

Highlights
-
•
Three bio macromolecules cathepsins B, H and L of physiological and pathological significance have been selected for the study.
-
•
The molecules have been designed by combining two important pharmacophores their cyclized analogues and were studied for their inhibitory effects on selected enzymes.
-
•
Two isomeric forms of chalconesemicarbazones are reported for the first time.
-
•
The synthesized compounds showed a competitive inhibition towards cathepsins B, H and L.
-
•
Docking experiments were run along with to relate with in vitro studies.
Keywords: Inhibitors, Cathepsin B, Cathepsin H, Cathepsin L
Abstract
Cathepsin B [EC 3.4.22.1], cathepsin H [EC 3.4.22.16] and cathepsin L [EC 3.4.22.15] are the most versatile lysosomal cysteine proteases and are responsible for intracellular protein degradation. These are involved in a number of pathological conditions including tissue degenerative processes. In the present work, we report the synthesis and systematic evaluation of differently substituted chalcones, chalconesemicarbazones, and diarylpyrazolines on cathepsins B, H and L activity. It was found that after a preliminary screening as cysteine protease inhibitors, chalconesemicarbazones were better inhibitors to these cysteine proteases than diarylpyrazolines followed by chalcones. All the synthesized compounds were identified as the best inhibitors to cathepsin L followed by cathepsin B and then cathepsin H. The results are compared with docking studies and it was found that all the compounds resulted in decrease in energy while interacting with the active site of the enzyme.
1. Introduction
Semicarbazones are important bioactive compounds possessing different biological activities such as anticonvulsant [1], antimicrobial [2], antiviral [3], antitumoral [4], antihypertensive [5] and antitubercular [6]. These compounds have also been found to be potent inhibitors to cruzain, cysteine protease of Trypanosoma cruzi and been shown to be trypanocidal [7]. The trypanocidal activity is related to cruzain inhibiting tendencies of the semicarbazones. Falcipain, another protozoal cysteine protease has also been reported to be inhibited by variety of semicarbazones. Therefore, semicarbazone inhibitors of cysteine proteases have potential use for prevention and treatment of protozoan infections such as trypanosomiasis, malaria and leishmaniasis. It has also been reported that semicarbazones of aryl and alkyl carbonyl compounds inhibit cysteine proteases of parasites more as compared to mammalian proteases and therefore indicate the possibility of their therapeutic use. The compounds also find use in inhibiting cysteine proteases associated with carcinogenesis, including cathepsins B, H and L [8].
Chalcones, another class of biologically active molecules, known to possess antimalarial [9], anticancer [10], antiprotozoal [11], anti-inflammatory [12], antibacterial [13], antioxidant [14], antifungal [15] activities are also reported to inhibit various enzymes like tyrosinase, alpha-amylase and beta-lectamase [16], [17], [18]. These, α,β-unsaturated compounds have been found to interact with serum albumin [19], [20], [21], [22], [23], [24], the protein responsible for the transportation of various molecules including drugs. In the present work we have synthesized chalcones semicarbazones by combining α,β-unsaturated carbonyl- and semicarbazone-pharmacophores, which resulted in formation of two isomers not reported earlier and have evaluated their effect on three important lysosomal cysteine proteases, cathepsin B [EC 3.4.22.1], cathepsin H [EC 3.4.22.16] and cathepsin L [EC 3.4.22.15]. Cysteine proteases are key factors in the pathogenesis of cancer invasion, arthritis, osteoporosis, and microbial infections [25]. Cathepsin B possesses exo as well as endopeptidase activity [26] and is also capable of peptidyl-dipeptidase [27], [28] and carboxypeptidase activities [29], [30]. The enzyme has been found to be involved in various pathological conditions such as chronic inflammatory diseases of airways and joints, cancer and pancreatitis [31], atherosclerosis [32], rheumatoid arthritis [33] Alzheimer's disease [34], etc. Cathepsin H an endo and amino-peptidase is also involved in many diseased conditions including breast carcinoma [35], melanoma and tumour metastasis [36], head and neck carcinoma [37], malignant prostate cancer [38]. Similarly, cathepsin L activity has also been reported to be involved in diseases such as osteoarthritis [39], [40], tumorigenesis [41], [42], [43], Ebola haemorrhagic fever, severe acute respiratory syndrome and Leishmaniasis [44], [45], [46]. Targeting these enzymes is therefore one of the strategies in the development of new chemotherapeutic agents for a number of diseases including tissue degenerative disorders.
We are working in the identification of small molecular weight compounds as inhibitors to endogenous proteolytic activities [47], [48], [49], [50], [51], [52]. In quest for the identification of some novel inhibitors to cathepsin B and cathepsin H, we have recently reported various non-peptidyl inhibitors such as bischalcones based quinazoline-2(1H)-ones, quinazoline-2(1H)-thiones (iv-vi) [53] and acyl hydrazides, triazoles (i-iii) [54].
Effects of hydrazones [55], hydroxyl chalcones [56] and their cyclized derivatives, formyl and benzoyl pyrazolines [57] have also been established on these enzymes. To explore further in this direction the present work is focused on chalcones, chalconesemicarbazones and their cyclized derivatives as inhibitors to cathepsins B, H and L. The intention to synthesize semicarbazones of chalcones was undertaken keeping in view that the semicarbazones possessing azomethine group in conjunction with α,β-unsaturation will have extended pharmacological activities. The compounds were further cyclized to pyrazolines which are also reported to possess diverse biological activities such as antimicrobial [58], antiamoebic [59], anti-inflammatory [60], anticancer [61], antidepressant [62] and antitubercular [63] activities. Effect of synthesized compounds was observed on cathepsins B, H and L to establish a structure activity relationship between the 1,3-diphenylprop-2-en-1-ones, substituted chalconesemicarbazones and substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide derivatives. Structure activity relationship (SAR) studies of open chain and cyclized derivatives can result in identification of potential inhibitors among related class of compounds [55], [56]. A comparative account of different moieties introduced in the synthesized compounds on cathepsin B, H and L, three lysosomal cysteine proteases inhibiting tendency has also been evaluated. The studies can lead to the development of new inhibitors of these enzymes.
2. Experimental
2.1. Materials
All the chemicals were of analytical grade. Fast Garnet GBC (o-aminoazotoluene diazonium salt), α-N-benzoyl-d,l-arginine-2-naphthylamide (BANA), Leu-βNA and ZPheArg-4mβNA were purchased from Bachern Feinchemikalien AG, Switzerland. The protein sample was concentrated using Amicon stirred cells with YM 10 membrane under nitrogen pressure of 4–5 psi. The source of enzyme was fresh goat liver obtained from local slaughter house.
2.2. Methods
Melting points were taken in open capillaries and are uncorrected. The progress of the reactions was monitored on silica gel G plates using iodine vapor as visualizing agent. Elisa plate reader was used for measuring absorbance in the visible range. The spectrofuge was used for centrifugation purpose. IR spectra were recorded on Horizon 300 MHz spectrometer. NMR spectra were recorded on Bruker 300 MHz instrument. The chemical shifts are expressed in ppm units from an internal TMS standard. All commercially available reagents were used as-received.
2.3. Synthesis
The title compounds were synthesized according to Scheme 1 .
Scheme 1.
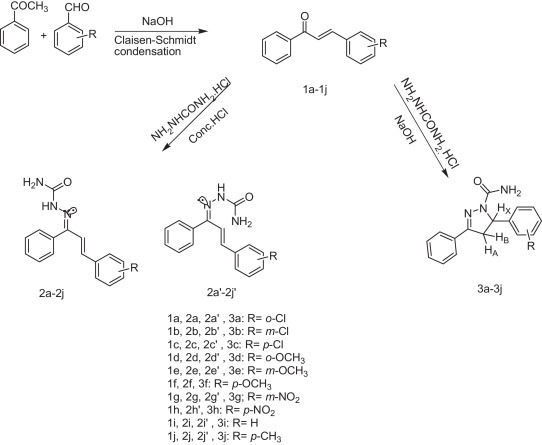
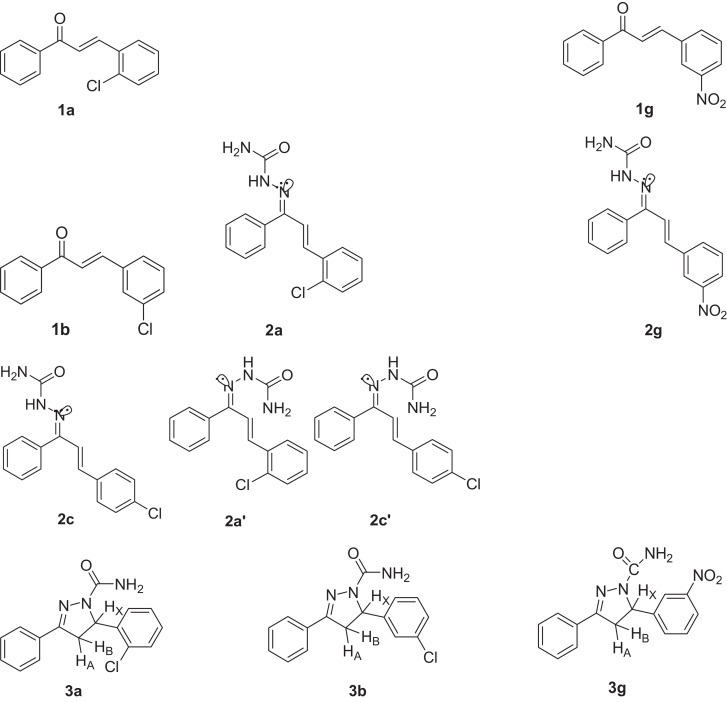
Synthesis of substituted chalcones (1a–1j), chalconesemicarbazones (2a–2j, 2a′–2j′) and pyrazolines (3a–3j).
2.3.1. Synthesis of substituted 1,3-diphenylprop-2-en-1-ones (1a–1j)
Acetophenone (0.01 mol) was added to equimolar quantity of substituted benzaldehyde (0.01 mol) dissolved in methanol (25 ml). To this solution equimolar NaOH pellets (0.01 mol) were added at once and the reaction mixture was stirred for 40 min at room temperature. Excess of methanol was again added and then again stirred for next 40 min at room temperature. It was then diluted with cold water. The product was separated, filtered and washed with water until neutral. The resulting chalcones were purified by recrystallization with methanol. The structures were characterized using melting point compared with literature melting point [19], [64], IR and NMR.
2.3.2. Synthesis of substituted chalconesemicarbazones (2a–2j, 2a′–2j′)
To an aqueous semicarbazide hydrochloride (0.02 mol), sodium acetate (0.03 mol) was dissolved. This solution was added to the ethanolic solution of substituted 1,3-diphenyl-2-en-1-ones (0.01 mol) (1a–1j). Then, few drops of concentrated hydrochloric acid were added. Reaction mixture was stirred at 40 °C and was monitored on TLC. After completion, the reaction mixture was poured into ice and precipitate, so obtained was filtered, washed with water, dried and recrystallized with ethanol. The product thus obtained was found to possess two stereoisomeric forms (Fig. 1 ) which were separated on column. The two isomers have been characterized using 1H NMR.
Fig. 1.
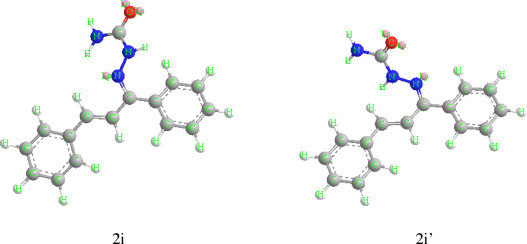
3-D structures (http://media.cambridgesoft.com/cbou130/cbou1302.exe) of two isomers 2i and 2i′ from chalconesemicarbazones showing shielding and desheilding of C-H proton with lone pair of nitrogen, respectively.
2.3.3. Synthesis of substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide derivatives (3a–3j)
To an aqueous sodium hydroxide (0.017 mol), semicarbazide hydrochloride (0.0085 mol) was dissolved. This solution was added to the solution of substituted 1,3-diphenyl-2-en-1-ones (0.005 mol) (1a–1j) in ethanol (25 ml) and then, refluxed for 6–8 h. TLC monitoring was extensively done. The solvent was evaporated and the solid so obtained was filtered, washed with water to neutral reaction, dried and recrystallized from ethanol. The structure was characterized using melting point compared with literature melting point [65], IR and NMR spectroscopic techniques.
2.4. Pharmacology
2.4.1. Purification of cathepsins B, H and L
All the purification steps were carried out at 4 °C. Cathepsins B, H and L were extracted and purified from goat liver by the established procedure reported previously [53] including the steps of acetone powder preparation, homogenization, acid autolysis, 30–70% (NH4)2SO4 fractionation, molecular sieve chromatography on sephadex G-100 and ion exchange chromatographies on CM Sephadex C-50 and DEAE A-50 sephadex. The specific activities of the cathepsin B, H and L were equal to ∼11.28 nmol/min/mg, ∼24.01 nmol/min/mg and ∼16.78 nmol/min/mg, respectively.
2.4.2. Autoproteolytic studies
The experiments were conducted in triplicates as described previously for endogenous proteolytic studies [47], [48], [49], [50], [51], [52].
2.4.3. Enzyme inhibition studies
Cathepsin B and L activity was determined using BANA [66] and ZPheArg-4mβNA [67] substrate at pH 6.0, respectively whereas cathepsin H activity was determined using Leu-βNA [68] substrate at pH 7.0. Effect of synthesized compounds was observed on the activities of cathepsins B, H and L at 1 × 10−5 M, 1 × 10−4 M and 1 × 10−8 M final concentration of each compound, respectively. First of all, enzyme was equilibrated in buffer of appropriate pH at 37 °C. Then 20 μl of individual compound was added in the reaction mixture separately to effect the final concentration of each compound as 0.1 mM concentration. After an incubation time of 30 min residual enzyme activity was estimated by the usual enzyme assay using the respective substrates. The experiments were performed in triplicate for each concentration and % activity has been calculated with respect to the control where no compound was added but an equivalent amount of solvent was present. Enzyme assays were similarly conducted at lower concentrations of each compound to observe the inhibitory effect of compounds at varying concentrations. The results are presented in Fig. 2, Fig. 3, Fig. 4 for cathepsins B, H and L, respectively.
Fig. 2.
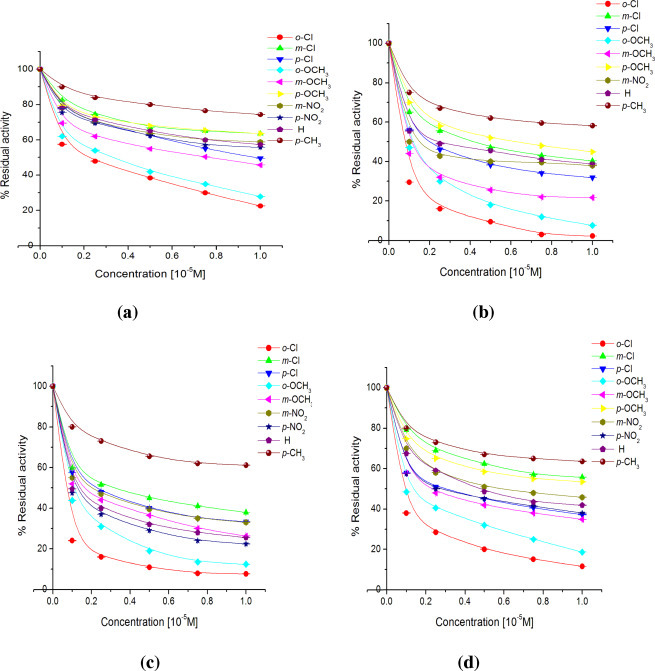
The results are depicted as % residual activities of cathepsin B in presence of different concentrations (0.1, 0.25, 0.50, 0.75 and 1.0 × 10−5 M) of various chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and diarylpyrazolines (3a–3j; d) after an interaction time of 30 min. The experiments were conducted in triplicate and were calculated w.r.t. the control having no compound but an equivalent amount of solvent was added.
Fig. 3.
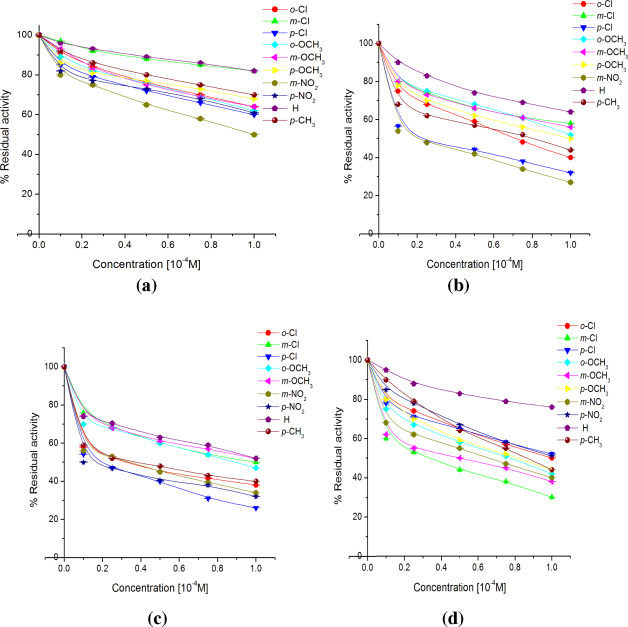
The results are depicted as % residual activities of cathepsin H in presence of different concentrations (0.1, 0.25, 0.50, 0.75 and 1.0 × 10−4 M) of various chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and diarylpyrazolines (3a–3j; d) after an interaction time of 30 min. The experiments were conducted in triplicate and were calculated w.r.t. the control having no compound but an equivalent amount of solvent was added.
Fig. 4.
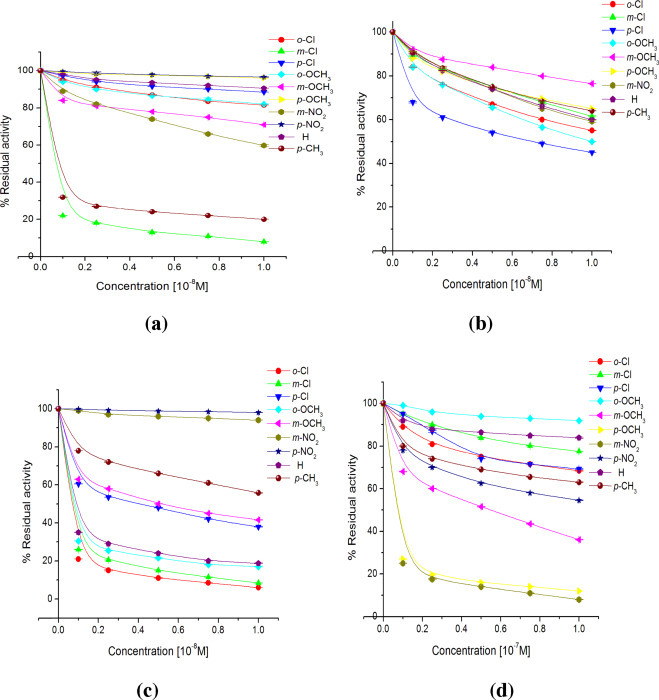
The results are depicted as % residual activities of cathepsin L in presence of different concentrations (0.1, 0.25, 0.50, 0.75 and 1.0 × 10−8 M) of various chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and (0.1, 0.25, 0.50, 0.75 and 1.0 × 10−7 M) of various diarylpyrazolines (3a–3j; d) after an interaction time of 30 min. The experiments were conducted in triplicate and were calculated w.r.t. the control having no compound but an equivalent amount of solvent was added.
2.4.4. Enzyme kinetic studies
After establishing the inhibitory action of 1,3-diphenylprop-2-en-1-ones (1a–1j), substituted chalconesemicarbazones (2a–2j, 2a′–2j′) and substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide derivatives (3a–3j) on cathepsins B, H and L experiments were designed to evaluate the type of inhibition and to determine the K i value of these compounds on cathepsin B, H and L. For that, enzyme activity was evaluated at different substrate concentration in presence and absence of a fixed concentration of inhibitor. Line-weaver Burk plots were drawn between 1/[S] and 1/V (Fig. 5, Fig. 6, Fig. 7 ). The K m value of cathepsin B, H and L for BANA, Leu βNA and ZPheArg-4mβNA was found to be 4.3 × 10−4 M, 5.0 × 10−4 M and 0.5 × 10−4 M, respectively. The K i values have been summarized in Table 1 .
Fig. 5.
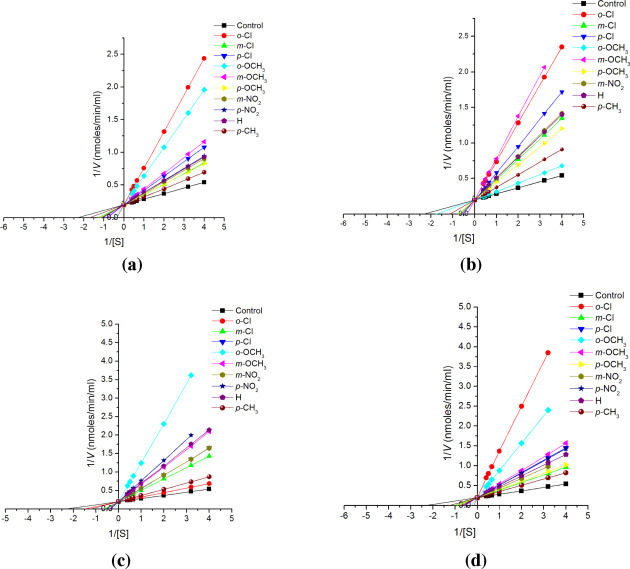
Lineweaver–Burk plots for inhibition of substituted chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and diarylpyrazolines (3a–3j; d) on cathepsin B at fixed concentration (10−5 M) of inhibitor and varying substrate i.e. BANA concentration (2.5, 2.0, 1.5, 1.0, 0.5, 0.3, 0.25 mM).
Fig. 6.
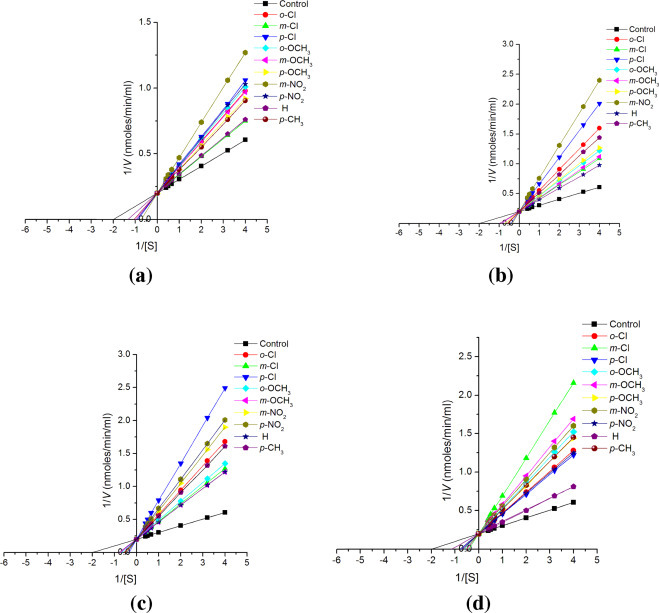
Lineweaver–Burk plots for inhibition of substituted chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and diarylpyrazolines (3a–3j; d) on cathepsin H at fixed concentration (10−4 M) of inhibitor and varying substrate i.e. Leu βNA concentration (2.5, 2.0, 1.5, 1.0, 0.5, 0.3, 0.25 mM).
Fig. 7.
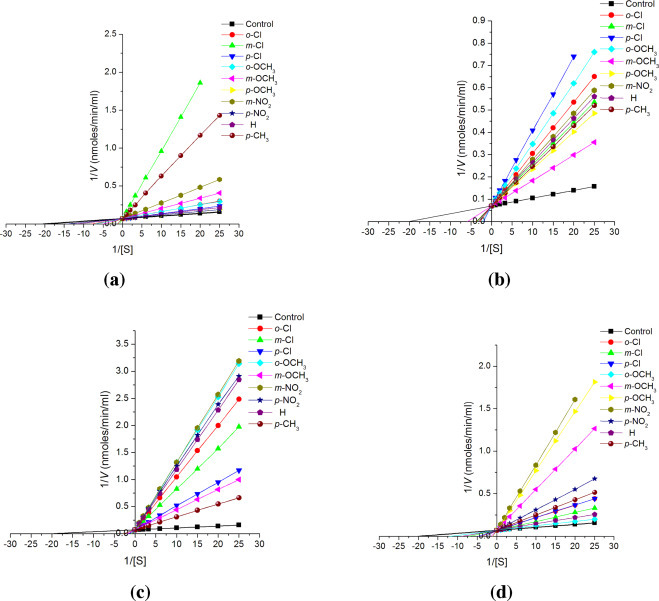
Lineweaver–Burk plots for inhibition of substituted chalcones (1a–1j; a), chalconesemicarbazone isomers (2a–2j; b), (2a′–2j′; c) and diarylpyrazolines (3a–3j; d) on cathepsin L at fixed concentration (10−8 M of chalcones (1a–1j), chalconesemicarbazone (2a–2j, 2a′–2j′) and 10−7 M of diarylpyrazolines (3a–3j)) of inhibitor and varying substrate i.e. ZPheArg-4mβNA concentration (1.0, 0.5, 0.3, 0.16, 0.1, 0.06, 0.05, 0.04 mM).
Table 1.
Ki values of various chalcones (1a–1j), chalconesemicarbazones (2a–2ja, 2a′–2j′) and diarylpyrazolines (3a–3j) for cathepsins B, H and L.
| Compounds | Cathepsin B | Cathepsin H | Cathepsin L |
|---|---|---|---|
|
Ki ± S.M.D [1 × 10−5 M] |
Ki ± S.M.D [1 × 10−5 M] |
Ki ± S.M.D [1 × 10−9 M] |
|
| 1a | 1.95 ± 0.02 | 11.0 ± 0.1 | 6.00 ± 0.02 |
| 1b | 13.0 ± 0.2 | 31.2 ± 0.2 | 0.04 ± 0.0001 |
| 1c | 6.61 ± 0.03 | 8.30 ± 0.03 | 12.50 ± 0.08 |
| 1d | 2.40 ± 0.06 | 10.00 ± 0.02 | 6.20 ± 0.02 |
| 1e | 6.32 ± 0.03 | 11.00 ± 0.007 | 3.50 ± 0.08 |
| 1f | 13.0 ± 0.1 | 13.80 ± 0.01 | 26.30 ± 0.07 |
| 1g | 10.0 ± 0.1 | 6.00 ± 0.02 | 2.05 ± 0.03 |
| 1h | 8.77 ± 0.04 | 10.00 ± 0.005 | 27.7 ± 0.3 |
| 1i | 9.14 ± 0.03 | 26.3 ± 0.2 | 15.15 ± 0.1 |
| 1j | 22.67 ± 0.3 | 15.10 ± 0.03 | 0.06 ± 0.0002 |
| Compounds | Cathepsin B | Cathepsin H | Cathepsin L |
|---|---|---|---|
|
Ki ± S.M.D [1 × 10−6 M] |
Ki ± S.M.D [1 × 10−5 M] |
Ki ± S.M.D [1 × 10−9 M] |
|
| 2a | 0.207 ± 0.001 | 4.30 ± 0.02 | 1.78 ± 0.02 |
| 2b | 43.0 ± 0.4 | 9.60 ± 0.06 | 2.42 ± 0.01 |
| 2c | 27.3 ± 0.4 | 2.90 ± 0.05 | 1.17 ± 0.02 |
| 2d | 2.70 ± 0.02 | 6.60 ± 0.07 | 1.56 ± 0.02 |
| 2e | 17.0 ± 0.2 | 7.40 ± 0.06 | 4.71 ± 0.02 |
| 2f | 52.4 ± 0.8 | 5.40 ± 0.03 | 2.66 ± 0.03 |
| 2g | 41.3 ± 0.5 | 2.30 ± 0.03 | 2.05 ± 0.02 |
| 2i | 42.5 ± 0.2 | 10.00 ± 0.04 | 2.27 ± 0.04 |
| 2j | 91.4 ± 0.9 | 4.50 ± 0.05 | 2.50 ± 0.02 |
| Compounds | Cathepsin B | Cathepsin H | Cathepsin L |
|---|---|---|---|
|
Ki ± S.M.D [1 × 10−6 M] |
Ki ± S.M.D [1 × 10−5 M] |
Ki ± S.M.D [1 × 10−10 M] |
|
| 2a′ | 2.70 ± 0.04 | 3.30 ± 0.04 | 0.4 ± 0.003 |
| 2b′ | 39.0 ± 0.5 | 6.00 ± 0.01 | 0.5 ± 0.005 |
| 2c′ | 36.4 ± 0.5 | 2.10 ± 0.02 | 9.3 ± 0.07 |
| 2d′ | 9.40 ± 0.05 | 5.40 ± 0.03 | 3.1 ± 0.05 |
| 2e′ | 24.0 ± 0.2 | 6.60 ± 0.03 | 11.0 ± 0.13 |
| 2g′ | 34.9 ± 0.2 | 3.30 ± 0.01 | 149.3 ± 2.0 |
| 2h′ | 19.50 ± 0.02 | 3.20 ± 0.02 | 168.0 ± 0.6 |
| 2i′ | 22.00 ± 0.04 | 6.60 ± 0.02 | 3.6 ± 0.04 |
| 2j′ | 104.0 ± 1.0 | 4.00 ± 0.04 | 16.9 ± 0.2 |
| Compounds | Cathepsin B | Cathepsin H | Cathepsin L |
|---|---|---|---|
|
Ki ± S.M.D [1 × 10−6 M] |
Ki ± S.M.D [1 × 10−5 M] |
Ki ± S.M.D [1 × 10−8 M] |
|
| 3a | 9.40 ± 0.03 | 6.00 ± 0.03 | 3.16 ± 0.04 |
| 3b | 82.7 ± 1.0 | 2.50 ± 0.05 | 5.05 ± 0.03 |
| 3c | 39.0 ± 0.2 | 6.60 ± 0.04 | 3.30 ± 0.01 |
| 3d | 14.80 ± 0.07 | 4.00 ± 0.01 | 21.74 ± 0.40 |
| 3e | 34.9 ± 0.1 | 4.30 ± 0.01 | 0.82 ± 0.006 |
| 3f | 69.30 ± 0.07 | 4.80 ± 0.02 | 0.05 ± 0.0002 |
| 3g | 58.10 ± 0.05 | 3.80 ± 0.02 | 0.04 ± 0.0005 |
| 3h | 39.00 ± 0.03 | 6.00 ± 0.03 | 1.69 ± 0.03 |
| 3i | 52.40 ± 0.01 | 19.20 ± 0.04 | 8.62 ± 0.02 |
| 3j | 116.2 ± 2.0 | 4.80 ± 0.05 | 2.34 ± 0.02 |
The experiments were conducted in triplicate using different concentrations of bana, leu βna and zphearg-4mβna as substrate for cathepsins b, h and l, respectively. The results are calculated using Lineweaver–Burk equation for competitive inhibitors.
2.4.5. Drug modelling studies
Docking studies were performed using iGemdock software. To conduct these, small molecular weight ligands were prepared using marvin sketch and were saved as MDL Mol File. Enzyme structure active site was retrieved from the Protein Data Bank (http://www.rcsb.org/) as cav2IPP B_PYS.pdb, cav8PCH H_NAG.pdb and cav3BC3L_CSW [69], [70], [71]. The prepared ligands and the binding site was loaded in the iGemdock programme and docking was run by setting GA parameters for Standard Docking Accuracy Settings, Docking experiments show a decrease in energy when enzyme and ligands interact. The E total resulting after H-bonding and van der Waals interactions are presented in Tables S1–S3 (Supplementary data). The docking poses of the most inhibitory compounds 1a, 2a, 2a′, 3a for cathepsin B, 1g, 2g, 2c′, 3b for cathepsin H and 1b, 2c, 2a′, 3g for cathepsin L are shown in Fig. 8, Fig. 9, Fig. 10 .
Fig. 8.
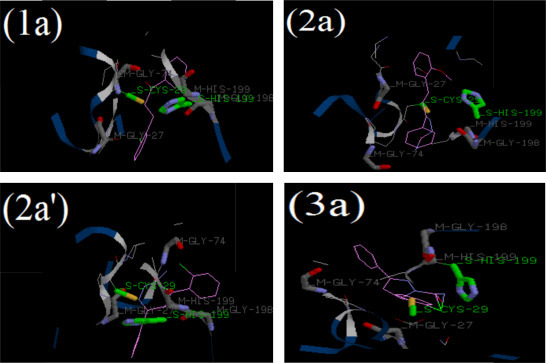
Binding of most inhibitory chalcones 1a, chalconesemicarbazone isomer 2a, chalconesemicarbazone isomer 2a′ and pyrazoline 3a into the binding site of cathepsin B (cav2IPP B_PYS.pdb).
Fig. 9.
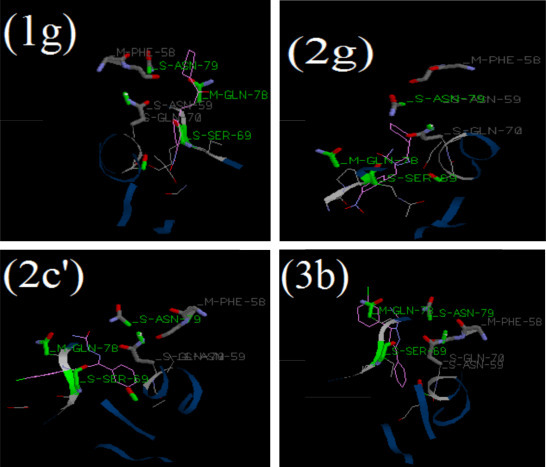
Binding of most inhibitory chalcones 1g, chalconesemicarbazone isomer 2g, chalconesemicarbazone isomer 2c′ and pyrazoline 3b into the binding site of cathepsin H (cav 8PCH H_NAG).
Fig. 10.
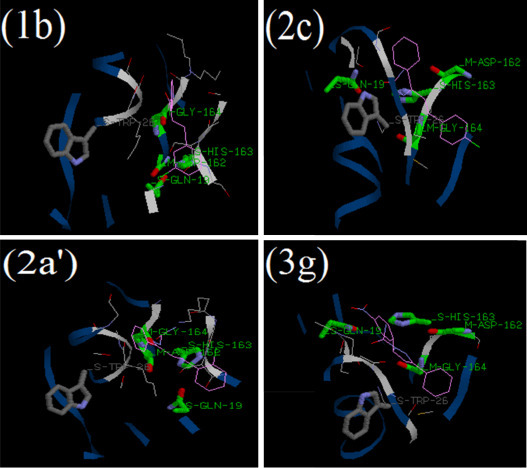
Binding of most inhibitory chalcones 1b, chalconesemicarbazone isomer 2c, chalconesemicarbazone isomer 2a′ and pyrazoline 3g into the binding site of cathepsin L (cav3BC3L_CSW).
3. Result and discussion
The synthesis of chalcones (1a–1j) from substituted benzaldehyde and acetophenone was carried out using Claisen-Schmidt condensation. The substituted Chalconesemicarbazones (2a–2j, 2a′–2j′) and substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide (3a–3j) were synthesized [65] from these chalcones using semicarbazide hydrochloride (Scheme 1). After establishing the structure of chalcones their corresponding semicarbazones were synthesized and the structures were elucidated using melting point, IR and NMR spectra.
The synthesis of chalconesemicarbazones resulted in two isomeric form 2a–2j and 2a′–2j′. These two isomeric forms were separated by column chromatography on silica gel using petroleum ether and ethyl acetate as eluent. The separated isomers were characterized by 1H NMR data. The isomers obtained were characterized on the basis of interaction of lone pair on nitrogen with C—H proton as explained by Curphy-Morrison chemical shift [72]. When lone pairs on nitrogen are syn to C—H bonds (2a–2j), the proton is shifted upfield showing the lower δ value. When lone pairs on nitrogen are anti to C—H bonds (2a′–2j′), the proton is shifted downfield. The conclusion was arrived on the basis of 3D molecular modelling studies. In 2i where lone pair on nitrogen is syn to C2-H (Scheme 1) on minimization of energy in 3D molecular modelling (Fig. 1) [73] clearly indicate that lone pair on nitrogen is in optimal position to affect shielding of C2-H and also to C3-H to some extent because of extension of conjugation. In 2i′ the lone pair on nitrogen is anti to C2-H and after 3D molecular modelling in minimized energy structure shown in Figure 1, it is clear that these can’t have any effect on CH CH protons. On comparing the 1H NMR, peaks for C2-H and C3-H for chalcones and chalconesemicarbazones, distinctive shielding effect is observed due to substitution of adjacent C O group with C N—NHCONH2. These isomeric forms are reported here for the first time, although a large number of reports are available in literature on the synthesis of these compounds [74], [75], [76]. In case of p-OCH3 and p-NO2 substituted isomers only one isomer was formed i.e. 2f and 2h, respectively.
On cyclization of chalconesemicarbazones, diarylpyrazolines are obtained and each of the two isomeric forms (2a–2j) and (2a′–2j′) resulted in single product (3a–3j). In contrast, two isomeric forms of pyrazolines have been reported by Shekarchi et al. [77]. In the 1H NMR spectra of (3a–3j), the characteristic ABX pattern has been obtained.
3.1. Effect of synthesized compounds on in vitro endogenous proteolysis in liver homogenate
Proteolytic activity is inhibited appreciably in presence of these compounds. In some cases ∼100% inhibition is achieved at 1 × 10−4 M concentration. Moreover the inhibition was more at 3.0 h and less at 24.0 h, emphasizing that the compounds inhibit the proteolytic activity reversibly. In case of chalcones, chloro and unsubstituted compounds showed 100% inhibition. For chalconesemicarbazones and diaryl pyrazolines, chloro and nitro substituted compounds showed 100% inhibition. In addition, o- and p-methoxy substituted chalconesemicarbazone and o-methoxy pyrazoline also exhibited 100% inhibition while unsubstituted and methyl substituted chalconesemicarbazones and pyrazolines exerted inhibition to lesser extent.
After establishing the inhibition of endogenous proteolytic activity in presence of chalcones and their derivatives at pH 5.0, where most of the proteolytic activity is attributed to cysteine proteases it was thought proper to study the effect of synthesized compounds on purified cathepsins B, H and L.
3.2. Effect of synthesized compounds on the activity of cathepsins B, H and L
The effect of differently substituted chalcones (1a–1j), chalconesemicarbazones (2a–2j, 2a′–2j′) and diarylpyrazolines (3a–3j) on the activity of cathepsins B, H and L at varying concentrations is shown in Fig. 2, Fig. 3, Fig. 4. From these plots of % residual activities versus the concentrations of different compounds, it can be observed that at a particular concentration all the synthesized compounds inhibited cathepsin L activity more than cathepsin B and H.
3.3. The inhibition type and Ki values
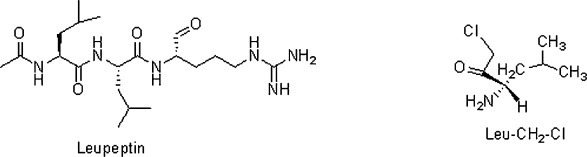
The type of inhibition caused by various compounds was determined through Lineweaver–Burk double reciprocal plot. In order to establish inhibition ability of the under consideration compounds results were compared with potent inhibitors of cathepsin B, e.g. Leupeptin and cathepsin H e.g. Leu-CH2-Cl, respectively. As reported in literature, Leupeptin being a potent peptide inhibitor of cathepsin B, inhibited the goat brain cathepsin B competitively with K i value of 12.5 × l0−9 M [66] whereas K i value for human liver cathepsin B [78] was reported to be 7.0 × 10−9 M. In contrast, K i value for human liver cathepsin H was reported to be 9.2 × 10−6 M [79] and K i value for goat brain cathepsin L was reported to be 1.45 × 10−9 M [67].
For evaluating the type of inhibition caused by different chalcones, chalconesemicarbazones and diarylpyrazolines, cathepsins B, H and L activity was measured at varying substrate i.e. BANA, Leu βNA and ZPheArg-4mβNA concentration in presence and absence of a fixed concentration of compound. The plots of 1/V and 1/[S] were straight lines intersecting at the Y-axis and shows that value of V max remains constant in all the compounds whereas the value of K m′ change with each compound. These studies suggested that chalcones, chalconesemicarbazones and diarylpyrazolines are competitive inhibitors to cathepsins B, H and L. Using the Lineweaver–Burk equation of competitive inhibition the K i values were calculated, which has been presented in Table 1.
Lineweaver–Burk plots of different chalcones (1a–1j), chalconesemicarbazones (2a–2j, 2a′–2j′) and diarylpyrazolines (3a–3j) for cathepsins B, H and L are shown in Fig. 5, Fig. 6, Fig. 7.
3.4. Structure activity relationship
Proteases contribute to tumour cell invasion and angiogenesis and are commonly associated with metastasis. It is now recognized that cysteine proteases play pivotal roles in cancer progression. Of all the cathepsins, studies have shown that cathepsins B, H and L are of significant importance as these are involved in various pathologies and oncogenic processes. Though these enzymes have been intensively studied as valuable targets for drug discovery and development, a number of peptide [80] and non-peptide [53], [54] inhibitors have been described in literature. Towards this endevour we have now synthesized chalcones and their semicarbazone and pyrazoline derivatives with different functionalities in order to explore their inhibitory potential on these important enzymes keeping in view the inhibitory potential of semicarbazones and pyrazolines as well as chalcones on cysteine proteases. The work has been envisaged by combining the two active pharmacophores.
Out of various synthesized compounds, (E)-3-(2-chlorophenyl)-1-phenylprop-2-en-1-one (1a), (1Z)-1-((E)-3-(2-chlorophenyl)-1-phenylallylidene)semicarbazide (2a), (1E)-1-((E)-3-(2-chlorophenyl)-1-phenylallylidene)semicarbazide (2a′) and 5-(2′-chlorophenyl)-3-phenyl-2-pyrazoline-1-carboxamide (3a) with K i values (1.95 ± 0.02) × 10−5 M, (0.207 ± 0.001) × 10−6 M, (2.70 ± 0.04) × 10−6 M and (9.40 ± 0.03) × 10−6 M, respectively showed maximum inhibition on the activity of cathepsin B. However, activity of cathepsin H was maximally inhibited by (E)-3-(3-nitrophenyl)-1-phenylprop-2-en-1-one (1g), (1Z)-1-((E)-3-(3-nitrophenyl)-1-phenylallylidene)semicarbazide (2g), (1E)-1-((E)-3-(4-chlorophenyl)-1-phenylallylidene)semicarbazide (2c′) and 5-(3′-chlorophenyl)-3-phenyl-2-pyrazoline-1-carboxamide (3b) with K i values (6.00 ± 0.02) × 10−5 M, (2.30 ± 0.03) × 10−5 M, (2.10 ± 0.02) × 10−5 M and (2.50 ± 0.05) × 10−5 M, respectively and were found to be the best inhibitors for cathepsin H. Similarly, (E)-3-(3-chlorophenyl)-1-phenylprop-2-en-1-one (1b), (1Z)-1-((E)-3-(4-chlorophenyl)-1-phenylallylidene)semicarbazide (2c), (1E)-1-((E)-3-(2-chlorophenyl)-1-phenylallylidene)semicarbazide (2a′) and 5-(3′-nitrophenyl)-3-phenyl-2-pyrazoline-1-carboxamide (3g) with K i values (0.04 ± 0.0001) × 10−9 M, (1.17 ± 0.02) × 10−9 M, (0.4 ± 0.003) × 10−10 M and (0.04 ± 0.0005) × 10−8 M, respectively showed maximum inhibition on the activity of cathepsin L. Followed by these results it was concluded that the synthesized compounds showed more inhibition on activity of cathepsin L than on cathepsins B and H. This may lead to the development of selective inhibitors to cathepsins B, H and L.
3.5. Small molecular weight representative molecules
Further it can be observed that chalconesemicarbazones showed maximum inhibition than their cyclized precursor pyrazolines followed by chalcones on the activity of cathepsin B and cathepsin H. Whereas, for cathespin L chalcones and chalconesemicarbazones were found to show more inhibition than pyrazolines. The results are discussed later in the next section along with the results obtained after molecular docking.
Among the substituted chalcones, chalconesemicarbazones and pyrazolines, the compounds bearing electron withdrawing moiety were found to be more inhibitory probably inducing more nucleophilic character at the site of attack shown in Scheme 2 .
Scheme 2.
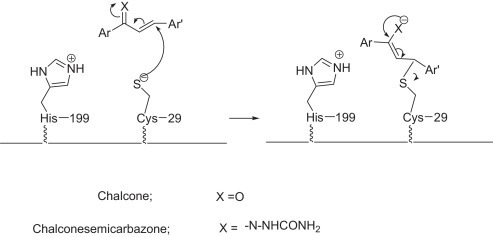
Proposed mechanism for inhibition of cathepsin B with chalcone and chalconesemicarbazone analogues.
The results obtained were compared with potential inhibitors of cathepsin B and L e.g. Leupeptin and cathepsin H, e.g. Leu-CH2-Cl, respectively [54]. It can be observed that Leupeptin showed ∼98.8% inhibition at 10−6 M concentration for cathepsin B whereas it showed ∼52.0% inhibition at 10−5 M concentration for cathepsin H and 89.2% inhibition at 10−9 M concentration for cathepsin L which is in accordance with the previously reported results. Similarly, Leu-CH2-Cl showed ∼9.5% inhibition at 10−5 M concentration for cathepsin B whereas it showed ∼93.5% inhibition at 10−5 M concentration for cathepsin H and 1.5% inhibition at 10−5 M concentration for cathepsin L. The results obtained are comparable with earlier results reported for brain cathepsin H [68], cathepsin B and cathepsin L [81].
3.6. Molecular docking experiment
The docking approach was used to study the interaction of compounds with the active site of cathepsin B, H and L to observe binding poses of individual compounds. Individual binding poses of each compound was assessed and their interactions in the active site of the enzyme were analyzed. The empirical scoring function of iGemDOCK is the estimated sum total of van der Waals, H-bonding and electrostatic energy.
Table S1 (Supplementary data) presents the data of docking studies of different chalcones, their acyclic and cyclic semicarbazone derivatives in cathepsin B active site (cav2IPP B_PYS.pdb). The results clearly indicate that binding energy of chalcones is less than pyrazoline derivatives which is less than the open chain semicarbazones. The binding energy of chalcones (1a–1j), chalconesemicarbazones (2a–2j, 2a′–2j′) and diarylpyrazolines (3a–3j) vary in the range −71.00 to −80.46, −83.11 to −99.13, −88.75 to −95.97 and −82.51 to −91.78, respectively. The most inhibitory compound in each series was 1a, 2a, 2a′ and 3a having E total as −71.76, −95.42, −90.54 and −84.27, respectively. These binding energies when compared to the binding energies of leupeptin with cathepsin B are lesser to approximately 20 units. The docking results explain that the title compounds may not be potent inhibitors to the protease activity to an extent of specific pepidyl inhibitors but certainly suggest a differential inhibitory pattern of these compounds on cathepsin B activity. However, maximum interaction is observed for BANA with a score of −129.83. Decrease in total energy for leupeptin-cathepsin B has come out be −116.42 of which the contribution of van der Waals interactions are more with a score of −86.37 as compared to H-bonds with a score of −29.44. Leupeptin-cathepsin B binding energy is due to peptide–protein interaction. Leupeptin is peptidyl in nature and therefore being a flexible molecule binds effectively with the enzyme active site resulting in higher binding energy. iGemDOCK provide algorithms for flexible docking approach for both ligands and proteins [82] therefore flexible ligands like leupeptin will show a larger decrease in total energy as compared to the molecules under study as these are smaller in structure and possess lesser flexibility compared to leupeptin. Therefore, the binding energy of title compounds is less than leupeptin. In the quest of synthesizing potent inhibitors of enzymes a structure-activity relationship is required where cyclic and acyclic analogues are to be studied. There are various pharmaco-dynamic reactions which molecule acting as drug encounters in vivo such as cyclization, oxidation, reduction etc. so we have here, provided the comparative account of in vitro studies and in-silico study on the structurally related compounds or their isomers. These compounds are synthesized from the same starting material. The open chain analogues are the intermediates therefore there are a need to screen their inhibitory potential along with the products i.e. the cyclized derivatives, pyrazolines in order to establish structure activity relationship. In this direction the present study has been undertaken to evaluate the effect of 1, 3-diphenylprop-2-en-1-ones (1a–1j), substituted chalconesemicarbazones (2a–2j, 2a′–2j′) which are open chain compounds and their cyclic analogues i.e. substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide derivatives (3a–3j) on the activity of cathepsin B, a protease of immense importance. During the synthetic studies of semicarbazones we came across the presence of two stereoisomers in the preparation. Docking experiments were conducted on these two isomers also and a differential effect was observed (Table S1-Supplementary data). The results of the docking studies support the in vitro experimental studies conducted on liver cathepsin B. It has been observed that chalcones were less inhibitory to the enzyme activity whereas semicarbazones and pyrazoline derivatives. This was quite surprising as chalcones having an α-β unsaturated carbonyl group seems to be more responsive towards thiol attack of cysteine present at the active site of enzyme but both the results after, in-silico docking experiments as well as in vitro solution studies clearly indicate that presence of azomethine carboxamide might have provided larger interaction to the binding of compound with the enzyme active site and there by exerting more inhibition than its precursor chalcones, which is further confirmed by the enzyme–ligand interaction study.
Fig. 8 shows the binding of most inhibitory compound 1a, 2a, 2a′ and 3a in the active site of cathepsin B. The ligands are shown in pink, residues involved in H-bonding and van-der Waals interactions are shown as green and grey, respectively. It is clearly observed that cys-29 and His-199 residues present at the catalytic site of the enzyme are involved in the binding of compounds. Based on these interactions, a mechanism of inhibition (Scheme 2) by chalcones and chalconesemicarbazones is proposed on the basis of a previously reported inhibition of cathepsin L, another important cysteine protease by thiosemicarbazones [83]. The thiolate of cys-29 attacks on the nucleophilic site in chalcones and chalconesemicarbazones, where the later is more stabilized to the binding site. In addition, Gly-198, Ala-200 and Trp-30 amino acids residues are also involved in the stabilization of compounds in binding site. Pyrazolines are also better inhibitors to cathepsin B activity than chalcones again emphasizes on the importance of binding of compound with the active site. Though pyrazolines lack any nucleophilic site in their structure but still they show better inhibitory potential probably due to the presence of side chain carboxamide and nitrogen in the ring altogether providing a larger binding area to the enzyme site. Among themselves the cyclized pyrazolines were less inhibitory than open chain semicarbazones. It can be interpreted that acyclic compounds interact more with the active site being straight in nature not to the extent of peptidyl inhibitors but certainly greater than the cyclized pyrazoline derivatives. This gives an understanding of the inhibition caused by the target compounds on structural basis.
Similar trends have been observed for cathepsin H. However, in cathepsin H, the most inhibitory compound in each series have been found to be 1g, 2g, 2c′ and 3b. Fig. 9 shows the binding of the most inhibitory compounds in the active site of cathepsin H. The binding energies of title compounds in the amino acyl binding site of cathepsin H (cav8PCHH_NAG) is presented in Table S2 (Supplementary data). Experimental results obtained can be correlated with the ligand-binding interactions. It is observed that for 1g, 2g, 2c′ and 3b, the binding energies computed come out to be −82.21, −96.59, −93.63 and −84.78. In each series, these most inhibitory compounds show a decrease in binding energy towards higher side. The binding energies show effective interaction between the enzyme binding site and inhibitory compounds may be responsible for these inhibition patterns.
Fig. 10 shows the binding of most inhibitory compounds 1b, 2c, 2a′ and 3g in the active site of cathepsin L. The results of the docking studies support the in vitro experimental studies conducted on goat liver cathepsin L. The binding energies of title compounds in the amino acyl binding site of cathepsin L (cav3BC3L_CSW) is presented in Table S3 (Supplementary data). The binding energies of 1b, 2c, 2a′ and 3g were found to be −77.07, −92.93, −104.04 and −114.94, respectively.
4. Conclusion
One of the important aspect of the present work comprise of synthesis and isolation of two stereoisomers of chalconesemicarbazones not reported earlier. Synthesis of these types of molecules has been largely reported in literature keeping in view of their vast biological activities and as precursors of pyrazolines and pyrazoles. The synthesized title compounds have been evaluated as better inhibitors for cathepsin L than cathepsin B followed by cathepsin H. Best inhibitor for cathepsin B has been evaluated as (1Z)-1-((E)-3-(2-chlorophenyl)-1-phenylallylidene) semicarbazide, (2a) with the K i value of (0.207 ± 0.001) × 10−6 M, for cathepsin H (1E)-1-((E)-3-(4-chlorophenyl)-1-phenylallylidene)semicarbazide (2c′) showed maximum inhibition with a K i value of (2.10 ± 0.02) × 10−5 M and for cathepsin L, (1E)-1-((E)-3-(2-chlorophenyl)-1-phenylallylidene)semicarbazide (2a′) showed maximum inhibition with the K i value of (0.4 ± 0.003) × 10−10 M Chalconesemicarbazones inhibited all the three enzymes effectively followed by pyrazolines and chalcones.
Conflict of interest
The authors have declared no conflict of interest.
Acknowledgment
Among the authors, Ravinder Kaur acknowledges Kurukshetra University, Kurukshetra for providing URS and other lab facilities.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ijbiomac.2015.07.029.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Yogeeswari P., Ragavendran J.V., Thirumurugan R., Induja S., Sriram D., Stables J.P. Synthesis and structure–activity relationship on anticonvulsant aryl semicarbazones. Med. Chem. 2006;2:55–62. doi: 10.2174/157340606775197778. [DOI] [PubMed] [Google Scholar]
- 2.Veerapandian M., Marimuthu M., Ilangovan P., Ganguly S., Yun K.S., Kim S., An J. Analytical and biological characterization of quinazoline semicarbazone derivatives. Med. Chem. Res. 2010;19:283–298. [Google Scholar]
- 3.Fabian L., Caputto M.E., Finkielsztein L.M., Moltrasio G.Y., Moglioni A.G. Semicarbazones and copper complexes of semicarbazones and thiosemicarbazones derived from 1-indanones: synthesis, structure and spectroscopy. Mol. Med. Chem. 2007;12:70–72. [Google Scholar]
- 4.Hall I.H., Lackey C.B., Kistler T.D., Durham R.W., Jouad E.M., Khan M., Thanh X.D., Diebbar-Sid S., Benali-Baitich O., Bouet G.M. Cytotoxicity of copper and cobalt complexes of furfural semicarbazone and thiosemicarbazone derivatives in murine and human tumor cell lines. Pharmazie. 2000;55:937–941. [PubMed] [Google Scholar]
- 5.Warren J.D., Woodward D.L., Hargreaves R.T. 4-Substituted semicarbazones of mono and dichlorobenzaldehydes as antihypertensive agents. J. Med. Chem. 1977;20:1520–1521. doi: 10.1021/jm00221a034. [DOI] [PubMed] [Google Scholar]
- 6.Fernando R.P., Pedro I.d.S.M., Leite Sergio R.A., Victor M.D., Alzir A.B., Daisy N.S., Scott G.F., Leite-Clarice Q.F. Thiosemicarbazones, semicarbazones, dithiocarbazates and hydrazide/hydrazones: anti-Mycobacterium tuberculosis activity and cytotoxicity. Eur. J. Med. Chem. 2010;45:1898–1905. doi: 10.1016/j.ejmech.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 7.Soares R.O., Echevarria A., Bellieny M.S., Pinho R.T., de Leo R.M., Seguins W.S., Machado G.M., Canto-Cavalheiro M.M., Leon L.L. Evaluation of thiosemicarbazones and semicarbazones as potential agents anti-Trypanosoma cruzi. Exp. Parasitol. 2011;129:381–387. doi: 10.1016/j.exppara.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 8.F.E. Cohen, X. Du, C. Guo, J.H. Mckerrow, Thiosemicarbazone and semicarbazone inhibitors of cysteine proteases and methods of their use, US Patent (2009) 7,495,023.
- 9.Li R., Kenyon G.L., Cohen F.E., Chen X., Gonq B., Dominquez J.N., Davidson E., Kurzban G., Miller R.E., Nuzum E.O. In vitro antimalarial activity of chalcones and their derivatives. J. Med. Chem. 1995;38:5031–5037. doi: 10.1021/jm00026a010. [DOI] [PubMed] [Google Scholar]
- 10.Szliszka E., Czuba Z.P., Mazur B., Paradysz A., Krol W. Chalcones and dihydrochalcones augment TRAIL-Mediated apoptosis in prostate cancer cells. Molecules. 2010;15:5336–5353. doi: 10.3390/molecules15085336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lunardi F., Guzela M., Rodrigues A.T., Corre R., Eger-Mangrich I., Steindel M., Grisard E.C., Assreuy J., Calixto J.B., Santos A.R. Trypanocidal and leishmanicidal properties of substitution containing chalcones. Antimicrob. Agents Chemother. 2003;47:1449–1451. doi: 10.1128/AAC.47.4.1449-1451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav H.L., Gupta P., Pawar P.S., Singour P.K., Patil U.K. Synthesis and biological evalution of anti-inflammatory activity of 1,3-diphenylpropenone derivatives. Med. Chem. Res. 2010;19:1–8. [Google Scholar]
- 13.Bhatia N.M., Mahadik K.R., Bhatia M.S. QSAR analysis of 1,3-diaryl-2-propen-1-ones and their indole analogs for designing potent antibacterial agents. Chem. Pap. 2009;63:456–463. [Google Scholar]
- 14.Sivakumar P.M., Prabhakar P.K., Doble M. Synthesis, antioxidant evalution and quantitative structure-activity relationship studies of chalcones. Med. Chem. Res. 2010;19:1–17. [Google Scholar]
- 15.Lahtchev K.L., Batovska D.I., Parushev St.P., Ubiyvovk V.M., Sibirny A.A. Antifungal activity of chalcones: a mechanistic study using various yeast strains. Eur. J. Med. Chem. 2008;43:2220–2228. doi: 10.1016/j.ejmech.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 16.Nerya O., Musa R., Khatib S., Tamir S., Vaya J. Chalcones as potent tyrosinase inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry. 2004;65:1389–1395. doi: 10.1016/j.phytochem.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Najafian M., Ebrahim-Habibi A., Hezareh N., Yaghmaei P., Parivar K., Larijani B. Trans-chalcone: a novel small molecule inhibitor of mammalian alpha-amylase. Mol. Biol. Rep. 2011;38:1617–1620. doi: 10.1007/s11033-010-0271-3. [DOI] [PubMed] [Google Scholar]
- 18.Jaramillo M.C., Mora C., Velez L.E., Quijano J. Kinetic and theoretical study of the chalcones as inhibitors of beta-lectamase enzyme. Med. Chem. 2009;5:434–439. doi: 10.2174/157340609789117895. [DOI] [PubMed] [Google Scholar]
- 19.Raghav N., Malik P. Solvent free synthesis of some chalcones and their effect on bovine serum albumin. Adv. Appl. Sci. Res. 2011;2:410–415. [Google Scholar]
- 20.Raghav N., Malik P. Spectrophotometric analysis of bovine serum albumin in presence of synthesized 1-(2′-furyl)-3(substituted phenyl)-2-propen-1-ones. Res. J. Pharm. Biol. Chem. 2011;2:755–760. [Google Scholar]
- 21.Raghav N., Malik P. Spectrophotometric analysis of bovine serum albumin in presence of synthesized 1-(2′-thienyl)-3(substituted phenyl)-2-propen-1-ones. Int. J. Appl. Biol. Pharm. Technol. 2011;2:218–223. [Google Scholar]
- 22.Garg S., Raghav N. Spectrophotometric analysis of bovine serum albumin in presence of some hydroxy- and nitro-substituted chalcones. Int. J. Pharm. Pharm. Sci. 2012;4:264–268. [Google Scholar]
- 23.Garg S., Ravish I., Raghav N. Analysis of bovine serum albumin in presence of some phenyl substituted chalcones. Int. J. Pharm. Pharm. Sci. 2013;5:372–375. [Google Scholar]
- 24.Garg S., Singh M., Raghav N. Spectrophotometric analysis of bovine serum albumin in presence of some bischalcones. Int. J. Appl. Biol. Pharm. Technol. 2013;4:87–91. [Google Scholar]
- 25.Selzer P.M., Pingel S., Hsieh I., Ugles B., Chan V.J., Engel J.C., Bogyo M., Russell D.G., Sakanari J.A., Mckerrow J.H. Cysteine protease inhibitors as chemotherapy: lessons from a parasite target. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11015–11022. doi: 10.1073/pnas.96.20.11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renko M., Pozgan U., Majera D., Turk D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010;277:4338–4345. doi: 10.1111/j.1742-4658.2010.07824.x. [DOI] [PubMed] [Google Scholar]
- 27.Aronson N.N., Jr., Barrett A.J. The specificity of cathepsin B. Hydrolysis of glucagon at the c-terminus by a peptidyldipeptidase mechanism. Biochem. J. 1978;171:759–765. doi: 10.1042/bj1710759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bond J.S., Barrett A.J. Degradation of fructose-1,6-bisphosphate aldolase by cathepsin B. A further example of peptidyldipeptidase activity of this proteinase. Biochem. J. 1980;189:17–25. doi: 10.1042/bj1890017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi T., Dehdarani A.H., Yonezawa S., Tang J. Porcine spleen cathepsin B is an exopeptidase. J. Biol. Chem. 1986;261:9375–9381. [PubMed] [Google Scholar]
- 30.Rowan A.D., Feng R., Konishi Y., Mort J.S. Demonstration by electrospray mass spectrometry that the peptidylpeptidase activity of cathepsin B is capable of rat cathepsin B c-terminal processing. Biochem. J. 1993;294:923–927. doi: 10.1042/bj2940923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mort J.S., Buttle D.J., Cathepsin B. Int. J. Biochem. Cell Biol. 1997;29:715–720. doi: 10.1016/s1357-2725(96)00152-5. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Sukhova G., Sun J., Xu W., Libby P., Shi G. Lysosomal cysteine proteases in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004;24:1359–1366. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- 33.Werb Z. In: Textbook of Rheumatology. Keller W.N., Harris E.D., Ruddy S., Sledge C.B., editors. W.B. Saunders; Philadelphia: 1989. Proteinases and matrix degradation; pp. 300–320. [Google Scholar]
- 34.Schechter I., Ziv E. Cathepsin S, B and L with aminopeptidases display β-secretase activity associated with the pathogenesis of Alzheimer's disease. Biol. Chem. 2011;392:555–569. doi: 10.1515/BC.2011.054. [DOI] [PubMed] [Google Scholar]
- 35.Gabrijelcic D., Svetic B., Spaic D., Skrk J., Budihna M., Dolenc I., Popovic T., Cotic V., Turk V. Cathepsin B, H and L in human breast carcinoma. Eur. J. Clin. Chem. Clin. Biochem. 1992;30:69–74. [PubMed] [Google Scholar]
- 36.Schweiger A., Stabuc B., Popovic T., Turk V., Kos J. Enzyme-linked immunosorbent assay for the detection of total cathepsin H in human tissue cytosols and sera. J. Immunol. Methods. 1997;201:165–232. doi: 10.1016/s0022-1759(96)00218-9. [DOI] [PubMed] [Google Scholar]
- 37.Budihna M., Strojan P., Smid L., Skrk J., Vrhovec I., Zupevc A., Rudolf Z., Zargi M., Krasover M., Svetic B., Kopitar-Jerala N., Kos J. Prognostic value of cathepsin B, H, L, D and their endogenous inhibitors stefins A and B in head and neck carcinoma. Biol. Chem. Hoppe-Seyler. 1996;377:385–390. doi: 10.1515/bchm3.1996.377.6.385. [DOI] [PubMed] [Google Scholar]
- 38.Friedrich B., Jung K., Lein M., Turk I., Rudolph B., Hampel G., Schnorr D., Loening S.A. Cathepsin B, H, L and cysteine protease inhibitors in malignant prostate cell lines, primary cultured prostatic cells and prostatic tissue. Eur. J. Cancer. 1999;35:138–144. doi: 10.1016/s0959-8049(98)00273-1. [DOI] [PubMed] [Google Scholar]
- 39.Maciewicz R.A., Wardale R.J., Wotton S.F., Duance V.C., Etherington D.J. Mode of activation of the precursor to cathepsin L: implication for matrix degradation in arthritis. Biol. Chem. Hoppe-Seyler. 1990;371:223–228. [PubMed] [Google Scholar]
- 40.Van Noorden C.J., Smith R.E., Rasnick D. Cysteine proteinase activity in arthritic rat knee joints and the effects of a selective systemic inhibitor, Z-Phe-AlaCH2F. J. Rheumatol. 1993;15:1525–1535. [PubMed] [Google Scholar]
- 41.Werle B., Ebert W., Klein W., Spiess E. Assessment of cathepsin L activity by use of the inhibitor CA-074 compared to cathepsin B activity in human lung tumor tissue. Biol. Chem. Hoppe-Seyler. 1995;376:157–164. doi: 10.1515/bchm3.1995.376.3.157. [DOI] [PubMed] [Google Scholar]
- 42.Scaddan P.B., Dufresne M.J. Characterization of cysteine proteases and their endogenous inhibitors in MCF-7 and adriamycin resistant MCF-7 human breast cancer cells. Invasion Metastasis. 1993;13:301–313. [PubMed] [Google Scholar]
- 43.Solovyeva N.I., Balayevskaya T.O., Dilakyan E.A., Zakamaldina-Zama T.A., Pozdnev V.F., Topol L.Z., Kisseljov F.L. Proteolytic enzymes at various stages of oncogenic transformation of rat fibroblasts I Aspartyl and cysteine proteinases. Int. J. Cancer. 1995;60:495–500. doi: 10.1002/ijc.2910600412. [DOI] [PubMed] [Google Scholar]
- 44.Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafati S., Salmanian A.H., Hashemi K., Schaff C., Belli S., Fasel N. Identification of Leishmania major cysteine proteinases targets of the immune response in humans. Mol. Biochem. Parasitol. 2001;113:35–43. doi: 10.1016/s0166-6851(00)00377-7. [DOI] [PubMed] [Google Scholar]
- 47.Raghav N., Kaur R., Singh M., Suman, Priyanka Effect of semicarbazones on endogenous protein hydrolysis in liver homogenate. Asian J. Chem. 2010;22:7097–7101. [Google Scholar]
- 48.Raghav N., Singh M., Kaur R., Suman, Priyanka Proteolytic studies in liver homogenate in presence of phenylhydrazone. Int. J. Pharm. Technol. 2010;2:743–749. [Google Scholar]
- 49.Raghav N., Singh M., Kaur R., Suman, Priyanka Proteolytic studies in liver homogenate in presence of arylhydrazone. Asian J. Chem. 2011;23:1409–1410. [Google Scholar]
- 50.Kaur R., Singh M., Jangra S., Raghav N. Proteolytic studies in liver homogenate in presence of substituted thiosemicarbazones. Int. J. Chem. Sci. 2012;10:1698–1704. [Google Scholar]
- 51.Singh M., Raghav N. In-Vitro inhibition studies on endogenous proteolysis of liver homogenate in presence of synthesized pyrazolines. Int. J. Pharm. Pharm. Sci. 2013;5:80–86. [Google Scholar]
- 52.Singh M., Raghav N. In-Vitro inhibition studies on endogenous proteolysis of liver homogenate in presence of synthesized pyrazoles. Int. J. Pharm. Pharm. Sci. 2013;5:365–368. [Google Scholar]
- 53.Raghav N., Singh M. Design synthesis and docking studies of bischalcones based quinazoline-2(1H)-ones and quinazoline-2(1H)-thiones derivatives as novel inhibitors of cathepsin B and cathepsin H. Eur. J. Pharm. Sci. 2014;54:28–39. doi: 10.1016/j.ejps.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 54.Raghav N., Singh M. Acyl hydrazide and triazoles as novel inhibitors of mammalian cathepsin B and cathepsin H. Eur. J. Med. Chem. 2014;77:231–242. doi: 10.1016/j.ejmech.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 55.Raghav N., Singh M. SAR studies of differently functionalized chalcones based hydrazones and their cyclized derivatives as inhibitors of mammalian cathepsin B and cathepsin H. Bioorg. Med. Chem. 2014;22:4233–4245. doi: 10.1016/j.bmc.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 56.Raghav N., Garg S. SAR studies of o-hydroxychalcones and their cyclised analogs and study them as novel inhibitors of cathepsin b and cathepsin H. Eur. J. Pharm. Sci. 2014;60:55–63. doi: 10.1016/j.ejps.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 57.Raghav N., Garg S. N-formylpyrazolines and N-benzoylpyrazolines as novel inhibitors of mammalian cathepsin B and cathepsin H. Bioorg. Chem. 2014;57:43–50. doi: 10.1016/j.bioorg.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 58.Ozdemir A., TuranZitouni G., Kaplanciklien Z.A., Revial G., Guven K. Synthesis and antimicrobial activity of 1-(4-aryl-2-thiazolyl)-3-(2-thienyl)-5-aryl-2-pyrazoline derivatives. Eur. J. Med. Chem. 2007;42:403–409. doi: 10.1016/j.ejmech.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Abid M., Bhat A.R., Athar F., Azam A. Synthesis, spectral studies and antiamoebic activity of new 1-N-substituted thiocarbamoyl-3-phenyl-2-pyrazolines. Eur. J. Med. Chem. 2007;42:1–9. doi: 10.1016/j.ejmech.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 60.Amir M., Kumar H., Khan S.A. Synthesis and pharmacological evaluation of pyrazoline derivatives as new anti-inflammatory and analgesic agents. Bioorg. Med. Chem. Lett. 2008;18:918–922. doi: 10.1016/j.bmcl.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 61.Havrylyuk D., Zimenkovsky B., Vasylenko O., Zaprutko L., Gzella A., Lesyk R. Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evalution of their anticancer activity. Eur. J. Med. Chem. 2008;42:1–9. doi: 10.1016/j.ejmech.2008.09.032. [DOI] [PubMed] [Google Scholar]
- 62.Palaskaa E., Aytemira M., Uzbay I.T., Erola D. Synthesis and antidepressant activities of some 3,5-diphenyl-2-pyrazolines. Eur. J. Med. Chem. 2001;36:539–543. doi: 10.1016/s0223-5234(01)01243-0. [DOI] [PubMed] [Google Scholar]
- 63.Ali M.A., Shaharyar M., Siddiqui A.A. Synthesis, structural activity relationship and anti-tubercular activity of novel pyrazoline derivatives. Eur. J. Med. Chem. 2007;42:268–275. doi: 10.1016/j.ejmech.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 64.Rueping M., Bootwicha T., Baars H., Sugiono E. Continous-flow hydration-condensation reaction: synthesis of α,β-unsaturated ketones from alkynes and aldehydes by using a heterogeneous solid acid catalyst. Beilstein J. Org. Chem. 2011;7:1680–1687. doi: 10.3762/bjoc.7.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siddiqui A.A., Rahman Md.A., Shaharyar Md., Mishra R. Synthesis and anticonvulsant activity of some substituted 3,5-diphenyl-2-pyrazoline-1-carboxamide derivatives. Chem. Sci. J. 2010;8:1–10. [Google Scholar]
- 66.Kamboj R.C., Pal S., Singh H. Purification and characterization of cathepsin B from goat brain. J. Biosci. 1990;15:397–408. [Google Scholar]
- 67.Kamboj R.C., Pal S., Raghav N., Singh H. Selective colorimetric assay for cathepsin L using Z-Phe-Arg-4-ethoxy-β-naphthylamide. Biochimie. 1993;75:873–878. doi: 10.1016/0300-9084(93)90042-q. [DOI] [PubMed] [Google Scholar]
- 68.Raghav N., Kamboj R.C., Parnami S., Singh H. Physico-chemical properties of brain cathepsin H. Indian J. Biochem. Biophys. 1995;32:279–285. [PubMed] [Google Scholar]
- 69.Huber C.P., Campbell R.L., Hasnain S., Hirama T., To R. 2013. Crystal Structure of the Tetragonal Form of Human Liver Cathepsin B.http://www.ebi.ac.uk/pdbe-srv/view/entry/2ipp/citation.html (31.05.13) [Google Scholar]
- 70.Guncar G., Podobnik M., Pungercar J., Strukelj B., Turk V., Turk D. Crystal structure of porcine cathepsin H determined at 2.1 Å resolution: location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure. 1998;6:51–61. doi: 10.1016/s0969-2126(98)00007-0. [DOI] [PubMed] [Google Scholar]
- 71.Chowdhary S.F., Joseph L., Kumar S., Tulsidas S.R., Bhat S., Ziomek E., Menard R., Sivaraman J., Purisima E.O. Exploring inhibitor binding at the S′ subsites of cathepsin L. J. Med. Chem. 2008;51:1361–1368. doi: 10.1021/jm701190v. [DOI] [PubMed] [Google Scholar]
- 72.J.R. Hans, University of Wisconsin. http://www.Chem.Wisc.edu/areas/reich/nmr/notes-5-hmr-2-shift.pdf (14.08.13).
- 73.http://media.cambridgesoft.com/cbou130/cbou1302.exe (04.09.13).
- 74.Sengupta S.K., Pandeya O.P., Rao G.P., Singh P. Efficacy of organophosphorus derivatives containing chalcones/chalconesemicarbazones against fungal pathogens of sugarcane. Metal-Based Drugs. 2002;8:293–302. doi: 10.1155/MBD.2002.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singhal M., Paul A., Singh H.P. Evaluation of anti-phlogistic activity of synthesized chalconesemicarbazone derivatives. J. Chem. Pharm. Res. 2010;2:90–98. [Google Scholar]
- 76.Singhal M., Paul A. Synthesis characterization and antipyretic evaluation of some novel chalconesemicarbazone derivatives. Res. J. Pharmacol. 2011;5:47–52. [Google Scholar]
- 77.Shekarchi M., Pirali-Hamedani M., Navidpour L., Adib N., Shafiee A. Synthesis, antibacterial and antifungal activities of 3-Aryl-5-(pyridine-3-yl)-4,5-dihydropyrazole-1-carbothioamide derivatives. J. Iran. Chem. Soc. 2008;5:150–158. [Google Scholar]
- 78.Knight C.G. Application of the substrate N-benzyloxycarbonyl-l-arginyl-l-arginine 2-naphthylamide to a study of the inhibition by Leupeptin. Biochem. J. 1980;189:447–453. doi: 10.1042/bj1890447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azaryan A., Galoyan A. Human and bovine brain cathepsin L and cathepsin H: purification, physic-chemical properties and specificity. Neurochem. Res. 1987;12:207–213. doi: 10.1007/BF00979539. [DOI] [PubMed] [Google Scholar]
- 80.Vicik R., Busemann M., Baumann K., Schirmeister T. Inhibitors of cysteine proteases. Curr. Top. Med. Chem. 2006;6:331–353. doi: 10.2174/156802606776287081. [DOI] [PubMed] [Google Scholar]
- 81.Kamboj R.C. Kurukshetra University; Kurukshetra, India: 1989. Proteolytic enzymes in brain tissue. (Ph.D. thesis) [Google Scholar]
- 82.Yang J.M., Chen C.C. GemDOCK: a generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinformatics. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- 83.Kishore Kumar G.D., Chavarria G.E., Charlton-Sevcik A.K., Yoo G.K., Song J., Strecker T.E., Siim B.G., Chaplin D.J., Trawick M.L., Pinney K.G. Functionalized benzophenone, thiophene, pyridine, and fluorine thiosemicarbazone derivatives as inhibitors of cathepsin L. Bioorg. Med. Chem. Lett. 2010;20:6610–6615. doi: 10.1016/j.bmcl.2010.09.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.