

A selfish B chromosome in the jewel wasp actively mediates its own drive by expression of the haploidizer gene.
Abstract
Numerous plants and animals harbor selfish B chromosomes that “drive” or transmit themselves at super-Mendelian frequencies, despite long-term fitness costs to the organism. Currently, it is unknown how B chromosome drive is mediated, and whether B-gene expression plays a role. We used modern sequencing technologies to analyze the fine-scale sequence composition and expression of paternal sex ratio (PSR), a B chromosome in the jewel wasp Nasonia vitripennis. PSR causes female-to-male conversion by destroying the sperm’s hereditary material in young embryos to drive. Using RNA interference, we demonstrate that testis-specific expression of a PSR-linked gene, named haploidizer, facilitates this genome elimination–and–sex conversion effect. haploidizer encodes a putative protein with a DNA binding domain, suggesting a functional link with the sperm-derived chromatin.
INTRODUCTION
Thousands of plants and animals harbor extra, nonessential chromosomes termed B chromosomes, which consist of both coding and noncoding sequences thought to be copied from the essential, or A, chromosomes. Because they are not needed, B chromosomes presumably carry no essential genes and therefore can be lost (1–4). To counter being lost, many B chromosomes have the extraordinary ability to “drive” or transmit themselves at frequencies above those predicted by Mendelian rules [reviewed in (5–7)]. Although drive is necessary for B chromosome transmission, it can impose deleterious effects on the inheritance of other genomic regions, thus having potentially strong influences on organismal fitness (5, 7, 8). Currently, it is not understood how B chromosomes can behave so differently in their transmission from other nondriving chromosomes.
One of the most extreme examples of drive is caused by a B chromosome known as PSR (paternal sex ratio), which has been detected at moderate frequencies (6 to 11%) within certain populations of the jewel wasp Nasonia vitripennis (9, 10). PSR is transmitted strictly paternally (i.e., via sperm), and its presence leads to the complete elimination of the sperm’s essential chromosomes, but not PSR itself, during the mitotic division immediately following fertilization (11). Paternal genome elimination (PGE) results directly from failure of the sperm’s chromatin to resolve into individualized chromosomes upon entry into mitosis, and it is preceded by the appearance of distinct chemical markers that indicate a disrupted chromatin state (1, 12–14). This effect is critical for the transmission of PSR. Wasps, like all hymenopteran insects (also including ants and bees), reproduce through haplodiploidy, in which unfertilized eggs develop into haploid males, while fertilized eggs become diploid females. Therefore, elimination of the sperm’s hereditary material by PSR converts all diploid, female-destined eggs into haploid males, the PSR-transmitting sex (1). Broadly, this effect results in severely male-biased sex ratios at the deme level, which can negatively affect wasp metapopulations (9, 10).
A fundamental biological question is how PSR and, more broadly, other B chromosomes are capable of interacting uniquely with the cellular environment to drive. Previous studies identified three distinct ~180 DNA base pair (bp) repeats, termed PSR2, PSR18, and PSR22, which are abundant in copy number on PSR, but not present on any of the five essential wasp chromosomes (15, 16). Each of these repeats contains a highly conserved, 8-bp palindromic motif that is reminiscent of those found in some transcription factor binding sequences (15, 16). These characteristics have led to speculation that PSR-specific sequences may act as a sink for some limited chromatin factor(s), drawing them away from the rest of the genome and causing defective chromatin structure (15). An underlying aspect of this scenario is that the driving effect of PSR is the result of intrinsic properties that are unique to its sequence composition. For this reason, we refer to such an effect as passive. Alternatively, PSR’s drive may involve the active expression of B-linked sequences. Recent studies have begun to identify individual sequences that are expressed from a number of different B chromosomes, including PSR (17, 18). While no studies have, to date, demonstrated functionality for any B-linked sequence, it is plausible that an RNA or protein expressed by PSR functions as an effector of drive by disrupting normal transmission of the paternal chromatin.
Previous transcriptional profiling uncovered a handful of long, polyadenylated RNAs expressed uniquely by PSR in the wasp testis (18). To further this work, we have used a combination of whole-genome sequencing technologies to generate large scaffolds of the PSR chromosome, onto which we mapped these PSR-expressed transcripts. These efforts produced a comprehensive portrayal of PSR’s sequence composition, models for its individual genes, and its transcribed loci. Using RNA interference (RNAi), we demonstrate that PSR-induced PGE requires the expression of one of these PSR-linked loci, demonstrating an active (i.e., gene expression based) involvement of this B chromosome in its own drive.
RESULTS
Whole-genome sequencing of PSR and the essential N. vitripennis chromosomes
To deduce PSR’s sequence composition, we separately sequenced the genome of both wild-type (WT) and PSR-carrying wasps using a combination of PacBio, Oxford Nanopore, and Illumina Technologies. This approach generated a 297-Mb assembly of 444 scaffolds with an N50 length of 6.6 Mb, resulting in a 13.9-fold decrease in scaffold number and a 9.3-fold increase in N50 length compared with the current N. vitripennis genome assembly (19). Using the Hymenoptera-specific set of universal single-copy orthologs, the genome completeness is estimated to be 96.5% (table S1) (20). Using the chromosome quotient method (table S2 and fig. S1) (21)), we identified 120 scaffolds (9.2 Mb in total, N50 of 124 kb) that were specific to PSR (table S3). In addition, 73 scaffolds totaling 272 Mb (91.58% of total genome) were assigned to the five essential N. vitripennis chromosomes using a set of previously developed genetic markers (fig. S2, table S4, and data S1 to S3) (22).
To better understand the sequence composition and organization of PSR (table S5), we performed a detailed computational assessment of PSR’s repetitive content using RepeatModeler and RepeatMasker (23, 24). These analyses revealed that 89.80% of PSR is composed of repetitive DNAs (Fig. 1A and table S6). The most abundant repeats (70.32%) are complex satellites belonging to four main families. Three of these satellite families—PSR2 (49.54%), PSR18 (42.64%), and PSR22 (17.22%)—are specific to PSR. PSR2 and PSR18 are typically found together on gene-coding scaffolds and they mostly overlap, whereas PSR22 is found on different sets of scaffolds without coding genes. A fourth repeat, NV79 (2.71%), is located on four PSR scaffolds, in which it is not overlapping with other repeats, and also on all five essential chromosomes (Fig. 1B). In addition, PSR contains DNA and RNA (retro-) transposable elements (TEs) (13.97%), simple repeats (0.36%) and other low-complexity regions (0.03%), and uncharacterized sequences (3.81%) (Fig. 1A and tables S5 and S6). Two PSR scaffolds contain telomeric sequences that cover 3.5 to 3.9 kb at one of the ends of each scaffold. The telomeric repeat found on PSR and at the ends of all five A chromosomes of N. vitripennis is “TTATTGGG” (fig. S3), which is different from the canonical sequence “TTAGGG” that is found in other insects (25).
Fig. 1. Composition and origin of PSR chromosome.
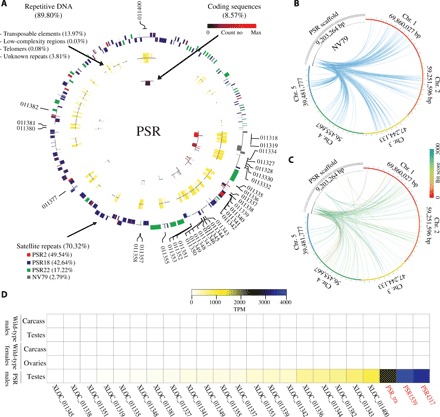
(A) Circular portrayal of the PSR chromosome scaffolds. The scaffolds are organized from largest to smallest (their natural order with respect to each other could not be determined with current data). The outermost track represents repetitive satellites with outer bars representing the positive DNA strand, and the inner bars represent the negative DNA strand. The colors represent the four major (70.32%) satellite families. The middle track represents other repetitive sequences (17.89%) including TEs, low-complexity regions, and telomeric sequences. The innermost track represents protein coding sequences (8.57%), and the color ranges from black to red for low to high expression, respectively. (B) The relationship of NV79 repeats between PSR scaffolds and host chromosomes 1 to 5, indicating that PSR repeats are homologous to sequences found on all chromosomes. (C) The relationship of PSR protein scaffolds sequences and host chromosomes. The links are colored by blast bit scores from red (highest) to blue (lowest). (D) Heat map of PSR gene with expression values higher than 10 TPM (table S8, columns M to Q). Expression levels are portrayed in colors ranging from white (low to no expression) to blue (highest expression). PSR genes that were disrupted with RNAi in this study are shown in red.
PSR expresses a small subset of genes in the N. vitripennis testis
Using Nanopore sequencing of full-length complementary DNA (cDNA) derived from PSR-carrying testes and whole animals, we identified 68 transcripts (table S7) encoded by 44 loci located on 14 of the 120 scaffolds and covering 8.57% of the total length of PSR (table S8). Most of the intronic regions are represented by TEs and low-complexity regions. Thus, the coding sequences corresponding to exonic regions represent only 0.51% of the entire PSR chromosome. Fifty of the PSR-encoded transcripts have identifiable open reading frames (ORFs) longer than 100 amino acids, and 34 of them have Pfam domains suggestive of certain molecular functions performed by these genes (table S7). Fifty-eight transcripts produced significant blast hits when searched against the nonredundant protein database. Thirty-one sequences had matches to proteins encoded by the WT N. vitripennis genome, although N. vitripennis genes were the best hits for only 13 of them. The corresponding genes are located in different regions across all five essential chromosomes (Fig. 1C) and span a range of functional groups, including transposon activity, transcription regulation, DNA binding, and protein binding. Twenty transcripts were best matched to genes found in the genome of a closely related parasitoid wasp, Trichomalopsis sarcophagae, which represents the most common lineage identified by blast searches. The remaining 25 best matches are to genes of other insects, although other noninsect lineages, including bacteria, are represented as well (table S8), and they mainly correspond to protein-coding regions of transposons. Thus, unlike some B chromosomes, whose sequences derived largely from one or a few large regions of essential chromosomes within their resident genomes (26–28), PSR consists primarily of three complex repeats (70.32%) and other sequences that are undetectable in the N. vitripennis genome and, in some cases, have strong similarity with genes from other organisms, as has been previously noted (29).
Quantification of the RNA sequencing (RNA-seq) data revealed that 25 of the 44 PSR-encoded loci are expressed at levels greater than 10 transcripts per million (TPM) in the testis (table S8). The three highest-expressed loci were selected for further analysis (Fig. 1D, table S8, and data S4). Two of these sequences were previously identified as PSR-4317 and PSR-1539 in an earlier transcriptome profiling study (17), whereas the third highest-expressed gene, PSR-tra, has a predicted ORF with limited homology to part of the wasp’s sex-determining gene, transformer. On the basis of their transcript sequences and chromosomal gene models (fig. S4), PSR-4317 and PSR-1539 contain putative translational ORFs of 279 and 242 amino acids, respectively (17). The putative ORF of PSR-4317 contains two adjacent nuclear receptor DNA binding domains, each composed of two C4-type zinc fingers [CDD (conserved domain database) accession cl02596], although one of the domains appears to be partial. However, no ligand-binding domain or other structured regions that are typically found in nuclear receptors could be detected (fig. S5 and data S5). Neither PSR-4317 nor PSR-1539 matches any gene present in the N. vitripennis genome. Instead, PSR-4317 strongly matches genes including a bacterium, several invertebrates, and a fish (fig. S5). These genes contain a similar DNA binding domain, which appears to underlie the similarity to PSR-4317. Outside of this region, there is little substantial sequence similarity, with one notable exception: a putative protein from an insect bacterial symbiont, Candidatus cardinium. The similarity of this protein extends across most of PSR-4317’s ORF (81%), including both DNA binding domains (fig. S5B). However, the overall similarity to the C. cardinium protein is low enough to prevent a definitive conclusion of their possible relatedness.
Expression of a PSR gene, haploidizer, is necessary for PGE
Using reverse transcription quantitative polymerase chain reaction (RT-qPCR), we detected expression of PSR-4317, PSR-1539, and PSR-tra in the male germ line, as well as in all tested embryonic and adult male somatic tissues, demonstrating that their expression is not restricted to a specific developmental period (fig. S6). If any of these genes play a role in PSR’s drive, then their action on the paternally inherited chromatin could, in principle, occur either during sperm formation or instead in the egg’s cytoplasm immediately following fertilization. To address these possibilities, we used RNAi to transiently reduce transcript levels for each of these genes in the male germ line and assessed for an effect on drive by measuring the proportion of female G1 progeny (Fig. 2, A, and B). Knockdown of PSR-4317 transcripts in the testes yielded a striking effect on the proportion of G1 females: 46% of treated males produced broods that ranged between 10 and 90% females (Fig. 2B). In contrast, RNAi targeting of the other two genes resulted in all-male broods similar to those from control (untreated) PSR+ wasps, despite the fact that their transcript levels were effectively reduced to levels comparable to RNAi-targeted PSR-4317 transcript levels (Fig. 2, A and B, and fig. S7). Maternal RNAi (30) targeting of PSR-4317 in the egg’s cytoplasm had no effect on the proportion of G1 females (fig. S8; see Materials and Methods). However, we cannot conclusively determine that maternal RNAi effectively targeted paternally contributed PSR-4317 transcripts in the egg.
Fig. 2. Effects of haploidizer targeted by RNAi in early embryos.
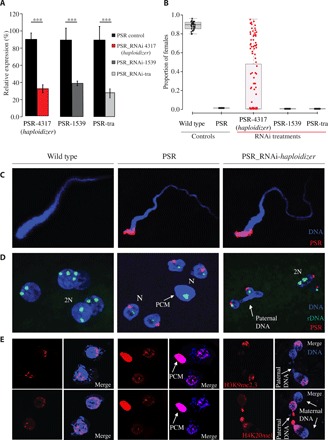
(A) Efficiency of RNAi measured by relative gene expression of the targeted genes. Asterisks indicate significant expression differences between control and RNAi-treated individuals [n = 5, P < 0.05, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test]. (B) Proportion of G1 females was measured for control and RNAi-treated wasps. The box plots depict the median (thick horizontal line within the box), the 25th and 75th percentiles (box margins), and the 1.5 interquartile range (thin horizontal line). (C) Confocal imaging of sperm from WT, PSR+, and haploidizer RNAi-treated PSR+ males. (D) Nuclei of individual embryos from WT, PSR+, and haploidizer RNAi-treated PSR+ fathers, stained for PSR (red) and rDNA (green). (E) Nuclei from individual embryos from WT, PSR+, and haploidizer RNAi-treated PSR+ fathers, stained for the histone marks, H3K9me2,3 (top panels, red), and H4K20me1 (bottom panels, also red). DNA is blue in all panels. PCM, paternal chromatin mass; 2N, diploid nuclei; N, haploid nuclei.
The production of female G1 progeny by RNAi-treated fathers could result from suppression of the genome-eliminating activity, but it could also occur from the destabilization and loss of PSR before the genome-eliminating activity has occurred. Several observations strongly argue against this latter possibility. First, individual sperm produced by RNAi-treated males contained a single copy of the PSR chromosome (Fig. 2C and fig. S9), and PSR was present in the nuclei of young fertilized embryos sired by these males (Fig. 2D and fig. S10). In addition, using PCR, we confirmed that the female G1 adults produced in our crosses were positive for multiple different PSR-specific sequences, suggesting that they inherited the B chromosome from their fathers (figs. S8C and S11 and table S9).
In light of these findings, the appearance of female G1 progeny may result instead from failure of PGE when PSR-4317 transcripts are knocked down. To test this possibility, we examined the mitotic behavior of the sperm- and egg-derived nuclei and their mitotic descendants in young embryos produced by RNAi-treated fathers. Normally, in embryos from control PSR+ fathers, the sperm-derived nuclear material fails to resolve into individual chromosomes upon entry into the first mitosis, forming a mass of unresolved chromatin that remains distinct from the maternally derived nuclei (Fig. 2D) (1, 14). This paternal chromatin mass (PCM) fails to segregate, eventually becoming lost within the embryo, as the egg-derived nuclei continue to divide (1, 14). In nearly all of the embryos from RNAi-treated PSR+ fathers for PSR-4317, there was no distinct PCM (Fig. 2D and fig. S10). Instead, in about 68% (26 of 38) of these embryos, the paternal chromatin formed bridges that spanned between dividing nuclei (Fig. 2D and fig. S10). These bridges were not observed in control PSR+ embryos (Fig. 2D). In addition, some embryos from RNAi-treated fathers contained multiple nuclei that varied widely in size, suggesting that they were mosaics of nuclei with varying ploidy levels. In support of these observations, we visualized one, two, and, in some cases, more ribosomal DNA (rDNA) foci per nucleus in these embryos (Fig. 2D). We also examined two histone posttranslational modifications (PTMs), H3K9me3 (trimethylation of histone 3 lysine 9) and H4K20me1 (methylation of histone 4 lysine 20), which, in control (PSR+) conditions, become abnormally distributed across the paternal chromatin (12). In young embryos from RNAi-treated fathers, however, both of these histone PTMs appeared more similar to patterns that are present in WT (non-PSR) embryos (Fig. 2E). Together, these observations suggest that RNAi targeting of PSR-4317 alleviates the defective state of the sperm-derived chromatin caused by PSR, thereby allowing the sperm-derived chromatin to partially segregate with the egg-derived chromatin, giving rise to embryos that are mosaics of nuclei with differing amounts of hereditary material derived from the two parents. Thus, we named this gene haploidizer because its expression is required for conversion of diploid embryos into haploids by causing PGE.
PSR-induced PGE alters embryonic sex by misregulation of key sex-determining genes
We further speculated that the PSR+ females generated by RNAi-treated PSR+ fathers arise from a portion of the mosaic embryos in which PGE is partially suppressed. Consistent with this hypothesis, using RT-PCR, we were able to amplify both female- and male-specific transcript isoforms of the sex-determining genes, transformer (tra) and doublesex (dsx), in all tested PSR+ G1 females, but only male-specific isoforms in control PSR+ and WT males (fig. S11). In N. vitripennis, initiation of female sex determination is regulated epigenetically (31) and requires an unknown trans-acting factor termed womanizer (wom). This factor promotes female-specific splicing of transformer (traF), whose presence, in turn, directs female-specific splicing of doublesex (dsxF), thereby promoting female development (31). It has been hypothesized that wom is epigenetically silenced during oogenesis, and its absence in unfertilized eggs ensures male-specific development (31). We therefore propose that the PSR-induced alteration of the sperm-derived chromatin in control (PSR+) embryos is sufficient to block the production of dsxF, thereby facilitating male development. However, in fertilized embryos from RNAi-treated PSR+ fathers, a less defective state of the paternal chromatin may allow some expression of wom and, subsequently, of traF and dsxF (Fig. 3). We note that PSR+ G1 females were unable to transmit PSR to their progeny, unlike their male siblings (Fig. 3). It is unlikely that the mosaic nature of these PSR+ females is the cause of this transmission failure because these individuals produce viable offspring, thus indicating normal transmission of the five essential chromosomes. Instead, this effect may reflect an intrinsic block to PSR transmission through the female germ line.
Fig. 3. Genetics and active model of PSR-induced genome elimination.
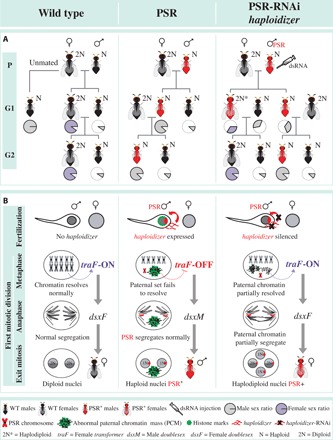
(A) The effects of RNAi knockdown of haploidizer on PSR transmission and the proportion of female offspring. The pie charts indicate the proportion of males (gray) and females (purple). (B) Model for involvement of haploidizer in PGE. Expression of PSR’s haploidizer gene leads to failure of the paternal chromatin to resolve into chromosomes. This action inhibits the activation of female transformer (traF) and, thus, female development. As a result, these fertilized eggs develop into haploid male offspring that carry PSR. RNAi treatment of haploidizer mitigates the chromatin defects, allowing partial expression of traF from the paternal set. This action reestablishes the female developmental pathway.
DISCUSSION
A longstanding question has been how PSR and other B chromosomes mediate their own drive. We have shown here that a PSR-expressed gene, named haploidizer, is necessary for elimination of the sperm-derived chromatin in young N. vitripennis embryos. RNAi knockdown of haploidizer in the testis suppresses PSR’s genome-eliminating activity, thereby blocking its female-to-male conversion effect. It is currently unknown how haploidizer functions in PGE. However, the presence of a C4-type zinc finger DNA binding domain in its putative ORF suggests that if produced, the encoded protein may directly associate with the sperm’s chromatin. A previous study showed that when PSR is present, several different posttranslational histone modifications appear abnormally across the paternal nuclear material, instead of in discrete regions (12). This result suggests that a defective chromatin state, reflected by these abnormal histone PTM patterns, leads to PGE (12). We speculate that haploidizer-encoded protein, if produced, may disrupt the state of the paternal chromatin by directly or indirectly altering the behavior of certain enzymes that establish these histone PTMs when the sperm-derived DNA undergoes repackaging from protamines to histones (12). Testing this hypothesis, and, more generally, discerning whether expression of haploidizer alone is sufficient to induce PGE, will have to await transgenic experimentation.
Another uncertain aspect of haploidizer is its origin. Most B-linked genes in other organisms arose as copies of essential genes located within their respective genomes (32–35). haploidizer does not match any N. vitripennis sequence. Instead, its best matches include a handful of zinc finger domain–containing genes from a wide range of organisms (fig. S5), and the similarity is confined to the DNA binding regions of these genes, with the exception of a putative Cardinium protein. Thus, at this point, we cannot conclusively determine the origin of haploidizer, although it is likely to lie outside of N. vitripennis. This scenario is consistent with the fact that other PSR-linked sequences also are most similar to genes from other organisms. Broadly, our findings demonstrate that B chromosomes can actively mediate their own drive, thus deepening our understanding of the strong influence that these selfish genetic elements can have on genome transmission.
MATERIALS AND METHODS
Sequencing and assembly of the N. vitripennis genome
Genomic DNA from 50 N. vitripennis individuals of the AsymCx strain, either with or without PSR, was isolated using the Blood & Cell Culture DNA Midi Kit (Qiagen, Cat# 13343) according to the manufacturer’s protocol. DNA integrity was assessed using the Genomic DNA ScreenTape assay (part number 5067-5365) for 4200 TapeStation System (Agilent Inc., Santa Clara, CA), quantified with the Qubit dsDNA HS Kit (Thermo Fisher Scientific, #Q32854), and used for PacBio whole-genome sequencing. Genomic libraries were sequenced on a PacBio RSII instrument using 30 P6-C4 flow cells, generating 3.47 million reads with a total yield of 30.98 Gb. Genomic libraries for Nanopore sequencing were prepared using the one-dimensional Genomic DNA by a ligation library construction kit (SQK-LSK109) and sequenced on two R9.4 flow cells using the MinION desktop sequencer (Oxford Nanopore Technologies Ltd., Oxford, UK). This approach generated 1.41 million reads with a total yield of 11.66 Gb (table S3). Base calling was performed with Albacore v2.3.1. Illumina libraries were constructed from WT and PSR-carrying samples using the NEBNext Ultra II DNA Library Prep Kit [New England Biolab (NEB), #E7645] and sequenced on Illumina HiSeq2500 in paired-end mode with the read length of 250 nucleotides (nt) and a sequencing depth of 30 million reads per sample. PacBio reads were assembled using Canu v1.6 (36) and polished with Quiver (Pacific Biosciences Inc.). The PacBio assembly was further scaffolded with Nanopore reads using npScarf v1.7-05b (37) and polished with Pilon v1.22 (38) using Illumina sequencing reads. PSR-specific scaffolds were identified by mapping Illumina reads generated from WT and PSR+ genomic DNA and calculating using the chromosome quotient, which is a normalized ratio of the number of WT and PSR+ reads mapping to a scaffold (table S2 and fig. S1) (21). The resulting assembly comprised 444 scaffolds with an N50 of 6.60 Mb and a total length of 297.31 Mb. One-hundred twenty of these scaffolds were PSR specific, with an N50 of 124 kb and a total length of 9.2 Mb (table S3).
Genetic map placement
Genetic markers developed based on differential hybridization of species-specific oligos between N. vitripennis and Nasonia giraulti (22) were used to place scaffolds on the genetic map. Seventy-three scaffolds totaling 272,286,400 bp (91.58%) were assigned to five linkage groups. Of these scaffolds, 13 (159,467,447 bp; 53.64%) were properly oriented, while 60 (112,818,953 bp; 37.95%) were located in pericentromeric regions without recombination between markers and were placed without orientation or precise relative position (fig. S2 and data S1). Genome completeness was assessed using the BUSCO pipeline (table S1) (20). Repetitive elements were discovered and masked by using RepeatModeler and RepeatMasker (23, 24). Furthermore, the current National Center for Biotechnology Information (NCBI) and OGS2 gene models (39) were mapped to our assembly (data S2 and S3, respectively).
Genome annotation with Nanopore RNA-seq
PSR+ testis and whole-animal RNA-seq libraries for Nanopore sequencing were constructed with the cDNA-PCR Sequencing Kit (SQK-PCS109) and sequenced on two R9.4 flow cells for 48 hours each using the MinION desktop sequencer (Oxford Nanopore Technologies Ltd., Oxford, UK). Base calling was performed with Guppy v3.0.3 using the high-accuracy mode. The libraries generated 16.05 and 11.43 million reads with yields of 12.16 and 12.01 Gb, respectively (table S4). Illumina RNA-seq libraries were constructed using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB, #E7770) and sequenced on Illumina HiSeq2500 in paired-end mode with a read length of 100 nt to the sequencing depth of 30 million reads each. Full-length cDNA reads were identified with Pychopper (Oxford Nanopore Technologies) and aligned to the genome with Minimap2 (40). The Pinfish pipeline (Oxford Nanopore Technologies) was used to generate gene models supported by at least 10 full-length aligned Nanopore reads. The Pinfish pipeline generated 18,593 transcripts corresponding to 11,482 genes (data S6). Sixty-eight transcripts (44 genes) are located on PSR-specific scaffolds. Nanopore reads were remapped to Pinfish transcripts with Minimap2, and transcript expression levels were quantified using Salmon (41). Illumina RNA-seq data were quantified against Pinfish transcripts with FeatureCounts (42).
Expression of PSR candidate genes in early development, carcass and testes
Twenty females from the AsymC strain were individually mated to a PSR male and given one Sarcophaga bullata pupa every other day for 4 days. Thereafter, these females were allowed to oviposit for 1 hour at 25°C. Five replicates of 30 embryos per time point were collected in TRIzol reagent (Invitrogen, USA) and stored at −20°C until extraction. For the 2- and 15-hour time points, embryos were incubated, respectively, 1 and 14 hours before collection to reach the right developmental time. Testes and carcasses were dissected from adult PSR+ males at 1 day after eclosion. Five replicates with three pairs of testes each and five replicates with three PSR carcasses each were collected in TRIzol reagent and stored at −20°C until extraction. Total RNA was extracted from each sample with TRIzol reagent according to the manufacturer’s instructions. Each sample was subjected to deoxyribonuclease treatment to eliminate any contaminating DNA, and approximately 1 μg of total RNA was reverse transcribed with oligo-dT and hexamer primers in a 1:6 ratio with the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD, USA). The cDNA was then diluted 30-fold before being used for qPCR. qPCR was performed with SYBR Green (Genesee Scientific). Diluted cDNA (4 μl) was used for each reaction of 20 μl of total containing primers at a final concentration of 0.2 μM and 10 μl of SYBR Green buffer solution. Three technical replicates for each reaction were performed to correct for experimental errors. Elongation factor 1α (ef1α) and adenylate kinase 3 (ak3) were used as reference genes for normalization of the data after the confirmation that their expression levels were constant throughout development and between tissues (fig. S11) (43). Reactions were run on a Roche LightCycler System with the following qPCR profile: 120-s activation phase at 95°C, 40 cycles of 15 s at 95°C, and 30 s at 62°C. The primers are listed in table S9. Expression levels of the three PSR genes relative to the reference genes were calculated by normalizing the expression data with LightCycler 96 software. Gene expression between samples was then compared by two-way analysis of variance (ANOVA) and Tukey HSD (Honestly Significant Difference) for the multiple comparison test in R statistical software (R Development Core Team 2012).
Parental RNAi of PSR candidate genes
RNAi knockdown of the three candidate genes was induced in early PSR male pupae by microinjecting double-stranded RNA (dsRNA) that is complementary to the candidate PSR genes. Fragments of 660, 400, and 601 bp were produced for the genes PSR-4317, PSR-1539, and PSR-tra, respectively. A T7 promoter was placed at either the 5′ or 3′ end of a given template PCR fragment using the primer shown in table S10. Once produced, these fragments were transcribed in each direction separately using the Megascript RNAi Kit (Ambion, Austin, Texas, USA). After 6 hours of incubation at 37°C, the two reactions were mixed and heated at 75°C for 5 min, followed by a slow cool down overnight. Exonuclease digestion was conducted to remove DNA and single-stranded RNA, and the dsRNA was subsequently purified according to the protocol kit. Last, dsRNAs were precipitated with ethanol, redissolved in water, and stored at −20°C.
PSR+ male pupae were injected in the abdomen following a previously described procedure (30), either with dsRNA (4 μg/μl) for PSR-4317, PSR-1539, or PSR-tra that was mixed with red dye. Injections were performed under continuous injection flow with the Femtojet Express system (Eppendorf) using aluminosilicate glass filaments pulled with a Sutter Instrument. Pupae were injected at their posterior ends until the abdomen turned pink. Slides with injected wasp pupae were incubated in an agar/phosphate-buffered saline (PBS) petri dish at 25°C. Control pupae were injected with red dye mixed with water in a 1:4 ratio. After emergence, each adult male was mated individually with one WT female for 24 hours and then stored at −80°C for further processing.
Genetic crosses
Females mated with RNAi-treated males were hosted with two S. bullata pupae and allowed to oviposit for 2 days. After oviposition, females were allowed to lay embryos into host pupae for fluorescence in situ hybridization (FISH) experiments (see below); host pupae were incubated at 25°C. Ten days later, the hosts were opened, and Nasonia pupae were scored by sex to assess the proportion of G1 females. DNA extracted from 35 G1 females produced by RNAi-treated fathers was tested for the presence of PSR by using PCR with PSR-specific primers (fig. S5A).
To test whether PSR could be transmitted by G1 females and males, about 35 PSR+ G1 females and 35 males PSR+ were singularly crossed with WT wasps of the opposite sex. The proportion of G2 female offspring was scored 10 days after oviposition (table S9). The presence of PSR in the subsequent generations was assessed by testing individual progeny using PCR- with PSR-specific primers (table S10).
Expression analysis of PSR genes after RNAi
To assess the efficiency of RNAi knockdown, total RNA was extracted from control and RNAi injected males (one male per sample), and cDNA conversion was performed as described above. Expression data were first analyzed as described above using Ef1α and ak3as reference genes. Relative expression levels of the three PSR genes to the reference genes were calculated by normalizing the expression data with LightCycler 96 software. Gene expression between samples was then compared by two-way ANOVA and Tukey HSD with the multiple comparison test in R statistical software (R Development Core Team 2012).
Embryo collection and fixation
WT-mated females, crossed with RNAi-treated PSR+ males, were allowed to oviposit into individual blowfly pupae for a certain length of time. Specifically, a 45-min to 1-hour laying time was used for obtaining embryos at the time of development between fertilization and immediately before the first mitotic division, whereas a laying time of 1 hour 15 min was used for obtaining embryos between the first mitotic division and until the second interphase. Embryos were carefully removed from host pupae with ultrafine forceps and placed into a 10-ml screw top glass vial. The following solutions were quickly placed into the vial with embryos in the following order: 3 ml of heptane, 1.5 ml of 1× PBS, and 600 μl of 37% formaldehyde. The vial with embryos in fixative was placed onto a platform rocker, and the embryos were fixed for exactly 28 min. Following this time, embryos were removed from the fixative with a micropipette and placed onto a small piece of Whatman paper and allowed to dry for ~30 s to 1 min. The embryos were lightly pressed onto double-sided adhesive tape secured to the surface of a clean 22-mm plastic petri dish. To hydrate the embryos, 1.5 ml of 1× PBT (1× PBS with 0.1%Triton X-100) was then placed into the petri dish. Subsequently, the embryos were carefully devitellinized under a dissecting microscope by using a 28-gauge hypodermic needle. The devitellinized embryos were transferred to a 0.6-ml microfuge tube, washed three times with 1× PBT, and stored at 4° to 6°C before staining.
DNA fluorescence in situ hybridization (FISH)
A small, single-stranded DNA (ssDNA) probe used to detect PSR in fixed embryos was based on the following sequence that is exclusive to the PSR chromosome: 5′-CACTGAAAACCAGAGCAGCAGTTGAGA-3′. The telomere was detected by using an ssDNA probe with the following sequence: 5′-TTATTGGGTTATTGGGTTATTGGGTTATTG-3′. The rDNA locus was marked with a cocktail of the following two ssDNA probes, each recognizing a different part of the N. vitripennis 18S Intergenic Spacer IGS repeat: 5′-TTAGACTTTTTCGAGCCTCCGAGA-3′ and 5′-ATTGACGCTCGCACATCACTCATT-3′. These probes were chemically synthesized by IDT Inc. (USA) and fluorescently labeled at their 5′ ends with Alexa-488, Cy3, or Cy5. Before DNA FISH, immunostained embryos were postfixed in the dark for 45 min in 4% paraformaldehyde and then washed three times in 2× saline-sodium citrate and Tween 20. From this point, whole mount DNA FISH was conducted exactly as previously described (44). For DNA FISH of squashed sperm, we followed a previously described protocol (45) using testis from adult males as a tissue source.
Immunostaining
To visualize H3K9me3 and H4K20me1, we stained fixed embryos with primary antibodies (rabbit anti-H3K9me3 and mouse anti-H4K20me1, Active Motif Inc.) diluted at 1:500 in 1× PBT overnight at 4°C on a platform rocker. Embryos were then washed three times at 10 min each with 1× PBT and then stained with fluorescently conjugated secondary antibodies at room temperature for 1 hour on a platform rocker in the dark. Secondary antibodies used in this study were anti-rabbit Cy3 and anti-mouse Cy5 (both at 1:300; Invitrogen, Thermo Fisher Scientific Inc., USA). Embryos were then washed as stated above and then mounted on a slide with VECTASHIELD mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., USA).
Confocal microscopy and image processing
Fluorescence microscopic imaging was conducted with a Leica TCS SPE confocal microscope. Images were collected as Z-series for each laser channel and subsequently merged for visual capture of cellular features within the same nucleus that were not in the same focal plane. Merged images were exported in the highest quality JPEG format and processed with Adobe Photoshop CS5 v.12.
Supplementary Material
Acknowledgments
We thank V. Kumar for help with genomic library preparations. Sequencing was performed at the Millard and Muriel Jacobs Genetics and Genomics Laboratory at the California Institute of Technology. Funding: This work was supported in part by funding from generous UCSD laboratory startup funds awarded to O.S.A. and NSF funding awarded to P.M.F. (NSF-1451839). Author contributions: O.S.A. and P.M.F. conceptualized the study. E.D.B., I.A., T.Y., P.M.F., and H.Q.M.N. performed the various genetic, cytological, and molecular experiments and the bioinformatic analyses in the study. All authors contributed to the writing, analyzed the data, and approved the final manuscript. Competing interests: The authors declare that they have no significant competing financial, professional, or personal interests that might have influenced the performance or presentation of the work described. Data and materials availability: All Illumina, PacBio, and Oxford Nanopore sequencing data can be found at NCBI under accession number PRJNA575073 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA575073?reviewer=uslhbl0iop93gab77lb31ts0n). Previously published RNA-seq data (18) were also used in this study. They can be found under the accession number PRJNA212516 (www.ncbi.nlm.nih.gov/bioproject/PRJNA212516). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz9808/DC1
REFERENCES AND NOTES
- 1.Werren J. H., Nur U., Eickbush D., An extrachromosomal factor causing loss of paternal chromosomes. Nature 327, 75–76 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Nur U., Werren J. H., Eickbush D. G., Burke W. D., Eickbush T. H., A “selfish” B chromosome that enhances its transmission by eliminating the paternal genome. Science 240, 512–514 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Jones R. N., B chromosomes in plants. New Phytol. 131, 411–434 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Camacho J. P., Sharbel T. F., Beukeboom L. W., B-chromosome evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 163–178 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones R. N., B-chromosome drive. Am. Nat. 137, 430–442 (1991). [Google Scholar]
- 6.Dalla Benetta E., Akbari O. S., Ferree P. M., Sequence expression of supernumerary B chromosomes: Function or fluff? Genes 10, 123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurst G. D. D., Werren J. H., The role of selfish genetic elements in eukaryotic evolution. Nat. Rev. Genet. 2, 597–606 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Kimura M., Kayano H., The maintenance of super-numerary chromosomes in wild populations of Lilium callosum by preferential segregation. Genetics 46, 1699–1712 (1961). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werren J. H., Beukeboom L. W., Population genetics of a parasitic chromosome: Theoretical analysis of PSR in subdivided populations. Am. Nat. 142, 224–241 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Werren J. H., Stouthamer R., PSR (paternal sex ratio) chromosomes: The ultimate selfish genetic elements. Genetica 117, 85–101 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Reed K. M., Werren J. H., Induction of paternal genome loss by the paternal-sex-ratio chromosome and cytoplasmic incompatibility bacteria (Wolbachia): A comparative study of early embryonic events. Mol. Reprod. Dev. 40, 408–418 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Aldrich J. C., Leibholz A., Cheema M. S., Ausiό J., Ferree P. M., A “selfish” B chromosome induces genome elimination by disrupting the histone code in the jewel wasp Nasonia vitripennis. Sci. Rep. 7, 42551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldrich J. C., Ferree P. M., Genome silencing and elimination: insights from a “selfish” B chromosome. Front. Genet. 8, 50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swim M. M., Kaeding K. E., Ferree P. M., Impact of a selfish B chromosome on chromatin dynamics and nuclear organization in Nasonia. J. Cell Sci. 125, 5241–5249 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Eickbush D. G., Eickbush T. H., Werren J. H., Molecular characterization of repetitive DNA sequences from a B chromosome. Chromosoma 101, 575–583 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Beukeboom L. W., Werren J. H., Deletion analysis of the selfish B chromosome, Paternal Sex Ratio (PSR), in the parasitic wasp Nasonia vitripennis. Genetics 133, 637–648 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akbari O. S., Antoshechkin I., Hay B. A., Ferree P. M., Transcriptome profiling of Nasonia vitripennis testis reveals novel transcripts expressed from the selfish B chromosome, paternal sex ratio. G3 3, 1597–1605 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Jing X. A., Aldrich J. C., Clifford C., Chen J., Akbari O. S., Ferree P. M., Unique sequence organization and small RNA expression of a “selfish” B chromosome. Chromosoma 126, 753–768 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Werren J. H., Richards S., Desjardins C. A., Niehuis O., Gadau J., Colbourne J. K.; Nasonia Genome Working Group , Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Hall A. B., Qi Y., Timoshevskiy V., Sharakhova M. V., Sharakhov I. V., Tu Z., Six novel Y chromosome genes in Anopheles mosquitoes discovered by independently sequencing males and females. BMC Genomics 14, 273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desjardins C. A., Gadau J., Lopez J. A., Niehuis O., Avery A. R., Loehlin D. W., Richards S., Colbourne J. K., Werren J. H., Fine-scale mapping of the Nasonia genome to chromosomes using a high-density genotyping microarray. G3 3, 205–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.A. F. A. Smit, R. Hubley, RepeatModeler Open-1.0 (2008–2015); www.repeatmasker.org.
- 24.A. F. A. Smit, R. Hubley, P. Green, RepeatMasker Open-4.0 (2013–2015); www.repeatmasker.org.
- 25.Vítková M., Král J., Traut W., Zrzavý J., Marec F., The evolutionary origin of insect telomeric repeats, (TTAGG)N. Chromosome Res. 13, 145–156 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Hanlon S. L., Miller D. E., Eche S., Scott Hawley R., Origin, composition, and structure of the Supernumerary B chromosome of Drosophila melanogaster. Genetics 210, 1197–1212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marques A., Klemme S., Houben A., Evolution of plant B chromosome enriched sequences. Genes 9, 515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jetybayev I. Y., Bugrov A. G., Dzuybenko V. V., Rubtsov N. B., B chromosomes in grasshoppers: Different origins and pathways to the modern Bs. Genes 9, 509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAllister B. F., Werren J. H., Hybrid origin of a B chromosome (PSR) in the parasitic wasp Nasonia vitripennis. Chromosoma 106, 243–253 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Lynch J. A., Desplan C., A method for parental RNA interference in the wasp Nasonia vitripennis. Nat. Protoc. 1, 486–494 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Verhulst E. C., Lynch J. A., Bopp D., Beukeboom L. W., van de Zande L., A new component of the Nasonia sex determining cascade is maternally silenced and regulates transformer expression. PLOS ONE 8, e63618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banaei-Moghaddam A. M., Meier K., Karimi-Ashtiyani R., Houben A., Formation and expression of pseudogenes on the B chromosome of rye. Plant Cell 25, 2536–2544 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker S. E. D., Duke Becker S. E., Thomas R., Trifonov V. A., Wayne R. K., Graphodatsky A. S., Breen M., Anchoring the dog to its relatives reveals new evolutionary breakpoints across 11 species of the Canidae and provides new clues for the role of B chromosomes. Chromosome Res. 19, 685–708 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Martis M. M., Klemme S., Banaei-Moghaddam A. M., Blattner F. R., Macas J., Schmutzer T., Scholz U., Gundlach H., Wicker T., Šimková H., Novák P., Neumann P., Kubaláková M., Bauer E., Haseneyer G., Fuchs J., Doležel J., Stein N., Mayer K. F. X., Houben A., Selfish supernumerary chromosome reveals its origin as a mosaic of host genome and organellar sequences. Proc. Natl. Acad. Sci. U.S.A. 109, 13343–13346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruban A., Schmutzer T., Scholz U., Houben A., How next-generation sequencing has aided our understanding of the sequence composition and origin of B chromosomes. Genes 8, 294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M., Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao M. D., Nguyen S. H., Ganesamoorthy D., Elliott A. G., Cooper M. A., Coin L. J. M., Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 8, 14515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C. A., Zeng Q., Wortman J., Young S. K., Earl A. M., Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rago A., Gilbert D. G., Choi J.-H., Sackton T. B., Wang X., Kelkar Y. D., Werren J. H., Colbourne J. K., OGS2: Genome re-annotation of the jewel wasp Nasonia vitripennis. BMC Genomics 17, 678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Dalla Benetta E., Beukeboom L. W., van de Zande L., Adaptive differences in circadian clock gene expression patterns and photoperiodic diapause induction in Nasonia vitripennis. Am. Nat. 193, 881–896 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Martin J., Chong T., Ferree P. M., Male killing Spiroplasma preferentially disrupts neural development in the Drosophila melanogaster embryo. PLOS ONE 8, e79368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larracuente A. M., Ferree P. M., Simple method for fluorescence DNA in situ hybridization to squashed chromosomes. J. Vis. Exp. 2015, 52288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz9808/DC1