the newly synthesized DNA. RT creates a double-stranded DNA, and the autocatalytic cycle repeats, resulting in a billion-fold amplification. (Figure reprinted with permission of Gen-Probe Inc., San Diego, CA.)
Biography
A Special Invitation to Authors
The editors of Clinical Microbiology Newsletter extend an invitation to authors who may wish to contribute an article or editorial for publication in CMN. Of special interest are timely topics of interest to clinical microbiologists and infectious disease physicians. Persons with suggestions for articles or editorials should contact the Editor listed below to discuss the details of their potential submission.
Paul A. Granato, Ph.D. Dept. of Microbiology & Immunology
WH 2204
SUNY Upstate Medical University
750 E. Adams St.
Syracuse NY 13210 USA
Tel.: 315-464-7653
e-mail: granatop@upstate.edu
Footnotes
Editor's Note: Part II of this article will be published in the September 1 issue of CMN (vol. 26, no. 17).
References
References
- 1.Urdea M.S. Branched amp-lification multimers for the sensitive, direct detection of human hepatitis virus. Nucleic Acids Symp. Ser. 1991;24:197–200. [PubMed] [Google Scholar]
- 2.Barany F. The ligase chain reaction (LCR) in a PCR world. PCR Methods Appl. 1991;1:5–16. doi: 10.1101/gr.1.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Wharam S.D. Specific detection of DNA and RNA targets using a novel isothermal nucleic acid amplification assay based on the formation of a three-way junction. Nucleic Acids Res. 2001;29:e54. doi: 10.1093/nar/29.11.e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang D.Y. Detection of rare DNA targets by isothermal ramification amplification. Gene. 2001;274:209–216. doi: 10.1016/s0378-1119(01)00607-2. [DOI] [PubMed] [Google Scholar]
- 5.Kwiatkowski R.W. Clinical, genetic, and pharmacogenetic applications of the Invader Assay. Mol. Diagn. 1999;4:353–364. doi: 10.1016/s1084-8592(99)80012-5. [DOI] [PubMed] [Google Scholar]
- 6.Compton J. Nucleic-acid sequence-based amplification. Nature (London) 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 7.Romano J.W. NASBA technology: isothermal amplification in qualitative and quantitative diagnostics. Immunol. Investig. 1997;26:15–28. doi: 10.3109/08820139709048912. [DOI] [PubMed] [Google Scholar]
- 8.Hill C.S. Molecular diagnostic testing for infectious diseases using TMA technology. Exp. Rev. Mol. Diagn. 2001;1:445–455. doi: 10.1586/14737159.1.4.445. [DOI] [PubMed] [Google Scholar]
- 9.Walker G.T. Strand displacement amplification – an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992;20:1691–1696. doi: 10.1093/nar/20.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellyer T.J., Nadeau J.G. Strand displacement amplification: a versatile tool for molecular diagnostics. Exp. Rev. Mol. Diagn. 2004;4:251–261. doi: 10.1586/14737159.4.2.251. [DOI] [PubMed] [Google Scholar]
- 11.Boom R. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyagi S., Kramer R. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 13.Kievits T. NASBA isothermal enzymatic in vitro nucleic acid amplification optimized for the diagnosis of HIV-1 infection. J. Virol. Methods. 1991;35:273–286. doi: 10.1016/0166-0934(91)90069-c. [DOI] [PubMed] [Google Scholar]
- 14.Ginocchio C.C. Multicenter evaluation of the performance characteristics of the NucliSens HIV-1 QT assay used for the quantitation of human immunodeficiency virus type 1 RNA. J. Clin. Microbiol. 2003;41:164–173. doi: 10.1128/JCM.41.1.164-173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McClernon, D. et al. 2004. Quantification of HIV-1 subtypes and clinically relevant recombinant virus using new molecular beacon viral load assay, abstr. 960. Abstr. 11th Conference on Retroviruses and Opportunistic Infections. 2004. San Francisco, CA.
- 16.Witt D.J. Analytical performance and clinical utility of a nucleic acid sequence-based amplification assay for detection of cytomegalovirus infection. J. Clin. Microbiol. 2000;38:3994–3999. doi: 10.1128/jcm.38.11.3994-3999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerna G. Clinical signi-ficance of expression of human cytomegalovirus pp67 late transcript in heart, lung, and bone marrow transplant recipients as determined by nucleic acid sequence-based amplification. J. Clin. Microbiol. 1999;37:902–911. doi: 10.1128/jcm.37.4.902-911.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F. Detection of human cytomegalovirus pp67 late gene transcripts in cerebrospinal fluid of human immunodeficiency virus type-1-infected patients by nucleic acid sequence-based amplification. J. Clin. Microbiol. 2000;38:1920–1925. doi: 10.1128/jcm.38.5.1920-1925.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F., Ginocchio C.C., Sillekens P. Nucleic acid sequence based amplification (NASBA) and molecular beacon detection for the identification of enterovirus in cerebrospinal fluid, abstr. T41. Abstr. 19th Annual Clinical Virology Symposium. Pan American Society for Clinical Virology; Clearwater, FL: 2003. [Google Scholar]
- 20.Zhang F. Evaluation of a modified NucliSens Basic Kit application for the detection of enterovirus, abstr. S35. Abstr. 17th Annual Clinical Virology Symposium. Pan American Society for Clinical Virology; Clearwater, FL: 2001. [Google Scholar]
- 21.Zhang F. Clinical performance and financial impact of rapid enterovirus detection in cerebrospinal fluid using the NucliSens Basic Kit and nucleic acid sequenced based amplification, abstr. T5. Abstr. 18th Annual Clinical Virology Symposium. Pan American Society for Clinical Virology; Clearwater, FL: 2002. [DOI] [PubMed] [Google Scholar]
- 22.Patel, U.A., F. Zhang, and C.C. Ginocchio. 2003. Rapid real-time detection of Plasmodium spp. using nucleic acid sequenced-based amplification (NASBA) and molecular beacons, abstr. C-285. Abstr. 103rd General Meeting of the American Society for Microbiology. Washington, D.C.