

Abstract
Developed over the past two decades, the antisense strategy has become a technology of recognised therapeutic potential, and many of the problems raised earlier in its application have been solved to varying extents. However, the adequate delivery of antisense oligodeoxynucleotides to individual cells remains an important and inordinately difficult challenge. Synthetic polymers appeared on this scene in the middle 1980s, and there is a surprisingly large variety used or proposed so far as agents for delivery of oligodeoxynucleotides. After discussing the principles of antisense strategy, certain aspects of the ingestion of macromolecules by cells, and the present situation of delivery procedures, this article analyses in detail the attempts to use synthetic polymers as carrier matrices and/or cell membrane permeabilisation agents for delivery of antisense oligodeoxynucleotides. Structural aspects of various polymers, as well as the results, promises and limitations of their use are critically evaluated.
Abbreviations: A, adenine; AS, antisense; C, cytosine; CD, cyclodextrin; DNA, deoxyribonucleic acid; EVAc, poly(ethylene-co-vinyl acetate); G, guanine; HART, hybrid-arrested translation; HELP, high-efficiency liquid phase; HEMA, 2-hydroxyethyl methacrylate; HPMA, N-(2-hydroxypropyl)methacrylamide; IPEC, interpolyelectrolyte complex; ODN, oligodeoxyribonucleotide, oligodeoxynucleotide; PAMAM, polyamidoamines; PCA, polycyanoacrylates; PDTEMA, N-[2-(2-pyridyldithio)]ethylmethacrylamide; PEDOT, poly(3,4-ethylenedioxythiophene); PEG, poly(ethylene glycol); PEI, polyethyleneimine; PEO, poly(ethylene oxide); PGA, poly(glycolic acid); PL, polylysine; PLA, poly(lactic acid); PLL, poly(l-lysine); POR, polyornithine; PS, polyspermine; RME, receptor-mediated endocytosis; RNA, ribonucleic acid; mRNA, messenger ribonucleic acid; RNase, ribonuclease; SNAIGE, synthetic or small nucleic acids interfering with gene expression; T, thymine; VP, 1-vinyl-2-pyrrolidinone
Keywords: Antisense strategy, Antisense oligodeoxynucleotides, Endocytosis, Drug delivery, Charged polymers, Neutral polymers
1. Introduction
Treatment with traditional drugs is based on molecular substitution, which involves the chemical modification of various compounds that are then subjected to screening programmes. An increasing array of inherited and acquired diseases is now understood by an ‘inside out’ approach involving the identification and targeting of the underlying genetic cause. As a result, therapeutic strategies based on the addition of genes to compensate for faulty genes, addition of genes with new functions, or disruption of gene expression have been developed and they are rapidly growing in diversity and scope. Gene therapy treats diseases caused by the malfunction of a gene by transferring genetic material into cells either to correct a genetic error (gene targeting) or to introduce a new function with therapeutic benefit (gene augmentation) [1], [2], [3], [4], [5], [6], [7].
Recent years have witnessed the development of gene disruption technologies. One of such approaches is the antigene strategy [8], [9], which aims either at the inactivation of genes by homologous recombination with DNA constructs or at disturbing gene function by combination with short fragments of DNA, i.e. oligodeoxynucleotides (ODNs), able to form triple helix structures. Differing in its approach from both traditional drug therapy and gene therapy, and—to a certain extent—from antigene strategy, is the antisense (AS) strategy (sometimes called ‘anti-mRNA’ or ‘antimessenger’). The ODNs used in AS strategy target the molecule acting as an intermediary in genetic expression, i.e. the messenger ribonucleic acid (mRNA), with the aim to inhibit the production of the proteins. This technology makes use of very short fragments of nucleic acid molecules, most commonly ODNs (typically 15–30 nucleotides long) (Fig. 1 ) that display a complementary base sequence to the target mRNA. They are able to interact in a rational way with the mRNA transcripts of a mutant or overexpressed gene, preventing their translation into disease-related proteins. In an ideal situation, the mRNA molecule is vulnerable to selective attack, resulting in the arrest of its processing and the suppression of protein biosynthesis. In AS strategy, the target is not the gene, and there is no transfer of genetic material involved. Compared to the traditional drug strategies that are usually nonspecific, the use of AS ODNs provides high specificity and selectivity. Ideally, an AS ODN of a precise length has only one target (the mRNA), and the binding to target is sequence specific. Considering that every single mRNA molecule would be able to generate a large multitude of protein copies unless its processing is arrested, the inhibition of gene expression has to be more efficient and specific than the inhibition of resulting proteins.
Fig. 1.
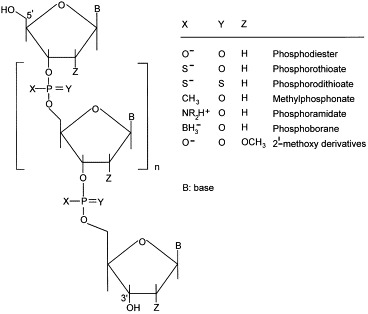
Structure of oligodeoxynucleotides used in antisense strategy.
Zamecnik and Stephenson were first to propose in 1978 the use of synthetic AS ODNs for therapeutic purpose [10], [11], but the term ‘antisense’ came into use in 1984 when it was coined by Izant and Weintraub [12], [13]. Over the last two decades, the AS strategy has received special attention due to the rationality of its foundation and the apparent ease in synthesising and using ODNs as potential drugs. Recent reports [14], [15] suggest that at least a dozen human clinical trials with AS ODNs have been initiated since 1992, directed at targets with roles in cancers, AIDS, inflammatory disorders, psoriasis, cytomegalovirus retinitis, and renal transplant rejections. A commercial antisense therapeutic product (Fomivirsen®) was approved by the FDA in 1998 for the treatment of cytomegalovirus retinitis. Although AS technology is becoming an established strategy of undisputed potential therapeutic power, controversies regarding the mechanism of action and experimental reproducibility, as well as some issues that are presently not well understood, are slowing down the progress in this field. The difficulty in finding an adequate delivery method for AS ODNs in relation to their cellular (or nuclear) uptake is currently perceived as one of the major drawbacks. A number of ingenious stratagems have been developed to deal with this problem, and the use of synthetic polymers as carriers for AS ODNs is one of them. This article attempts to provide a detailed account of the structure, suitability, limitations, and outcomes of the polymers proposed so far for the delivery of AS ODNs.
One expects that the problems underlying the delivery of ODNs in AS strategy should not be significantly different from those underlying the nonviral delivery of DNA in gene therapy. The latter topic has been extensively reviewed [16], [17], [18]. Indeed, many polymer carriers presented here have been also tried for delivery of DNA. However, the size of DNA (commonly a plasmid) is considerably larger than that of an oligonucleotide, which makes cellular and nuclear uptake of plasmids more difficult and creates an additional technical challenge that may limit the range of potential carriers. The nonviral delivery methods employed in gene therapy appear to be less efficient than the virus-mediated delivery [17], [19].
2. Antisense strategy: principles and mechanism
The principle of AS therapeutic action follows the central dogma of molecular biology in eukaryotic cells, and was comprehensively discussed in some major reviews [20], [21], [22], [23], [24], [25], [26]. The basic assumption is simple: if a single-stranded nucleic acid could hybridise to a nucleic acid, the result should be blockage of the normal utilisation in cells of the latter.
Every gene (a double-stranded DNA), carrying the genetic information for a particular protein, is transcribed in the cell nucleus to copies of single-stranded mRNA, the intermediary carrier of information. The primary transcript, the so-called pre-mRNA, undergoes a number of maturation processes to become mature mRNA, which is then translocated into the cytoplasm. Each copy of mRNA then serves as a template for the biosynthesis of a large number of molecules of the particular protein by translation on ribosomes (the protein biosynthesis apparatus in cell), leading to the appearance of the phenotype determined by the original gene. A description of an ideal AS mechanism would be that an AS ODN enters the cell and interacts specifically and directly with molecules of its complementary mRNA. These ODNs are called ‘antisense’ because they constitute a complementary strand to a portion of a coding sequence, generally designated the ‘sense’ strand. Single-stranded mRNA binds to the AS ODN by hydrogen bonds, usually by Watson–Crick base pairing (Fig. 2 ), and forms a double-helix hybrid (duplex). Regarding AS ODNs as drugs, their affinity for receptors (the mRNA sequences) is the result of two processes, the hybridisation by base pairing and the base stacking in the double helix that is formed. The specificity of AS ODNs as drugs results from the highly selective nature of the base pairing itself.
Fig. 2.
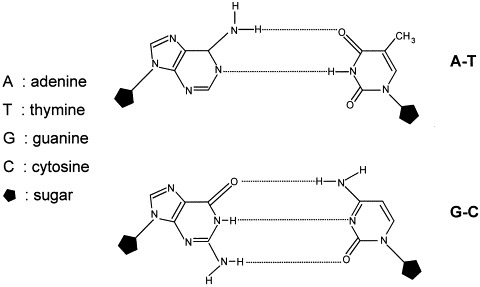
Watson–Crick base pairing.
The hybridised mRNA species become obsolete for translation from several possible causes. For instance, binding of an AS ODN to the mRNA at the site where translation starts prevents the binding of factors that initiate or modulate the translation. Also, the hybrid formation may block the movement of the ribosomes along the mRNA molecule; their progress is held up for a while until they finally drop off. Translation being suppressed, the biosynthesis of the protein for which the target mRNA served as a template is no longer possible (Fig. 3 ). Paterson et al. [27] demonstrated this mechanism for the first time in a cell-free system, and coined for it the term of ‘hybrid-arrested translation’ (HART). Its therapeutic consequence is obvious when the aim is the cancellation of a disease-related protein.
Fig. 3.
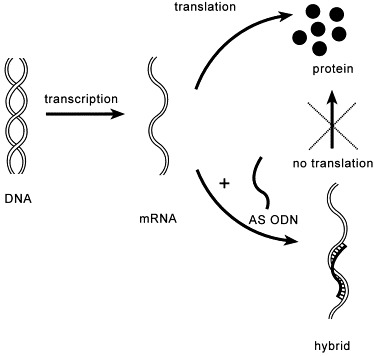
Inhibition of translation by an AS ODN.
Translational arrest is indeed the inhibitory mechanism for which the majority of AS ODNs have been initially designed. However, there is compelling experimental evidence that more than one mechanism can be operative besides HART. The mechanisms by which AS ODNs induce effects appear to be complex, many, and might even be more numerous than thought. In fact, only in a limited number of cases a true antisense inhibitory action has been rigorously demonstrated. Since rather little is presently understood about the contributions of various mechanisms to the events that happen after the ODNs were delivered and exposed to their nucleic acid receptors, a treatment of the operative inhibitory mechanisms involved in the so-called ‘antisense’ activity would remain largely theoretical and beyond the aim of this review. The specialists in the field have extensively discussed these mechanisms [4], [9], [20], [21], [23], [24], [26], [28], [29], [30], [31], [32], [33], [34]. Attempts to rationalise both proven and putative mechanisms led to classifications which take into account that any of the events occurring in the processing of mRNA may be inhibited by intervening ODNs [21], [23], [26], [34]. For instance, AS ODNs can interfere with any of the events preceding translation and taking place in the nucleus, i.e. transcription (in which case the target is DNA), splicing, or maturation. These inhibitory pathways are sometimes classified as ‘occupancy-mediated mechanisms’ [23], a category that also includes HART. Other mechanisms, classified as ‘occupancy-mediated destabilisation’ [23], [26], refer to the inhibition of early post-transcriptional key steps in the processing of pre-mRNA. In order to assure a stable mature mRNA, the pre-mRNA must undergo both capping with a specific protein at the 5′-end and addition of polyadenylate chains at the 3′-end. By interacting with 5′- and 3′-regions of the pre-mRNA, the ODNs can inhibit these steps and thus destabilise the RNA species. Another important mechanism relies on the activation of ribonuclease H (RNase H), a group of ubiquitous enzymes that specifically cleave the RNA strand of an RNA–DNA duplex. The RNase H-mediated cleavage is very effective and appears to be the most commonly accepted mechanism for the activity of AS ODNs, although the recognition of the ODN substrates by RNase H is restricted due to its high sensitivity to the structural changes in ODNs.
Another mechanism is based on the chemical alteration of the target nucleic acid by oligonucleotides bearing reactive species that can induce alkylation or crosslinking. When the oligonucleotides are conjugated to complexes of redox-active metals able to generate activated oxygen species, they can specifically bind to, and then cleave, the target nucleic acid (hence the term ‘artificial endonucleases’ [21]). Related to the latter method, the use of ribozymes has attracted much attention [20], [34], [35]. Ribozymes are RNA species able to catalyse the cleavage of the RNA species within RNA–ribozyme duplexes. The therapeutic exploitation involves their attachment to oligonucleotides in order to be delivered as synthetic entities consisting of antisense domains and catalytic domains. Other single- or double-stranded nucleic acid species, known as ‘aptamers’ [15], [32], bind to proteins that have regulatory functions in the expression of genes. Finally, nonspecific effects of certain sense or homopolymeric ODNs have been observed in many instances [21], suggesting more intricate inhibitory mechanisms.
Strictly speaking, most of these mechanisms are not antisense processes. While, by definition, an AS mechanism is sequence specific and has the mRNA as the target of its action, many of the above mechanisms are sequence independent and/or are targeting DNA or proteins. In an attempt to reconcile this discrepancy and envelop more adequately the large variety of mechanisms and approaches commonly covered by the antisense umbrella term, Leonetti et al. [29] proposed the generic concept of ‘synthetic or small nucleic acids interfering with gene expression’ (SNAIGE). Apart from HART and the mechanisms mentioned above, SNAIGE also includes the disruption of gene expression by triple-helix formation through the hybridisation of oligonucleotides with double-stranded DNA, which is clearly an antigene strategy aimed at transcriptional arrest.
Regardless of the precise mechanism of action, which is still a matter of conjecture from case to case, the advances in AS strategy are afflicted by major problems. While significant progress has been lately recorded toward solving some of these problems, such as stability of ODNs and their hybrids to nuclease activity, target selection and specificity, sequence accessibility, biological activity in vivo, and technical feasibility, there remains a predominant challenge: the ability to suitably deliver the ODNs in order to assure maximum cellular permeability, effective internalisation, and improved efficiency in reaching the target.
3. Cellular internalisation of oligodeoxynucleotides and intracellular events
It is perhaps beneficial to briefly recapitulate what is known about the processes by which eukaryotic cells are able to ingest or eject macromolecules, a topic routinely treated in cell biology textbooks. The import and export of macromolecules (mainly proteins), other particulate matter, and low molecular weight solutes across the plasma membrane is a way by which the cells continuously communicate with the environment. Plasma membrane is the key factor in accumulation or elimination of various components, and it performs these tasks through transport (passive diffusion) and endocytosis. In the transport process, the membrane is stationary, and the solutes (mainly those with low molecular weight) diffuse through it. Endocytosis is the process of ingesting macromolecules or other particulate matter and involves formation of membrane-bounded vesicles. We will not discuss here the release of substances from cell (exocytosis), as it is not yet clear what role this process has in the intracellular trafficking of ODNs.
Endocytosis consists sequentially of inward folding of the plasma membrane (invagination), enveloping a droplet of extracellular medium, pinching off the membrane, and formation of an intracellular vesicle containing the ingested material. The vesicles fuse to form endosomes, which increase their size (to maximum 500 nm) by further fusing with each other or with other intracellular vesicles. This process is known as pinocytosis, and is different from phagocytosis, an endocytic process involving ingestion of bacteria or very large particles of food or foreign material by specialised cells. Most endosomes fuse with membrane-bounded organelles, known as lysosomes, which contain large amounts of hydrolytic enzymes. Endosomes fuse with primary lysosomes to form secondary lysosomes, which are the final destination of the internalised macromolecules targeted for degradation. While the ingested material is rapidly broken down by the lysosomal enzymes, the endocytic membranes are returned to the plasma membrane. Some endosomes bypass the lysosomes, traverse the cytoplasm (transcytosis), and either are incorporated into the plasma membrane or they release their contents by exocytosis. Macromolecules that do not have features recognisable by the membrane are ingested by nonspecific endocytosis, and some cells (e.g. those of the reticuloendothelial system) perform this process at a much higher rate than other cells (e.g. fibroblasts). The main idea to be retained is that the endosomal compartment of a cell performs the important task of sorting the internalised material for (a) degradation in lysosomes, (b) recycling to the plasma membrane, and (c) transcytosis.
Usually, the endocytic cycle begins with endosomes generated by small invaginations occurring in regions of the plasma membrane that are coated with electron-dense structures and are known as coated pits. The pits pinch off the membrane and form coated vesicles, which eventually shed their coats and fuse with early endosomes. The main protein of the coat is clathrin. As the clathrin-coated pits invaginate to become vesicles, the entrapped extracellular fluid (with its components) is internalised by a process called fluid-phase endocytosis. In most animal cells, clathrin-coated pits and vesicles provide a specialised pathway for ingestion of certain macromolecules by cells, termed receptor-mediated endocytosis (RME) or specific adsorptive endocytosis. This process takes place when a receptor at the plasma membrane binds to an extracellular macromolecule (ligand), and the receptor–ligand complex is internalised at a much higher rate than the macromolecules ingested through other pathways. The receptors are present in coated pits even in the absence of ligands but additional receptors accumulate after the binding of ligands. Models have been developed to explain the role of clathrin in the accumulation of receptors in coated pits [36]. While the clathrin-mediated pathways appear to be responsible for the majority of RME and for some pinocytic processes, clathrin-independent pathways for endocytosis have been demonstrated in several situations. Such pathways may proceed via small (50–100 nm) invaginations (caveolae) that contain a specific protein (caveolin) and certain receptors, or through smooth uncoated invaginations (150–300 nm). This process, which is termed ‘potocytosis’ [32], appears to deposit the macromolecules directly into cytosol, avoiding the lysosomal compartments, and some authors regard it as a nonendocytic process.
In order to perform their inhibitory tasks, the AS ODNs must pass through the plasma membrane, escape from cytoplasmic vesicles, and then reach the target in cytoplasm. (When nuclear events are targeted, the ODNs must also enter the nucleus.) There is, however, a serious difficulty: plasma membrane is a potent natural barrier both to large molecules and to negatively charged molecules. Surprisingly, although ODN molecules have at least one of these characteristics, implying little or no proficiency in diffusing across membranes, their ingestion takes place better than would have been expected. For instance, it was demonstrated [37] that about 7.5% of a radiolabeled AS ODN added as a solution in serum-free medium to human cultured cells was internalised by cells within 15 min. The uptake of ‘naked’ AS ODNs is generally too low for a telling biological effect in an in vitro setting. On the contrary, AS effects have been attained in the in vivo setting with naked ODNs, not only in animal experiments but also in some clinical trials, and tentative explanations have been advanced for this apparent dichotomy [15].
Although many aspects of the ingestion of ODNs by cells have yet to be clarified, there is compelling evidence that they enter the cells by an endocytic process [15], [29], [32], [36], [38], [39], [40], [41], [42], [43], [44], [45], [46], not through passive diffusion as earlier thought. Pinocytosis, RME, nonspecific adsorptive endocytosis, and endocytosis mediated by membrane destabilisation were amongst the pathways suggested or demonstrated in different experimental conditions. Non-endocytic processes (e.g. potocytosis) have been also proposed [32], [45], [46], [47]. The identification of receptors for ODNs and their role in RME remain to be elucidated. A number of cell surface ‘receptor-like’ proteins, yet to be characterised, have been associated with the internalisation of AS ODNs [39], [40], [45]; the results are sometimes conflicting and seem to be affected by the type of cells and experimental conditions. It was also shown [46] that the cellular uptake, intracellular trafficking, and activity of AS ODNs were related to the phases of the cell cycle, while the ingestion mechanism was apparently not influenced. According to some authors, two aspects of the internalisation of ODNs should be distinguished [48]: cellular uptake as such and release into the cytoplasm (or nucleus). Cellular uptake refers to binding of ODNs to the plasma membrane and their general ingestion by the cell. Release into the cytoplasm refers to the amount of ODNs that actually reach a compartment where they can be pharmacologically active.
After entering the cell, ODNs accumulate in endosomes and lysosomes. In lysosomes, their predictable fate is degradation. However, a fraction of ODNs escapes from endosomes into cytoplasm by mechanisms not completely understood. This efflux may take place through simple diffusion (unlikely), by transient destabilisation of endosomal membrane mediated by certain membrane proteins, or by leakage during a fusion event between two vesicles [39]. Regardless, it is to be assumed that the observed biological activity is due to those small amounts of AS ODNs that managed to escape from endosomes. Some of the released ODNs may be then transported to the nucleus and eventually enter the nucleus where they may interact with nuclear DNA and RNA species. This scenario is probably more complex than it appears and certainly raises additional problems. Pharmacologically, the endosomal sequestration of ODNs is a nonproductive detour, while lysosomal degradation is a dead end. Facilitation of endosomal escape was consequently an important aim of various stratagems to improve the efficacy of AS ODNs. The endosomal compartmentalisation can be completely bypassed by injecting ODNs directly into cytosol (microinjection), which leads to their rapid accumulation in the nucleus [29], [45]. (For this reason, microinjection experiments are of little significance for the study of compartmentalisation of the ODNs internalised by endocytosis.) The penetration into the nucleus can take place through passive diffusion through nuclear pores or through active signal-mediated diffusion [29], [32]. The former pathway is not possible for macromolecules exceeding 60 kDa in molecular weight, in which case the contribution of nuclear localisation signal molecules is necessary. As the AS ODNs seldom exceed 9 kDa, their nuclear uptake through the pores is unlikely to be obstructed. Although the intracellular fate of ODNs seems to favour a nuclear localisation, there is evidence that they are distributed both in the nucleus and in the cytosol [39], [40]. The conflicting results regarding the preferential distribution may rather come from the large variation of the experimental conditions and from the notoriously poor reproducibility of antisense experiments.
4. Delivery of AS ODNs: the current status
There have been many strategies developed for the enhancement of rate and/or selectivity of cellular internalisation of AS ODNs. In an attempt to make more intelligible the vast literature currently available on this topic, a recent review [48] classifies the delivery strategies into two groups. The first group concerns the cellular uptake process and includes systems able to promote the binding of ODNs to the plasma membrane by either protecting them from degradation or by enhancing their interaction with the membrane. Such systems include conjugation of various compounds (e.g. cholesterol) to ODNs, complexation of ODNs with cationic compounds (e.g. cationic polymers), encapsulation of ODNs (e.g. within liposomes, cyclodextrins), and labeling targets (e.g. intercalating agents, ligands for cell-surface receptors) to ODNs. The second group concerns the entry into the cytoplasm (or nucleus) and includes systems able to facilitate the escape from endosomes, such as cytoplasmic transfer techniques (e.g. microinjection, electroporation) or endosomal membrane destabilising agents (e.g. viral peptides, fusogenic agents). The strategies are used alone or in combination, and every system has its own advantages and drawbacks. Only a brief overview of strategies that do not involve synthetic polymers is presented below.
Administration of naked AS ODNs by infusion or injection in various parts of the body is still the preferred method in vivo, however it requires multiple doses. Experiments in animals showed that nondamaging administration routes (nasally, orally, dermal patches, eye drops) are much less efficient [44]. The AS ODNs can be delivered as naked molecules by microinjection into the cytoplasm or nucleus [49], [50], [51], [52], [53]. Technically, this is a very complex procedure, mainly achievable in cell cultures, and is associated with poor reproducibility and unsatisfactory error range. Electroporation, an electrophoresis-mediated technique, was thought to be feasible only for cell lines in culture and also may induce damage to cells. Although the use of electroporation has been recently re-advocated in gene therapy, there are only a few reports on its application to delivery of ODNs [52], [54]. Viral delivery systems have been used to a little extent in AS strategy [55], [56], [57], [58].
Encapsulation or incorporation in liposomes is currently the preferred method for the delivery of AS ODNs [21], [29], [32], [41], [45], [59], [60], [61], [62], [63], [64], [65], [66], [67], [68], [69] and, besides the intravenous infusion and subcutaneous, intramuscular or intraocular injection of naked ODNs, probably the only other method used in human clinical trials. (Ultimately, the suspensions of liposomes are also administered by infusion or injection.) Liposomes are microscopic closed vesicles varying between 20 nm and 4 μm in diameter and consisting of unilamellar or multilamellar phospholipid membranes that surround aqueous spaces where ODNs can be entrapped. Enhancement of cellular uptake of AS ODNs and, in some cases, of antisense effects has been reported with a large variety of liposomal carrier systems including anionic liposomes, pH-sensitive liposomes, immunoliposomes (i.e. containing antibodies directed at specific cell-surface receptors), fusogenic liposomes (i.e. able to merge with the cellular or endosomal membranes), and cationic liposomes. By far, the cationic lipids are the most frequently used in AS strategies, to the extent that some preparations are now available commercially (e.g. Cytofectin® and Lipofectin® products), as they appear to be active permeabilisation agents and destabilisers of the endosomal membrane. When using cationic lipids, the encapsulation is not a necessary step, as they easily form complexes with ODNs. In spite of their popularity, it is perceived that the liposomes have not been investigated sufficiently as carriers for AS ODNs. Also, there are some limitations on their use. For instance, the basic tendency of cationic liposomes is to accumulate in the reticuloendothelial system [70], leading to a short life in the serum and reduced access to other tissues. Generally, the size of the liposomes of practical importance is too large and their surface charge density is too high. Finally, some types of liposomes display poor encapsulation efficiency. There is no wonder that the search for other carriers has ever continued, one of the results being the association of AS ODNs with synthetic polymer carriers, the topic of our present article.
There is, however, some skepticism about the need for delivery systems for AS ODNs [15]. Most permeabilisation adjuvants employed in delivery systems interact with the plasma membrane, which is the very reason why they are used in this capacity. Consequently, the receptors, signaling processes, and then other functions of the cell may be seriously affected. This aspect has not been properly investigated. Other important questions also arise. Since the AS ODNs can be delivered without any carrier and they still can display AS activity, is there any need to bother with developing delivery systems? In reply, there are many reports indicating enhanced therapeutic effects of AS ODNs when they are delivered in association with an adjuvant. Another issue is that the association of AS ODNs with carriers will increase the size and complexity of the particles to be delivered, thus limiting their access to certain targets or even contributing to their degradation. In reply, we can cite some of the reasons why delivery systems are employed in the first place, i.e. increased cellular uptake, protection against degradation, and improved specificity. Nevertheless, debate is still open about the large size of ODN/carrier pharmacophores and toxicity of carriers.
The improvement of delivery systems for AS ODNs must be therefore an ongoing challenge for those involved in AS strategy research. One reason is the recognition of the fact that the way in which AS ODNs are delivered has dramatic consequences on their fate, as demonstrated in vitro by Wagner et al. [71], [72], [73] in a series of elegant experiments. They found that naked AS ODNs incubated with cells in culture were accumulated in cytoplasmic vesicles (likely endosomes and lysosomes), but not in the nucleus. When the ODNs were microinjected directly into the cytoplasm, all cells showed nuclear localisation of ODNs. When delivery to the cell cultures took place via cationic liposomes, 90% of cells showed nuclear localisation of ODNs. Another reason, probably the most important albeit seldom acknowledged, is the need for a sustained delivery in vivo. Many studies indicate that although ODNs can gain access to the target tissue in vivo, they are eliminated rapidly and repeated administration is required to achieve therapeutic effects [9]. It should be also considered that biological activity of an AS ODN needs to be maintained for additional periods of time to allow for the decay of the preexisting levels of the disease-related protein. The in vivo delivery techniques chiefly used at the present, i.e. infusion or injection of naked molecules and liposomal systems, do not assure adequately long-term maintenance of ODNs in tissues.
5. Synthetic polymers as carriers for delivery of AS ODNs
A detailed overview of the polymer carriers used in AS strategy is attempted in this section. In most of the experiments to be discussed here, phosphorothioate and phosphodiester ODNs were employed as antisense agents, with a preference for the former due to their enhanced resistance to enzymatic attack. The nature of AS ODNs plays, however, a less significant role in the choice and performance of the carrier. Unless relevant to other issues, the phosphorothioate or phosphodiester nature of the AS ODNs was not specified in our exposition.
5.1. Cationic polymers
Since the cellular membrane is negatively charged, it was the cationic polymers that have received the most attention as potential carriers for AS ODNs. The coulombic forces governing the interaction between plasma membrane and polycations are so strong that the influence of other properties of the polymers (e.g. hydrophilicity) is drastically diminished. Indeed, a variety of cationic polymers were able to promote the internalisation of AS ODNs in some cell types, as will be described further. However, the cationic polymers do not constitute the ideal solution. Some cell lines do not take up the ODN–polymer conjugates for reasons yet to be elucidated. There are also serious concerns about the cytotoxicity of positively charged polymers. Cationic carriers must contain enough charge to neutralise the ODNs and also to provide sufficient residual charge for interaction with the membrane of cells. It is, nevertheless, difficult to ascertain how much residual charge is enough, and even more difficult to adjust it to levels that will not render the carrier–ODN conjugate cytotoxic. On the other hand, in conditions of contact with serum, the positive charge of the carrier might be never enough to counterbalance the negatively charged serum components. The internalisation of conjugates can be prohibited in such cases, as firstly demonstrated with a cationic liposome–plasmid DNA conjugate [74].
5.1.1. Synthetic peptides
The interaction of nucleic acids with basic polyelectrolytes is a process that has been known for a long time. In 1946, Kleczkowski showed [75] that the conjugation of proteins with nucleic acids is a general phenomenon that takes place whenever the pH allows them to be of opposite charges, and also suggested that the interaction of proteins with viral nucleoproteins may be responsible for reducing the infectivity of some viruses. At about the same time, other investigators found that the growth of Bacillus anthracis was inhibited by a fraction of the calf thymus [76], as well as by a detergent (a benzylalkylammonium chloride) and by a protein, histone [77]. Both histone and thymus fractions are polypeptides with a high content in the amino acid l-lysine. In further studies, polylysine (PL, Fig. 4 ) and, to a lesser extent, copolymers lysine–valine were shown to induce the agglutination of avian erythrocytes [78], to reduce the infectivity of type A influenza virus [79] and of tobacco mosaic virus [80], [81], and to induce the agglutination of bacterial cells [82]. Electrostatic binding of the cationic polypeptides to the surface of cells or viruses was suggested as the responsible mechanism. These findings were later confirmed [83] for a greater range of polypeptides and bacteria. As the neutral or anionic peptides did not show any effect, it was also suggested [83] that the effect of cationic peptides may be due to a more complex mechanism that includes the destabilisation of cellular membrane and penetration of polymers into the cell, the interaction between nucleic acids and polymers, or the blockage of some enzymatic processes. The effects of variously charged peptides on the agglutination of human erythrocytes were attributed to the same mechanism [84], [85].
Fig. 4.
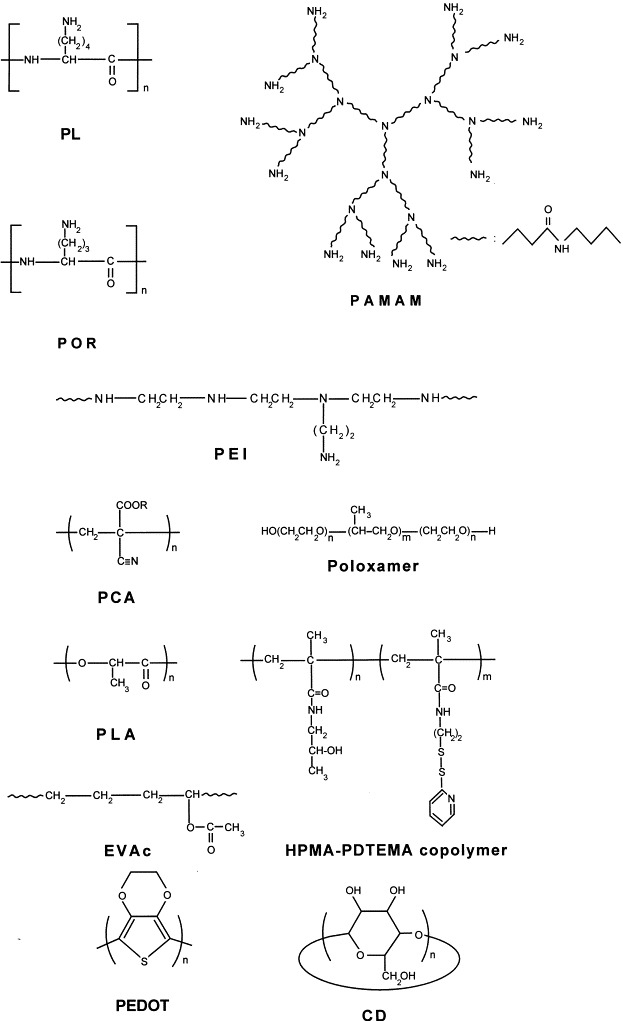
Structure of some of the polymers used as carriers for AS ODNs.
An early application of polycations was the development of a standard procedure for assaying infectious viral nucleic acids, based on the observation that cationic polyelectrolytes such as PL, polyornithine (POR, Fig. 4), or diethylaminoethyl dextran, enhance substantially the infectivity of poliovirus single- and multi-stranded RNAs by promoting their adsorption by cells [86], [87], [88], [89]. Peptide-induced enhancement of viral activity, noticed in other viruses too [90], was regarded as a result of the polycations acting as permeabilisation agents. The enhancement by cationic polyelectrolytes of polynucleotide-induced interferon production was explained by the same mechanism [91], [92], [93]. Another application of cationic polypeptides (PL, POR, polyarginine) included their use as model compounds in the investigation of interaction between nucleic acids and histone species, which plays an important role in the regulation of genetic process. These studies [94], [95], [96], [97], [98], [99] suggested that the binding reaction is more than a simple electrostatic neutralisation, involving steric selectivity and inducing stabilisation of DNA.
As expected, these findings eventually triggered the use of cationic polypeptides as carriers for transportation of bioactive agents. In 1965, Ryser's group at the Harvard Medical School found that the uptake of radiolabeled serum albumin by sarcoma-180 cell cultures is significantly enhanced by the presence of proteins rich in lysine or of synthetic peptides obtained from lysine (l, d or ld), l-ornithine, or l-histidine [100]. They concluded that the process must be more than a simple result of electrostatic interaction and suggested polycation-induced changes in the structure of cellular membrane, as surmised previously by others [85]. Subsequent investigations [101], [102] indicated that individual amino acids or diamines do not have promoting effects, and that the effect of polypeptides is related to their molecular weight and to the distance between amino groups in their molecule, from which it was concluded that their attachment to the cellular membrane must take place through multiple centres. In later studies [103], [104], [105], Ryser's group advocated the covalent binding of carrier polypeptides to the bioactive agent. The cellular uptake of a drug (methotrexate) was considerably enhanced by binding it to poly(l-lysine) (PLL), and the drug was released into the cell in a pharmacologically active form [104]. Its potency was further increased by complexation of the drug–PLL conjugate with heparin, which was believed to facilitate the endosomal escape [105].
Although seldom acknowledged, the first study of the effect of PLL on the cellular uptake of polynucleotides has to be credited to Schell [106], who demonstrated that both pretreatment of cells with PLL and complexation of the polynucleotides with PLL enhanced the uptake of radiolabeled synthetic polyhomoribonucleotides by Ehrlich ascites tumour cells. The mechanisms suggested were based on the interaction between PLL and cellular membrane or on the formation of polynucleotide–PLL complexes with a high affinity for the cellular membrane. It was, however, more than a decade later that PLL was eventually considered as a potential carrier for AS ODNs delivery, mainly due to the pioneering work of Lebleu's group at the University of Montpellier (France), who demonstrated the efficiency of PLL–ODN covalent conjugates in several in vitro biological models. Initially, a dose-dependent inhibition of virus growth was observed when conjugates of PLL and 2′–5′ oligoadenylates (mediators of antiviral activity of interferons) were administered to L1210 or L929 cell cultures infected with various viruses (vesicular stomatitis, vaccinia, etc.) [107], [108]. As cells do not take free oligoadenylates, this was a clear demonstration of PLL efficiency as a permeabilisation agent. Subsequently, the same group showed that AS ODNs complementary to certain regions of the vesicular stomatitis virus nucleocapsid protein mRNA were much more active in cell cultures when conjugated to PLL (10–50 times relative to free ODNs) [109], [110], [111], [112], providing a sequence-specific and dose-dependent antiviral activity. However, it was also noticed that PLL is cytotoxic in high doses or ineffective in some cell lines. By using fluorescein-labeled ODNs, these investigators demonstrated that the uptake of conjugates is both accelerated and enhanced as compared to the unconjugated ODNs. As a mechanism, it was suggested that the conjugates are taken up by cells by nonspecific adsorptive endocytosis, and the AS ODNs are then released through the enzymatic degradation of the lysine moiety after the conjugates were sequestered within acidic cytoplasmic compartments. In summarising their results [29], [41], the authors recommended the use of PLL in order to assure an efficient delivery of AS ODNs. PLL–ODN conjugates were also used to protect infected human MT4 cell lines from the pathologic effects of human immunodeficiency virus type 1 (HIV-1), by significantly reducing the production of viral proteins [113]. In other studies, the effect of PLL–ODN conjugates was further enhanced by complexation with a polyanion (heparin) [29], [114], and some tentative explanations were advanced (reduction of PLL cytotoxicity, modification of the ODN release from conjugate, and/or interference of heparin with virus multiplication). The use of PLL in AS strategy, partially reviewed in a number of articles [9], [29], [32], [41], [48], [115], is still advocated, and a large range of synthetic PLL products (3–60 kDa) are available commercially.
Other synthetic peptides were also investigated as carriers. Covalent conjugates of POR with AS ODNs were evaluated in a cell-free in vitro system by estimating their capacity to stimulate RNase H activity and the effect on the translation of complementary radiolabeled mRNA [116]. Both tests showed enhancement of the activity of POR–ODN conjugates as compared to unconjugated ODNs. Recently, a copolymer of lysine with serine, modified with poly(ethylene glycol), was used as a carrier for AS ODNs targeted against c-raf protein mRNA in pancreatic cancer cells in vitro [117]. The antiproliferative effect of the noncovalent complexes was evident, while the free AS ODNs had no such effect.
Target-labeling strategies using PLL were developed independently at the same time when PLL was proposed as a carrier. Targeting agents are tissue-specific ligands that are linked to PLL and are able to interact specifically with receptors on the target cells. Wu's group at the University of Connecticut was the first to develop a glycoprotein ligand (asialoglycoprotein) covalently coupled to PLL, resulting in conjugates able to function as carriers for delivery of DNA [118], [119] or AS ODNs [120] to hepatoma cells in vitro and in vivo. Not only were the soluble ODN (or DNA)–PLL–asialoglycoprotein conjugates effective in delivering the ODNs or DNA specifically to target cells, but their resistance to nuclease degradation was also enhanced substantially [121]. Although other investigators [122], [123], [124] confirmed the efficacy of PLL–glycoprotein conjugates, more recent research suggested that the direct covalent binding of asialoglycoprotein to ODNs, which excludes the use of PLL, might provide a more efficient delivery system [125]. Other PLL–ligand systems are currently investigated for delivery of AS ODNs [32], [48].
In another approach, noncovalent PLL–ODN complexes were incorporated by diffusion into alginate nanoparticles [126]. This system provided protection against nuclease degradation and also delivered efficiently into animal organs an AS ODN targeted to the mRNA of a retrovirus protein. PLL is also an essential component of the so-called ‘terplex system’ developed for the delivery of DNA and AS ODNs [127], [128]. The system is based on a balanced hydrophobicity and net surface charge between N-stearyl PLL, a lipoprotein and DNA (or ODN). When delivered in vitro by this system, a c-myb AS ODN (18 mer) showed distinctive antiproliferative effects in cultured smooth muscle cells and human lung fibroblasts.
Unlike PLL and other homopoly(amino acids), which are mainly employed because of their electropositive charge, the design and use of some heteropeptides was prompted by their translocation properties, i.e. the ability to destabilise the endosomal membrane and thus facilitate the release of ODN molecules into the cytoplasm before their degradation prevails [48]. These ‘fusogenic’ peptides perform the task through conformational changes induced by the increase of acidity that follows the cytoplasmic compartmentalisation. Most of such peptides were designed and synthesised using as a model the hemagglutinin envelop protein of the influenza virus [48], [129], [130], and the biological activity of their covalent conjugates with AS ODNs has been demonstrated in vitro in various cell lines. Another peptide that proved to be an effective permeabilisation agent was designed to contain a hydrophobic domain like in the fusion sequence of an HIV protein and a hydrophilic domain like in the nuclear localisation sequence of a T-antigen [131]. While the fusogenic peptides have the advantage of following a natural pathway for entering the cytoplasm, they are too expensive to produce and can also cause immunogenic reaction. The results of a recent study [132] provide an even more pessimistic prospect. Eighteen fusogenic peptides containing membrane translocation and nuclear localisation structural motifs were covalently bound to AS ODNs complementary to the initiation codon region of firefly luciferase mRNA and administered to a stable cell line in culture (CHO-AA8-Luc) expressing luciferase activity. None of the conjugates were able to inhibit significantly the luciferase expression, while a formulation based on liposomes (Cytofectin®) induced the expected inhibition. Studies by confocal fluorescein microscopy showed that the conjugates were entrapped in endosomes, and the authors concluded that additional structural motives in the fusogenic peptides are necessary to enable them to escape from endosomes.
In an attempt to rationalise the use of polycations as carriers for nucleic acids, Kabanov et al. [133], [134] extended the use of the term ‘interpolyelectrolyte complex’ (IPEC) to define the water-soluble product of the spontaneous assembling of DNA and a polycation upon mixing. In IPECs, the DNA molecules change their conformation becoming more compact, hence more resistant to nuclease attack. The polycationic component can release some DNA chains during competitive interaction with elements of the cellular membrane. A considerable range of polycations were tentatively used for delivery of DNA in gene therapy, including PLL and derivatives, poly(4-vinylpyridinium) salts, polyamidoamine dendrimers, polyethyleneimine, polymers of 2-alkylaminoethyl methacrylates, diethylaminoethyl dextran, and ‘oligocations’ such as spermidine derivatives. As shown in this article, only some of these polymers have been used so far in the AS strategy.
5.1.2. Dendritic polyamidoamines
The dendrimers are highly branched polymers with a globular architecture. Polyamidoamine (PAMAM) dendrimers (Fig. 4) can be synthesised with a well-defined diameter and a precise number of terminal amino groups at each branching level (‘generation’). Haensler and Szoka Jr [135] were the first to investigate their use as carriers for DNA in gene therapy. Subsequent studies [136], [137], [138] demonstrated that PAMAM–DNA complexes were able to mediate an efficient transfer of plasmid DNA into various cell types in vitro. So far, PAMAM dendrimers have been applied to a little extent as carriers for AS ODNs. In such a study [139], a 27-mer AS ODN complementary to the ATG region of pCMV luciferase plasmid inhibited luciferase expression when delivered as a complex with a PAMAM dendrimer to D5 mouse melanoma cells and Rat2 embryonic fibroblasts, while the AS ODN alone had a negligible effect. A generation 7 ethylenediamine-core dendrimer (512 surface amino groups, MW 90 kDa) was the most efficient carrier when complexed at 1 : 10 charge ratio (ODN to dendrimer). In another study [140], the uptake of a fluorescein-labeled phosphorothioate ODN by human astrocytoma cells was enhanced 50-fold by complexation with a generation 3 dendrimer at 1 : 20 charge ratio. The ODN was distributed both in cytoplasm and in nuclei.
5.1.3. Polyethyleneimine
Polyethyleneimine (PEI, Fig. 4) is produced by the acid-catalysed polymerisation of aziridine, involving propagation through cyclic immonium cations. Due to end-group reactions and to the high reactivity of the intermediate immonium species, the resulting polymer is usually branched. PEI is very soluble in water and has a high capacity to charge electropositively, as every third atom is ionisable nitrogen. PEI was proposed as a carrier for both AS ODNs and plasmid DNA in the same study [141]. A rhodamine-labeled AS ODN targeted at the translation region of the chicken α-thyroid hormone receptor was successfully delivered to embryonic chicken hypothalamic neurons as a complex with PEI, while the uptake of the free ODN could not be detected. Interestingly, the ingested ODN was almost entirely localised into cell nuclei. Since then, PEI has been episodically evaluated as a carrier for plasmid DNA into various cell types and experimental animals, but there was little reported on its use for the delivery of AS ODNs. It was shown [142] that complexation with PEI improved both the uptake and the biological activity of phosphodiester AS ODNs targeted to different regions of Ha-ras mRNA and to the 3′-untranslated region of C-raf kinase, when evaluated in cultures of human bladder carcinoma T24 cells. On the contrary, their liposomal delivery (Lipofectin®) did not elicit antisense activity probably because of nuclease degradation. However, the complexes of PEI with phosphorothioate AS ODNs were so stable that they were taken up by the cells without releasing the ODNs, which therefore were not able to hybridise and show antisense activity.
A recent report on the fate of PEI/DNA complexes in cells is of a general relevance to the use as carriers not only of PEI but, perhaps, of all cationic polymers. In carefully conducted experiments, Mikos et al. [143] followed the path of fluorescein-labeled PEI and PEI/DNA complexes when delivered to cultured EA.hy 926 cells, a line of mixed human immortalised cells. Using confocal microscopy, they found that both complexes and polymer particles attach to cells and are ingested by endocytosis as ‘clumps’. The cytoplasmic vesicles grew in number and size and some of them underwent disruption and released their contents. But the most significant finding was that PEI particles, either delivered as part of complexes with DNA or alone, were eventually localised into nuclei as ordered structures. This suggests the possibility that PEI or other cationic polymers may interfere adversely with host genes, raising serious concerns about their use in human therapies.
5.1.4. Cationic block copolymers
Cationic block copolymers have been developed as carriers for DNA or AS ODNs by Kabanov et al. [144], [145] from polyspermine (PS) and poly(ethylene oxide) (PEO), and by Kataoka et al. [146] from PLL and PEO. Conceptually, these amphiphilic polymers (consisting of a hydrophilic PEO shield surrounding a hydrophobic polycationic core) are able to form micellar structures with DNA or ODNs, which were termed ‘block ionomer complexes’ [145] or ‘polyion complex micelles’ [146]. The synthesis of such block copolymers is quite difficult and, as it is usually accompanied by a high polydispersion of the molecular weights, their purification is extremely laborious. In addition, their complexation with AS ODNs is a difficult problem because of the large difference between the size of the two macromolecules [145]. These drawbacks likely prevented a more extended use of the cationic block copolymers as carriers for AS ODNs. Nevertheless, some studies clearly indicated their efficiency. For instance, a PS–PEO block copolymer was used to deliver an AS ODN complementary to the splice junction of herpes virus type 1 early pre-mRNAs 4 and 5, to infected Vero cells in culture [144]. The inhibition of virus reproduction was markedly increased as compared to that induced by the free AS ODN. It was demonstrated [147] that both PS–PEO and PEI–PEO block copolymers form easily water-soluble complexes with a 24-mer AS ODN and increase its resistance to nuclease degradation. In another study [148], the delivery of AS ODNs, targeted at the suppression of amphiphysin I, as complexes with PS–PEO block copolymers was significantly successful in cultures of hippocampal neurons. In a recent study [149], PS–PEO block copolymers were used to deliver an AS ODN targeted to the fibronectin mRNA into the vitreous cavity of rat eyes, where downregulation of fibronectin expression was achieved successfully.
5.1.5. Polymers with induced surface charge
In some neutral polymers positive surface charges were induced deliberately in order to render them carriers for AS ODNs, based on the principle that cationic polymers will perform better due to the negative charge of the cell membrane. Positive charges were generated in polystyrene nanoparticles by performing the emulsion polymerisation in the presence of cationic initiators, such as 2,2′-azo-bis-(2-amidinopropane) dihydrochloride or 2,2′-azo-bis-[2-(2-imidazoline-2-yl)propane] dihydrochloride [150]. These nanoparticles were thoroughly characterised (size distribution, surface charge, colloidal stability, cytotoxicity, adsorption/desorption of ODNs) and complexed with four different ODNs. It was observed that the resistance to nuclease degradation of the complexed ODNs increased significantly when compared to that of free ODNs. Although a total desorption of ODNs from the particles was achieved only by adding an ionic surfactant, the authors concluded that the cationic polystyrene nanoparticles could function as a suitable delivery system for ODNs. However, no in vitro or in vivo experiments were reported to date.
Polycyanoacrylates (PCA, Fig. 4) are neutral polymers that, in the form of nanoparticles, have been made suitable to function as carriers for AS ODNs by generating positively charged entities on their surface which are able to mediate the electrostatic adsorption of AS ODNs by formation of ion pairs. Unlike the cationic polystyrene described above, the charges on the surface of PCAs are not part of the chemical structure of the polymer, as they are introduced in the system at the time of complexation with ODNs. These investigations were commenced by a group at the Centre National de la Recherche Scientifique in Paris, which provided most of the results currently available [151], [152], [153], [154], [155], [156]. As mediators for ion-pair formation, hydrophobic cations such as quaternary ammonium salts or tetraphenylphosphomium chloride were proposed; in the majority of experiments, cetyltrimethylammonium bromide was added to ODN prior to complexation to the polymer nanoparticles. It was also demonstrated that in the absence of such cations, only negligible amounts of ODNs are complexed to PCAs. It was suggested that the ion-paring cationic agents bind to the polymer through hydrophobic interactions, while their ionic moieties bind electrostatically to ODN. The experiments showed that both the resistance to nuclease degradation and the intracellular concentration of AS ODNs were considerably enhanced by complexation via ion-pairing agents to PCAs. Systems employed so far include poly(isobutyl cyanoacrylate) [151], [156] and poly(isohexyl cyanoacrylate) [151], [152], [154], [155], and this group has achieved significant success in delivery of certain AS ODNs. For instance [152], complexes of AS ODNs targeted to a point mutation in codon 12 of the Ha-ras mRNA inhibited the proliferation of cells expressing the mutated gene at concentrations 100 times lower than free ODNs, and inhibited tumour growth in mice after injection of oncogene-carrying cells. In another experiment [154], AS ODNs complementary to vesicular stomatitis virus nucleocapsid protein mRNA were ingested efficiently by human macrophage-like U937 cells, and their stability was greatly improved by complexation. Since at high concentrations the ion-pairing agents can elicit cytotoxic reactions, an attempt has been made to remove them from the system by covalently modifying the ODNs with hydrophobic moieties, such as cholesterol [155]. Up to 60% of the covalent conjugates were adsorbed onto PCA nanoparticles in the absence of ion-pairing cations, however the modification of ODNs reduced their AS activity. In a recent study [157], transient positive charges were generated on the surface of poly(n-hexyl cyanoacrylate) nanoparticles by using diethylaminoethyl dextran as an emulsion stabiliser during polymerisation. A fluorescein-labeled AS ODN complementary to the splice junction of herpes virus type 1 pre-mRNA 4 was complexed to these nanoparticles and delivered to cultured Vero cells. While the free AS ODN was almost completely degraded in the culture medium, the adsorbed AS ODN remained intact, and its effective ingestion by the cells was demonstrated by laser scanning confocal microscopy and flow cytometry.
5.2. Neutral polymers
The use of neutral polymers came into existence likely because the cationic polymers did not prove to be the ideal carriers for AS ODNs delivery, due especially to their intrinsic cytotoxicity. There are no interactions between the electronegative cellular membrane and a neutral polymer that may facilitate some internalisation process of polymer–ODN conjugates. The neutral polymers are used for their ability to act as carrier systems which assure a sustained release in the long term of the ODN molecules, rather than for a role they may play in the process of cellular uptake, the latter being essentially reduced in this case to the problem of ingestion of free ODN molecules. However, neutral polymers and their conjugates with ODNs can be, in principle, ingested by cells through a pinocytic process. Whether the physical or chemical bond between ODN and polymer is cleaved before or after ingestion of conjugates is an issue little investigated.
5.2.1. Synthetic hydrogels
The synthetic hydrogels are the epitome of soft polymeric biomaterials, which cause insignificant damage to the soft tissue. However, there are very few applications in the delivery of AS ODNs. A class of gels, known by the trade names of Pluronic® or Poloxamer®, has attracted the attention of some investigators due to their reversible thermal gelation properties. These water-soluble materials are uncrosslinked block copolymers of poly(ethylene oxide) and poly(propylene oxide) (Fig. 4), which for particular compositions and block sequences acquire the unusual property of being liquid at ambient or low temperatures while becoming gels at the physiological temperature of the living tissues. Such a block copolymer, supplied as F-127 by BASF Corporation, was the subject of two reports regarding its use as a carrier for AS ODNs. In a study of AS treatment of vasculoproliferative disease [158], AS ODNs targeted to c-myc and c-myb mRNAs were incorporated in a solution of Pluronic® F-127 and then administered to cultured vascular smooth muscle cells or surgically inserted around denuded carotid artery of rats. While the delivery system assured an efficient in vitro cellular uptake of the ODNs and reduced cellular proliferation, the inhibition in vivo of hyperplasia was not effective. Since another system offering prolonged delivery of the ODNs was efficient in vivo (see Section 5.2.5), it was concluded that the release duration from the gel was too short. In an attempt to improve the liposomal delivery of AS ODNs to the vitreous body of the eye, Couvreur et al. [159] dispersed liposomes in a mixture of ODN and aqueous solutions of copolymer F-127, the latter being chosen on the presumption that it will form a gel at the temperature in the eye. Only the influence of the copolymer concentration on the release profile of a radiolabeled model ODN (oligothymidylate, pdT16) in a cell-free in vitro setting was investigated in this study. It was found that the release of liposomes content could be controlled by varying the concentration of Pluronic® in the medium. However, this concentration eventually dropped, as the copolymer is soluble in water. Indeed, the solubility in water seems to be the major drawback of this class of polymers as carriers for ODNs. Ultimately, the carrier will be dispersed into the surrounding physiological medium at a rate that is too high for a proper sustained release, while it is unlikely that its presence affects the mechanism of ODN ingestion.
The crosslinked hydrogels, able to absorb large amounts of water while not dissolving, also attracted attention as possible carriers for ODNs. Aiming at producing artificial vitreous substitutes, a group at Lions Eye Institute (Western Australia) has developed crosslinked hydrogels by homopolymerisation of 1-vinyl-2-pyrrolidinone (VP) or its copolymerisation with 2-hydroxyethyl methacrylate (HEMA) [160], [161], [162], [163], [164], [165]. These materials, which displayed viscoelastic behaviour and could be injected through needles of common sizes, proved not only to lack cytotoxicity [166] but they also showed serum-like growth promoting effect on an anchorage-dependent cell line (3T3 mouse fibroblasts) in static cultures [167]. It was hypothesised that the latter is due to the physical protection of cells by hydrogels and to the ability of hydrogels to mimic the extracellular matrix. As physical protection may involve the interaction between polymer and cell membrane in a way that is not harmful to the cell, it is conceivable that the hydrogel particles can come very close to cells and even contact their membrane without destabilising it or affecting the cell functions. On this background, the group designed a crosslinked copolymer of VP with HEMA (5%), which was covalently bound to an AS ODN targeted to the human rhodopsin gene coding for a mutation at amino acid 351 of the protein [168]. The conjugate was subjected to cell-free in vitro release experiments over 46 days in the presence of natural bovine vitreous humour. Compared to the control (no enzyme present), a significant amount of ODN was released from the hydrogel–ODN conjugate. A possible explanation is that the presence of the enzymes contained in the natural vitreous humour may cleave the covalent bond and induce the release of the ODN. Being a directly injectable gel, this system is attractive for intravitreal delivery of AS ODNs, and further investigations are in progress.
5.2.2. Biodegradable lactone-based polymers
The polyesters produced from cyclic monomers containing lactone structural moieties have a long history of use as biodegradable biomaterials. This class mainly includes polyesters made by polycondensation of l-lactide, glycolide, caprolactone, dioxanone, cyclic carbonates and their derivatives. Polylactide and polyglycolide, respectively known also as poly(lactic acid) (PLA) and poly(glycolic acid) (PGA), are the most investigated biodegradable polymers, which also found applications as carriers for AS ODNs. Akhtar's group at the Aston University in Birmingham (UK) were the first to propose PLA (Fig. 4) for this purpose [169]. Radiolabeled AS ODNs, targeted to the splicing region of tat gene in HIV mRNA or to the AUG initiation codon of human c-myc oncogene exon 2, were incorporated within chloroform-cast films of PLA, and the release profile was monitored over 28 days in cell-free serum or buffer media. The release data corresponded kinetically to the case of diffusion from monolithic devices, and included an initial burst release. The PLA-entrapped ODNs were resistant to degradation by serum nucleases, while the free ODNs were rapidly degraded in serum. The same group developed P(LA-co-GA) microspheres (1–2 or 10–20 μm in size) and evaluated them in vitro [170]. The AS ODNs employed in the preceding study were incorporated in microspheres by a double-emulsion solvent evaporation method and their release profile was studied in buffer media. Microspheres containing radiolabeled AS ODNs were also administered to cultures of mouse macrophage cells (RAW 264.7 line). It was found that the cellular uptake was enhanced and a diffuse intracellular and intranuclear distribution of ODNs occurred when delivered through microspheres. The release duration from larger microspheres was much longer than that from the smaller ones. Further investigations in vitro [171] with a series of AS ODNs showed great advantages of the P(LA-co-GA) microspheres regarding the release characteristics and the enhancement of resistance to nuclease attack. The efficiency of this system was independently confirmed [172] when used to deliver an AS ODN complementary to the translation initiation start region of the rat tenascin mRNA, which led to the inhibition of proliferation and migration of vascular muscle cells in culture. Other investigators used cylindrical implants of P(LA-co-GA) in an attempt to improve the delivery of AS ODNs into the vitreous humour of the eye [173]. An AS ODN complementary to the translation initiation codon region of herpes simplex virus mRNA was incorporated into cylinders of P(LA-co-GA) by heating in an appropriate mould. Each implant contained about 0.5 mg of ODN. The release was studied both in buffer media and by incubation in bovine vitreous fluid (double-filtered natural humour). It was determined that the molecular weight of the polymer did not affect the duration of release. The amount of ODN released in the vitreous fluid was lower than in the buffer solutions, presumably due to the enzymatic degradation of the ODN.
5.2.3. Poly(ethylene glycol)
Poly(ethylene glycol) (PEG) is the term used for the polyoxyethylene polymers with low molecular weight, while the term poly(ethylene oxide) is preferred to designate polyoxyethylenes with high molecular weight. As this convention is rather obscure and generally overlooked, the acronym PEG will be used throughout this section regardless of molecular weight.
PEGs constitute a class of water-soluble polymers with many applications. When crosslinked, PEGs behave like typical hydrogels, a form in which they were employed as carriers in various drug delivery formulations. However, their use in the delivery of ODNs is not based on hydrogels. PEG chains were incorporated covalently as chemical modifiers in order to (a) mask the identity of ODNs, protecting them from recognition by nucleases, and (b) enhance cellular penetration, due probably to the chain structure of PEG. In oligonucleotide chemistry, short-chain PEGs have been inserted as nonnucleotidic flexible linkage in triplex-forming ODNs [174], synthetic ribozymes [175], and duplex- and triplex-forming DNA constructs [176]. Longer-chain PEGs have been used as agents in the solid-phase synthesis of ODNs [177], [178], [179]. The effect of PEG binding to AS ODNs on their cellular internalisation was investigated for the first time by Summerton et al. [180]. A series of short AS ODNs complementary to portions of the CCUCC sequence in prokaryotic 16S-like ribosomal RNA were tested in E. coli bacterial cell cultures in order to detect their inhibitory effects on protein synthesis. The unmodified ODNs manifested such effects only in the ML 308-225 cells, a strain lacking an outer cell wall, and they were ineffective in the D-10 cells, which have normal membrane and permeability properties. The PEG-modified AS ODNs showed a substantial inhibitory effect in the normal cell colonies. The authors surmised that the PEG chain assisted the penetration of ODNs into the outer cell wall, and then was removed by enzymatic cleavage. In another study [181], PEG chains were attached to a large number of ODNs, and the properties of the resulting conjugates were thoroughly investigated. Their hybridisation capacity was not altered by the presence of PEG chains, while their resistance to phosphodiesterase increased considerably. Similar results were reported in a study comparing the resistance to S1 nuclease and specific binding activity of a 15-mer ODN in three modifications, i.e. phosphodiester, phosphorothioate, and PEG-modified phosphodiester [182]. These authors also investigated the retention in plasma of the three ODNs after their intravenous administration in mice. While the PEG-modified phosphodiester ODN displayed retention 5 times longer than that of the phosphodiester, it was the unmodified phosphorothioate ODN which displayed by far the longest retention.
The development of a new solid-phase synthetic procedure for the large-scale synthesis of ODNs, using PEG as a soluble polymeric support, or ‘high-efficiency liquid phase’ (HELP) [183], made possible the production of ODNs (up to 20 mers) containing PEG moieties of variable molecular weight (5–20 kDa). Using this method, a PEG-modified 12-mer AS ODN targeted to HIV-1 mRNA was synthesised [184]. The ODN moiety in the covalent conjugate maintained normal hybridisation capacity and displayed enhanced resistance to phosphodiesterase and nucleotidase degradation, however its antisense activity has yet to be reported.
5.2.4. Copolymers of N-(2-hydroxypropyl)methacrylamide
The homopolymer of N-(2-hydroxypropyl)methacrylamide (HPMA) and its copolymers have been extensively studied as carriers for the site-specific and targeted delivery of drugs [185], [186], [187], [188], [189], [190]. It was shown that HPMA-based polymers exhibit little immunogenicity [191], [192], and they are both pinocytosed by cells [193], [194] and distributed in organs or excreted [195] in a molecular-weight-dependent manner. Few other polymers were subjected to such detailed investigations, however, we are aware of only one study [196] regarding the use of HPMA polymers as carriers for AS ODNs. In this study, a copolymer of HPMA and N-[2-(2-pyridyldithio)]ethylmethacrylamide (PDTEMA) (Fig. 4) was conjugated covalently through sulfhydryl groups to a fluorescein-labeled AS ODN (anti-HIV), and the uptake of the conjugate into cultured HeLa S3 cells was examined by confocal microscopy. Relative to the free AS ODN, the cellular uptake of the conjugate was slightly enhanced, and the conjugate was internalised mainly in endosomes. The presence of a peptide with translocation properties bound to the copolymer did not further enhance the cellular uptake, presumably because the electrostatic association of the peptide with ODN prevented its interaction with the cell membrane.
5.2.5. Hydrophobic elastomers
In a study [158] previously discussed (see Section 5.2.1), it was shown that AS ODNs, targeted to c-myc mRNA, incorporated in solutions of a water-soluble polymer (Pluronic®) and then implanted around the carotid artery of rats failed to show an antisense effect, i.e. the inhibition of hyperplasia, while the same ODN incorporated into poly(ethylene-co-vinyl acetate) (Fig. 4) maintained 99.6% inhibition for the duration of the experiment. The copolymer, known as EVAc or Elvax®, was a hydrophobic elastomer that released the incorporated agents very slowly due to its hydrophobic properties and low rate of aqueous diffusion. EVAc copolymers were the first nonhydrogel polymers to be used for the sustained delivery of biomacromolecules [197], [198], at a time when it was generally believed that large molecules could not be released from such polymers. Concerns were later expressed [199] regarding the loss of biological activity of some biomacromolecules, such as basic fibroblast growth factor, following incorporation in EVAc. This loss was attributed to contact with dichloromethane, a solvent that is mandatory in preparation of EVAc-based delivery systems.
5.2.6. Cyclodextrins
Cyclodextrins (CDs, Fig. 4) comprise a group of water-soluble cyclic polysaccharides containing 6–8 glucose units in α-(1,4) linkage. α-CD, β-CD, and γ-CD have respectively 6, 7, and 8 glucose units and they are all available commercially. Their cyclic structure provides an internal hydrophobic cone-shaped cavity that is surrounded by a hydrophilic shell containing a large number of hydroxyl groups. Due to this conformation, CDs and their derivatives readily form complexes with a large variety of compounds, and consequently they have been investigated as carriers for drugs. In the first study [200] on their application for AS ODNs delivery, complexes of hydroxypropyl and hydroxyethyl β-CDs with radiolabeled and fluorescein-labeled ODNs were prepared and their uptake into human T cell leukemia cell lines was investigated. As compared to the free ODN, the uptake increased 2–3 times within 48 h, and the presence of ODNs was detected intracellularly. The stability to nucleases was not affected by complexation with CDs. In another study [201], a series of derivatives of β-CD and γ-CD were produced and complexed with an AS ODN (phosphodiester or phosphorothioate) targeted to initiation region of the mRNA coding for the spike protein and containing the intergenic consensus sequence of a bovine enteric coronavirus. The antiviral activity of complexes was investigated in a human adenocarcinoma tumour cell line (HRT-18). Both AS ODNs showed antiviral activity when delivered as free molecules, but the phosphodiester induced no more than 34% viral inhibition, as compared to 90% for phosphorothioate. However, the activity of the phosphodiester was markedly improved by complexation with β-CDs. A complex of this AS ODN with a β-CD derivative, 6-deoxy-6-S-β-d-galactopyranosyl-6-thiocyclomaltoheptose, in a molar ratio of 1 : 100 led to a viral inhibition of 90%.
5.3. Anionic polymers
It is rather surprising that anionic polymers have not attracted much attention as potential carriers for AS ODNs. The most plausible reason lies in the misconception that the anionic polymers would be strongly repelled by the cell membrane as they are charged electronegatively too. This is not the case, however. Summarising his and others’ results, Pitha [202] has revealed a different situation, that anionic polymers are adsorbed by cells to a much greater extent than neutral polymers and they are able to create openings in the cell membranes. In quantitative terms, while the membrane of a single cell can bind 1 pg cationic polymer, it binds 0.1 pg anionic polymer, but only 0.001 pg neutral polymer. Nevertheless, it was also demonstrated [102] that anionic polymers do not increase the uptake of proteins into cells. The only applications to date of anionic polymers for delivery of DNA or ODNs involved the use of a crosslinked poly(acrylic acid) hydrogel (Hydro-Plus®). Using a standard angioplasty polyethylene balloon catheter coated with hydrogel, DNA was imbibed through diffusion into the hydrogel coat, and then delivered to the arterial wall in rabbits during balloon angioplasty [203]. Employing an identical device, other investigators incorporated into the hydrogel layer an AS ODN targeted to the c-myb mRNA and examined the effect on smooth muscle cell proliferation [204]. Porcine iliac or carotid arteries underwent angioplasty with these catheters. One week later, the immunohistochemical analysis of isolated arterial segments revealed less cell proliferation in the antisense-treated arteries, as compared to sense ODN controls. Although relatively successful, these applications are no more than examples of sustained delivery of naked ODNs through aqueous diffusion from a hydrogel, and do not involve interaction between the negative charges of the polymer carrier and the plasma membrane of the target cells.
5.4. Electrically conductive polymers
Polymers possessing conjugated π-electron backbone chains have unusual electronic properties. For instance, they can be readily oxidised or reduced by doping with acceptors or donors of electrons, a process that turns them from insulating materials into electroconducting ones, in some cases approaching the conductivity levels of metals. The great potential of electrically conductive conjugated polymers for a large variety of practical applications led to a vast investigative literature regarding their synthesis and properties [205], [206], [207]. The application of these materials in oligonucleotide chemistry originated in the development of an electrochemically controlled synthesis of conductive polymer films containing ODNs [208], [209]. Copolymerisation of pyrrole and pyrrole–ODN covalent conjugates was performed electrochemically and resulted in deposition on an electrode of solid films containing grafted ODNs. It was shown that the ODNs maintained their hybridisation capacity and the films were efficient in detecting point mutations on a synthetic DNA construct. Other investigators went further [210] and produced electrically conductive polymer films containing ODNs with the sole intention of using them for the delivery of ODNs. Films of poly(3,4-ethylenedioxythiophene) (PEDOT, Fig. 4) produced on an electrode were subsequently electrooxidised in the presence of ODNs. The release of entrapped radiolabeled ODNs in buffer solution was monitored over several days. After an initial burst release of electrostatically surface-bound ODN, the remaining ODN was released very slowly, in a diffusion-controlled manner.
6. Conclusions
Perhaps it is not immediately obvious that the use of polymers to deliver AS ODNs to cells represents an example of the application of biomaterials. Indeed, the use of synthetic polymers as orthopaedic prostheses, heart valves, intraocular lenses, or contact lenses, to mention only a few major applications, seems to present problems that have little or nothing in common with the ingestion of the same (or similar) polymers by individual cells. In reality, the application of synthetic polymers in AS strategy is a particular instance where the principles of biomaterials science must be used to design an extremely specific set of properties enabling the polymers to perform the unique task of assisting certain biomacromolecules to reach the intracellular space with unaltered biological activity. A surprisingly large variety of synthetic polymers have been used or proposed as carriers in antisense therapeutic strategy, unlike in gene therapy where the viral delivery systems are still preferred. There are two aspects of the use of polymers as carriers for AS ODNs. One implicates the sustained release of physically incorporated or covalently bound ODNs, governed either by aqueous diffusion or by chemical degradation, which is relatively a classic field of study. The other aspect involves intricate cooperation between discrete polymeric matter and the plasma membrane of the cells, which brings about problems seldom encountered previously in the application of biomaterials. In spite of profuse research, none of the polymer carriers developed so far were able to replace convincingly as a delivery system the infusion (or injection) of free or liposome-encapsulated AS ODNs, although some of the proposed carriers (cationic polymers, biodegradable polymers) showed promising results. Perhaps the attention should be concentrated in the future on combinations between truly functional fusogenic peptides and cationic or neutral polymers. Also, more work is needed to clarify the influence of carrier biodegradation on the events taking place at the cell surface during the ingestion process. However, the fact that the outcome so far is not of decisive help in establishing which polymers shall be the carriers of choice is rather a moot point. This review clearly demonstrates a consistent and ever increasing interest in the polymer-aided delivery of therapeutic AS ODNs, which brings hopes that this biomaterials application will be successful in the forthcoming future. As a new generation of drug therapy in an advanced stage of development, the antisense strategy only awaits a suitable delivery system in order to live up to its promise.
Acknowledgements
This work was supported in part by grants from the Raine Medical Research Foundation (Western Australia) and the Ophthalmic Research Institute of Australia. The expert assistance of the staff in the Medical Library of the University of Western Australia is kindly acknowledged. We thank Gillian Northcott for executing the graphic material.
References
- 1.Anderson W.F. Prospects for human gene therapy. Science. 1984;226:401–409. doi: 10.1126/science.6093246. [DOI] [PubMed] [Google Scholar]
- 2.Friedmann T. Progress toward human gene therapy. Science. 1989;244:1275–1281. doi: 10.1126/science.2660259. [DOI] [PubMed] [Google Scholar]
- 3.Capecchi M.R. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 4.Schreier H. The new frontier: gene and oligonucleotide therapy. Pharm Acta Helv. 1994;68:145–159. doi: 10.1016/0031-6865(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 5.Verma I.M, Somia N. Gene therapy – promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 6.Friedmann T. Overcoming the obstacles to gene therapy. Sci Am. 1997;276(6):80–85. [PubMed] [Google Scholar]
- 7.Yanez R.J, Porter A.C.G. Therapeutic gene targeting. Gene Ther. 1998;5:149–159. doi: 10.1038/sj.gt.3300601. [DOI] [PubMed] [Google Scholar]
- 8.Gewirtz A.M, Stein C.A, Glazer P.M. Facilitating oligonucleotide delivery: helping antisense deliver on its promise. Proc Natl Acad Sci USA. 1996;93:3161–3163. doi: 10.1073/pnas.93.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akhtar S. Antisense technology: selection and delivery of optimally acting antisense oligonucleotides. J Drug Targeting. 1998;5:225–234. doi: 10.3109/10611869808995877. [DOI] [PubMed] [Google Scholar]
- 10.Zamecnik P.C, Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephenson M.L, Zamecnik P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izant J.G, Weintraub H. Inhibition of thymidine kinase gene expression by anti-sense RNA: a molecular approach to genetic analysis. Cell. 1984;36:1007–1015. doi: 10.1016/0092-8674(84)90050-3. [DOI] [PubMed] [Google Scholar]
- 13.Izant J.G, Weintraub H. Constitutive and conditional suppression of exogenous and endogenous genes by anti-sense RNA. Science. 1985;229:345–352. doi: 10.1126/science.2990048. [DOI] [PubMed] [Google Scholar]
- 14.Temsamani J, Guinot P. Antisense oligonucleotides: a new therapeutic approach. Biotechnol Appl Biochem. 1997;26:65–71. [PubMed] [Google Scholar]
- 15.Juliano R.L, Alahari S, Yoo H, Kole R, Cho M. Antisense pharmacodynamics: critical issues in the transport and delivery of antisense oligonucleotides. Pharm Res. 1999;16:494–502. doi: 10.1023/a:1011958726518. [DOI] [PubMed] [Google Scholar]
- 16.Ledley F.D. Non-viral gene therapy. Curr Opin Biotechnol. 1994;5:626–636. doi: 10.1016/0958-1669(94)90085-x. [DOI] [PubMed] [Google Scholar]
- 17.Ledley F.D. Nonviral gene therapy: the promise of genes as pharmaceutical products. Hum Gene Ther. 1995;6:1129–1144. doi: 10.1089/hum.1995.6.9-1129. [DOI] [PubMed] [Google Scholar]
- 18.Domb A.J, Levy M.Y. Polymers in gene therapy. In: Ottenbrite R.M, editor. vol. 2. Technomic; Lancaster, PA: 1999. pp. 1–15. (Frontiers in biomedical polymer applications). [Google Scholar]
- 19.Felgner P.L. Nonviral strategies for gene therapy. Sci Am. 1997;276(6):86–90. doi: 10.1038/scientificamerican0697-102. [DOI] [PubMed] [Google Scholar]
- 20.Colman A. Antisense strategies in cell and developmental biology. J Cell Sci. 1990;97:399–409. doi: 10.1242/jcs.97.3.399. [DOI] [PubMed] [Google Scholar]
- 21.Uhlmann E, Peyman A. Antisense oligonucleotides: a new therapeutic principle. Chem Rev. 1990;90:543–584. [Google Scholar]
- 22.Cazenave C, Helene C. Antisense oligonucleotides. In: Mol J.N.M, van der Krol A.R, editors. Antisense nucleic acids and proteins. Fundamentals and applications. Marcel Dekker; New York: 1991. pp. 47–93. [Google Scholar]
- 23.Crooke S.T. Therapeutic applications of oligonucleotides. Ann Rev Pharmacol Toxicol. 1992;32:329–376. doi: 10.1146/annurev.pa.32.040192.001553. [DOI] [PubMed] [Google Scholar]
- 24.Toulme J.J. Artificial regulation of gene expression by complementary oligonucleotides – an overview. In: Murray J.A.H, editor. Antisense RNA and DNA. Wiley-Liss; New York: 1992. pp. 175–194. [Google Scholar]
- 25.De Mesmaeker A, Haner R, Martin P, Moser H.E. Antisense oligonucleotides. Acc Chem Res. 1995;28:366–374. [Google Scholar]
- 26.Crooke S.T. Progress in antisense therapeutics. Med Res Rev. 1996;16:319–344. doi: 10.1002/(SICI)1098-1128(199607)16:4<319::AID-MED2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Paterson B.M, Roberts B.E, Kuff E.L. Structural gene identification and mapping by DNA mRNA hybrid-arrested cell-free translation. Proc Natl Acad Sci USA. 1977;74:4370–4374. doi: 10.1073/pnas.74.10.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray J.A.H, Crockett N. Antisense techniques: an overview. In: Murray J.A.H, editor. Antisense RNA and DNA. Wiley-Liss; New York: 1992. pp. 1–49. [Google Scholar]
- 29.Leonetti J.P, Degols G, Clarenc J.P, Mechti N, Lebleu B. Cell delivery and mechanisms of action of antisense oligonucleotides. Prog Nucleic Acid Res. 1993;44:143–166. doi: 10.1016/s0079-6603(08)60219-6. [DOI] [PubMed] [Google Scholar]
- 30.Brysch W, Schlingensiepen K.H. Design and application of antisense oligonucleotides in cell culture, in vivo, and as therapeutic agents. Cell Mol Neurobiol. 1994;14:557–568. doi: 10.1007/BF02088837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kregenow D, Ratajczak M.Z, Gewirtz A.M. Disrupting the flow of genetic information with antisense oligodeoxynucleotides: research and therapeutic applications. In: Akhtar S, editor. Delivery strategies for antisense oligonucleotide therapeutics. CRC Press; Boca Raton, FL: 1995. pp. 1–15. [Google Scholar]
- 32.Mahato R.I, Takakura Y, Hashida M. Development of targeted delivery systems for nucleic acid drugs. J Drug Targeting. 1997;4:337–357. doi: 10.3109/10611869709017892. [DOI] [PubMed] [Google Scholar]
- 33.Alama A, Barbieri F, Cagnoli M, Schettini G. Antisense oligonucleotides as therapeutic agents. Pharmacol Res. 1997;36:171–178. doi: 10.1006/phrs.1997.0227. [DOI] [PubMed] [Google Scholar]
- 34.Lavrovsky Y, Chen S, Roy A.K. Therapeutic potential and mechanism of action of oligonucleotides and ribozymes. Biochem Mol Med. 1997;62:11–22. doi: 10.1006/bmme.1997.2631. [DOI] [PubMed] [Google Scholar]
- 35.Woolf T.M. To cleave or not to cleave: ribozymes and antisense. Antisense Res Dev. 1995;5:227–232. doi: 10.1089/ard.1995.5.227. [DOI] [PubMed] [Google Scholar]
- 36.Slepnev V.I, De Camilli P. Endocytosis: an overview. In: Kabanov A.V, Felgner P.L, Seymour L.W, editors. Self-assembling complexes for gene delivery. Wiley; Chichester: 1998. pp. 71–88. [Google Scholar]
- 37.Zamecnik P.C, Goodchild J, Taguchi Y, Sarin P.S. Inhibition of replication and expression of human T-cell lymphotrophic virus type III in cultured cells by exogenous synthetic oligonucleotides complementary to viral RNA. Proc Natl Acad Sci USA. 1986;83:4143–4146. doi: 10.1073/pnas.83.12.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loke S.L, Stein C.A, Zhang X.H, Mori K, Nakanishi M, Subasinghe C, Cohen J.S, Neckers L.M. Characterization of oligonucleotide transport into living cells. Proc Natl Acad Sci USA. 1989;86:3474–3478. doi: 10.1073/pnas.86.10.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akhtar S, Shoji Y, Juliano R.L. Pharmaceutical aspects of the biological stability and membrane transport characteristics of antisense oligonucleotides. In: Erickson R.P, Izant J.G, editors. Gene regulation: biology of antisense RNA and DNA. Raven Press; New York: 1992. pp. 133–145. [Google Scholar]
- 40.Bennett R.M. As nature intended? The uptake of DNA and oligonucleotides by eukaryotic cells. Antisense Res Dev. 1993;3:235–241. doi: 10.1089/ard.1993.3.235. [DOI] [PubMed] [Google Scholar]
- 41.Clarenc J.P, Degols G, Leonetti J.P, Milhaud P, Lebleu B. Delivery of antisense oligonucleotides by poly(l-lysine) conjugation and liposome encapsulation. Anti-Cancer Drug Des. 1993;8:81–94. [PubMed] [Google Scholar]
- 42.Crooke R.M. Cellular uptake, distribution and metabolism of phosphorothioate, phosphodiester, and methylphosphonate oligonucleotides. In: Crooke S.T, Lebleu B, editors. Antisense research and applications. CRC Press; Boca Raton, FL: 1993. pp. 427–449. [Google Scholar]
- 43.Neckers L.M. Cellular internalization of oligodeoxynucleotides. In: Crooke S.T, Lebleu B, editors. Antisense research and applications. CRC Press; Boca Raton, FL: 1993. pp. 451–460. [Google Scholar]
- 44.Vlassov V.V, Yakubov L.A, Karamyshev V, Pautova L, Rykova E, Nechaeva M. In vivo pharmacokinetics of oligonucleotides following administration by different routes. In: Akhtar S, editor. Delivery strategies for antisense oligonucleotide therapeutics. CRC Press; Boca Raton, FL: 1995. pp. 71–83. [Google Scholar]
- 45.Zelphati O, Szoka Jr F.C. Liposomes as a carrier for intracellular delivery of antisense oligonucleotides: a real or magic bullet? J Control Rel. 1996;41:99–119. [Google Scholar]
- 46.Wu-Pong S, Bard J, Huffman J, Jimerson J. Oligonucleotide biological activity: relationship to the cell cycle and nuclear transport. Biol Cell. 1997;89:257–261. [PubMed] [Google Scholar]
- 47.Zamecnik P, Aghajanian J, Zamecnik M, Goodchild J, Witman G. Electron micrographic studies of transport of oligodeoxynucleotides across eukaryotic cell membranes. Proc Natl Acad Sci USA. 1994;91:3156–3160. doi: 10.1073/pnas.91.8.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang E, Ajmani P.S, Hughes J.A. Oligonucleotide delivery: a cellular prospective. Pharmazie. 1999;54:559–566. [PubMed] [Google Scholar]
- 49.Sagata N, Oskarsson M, Copeland T, Brumbaugh J, Vande Woude G.F. Function of c-mos proto-oncogene product in meiotic maturation in Xenopus oocytes. Nature. 1988;335:519–525. doi: 10.1038/335519a0. [DOI] [PubMed] [Google Scholar]
- 50.Blondel D, Harmison G.G, Schubert M. Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J Virol. 1990;64:1716–1725. doi: 10.1128/jvi.64.4.1716-1725.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fenster S.D, Wagner R.W, Froehler B.C, Chin D.J. Inhibition of human immunodeficiency virus type-1 env expression by C-5 propyne oligonucleotides specific for rev-response element stem-loop V. Biochemistry. 1994;33:8391–8398. doi: 10.1021/bi00194a002. [DOI] [PubMed] [Google Scholar]
- 52.Schaal H, Klein M, Gehrmann P, Adams O, Scheid A. Requirement of N-terminal amino acid residues of gp41 for human immunodeficiency virus type 1-mediated cell fusion. J Virol. 1995;69:3308–3314. doi: 10.1128/jvi.69.6.3308-3314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamprecht R, Hazvi S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long-but not short-term conditioned taste aversion memory. J Neurosci. 1997;17:8443–8450. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergan R, Hakim F, Schwartz G.N, Kyle E, Cepada R, Szabo J.M, Fowler D, Gress R, Neckers L. Electroporation of synthetic oligonucleotides: a novel technique for ex vivo bone marrow purging. Blood. 1996;88:731–741. [PubMed] [Google Scholar]
- 55.Martiat P, Lewalle P, Taj A.S, Philippe M, Larondelle Y, Vaerman J.L, Wildmann C, Goldman J.M, Michaux J.L. Retrovirally transduced antisense sequences stably suppress P210 BCR-ABL expression and inhibit the proliferation of BCR/ABL-containing cell lines. Blood. 1993;81:502–509. [PubMed] [Google Scholar]
- 56.Morishita R, Gibbons G.H, Ellison K.E, Nakajima M, Zhang L, Kaneda Y, Ogihara T, Dzau V.J. Single intraluminal delivery of antisense cdc2 kinase and proliferating-cell nuclear antigen oligonucleotides results in chronic inhibition of neointimal hyperplasia. Proc Natl Acad Sci USA. 1993;90:8474–8478. doi: 10.1073/pnas.90.18.8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aoki M, Morishita R, Higaki J, Moriguchi A, Kida I, Hayashi S, Matsushita H, Kaneda Y, Ogihara T. In vivo transfer efficiency of antisense oilgonucleotides into the myocardium using HVJ-liposome method. Biochem Biophys Res Commun. 1997;231:540–545. doi: 10.1006/bbrc.1996.5762. [DOI] [PubMed] [Google Scholar]
- 58.Yonemitsu Y, Kaneda Y, Muraishi A, Yoshizumi T, Sugimachi K, Sueishi K. HVJ (Sendai virus)-cationic liposomes: a novel and potentially effective liposome-mediated technique for gene transfer to the airway epithelium. Gene Ther. 1997;4:631–638. doi: 10.1038/sj.gt.3300463. [DOI] [PubMed] [Google Scholar]
- 59.Juliano R.L, Akhtar S. Liposomes as a drug delivery system for antisense oligonucleotides. Antisense Res Dev. 1992;2:165–176. doi: 10.1089/ard.1992.2.165. [DOI] [PubMed] [Google Scholar]
- 60.Bennett C.F, Chiang M.Y, Chan H, Grimm S. Use of cationic lipids to enhance the biological activity of antisense oilgonucleotides. J Liposome Res. 1993;3:85–102. [Google Scholar]
- 61.Thierry A.R, Takle G.B. Liposomes as a delivery system for antisense and ribozyme compounds. In: Akhtar S, editor. Delivery strategies for antisense oligonucleotide therapeutics. CRC Press; Boca Raton, FL: 1995. pp. 199–221. [Google Scholar]
- 62.Bennett C.F. Intracellular delivery of oligonucleotides with cationic liposomes. In: Akhtar S, editor. Delivery strategies for antisense oligonucleotide therapeutics. CRC Press; Boca Raton, FL: 1995. pp. 223–232. [Google Scholar]
- 63.Ropert C, Couvreur P, Malvy C. The delivery of oilgonucleotides using pH sensitive liposomes. In: Akhtar S, editor. Delivery strategies for antisense oligonucleotide therapeutics. CRC Press; Boca Raton, FL: 1995. pp. 233–245. [Google Scholar]
- 64.Tari A, Khodadadian M, Ellerson D, Deisseroth A, Lopez-Berestein G. Liposomal delivery of oligodeoxynucleotides. Leukemia Lymphoma. 1996;21:93–97. doi: 10.3109/10428199609067585. [DOI] [PubMed] [Google Scholar]
- 65.Bennett C.F, Zuckerman J.E, Kornbrust D, Sasmor H, Leeds J.M, Crooke S.T. Pharmacokinetics in mice of a [3H]-labeled phosphorothioate oligonucleotide formulated in the presence and absence of a cationic lipid. J Control Rel. 1996;41:121–130. [Google Scholar]
- 66.Lavigne C, Thierry A.R. Enhanced antisense inhibition of human immunodeficiency virus type 1 in cell cultures by DLS delivery system. Biochem Biophys Res Commun. 1997;237:566–571. doi: 10.1006/bbrc.1997.7191. [DOI] [PubMed] [Google Scholar]
- 67.Hangai M, Tanihara H, Honda Y, Kaneda Y. In vivo delivery of phosphorothioate oligonucleotides into murine retina. Arch Ophthalmol. 1998;116:342–348. doi: 10.1001/archopht.116.3.342. [DOI] [PubMed] [Google Scholar]
- 68.Marcusson E.G, Bhat B, Manoharan M, Bennett C.F, Dean N.M. Phosphorothioate oligodeoxyribonucleotides dissociate from cationic lipids before entering the nucleus. Nucleic Acids Res. 1998;26:2016–2023. doi: 10.1093/nar/26.8.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bochot A, Couvreur P, Fattal E. Intravitreal administration of antisense oligonucleotides: potential of liposomal delivery. Prog Retinal Eye Res. 2000;19:131–147. doi: 10.1016/s1350-9462(99)00014-2. [DOI] [PubMed] [Google Scholar]
- 70.Litzinger D.C. Limitations of cationic liposomes for antisense oligonucleotide delivery in vivo. J Liposome Res. 1997;7:51–61. [Google Scholar]
- 71.Fisher T.L, Terhorst T, Cao X, Wagner R.W. Intracellular disposition and metabolism of fluorescently-labeled unmodified and modified oligonucleotides microinjected into mammalian cells. Nucleic Acids Res. 1993;21:3857–3865. doi: 10.1093/nar/21.16.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wagner R.W, Matteucci M.D, Lewis J.G, Gutierrez A.J, Moulds C, Froehler B.C. Antisense gene inhibition by oligonucleotides containing C-5 propyne pyrimidines. Science. 1993;260:1510–1515. doi: 10.1126/science.7684856. [DOI] [PubMed] [Google Scholar]
- 73.Wagner R.W. Gene inhibition using antisense oligodeoxynucleotides. Nature. 1994;372:333–335. doi: 10.1038/372333a0. [DOI] [PubMed] [Google Scholar]
- 74.Felgner P.L, Gadek T.R, Holm M, Roman R, Chan H.W, Wenz M, Northrop J.P, Ringold G.M, Danielsen M. Lipofectin: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci USA. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kleczkowski A. Combination between different proteins and between proteins and yeast nucleic acid. Biochem J. 1946;40:677–687. doi: 10.1042/bj0400677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bloom W.L, Watson D.W, Cromartie W.J, Freed M. Studies on infection with Bacillus anthracis. IV. Preparation and characterization of an anthracidal substance from various animal tissues. J Infect Dis. 1947;80:41–52. doi: 10.1093/infdis/80.1.41. [DOI] [PubMed] [Google Scholar]
- 77.Weissman N, Graf L.H. Studies on infection with Bacillus anthracis. VII. A comparison of the antibacterial effects of calf thymus histone and a quaternary ammonium cationic detergent on B. anthracis. J Infect Dis. 1947;80:145–153. doi: 10.1093/infdis/80.2.145. [DOI] [PubMed] [Google Scholar]
- 78.Rubini J.R, Stahmann M.A, Rasmussen A.F. Agglutination of red cells by synthetic lysine polypeptides. Proc Soc Exp Biol Med. 1951;76:659–662. doi: 10.3181/00379727-76-18587. [DOI] [PubMed] [Google Scholar]
- 79.Rubini J.R, Rasmussen A.F, Stahmann M.A. Inhibitory effect of synthetic lysine polypeptides on growth of influenza virus in embryonated eggs. Proc Soc Exp Biol Med. 1951;76:662–665. doi: 10.3181/00379727-76-18588. [DOI] [PubMed] [Google Scholar]
- 80.Stahmann M.A, Graf L.H, Patterson E.L, Walker J.C, Watson D.W. The inhibition of tobacco mosaic virus by synthetic lysine polypeptides. J Biol Chem. 1951;189:45–52. [PubMed] [Google Scholar]
- 81.Burger W.C, Stahmann M.A. The combination of lysine polypeptides with tobacco mosaic virus. J Biol Chem. 1951;193:13–22. [PubMed] [Google Scholar]
- 82.Burger W.C, Stahmann M.A. The agglutination and growth inhibition of bacteria by lysine polypeptides. Arch Biochem Biophys. 1952;39:27–36. doi: 10.1016/0003-9861(52)90257-9. [DOI] [PubMed] [Google Scholar]
- 83.Katchalski E, Bichowski-Slomnitzki L, Volcani B.E. The action of some water-soluble poly-α-amino acids on bacteria. Biochem J. 1953;55:671–680. doi: 10.1042/bj0550671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nevo A, de Vries A, Katchalsky A. Interaction of basic polyamino acids with the red blood cell. I. Combination of polylysine with single cells. Biochim Biophys Acta. 1955;17:536–547. doi: 10.1016/0006-3002(55)90416-9. [DOI] [PubMed] [Google Scholar]
- 85.Katchalsky A, Danon D, Nevo A. Interactions of basic polyelectrolytes with the red blood cell. II. Agglutination of red blood cells by polymeric bases. Biochim Biophys Acta. 1959;33:120–138. doi: 10.1016/0006-3002(59)90505-0. [DOI] [PubMed] [Google Scholar]
- 86.Koch G, Quintrell N, Bishop J.M. An agar cell-suspension plaque assay for isolated viral RNA. Biochem Biophys Res Commun. 1966;24:304–309. doi: 10.1016/0006-291x(66)90155-0. [DOI] [PubMed] [Google Scholar]
- 87.Koch G, Bishop J.M. The effect of polycations on the interaction of viral RNA with mammalian cells: studies on the infectivity of single- and double-stranded poliovirus RNA. Virology. 1968;35:9–17. doi: 10.1016/0042-6822(68)90300-0. [DOI] [PubMed] [Google Scholar]
- 88.Bishop J.M, Koch G. Plaque assay for poliovirus and poliovirus specific RNAs. In: Habel K, Salzman N.P, editors. Fundamental techniques in virology. Academic Press; New York: 1969. pp. 131–145. [Google Scholar]
- 89.Wentzky P, Koch G. Influence of polycations on the interaction between poliovirus multistranded ribonucleic acid and HeLa cells. J Virol. 1971;8:35–40. doi: 10.1128/jvi.8.1.35-40.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takebe I, Otsuki Y. Infection of tobacco mesophyll protoplats by tobacco mosaic virus. Proc Natl Acad Sci USA. 1969;64:843–848. doi: 10.1073/pnas.64.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dianzani F, Cantagalli P, Gagnoni S, Rita G. Effect of DEAE-dextran on production of interferon induced by synthetic double-stranded RNA in L cell cultures. Proc Exp Biol Med. 1968;128:708–710. doi: 10.3181/00379727-128-33105. [DOI] [PubMed] [Google Scholar]
- 92.Bausek G.H, Merigan T.C. Cell interaction with a synthetic polynucleotide and interferon production in vitro. Virology. 1969;39:491–498. doi: 10.1016/0042-6822(69)90097-x. [DOI] [PubMed] [Google Scholar]
- 93.Pitha P.M, Pitha J. Interferon induction by single-stranded polynucleotides modified with polybases. J Gen Virol. 1974;24:385–390. doi: 10.1099/0022-1317-24-2-385. [DOI] [PubMed] [Google Scholar]
- 94.Akinrimisi E.O, Bonner J, Ts’o P.O.P. Binding of basic proteins to DNA. J Mol Biol. 1965;11:128–136. doi: 10.1016/s0022-2836(65)80178-4. [DOI] [PubMed] [Google Scholar]
- 95.Tsuboi M, Matsuo K, Ts’o P.O.P. Interaction of poly-l-lysine and nucleic acids. J Mol Biol. 1966;15:256–267. doi: 10.1016/s0022-2836(66)80225-5. [DOI] [PubMed] [Google Scholar]
- 96.Shih T.Y, Bonner J. Template properties of DNA–polypeptide complexes. J Mol Biol. 1970;50:333–344. doi: 10.1016/0022-2836(70)90196-8. [DOI] [PubMed] [Google Scholar]
- 97.Leng M, Felsenfeld G. The preferential interactions of polylysine and polyarginine with specific base sequences in DNA. Proc Natl Acad Sci USA. 1966;56:1325–1332. doi: 10.1073/pnas.56.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Olins D.E, Olins A.L, von Hippel P.H. Model nucleoprotein complexes: studies on the interaction of cationic homopolypeptides with DNA. J Mol Biol. 1967;24:157–176. doi: 10.1016/0022-2836(67)90324-5. [DOI] [PubMed] [Google Scholar]
- 99.Li H.J, Chang C, Weiskopf M. Helix–coil transition in nucleoprotein-chromatin structure. Biochemistry. 1973;12:1763–1772. doi: 10.1021/bi00733a016. [DOI] [PubMed] [Google Scholar]
- 100.Ryser H.J.P, Hancock R. Histones and basic polyamino acids stimulate the uptake of albumin by tumor cells in culture. Science. 1965;150:501–503. doi: 10.1126/science.150.3695.501. [DOI] [PubMed] [Google Scholar]
- 101.Ryser H.J.P. A membrane effect of basic polymers dependent on molecular size. Nature. 1967;215:934–936. doi: 10.1038/215934a0. [DOI] [PubMed] [Google Scholar]
- 102.Ryser H.J.P. Uptake of protein by mammalian cells: an underdeveloped area. Science. 1968;159:390–396. doi: 10.1126/science.159.3813.390. [DOI] [PubMed] [Google Scholar]
- 103.Shen W.C, Ryser H.J.P. Conjugation of poly-l-lysine to albumin and horseradish peroxidase: a novel method of enhancing the cellular uptake of proteins. Proc Natl Acad Sci USA. 1978;75:1872–1876. doi: 10.1073/pnas.75.4.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ryser H.J.P, Shen W.C. Conjugation of methotrexate to poly(l-lysine) increases drug transport and overcomes drug resistance in cultured cells. Proc Natl Acad Sci USA. 1978;75:3867–3870. doi: 10.1073/pnas.75.8.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shen W.C, Ryser H.J.P. Poly(l-lysine) has different membrane transport and drug-carrier properties when complexed with heparin. Proc Natl Acad Sci USA. 1981;78:7589–7593. doi: 10.1073/pnas.78.12.7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schell P.L. Uptake of polynucleotides by mammalian cells. XIV. Stimulation of the uptake of polynucleotides by poly(l-lysine) Biochim Biophys Acta. 1974;340:323–333. doi: 10.1016/0005-2787(74)90277-9. [DOI] [PubMed] [Google Scholar]
- 107.Bayard B, Bisbal C, Lebleu B. Activation of ribonuclease L by (2′–5′)(A)4–poly(l-lysine) conjugates in intact cells. Biochemistry. 1986;25:3730–3736. doi: 10.1021/bi00360a038. [DOI] [PubMed] [Google Scholar]
- 108.Bisbal C, Silhol M, Lemaitre M, Bayard B, Salehzada T, Lebleu B, Percee T.D, Blackburn M.G. 5′-Modified agonist and antagonist (2′–5′) (A)n analogues: synthesis and biological activity. Biochemistry. 1987;26:5172–5178. doi: 10.1021/bi00390a041. [DOI] [PubMed] [Google Scholar]
- 109.Lemaitre M, Bayard B, Lebleu B. Specific antiviral activity of a poly(l-lysine)-conjugated oligodeoxyribonucleotide sequence complementary to vesicular stomatitis virus N protein mRNA initiation site. Proc Natl Acad Sci USA. 1987;84:648–652. doi: 10.1073/pnas.84.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Leonetti J.P, Rayner B, Lemaitre M, Gagnor C, Milhaud P.G, Imbach J.L, Lebleu B. Antiviral activity of conjugates between poly(l-lysine) and synthetic oligodeoxyribonucleotides. Gene. 1988;72:323–332. doi: 10.1016/0378-1119(88)90159-x. [DOI] [PubMed] [Google Scholar]
- 111.Degols G, Leonetti J.P, Gagnor C, Lemaitre M, Lebleu B. Antiviral activity and possible mechanisms of action of oligonucleotides–poly(l-lysine) conjugates targeted to vesicular stomatitis virus mRNA and genomic mRNA. Nucleic Acids Res. 1989;17:9341–9350. doi: 10.1093/nar/17.22.9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leonetti J.P, Degols G, Lebleu B. Biological activity of oligonucleotide–poly(l-lysine) conjugates: mechanism of cell uptake. Bioconjugate Chem. 1990;1:149–153. doi: 10.1021/bc00002a010. [DOI] [PubMed] [Google Scholar]
- 113.Stevenson M, Iversen P.L. Inhibition of human immunodeficiency virus type 1-mediated cytopathic effects by poly(l-lysine)-conjugated synthetic antisense oligodeoxyribonucleotides. J Gen Virol. 1989;70:2673–2682. doi: 10.1099/0022-1317-70-10-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Degols G, Leonetti J.P, Lebleu B. Sequence-specific activity of antisense oligonucleotides conjugated to poly(l-lysine) carriers. Ann NY Acad Sci. 1992;660:331–333. doi: 10.1111/j.1749-6632.1992.tb21104.x. [DOI] [PubMed] [Google Scholar]
- 115.Boado R.J. Antisense drug delivery through the blood–brain barrier. Adv Drug Del Rev. 1995;15:73–107. [PubMed] [Google Scholar]
- 116.Zhu T, Wei Z, Tung C.H, Dickerhof W.A, Breslauer K.J, Georgopoulos D.E, Leibowitz M.J, Stein S. Oligonucleotide–poly-l-ornithine conjugates: binding to complementary DNA and RNA. Antisense Res Dev. 1993;3:265–275. doi: 10.1089/ard.1993.3.265. [DOI] [PubMed] [Google Scholar]
- 117.Aoki Y, Kawa S, Karasawa Y, Horiuchi A, Kiyosawa K. Anti-proliferative effects of unmodified antisense oligodeoxynucleotides targeted against c-raf mRNA: use of poly(lysine/serine) copolymers or cationic lipopolyamines. Clin Exp Pharmacol Physiol. 1998;25:702–705. doi: 10.1111/j.1440-1681.1998.tb02279.x. [DOI] [PubMed] [Google Scholar]
- 118.Wu G.Y, Wu C.H. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J Biol Chem. 1987;262:4429–4432. [PubMed] [Google Scholar]
- 119.Wu G.Y, Wu C.H. Receptor-mediated gene delivery and expression in vivo. J Biol Chem. 1988;263:14621–14624. [PubMed] [Google Scholar]
- 120.Wu G.Y, Wu C.H. Specific inhibition of hepatitis B viral gene expression in vitro by targeted antisense oligonucleotides. J Biol Chem. 1992;267:12436–12439. [PubMed] [Google Scholar]
- 121.Chiou H.C, Tangco M.V, Levine S.M, Robertson D, Kormis K, Wu C.H, Wu G.Y. Enhanced resistance to nuclease degradation of nucleic acids complexed to asialoglycoprotein–polylysine carriers. Nucleic Acids Res. 1994;22:5439–5446. doi: 10.1093/nar/22.24.5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bunnell B.A, Askari F.K, Wilson J.M. Targeted delivery of antisense oligonucleotides by molecular conjugates. Somat Cell Mot Genet. 1992;18:559–569. doi: 10.1007/BF01232652. [DOI] [PubMed] [Google Scholar]
- 123.Lu X.M, Fischman A.J, Jyawook S.L, Hendricks K, Tompkins R.G, Yarmush M.L. Antisense DNA delivery in vivo: liver targeting by receptor-mediated uptake. J Nucl Med. 1994;35:269–275. [PubMed] [Google Scholar]
- 124.Roth C.M, Reiken S.R, Le Doux J.M, Rajur S.B, Lu X.M, Morgan J.R, Yarmush M.L. Targeted antisense modulation of inflammatory cytokine receptors. Biothechnol Bioengng. 1997;55:72–81. doi: 10.1002/(SICI)1097-0290(19970705)55:1<72::AID-BIT9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 125.Rajur S.B, Roth C.M, Morgan J.R, Yarmush M.L. Covalent protein–oligonucleotide conjugates for efficient delivery of antisense molecules. Bioconjugate Chem. 1997;8:935–940. doi: 10.1021/bc970172u. [DOI] [PubMed] [Google Scholar]
- 126.Aynie I, Vauthier C, Chacun H, Fattal E, Couvreur P. Spongelike alginate nanoparticles as a new potential system for the delivery of antisense oligonucleotides. Antisense Nucleic Acid Drug Dev. 1999;9:301–312. doi: 10.1089/oli.1.1999.9.301. [DOI] [PubMed] [Google Scholar]
- 127.Kim J.S, Kim B.I, Maruyama A, Akaike T, Kim S.W. A new non-viral DNA delivery vector: the terplex system. J Control Rel. 1998;53:175–182. doi: 10.1016/s0168-3659(97)00251-4. [DOI] [PubMed] [Google Scholar]
- 128.Lentz M.J, Kim S.W. Terplex system for delivery of DNA and oligonucleotides. In: Kabanov A.V, Felgner P.L, Seymour L.W, editors. Self-assembling complexes for gene delivery. Wiley; Chichester: 1998. pp. 149–166. [Google Scholar]
- 129.Bongartz J.P, Aubertin A.M, Milhaud P.G, Lebleu B. Improved biological activity of antisense oligonucleotides conjugated to a fusogenic peptide. Nucleic Acids Res. 1994;22:4681–4688. doi: 10.1093/nar/22.22.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pichon C, Freulon I, Midoux P, Mayer R, Monsigny M, Roche A.C. Cytosolic and nuclear delivery of oligonucleotides mediated by an amphiphilic anionic peptide. Antisense Nucleic Acid Drug Dev. 1997;7:335–343. doi: 10.1089/oli.1.1997.7.335. [DOI] [PubMed] [Google Scholar]
- 131.Morris M.C, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Antopolsky M, Azhayeva E, Tengvall U, Auriola S, Jaaskelainen I, Ronkko S, Honkakoski P, Urtti A, Lonnberg H, Azhayev A. Peptide–oligonucleotide phosphorothioate conjugates with membrane translocation and nuclear localization properties. Bioconjugate Chem. 1999;10:598–606. doi: 10.1021/bc980133y. [DOI] [PubMed] [Google Scholar]
- 133.Kabanov A.V, Kabanov V.A. DNA complexes with polycations for the delivery of genetic material into cell. Bioconjugate Chem. 1995;6:7–20. doi: 10.1021/bc00031a002. [DOI] [PubMed] [Google Scholar]
- 134.Kabanov A.V, Szoka Jr F.C, Seymour L.W. Interpolyelectrolyte complexes for gene delivery: polymer aspects of transfection activity. In: Kabanov A.V, Felgner P.L, Seymour L.W, editors. Self-assembling complexes for gene delivery. Wiley; Chichester: 1998. pp. 197–218. [Google Scholar]
- 135.Haensler J, Szoka Jr F.C. Polyamidoamine cascade polymers mediate efficient transfection of cells in culture. Bioconjugate Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 136.Tang M.X, Redemann C.T, Szoka Jr F.C. In vitro gene delivery by degraded polyamidoamine dendrimers. Bioconjugate Chem. 1996;7:703–714. doi: 10.1021/bc9600630. [DOI] [PubMed] [Google Scholar]
- 137.Bielinska A, Kukowska-Latallo J, Piehler L.T, Tomalia D.A, Spindler R, Yin R, Baker Jr J.R. Starburst® PAMAM dendrimers: a novel synthetic vector for the transfection of DNA into mammalian cells. Polym Mater Sci Engng. 1995;73:273–274. [Google Scholar]
- 138.Kukowska-Latallo J.F, Bielinska A.U, Johnson J, Spindler R, Tomalia D.A, Baker Jr J.R. Efficient transfer of genetic material into mammalian cells using Starburst polyamidoamine dendrimers. Proc Natl Acad Sci USA. 1996;93:4897–4902. doi: 10.1073/pnas.93.10.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bielinska A, Kukowska-Latallo J.F, Johnson J, Tomalia D.A, Baker Jr J.R. Regulation of in vitro gene expression using antisense oligonucleotides or antisense expression plasmids transfected using starburst PAMAM dendrimers. Nucleic Acids Res. 1996;24:2176–2182. doi: 10.1093/nar/24.11.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.DeLong R, Stephenson K, Loftus T, Fisher M, Alahari S, Nolting A, Juliano R.L. Characterization of complexes of oligonucleotides with polyamidoamine starburst dendrimers and effects on intracellular delivery. J Pharm Sci. 1997;86:762–764. doi: 10.1021/js960409f. [DOI] [PubMed] [Google Scholar]
- 141.Boussif O, Lezoualch F, Zanta M.A, Mergny M.D, Scherman D, Demeneix B, Behr J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dheur S, Dias N, van Aerschot A, Herdewijn P, Bettinger T, Remy J.S, Helene C, Saison-Behmoaras E.T. Polyethylenimine but not cationic lipid improves antisense activity of 3′-capped phosphodiester oligonucleotides. Antisense Nucleic Acid Drug Dev. 1999;9:515–525. doi: 10.1089/oli.1.1999.9.515. [DOI] [PubMed] [Google Scholar]
- 143.Godbey W.T, Wu K.K, Mikos A.G. Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci USA. 1999;96:5177–5181. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kabanov A.V, Vinogradov S.V, Suzdaltseva Y.G, Alakhov V.Y. Water-soluble block polycations as carriers for oligonucleotide delivery. Bioconjugate Chem. 1995;6:639–642. doi: 10.1021/bc00036a001. [DOI] [PubMed] [Google Scholar]
- 145.Seymour L.W, Kataoka K, Kabanov A.V. Cationic block copolymers as self-assembling vectors for gene delivery. In: Kabanov A.V, Felgner P.L, Seymour L.W, editors. Self-assembling complexes for gene delivery. Wiley; Chichester: 1998. pp. 221–239. [Google Scholar]
- 146.Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymers in physiological saline. Macromolecules. 1996;29:8556–8557. [Google Scholar]
- 147.Vinogradov S.V, Bronich T.K, Kabanov A.V. Self-assembly of polyamine–poly(ethylene glycol) copolymers with phosphorothioate oligonucleotides. Bioconjugate Chem. 1998;9:805–812. doi: 10.1021/bc980048q. [DOI] [PubMed] [Google Scholar]
- 148.Mundigi O, Ochoa G.C, David C, Slepnev V.I, Kabanov A, De Camilli P. Amphiphysin I antisense oligonucleotides inhibit neurite outgrowth in cultured hippocampal neurons. J Neurosci. 1998;18:93–103. doi: 10.1523/JNEUROSCI.18-01-00093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Roy S, Zhang K, Roth T, Vinogradov S, Kao R.S, Kabanov A. Reduction of fibronectin expression by intravitreal administration of antisense oligonucleotides. Nature Biotechnol. 1999;17:476–479. doi: 10.1038/8654. [DOI] [PubMed] [Google Scholar]
- 150.Fritz H, Maier M, Bayer E. Cationic polystyrene nanoparticles: preparation and characterization of a model drug carrier system for antisense oligonucleotides. J Colloid Interface Sci. 1997;195:272–288. doi: 10.1006/jcis.1997.5172. [DOI] [PubMed] [Google Scholar]
- 151.Chavany C, Doan T.L, Couvreur P, Puisieux F, Helene C. Polyalkylcyanoacrylate nanoparticles as polymeric carriers for antisense oligonucleotides. Pharm Res. 1992;9:441–449. doi: 10.1023/a:1015871809313. [DOI] [PubMed] [Google Scholar]
- 152.Schwab G, Chavany C, Duroux I, Goubin G, Lebeau J, Helene C, Saison-Behmoaras T. Antisense oligonucleotides adsorbed to polyalkylcyanoacrylates nanoparticles specifically inhibit mutated Ha-ras-mediated cell proliferation and tumorigenicity in nude mice. Proc Natl Acad Sci USA. 1994;91:10460–10464. doi: 10.1073/pnas.91.22.10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schwab G, Duroux I, Chavany C, Helene C, Saison-Behmoaras E. An approach for new anticancer drugs: oncogene-targeted antisense DNA. Ann Oncol. 1994;5:S55–S58. doi: 10.1093/annonc/5.suppl_4.s55. [DOI] [PubMed] [Google Scholar]
- 154.Chavany C, Saison-Behmoaras T, Doan T.L, Puisieux F, Couvreur P, Helene C. Adsorption of oligonucleotides onto polyisohexylcyanoacrylate nanoparticles protects them against nucleases and increases their cellular uptake. Pharm Res. 1994;11:1370–1378. doi: 10.1023/a:1018923301967. [DOI] [PubMed] [Google Scholar]
- 155.Godard G, Boutorine A.S, Saison-Behmoaras E, Helene C. Antisense effects of cholesterol–oligodeoxynucleotide conjugates associated with poly(alkylcyanoacrylate) nanoparticles. Eur J Biochem. 1995;232:404–410. [PubMed] [Google Scholar]
- 156.Nakada Y, Fattal E, Foulquier M, Couvreur P. Pharmacokinetics and biodistribution of oligonucleotide adsorbed onto poly(isobutylcyanoacrylate) nanoparticles after intravenous administration in mice. Pharm Res. 1996;13:38–43. doi: 10.1023/a:1016017014573. [DOI] [PubMed] [Google Scholar]
- 157.Zobel H.P, Kreuter J, Werner D, Noe C.R, Kumel G, Zimmer A. Cationic polyhexylcyanoacrylate nanoparticles as carriers for antisense oligonucleotides. Antisense Nucleic Acid Drug Dev. 1997;7:483–493. doi: 10.1089/oli.1.1997.7.483. [DOI] [PubMed] [Google Scholar]
- 158.Edelman E.R, Simons M, Sirois M.G, Rosenberg R.D. c-myc in vasculoproliferative disease. Circ Res. 1995;76:176–182. doi: 10.1161/01.res.76.2.176. [DOI] [PubMed] [Google Scholar]
- 159.Bochot A, Fattal E, Gulik A, Couarraze G, Couvreur P. Liposomes dispersed within a thermosensitive gel: a new dosage form for ocular delivery of oligonucleotides. Pharm Res. 1998;15:1364–1369. doi: 10.1023/a:1011989202488. [DOI] [PubMed] [Google Scholar]
- 160.Vijayasekaran S, Chirila T.V, Hong Y, Tahija S.G, Dalton P.D, Constable I.J, McAllister I.L. Poly(1-vinyl-2-pyrrolidinone) hydrogels as vitreous substitutes: histopathological evaluation in the animal eye. J Biomater Sci Polym Edn. 1996;7:685–696. doi: 10.1163/156856296x00453. [DOI] [PubMed] [Google Scholar]
- 161.Hong Y, Chirila T.V, Vijayasekaran S, Dalton P.D, Tahija S.G, Cuypers M.J.H, Constable I.J. Crosslinked poly(1-vinyl-2-pyrrolidinone) as a vitreous substitute. J Biomed Mater Res. 1996;30:441–448. doi: 10.1002/(SICI)1097-4636(199604)30:4<441::AID-JBM2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 162.Hong Y, Chirila T.V, Cuypers M.J.H, Constable I.J. Polymers of 1-vinyl-2-pyrrolidinone as potential vitreous substitutes: physical selection. J Biomater Appl. 1996;11:135–181. doi: 10.1177/088532829601100202. [DOI] [PubMed] [Google Scholar]
- 163.Hong Y, Chirila T.V, Vijayasekaran S, Shen W, Lou X, Dalton P.D. Biodegradation in vitro and retention in the rabbit eye of crosslinked poly(1-vinyl-2-pyrrolidinone) hydrogel as a vitreous substitute. J Biomed Mater Res. 1998;39:650–659. doi: 10.1002/(sici)1097-4636(19980315)39:4<650::aid-jbm21>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 164.Chirila T.V, Hong Y. Poly(1-vinyl-2-pyrrolidinone) hydrogels as vitreous substitutes: a rheological study. Polym Int. 1998;46:183–195. [Google Scholar]
- 165.Chirila T.V, Hong Y, Dalton P.D, Constable I.J, Refojo M.F. The use of hydrophilic polymers as artificial vitreous. Prog Polym Sci. 1998;23:475–508. [Google Scholar]
- 166.Hong Y, Chirila T.V, Vijayasekaran S, Davies M.J, Maley M.A.L. In vitro cytotoxicity assay of synthetic hydrogels as potential vitreous substitutes. In Vitro Toxicol. 1996;9:73–82. [Google Scholar]
- 167.Hong Y, Chirila T.V, Fitton J.H, Ziegelaar B.W, Constable I.J. Effect of crosslinked poly(1-vinyl-2-pyrrolidinone) gels on cell growth in static cell cultures. Bio-Med Mater Engng. 1997;7:35–47. [PubMed] [Google Scholar]
- 168.Lou X, Garrett KL, Rakoczy PE, Chirila TV. Synthetic hydrogels as carriers in antisense therapy: preliminary evaluation of an oligodeoxynucleotide covalent conjugate with a copolymer of 1-vinyl-2-pyrrolidinone and 2-hydroxyethyl methacrylate. J Biomater Appl 2001;15:in press. [DOI] [PubMed]
- 169.Lewis K.J, Irwin W.J, Akhtar S. Biodegradable poly(l-lactic acid) matrices for the sustained delivery of antisense oligonucleotides. J Control Rel. 1995;37:173–183. [Google Scholar]
- 170.Akhtar S, Lewis K.J. Antisense oligonucleotide delivery to cultured macrophages is improved by incorporation into sustained-release biodegradable polymer microspheres. Int J Pharm. 1997;151:57–67. [Google Scholar]
- 171.Lewis K.J, Irwin W.J, Akhtar S. Development of a sustained-release biodegradable polymer delivery system for site-specific delivery of oligonucleotides: characterization of P(LA–GA) copolymer microspheres in vitro. J Drug Targeting. 1998;5:291–302. doi: 10.3109/10611869808995882. [DOI] [PubMed] [Google Scholar]
- 172.Cleek R.L, Rege A.A, Denner L.A, Eskin S.G, Mikos A.G. Inhibition of smooth muscle cell growth in vitro by an antisense oligodeoxynucleotide released from poly(dl-lactic-co-glycolic acid) microparticles. J Biomed Mater Res. 1997;35:525–530. doi: 10.1002/(sici)1097-4636(19970615)35:4<525::aid-jbm12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 173.Yamakawa I, Ishida M, Kato T, Ando H, Asakawa N. Release behavior of poly(lactic acid-co-glycolic acid) implants containing phosphorothioate oligodeoxynucleotide. Biol Pharm Bull. 1997;20:455–459. doi: 10.1248/bpb.20.455. [DOI] [PubMed] [Google Scholar]
- 174.Kessler D.J, Pettitt B.M, Cheng Y.K, Smith S.R, Jayaraman K, Vu H.M, Hogan M.E. Triple helix fromation at distant sites: hybrid oligonucleotides containing a polymeric linker. Nucleic Acids Res. 1993;21:4810–4815. doi: 10.1093/nar/21.20.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thomson J.B, Tuschl T, Eckstein F. Activity of hammerhead ribozymes containing non-nucleotidic linkers. Nucleic Acids Res. 1993;21:5600–5603. doi: 10.1093/nar/21.24.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rumney S, Kool E.T. Structural optimization of non-nucleotide loop replacements for duplex and triplex DNAs. J Am Chem Soc. 1995;117:5635–5646. doi: 10.1021/ja00126a004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bayer E, Maier M, Bleicher K, Gaus H.J. Synthesis of 3′-PEG-modified oligonucleotides on PS-PEG tentacle polymers. Z Naturforsch B. 1995;50:671–676. [Google Scholar]
- 178.De Napoli L, Di Fabio G, Messere A, Varra M, Piccialli G. New solid-phase approach to obtain 3′-derivatized oligonucleotides. Gazz Chim Ital. 1996;126:755–759. [Google Scholar]
- 179.Tarasow T.M, Tinnermeier D, Zyzniewski C. Characterization of oligodeoxyribonucleotide–polyethylene glycol conjugates by electrospray mass spectrometry. Bioconjugate Chem. 1997;8:89–93. doi: 10.1021/bc960082+. [DOI] [PubMed] [Google Scholar]
- 180.Rahman M.A, Summerton J, Foster E, Cunningham K, Stirchak E, Weller D, Schaup H.W. Antibacterial activity and inhibition of protein synthesis in Escherichia coli by antisense DNA analogs. Antisense Res Dev. 1991;1:319–327. doi: 10.1089/ard.1991.1.319. [DOI] [PubMed] [Google Scholar]
- 181.Jaschke A, Furste J.P, Nordhoff E, Hillenkamp F, Cech D, Erdmann V.A. Synhthesis and properties of oligodeoxyribonucleotide–polyethylene glycol conjugates. Nucleic Acids Res. 1994;22:4810–4817. doi: 10.1093/nar/22.22.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kawaguchi T, Asakawa H, Tashiro Y, Juni K, Sueishi T. Stability, specific binding activity, and plasma concentration in mice of an oligodeoxynucleotide modified at 5′-terminal with poly(ethylene glycol) Biol Pharm Bull. 1995;18:474–476. doi: 10.1248/bpb.18.474. [DOI] [PubMed] [Google Scholar]
- 183.Bonora G.M. Polyethylene glycol. A high-efficiency liquid phase (HELP) for the large-scale synthesis of the oligonucleotides. Appl Biochem Biotechnol. 1995;54:3–15. [Google Scholar]
- 184.Bonora G.M, Ivanova E, Zarytova V, Burcovich B, Veronese F.M. Synthesis and characterization of high-molecular mass polyethylene glycol-conjugated oligonucleotides. Bioconjugate Chem. 1997;8:793–797. doi: 10.1021/bc970082p. [DOI] [PubMed] [Google Scholar]
- 185.Duncan R, Kopeckova P, Strohalm J, Hume I.C, Lloyd J.B, Kopecek J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. II. Evaluation of daunomycin conjugates in vivo against L1210 leukaemia. Br J Cancer. 1988;57:147–156. doi: 10.1038/bjc.1988.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Flanagan P.A, Kopeckova P, Kopecek J, Duncan R. Evaluation of protein-N-(2-hydroxypropyl)methacrylamide copolymer conjugates as targetable drug carriers. 1. Binding, pinocytic uptake and intracellular distribution of transferrin and anti-transferrin receptor antibody conjugates. Biochim Biophys Acta. 1989;993:83–91. doi: 10.1016/0304-4165(89)90146-3. [DOI] [PubMed] [Google Scholar]
- 187.Subr V, Duncan R, Kopecek J. Release of macromolecules and daunomycin from hydrophilic gels containing enzymatically degradable bonds. J Biomater Sci Polym Edn. 1990;1:261–278. doi: 10.1163/156856289x00145. [DOI] [PubMed] [Google Scholar]
- 188.Brondsted H, Kopecek J. Hydrogels for site-specific oral drug delivery: synthesis and characterization. Biomaterials. 1991;12:584–592. doi: 10.1016/0142-9612(91)90056-g. [DOI] [PubMed] [Google Scholar]
- 189.Kopecek J, Kopeckova P, Brondsted H, Rathi R, Rihova B, Yeh P.Y, Ikesue K. Polymers for colon-specific drug delivery. J Control Rel. 1992;19:121–130. [Google Scholar]
- 190.Peterson C.M, Lu J.M, Sun Y, Peterson C.A, Shiah J.G, Straight R.C, Kopecek J. Combination chemotherapy and photodynamic therapy with N-(2-hydroxypropyl)methacrylamide copolymer-bound anticancer drugs inhibit human ovarian carcinoma heterotransplanted in nude mice. Cancer Res. 1996;56:3980–3985. [PubMed] [Google Scholar]
- 191.Rihova B, Kopecek J, Ulbrich K, Pospisil M, Mancal P. Effect of the chemical structure of N-(2-hydroxypropyl)methacrylamide copolymers on their ability to induce antibody formation in inbred strains of mice. Biomaterials. 1984;5:143–148. doi: 10.1016/0142-9612(84)90048-6. [DOI] [PubMed] [Google Scholar]
- 192.Rihova B, Kopecek J, Ulbrich K, Chytry V. Immunogenicity of N-(2-hydroxypropyl)methacrylamide. Makromol Chem Suppl. 1985;9:13–24. [Google Scholar]
- 193.Duncan R, Cable H.C, Rejmanova P, Kopecek J, Lloyd J.B. Tyrosinamide residues enhance pinocytic capture of N-(2-hydroxypropyl)methacrylamide copolymers. Biochim Biophys Acta. 1984;799:1–8. doi: 10.1016/0304-4165(84)90320-9. [DOI] [PubMed] [Google Scholar]
- 194.Cartlidge S.A, Duncan R, Lloyd J.B, Rejmanova P, Kopecek J. Soluble, crosslinked N-(2-hydroxypropyl)methacrylamide copolymers as potential drug carriers. 1. Pinocytosis by rat visceral yolk sacs and rat intestine cultured in vitro. Effect of molecular weight on uptake and intracellular degradation. J Control Rel. 1986;3:55–66. [Google Scholar]
- 195.Seymour L.W, Duncan R, Strohalm J, Kopecek J. Effect of molecular weight (Mw) of N-(2-hydroxypropyl)methacrylamide copolymers on body distribution and rate of excretion after subcutaneous, intraperitoneal, and intravenous administration to rats. J Biomed Mater Res. 1987;21:1341–1358. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]
- 196.Wang L, Kristensen J, Ruffner D.E. Delivery of antisense oligonucleotides using HPMA polymer: synthesis of a thiol polymer and its conjugation to water-soluble molecules. Bioconjugate Chem. 1998;9:749–757. doi: 10.1021/bc980034k. [DOI] [PubMed] [Google Scholar]
- 197.Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 198.Tsong-Pin T, Langer R. Polymers for the controlled release of macromolecules: effect of molecular weight of ethylene–vinyl acetate copolymer. J Biomed Mater Res. 1985;19:445–460. doi: 10.1002/jbm.820190409. [DOI] [PubMed] [Google Scholar]
- 199.Edelman E.R, Mathiowitz E, Langer R, Klagsburn M. Controlled and modulated release of basic fibroblast growth factor. Biomaterials. 1991;12:619–626. doi: 10.1016/0142-9612(91)90107-l. [DOI] [PubMed] [Google Scholar]
- 200.Zhao Q, Temsamani J, Agrawal S. Use of cyclodextrin and its derivatives as carriers for oligonucleotide delivery. Antisense Res Dev. 1995;5:185–192. doi: 10.1089/ard.1995.5.185. [DOI] [PubMed] [Google Scholar]
- 201.Abdou S, Collomb J, Sallas F, Marsura A, Finance C. Beta-cyclodextrin derivatives as carriers to enhance the antiviral activity of an antisense oligonucleotide directed toward a coronavirus intergenic consensus sequence. Arch Virol. 1997;142:1585–1602. doi: 10.1007/s007050050182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pitha J. Polymer–cell surface interactions and drug targeting. In: Goldberg E.P, editor. Targeted drugs. Wiley; New York: 1983. pp. 113–126. [Google Scholar]
- 203.Riessen R, Rahimizadeh H, Blessing E, Takeshita S, Barry J.J, Isner J.M. Arterial gene transfer using pure DNA applied directly to a hydrogel-coated angioplasty balloon. Hum Gene Ther. 1993;4:749–758. doi: 10.1089/hum.1993.4.6-749. [DOI] [PubMed] [Google Scholar]
- 204.Azrin M.A, Mitchel J.F, Bow L.M, Pedersen C.A, Cartun R.W, Aretz T.H, Waters D.D, McKay R.G. Local delivery of c-myb antisense oligonucleotides during balloon angioplasty. Cathet Cardiovasc Diagn. 1997;41:232–240. doi: 10.1002/(sici)1097-0304(199707)41:3<232::aid-ccd2>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 205.Frommer J.E, Chance R.R. Electrically conductive polymers. In: Mark H.F, Bikales N.M, Overberger C.G, Menges G, Kroschwitz J.I, editors. vol. 5. Wiley-Interscience; New York: 1986. pp. 462–507. (Encyclopedia of polymer science and technology). [Google Scholar]
- 206.Chen S.A, Hua M.Y, Hwang G.W. Conducting polymers (self-acid-doped, conjugated) In: Salamone J.C, editor. vol. 2. CRC Press; Boca Raton, FL: 1996. pp. 1447–1455. (Polymeric materials encyclopedia). [Google Scholar]
- 207.De Paoli M.A, Zoppi R.A, Felisberti M.I. Conductive elastomeric blends. In: Salamone J.C, editor. vol. 2. CRC Press; Boca Raton, FL: 1996. pp. 1455–1461. (Polymeric materials encyclopedia). [Google Scholar]
- 208.Livache T, Roget A, Dejean E, Barthet C, Bidan G, Teoule R. Preparation of a DNA matrix via an electrochemically directed copolymerization of pyrrole and oligonucleotides bearing a pyrrole group. Nucleic Acids Res. 1994;22:2915–2921. doi: 10.1093/nar/22.15.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Roget A, Livache T, Marchand J, Teoule R. Electrochemically directed copolymerization of pyrrole and oligonucleotides. Nucleosides Nucleotides. 1995;14:943–946. [Google Scholar]
- 210.Piro B, Pham M.C, Ledoan T. Electrochemical method for entrapment of oligonucleotides in polymer-coated electrodes. J Biomed Mater Res. 1999;46:566–572. doi: 10.1002/(sici)1097-4636(19990915)46:4<566::aid-jbm15>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]