

Gastritis—inflammation of the stomach—is a frequently cited differential yet rarely characterized diagnosis in cases of canine anorexia and vomiting. Although the list of rule-outs for acute or chronic gastritis is extensive (Box 1 ) [1], a review of the veterinary literature reveals fewer than 15 articles that have focused on clinical cases of canine gastritis over the last 25 years [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14]. The dog frequently appears in the human literature as an experimentally manipulated model for the study of endoscopic techniques or the effect of medications on gastric mucosa [15], [16], [17], [18], [19], [20]. In the veterinary patient, cases of acute gastritis are rarely pursued with the complete diagnostic armamentarium, and cases of chronic gastritis are rarely found to occur as an entity isolated from the rest of the gastrointestinal tract. This article focuses on those findings most clinically relevant to cases of canine gastritis in veterinary medicine.
Box 1. Differential diagnosis for cases of acute or chronic gastritis in dogs.
- Breed-associated gastritis
- Basenji, Norwegian Lundehund (atrophic gastritis)
Dietary indiscretion, foreign bodies
- Drugs
- Nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, antibiotics, chemotherapeutics
Eosinophilic gastritis
Food allergy, food sensitivity
- Granulomatous gastritis
- Idiopathic, infectious, neoplasia, foreign body, systemic granulomatous disease
Immune-mediated gastritis
Infectious gastritis
Viral, bacterial, fungal
Lymphocytic/plasmacytic gastritis
- Motility disorders, reflux disease
- Bilious vomiting syndrome
Neoplasia
Parasitic gastritis
- Secondary gastritis (systemic disease)
- Central nervous system disease, renal failure, liver failure, endocrine disease
Toxins, plants, chemicals
Pathophysiology
The mucosal lining of the stomach normally acts as an effective defensive barrier against acidity, detergents, bacteria, and changes in temperature. That mucosal defense consists of secretions, cells, and blood. Normal gastric secretions represent the first line of defense and include acid, mucus, bicarbonate, and antibacterial substances. The gastric epithelium serves as a barrier to the back-diffusion of acid and is quickly repaired by restitution after injury. The gastric microvasculature is exquisitely responsive to neuronal, hormonal, and inflammatory signals. This blood supply is central to the maintenance of gastric mucosal integrity, the elimination of noxious substances, and gastric epithelial turnover [21], [22], [23]. Macrophages and mast cells are part of the innate immune system that coordinates the gastric inflammatory response when challenged by antigenic stimulation [24]. Finally, like much of the gastrointestinal tract, the gastric mucosa has a large capacity for quickly repairing damaged tissue (ie, restitution of ulcerated mucosal epithelium) [25].
In cases of excessive or inappropriate gastric inflammation, although a cause or causative agent is rarely determined, many of the pathologic changes have been elucidated [26], [27]. Chemical injury, ischemia, infection, or antigens can stimulate the release of inflammatory mediators and vasoactive compounds from a variety of cell types (eg, neutrophils, mast cells, platelets, endothelial cells, neurons) (Box 2 ) [28], [29]. Subsequent exfoliation of surface gastric epithelial cells and disruption of the normal mucosal barrier result in back-diffusion of gastric acid, pepsin, and gastric lipase. This inflammatory cascade stimulates further acid secretion and mucosal damage, increases cell membrane permeability, and alters microvascular blood flow. The continued interplay between ischemia and inflammation results in gastric erosion, ulceration, hypoxia, hemorrhage, edema, and necrosis [30].
Box 2. Inflammatory and vasoactive mediators of gastritis.
- Cytokines
- Interleukin-1β
- Tumor necrosis factor-α
- Chemokines
- Leukotriene B4
Endothelin-1
Histamine
Nitric oxide
- Neuropeptides
- Calcitonin gene-related peptide
- Substance P
- Oxygen free radicals
- Platelet-activating factor
Peroxidases
- Proteinases
- Trypsin
Thromboxane
Differentials and diagnosis
Food and foreign bodies
“Garbage gut” is a catch-all diagnosis for cases of acute gastritis, where dogs are likely to have ingested actual garbage, molds, fungi, spoiled or raw food, leftovers, or cat litter. Beyond the radiographic demonstration of gastric distention secondary to overindulgence, these cases are infrequently subjected to extensive diagnostic effort. These patients usually respond to a brief period of gastric inactivity and dietary counseling, although acute pancreatitis is a serious potential sequela. Persistent or repeat offenders should be examined for causes of polyphagia and pica (eg, malnourishment, maldigestion, malabsorption, hyperadrenocorticism, behavioral issues). Outbreaks of food poisoning such as are periodically seen in the human population seem to be either rare or underappreciated events in our canine companions.
Foreign bodies may cause direct physical damage to the mucosal barrier on their way through, or they may lodge in the pylorus, resulting in acute gastritis, vomiting, gastric ulceration, and biochemical changes consistent with an upper gastrointestinal obstruction. The diagnosis can be straightforward when the foreign object is radiographically distinct; however, the pylorus can be a difficult region to elucidate, and tumors, pyloric hyperplasia or stenosis, and gastric atony must be considered on the list of differentials. Progression of the disease, repeat radiographic images, contrast studies, or ultrasound examination may provide additional clues in cases of acute gastritis that do not respond to conservative management as anticipated. Uncomplicated cases of acute gastritis should resolve without the use of antiemetics, H2-receptor blockers, or gastrointestinal protectants, and their indiscriminant use may mask symptoms that would otherwise prompt a more in-depth examination of the patient.
The onset of gastrointestinal symptoms related to the ingestion of specific food items, where the underlying mechanism is an immune-mediated reaction, defines a food allergy. Pruritus rather than gastritis is the most common clinical sign of a food allergy, and the stomach may not be the portion of the gastrointestinal tract most commonly afflicted. In fact, gastrointestinal symptoms may be present in only 10% to 15% of cases of canine food allergy, although up to 50% of cats with chronic idiopathic gastrointestinal symptoms may respond to manipulation of the dietary protein source [31].
In many cases, human beings diagnosed as being allergic to certain foods are also found to be suffering from Helicobacter pylori infection, complicating the interpretation of gastric pathologic findings [32]. In children with confirmed cases of food allergy, a close relation was found to duodenal pathologic findings, whereas no significant association was seen with gastric lesions [33]. A similar lack of gastric pathologic findings was demonstrated in human adults suffering from food allergies without concurrent H pylori infection [34]. Proteins are the foodstuff most commonly incriminated in food-allergic dogs, and the gastric mucosa is not normally a site of absorption for these polypeptides (Box 3 ). The age of onset can be anywhere between puppy and adulthood, although many reports identify a significant number of young animals (<1 year of age).
Box 3. Foodstuffs thought to induce an adverse immune response in the dog.
Milk
Oatmeal
Dog biscuits
Eggs
Wheat
Commercial dog foods
Beef
Kidney beans
Flavorings
Mutton
Corn
Additives
Pork
Soy
Preservatives
Chicken
Rice
Supplements
Horse
Potato
Dyes
Rabbit
Maize
The pathophysiology of a food-allergic reaction is complex and not yet completely understood. The adverse response may involve immediate, delayed, or mixed hypersensitivity reactions as well as multiple inflammatory cells and mediators. Gut-associated lymphoid tissue can present intact material to the host immune system through specialized gastrointestinal antigen-presenting cells, M cells, and macrophages. IgA-producing B cells, IgE antibodies, helper T cells, eosinophils, and mast cells are all located in the lamina propria of the digestive tract as potential contributors to the antigen-driven response. Histamine, serotonin, vasoactive intestinal polypeptide, proteinases, prostaglandins, leukotrienes, and interleukins are just a few of the inflammatory mediators released by the complex interplay of the various cell types present [35], [36], [37].
Ideally, the diagnosis of a food allergy would include identification of the offending antigen; demonstration of the correlation between antigen exposure, clinical signs, and pathologic changes; and elucidation of the immunologic mechanism. If the symptoms are eliminated in response to an appropriate diet trial, it should be demonstrated that they reappear with the subsequent reintroduction of the incriminated antigen—a diagnostic step usually declined by owners.
In contrast, food intolerance is a nonimmune, idiosyncratic, physiologic, metabolic, or toxic response to a food item. Symptoms of food intolerance may mimic any abnormal gastrointestinal reaction; therefore, it is a particularly difficult condition to diagnose [37]. Food intolerance may be the result of a deficiency in a specific digestive enzyme, with the most often cited example being lactose intolerance secondary to a deficiency in the enzyme lactase.
Drugs, toxins, and chemicals
More than 30 varieties of plants and innumerable household chemicals are potential causes of canine gastritis (Box 4 ) [1]. Although not a toxin by itself, the urease activity of H pylori is in part responsible for the pathogenicity of this organism in people. Many plants contain the same enzyme, which may contribute indirectly to their role in gastritis [38]. Chemicals may be directly caustic to the gastric mucosa or may affect gastric function (ie, increased acid secretion, decreased bicarbonate secretion, change in motility) and result in secondary inflammation.
Box 4. Common household plants and chemicals associated with gastritis.
Daffodil
Ethylene glycol
Mushrooms
Deodorants
Ivy
Detergents
Azalea
Nitrates
Rhododendron
Heavy metals
Poinsettia
Acids
Holly
Bleach
Honeysuckle
Pine oil
Mistletoe
Rubbing alcohol
Jasmine
Driveway salt
NSAIDs are one of the most common causes of acute gastric erosion and chronic gastric ulceration leading to hospitalization and even death in human beings [39]. These compounds work by blocking the conversion of arachidonic acid into inflammatory mediators via the cyclooxygenase (COX) enzyme. Despite the advent of NSAIDs manufactured specifically for veterinary patients (eg, carprofen [Rimadyl], etodolac [Etogesic]), the use of aspirin is still commonplace for dogs with occasional stiffness or chronic osteoarthritis. Ibuprofen, acetaminophen, and indomethacin are other common over-the-counter NSAIDs administered by owners or ingested by opportunistic pets [40], [41]. The depletion of endogenous protective prostaglandins, a decrease in mucus and bicarbonate secretions, a disruption of the epithelial cell layer, a reduction of the surface epithelial cell hydrophobicity, a reduction in mucosal blood flow, an increase in neutrophil adherence, and direct mucosal injury are all components of the deleterious effects of NSAIDs on the gastric mucosa [42], [43]. The antrum and pylorus are the portions of the stomach most susceptible to NSAID-induced lesions. Those lesions can range from surface erythema to erosions to full-thickness ulcerations [44]. The greater the severity of mucosal damage, the greater is the volume of blood entering the gastric lumen and the more likely it is that hematemesis or melena is part of the presenting complaint.
Because of the prevalence of NSAID-induced gastric ulceration, the development of increasingly specific COX-2 inhibitors has burgeoned into a multibillion dollar pharmaceutic industry, and COX-2 selective inhibitors are now some of the most frequently prescribed drugs in human medicine [45]. The theory behind the use of COX-2 inhibitors is illustrated in Fig. 1 [46]. Although not entirely COX-2 selective, Rimadyl and Etogesic are two NSAIDs approved for use in dogs and designed to reduce ulcer formation relative to aspirin. Endoscopic examination used to compare the gastric mucosa of dogs given aspirin with that of dogs treated with etodolac found that dogs given aspirin invariably had mucosal erosions by day 17 of treatment, whereas none of the dogs given etodolac were found to have any gastric lesions [47]. Regardless of the NSAID administered, none of the dogs in this study were found to have any biochemical abnormalities, vomiting, anorexia, or melena. Other similar studies have confirmed the ubiquity of gastric lesions in dogs receiving aspirin as well as illustrating only the most minor changes in dogs receiving either etodolac or carprofen [48], [49].
Fig. 1.
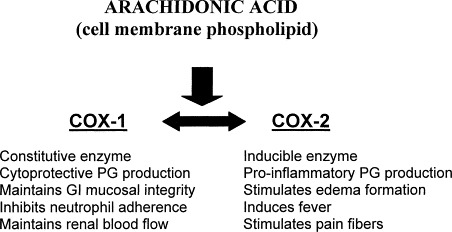
Function of prostaglandin products formed from arachidonic acid through cyclooxygenase COX-1 and COX-2 enzymatic pathways.
Interestingly, the COX selectivity of these NSAIDs seems to depend in part on the specific assay conditions used to determine the COX-2/COX-1 ratio. Using an in vitro canine monocyte/macrophage cell line, carprofen was found to be only 1.75 times more active against COX-2 than COX-1 [50]. In a separate study using an enzymatic assay, carprofen inhibited canine COX-2 activity 100 times more effectively than COX-1 activity [51]. Regardless of the molecular mechanism of action, both carprofen and etodolac seem to be significantly less ulcerogenic than aspirin.
New and more specific COX-2 inhibitors are being continually developed and made available to the veterinary practitioner. For example, deracoxib (Deramaxx) is a recently released NSAID designed to act as a specific COX-2 antagonist. Novartis claims the in vitro COX-1/COX-2 IC50 (amount of drug required to inhibit 50% of enzyme activity) ratio is 1275, consistent with a COX-2 specific medication. Deracoxib is approved by the US Food and Drug Administration (FDA) for use in dogs to help control postoperative pain after orthopedic procedures. The manufacturer reports no gastrointestinal, renal, or hepatic toxicity and no blood clotting abnormalities or drop in plasma protein. At the time of publication, no information was available regarding the use of Deramaxx in clinical cases. Despite the theoretic advantages of COX-2 inhibitors, this class of drug is not without potential side effects and should not be prescribed without appropriate client education [46], [52], [53].
In addition to NSAIDs, corticosteroids, antibiotics, and chemotherapeutics are potential causes of acute gastritis, anorexia, and vomiting. The deleterious mechanisms behind bouts of gastritis induced by these medications remain unclear (ie, direct mucosal injury, alterations in gastric pH, stimulation of innate immunity) and seem to be highly variable and host dependent. Corticosteroids decrease and alter the composition of gastric mucus and decrease mucosal cell turnover. The association between dexamethasone therapy and melena has been appreciated for some time, and the combination of corticosteroids and NSAIDs creates an extremely ulcerogenic gastric environment [54]. Gastric lesions can appear as soon as 36 hours after dexamethasone administration alone [55].
Infectious agents
Mycotic gastritis has been rarely reported (ie, pythiosis), and the acidic environment of the empty stomach is usually free of bacteria. Salmonella spp, Campylobacter jejuni, and Clostridium perfringens are differentials for gastroenteritis, with diarrhea as the most common presenting complaint, and are not discussed further in this article [56]. H pylori is a well-established cause of chronic gastritis and gastric ulceration in people, but whether these spiral-shaped organisms play a role in the pathologic changes of canine gastritis remains to be established [57], [58]. A variety of Helicobacter spp (although not H pylori) [59], [60] can be found in the stomachs of upward of 80% of dogs whether they are vomiting or not [61], [62], [63]. The possible modes of transmission may include fecal-oral, oral-oral, water-borne infection, or through nursing. Urease production, cytology, histopathology, culture, serology, and polymerase chain reaction (PCR) analysis can be used to diagnose Helicobacter infection, but in most naturally infected dogs, these species seem to cause no clinically significant change in gastric physiology or function [63], [64]. In addition, one treatment regimen commonly used in cases of human H pylori gastritis (the combination of amoxicillin, metronidazole, and famotidine) proved effective at suppressing canine gastric Helicobacter inhabitants for only a brief period of time [64], [65]. The differences in Helicobacter spp pathogenicity between people and dogs may be related to differences in the infective Helicobacter species themselves or to differences in the host immune response to the infective organisms. In fact, some cases of gastritis in dogs with Helicobacter spp do respond favorably to treatment directed at this organism. Although Helicobacter spp can readily be found in the stomachs of vomiting dogs, it would seem unwise to cite that discovery as reason for ending the search for cause in cases of canine gastritis [66].
Pathogenic enteric viruses in dogs include parvovirus, distemper virus, rotavirus, and coronavirus. Gastritis is rarely the primary concern in these diseases, with intestinal or systemic involvement being most responsible for patient morbidity and mortality. Parvovirus, in particular, is an ongoing area of active research, and the reader is referred to a review on the subject [67].
Gastric ulceration
The incidence of canine gastric ulcer disease are undetermined. Although vomiting and anorexia would be the expected symptoms of this condition, human gastric ulcer disease can remain clinically “silent” for a substantial period during the progression of the disease.
The cause of gastric ulcer formation is most likely multifactorial, involving mucosal, vascular, endocrine, and neurologic variables. For example, stress is an accepted cause of gastric ulceration in people, and there is an increased incidence of ulcerogenesis in stressed hypothyroid rats mediated by gastric acid hypersecretion [68].
Septic patients, postoperative patients, and patients that have been burned or experienced head trauma are predisposed to developing gastric ulcers [69]. Increased vagal activity, increased gastric acid secretion, histamine release, decreased mucosal barrier function, decreased prostaglandin synthesis, and decreased mucosal blood flow are all potential causative factors [70]. The vagus nerve mediates excitatory input for increased acid secretion by parietal cells and increased bowel motility. In addition to the vagal release of acetylcholine, a wide variety of neuromodulators are known to be active in the gut (ie, serotonin, norepinephrine, gastrin, somatostatin, substance P, vasointestinal active peptide [VIP]), and both VIP and thyrotropin-releasing hormone (TRH) have been shown to induce or aggravate gastric ulcers. Although perhaps difficult to quantify, it is easy to appreciate the fact that our veterinary patient population is also subjected to stress, whether it be from illness, surgery, hospitalization, or even more subtle factors. The role of drugs in gastric ulceration has already been eluded to, and in 65% of the complications seen secondary to peptic ulcer disease in people (ie, hemorrhage, perforation), the episode can be linked to recent NSAID ingestion [71]. Altered gastric motility and disorganized myoelectric complex activity have been demonstrated secondary to indomethacin administration in dogs, resulting in gastric ulceration [72]. Although excessive corticosteroids can damage the gastric mucosa, glucocorticoids have a permissive role in the gastric mucosal protection induced by prostaglandins. This aspect of mucosal protection is lost in hypoadrenocorticism, and gastric ulceration is likely an attendant complication in many cases of canine Addison's disease.
Although the presence of melena, hematemesis, positive fecal occult blood, or an elevated blood urea nitrogen (BUN)/creatinine ratio suggests the presence of significant gastric ulceration, the definitive diagnosis relies on visualization (endoscopy) and histopathology. Once a presumptive or definitive diagnosis is made, treatment begins with the cessation of any potentially ulcerogenic substances, followed by any number or combination of medications described in the next section. The general goals of gastric ulcer therapy are to eliminate any identified inciting agent or condition, protect already damaged mucosal tissue, decrease gastric acidity, and promote rapid restitution of the normal mucosal barrier and defense functions.
Other causes
Malignant gastric neoplasia in the dog includes carcinoma, leiomyosarcoma, and lymphoma. Benign gastric tumors include adenomas and leiomyomas. The reader is referred to a recent excellent review for further information on neoplasia as a rule-out for gastritis in the dog [73].
The nematode Physaloptera is the classic parasitic rule-out for chronic gastritis [74]. Intermediate hosts include beetles, crickets, and cockroaches. Adult worms usually occupy the fundus of the stomach or pyloric antrum and, unfortunately, are often diagnosed during the endoscopic search for a more ominous cause of vomition. Because Physaloptera eggs are difficult to find with examination of the feces, a single dose of pyrantel pamoate (4.5 mg/lb) before endoscopy is a simple, inexpensive, and noninvasive strategy for removing this parasite from the rule-out list. Pyrantel is also effective against roundworms, which may cause gastritis during their migratory trek through the stomach [75].
Lymphocytic/plasmacytic gastritis, eosinophilic gastritis, and granulomatous gastritis are best used as histologic descriptions of immune-mediated gastric pathologic findings. Although the term idiopathic may be used in each case to imply that the infiltrating inflammatory cells are the primary causative agent, these cells are most often present in response to a distinct pathologic disturbance, such as neoplastic transformation, parasite infestation, foreign antigens, or infectious agents. If no causative agent can be identified and trial therapy has been attempted where appropriate (ie, treatment for parasites and allergies in eosinophilic gastritis), these cases are treated as primary immune-mediated disturbances with nonspecific but often effective immunosuppression.
Duodenal-gastric reflux (bilious vomiting syndrome) is a component of a variety of human diseases often seen in children or after intestinal surgery [76], [77], [78]. The syndrome is thought to result from abnormalities in the motor function of the stomach and changes in the speed of gastric emptying [79]. In dogs, the diagnosis is one of exclusion to account for vomiting secondary to bile-induced gastric inflammation. Bile salts acting as detergents dissolve the mucosal lipids that help to form the gastric mucosal barrier, allowing for back-diffusion of hydrogen ions and subsequent gastritis [80], [81]. Dogs with the syndrome usually vomit in the morning after an overnight fast and often respond to late night feedings, a prokinetic drug, an H2-receptor antagonist, or some combination thereof.
Secondary gastritis
Amine precursor uptake and decarboxylation (APUD) tumors (ie, gastrinoma), endocrinopathies, and organ failure can all result in gastric hyperacidity and inflammation.
In people, peptic ulcer formation following gastric hyperacidity secondary to excessive gastrin production by a gastrinoma is known as Zollinger-Ellison syndrome. Gastrin not only stimulates excessive acid secretion but seems to decrease the tone of the pyloric sphincter, allowing for duodenal-gastric reflux of bile [82]. The first case report of the canine version of Zollinger-Ellison syndrome appeared in 1977 in a dog with esophagitis, gastritis, and a duodenal ulcer [83]. Gastrinomas are rare in dogs and usually result in vomiting, weight loss, anorexia, and intermittent diarrhea. The biochemistry panel in these dogs may be consistent with a pyloric outflow obstruction (ie, hypokalemia, hypochloremia, metabolic acidosis). Plasma gastrin levels can be measured using a radioimmunoassay kit, and the laboratory should be contacted for proper sample-handling ins tructions. A significant elevation in gastrin should prompt an effort toward tumor localization (eg, ultrasound, CT, MRI, radiolabeled-somatostatin analogues), although that effort may ultimately depend on intraoperative pancreatic palpation. A more complete discussion of the diagnosis and treatment of canine gastrinomas can be found in an excellent recent review [84].
Liver disease can also result in hypergastrinemia, although not usually to the degree seen with a gastrinoma. The loss of hepatic function also results in an increase in a variety of metabolic byproducts and toxins that may directly affect gastric function or stimulate symptoms of gastritis as a component of hepatic encephalopathy. People with chronic renal failure often bleed into their stomachs. Increased gastric mucosal permeability, a decrease in gastric mucosal blood flow, and mucosal ischemia lead to a more acidic intramucosal environment [85].
Gastric ulceration is a frequent complication in dogs with hypoadrenocorticism, contributing to the symptoms of anorexia and vomiting. Systemic hypovolemia with an attendant decrease in gastric mucosal blood flow, loss of the permissive effect of glucocorticoids on mucosal defense, and significant electrolyte abnormalities are all likely contributors to the gastritis seen with this endocrinopathy.
Treatment of gastritis
Because of the established importance of H pylori in human beings, most of the literature directed toward the treatment of gastritis in people addresses the eradication of that causative agent [86]. Treatment of Helicobacter spp in dogs usually entails a 2- to 3-week course of triple therapy: amoxicillin, metronidazole, and famotidine, with azithromycin, clarithromycin, omeprazole, or ranitidine as an alternative substitution. The treatment of bleeding gastric ulcers is also extensively researched but almost invariably involves endoscopy and laser coagulation or similar therapy. Ironically, in one of the few studies looking specifically at the treatment of canine gastric ulceration secondary to neurosurgery and steroid administration, it was concluded that neither omeprazole nor misoprostol was effective in healing or preventing the development of gastric mucosal lesions [87]. This is in contrast to the prevention of gastric ulceration in human beings using NSAIDs, where the use of either a prostaglandin analogue or proton pump inhibitor proved beneficial [88]. Thus, unfortunately, attempting to draw conclusions regarding the treatment of canine gastritis from the current literature is a precarious exercise at best. This is further complicated by the inherent variability in what constitutes appropriate treatment for the myriad of conditions falling under the heading of “gastritis.” The correct therapy may range from emergency exploratory laparotomy to simply the withholding of food on an outpatient basis. Assuming appropriate steps are taken to rule out gastrointestinal obstruction, a brief period of gastric “rest” (withholding food but not water for 24–48 hours) is usually sufficient therapy for resolution in cases of simple acute gastritis. If symptoms persist or worsen during the period of gastric rest or return shortly after the reintroduction of food, further treatment should be superseded by more extensive diagnostics (eg, complete blood work with appropriate ancillary tests, repeat radiographs, or more advanced imaging).
The most effective treatment for canine gastritis is quite obviously that treatment directed toward a specific identified cause (eg, antiparasitic agents, surgical removal of a gastrinoma, discontinuation of an offending drug, removal of an inciting allergen). In lieu of or in addition to specific treatments, there are a large number of agents that can be used in a nonspecific manner, all directed toward the relief of gastritis and its symptoms. Table 1 is a brief summary of those treatments used most commonly in veterinary medicine. The appropriate choice of medication is based on knowledge of the derangement most likely underlying the symptoms (eg, increased gastric acidity in uremic gastritis, gastric hypomotility in bilious vomiting syndrome) and an understanding of the mechanism of action for each drug.
Table 1.
Drugs frequently used in the treatment of canine gastritis
| Drug | Class | Mechanism of action |
|---|---|---|
| Cimetidine | H2-receptor antagonist | Competitively inhibits histamine binding to parietal cell H2 receptors—inhibits gastric acid secretion |
| Ranitidine | H2-receptor antagonist | Inhibits acid secretion to greater extent than cimetidine, prokinetic (acetylcholinesterase inhibition), increase LES pressure |
| Famotidine | H2-receptor antagonist | Similar in potency to ranitidine, no prokinetic or LES effects |
| Nizatidine | H2-receptor antagonist | Similar to ranitidine |
| Misoprostol | Prostaglandin E1 analogue | Inhibits adenylate cyclase, reducing cAMP and protein kinase-dependent H+ production, cytoprotective, increases bicarbonate secretion and mucosal blood flow |
| Omeprazole | Proton pump inhibitor | Inhibits patients cell H+/K+ ATPase enzyme—more potent gastric acid inhibition than with H2-receptor antagonists |
| Sucralfate | Basic aluminum salt of sulfated sucrose | Cytoprotective; selectively binds to ulcerated tissue, binds bile and pepsin, stimulates bicarbonate and prostaglandin E2 secretion, reduces parietal cell responsiveness, preserves mucosal blood flow. |
| Metoclopramide | Antiemetic, prokinetic | Stimulates upper gastrointestinal motility (acetylcholine sensitization), increases LES pressure; dopamine antagonist in CRTZ |
| Domperidone (not yet approved in United States) | Antiemetic, prokinetic | Dopamine antagonist, similar in action to metoclopramide |
| Diphenhydramine | Antihistamine, Antiemetic | Completively inhibits histamine binding to H1 receptors |
| Chlorpromazine | Antiemetic | Phenothiazine derivative, inhibition of CRTZ and emetic center |
| Prochlorperazine | Antiemetic | Phenothiazine derivative, inhibition of CRTZ and emetic center |
| Ondansetron | Antiemetic | 5-HT3 receptor antagonist at periphery and CRTZ |
| Cispride (if available) | Prokinetic | Accelerates gastric emptying, increases LES pressure (enhanced acetylcholine release) |
| Colloidal bismuth | Protectant | Chelates proteinaceous material at the base of an ulcer, complexes with mucoglycoproteins to provide additional diffusion barrier to acid |
Abbreviations: ATPase, adenosine triphosphatase; cAMP, cyclic adenosine monophosphate; CRTZ, chemoreceptor trigger zone; LES, lower esophageal sphincter.
References
- 1.Guilford W.G, Center S.A, Strombeck D.R, Williams D.A, Meyer D.J, editors. Strombeck's small animal gastroenterology. 3rd edition. WB Saunders; Philadelphia: 1996. pp. 261–302. [Google Scholar]
- 2.Neiger R, Simpson K.W. Helicobacter infection in dogs and cats: fact and fiction. J Vet Intern Med. 2000;14:125–133. doi: 10.1892/0891-6640(2000)014<0125:iidacf>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 3.Simpson K.W, Strauss-Avali D, McDonough P.L. Gastric function in dogs with naturally acquired Helicobacter spp. infection. J Vet Intern Med. 1999;13:507–515. doi: 10.1892/0891-6640(1999)013<0507:gfidwn>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 4.Cattoli G, van Vugt R, Zanoni R.G. Occurrence and characterization of gastric Helicobacter spp. in naturally infected dogs. Vet Microbiol. 1999;70:239–250. doi: 10.1016/s0378-1135(99)00150-9. [DOI] [PubMed] [Google Scholar]
- 5.Baba A.L, Catoi C, Basea I. Canine gastritis, associated with Helicobacter spp. infection. Buletinul Universitatii de Stiinte Agricole Cluj Napoca Seria Zootehnie si Medicina Veterinara. 1995;49:527–531. [Google Scholar]
- 6.Haziroglu R, Diker K.S, Guyenc T. Canine gastritis associated with Helicobacter felis. Deutsche Tieraztliche Wochenschrift. 1995;102:474–476. [PubMed] [Google Scholar]
- 7.Lee A, Krakowka S, Fox J.G. Role of Helicobacter felis in chronic canine gastritis. Vet Pathol. 1992;29:487–494. doi: 10.1177/030098589202900601. [DOI] [PubMed] [Google Scholar]
- 8.Patra P.C, Tripathy S.B. Gastritis in dogs and its therapy. Indian Vet J. 1986;63:940–943. [Google Scholar]
- 9.Burrows C.F. Diseases of the canine stomach. Vet Annu. 1986;26:270–282. [Google Scholar]
- 10.Clark W.A. Canine gastric hyperplasia. Vet Annu. 1985;25:245–247. [Google Scholar]
- 11.Happe R.P, van den Brom W.E, van der Gaag I. Duodenogastric reflux in the dog, a clinicopathological study. Res Vet Sci. 1982;33:280–286. [PubMed] [Google Scholar]
- 12.English R.E, Breitschwerdt E.B, Grindem C.B. Zollinger-Ellison syndrome and myelofibrosis in a dog. JAVMA. 1988;192:1430–1434. [PubMed] [Google Scholar]
- 13.Eaton K.A, Paola J.P, Johnson S.E. Gastritis associated with gastric bacteria in asymptomatic, random source dogs. Vet Pathol. 1992;29:454. [abstract] [Google Scholar]
- 14.Lee A, Krakowka S, Fox J.G. Role of Helicobacter felis in chronic canine gastritis. Vet Pathol. 1992;29:487–494. doi: 10.1177/030098589202900601. [DOI] [PubMed] [Google Scholar]
- 15.Chung S.C, Sung J.Y, Suen M.W. Endoscopic assessment of mucosal hemodynamic changes in a canine model of gastric ulcer. Gastrointest Endosc. 1991;37:310–313. doi: 10.1016/s0016-5107(91)70721-9. [DOI] [PubMed] [Google Scholar]
- 16.Whitford G.M, Gashley D.H, Garman R.H. Effects of fluoride on structure and function of canine gastric mucosa. Dig Dis Sci. 1997;42:2146–2155. doi: 10.1023/a:1018895207333. [DOI] [PubMed] [Google Scholar]
- 17.Nagamine K, Inokuchi K, Sakata H. Development of erosive gastritis in a canine model of esophageal varices. Jpn J Surg. 1986;16:218–224. doi: 10.1007/BF02471096. [DOI] [PubMed] [Google Scholar]
- 18.Duane W.C, McHale A.P, Sievert C.E. Lysolecithin-lipid interactions in disruption of the canine gastric mucosal barrier. Am J Physiol. 1986;250:G275–G279. doi: 10.1152/ajpgi.1986.250.3.G275. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan T.R, Jr., Dempsey D.T, Milner R. Effect of local acid-base status on gastric mucosal blood flow and surface cell injury by bile acid. J Surg Res. 1994;56:112–116. doi: 10.1006/jsre.1994.1019. [DOI] [PubMed] [Google Scholar]
- 20.Gana T.J, MacPherson B.R, Koo J. Gastric mucosal blood flow in misoprostol pretreated aspirin-induced ulceration. Ann Surg. 1988;207:327–334. doi: 10.1097/00000658-198803000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdel-Salam O.M, Czimmer J, Debreceni A. Gastric mucosal integrity: gastric mucosal blood flow and microcirculation. An overview. J Physiol (Paris) 2001;95:105–127. doi: 10.1016/s0928-4257(01)00015-8. [DOI] [PubMed] [Google Scholar]
- 22.Kawano S, Tsuji S. Role of mucosal blood flow: a conceptional review in gastric mucosal injury and protection. J Gastroenterol Hepatol. 2000;15(Suppl):D1–D6. doi: 10.1046/j.1440-1746.2000.02142.x. [DOI] [PubMed] [Google Scholar]
- 23.Sorbye H, Svanes K. The role of blood flow in gastric mucosal defense, damage and healing. Dig Dis. 1994;12:305–317. doi: 10.1159/000171465. [DOI] [PubMed] [Google Scholar]
- 24.Wallace J.L, Granger D.N. The cellular and molecular basis of gastric mucosal defence. FASEB J. 1996;10:731–740. doi: 10.1096/fasebj.10.7.8635690. [DOI] [PubMed] [Google Scholar]
- 25.Paimela H, Goddard P.J, Silen W. Present views on restitution of gastrointestinal epithelium. Dig Dis Sci. 1995;40:2495–2496. doi: 10.1007/BF02063263. [DOI] [PubMed] [Google Scholar]
- 26.Szabo S, Nagy L. Pathways, mediators and mechanisms of gastroduodenal mucosal injury. Acta Physiol Hung. 1992;80:9–21. [PubMed] [Google Scholar]
- 27.Szabo S. Mechanisms of gastric mucosal injury and protection. J Clin Gastroenterol. 1991;13(Suppl):S21–S34. [PubMed] [Google Scholar]
- 28.Wallace J.L, Ma L. Inflammatory mediators in gastrointestinal defense and injury. Exp Biol Med. 2001;226:1003–1015. doi: 10.1177/153537020122601107. [DOI] [PubMed] [Google Scholar]
- 29.Perry M.A, Wadhwa S, Parks D.A. Role of oxygen radicals in ischemia-induced lesions in the cat stomach. Gastroenterology. 1986;90:362–367. doi: 10.1016/0016-5085(86)90933-9. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson E.D. Circulatory mechanisms of gastric mucosal damage and protection. Gastroenterology. 1992;102:1788–1800. doi: 10.1016/0016-5085(92)91745-p. [DOI] [PubMed] [Google Scholar]
- 31.Guilford W.G, Jones B.R, Markwell P.J. Food sensitivity in cats with chronic idiopathic gastrointestinal problems. J Vet Intern Med. 2001;15:7–13. doi: 10.1892/0891-6640(2001)015<0007:fsicwc>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Bartuzi Z, Korenkiewicz J, Romanski B. Correlation between Helicobacter pylori infection and food allergy in chronic gastritis. Med Sci Monit. 2000;6:530–538. [PubMed] [Google Scholar]
- 33.Kokkonen J, Ruuska T, Karttunen T.J. Mucosal pathology of the foregut associated with food allergy and recurrent abdominal pains in children. Acta Paediatr. 2001;90:16–21. doi: 10.1080/080352501750064824. [DOI] [PubMed] [Google Scholar]
- 34.Bartuzi Z, Gawronska-Ukleja E, Romanski B. Macroscopic picture of gastric mucosa in endoscopic examination of patients with chronic gastritis and food allergy. Med Sci Monit. 2000;6:1203–1208. [PubMed] [Google Scholar]
- 35.Proujansky R, Winter H.S, Walker W.A. Gastrointestinal syndromes associated with food sensitivity. Adv Pediatr. 1988;35:219–237. [PubMed] [Google Scholar]
- 36.Bischoff S.C, Mayer J.H, Manns M.P. Allergy and the gut. Int Arch Allergy Immunol. 2000;121:270–283. doi: 10.1159/000024340. [DOI] [PubMed] [Google Scholar]
- 37.Blakemore J.C. Gastrointestinal allergy. Vet Clin North Am Small Anim Pract. 1994;24:655–695. doi: 10.1016/s0195-5616(94)50077-9. [DOI] [PubMed] [Google Scholar]
- 38.LeVeen H.H, LeVeen E.G, LeVeen R.F. Awakenings to the pathogenicity of urease and the requirement for continuous long term therapy. Biomed Pharmacother. 1994;48:157–166. doi: 10.1016/0753-3322(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 39.Hawkey C.J. NSAID toxicity: where are we and how do we go forward? J Rheumatol. 2002;29:650–652. [PubMed] [Google Scholar]
- 40.Jones R.D, Baynes R.E, Nimitz C.T. Nonsteroidal anti-inflammatory drug toxicosis in dogs and cats: 240 cases (1989–1990) JAVMA. 1992;201:475–477. [PubMed] [Google Scholar]
- 41.Nicoloff D.M. Indomethacin. Effect on gastric secretion, parietal cell populations, and ulcer provocation in the dog. Arch Surg. 1968;97:809–815. doi: 10.1001/archsurg.1968.01340050149024. [DOI] [PubMed] [Google Scholar]
- 42.Shorrock C.J, Rees W.D. Mechanisms of gastric damage by non-steroidal anti-inflammatory drugs. Scand J Rheumatol Suppl. 1989;78:5–11. doi: 10.3109/03009748909101457. [DOI] [PubMed] [Google Scholar]
- 43.Konturek J.W, Dembinski A, Stoll R. Gastric mucosal blood flow and neutrophil activation in aspirin-induced gastric mucosal damage in man. Scand J Gastroenterol. 1993;28:767–771. doi: 10.3109/00365529309104006. [DOI] [PubMed] [Google Scholar]
- 44.Dow S.W, Rosychuck A.W, McChesney A.E. Effects of flunixin and flunixin plus prednisone on the gastrointestinal tract of dogs. Am J Vet Res. 1990;51:1131–1138. [PubMed] [Google Scholar]
- 45.Green G.A. Understanding NSAIDs: from aspirin to COX-2. Clin Cornerstone. 2001;3:50–60. doi: 10.1016/s1098-3597(01)90069-9. [DOI] [PubMed] [Google Scholar]
- 46.Wallace J.L, Ma L. Inflammatory mediators in gastrointestinal defense and injury. Exp Biol Med. 2001;226:1003–1015. doi: 10.1177/153537020122601107. [DOI] [PubMed] [Google Scholar]
- 47.Nishihara K, Kikuchi H, Kanno T. Comparison of the upper gastrointestinal effects of etodolac and aspirin in healthy dogs. J Vet Med Sci. 2001;63:1131–1133. doi: 10.1292/jvms.63.1131. [DOI] [PubMed] [Google Scholar]
- 48.Reimer M.E, Johnston S.A, Leib M.S. The gastroduodenal effects of buffered aspirin, carprofen, and etodolac in healthy dogs. J Vet Intern Med. 1999;13:472–477. doi: 10.1892/0891-6640(1999)013<0472:tgeoba>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 49.Forsyth S.F, Guilford W.G, Haslett S.J. Endoscopy of the gastroduodenal mucosa after carprofen, meloxicam and ketoprofen administration in dogs. J Small Anim Pract. 1998;39:421–424. doi: 10.1111/j.1748-5827.1998.tb03748.x. [DOI] [PubMed] [Google Scholar]
- 50.Kay-Mugford P, Benn S.J, LaMarre J. In vitro effects of nonsteroidal anti-inflammatory drugs on cyclooxygenase activity in dogs. Am J Vet Res. 2000;61:802–810. doi: 10.2460/ajvr.2000.61.802. [DOI] [PubMed] [Google Scholar]
- 51.Ricketts A.P, Lundy K.M, Seibel S.B. Evaluation of selective inhibition of canine cyclooxygenase 1 and 2 by carprofen and other nonsteroidal anti-inflammatory drugs. Am J Vet Res. 1998;59:1441–1446. [PubMed] [Google Scholar]
- 52.Peskar B.M, Maricic N, Gretzera B. Role of cyclooxygenase-2 in gastric mucosal defense. Life Sci. 2001;69:2993–3003. doi: 10.1016/s0024-3205(01)01407-2. [DOI] [PubMed] [Google Scholar]
- 53.Bjorkman D.J. One hundred years of NSAID gastropathy: are coxibs the answer? Rev Gastroenterol Disord. 2001;1:121–127. [PubMed] [Google Scholar]
- 54.Dow S.W, Rosychuk R.A.W, McChesney A.E. Effects of flunixin and flunixin plus prednisone on the gastrointestinal tract of dogs. Am J Vet Res. 1990;51:1131–1138. [PubMed] [Google Scholar]
- 55.Sorjonen D.C, Dillon A.R, Powers R.D. Effects of dexamethasone and surgical hypotension on the stomach of dogs: clinical, endoscopic, and pathologic evaluations. Am J Vet Res. 1983;44:1233–1237. [PubMed] [Google Scholar]
- 56.McDonough P.L, Simpson K.W. Diagnosing emerging bacterial infections: salmonellosis, campylobacteriosis, clostridial toxicosis, and helicobacteriosis. Semin Vet Med Surg. 1996;11:187–197. doi: 10.1016/s1096-2867(96)80032-6. [DOI] [PubMed] [Google Scholar]
- 57.Covacci A, Telford J.L, Del Giudice G. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 58.Cave D.R. Chronic gastritis and Helicobacter pylori. Semin Gastrointest Dis. 2001;12:196–202. [PubMed] [Google Scholar]
- 59.Jalava K, Kaartinen M, Utriainen M. Helicobacter salomonis sp. nov., a canine gastric Helicobacter sp. related to Helicobacter felis and Helicobacter bizzozeronii. Int J Syst Bacteriol. 1997;47:975–982. doi: 10.1099/00207713-47-4-975. [DOI] [PubMed] [Google Scholar]
- 60.Strauss-Ayali D, Simpson K.W, Schein A.H. Serological discrimination of dogs infected with gastric Helicobacter spp. and uninfected dogs. J Clin Micro. 1999;37:1280–1287. doi: 10.1128/jcm.37.5.1280-1287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eaton K.A, Dewhirst F.E, Paster B.J. Prevalence and varieties of Helicobacter species in dogs from random sources and pet dogs: animal and public health implications. J Clin Microbiol. 1996;34:3165–3170. doi: 10.1128/jcm.34.12.3165-3170.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hermanns W, Kregel K, Breuer W. Helicobacter-like organisms: histopathological examination of gastric biopsies from dogs and cats. J Comp Path. 1995;112:307–318. doi: 10.1016/s0021-9975(05)80083-0. [DOI] [PubMed] [Google Scholar]
- 63.Yamasaki K, Suematsu H, Takahashi T. Comparison of gastric lesions in dogs and cats with and without gastric spiral organisms. JAVMA. 1998;212:529–533. [PubMed] [Google Scholar]
- 64.Simpson K.W, Strauss-Ayali D, McDonough P.L. Gastric function in dogs with naturally acquired gastric Helicobacter spp. infection. J Vet Intern Med. 1999;13:507–515. doi: 10.1892/0891-6640(1999)013<0507:gfidwn>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 65.Walsh J.H, Peterson W.L. The treatment of Helicobacter pylori infection in the management of peptic ulcer disease. N Engl J Med. 1995;333:984–991. doi: 10.1056/NEJM199510123331508. [DOI] [PubMed] [Google Scholar]
- 66.Simpson K, Neiger R, DeNovo R. The relationship of Helicobacter spp. infection to gastric disease in dogs and cats. J Vet Intern Med. 2000;14:223–227. [PubMed] [Google Scholar]
- 67.Pollock R.V.H, Coyne M.J. Canine parvovirus. Vet Clin North Am Small Anim Pract. 1993;23:555–568. doi: 10.1016/S0195-5616(93)50305-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hernandez D.E, Walker C.H, Mason G.A. Influence of thyroid states on stress gastric ulcer formation. Life Sci. 1988;42:1757–1764. doi: 10.1016/0024-3205(88)90042-2. [DOI] [PubMed] [Google Scholar]
- 69.Cho C.H, Koo M.W.L, Garg G.P. Stress-induced gastric ulceration: its aetiology and clinical implications. Scand J Gastroenterol. 1992;27:257–262. doi: 10.3109/00365529209000071. [DOI] [PubMed] [Google Scholar]
- 70.Konturek P.K, Brzozowski T.Z, Konturek S.J. Role of epidermal growth factor, prostaglandin, and sulfhydryls in stress-induced gastric lesions. Gastroenterology. 1990;99:1607–1615. doi: 10.1016/0016-5085(90)90464-c. [DOI] [PubMed] [Google Scholar]
- 71.Wallace J.L. Gastric ulceration: critical events at the neutrophil-endothelium interface. Can J Physiol Pharmacol. 1993;71:98–102. doi: 10.1139/y93-014. [DOI] [PubMed] [Google Scholar]
- 72.Fioramonti J, Bueno L. Gastrointestinal myoelectric activity disturbances in gastric ulcer disease in rats and dogs. Dig Dis Sci. 1980;25:575–580. doi: 10.1007/BF01318869. [DOI] [PubMed] [Google Scholar]
- 73.Gualtieri M, Monzeglio M.G, Scanziani E. Gastric neoplasia. Vet Clin North Am Small Anim Pract. 1999;29:415–440. [PubMed] [Google Scholar]
- 74.Burrows C.F. Infection with the stomach worm Physaloptera as a cause of chronic vomiting in the dog. J Am Anim Hosp Assoc. 1983;19:947–950. [Google Scholar]
- 75.Hall J.A. Diseases of the stomach. In: Ettinger S.J, Feldman E.C, editors. Textbook of veterinary internal medicine. 5th edition. WB Saunders; Philadelphia: 2000. pp. 1154–1182. [Google Scholar]
- 76.Philip P.A. Superior mesenteric artery syndrome: an unusual cause of intestinal obstruction in brain-injured children. Brain Inj. 1992;6:351–358. doi: 10.3109/02699059209034949. [DOI] [PubMed] [Google Scholar]
- 77.Fang S.B, Lee H.C, Huang F.Y. Intestinal pseudo-obstruction followed by major clinical features of Kawasaki disease: report of one case. Acta Paediatr Taiwan. 2001;42:111–114. [PubMed] [Google Scholar]
- 78.Bondurant F.J, Maull K.I, Nelson H.S. Bile reflux gastritis. South Med J. 1987;80:161–165. doi: 10.1097/00007611-198702000-00005. [DOI] [PubMed] [Google Scholar]
- 79.Eagon J.C, Miedema B.W, Kelly K.A. Postgastrectomy syndromes. Surg Clin North Am. 1992;72:445–465. doi: 10.1016/s0039-6109(16)45689-6. [DOI] [PubMed] [Google Scholar]
- 80.Duane W.C, Wiegand D.M. Mechanism by which bile salt disrupts the gastric mucosal barrier in the dog. J Clin Invest. 1980;66:1044–1049. doi: 10.1172/JCI109932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ritchie W.P., Jr. Alkaline reflux gastritis. Late results on a controlled trial of diagnosis and treatment. Ann Surg. 1986;203:537–544. doi: 10.1097/00000658-198605000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGuigan J.E. The endocrine ulcer concept. Dig Dis. 1976;21:144–147. doi: 10.1007/BF01072061. [DOI] [PubMed] [Google Scholar]
- 83.Straus E, Johnson G.F, Yalow R.S. Canine Zollinger-Ellison syndrome. Gastroenterology. 1977;72:380–381. [PubMed] [Google Scholar]
- 84.Simpson K.W. Gastrinoma in dogs. In: Bonagura J.D, editor. Current veterinary therapy XIII. WB Saunders; Philadelphia: 2000. pp. 617–621. [Google Scholar]
- 85.Diebel L, Kozol R, Wilson R.F. Gastric intramucosal acidosis in patients with chronic kidney failure. Surgery. 1993;113:520–526. [PubMed] [Google Scholar]
- 86.Ruggiero P, Peppoloni S, Berti D. New strategies for the prevention and treatment of Helicobacter pylori infection. Expert Opin Invest Drugs. 2002;11:1127–1138. doi: 10.1517/13543784.11.8.1127. [DOI] [PubMed] [Google Scholar]
- 87.Neiger R, Gaschen F, Jaggy A. Gastric mucosal lesions in dogs with acute intervertebral disc disease: characterization and effects of omeprazole or misoprostol. J Vet Intern Med. 2000;14:33–36. doi: 10.1892/0891-6640(2000)014<0033:gmlidw>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 88.Graham D.Y, Agrawal N.M, Roth S.H. Prevention of NSAID-induced gastric ulcer with misoprostol: multicentre, double-blind, placebo-controlled trial. Lancet. 1988;2:1277–1280. doi: 10.1016/s0140-6736(88)92892-9. [DOI] [PubMed] [Google Scholar]