

Hypertrophic cardiomyopathy–causing mutations disrupt a key regulatory off state of myosin in thick filaments.
Abstract
Hypertrophic cardiomyopathy (HCM) mutations in β-cardiac myosin and myosin binding protein-C (MyBP-C) lead to hypercontractility of the heart, an early hallmark of HCM. We show that hypercontractility caused by the HCM-causing mutation R663H cannot be explained by changes in fundamental myosin contractile parameters, much like the HCM-causing mutation R403Q. Using enzymatic assays with purified human β-cardiac myosin, we provide evidence that both mutations cause hypercontractility by increasing the number of functionally accessible myosin heads. We also demonstrate that the myosin mutation R403Q, but not R663H, ablates the binding of myosin with the C0-C7 fragment of MyBP-C. Furthermore, addition of C0-C7 decreases the wild-type myosin basal ATPase single turnover rate, while the mutants do not show a similar reduction. These data suggest that a primary mechanism of action for these mutations is to increase the number of myosin heads functionally available for interaction with actin, which could contribute to hypercontractility.
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is a prevalent genetic cardiac disease affecting ~1 in every 200 to 500 people (1). HCM results in hypercontractility of the heart, followed by hypertrophy, fibrosis, myocardial disarray, diastolic dysfunction, and sometimes sudden death among HCM patients (1, 2). Because hypercontractility precedes hypertrophy, it is hypothesized that hypercontractility at the molecular scale is a proximal cause of disease. This is supported by the observation that most of the known mutations implicated in genetic HCM are found in two sarcomeric proteins, myosin and myosin binding protein-C (MyBP-C), which together account for ~70% of all known HCM mutations, ~35% in each (3).
Sarcomeric contraction is driven by the cyclic interaction between actin and myosin in the presence of adenosine 5′-triphosphate (ATP). The resultant power generation is equated to the ensemble force generated by myosin heads on actin multiplied by the velocity of actin filaments driven by myosin heads. Ensemble force is the product of the intrinsic force (Fi) generated by a single myosin head on actin, the duty ratio, which is the fraction of time a myosin molecule remains bound to actin, and the total number of functionally accessible myosin heads (Na) available for interaction with the actin filaments (4). It was initially thought that hypercontractility due to HCM-causing mutations might increase power output by increasing the intrinsic force, duty ratio, or velocity of the motor. However, in previous work, we and others have demonstrated that most HCM-causing mutations in myosin appear to primarily increase ensemble force by increasing Na (5–10).
How Na is modulated in response to the dynamic needs of the heart is an open question for investigation. Cardiac myosin has two N-terminal globular head, or motor, domains [subfragment 1 (S1)] and a long coiled-coil tail. The N-terminal ~40% of the tail is called subfragment 2 (S2). The two S1 domains attached to the S2 domain constitute heavy meromyosin (HMM). The heads of myosin in cardiac muscle thick filaments can exist in at least two different states: an open state, which is able to interact with actin, and a closed state, where the myosin heads containing the mesa domains (5) are folded back onto their own S2 tail and are not accessible for interaction with actin. The best structural model of this folded-back sequestered state is the interacting heads motif (IHM) (7, 10–15). These two structural states have been correlated to distinct biochemical adenosine triphosphatase (ATPase) rates: In relaxed muscle (low Ca2+ concentration) where the heads are unable to bind to the tropomyosin.troponin (Tm.Tn)–regulated thin filaments, the open state heads are believed to be turning over ATP at a rate of ~0.03 s−1 [disordered relaxed state (DRX), two orders of magnitude slower than the actin-activated rate of ~3 s−1], while the heads in the IHM state are believed to be in a super relaxed state (SRX) with an even lower ATP turnover of ~0.003 s−1 (16, 17). Thus, SRX is defined as a very slow ATP turnover state of myosin as measured by a fluorescent-ATP turnover assay (16), while IHM is a structural state in which the myosin heads fold back on their own tail, leading to an off state of the thick filament. The SRX and IHM states are correlated, but there can be circumstances where a non-IHM state can also lead to SRX-like rates (17). Thus, an attractive model is that the equilibrium of myosin between the IHM and the open state in cardiac fibers regulates the power generation of the sarcomere by controlling Na (5, 6). This fits with the finding that a large fraction of pathogenic HCM mutations are found on the myosin mesa, proximal S2, the converter domain, and other key intramolecular interaction sites in the IHM structure and are predicted to disrupt the ability of myosin to form the IHM and thus increase Na (5–7, 10, 14, 18).
MyBP-C has been posited to contribute to this regulation of Na via an interaction with myosin. MyBP-C is an elongated multidomain protein consisting of immunoglobulin G (IgG)– and fibronectin-like domains (19). There is one MyBP-C molecule for every three myosin molecules in the C-zone of the myosin thick filament, or about one for every six myosin molecules when considering the entire thick filament (20, 21). Biochemical studies have shown that the N-terminal domains of MyBP-C can interact with actin (22, 23), the myosin regulatory light chain (RLC) (24), and the proximal part of S2 (proximal S2) (25), as well as with short S1 [sS1; the myosin head fragment ending just after the essential light chain (ELC) and therefore missing the RLC] (7). The interaction of MyBP-C with myosin has been postulated to sequester heads into a state not accessible for binding to actin, perhaps the IHM state, thus reducing Na (6, 7). Relatedly, it was shown that mice lacking the cardiac isoform of MyBP-C (cMyBP-C) exhibit a lesser fraction of SRX than wild-type (WT) mice (26). In addition, patient samples from patients with confirmed cMyBP-C mutations also show a decrease in the SRX (27). However, direct biochemical evidence of an effect of MyBP-C on purified myosin molecules has been lacking.
Here, we investigate two myosin HCM mutations, R403Q and R663H, that lie on the myosin mesa surface (5) using a variety of biochemical and biophysical assays to examine the effects of the mutations on the parameters that affect the power output of the motor, including its interaction with MyBP-C. A previous study showed that the R403Q HCM mutation has minimal effects on the basic contractility parameters of human ß-cardiac sS1 that are unlikely to account for the hypercontractility seen clinically (28). Here, we show that this is even more dramatically the case for the R663H HCM mutation, which leads to virtually no changes in the fundamental contractile behaviors of human β-cardiac myosin. Thus, we used biochemical assays to test whether these mutations increase the number of heads functionally accessible for interaction with actin. In a previous study, we showed that these mutations decrease SRX at the level of muscle fibers (17); this study expands on that work by demonstrating that these effects extend to purified myosin and MyBP-C molecules in a tightly controlled system. We found that these mutations cause a marked increase in the proportion of myosin that adopts the DRX in the absence of actin, and that while R663H had no effect on MyBP-C binding, R403Q showed a notable decrease in MyBP-C binding, which may contribute to an overall increase in Na. In addition, we show that MyBP-C increases the percentage of WT myosin in the SRX, but not when the R403Q or R663H mutations are present. To our knowledge, this is the first molecular evidence that purified MyBP-C can act directly on myosin HMM to increase its fraction of SRX heads. Thus, we conclude that the primary mode by which these mutations cause hypercontractility is by increasing the number of heads functionally available for interaction with actin.
RESULTS
The HCM mutation R663H has no effect on fundamental parameters of myosin-based contractility
The R663H HCM mutation is nearly unique among the HCM mutations that have been studied at the molecular level in that it has no significant effect on the fundamental parameters of myosin-based contractility (Fig. 1 and table S1). The kcat of the actin-activated ATPase activity of R663H human β-cardiac sS1 (2.9 ± 0.1 s−1) is nearly identical to that of WT human β-cardiac sS1 (WT, 3.0 ± 0.2 s−1) (Fig. 1A), and the intrinsic force of the mutant and WT forms is within 10% of one another (WT, 1.4 ± 0.1 pN; R663H, 1.5 ± 0.1 pN; Fig. 1B and fig. S1). The velocities in the in vitro motility assay with both actin alone (WT, 820 ± 39 nm/s; R663H, 834 ± 31 nm/s) (Fig. 1C) and with regulated thin filaments at pCa = 4 (Fig. 1D) (WT, 1019 ± 25; R663H, 944 ± 42 nm/s) are not significantly different from the WT human b-cardiac sS1. In addition, the Ca2+ concentration dependence of regulated thin filament velocities of the WT (Fig. 1E) and R663H (Fig. 1F) human ß-cardiac sS1 constructs is nearly identical. Fitting the data with the Hill equation yielded the pCa50 values of 6.3 and 6.4 (obtained from the fitting of data averaged from four different experiments) for WT and R663H sS1, respectively.
Fig. 1. Fundamental contractile parameters of WT (black) and R663H (dark red) human β-cardiac sS1.
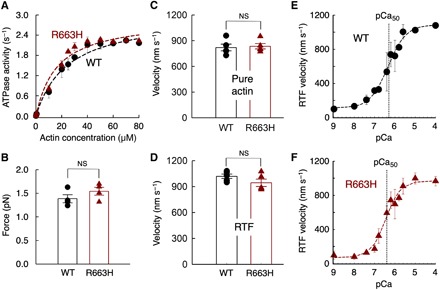
(A) Actin-activated ATPase data for WT and R663H sS1. Fitting the traces yielded kcat values of 3.0 ± 0.2 and 2.9 ± 0.1 s−1 for WT and R663H sS1, respectively. Dashed lines are the fitted lines. Representative data (average of two experiments from single preparations of both proteins) are shown. (B) Intrinsic force measurements of WT (black) and R663H (dark red) human β-cardiac sS1 using an optical trap. The individual force values were averaged from force measurements of four molecules of each motor. (C) Comparison of in vitro motility of actin filaments for WT (black) and R663H (dark red) human β-cardiac sS1 (five different experiments from five independent protein preparations for each of WT and R663H sS1). (D) Comparison of in vitro motility of regulated thin filaments at pCa = 4.0 for WT (black) and R663H (dark red) human β-cardiac sS1 (five different experiments from four independent protein preparations for each of WT and R663H sS1). (E) Ca2+ sensitivity measurements for WT human β-cardiac sS1 (four different experiments were averaged). (F) Ca2+ sensitivity measurements for R663H human β-cardiac sS1 (four different experiments were averaged). The data for (E) and (F) were fitted (dashed lines) to the Hill equation to estimate the pCa50. When each of the four individual experiments was fitted to the Hill equation separately, mean pCa values of 6.3 ± 0.2 and 6.5 ± 0.2 were obtained for WT and R663H, respectively. For all panels, error bars denote SEM. NS, not significant, P > 0.05. (B) P = 0.24; (C) P = 0.78; (D) P = 0.18; (E and F) P = 0.46 (four individual experiments were used for calculation).
Because we did not see changes in the fundamental contractile parameters of myosin, we investigated whether the IHM-forming sS1-S2 interaction was affected by the R663H mutation. The IHM consists of a blocked head (the actin-binding domain is blocked) and a free head (the actin-binding domain is available). The blocked head interacts with proximal S2 and with the free head. The positions of residue Arg663 (light blue) on the IHM homology model of human β-cardiac myosin constructed by Robert-Paganin et al. (14) are shown on the blocked head mesa (dark red) and free head mesa (light brown) in Fig. 2A. The blocked head Arg663 (bh R663) is near but not interacting with the proximal S2, and the free head Arg663 (fh R663) is near the interface of a head-head interaction site (Fig. 2, A and B). Given the closeness of Arg663 to the proximal S2 in the model, we measured the KD for binding of proximal S2 to the WT (62 ± 8 μM obtained from six different measurements of two different protein preparations; representative data shown in Fig. 2C) and R663H (58 ± 4 μM obtained from six different measurements of two different protein preparations; representative data shown in Fig. 2D) human β-cardiac sS1. They were not significantly different (P = 0.62). This is consistent with the IHM model, where Arg663 is not in direct contact with proximal S2 (Fig. 2B). We previously demonstrated that R403Q human β-cardiac sS1, which is also not in direct contact with proximal S2 in the IHM model, did not show a change in KD for binding to proximal S2 (7). In contrast, Arg249, His251, and Arg453 are all directly underneath the proximal S2 in the IHM model (Fig. 2B), and the HCM mutations R249Q, H251N, and R453C all significantly weaken the binding affinity of human ß-cardiac sS1 to proximal S2 (7, 29).
Fig. 2. The affinity of WT and R663H human ß-cardiac sS1 binding to proximal S2.
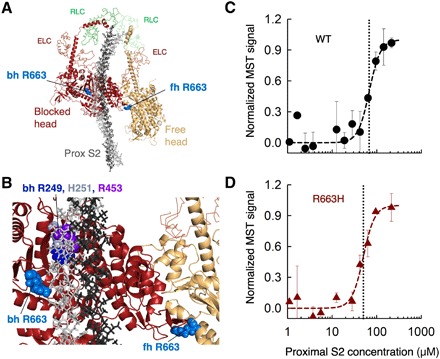
(A) Structural model of the IHM state for human β-cardiac myosin [human sequestered state model; from Robert-Paganin et al. (14)]. The S1 portion of the heavy chain is shown in cartoon mode (PyMOL), the light chains are in ribbon mode, and the proximal S2 is in stick mode. The positions of the blocked head (bh) and free head (fh) Arg663 residues are shown as spheres in light blue. (B) Blowup of the IHM model showing the relationships between the Arg663 residues and the proximal S2 position and the head-head interaction zone. Bh R249 (blue), H251 (gray), and R453 (purple) are shown for reference. (C) MST binding data for WT human ß-cardiac sS1 and proximal S2 in 100 mM KCl. (D) MST binding data for R663H human ß-cardiac sS1 and proximal S2in 100 mM KCl. Data in (C) and (D) are representative data from two measurements from a single set of protein preparations.
We conclude that the hypercontractility caused by the HCM mutation R663H, like R403Q (28), cannot be explained by changes in ATPase, velocity or intrinsic force, or the direct binding interaction between the sS1 domain and proximal S2. We therefore explored whether an alternate mechanism could be that the R663H and R403Q mutations release myosin heads from their SRX, which would provide more heads available for interaction with actin and thereby cause the hypercontractility seen clinically.
The HCM mutations R403Q and R663H decrease the percentage of SRX in preparations of human b-cardiac 25-hep HMM
One hallmark of the ability for a population of myosin molecules to form the IHM is a reduction in the apparent kcat of the actin-activated ATPase activity of 25-hep HMM (which contains proximal S2; fig. S2) compared to that of 2-hep HMM (which does not contain proximal S2; fig. S2) (30). Thus, WT human β-cardiac 25-hep HMM (kcat = 1.4 ± 0.1 s−1) has 58 ± 3% of the ATPase activity of WT 2-hep HMM (kcat = 2.4 ± 0.1 s−1) (table S2) (7, 30). Both R403Q and R663H human ß-cardiac 2-hep HMM have the same activity as the WT 2-hep HMM (R403Q, 2.7 ± 0.2 s−1; R663H, 2.8 ± 0.2 s−1) (Fig. 3, A and B). The 25-hep HMM versions of the R403Q and R663H human β-cardiac myosins do not show the same ~40% decrease in activity compared to the 2-hep versions seen for the WT construct (Fig. 3, A and B, and table S2). Both the R403Q and R663H human β-cardiac 25-hep HMMs show higher activity (R403Q, 2.2 ± 0.2 s−1; R663H, 1.9 ± 0.1 s−1) than the WT 25-hep HMM (1.4 ± 0.1 s−1) (30), suggesting that these mutations cause heads to be released from the IHM state. These results suggest that there is a proximal S2–dependent stabilization of the IHM for WT human ß-cardiac 25-hep HMM that is destabilized by the presence of either the R403Q or R663H mutation. Because our results did not show a mutation-dependent change in the direct binding of the myosin head to the S2 tail, we speculate that these effects may be a result of a weakening of the interaction between the blocked head with the free head in the IHM structure, where the bh R403 and fh R663 residues may play a role (Fig. 3, G and H). Thus, these results suggest that appropriate interaction of the blocked and free heads depends on the presence of proximal S2 to form the IHM structure.
Fig. 3. Functional assays for ATP turnover by WT, R403Q, and R663H human b-cardiac 25-hep HMM and 2-hep HMM.

(A) Actin-activated ATPase of R403Q 2-hep HMM (light blue) and R403Q 25-hep HMM (dark blue). (B) Actin-activated ATPase of R663H 2-hep HMM (orange) and R663H 25-hep HMM (dark red). For (A) and (B), data are combined from two experiments from one protein preparation. Each point is an average, with the error bar as SEM. (C) Fluorescence decay of mant-nucleotide release from WT human ß-cardiac 25-hep HMM in 25 mM KAc (solid black curve). Fitting the traces (dashed cyan curve) to a double-exponential equation yielded the DRX and SRX rates of 0.030 and 0.0034 s−1. The simulated single-exponential orange dashed curve is the SRX rate (0.0034 s−1), and the simulated single-exponential dashed green curve is the DRX rate (0.030 s−1). The simulated orange and green dashed curves act as visual references for the single exponential fits of slow and fast phases, respectively, that derive from fitting the solid black data curves with the best two exponential fits. Thus, the data (black lines) were fit with a combination of these two single exponentials. (D) Fluorescence decay of mant-nucleotide release from R403Q human ß-cardiac 25-hep HMM in 25 mM KAc, where the fast and slow fitting rates were 0.017 and 0.0017 s−1, respectively. (E) Fluorescence decay of mant-nucleotide release from R663H human ß-cardiac 25-hep HMM in 25 mM KAc, where the fast and slow fitting rates were 0.047 and 0.0031 s−1, respectively. (C) and (E) show representative data from one preparation of each protein. (F) Percentage of myosin heads in the SRX (orange) versus DRX (olive green) states calculated from the amplitudes of the double-exponential fits of the fluorescence decays corresponding to the mant-nucleotide release from the 25-hep HMMs shown in (C) to (E). The data are from nine measurements (from six individual protein preparations done on different days) of WT 25-hep HMM, five measurements of R403Q 25-hep HMM (from four individual protein preparations done on different days), and seven measurements of R663H 25-hep HMM (from five individual protein preparations done on different days). Error bars, SEM. **P ≤ 0.01; ****P ≤ 0.0001. WT versus R403Q, P = 0.01; WT versus R663H, P = 0.00003. (G) Homology model of the IHM state of human β-cardiac 25-hep HMM [human sequestered state model from Robert-Paganin et al. (14)] showing the positions of the blocked head (bh) Arg403 residue (dark blue) and the free head (fh) Arg663 residue (light blue) at a head-head interaction site. (H) Blowup of the IHM model showing the positions of the bh Arg403 and fh Arg663 residues at the interface between the blocked head (dark red) and the free head (light brown).
The definition of the SRX is a population of myosin molecules with a reduced level of single turnover basal ATPase rate (16). These rates (DRX, ~0.03 s−1; SRX, ~0.003 s−1) are measured by loading a fluorescent analog of ATP (mant-ATP) onto the myosin, which results in a higher fluorescence, and then chasing with an excess of unlabeled ATP and watching the decrease in fluorescence as the mant-nucleotide dissociates from the myosin (Fig. 3C, WT myosin, black curve; table S3). The decay rate shown in Fig. 3C cannot be fit by a single exponential, indicating a mixture of DRX and SRX turnover rates in the WT human ß-cardiac 25-hep HMM population. The amplitudes provide a measure of the percentages of DRX (58 ± 2%) and SRX (42 ± 2%) in the population at a particular ionic strength (Fig. 3F). In contrast to the WT myosin, R403Q and R663H human ß-cardiac 25-hep HMM showed only 14 ± 7% and 10 ± 5% SRX in the population, respectively (Fig. 3, D to F, and table S3).
We have shown previously that SRX is correlated with formation of a folded-back sequestered state (17), probably the IHM. Thus, the decreased SRX that arises from these mutations suggests that they prevent formation of the IHM, in agreement with the actin-activated ATPase measurements. Together, these data support our hypothesis that bhR403 and fhR663 residues may play a role in stabilizing the IHM structure, and that the HCM mutations at these residues weaken the blocked head interaction with the free head (Fig. 3, G and H), providing more myosin heads for interaction with actin.
R403Q, but not R663H, disrupts the interaction of MyBP-C with human b-cardiac 25-hep HMM
We and others have hypothesized that the N-terminal C0-C2 fragment of MyBP-C binds to a folded state of myosin (7). Previous binding data showed that both full-length MyBP-C and C0-C2 bind to WT sS1 in a phosphorylation-dependent manner (7). Here, we examined the binding of the C0-C7 fragment of human cardiac MyBP-C to human ß-cardiac 25-hep HMM using microscale thermophoresis (MST). We used this longer C0-C7 fragment to account for any and all effects that the myosin mutations may have in changing MyBP-C binding. We have left off the C8-C10 region, as it is known to interact with light meromyosin (LMM) (31, 32).
The KD for binding of R663H 25-hep HMM to C0-C7, as determined by MST by following the HMM enhanced green fluorescent protein (eGFP) fluorescence and titrating with C0-C7, was 1.1 ± 0.2 μM (n = 6 replicates from two independent protein preparations), which was not statistically different from that of WT 25-hep HMM (KD = 1.5 ± 0.1 μM, n = 9 replicates from two independent protein preparations, P = 0.051; Fig. 4A). Similar results were obtained when we used fluorescently labeled C0-C7 and titrated with 25-hep HMM (WT KD = 1.1 ± 0.2 μM, n = 15 replicates from five independent protein preparations; R663H KD = 0.5 ± 0.2 μM, n = 7 replicates from two independent protein preparations; P = 0.085; fig. S3). In contrast, the R403Q 25-hep HMM showed no binding to C0-C7 up to 50 μM C0-C7 (n = 6 independent protein preparations; Fig. 4A). One highly speculative model that fits all the previously known data for binding of C0-C7 to the IHM state of human ß-cardiac myosin is shown in Fig. 4C (only C0-C2 is shown because the position of C3-C7 is uncertain). This is an updated version of our previous model, which is based on existing data about the binding of MyBP-C fragments to myosin (7). In this model, the Arg403 residue on the blocked head of the IHM may interact with the M domain, and the Arg403 residue on the free head may interact with the C2 domain.
Fig. 4. Effect of R403Q and R663H mutations on C0-C7 binding and activity.

(A) Binding of human ß-cardiac 25-hep HMM with the C0-C7 fragment of MyBP-C for WT (black), R663H (dark red), and R403Q (blue). Data points represent the mean at each C0-C7 concentration with the SEM shown. These representative data are the average of five measurements from a single set of protein preparations. (B) Homology model showing the N-terminal residues C0 to C2 of cardiac MyBP-C. (C) Backside view of a structural model of the IHM state for human β-cardiac myosin [human sequestered state model from Robert-Paganin et al. (14)] with C0-C2 bound in the groove between proximal S2 and the mesa of the free head. (D) Fluorescence decay of mant-nucleotide release from WT human b-cardiac 25-hep HMM in the presence of 10 μM C0C7 in 25 mM KAc (solid black curve). Fitting the traces (dashed orange curve) to a single-exponential equation yielded the SRX rate of 0.0052 s−1. (E) Fluorescence decay of mant-nucleotide release from R403Q human ß-cardiac 25-hep HMM in the presence of 10 μM C0C7 in 25 mM KAc, where the fast and slow rates were 0.016 and 0.0027 s−1, respectively. (F) Fluorescence decay of mant-nucleotide release from R663H human b-cardiac 25-hep HMM in the presence of 10 μM C0C7 in 25 mM KAc, where the fast and slow rates were 0.032 and 0.0056 s−1, respectively. The simulated orange and green dashed curves in (E) and (F) act as visual references for the double exponential fits of slow and fast phases, respectively, that derive from fitting the solid black data curves with the best two exponential fits shown in dashed cyan curves. (D) to (F) show representative data from one preparation of each protein. (G) Percentage of myosin heads in the SRX (orange) versus DRX (olive green) calculated from the amplitudes of the double-exponential fits of the fluorescence decays corresponding to the mant-nucleotide release from the 25-hep HMMs in the presence of 10 μM C0C7 shown in (D) to (F). The data are from seven measurements (from four individual protein preparations done on different days) of WT 25-hep HMM, five measurements (from three individual protein preparations done on different days) of R403Q 25-hep HMM, and seven measurements (from three individual protein preparations done on different days) of R663H 25-hep HMM. Error bars, SEM. ****P ≤ 0.0001. WT versus R403Q, P = 0.00006; WT versus R663H, P = 0.00005.
MyBP-C increases the SRX population of WT 25-hep HMM, but not R403Q or R663H 25-hep HMM
The binding of C0-C7 to 25-hep HMM was tested with the SRX assay to understand the functional consequences of its binding. The population of SRX for WT 25-hep HMM increases with the addition of 10 μM C0-C7 (Fig. 4, D and G). WT 25-hep HMM had 42 ± 2% (Fig. 3F and table S3) SRX, which increased to 81 ± 7% (Fig. 4G and table S4) with the addition of 10 μM C0-C7 (P = 0.001 for WT 25-hep HMM with and without C0-C7), indicating that the binding of C0-C7 to myosin leads to an increase in the number of myosin heads with the SRX ATP turnover rate. The amount of SRX remains ~25 to 30% (Fig. 4G and table S4) for both R403Q (Fig. 4E) and R663H (Fig. 4F) in the presence of 10 μM C0-C7. The change in the amount of SRX is statistically insignificant in the absence and presence of C0-C7 for both the mutants (P = 0.31 for R403Q 25-hep HMM with and without C0-C7; P = 0.07 for R663H 25-hep HMM with and without C0-C7). This is expected in the case of the R403Q mutation, which we showed could not bind C0-C7; however, it appears that despite the fact that the R663H mutant remains able to bind to C0-C7, this binding does not significantly increase the SRX population.
DISCUSSION
Both the R403Q (28) and R663H mutations have marginal effects on fundamental parameters of the actin-activated chemomechanical cycle. A previous study, however, showed that the R403Q and R663H mutations destabilize the SRX in functional studies of cardiac fibers, consistent with causing an increase in Na (17). The results we present here complement and build on that work by showing that the R403Q and R663H mutations disrupt the SRX of purified human ß-cardiac 25-hep HMM, again consistent with their causing an increase in Na. Similar results were recently reported for the myosin HCM mutations R249Q, H251N, D382Y, and R719W (30). These data all support a unifying hypothesis that the primary effect of most HCM mutations may be to increase the number of functionally available heads in the sarcomere, leading to the hypercontractility seen clinically. Furthermore, the locations of all these mutations are consistent with the IHM structural model for the folded-back sequestered state of human ß-cardiac myosin. The residue Arg403 is located at a particularly interesting location of the mesa region. It sits at a junction of three surface domains: the actin-binding domain, the mesa, and the primary head-head interaction site (6), the latter two of which are important for IHM formation. The mutation does cause a three- to fivefold increase in the KM for actin binding (table S2), but all other actin interaction parameters studied using sS1 show very little change by the R403Q mutation (28). The location of the R403Q mutation, however, may be expected to affect the stability of the IHM. This is consistent with what we report here. The free head R663H mutation similarly lies at a head-head interaction site within the IHM, and we propose that it therefore weakens the IHM complex.
The possible role of MyBP-C in sequestering heads is a pivotal area of current and future research. Previously, it has been shown that both full-length MyBP-C and the N-terminal fragment C0-C2 can bind to sS1 in a phosphorylation-dependent manner (7). In addition, MyBP-C fragments have been demonstrated to bind the proximal region of S2 and the myosin RLC (24, 25). Here, we demonstrate a tighter binding of a near-native fragment of MyBP-C, C0-C7, to the purified 25-hep HMM, which contains both the myosin head and proximal tail. We also show that the R403Q mutation ablates this binding, while the R663H mutation maintains WT-like binding to C0-C7. In our hypothetical model of a C0-C2-HMM complex (Fig. 4C), both the free and blocked head Arg403 residues are potentially stabilizing the complex, and it therefore might not be surprising that the R403Q mutation so dramatically disrupts the complex. It must be emphasized, however, that this is a highly hypothetical model, and the structure of the MyBP-C–HMM complex is entirely unknown. While we have depicted the C0-C2-HMM complex as an IHM complex, it is entirely possible that MyBP-C is sequestering myosin heads away from interaction with actin by binding the heads in a different folded conformation or even in a more open configuration (33). Thus, a high priority in the field is to obtain even a low-resolution structure of the MyBP-C–HMM complex.
In orthogonal work, ablation or phosphorylation of cMyBP-C has been shown to reduce the SRX population in mouse muscle fibers (26, 34, 35). In addition, patient-derived samples with confirmed MyBP-C mutations have reduced SRX populations (27). Here, we have used a bottom-up approach to show that purified C0-C7 can induce an increased SRX population with purified WT 25-hep HMM, which is consistent with these previous studies. In addition, we have shown that the C0-C7 is unable to induce an increase in SRX when the R403Q and R663H mutations are present in the 25-hep HMM. This implies that these mutants, which reduce the fraction of SRX to nearly the levels of sS1 and 2-hep (17), prevent C0-C7 from inducing SRX rates, despite the fact that R663H is able to bind C0-C7 with a similar affinity to WT. Therefore, binding of C0-C7 is not necessarily indicative of its ability to induce SRX. Instead, because both of these mutations might affect the head-head interface, it is possible that the ability of the heads to interact with one another is an important prerequisite for the MyBP-C to increase the SRX population. Any interaction with MyBP-C would only influence the sequestered population of one-third of the heads in the C-zone, or approximately one-sixth of heads overall (20, 21). Therefore, our results in the absence of MyBP-C would apply to a large fraction of heads in the thick filament and the inability of MyBP-C to increase SRX for either mutant compounds that affect an additional one-sixth of heads.
The potential importance of a folded-back sequestered state of cardiac myosin has recently been highlighted in diseases like HCM, and also in the mechanism of action of cardiac inhibitors like mavacamten (17, 35, 36). The cardiac activator omecamtiv mecarbil has also been shown to stabilize the open state of cardiac myosin while also affecting the kinetics of actin-bound myosin motors (37–39). Thus, further research into the determinants of the sequestered state of myosin and the role of MyBP-C in holding myosin heads in a reserved state can lead to insights into drug discovery for cardiac activators and inhibitors.
MATERIALS AND METHODS
Homology modeling of the human cardiac C0-C2 fragment of MyBP-C
Template identification
The complete MyBP-C protein sequence, which consists of 1274 amino acids and has a calculated molecular weight of 140.78 kDa, was retrieved from the UniProtKB database (http://www.uniprot.org/) (accession number Q14896). Searching the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) (http://www.rcsb.org/) confirmed that the complete tertiary structure of MyBP-C was not publicly available (40). The BLASTP search of the N-terminal region consisting of domains C0-C2 resulted in homologs with low sequence identity and subsequently suboptimal structure from prediction servers. This led us to fragment the sequence, keeping functional regions intact based on SMART (41) and PFAM (42) domain definitions. The fragmentation of the MyBP-C sequence based on functional regions showed some optimal identical structural templates for C0, C1, M-domain, and C2 regions from PDB (40).
Multi-template homology modeling of the N terminus of MyBP-C
The above structural templates were used to construct the homology model of the N terminus of MyBP-C by stitching the structures of C0, C1, M-domain, and C2 regions together. The model was constructed using the multi-template modeling method of the Modeller program (Modeller V9.20) (43). The modeled structures were energy-minimized to convergence using SYBYL software and then finally passed through the validation process using PROSA (44) and Procheck (45).
Expression and purification of human β-cardiac myosin constructs
A modified AdEasy Vector System (Qbiogene Inc.) was used to generate WT and mutant (R403Q and R663H) human β-cardiac constructs as described in detail elsewhere (7, 46, 47). Three different truncated versions of myosin were created: sS1, 2-hep HMM, and 25-hep HMM, consisting of residues 1 to 808, 1 to 855, and 1 to 1016, respectively.
The complementary DNAs (cDNAs) of two different truncated versions of MYH7 (residues 1 to 808), corresponding to an sS1, followed by a flexible GSG (Gly-Ser-Gly) linker were made: (i) a C-terminal eGFP linker or (ii) a C-terminal eight-residue (RGSIDTWV) PDZ-binding peptide. The 2-hep cDNA consists of residues 1 to 855 of MYH7 followed by a GCN4 leucine zipper to ensure dimerization. The GCN4 is followed by an eGFP moiety flanked by flexible GSG linkers and a C-terminal eight-residue (RGSIDTWV) PDZ-binding peptide. The 25-hep HMM construct is similar to the 2-hep HMM construct, except that it has 25 heptad repeats (175 amino acids) of the S2 region (1 to 1016 residues). Human ventricular ELC with an N-terminal FLAG tag (DYKDDDDK) and Tobacco Etch Virus (TEV) protease site was coexpressed with the heavy chain using an adenoviral vector/mouse myoblast C2C12 system. Expression and purification of all three constructs were done as described previously (7).
Purification of C0-C7
A C0-C7 construct with a C-terminal His-tag was codon-optimized for expression in Escherichia coli and was expressed using the pET-21 a (+) expression vector. Cells containing the C0-C7 plasmid were grown in Rosetta (DE3) pLysS cells, induced, and harvested as described by the manufacturer (Qiagen, Germany). The cells were then lysed in a lysis buffer containing 10 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, 1 mM β-mercaptoethanol, leupeptin (0.01 mg/ml), 1 mM phenylmethylsulfonyl fluoride, and Roche cOmplete protease inhibitor cocktail. Lysis was carried out on an emulsiflex or a sonicator, and the lysate was clarified by centrifugation at 35,000g for 35 min. The supernatant was loaded on a nickel–nitrilotriacetic acid (Ni-NTA) column (GE) on a fast protein liquid chromatography. The column was washed with 10 and 15 mM imidazole followed by a linear elution gradient of 15 to 400 mM imidazole. After analyzing the fractions on SDS–polyacrylamide gel electrophoresis (PAGE), pure fractions were pooled together and concentrated using an Amicon 10-kDa molecular weight cutoff filter. These concentrated fractions were then diluted into a buffer containing 10 mM imidazole (pH 7.5), 4 mM MgCl2, and 1 mM dithiothreitol (DTT) and loaded on an ion exchange Q-column (GE). The dilution step was essential to bring down the ionic strength to 60 to 70 mM for efficient attachment of the C0-C7 to the Q-column. The protein was eluted by a 0 to 100% linear NaCl gradient, and the peak fractions were analyzed by SDS-PAGE. Pure fractions were pooled, concentrated, and finally loaded on a preparatory grade Superdex S200 (16/600) size exclusion column. The C0-C7 eluted as a peak from the size exclusion chromatography column with minor shoulder peaks. Only the major peak fractions were collected, analyzed by SDS-PAGE, pooled, and subsequently used for MST experiments. The protein was eluted in a buffer containing 10 mM imidazole (pH 7.5), 100 mM potassium acetate, 4 mM MgCl2, 1 mM EDTA, and 1 mM DTT.
In vitro motility assay
The motility assay was performed as described in detail previously (47, 48). Both WT and mutant protein(s) were purified simultaneously and studied on the same day to minimize variability. All the experiments were performed at 23°C. Glass coverslips (VWR micro cover glass) were coated with a mixture of 0.2% nitrocellulose (Ernest Fullam Inc.) and 0.2% collodion (Electron Microscopy Sciences) dissolved in amyl acetate (Sigma) and air-dried before use. A permanent double-sided tape (Scotch) was used to construct four channels in each slide, and four different experiments were performed on the same slide. Partially inactivated myosin heads in S1 preparations were removed by the “dead-heading” process before performing the motility assay. The process of dead-heading had the following steps: A 10-fold molar excess of F-actin was added to myosin in the presence of 2 mM ATP, the mixture was incubated for 15 min in an ice bucket, 50 mM MgCl2 was added to form F-actin Mg2+-paracrystals and incubated for 5 min, the dead heads bound to actin paracrystals were sedimented at 350,000g for 15 min, the supernatant was collected, and the sS1 concentration was measured using the Bradford reagent (Bio-Rad). Before any experiments, dead-headed sS1 was diluted in 10% ABBSA {assay buffer [AB; 25 mM imidazole (pH 7.5), 25 mM KCl, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT] with bovine serum albumin (BSA; 0.1 mg/ml) diluted in AB}, unless otherwise stated.
For motility experiments using pure actin, reagents were sequentially flowed through the channels in the following order: 10 μl of 4 μM SNAP-PDZ18 diluted in AB and incubated for 3 min, 20 μl of ABBSA to block the surface from nonspecific attachments and incubated for 2 min, 10 μl of a mixture of eight-residue (RGSIDTWV)–tagged human b-cardiac sS1 (~0.05 to 0.1 mg/ml) and incubated for 3 min, 20 μl of AB to wash any unattached proteins, and finally 10 μl of the GO solution [5 to 10 nM tetramethylrhodamine (TMR)–phalloidin (Invitrogen)–labeled bovine actin; 2 mM ATP (Calbiochem); an oxygen-scavenging system consisting of 0.2% glucose, glucose oxidase (0.11 mg/ml; Calbiochem), and catalase (0.018 mg/ml; Calbiochem); and an ATP regeneration system consisting of 1 mM phosphocreatine (Calbiochem) and creatine phosphokinase (0.1 mg/ml; Calbiochem) in ABBSA].
For regulated thin filament (RTF) motility experiments, reagents were sequentially flowed through the channels in the following order: 10 μl of 4 μM SNAP-PDZ18 diluted in AB and incubated for 3 min, 20 μl of ABBSA to block the surface from nonspecific attachments and incubated for 2 min, 10 μl of a mixture of eight-residue (RGSIDTWV)–tagged human ß-cardiac sS1 (~0.05 to 0.1 mg/ml) and incubated for 3 min, 10 μl of AB to wash any unattached proteins, 10 μl of 5 to 10 nM TMR-phalloidin (Invitrogen)–labeled bovine actin and incubated for 3 min, 10 μl of AB buffer to wash, 10 μl of 400 nM tropomyosin. troponin complex and incubated for 6 min, and 10 μl of the GO solution {2 mM ATP (Calbiochem), an oxygen-scavenging system [0.2% glucose, glucose oxidase (0.11 mg/ml; Calbiochem), and catalase (0.018 mg/ml; Calbiochem)], and an ATP regeneration system [1 mM phosphocreatine (Calbiochem) and creatine phosphokinase (0.1 mg/ml; Calbiochem)] in ABBSA}.
For all experiments, movies were obtained using a Nikon Ti-E inverted microscope with an AndoriXon+EMCCD camera (model DU885). All experiments were repeated with at least four different fresh protein preparations. At each condition, at least three different movies with duration of 30 s were recorded. Filament tracking and analysis of movies were performed using the previously published Fast Automated Spud Trekker method, as described earlier (48).
Single-molecule force measurements
The experimental setup of the dual-beam optical trap is described in detail elsewhere (49). All experiments were performed at 23°C. Dead heads of purified myosins were eliminated as described above. The eGFP tag at the C-terminal end of the myosin was used for surface attachment through binding with anti-GFP antibody. The nitrocellulose-coated glass surface of the sample chamber was also coated with 1.5-μm-diameter silica beads that acted as platforms. The steps followed for preparing the chamber were as follows: 20 μl of anti-GFP antibody (~0.01 mg/ml; Abcam) was flowed through the chamber; 20 μl of AB buffer containing BSA (1 mg/ml) (ABBSA buffer) was flowed through to block the exposed surface; the surface was sparsely coated with myosin by flowing through ~50 to 200 pM of human β-cardiac sS1; the chamber was washed with 20 μl of AB buffer; 20 μl of ABBSA buffer containing 250 to 500 pM ATP, TMR-phalloidin–labeled biotin-actin filaments, neutravidin-coated polystyrene beads (Polysciences), and the oxygen-scavenging and ATP regeneration systems described above was flowed through the chamber; and the chamber was sealed with vacuum grease to stop evaporation of the solution. Two neutravidin-coated polystyrene beads (1 μm diameter) were trapped in two different laser beams. Bead-actin-bead assembly, also known as a dumbbell, was formed using the trapped beads bound to each end of a TMR-phalloidin– and biotin-labeled actin filament. The dumbbell was stretched to remove compliance in the actin filament and brought close to the bead pedestal on the surface for interaction with myosin. A trap stiffness of ~0.1 pN/nm was used.
The data collected from individual molecules were analyzed for force measurements, and such experiments were performed using myosins from at least two protein purifications. The number of total force events from individual molecules on average was 20 to 300. We have always observed that the force distribution of all myosin constructs is accompanied with a long tail (fig. S1), as reported previously (49). Each force distribution was fitted to a double-Gaussian function to take account of the smaller population of higher force events in the analysis (fig. S1). The major peak of the fit yielded the intrinsic force of an individual molecule reported here. Such intrinsic force values of multiple molecules were used to calculate the mean force value (Fig. 1B).
MST measurements
A detailed method of MST for determination of the dissociation constant for the binding of two proteins was described previously (7). For all interactions, the unlabeled protein partner was titrated into a fixed concentration of the fluorescently labeled partner (42 nM). Binding experiments between sS1 and proximal S2 were performed as described before (7). We carried out binding experiments between 25-hep HMM and C0-C7 by two methods. In one case, C0-C7 was titrated against 42 nM 25-hep myosin, and in the other, 25-hep HMM was titrated against 42 nM fluorescently labeled C0-C7. Sixteen such serially diluted concentrations of the unlabeled protein partner were prepared to generate one full binding isotherm. 25-hep and C0-C7 binding reactions were carried out in a buffer containing 10 mM imidazole (pH 7.5), 4 mM MgCl2, 1 mM EDTA, 1 mM DTT, 50 mM KAc, 1 mM ATP, and 0.05% Tween 20.
Samples were loaded into NT.115 premium treated capillaries (NanoTemper Technologies) after the reaction was incubated in the dark at 23°C for 30 min. The samples were then mounted in the Monolith NT.115 apparatus (NanoTemper Technologies) for binding measurements. All the data were recorded at 23°C. When myosin was held constant and C0-C7 was titrated, the myosin’s GFP fluorescence was measured by a blue light-emitting diode (LED) at 30% excitation power (blue filter; excitation, 460 to 480 nm; emission, 515 to 530 nm) and IR-Laser power at 60% was used. C0-C7 was fluorescently labeled with a commercial His-tag labeling kit according to the manufacturer’s instructions (NanoTemper Technologies). His-labeled C0-C7 fluorescence was measured by a red LED at 60 to 90% excitation power (red filter; excitation, 605 to 645 nm; emission, 680 to 685 nm) and IR-Laser power at 60 or 80% was used. Data analysis was performed with software NTAffinity Analysis (NanoTemper Technologies), where the binding isotherms were derived from the raw fluorescence data. At least two independent measurements each from at least two different preparations of protein were carried out in each case. Representative binding curves are shown in Figs. 2 and 4.
Actin-activated ATPase assay
All the experiments were performed using freshly prepared myosin at 23°C as described previously (7). WT and R663H sS1, 2-hep HMM, and 25-hep HMM mutant proteins were prepared simultaneously to minimize the effect of preparation variability. Myosin concentration was measured using eGFP absorbance. G-actin was prepared as described previously (7). F-actin free of ATP was prepared by extensively dialyzing G-actin into ATPase buffer to remove any residual ATP. Actin concentration was then measured using absorbance at 290 nm in a spectrophotometer. This eliminates any contribution from the nucleotide at 280 nm.
The steady-state actin-activated ATPase activities of the human β-cardiac myosin preparations were determined using a colorimetric assay to measure inorganic phosphate production at various time points (0 to 30 min) from a solution containing myosin (0.01 mg/ml), ATP and increasing amounts of actin filaments (0 to 100 μM), and ATPase buffer [10 mM imidazole (pH 7.5), 5 mM KCl, 1 mM DTT, and 3 mM MgCl2]. The time-dependent rate for each actin concentration was calculated by fitting the phosphate signal as a function of time to a linear function. The slope was then converted to activity units normalized to a single myosin head. The kcat was extracted from the data by fitting the activity at each actin concentration to the Michaelis-Menten equation to determine maximal activity using the curve fitting toolbox in MatLab. The errors in the fitted values were determined using 100 bootstrap iterations.
Single turnover experiments
Single turnover experiments and the analysis of data were described in detail previously (17). A plate-based fluorescence instrument (Tecan model, Infinite M200 PRO) was used to collect fluorescence data. Single turnover experiments were performed in a 96-well plate (Greiner polypropylene microplate). Experiments were performed with three different human ß-cardiac myosin constructs: WT, R403Q, and R663H 25-hep HMM. Myosin (200 nM) in a buffer containing 10 mM tris (pH 7.5), 4 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 25 mM KAc was mixed with 2′-(or-3′)-O-(N-methylanthraniloyl) adenosine 5′-triphosphate (mant-ATP, Thermo Fisher Scientific) at a final concentration of 200 nM. After 10 s, 4 mM ATP was added, followed by measuring the fluorescence signal at 470 nm after excitation at 405 nm for every ~2 s for 16 min. The fluorescence signals at time = 0 s and time = infinite were used to normalize the kinetic traces. Separate experiments were performed to get the fluorescence signal at time = 0 s by mixing 200 nM myosin and 200 nM mant-ATP and collecting the fluorescence signal. The fluorescence signal at time = infinite was obtained from the fluorescence measurement of 200 nM mant-ADP. The kinetic traces fitted to a biexponential decay function yielded the amplitudes and rates of fast (DRX rate, ~0.03 s−1) and slow (SRX rate, ~0.003 s−1) phases. For experiments involving C0-C7, 10 μM C0-C7 was preincubated with 200 nM 25-hep HMM for 15 min before adding 200 nM mant-ATP and chasing with 4 mM ATP. The rates are reported with the mean and its standard error. All experiments were performed with at least three fresh myosin purifications for each construct.
Supplementary Material
Acknowledgments
We thank C. Liu and other members of Spudich laboratory for their comments on the paper. Funding: This work was funded by NIH grants GM33289 and HL117138 to J.A.S. D.V.T. is supported by a Stanford Lucile Packard CHRI Postdoctoral Award (UL1 TR001085) and an American Heart Association Postdoctoral Fellowship (17POST33411070). M.M.M. was supported by the Stanford Cellular and Molecular Biology training grant. A.S.A. was support by a Stanford Lucile Packard CHRI Postdoctoral Award (UL1 TR001085), Stanford CVI Postdoctoral Award, Stanford ChEM-H Postdocs at the Interface Award, and American Heart Association Postdoctoral Fellowship (16POST30890005). Author contributions: S.S.S. and K.M.R. purified human ß-cardiac myosins. D.V.T. and M.M.M. purified MyBP-C. S.S.S. performed the experiments for measuring basic contractile parameters of sS1. S.S.S., D.V.T., and M.M.M. performed single ATP turnover and MST binding experiments with purified human ß-cardiac myosins and MyBP-C and analyzed the data. A.S.A. performed actin-activated ATPase assays and the analysis of data. S.N.P. constructed the homology model of the C0-C2 domain of MyBP-C. S.S.S., D.V.T., M.M.M., K.M.R., and J.A.S. wrote the paper. All authors contributed to data analysis/interpretation and editing of the manuscript. Competing interests: J.A.S. is a co-founder of Cytokinetics and MyoKardia, biotechnology companies developing small molecules that target the sarcomere for a variety of diseases, and a member of their scientific advisory boards. K.M.R. is a member of the MyoKardia scientific advisory board. All other authors declare no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaax0069/DC1
REFERENCES AND NOTES
- 1.Semsarian C., Ingles J., Maron M. S., Maron B. J., New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Harvey P. A., Leinwand L. A., The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konno T., Chang S., Seidman J. G., Seidman C. E., Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 25, 205–209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spudich J. A., Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spudich J. A., The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 43, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trivedi D. V., Adhikari A. S., Sarkar S. S., Ruppel K. M., Spudich J. A., Hypertrophic cardiomyopathy and the myosin mesa: Viewing an old disease in a new light. Biophys Rev. 10, 27–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nag S., Trivedi D. V., Sarkar S. S., Adhikari A. S., Sunitha M. S., Sutton S., Ruppel K. M., Spudich J. A., The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 24, 525–533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhikari A. S., Trivedi D. V., Sarkar S. S., Song D., Kooiker K. B., Bernstein D., Spudich J. A., Ruppel K. M., β-Cardiac myosin hypertrophic cardiomyopathy mutations release sequestered heads and increase enzymatic activity. Nat. Commun. 10, 2685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore J. R., Leinwand L., Warshaw D. M., Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alamo L., Ware J. S., Pinto A., Gillilan R. E., Seidman J. G., Seidman C. E., Padrón R., Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 6, e24634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendt T., Taylor D., Trybus K. M., Taylor K., Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. U.S.A. 98, 4361–4366 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodhead J. L., Zhao F.-Q., Craig R., Egelman E. H., Alamo L., Padrón R., Atomic model of a myosin filament in the relaxed state. Nature 436, 1195–1199 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Al-Khayat H. A., Kensler R. W., Squire J. M., Marston S. B., Morris E. P., Atomic model of the human cardiac muscle myosin filament. Proc. Natl. Acad. Sci. U.S.A. 110, 318–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert-Paganin J., Auguin D., Houdusse A., Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat. Commun. 9, 4019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoghbi M. E., Woodhead J. L., Moss R. L., Craig R., Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc. Natl. Acad. Sci. U.S.A. 105, 2386–2390 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooijman P., Stewart M. A., Cooke R., A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys J. 100, 1969–1976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson R. L., Trivedi D. V., Sarkar S. S., Henze M., Ma W., Gong H., Rogers C. S., Gorham J. M., Wong F. L., Morck M. M., Seidman J. G., Ruppel K. M., Irving T. C., Cooke R., Green E. M., Spudich J. A., Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. U.S.A. 115, E8143–E8152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homburger J. R., Green E. M., Caleshu C., Sunitha M. S., Taylor R. E., Ruppel K. M., Metpally R. P. R., Colan S. D., Michels M., Day S. M., Olivotto I., Bustamante C. D., Dewey F. E., Ho C. Y., Spudich J. A., Ashley E. A., Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. U.S.A. 113, 6701–6706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris S. P., Lyons R. G., Bezold K. L., In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 108, 751–764 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craig R., Offer G., The location of C-protein in rabbit skeletal muscle. Proc. R. Soc. Lond. B Biol. Sci. 192, 451–461 (1976). [DOI] [PubMed] [Google Scholar]
- 21.Bennett P., Craig R., Starr R., Offer G., The ultrastructural location of C-protein X-protein and H-protein in rabbit muscle. J. Muscle Res. Cell Motil. 7, 550–567 (1986). [DOI] [PubMed] [Google Scholar]
- 22.Shaffer J. F., Kensler R. W., Harris S. P., The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 284, 12318–12327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kensler R. W., Shaffer J. F., Harris S. P., Binding of the N-terminal fragment of C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 174, 44–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ratti J., Rostkova E., Gautel M., Pfuhl M., Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 286, 12650–12658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruen M., Gautel M., Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 286, 933–949 (1999). [DOI] [PubMed] [Google Scholar]
- 26.McNamara J. W., Li A., Smith N. J., Lal S., Graham R. M., Kooiker K. B., van Dijk S. J., Dos Remedios C. G., Harris S. P., Cooke R., Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 94, 65–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNamara J. W., Li A., Lal S., Bos J. M., Harris S. P., van der Velden J., Ackerman M. J., Cooke R., Dos Remedios C. G., MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLOS ONE 12, e0180064 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nag S., Sommese R. F., Ujfalusi Z., Combs A., Langer S., Sutton S., Leinwand L. A., Geeves M. A., Ruppel K. M., Spudich J. A., Contractility parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci. Adv. 1, e1500511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adhikari A. S., Kooiker K. B., Sarkar S. S., Liu C., Bernstein D., Spudich J. A., Ruppel K. M., Early-onset hypertrophic cardiomyopathy mutations significantly increase the velocity, force, and actin-activated ATPase activity of human β-cardiac myosin. Cell Rep. 17, 2857–2864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.A. S. Adhikari, D. V. Trivedi, S. S. Sarkar, D. Song, K. B. Kooiker, D. Bernstein, J. A. Spudich, K. M. Ruppel, Hypertrophic cardiomyopathy mutations at the folded-back sequestered β-cardiac myosin S1-S2 and S1-S1 interfaces release sequestered heads and increase myosin enzymatic activity. bioRxiv 537159 [Preprint]. 31 January 2019. 10.1101/537159. [DOI] [PMC free article] [PubMed]
- 31.Miyamoto C. A., Fischman D. A., Reinach F. C., The interface between MyBP-C and myosin: Site-directed mutagenesis of the CX myosin-binding domain of MyBP-C. J. Muscle Res. Cell Motil. 20, 703–715 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Flashman E., Watkins H., Redwood C., Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem. J. 401, 97–102 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spudich J. A., Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflügers Arch. 471, 701–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNamara J. W., Singh R. R., Sadayappan S., Cardiac myosin binding protein-C phosphorylation regulates the super-relaxed state of myosin. Proc. Natl. Acad. Sci. U.S.A. 116, 11731–11736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toepfer C. N., Wakimoto H., Garfinkel A. C., McDonough B., Liao D., Jiang J., Tai A. C., Gorham J. M., Lunde I. G., Lun M., Lynch T. L. IV, McNamara J. W., Sadayappan S., Redwood C. S., Watkins H. C., Seidman J. G., Seidman C. E., Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 11, eaat1199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohde J. A., Roopnarine O., Thomas D. D., Muretta J. M., Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. U.S.A. 115, E7486–E7494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kampourakis T., Zhang X., Sun Y.-B., Irving M., Omecamtiv mercabil and blebbistatin modulate cardiac contractility by perturbing the regulatory state of the myosin filament. J. Physiol. 596, 31–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C., Kawana M., Song D., Ruppel K. M., Spudich J. A., Controlling load-dependent kinetics of beta-cardiac myosin at the single-molecule level. Nat. Struct. Mol. Biol. 25, 505–514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woody M. S., Greenberg M. J., Barua B., Winkelmann D. A., Goldman Y. E., Ostap E. M., Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat. Commun. 9, 3838 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., The protein data bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Letunic I., Bork P., 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L. L., Tate J., Punta M., Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb B., Sali A., Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiederstein M., Sippl M. J., ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M., AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K., Spudich J. A., Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. U.S.A. 110, 12607–12612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawana M., Sarkar S. S., Sutton S., Ruppel K. M., Spudich J. A., Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci. Adv. 3, e1601959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aksel T., Choe Yu E., Sutton S., Ruppel K. M., Spudich J. A., Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung J., Sivaramakrishnan S., Dunn A. R., Spudich J. A., Single-molecule dual-beam optical trap analysis of protein structure and function. Methods Enzymol. 475, 321–375 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaax0069/DC1