

Abstract
Successful establishment of pregnancy depends on steroid hormone-driven cellular changes in the uterus during the peri-implantation period. To become receptive to embryo implantation, uterine endometrial stromal cells (ESCs) must transdifferentiate into decidual cells that secrete factors necessary for embryo survival and trophoblast invasion. Autophagy is a key homeostatic process vital for cellular homeostasis. Although the uterus undergoes major cellular changes during early pregnancy, the precise role of autophagy in uterine function is unknown. Here, we report that conditional knockout of the autophagy protein FIP200 in the reproductive tract of female mice results in reduced fecundity due to an implantation defect. In the absence of FIP200, aberrant progesterone signaling results in sustained uterine epithelial proliferation and failure of stromal cells to decidualize. Additionally, loss of FIP200 impairs decidualization of human ESCs. We conclude that the autophagy protein FIP200 plays a crucial role in uterine receptivity, decidualization, and fertility. These data establish autophagy as a major cellular pathway required for uterine receptivity and decidualization in both mice and human ESCs.
Keywords: endometrium, fertility, hESCs, implantation, pregnancy, PR-Cre
Uterine specific knock out of the autophagy protein FIP200 in the female reproductive tract reduces fertility due to impaired uterine receptivity and stromal decidualization.
Introduction
Post-implantation pregnancy loss occurs in 30% of women [1–3]. Although between 30 and 60% of miscarriages are attributed to poor embryo quality, the remaining are unexplained and possibly due to a non-receptive endometrium [4]. Endometrial receptivity is intricately orchestrated by the ovarian hormones estrogen and progesterone which signal to uterine endometrial cells to remodel and become receptive to embryo implantation. During the follicular phase of each menstrual cycle, high levels of estrogen stimulate endometrial epithelial proliferation. Conversely, during the secretory phase, progesterone signals the endometrial stromal cells (ESCs) to transdifferentiate from elongated fibroblast-like cells to rounded secretory decidual cells. Decidualized ESCs provide a suitable environment for trophoblast invasion and secrete factors necessary for embryo survival. Thus, defining the molecular mechanisms underlying decidualization is important for developing strategies to prevent early pregnancy loss.
One mechanism that appears to be important for decidualization is the cellular recycling process autophagy. In the mouse uterus, autophagy is regulated by estrogen and progesterone [5] and multiple pieces of evidence indicate that autophagy induction is important for ESC decidualization. For example, the autophagy marker LC3b is expressed in human ESCs during the secretory phase of endometrial remodeling when decidualization occurs [6] and in the maternal decidual cells of the early human placenta [7]. Additionally, ESCs from obese women have reduced autophagic flux and display poor decidualization [8]. Similarly, in rodents, autophagy is increased during ESC decidualization but is impaired in mice that consume a high-fat diet and have poor decidualization and implantation [8]. Although these data suggest autophagy may play a role in ESC decidualization, a direct mechanism remains to be elucidated.
Here, we addressed this question by examining the loss of a critical autophagy protein, focal adhesion kinase family interacting protein of 200 kDa (FIP200), in the female reproductive tract. FIP200, which is encoded by the RB1 Inducible Coiled-Coil 1 (RB1CC1) gene, is the main scaffolding protein for the unc-51 like autophagy activating kinase (ULK) complex. This complex works to induce autophagy in response to cellular stress such as bacterial infection [9, 10], amino acid starvation, or high AMP levels. Once activated, the ULK complex initiates formation of a double-membrane phagophore [11, 12], which elongates and engulfs targeted proteins and organelles into an autophagosome [11, 12]. Finally, the autophagosome fuses with a lysosome to degrade its contents and provide energy and building blocks for the cell [11, 12].
Given its role in scaffolding the ULK complex, FIP200 is essential for autophagy. However, its role in decidualization is unknown. Here, we examine the requirement of autophagy for embryo implantation by conditionally knocking out FIP200 in the female reproductive tract. We provide evidence that autophagy is necessary for uterine receptivity and decidualization, two key processes that govern the establishment of pregnancy.
Materials and methods
Animal care and use
All animal studies were approved by the Animal Care and Use committee at Washington University School of Medicine. Age-matched female mice on a C57/BL6 genetic background (The Jackson Laboratory, Bar Harbor, ME) were used in this study. FIP200flox/flox [13] mice, in which exons 4 and 5 are flanked by loxp sites, and progesterone receptor cre (PRcre/+) [14] mice were previously described. Mice from the two lines were mated together to generate female mice with FIP200 conditionally ablated in the reproductive tract (FIP200flox/flox PRcre/+ mice, hereafter referred to as FIP200 cKO). Genotyping was performed with RedTaq Polymerase (Sigma, St. Louis, MO, USA) mix according to manufacturer instructions and gene-specific primers listed in Table S1.
Fertility tests and timed matings
To determine fecundity, control (FIP200flox/flox) and FIP200 cKO female mice were mated to wild type C57/BL6 male mice (Jackson Laboratories, Bar Harbor MA, USA) of proven fertility for 6 months. Breeding cages were checked daily for pups, which were promptly removed from the cage. For timed mating, the morning on which the copulatory plug was first observed was considered 1 dpc. To measure the ovulation rate, blastocysts were flushed from the uterine horns with phosphate buffered saline at 4 dpc (just before implantation). To visualize implantation sites, mice received a tail vein injection of 50 μL of 1% Chicago Sky Blue dye (Sigma-Aldrich, St. Louis, MO, USA) at 5 and 6 dpc just prior to sacrifice.
Hormone treatments
Artificial hormone treatments were performed as previously described [15]. Control and FIP200 cKO mice were ovariectomized and allowed to recover 2 weeks to remove endogenous hormones. Mice were then injected with 100 ng of estrogen (E2) (Sigma-Aldrich, St. Louis, MO, USA) for two consecutive days, allowed to rest for 2 days, and then randomized into one of three groups: 1) Vehicle (Oil) mice received four consecutive days of sesame oil injections. 2) E2 mice received 3 days of sesame oil injections followed by a single injection of 50 ng E2 on the 4th day. 3) E2-P4 mice received 1 mg of progesterone (P4) (Sigma-Aldrich, St. Louis, MO, USA) for three consecutive days followed by a single injection of 1 mg P4 plus 50 ng E2 on the 4th day. Mice were sacrificed 16 h after the final hormone injection. All hormones were delivered by subcutaneous injection in a 90% sesame oil:10% ethanol vehicle.
Artificial decidualization
Oil-induced artificial decidualization was performed on 6–8-week-old female mice as previously described [15, 16]. Briefly, mice were ovariectomized and allowed to recover for 2 weeks to remove traces of endogenous hormones. Mice received 100 ng E2 on two consecutive days, rested for 2 days, and then received of 10 ng E2 plus 1 mg P4 on the next 2 days. About 6 h following the last hormone injection, 50 μL of sesame oil was injected into the lumen of the right uterine horn. Mice received daily injections of 1 mg P4 for four additional days. Uterine horns were collected and weighed 5 days after oil stimulation.
Vasectomy and induction of decidualization in pseudo-pregnant mice
Vasectomy of an 8–10 week male mouse was performed as previously described [17]. Briefly, the male mice were anesthetized and moderate pressure was applied to the abdomen to push the testes into the scrotal sac and a 10 mm midline skin incision was made. The vas deferens were isolated and held with forceps in such a way that a loop (~0.5 cm) was created and the looped region of the vas deferens was cauterized. The incision was closed with the help of suture clips and the animals were allowed to recuperate for ~ 2 weeks. Vasectomized males were mated with sexually mature females (8–10 week old) to obtain a pseudo-pregnancy that was confirmed by the presence of a whitish vaginal plug on the next day. The day of plug considered as day 1 of pseudo-pregnancy. Decidualization was induced in the pseudo-pregnant female mice at day 4 (09:00) through the infusion of 25 μL of sesame oil in one uterine horn and the contra-lateral uterine horn was not infused with oil, which served as a non-decidualized/control horn. Uterine tissues were collected after 48 h of oil injection and wet weight was calculated for decidualized and non-decidualized/control horn [18].
RNA isolation and real-time polymerase chain reaction
Tissue was immediately frozen in liquid nitrogen and stored at −80 °C. Total RNA was isolated with the Purelink RNA mini kit (Invitrogen, Carlsbad, CA, USA) and quantified with a NanoDrop 2000 (Thermo Scientific, Waltham, MA, USA). Then, 1 μg of RNA was DNase treated and reverse transcribed with the QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD, USA) to cDNA which was diluted and amplified. To quantify FIP200 (Rb1cc1) expression in FIP200 cKO mice, primers specific to the excised exons (Table S1) were used with Fast SYBR green mastermix (Applied Biosystems/Life Technologies, Grand Island, NY) on a 7500 Fast Real-time PCR system (Applied Biosystems/Life Technologies, Grand Island, NY). Cycling parameters were as follows: 95 °C for 20 s followed by 40 cycles of 95 °C for 3 s and 60 °C for 30 s. Efficiency of all SYBR green primer sets were determined and validated using serial dilutions to generate a five-point curve. Efficiencies of primer sets were 90, 98, and 91% for 18 s, Rb1cc1, and Prl8a2, respectively. Additionally, melt curve analysis was used to determine the amplification of a single PCR product. Other genes of interest were amplified with TaqMan 2× master mix (Applied Biosystems/Life Technologies, Grand Island, NY) and gene-specific assays (Table S1). Cycling parameters were as follows: 50 °C for 20 s and 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The delta–delta cycle threshold method was used to normalize expression to the reference gene 18 s [19].
Histological analysis
Tissues were immediately fixed in 10% neutral buffered formalin for 24 h and dehydrated to 70% ethanol. Specimens were embedded in paraffin, sectioned (5 μm), and stained with hematoxylin and eosin by standard protocols. For immunohistochemistry, sections were treated with sodium citrate for antigen retrieval, blocked in bovine serum albumin (Vector Laboratories, Burlinggame, CA, USA), incubated with primary antibodies (Table S2), processes with Vectastain Elite ABC HRP Kit and DAB (3,3′-Diaminobenzidine) peroxidase substrate kit (Vector Laboratories, Burlingame, CA, USA), and counterstained with hematoxylin (Richard-Allan Scientific, San Diego, CA, USA). To ensure specificity, isotype control was included with staining procedure as shown in Supplementary Figure S1. A Nikon Eclipse E800 microscope was used for imaging. Image processing for brightness, contrast, and threshold was performed using Adobe Photoshop. All processing was applied equally to the entire image and across all treatment groups.
Immunofluorescence
Formalin-fixed and paraffin-embedded sections were deparaffinized in xylene, rehydrated in an ethanol gradient, and boiled in citrate-buffer (Vector Laboratories Inc., CA, USA) for antigen retrieval for 20 min. After blocking with 2.5% goat-serum in PBS (Vector laboratories) for 1 h at room temperature, sections were incubated overnight at 4 °C with primary antibodies (Table S2) diluted in 2.5% normal goat serum. After washing with PBS, sections were incubated with Alexa Fluor 488-conjugated secondary antibodies (Life Technologies) for 1 h at room temperature, washed, and mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Scientific).
Chromogenic in-Situ hybridization
Formalin-fixed and paraffin-embedded sections were subjected to in-situ hybridization using the Thermo Fisher Scientific View RNA™ Tissue Assay Core Kit (cat no. 19931) as per manufacturer’s protocol. Briefly, sections were deparaffinized in xylene and ethanol, and boiled in pre-treatment solution for 20 min. Slides were cool down by washing with ddH2O and treated with Protease QF for 15 min at 40 °C. Subsequently, sections were washed with PBS and incubated with 1:20 diluted mouse Rb1cc1 probe (view RNA cell tissue Gene Expression Assays, cat. no. VB1–3029092-VT, Thermo Fisher Scientific) overnight at 40 °C. A Nikon Eclipse E800 microscope was used for imaging.
Human ESC isolation
Endometrial biopsies from healthy women of reproductive age were obtained during the proliferative phase (days 8–12) of the menstrual cycle. Volunteers provided written informed consent in accordance with an Institutional Review Board-approved protocol from Washington University School of Medicine and the guidelines of the Declaration of Helsinki [20]. Human ESCs were isolated as described previously [21], cultured, and propagated in DMEM/F-12 media containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 0.1 mg/mL streptomycin. Cells were stimulated to decidualize by treating for 6 days with 100 nM Estradiol (cat. no. E1024, Sigma-Aldrich), 10 μM Medroxyprogesterone17-acetate (cat. no. M1629, Sigma-Aldrich, and 50 μM N6, 2′-O-Dibutyryladenosine 3′ 5′–cyclic monophosphate sodium salt (cat. no. D0260, Sigma-Aldrich) in 1× Opti-MEM I reduced-serum media containing 2% charcoal-stripped FBS [22]. Unstimulated control cells received 0.2% ethanol vehicle. All experiments were conducted with early passage hESCs (no more than four passages) isolated from at least three subjects. Results are shown as mean ± standard error (SE) from three replicates of one patient-derived primary endometrial cell line.
Human ESC siRNA transfection and decidualization
For siRNA-mediated knockdown of RB1CC1, hESCs were plated in six-well culture plates and treated with Lipofectamine RNA iMAX reagent and 60 pmol of either non-targeting siRNA (D-001810-10-05) or siRNAs targeting RB1CC1 (L-021117-00-0005), (GE Healthcare Dharmacon Inc., Lafayette, CO, USA). After 48 h, decidualization was induced, and medium was changed every 2 days until day six. Cells were then harvested, and the total RNA isolation kit (Invitrogen/Life Technologies, Grand Island, NY, USA) was used to isolate RNA. Quantitative PCR analyses were performed with the gene-specific assays described in Table S1, and values were normalized to 18S ribosomal RNA. Transcript levels of decidualization markers PRL and IGFBP-1 were used as molecular readouts for hESC decidualization [23, 24].
Statistical analysis
All data are presented as mean ± SE. A two-tailed paired student’s t-test was used to analyze experiments with two experimental groups, and a two-way analysis of variance (ANOVA) with Sidak’s correction was used to analyze experiments containing four or more groups. Finally, a one-way ANOVA with Sidak’s correction was used to analyze time course data. P < 0.05 was considered significant. GraphPad Prism 7 software was used for all statistical analyses.
Results
Conditional knockout of FIP200 in the reproductive tract reduces fertility but does not impair ovarian function
Global loss of FIP200 in mice causes fetal heart failure and mortality in mid/late gestation [13]. Therefore, we conditionally knocked out FIP200 in the female reproductive tract by crossing FIP200flox/flox [13] mice with mice expressing cre recombinase under control of the PR promoter (PRcre/+) [14]. In FIP200 cKO mice, FIP200 (Rb1cc1) expression was significantly decreased in the uterus and ovary but not in the liver, where PR is not expressed (Figure 1A). To determine the impact of loss of FIP200 on reproductive outcomes, we carried out 6-month breeding trials. Over this time, FIP200 cKO female mice delivered fewer litters and fewer pups per litter than control mice (Table 1). As PR is expressed in granulosa and theca cells of the preovulatory follicles, we evaluated ovarian function in FIP200 cKO mice. Histological evaluation of the ovary at 4 days post-coital (dpc) showed normal morphology (Figure 1B). Additionally, at 4 dpc, we recovered a similar number of blastocysts from FIP200 cKO and control mice (Figure 1C). Together, these results suggest that loss of FIP200 in the reproductive tract impairs fertility but not ovulation.
Figure 1.
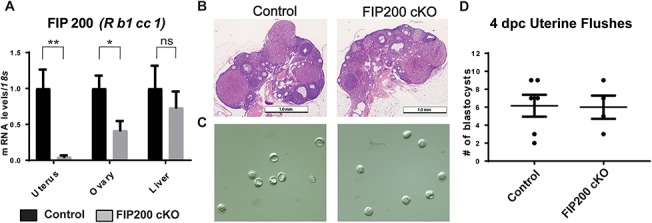
Loss of FIP200 in the female reproductive tract impairs fertility but not ovarian function. (A) Real-time polymerase chain reaction (qPCR) assessment of FIP200 (Rb1cc1) expression (normalized to 18 s) in tissues from virgin control and FIP200 cKO mice (n = 4 per group). (B) Ovarian histology of 4 dpc pregnant mice. (C) Representative images and (D) quantification of blastocysts retrieved from control and FIP200 cKO mice on 4 dpc (n = 8, 4 per group). *P < 0.05, **P < 0.01 (Two-tailed paired student’s t-test).
Table 1.
Results of six-month breeding trial.
| Genotype | n | Time to first litter (days) | Number of litters | Pups/litter | Pup weight (mg) |
|---|---|---|---|---|---|
| Control | 7 | 20 ± 0.5 | 6.2 ± 0.6 | 7.1 ± 0.4 | 1315 ± 66 |
| FIP200 cKO | 6 | 46.14 ± 6.9** | 2.3 ± 0.6*** | 2.8 ± 0.75** | 1441 ± 51 |
** P < 0.01.
*** P < 0.001.
Loss of FIP200 impairs uterine receptivity and embryo implantation
Given that ovarian function was normal in FIP200 cKO mice, we next wondered whether the implantation defect was because the uteri in FIP200 cKO mice failed to become receptive. Uterine receptivity requires estrogen-dependent epithelial proliferation, which is inhibited by progesterone just before embryo attachment and implantation. To evaluate receptivity, we stained the uteri from 4 dpc mice with an antibody to the proliferation marker Ki-67. In control mice, the stromal cells were positive for Ki-67, but the epithelial cells were not. In FIP200 cKO mice, the epithelial cells were positive for Ki-67, and the stromal cells were less proliferative than in control mice (Figure 2A). However, expression of estrogen receptor alpha (ERα) and PR appeared similar in uteri from control and FIP200 cKO mice (Figure 2B and C). Next, we determined whether sustained E2-resposnes in FIP200 cKO mice results in embryo implantation defect. As shown in Figure 2D, we found no implanting embryos in FIP200 cKO mice at 5 dpc. Histological analysis of day 5 implantation sites from control mice showed the typical formation of the implantation chamber and the attachment of embryonic trophectoderm with uterine luminal epithelium (Figure 2E). In contrast, we noted a failure to form implantation chambers in FIP200 cKO mice 5 dpc uteri. Further, we failed to observe the initial opposition of embryo onto luminal epithelium in FIP200 cKO mice 5 dpc uteri (Figure 2D and E). Consistent with defect in embryo implantation due to sustained E2 action, we found elevated expression of MUC1 in epithelial cells in 5 dpc uteri from FIP200 cKO mice (Figure 2F). Moreover, we also found reduced expression of LIF in 5 dpc uteri from FIP200 cKO mice (Figure 2G). To ensure specificity, isotype control was shown in Figure 2H. Further, the uteri of control mice at 6 dpc had multiple implantation sites, those of FIP200 cKO mice had no implantation sites (Figure S2A and C). Additionally, histological examination of uteri from control mice revealed that embryos had implanted into the uterus. The stromal cells of control uteri were rounded and polynucleated indicating that they were decidualized. In contrast, in FIP200 cKO mice, embryos were adhered to the intact uterine epithelium, and the uterine stromal cells maintained a fibroblast morphology (Figure S2B). Together, these findings suggest that retained expression of MUC1 in the luminal epithelium and reduced expression of LIF disrupted uterine receptivity, resulting in implantation defect in FIP200 cKO mice.
Figure 2.
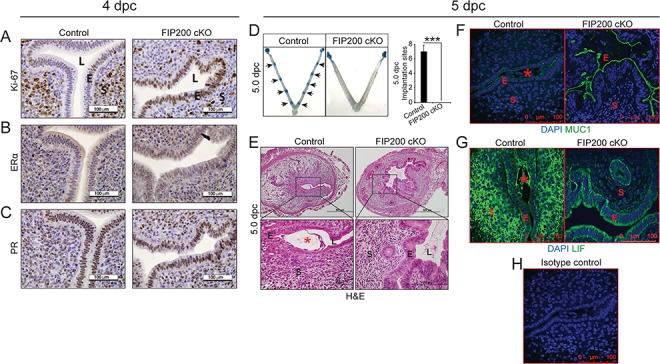
Loss of FIP200 in the mouse reproductive tract compromises embryo implantation and uterine receptivity. (A–C) Representative images of immunostaining for Ki-67 (A), ERα (B), and PR (C) of control and FIP200 cKO uteri at 4 dpc (n = 5, 4). (D) Embryo implantation sites at 5 dpc in control and FIP200 cKO mice. Black arrow denotes implantation sites. Quantitation of the number of implantation sites at 5 dpc as visualized by Chicago blue dye injection. (E-G) Representative cross-section images of the uterus at dpc 5 Control and FIP200 cKO mice stained for Hematoxylin and Eosin (E), MUC1 (F), LIF (G) and Isotype control (H) (n = 5,3). E indicates epithelium, S indicates stroma, L indicates the uterine lumen and asterisk indicates the embryo. ***P < 0.001 (Two-tailed paired student’s t-test).
Next, to determine whether loss of FIP200 led to sustained epithelial proliferation because of enhanced estrogen signaling or the failure of progesterone to inhibit proliferation [25], we used a controlled steroid hormone regimen to artificially induce the receptive uterus. In this model, ovariectomized mice were exposed to estrogen (E2) for 2 days, rested for 2 days, and then exposed to either vehicle (oil), E2, or E2 plus progesterone (E2 + P4). Alternatively, uterine epithelial cells from control mice were proliferative in the E2 group but not in the oil or E2 + P4 group (Figure 3A). Uterine epithelial cells in the FIP200 cKO mice were proliferative in both the E2 and E2 + P4 groups (Figure 3A). The ability of P4 to inhibit E2-driven epithelial proliferation was severely curtailed in FIP200 cKO uterus (Figure 3A), suggesting FIP200 is required for P4-dependent inhibition of epithelial proliferation.
Figure 3.
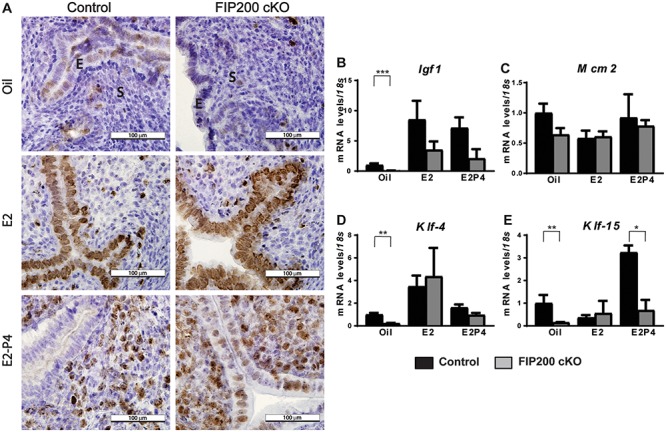
FIP200 is necessary for progesterone inhibition of E2-dependent epithelial proliferation. (A) Representative Ki-67 staining of uterine sections from control and FIP200 cKO mice treated as indicated. (B–E) qPCR analysis of relative expression (normalized to 18 s) of the indicated genes (n = 4–6 mice per group). E indicates epithelium, S indicates stroma and arrow heads indicate sustained epithelial proliferation. *P < 0.05, **P < 0.01, ***P < 0.001, (2-Way ANOVA with Sidak’s correction).
This result prompted us to measure the expression of genes involved in inhibiting estrogen-dependent epithelial proliferation. The key epithelial E2-responsive gene Igf1 was expressed similarly in uteri from FIP200 cKO and control mice treated with E2 or E2 + P4 (Figure 3B). Finally, we examined expression of the transcription factors Klf-4 and Klf-15, which compete for the Mcm2 promoter to drive cell cycle progression [26]. Normally, E2 stimulation causes Klf-4 to bind the Mcm2 promoter and stimulate its expression to drive proliferation, whereas P4 stimulation induces Klf-15 expression and inhibits Mcm2 and subsequent proliferation. Mcm2 expression was not affected by loss of FIP200 (Figure 3C). We found that Klf-4 expression was increased after E2 treatment and decreased after E2 + P4 treatment in uteri from both control and FIP200 cKO mice (Figure 3D). However, Klf15 expression was increased after E2 + P4 treatment in uteri from control mice but not in those from FIP200 cKO (Figure 3E). Together, these results suggest a distinct role for FIP200 in progesterone responses during endometrial receptivity.
Loss of FIP200 impairs decidualization
In our natural mating experiments, decidualization appeared to be impaired in FIP200 cKO mice (Figure 2B). To confirm this, we used an artificial decidualization model in which we ovariectomized mice and then injected one uterine horn with sesame oil to stimulate deciduoma formation. Whereas the stimulated horn in control mice was larger than the unstimulated horn and contained round polynucleated decidual cells, the stimulated horn in FIP200 cKO mice was not enlarged and contained mononucleated cells (Figure 4A). Following normalization to the loading horn of FIP200 cKO mice weighed less than the stimulated horns of controls (Figure 4B). Additionally, mRNA expression of the decidualization markers Prl8a2 (previously known as prolactin related protein), Wnt4, and Bmp2 was induced in the stimulated horns of control mice but not in those of FIP200 cKO mice (Figure 4C–E). Further, a blunted decidual response with FIP200 ablation was observed in day 4 pseudo-pregnant mice (Figure 4F and G). These results indicate that FIP200 was required for decidualization.
Figure 4.
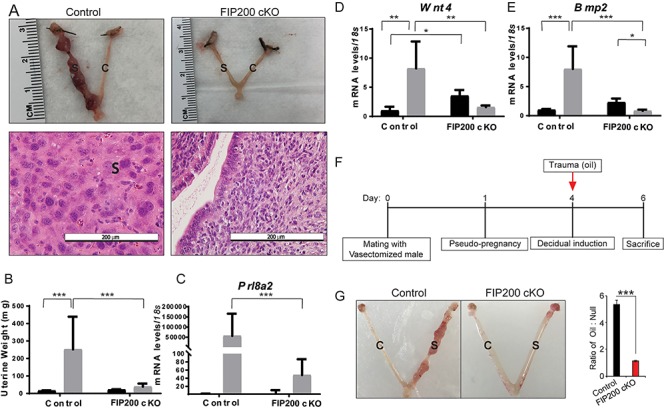
Loss of FIP200 in the reproductive tract impairs ESC decidualization. (A) Representative images of uteri and uterine histology after artificial decidualization. (B) Wet weight of unstimulated and stimulated uterine horns from control and FIP200 cKO mice (n = 5–8 per group). (D-E) qPCR expression of the decidualization markers, Prl8a2, Wnt4, and Bmp2 after artificial decidualization (n = 5–7 per group). *P < 0.05, **P < 0.01, ***P < 0.001 (2-Way ANOVA with Sidak’s correction). (F) Time-line for induction of decidualization in day 4 pseudo-pregnant female mice. (G) Representative images showing the gross morphology of control and stimulated uterine horns from control (n = 5) and FIP200 cKO (n = 3) mice collected 2 days after oil injection from the pseudo-pregnant female mice. Right side graph represents the ratio between the wet weight of the oil-treated horn to the wet weight of the untreated horn. ‘C’ denotes control horn and ‘S’ denotes stimulated horn; ***P < 0.001 (Two-tailed paired student’s t-test).
FIP200 is required for hESC decidualization
To determine whether FIP200 was required for human decidualization, we obtained ESCs from uterine biopsies and hormonally stimulated them to decidualize for 6 days in culture. Decidualization was confirmed by increased expression of the markers PRL and IGFBP1 (Figure 5A). As we saw, the expression of FIP200 (RB1CC1) increased during decidualization of human ESCs (Figure 5A). To determine if FIP200 was required for hESC decidualization, we compared decidualization in ESCs transfected with control siRNA or siRNA targeting RB1CC1. In cells transfected with control siRNA, 6 days of hormonal stimulation transformed the fibroblast-like stromal cells into rounded decidual cells (Figure 5B) and increased expression of PRL and IGFBP1 (Figure 5C). In cells transfected with RB1CC1 siRNA, hormonally stimulated cells maintained a fibroblast appearance and the expression of the decidualization markers PRL and IGFBP1 were not as highly upregulated (Figure 5B and C). Together, these results reveal that FIP200 is an evolutionarily conserved autophagy protein required for decidualization in both mouse and human ESCs.
Figure 5.
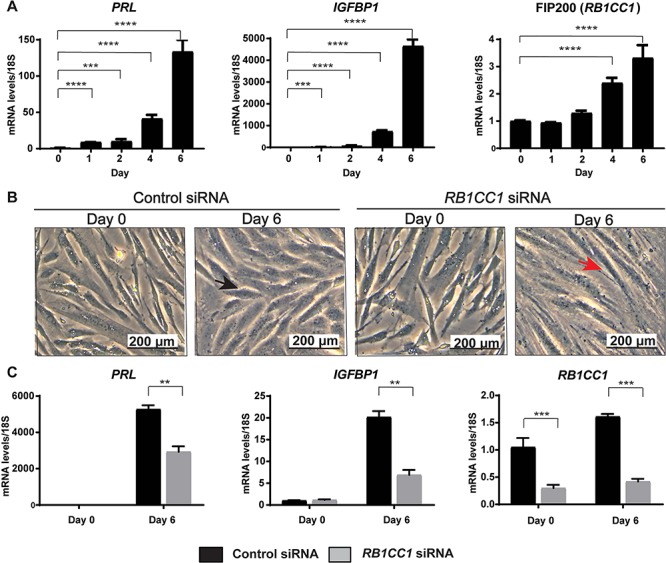
FIP200 is required for human ESC decidualization. (A) qPCR analysis of the decidualization markers (PRL and IGFPB1) and FIP200 (RB1CC1) at the indicated days after induction of decidualization. (B) Morphology of cells and (C) qPCR analysis of the indicated decidualization markers and FIP200 (RB1CC1) following transfection with control and RBCC1. Data is normalized to 18S and expressed as fold change over Day 0 controls. Black arrowhead indicates decidualizing morphology and red arrowhead indicates fibroblast morphology. *P < 0.05, **P < 0.01, ***P < 0.001. A 1-Way ANOVA with Sidak’s correction was used to analyze time course data while a 2-Way ANOVA with Sidak’s correction was used to evaluate the siRNA knock down experiment.
Discussion
Here, we provide several lines of evidence that the autophagy is required for decidualization in both mouse and human cells. First, loss of FIP200 in the mouse reproductive tract reduced fertility in naturally mated mice due to implantation failure. Second, we used two mouse models, one mimicking the hormonal changes of pregnancy and one in which we induced decidualization, to show that FIP200 is required for uterine receptivity and decidualization in vivo. Finally, we show that human ESCs in which FIP200 expression was decreased had impaired ability to decidualize in vitro. Together, these data indicate that FIP200 plays an evolutionarily conserved role in decidualization.
This study adds genetic support to existing evidence in both rodents and human cell lines [8] that autophagy is necessary for ESC decidualization and embryo implantation. Autophagy may be required in decidualization to help meet the high energetic demands of this process. Unlike many cells, decidualizing ESCs do not primarily rely on glycolysis but instead metabolize fatty acids in the β-oxidation pathway [27], increase expression of glucose transporters [28], and metabolize glucose in the pentose phosphate pathway. Although the pentose phosphate pathway generates nicotinamide adenine dinucleotide phosphate and ribose 5-phosphate, which are used to synthesize cholesterol, fatty acids, and nucleotides, this pathway produces less adenosine triphosphate (ATP) per molecule of glucose than glycolysis. The resulting decrease in cellular ATP likely triggers autophagy in the ESCs [8], enabling them to maintain energy homeostasis during decidualization.
Previous work has shown that autophagy is hormonally regulated in the endometrium, though its exact role in receptivity was unclear. Recent data has shown that autophagy is induced in the ovariectomized mouse uterus to facilitate glycogen breakdown in the absence of estrogen and is suppressed by estrogen and progesterone treatment [5]. During pregnancy, murine uterine autophagy is highest in the preimplantation window [5]. In human endometrial biopsies, autophagy occurred in both the glandular epithelium and the stroma throughout the menstrual cycle, with its highest activity occurring during the secretory phase when the stroma is decidualized [6]. Further work in human endometrial Ishikawa cells indicated that autophagy was induced following both progesterone treatment as well as hormonal withdraw [6]. These studies suggest that autophagy is hormonally regulated. Our study adds to the complex relationship between steroid hormone signaling and autophagy in the uterus by providing evidence that progesterone inhibition of estrogen dependent epithelial proliferation is impaired in FIP200 cKO mice with reduced autophagy. Together, these studies support a role for autophagy in hormone-induced endometrial receptivity.
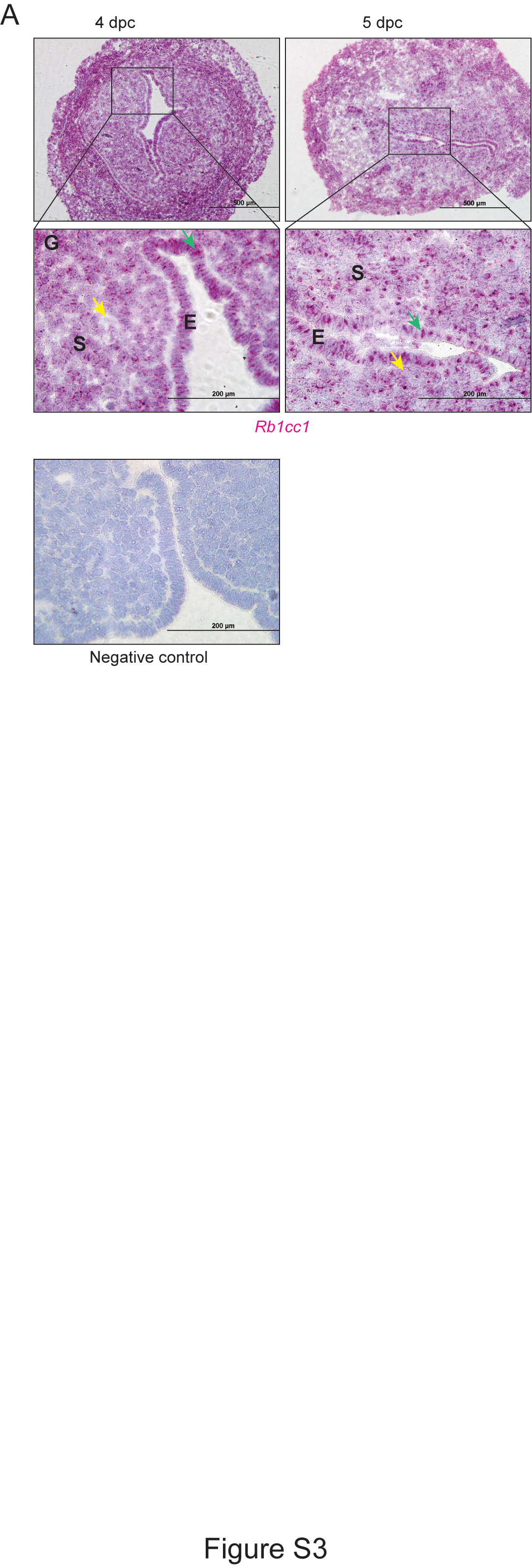
Uterine receptivity requires that the endometrial epithelial cells proliferate in response to estrogen and then cease proliferating in response to progesterone. Our data indicate that loss of FIP200 impairs this response. In the natural mating experiment, the epithelium of 4 dpc FIP200 cKO mice was proliferative, whereas the epithelium of control mice was not. Additionally, we observed increased epithelial proliferation in the ovariectomized cKO uteri treated with E2-P4 further suggesting a failure of progesterone signaling to inhibit E2 stimulated proliferation in the epithelium. This excess proliferation may be because Klf-15 expression is not activated in uteri from FIP200 cKO mice. Normally, as P4 rises and binds to its receptor (PR), the P4 target gene Klf-15 inhibits Mcm2 expression and prevents epithelial proliferation [26]. Thus, loss of FIP200 may impair this important step in progesterone regulation of estrogen-dependent epithelial proliferation. Interestingly, FIP200 is localized in both uterine epithelia and stroma during window of uterine receptivity (Figure S3). Thus, it is intriguing to test the relative contribution of epithelia and stromal FIP200 in mediating progesterone regulation of estrogen-dependent epithelial proliferation in near future.
Like many autophagy proteins, FIP200 interacts with multiple proteins and functions outside the autophagy pathway [29]. Although our data indicate that FIP200 plays an important role in uterine receptivity, we cannot rule out the possibility that it does so via a non-autophagic function. Consistent with our finding that FIP200 cKO mice have sustained uterine epithelial proliferation, loss of FIP200 promotes cell cycle progression in breast cancer cells [29]. This function appears to require the autophagy pathway. Chen and colleagues showed that a FIP200 mutant that could not interact with the autophagy protein Atg13 could not promote tumor proliferation [30]. Thus, it seems likely that the excess uterine epithelial proliferation we noted in FIP200 cKO mice was autophagy dependent. This result supports a recent study showing knock down of ATG5 and ATG7 also prevents proper decidualization in human ESCs [31]. Together these lines of evidence support a critical role of autophagy in decidualization. However, future studies knocking out other autophagy proteins in the uterus are required to confirm this conclusion.
Our study is the first to provide a direct link between autophagy and decidualization. Understanding the role of autophagy in uterine receptivity is critical to improving artificial reproduction success rates. For example, high body mass index negatively correlates with implantation rate [32, 33] in women undergoing in vitro fertilization, and recent human and mouse evidence suggests that aberrant autophagy in the endometrium plays a role [8]. Our data showing the importance of autophagy in uterine receptivity and stromal decidualization promotes the possibility of developing autophagy-stimulating strategies to improve reproductive outcomes for obese women and those experiencing recurrent implantation defects.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to Francesco DeMayo (National Institute of Environmental Health Sciences) and Herbert Virgin (Washington University School of Medicine) for providing the PRcre/+ and FIP200flox/flox mice, respectively. We sincerely thank Marina Rowan, Gwendalyn Kreleler, Alma Johnson, Wendy Zhang, Michaela Reid, Sara Arfania and Andrew Cusumano for their technical expertise, and Deborah Frank for her critical editorial review of this manuscript.
Footnotes
† Grant Support: This work was supported by the following National Institutes of Health grants: NICHD R01 HD065435 and R01 HD083895 (to KHM), NICHD R01 HD065435 and R00-HD080742 (to RK), and T32 DK007120 (to AKO).
Conflict of interest
The authors have declared that no conflict of interest exists.
Funding
This work was supported by the following National Institutes of Health grants: NICHD R01 HD065435 and R01 HD083895 (to KHM), NICHD R01 HD065435 and R00HD080742 (to RK), and T32 DK007120 (to AKO).
References
- 1. Macklon NS, Geraedts JP, Fauser BC. Conception to ongoing pregnancy: The 'black box' of early pregnancy loss. Hum Reprod Update 2002; 8:333–343. [DOI] [PubMed] [Google Scholar]
- 2. Chard T. Frequency of implantation and early pregnancy loss in natural cycles. Baillieres Clin Obstet Gynaecol 1991; 5:179–189. [DOI] [PubMed] [Google Scholar]
- 3. Teklenburg G, et al. The molecular basis of recurrent pregnancy loss: Impaired natural embryo selection. Mol Hum Reprod 2010; 16:886–895. [DOI] [PubMed] [Google Scholar]
- 4. Quenby S, et al. Recurrent miscarriage: A defect in nature's quality control? Hum Reprod 2002; 17:1959–1963. [DOI] [PubMed] [Google Scholar]
- 5. Choi S, et al. Suppression of autophagic activation in the mouse uterus by estrogen and progesterone. J Endocrinol 2014; 221:39–50. [DOI] [PubMed] [Google Scholar]
- 6. Choi J, et al. The role of autophagy in human endometrium. Biol Reprod 2012; 86:70. [DOI] [PubMed] [Google Scholar]
- 7. Avagliano L, et al. Autophagy in normal and abnormal early human pregnancies. Reprod Sci 2015; 22:838–844. [DOI] [PubMed] [Google Scholar]
- 8. Rhee JS, et al. Diet-induced obesity impairs endometrial stromal cell decidualization: A potential role for impaired autophagy. Hum Reprod 2016; 31:1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol 2016; 39:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: Origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013; 14:759–774. [DOI] [PubMed] [Google Scholar]
- 13. Gan B, et al. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol 2006; 175:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soyal SM, et al. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 2005; 41:58–66. [DOI] [PubMed] [Google Scholar]
- 15. Tsai JH, et al. Glucosamine inhibits decidualization of human endometrial stromal cells and decreases litter sizes in mice. Biol Reprod 2013; 89:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frolova AI, O'Neill K, Moley KH. Dehydroepiandrosterone inhibits glucose flux through the pentose phosphate pathway in human and mouse endometrial stromal cells, preventing decidualization and implantation. Mol Endocrinol 2011; 25:1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller AL, et al. Using the mouse grimace scale and behaviour to assess pain in CBA mice following vasectomy. Appl Anim Behav Sci 2016; 181:160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sroga JM, Ma X, Das SK. Developmental regulation of decidual cell polyploidy at the site of implantation. Front Biosci (Schol Ed) 2012; 4:1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 20. WMA Declaration of Helsinki Serves as Guide to Physicians Calif Med 1966; 105:149–150. [PMC free article] [PubMed] [Google Scholar]
- 21. Kommagani R, et al. Acceleration of the glycolytic flux by steroid receptor coactivator-2 is essential for endometrial decidualization. PLoS Genet 2013; 9:e1003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Camden AJ, et al. Growth regulation by estrogen in breast cancer 1 (GREB1) is a novel progesterone-responsive gene required for human endometrial stromal decidualization. Mol Hum Reprod 2017; 23:646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brosens JJ, Hayashi N, White JO. Progesterone receptor regulates decidual prolactin expression in differentiating human endometrial stromal cells. Endocrinology 1999; 140:4809–4820. [DOI] [PubMed] [Google Scholar]
- 24. Michalski SA, et al. Isolation of human endometrial stromal cells for in vitro Decidualization. J Vis Exp 2018; 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cha J, Sun X, Dey SK. Mechanisms of implantation: Strategies for successful pregnancy. Nat Med 2012; 18:1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ray S, Pollard JW. KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing. Proc Natl Acad Sci U S A 2012; 109:E1334–E1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsai JH, et al. The fatty acid beta-oxidation pathway is important for decidualization of endometrial stromal cells in both humans and mice. Biol Reprod 2014; 90:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frolova A, et al. Facilitative glucose transporter type 1 is differentially regulated by progesterone and estrogen in murine and human endometrial stromal cells. Endocrinology 2009; 150:1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gan B, Guan JL. FIP200, a key signaling node to coordinately regulate various cellular processes. Cell Signal 2008; 20:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen S, et al. Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev 2016; 30:856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mestre Citrinovitz AC, Strowitzki T, Germeyer A. Decreased autophagy impairs Decidualization of human endometrial stromal cells: A role for ATG proteins in endometrial physiology. Int J Mol Sci 2019; 20:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brewer CJ, Balen AH. The adverse effects of obesity on conception and implantation. Reproduction 2010; 140:347–364. [DOI] [PubMed] [Google Scholar]
- 33. Luke B, et al. Female obesity adversely affects assisted reproductive technology (ART) pregnancy and live birth rates. Hum Reprod 2011; 26:245–252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.