

Abstract
A novel Co(II)-catalyzed polyene cyclization was developed that is uniquely effective when performed in hexafluoroisopropanol as solvent. The process is presumably initiated by metal-catalyzed hydrogen atom transfer (MHAT) to 1,1-disubstituted or monosubstituted alkenes, and the reaction is remarkable for its tolerance of internal alkenes bearing either electron-rich methyl or electron-deficient nitrile substituents; electron-rich aromatic terminators are required in both cases. Terpenoid scaffolds with different substitution patterns are obtained with excellent diastereoselectivities, and the bioactive C20-oxidized abietane diterpenoid carnosaldehyde was made to showcase the utility of the nitrile-bearing products. We also provide the results of several mechanistic experiments that suggest that the process features an MHAT-induced radical bicyclization with late-stage oxidation to regenerate the aromatic terminator.
Keywords: terpenoids, metal-catalyzed hydrogen-atom-transfer, nitrile, bicyclization, cobalt
Graphical Abstract
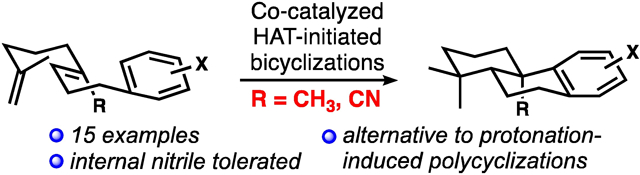
A cobalt-catalyzed hydrogen-atom-transfer initiates bicyclization reactions involving internal electron-rich or electron-poor alkenes and electron-rich arene terminators, providing a diversity of complex polycyclic products.
Introduction.
The polycyclic products of biosynthetic polyene-type cyclizations are endowed with a wealth of structural diversity and biological activity. Inspired by these compelling secondary metabolites and their fascinating biogeneses, chemists have engaged in decades of work emulating nature’s polyene cyclizations, starting with the seminal studies of Stork, Eschenmoser, Johnson, van Tamelen, and others.[1,2] As a result, chemists have accrued an excellent understanding of the reactivity and stereochemical outcomes of cation-initiated polycyclizations of polyenes as applied to bioinspired terpenoid synthesis. Many new polycyclization methods have been developed that have been tested in the total syntheses of these natural products. These advances include methods underpinned by either radical or organometallic intermediates.[3,4] However, gaps in this general area remain, including in the cyclization of systems that electronically deviate significantly from the natural terpenoid precursors.
Inspired in part by the structures of the atisane alkaloids (1, Figure 1) and their more complex hetisine and hetidine congeners,[5] as well as C19–C20 lactone-containing diterpenoids such as neotripterifordin (2)[6], we aimed to develop bicyclization reactions that would tolerate an electron-withdrawing group (ester or nitrile) in place of the ubiquitous, geraniol-derived C20 methyl group (see 3 to 4). Although biosynthetically the oxidation of this carbon surely occurs post-cyclization, it would be strategically valuable to incorporate these functional groups prior to laboratory cyclization; this idea is in line with our broader program to extend the range of functionalized substrates in stereocontrolled polycyclizations.[7] We presumed that this C20 “handle” would prove advantageous in the subsequent applications to molecules such as 1, 2, and the many other diterpenoids with an oxidized C20 that is frequently also part of a ring structure. These synthesis endeavors themselves were largely discouraged by recent attractive, closely related successes by the group of Li,[8] who included a secondary allylic ether function at this position in a Carreira-type enantioselective polycyclization,[4b] and the Ma group,[9] who used a different scaffold-building strategy to make nitrile compounds related to 4b. However, as our studies unfolded, we were surprised to discover conditions for Co-catalyzed HAT-initiated dialkenylarene bicyclizations with a rather broad scope, including the intriguing tolerance of either a C20 electron-donating methyl group (3a) or an electron-withdrawing nitrile (3b). These results and some preliminary mechanistic investigations are the subject of this report.
Figure 1.
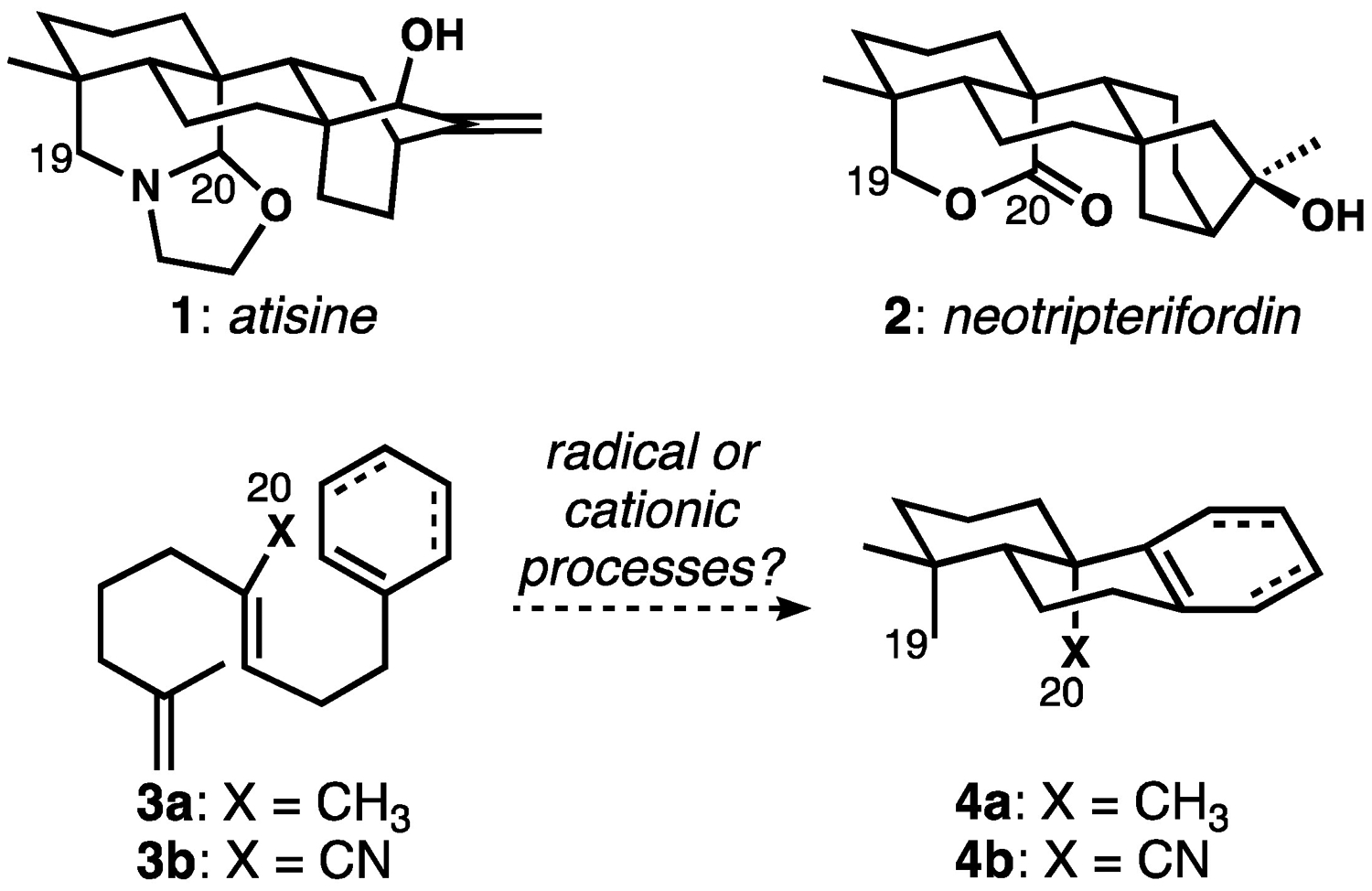
A. Atisane alkaloids and neotripterifordin inspired our efforts to develop bicyclization reactions with electron-withdrawing groups at C20.
Results and Discussion.
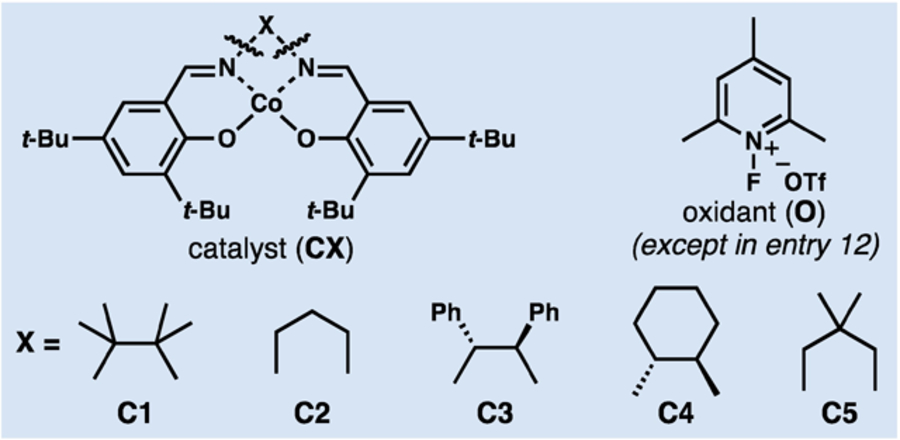
Building upon extensive recent reports in the area of metal-catalyzed hydrogen atom transfer reactions to alkenes (MHAT),[10] we surmised that a tertiary radical could be easily generated from 1,1-disubstituted alkenes of type 3, and that under appropriate oxidative conditions, such as those from the Shigehisa group,[11,12] cation generation could result by radical oxidation. Depending upon relative rates of oxidation and cyclization, either radical or cationic C–C bond-forming events would take place. Because we were most interested in developing cyclizations with C20 electron-withdrawing groups, we initiated our studies with diene 5a (Table 1), which was easily assembled by virtue of the oxygenation at C2.[13] We first evaluated a catalytic system similar to the one reported by Shigehisa for hydroarylation of alkenes.[11d] Treatment of substrate 5a in acetone with phenylsilane, N-fluoropyridinium oxidant O, and cobalt complex C1 yielded tricyclic framework 6a in moderate yield (entry 1). The use of substoichiometric quantities of oxidant was not tolerated (entry 2), and different silanes, including “Ruben-silane”[14] (entries 3 and 4) did not significantly improve the outcome. For its convenience and low price, TMDSO was chosen as the preferred silane. Other single electron oxidants, including Cu(OTf)2 and CAN, led to decomposition (not shown, see SI). Fortuitously, a small solvent screen revealed that carrying out the reaction in HFIP led to significantly improved yields (entries 4–8). A screen of a few different catalysts (entries 8–10) revealed that only complex C1 gave full conversion in 3 h. Decreasing the quantity of silane resulted in lower conversion and increased formation of unidentified side products (not shown). For practical purposes, using three equivalents of both silane and oxidant was found to be optimal as the reaction time was drastically reduced, and the reactivity was cleaner. Traces of the desired product were observed without any added oxidant (entry 11). Although we considered that this result might be due to adventitious oxygen, replacing the N-fluoropyridinium oxidant with an atmosphere of molecular oxygen led to a complex mixture, containing only traces of 6a (entry 12). In the context of the optimal solvent for this transformation, it is noteworthy that Shigehisa has reported a method for hydrofunctionalization of alkenes employing fluorinated alcohols as nucleophiles using the same catalytic system.[11e] Other conditions evaluated (not shown) but found to be ineffective included: (a) Shenvi’s Co-catalyzed conditions for alkene isomerization, which can also lead to cyclization and hydroarylation;[15] (b) Shenvi’s Mn-mediated conditions for intramolecular radical hydroarylation;[16] and (c) Baran’s Fe-catalyzed system,[17] which is excellent at promoting Giese-type reactions[18] that strongly resemble the first ring formation in the transformation of 5a to 6a.
Table 1.
Optimization of Bicyclization Reaction Conditions
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | silane | Y | Solventb | yield (%)c |
| 1 | C1 | PhSiH3 | 3.0 | Me2CO | 40 |
| 2 | C1 | PhSiH3 | 0.5 | Me2CO | <5d |
| 3 | C1 | Ph(i-PrO)SiH2 | 3.0 | Me2CO | 45 |
| 4 | C1 | TMDSO | 3.0 | Me2CO | 42 |
| 5 | C1 | PhSiH3 | 3.0 | i-PrOH | 0 |
| 6 | C1 | TMDSO | 2.0 | PhCF3 | 0 |
| 7 | C1 | TMDSO | 3.0 | CH2Cl2 | <5 |
| 8 | C1 | TMDSO | 3.0 | i-PrOH | <5 |
| 9 | C1 | TMDSO | 3.0 | HFIP | 87 |
| 10 | C2 | TMDSO | 3.0 | HFIP | 31d |
| 11 | C3 | TMDSO | 3.0 | HFIP | <5 |
| 12 | C1 | TMDSO | 0 | HFIP | <5d |
| 13e | C1 | TMDSO | O2 | HFIP | <5 |
| 14 | – | TMDSO | 3.0 | HFIP | 0 |
 | |||||
All reactions were carried out using 5a (0.02 mmol), oxidant, catalyst (0.002 mmol), silane (0.07 mmol), in solvent (0.5 M) at ambient temperature (rt) for 4h under Ar.
Reaction mixtures were purged with Ar for 10 min. before the addition of silane.
For entries 1, 3, 4, 8, 9 isolated yields are reported, for other entries yields were estimated via 1H NMR spectroscopic analysis.
Incomplete conversion was observed.
Reaction was run for 12h. TBS: terti-butyldimethylsilyl; TMDSO: tetramethyldisiloxane; HFIP: 1,1,1,3,3,3-hexafluoroisopropanol
With the optimized conditions in hand, we investigated the scope of this process (Figure 2). Substrates with alkoxy-substituted aromatic terminators underwent bicyclization yielding products 6a–6c in good yield, and excellent diastereoselectivity. Products bearing TBS-protected alcohols at C2 were each isolated as single stereoisomers, presumably owing to the preference for the bulky silyloxy substituent to assume a pseudoequatorial orientation in the transition structure for the first ring closure. A related system was used by Liu and co-workers in an elegant synthesis of hispidanin A; in this case the key iron-catalyzed, HAT-initiated bicyclization terminated onto an electron-poor alkene and was not fully stereoselective.[19] The meta-substituted anisole substrate gave a 1.7:1 para/ortho mixture of regioisomers (6b). The aryl bromide functionality in 5c was tolerated under the reaction conditions. Naphthyl and toluyl substrates underwent cyclization in moderate yields (products 6d and 6e), suggesting that electron-rich arenes are the best terminators for this process. A Boc-protected indole substrate cyclized to give an indoline product 6f along with a small quantity of the anticipated indole product; the same was observed for the corresponding N-tosyl substrate yielding 6g. The indoline product might be produced via the competitive silane reduction of a post-cyclization cationic intermediate (see below). Cyclization to generate tricyclic scaffold 6h was performed on a 1 mmol scale and constitutes a formal total synthesis of pisiferin (not shown).[20] Further, we converted 6h to the recently described bioactive aromatic abietane diterpenoid[21] carnosaldehyde in three steps,[13,22] demonstrating the utility of the C20-oxidized products. Ester- and trifluoromethyl-bearing arenes did not yield any of the desired cyclization products (6i and 6j, respectively), suggesting that electron-poor arenes are not suitable terminating groups for these reactions.
Figure 2.
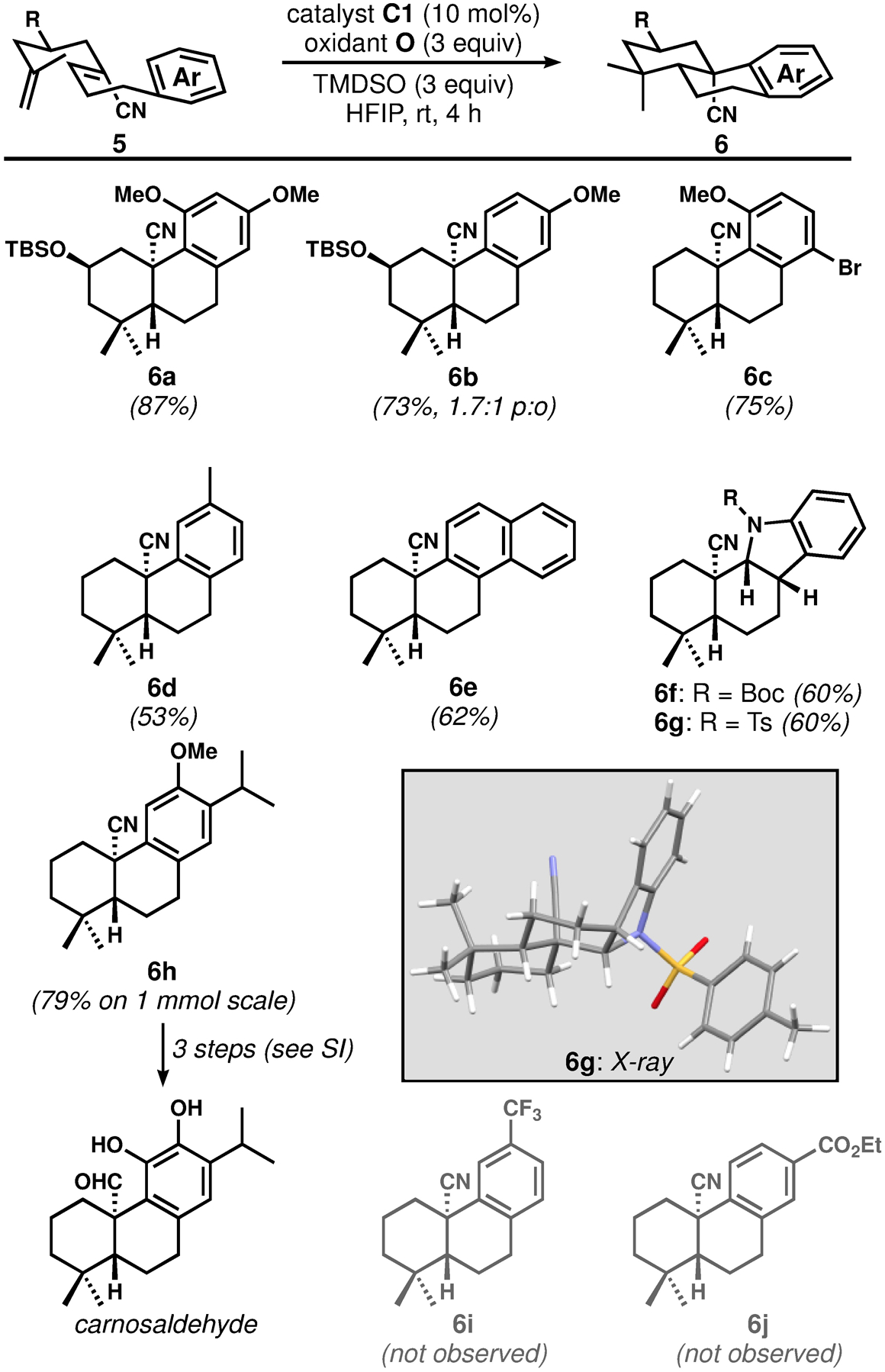
Scope of the bicyclization of nitrile-substituted substrates (yields shown are for isolated and purified compounds). Boc: tert-butoxycarbonyl; Ts: para-toluenesulfonyl.
In light of Shigehisa’s report on hydroarylation of unactivated alkenes,[11d] we changed the substrate to include the trisubstituted, methyl-bearing internal alkene that is more typical of bioinspired polyene cyclizations. Several of these substrates reacted productively, generating tricyclic compounds 8a–8d with reasonable efficiency[23] under nearly identical conditions used for the nitrile substrates (Figure 2); in these cases less sterically crowded catalyst C2 proved slightly superior to C1. Product 8c, a known compound,[24] was formed along with products of terminal to internal alkene isomerization of the substrate; owing to the formation of multiple products of similar polarity, it could only be obtained in about 90% purity. The product of furan termination (8e) was not observed, which was unexpected; however, Co-catalyzed MHAT to furans is known.[25] The failure to produce 8f and 8g via termination with electron-poor arenes mirrors our results with similar acrylonitrile substrates shown in Figure 2.
Preliminary Mechanistic Experiments.
We realized our initial goal to develop bicyclization reactions that tolerated both electron-deficient and electron-rich internal alkenes. With the exception of the reactions of indole substrates that gave indoline products 6f and 6g, these reactions are redox neutral with a presumed initial alkene reduction by MHAT, which therefore necessitates an oxidation at some point further along in the overall reaction mechanism. The three plausible mechanisms that follow from this assumption are shown in Scheme 1. We had considered the possibility that the first cyclizations in the acrylonitrile cases are 6-endo Giese-type,[18] but that the much slower radical cyclization in the substrates lacking the activating nitrile could allow for radical oxidation to compete, with the cyclizations proceeding via carbocationic manifolds. However, as discussed below, most of our results, along with some of our preliminary mechanistic experiments that we will describe, suggest the likelihood that the bicyclization reactions of both substrate types are radical in nature.
Scheme 1.
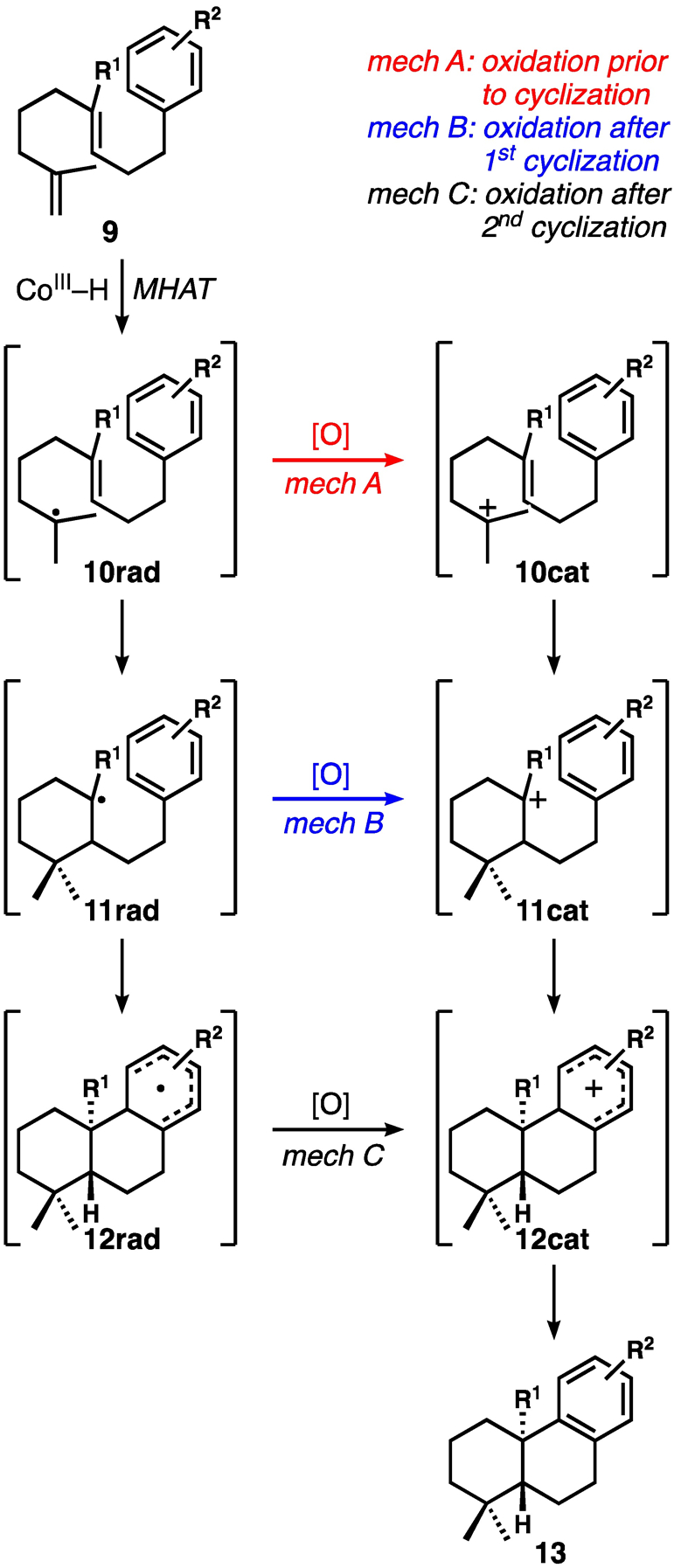
Plausible mechanistic options for bicyclization reactions
Initially designed to expand the scope with respect to electron-withdrawing group, the substitution of the nitrile group with an ester instead yielded surprising results (Scheme 2). Submitting 14-Z to standard reaction conditions yielded a complex mixture in which the expected product 15 was not observed (Scheme 2). Coincidentally, we found that the geometrical isomer 14-E underwent a 5-exo cyclization process to give 16. Products containing the expected decalin framework were not detected in this case, either. For this transformation, substoichiometric amounts of oxidant were sufficient for full conversion (conditions as shown, plausibly just enough oxidant to generate Co(III)), suggesting that the mechanism could be akin to that in the Co(III)-catalyzed, HAT-induced cycloisomerizations reported by Shenvi et al.[15] We currently have no explanation for the lack of productive reactivity of 14-Z. The desired bicyclization of the E-isomer likely suffers from severe non-bonded interactions in the transition structure;[26] we see the same reactivity with the E-unsaturated nitrile substrates, but have never been able to isolate the product corresponding to 16 in pure form. Another unexplained result is the lack of formation of a cyclopentane product analogous to 16 from 14-Z, in spite of the very similar steric demand on these two cyclizations. Finally, it is noteworthy that we have never seen cis-decalin structures resulting from the E-isomers of the nitrile substrates.[26[
Scheme 2.
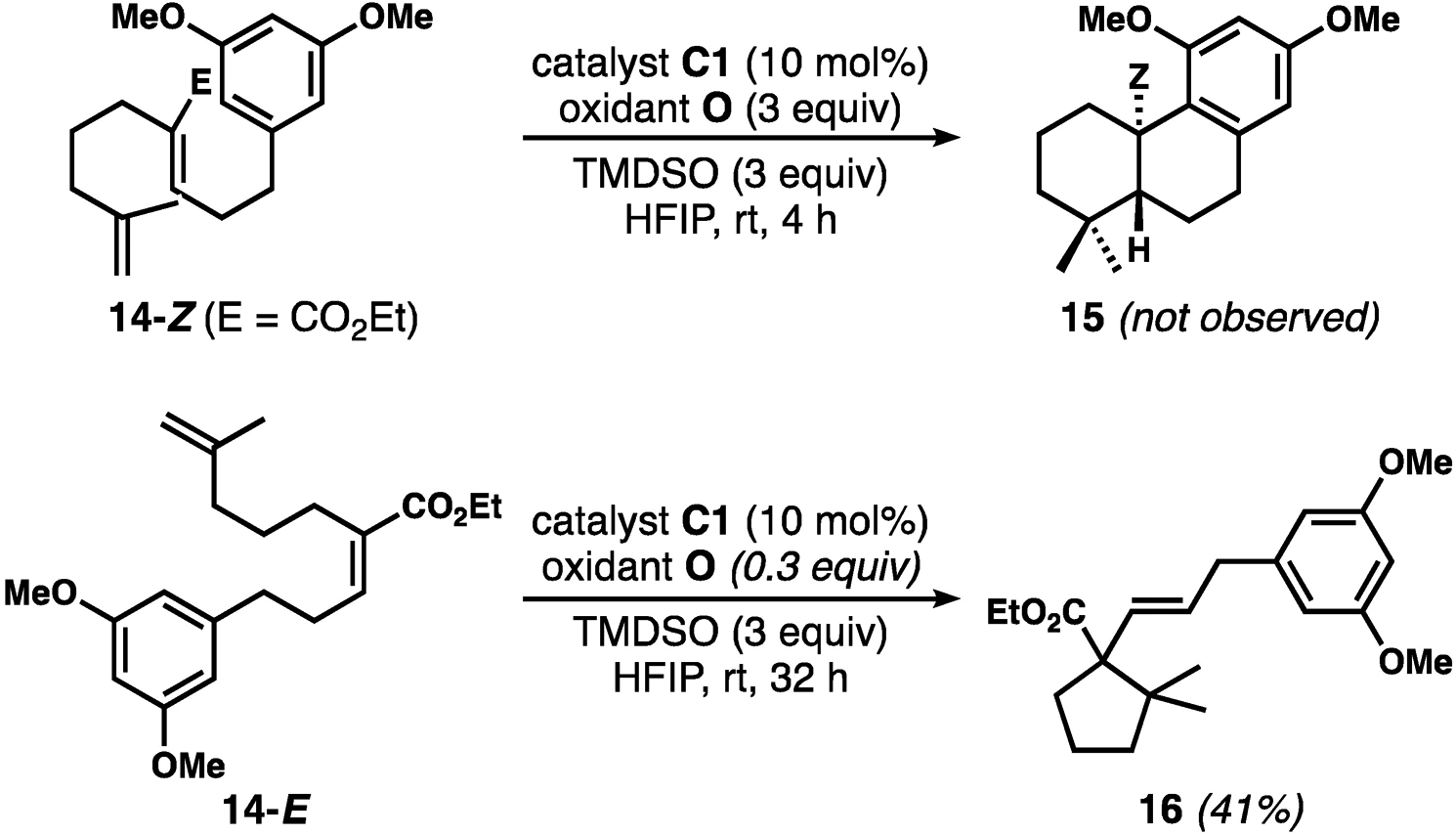
Attempted bicyclizations with acrylate esters as the internal alkene
Due to the high energy of secondary carbocations, we assumed that secondary radicals resulting from HAT to monosubstituted alkenes, such as 17 (Figure 4) could not be oxidized prior to cyclization; if the reaction worked it was expected to involve a Giese-type first cyclization. HAT to monosubstituted alkenes is normally slower than to 1,1-disubstituted alkenes of types 5 and 7 that we had been investigating up to this point;[10,27] nonetheless, the reaction of 17 under our standard conditions yielded a separable mixture of three stereoisomeric products. Surprisingly, the product ratios were found to be dependent on the structure of the cobalt complex. Secondary alkyl-cobalt(III) intermediates are thought to be in equilibrium with secondary radicals formed by MHAT[12,28] and it is therefore plausible that (some of) the catalysts might be associated with the activated substrate after the MHAT event to 17, thus providing a possible explanation for the catalyst-dependent difference in product ratios. We found that the less sterically encumbered cobalt complexes C2, C4, and C5 gave better stereoselectivity (ratio 18:20) for the formation of the first C–C bond, which could be consistent with the bulky catalyst C1 being more dissociated from the organic radical. Conversely, closure of the final ring proceeded with similarly modest selectivity regardless of the catalyst structure (ratio 18+20:19). It is likely that cis-decalin formation (19) is only kinetically feasible without an axial methyl group on C4, which is consistent with the sole formation of trans-decalins with the disubstituted alkene substrates used in Figure 2 and in many other related systems.[26] Collectively, these results suggest that the reaction of 17 is stereoselective but not stereospecific with respect to the trisubstituted alkene and is thus likely to involve radical intermediates; however, this tentative conclusion cannot be directly transferred to the reactions described in Figure 1, because in that case oxidation of the initially formed radical to a tertiary carbocation could reasonably occur. The same terminal alkene initiation site was used with the methyl-substituted internal alkene 20; no characterizable products were observed. Although a bicyclization reaction via radical intermediates might be expected to occur on the basis of our other results, the success of the reaction of 20 requires a competitive HAT to the terminal alkene over the plausibly more reactive electron-rich internal trisubstituted alkene.[10,27]
Figure 4.
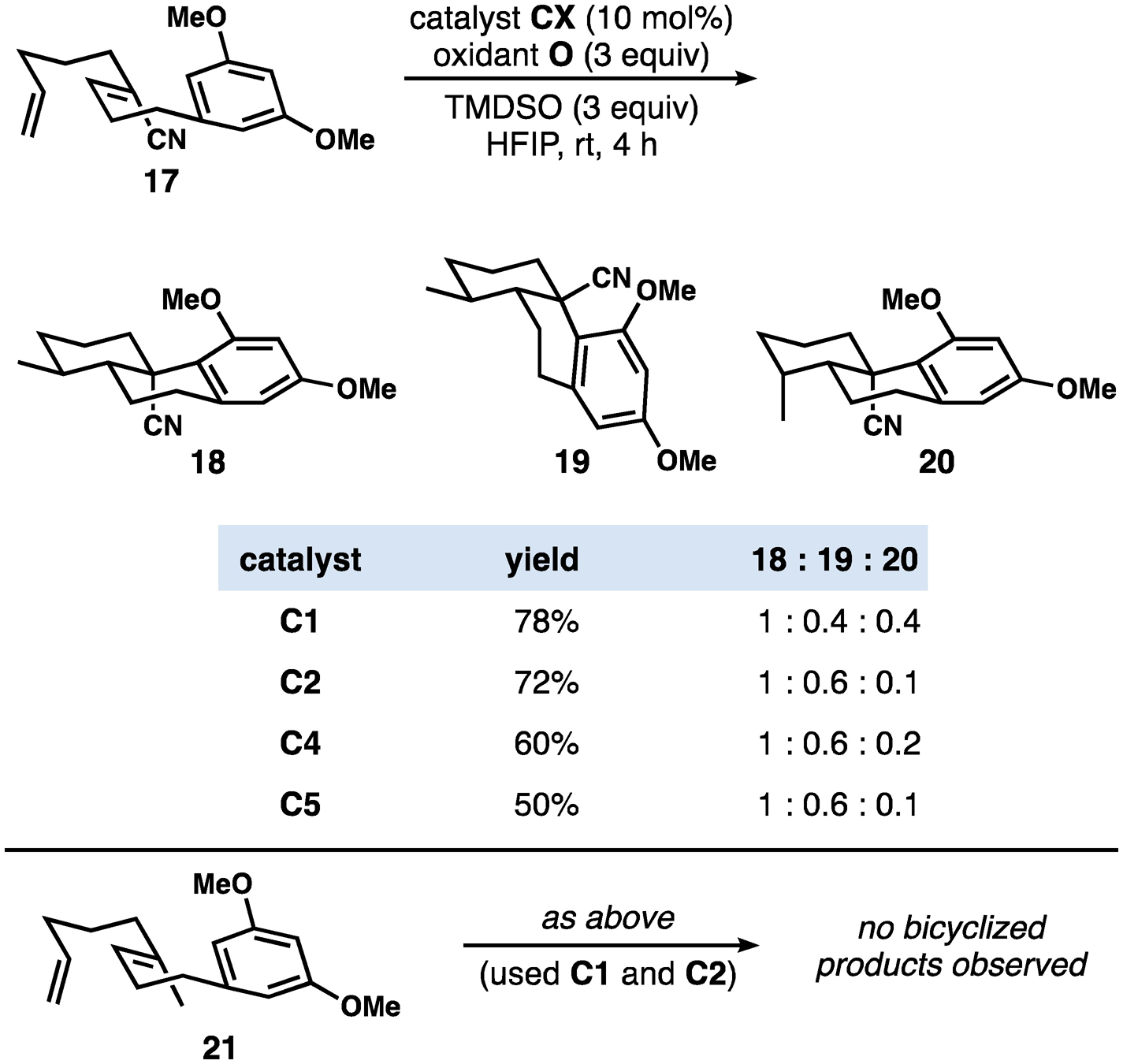
Stereochemical outcomes of bicyclizations initiated by HAT to monosubstituted alkenes (product ratios and yields determined using 1H NMR with an internal standard)
In a further attempt to probe whether or not carbocationic intermediates were involved in these reactions, we submitted carbonate 22 to standard reaction conditions (Scheme 3). No cyclic carbonate products of type 23 were detected; Instead, alkyl fluoride 24 was identified as the major product when catalyst C1 was used. The use of catalyst C2 gave a mixture of alkene isomers where 25 was the major isolated product. These results provide circumstantial evidence against carbocation intermediates; each result corresponds to radical-based transformations previously reported by the Shigehisa/Hiroya group[11a] and the Shenvi group,[15] respectively. Interestingly, we note that the catalysts used in our hydrofluorination and alkene isomerization results are in fact interchanged with those used in the original reports. We also investigated the cyclization of 26, the success of which should require the intermediacy of an α-nitrilo-cation for nucleophilic capture by the pendant hydroxyl group.[29] This reaction yielded a complex mixture of unidentifiable products from which 27 was not observed.
Scheme 3.
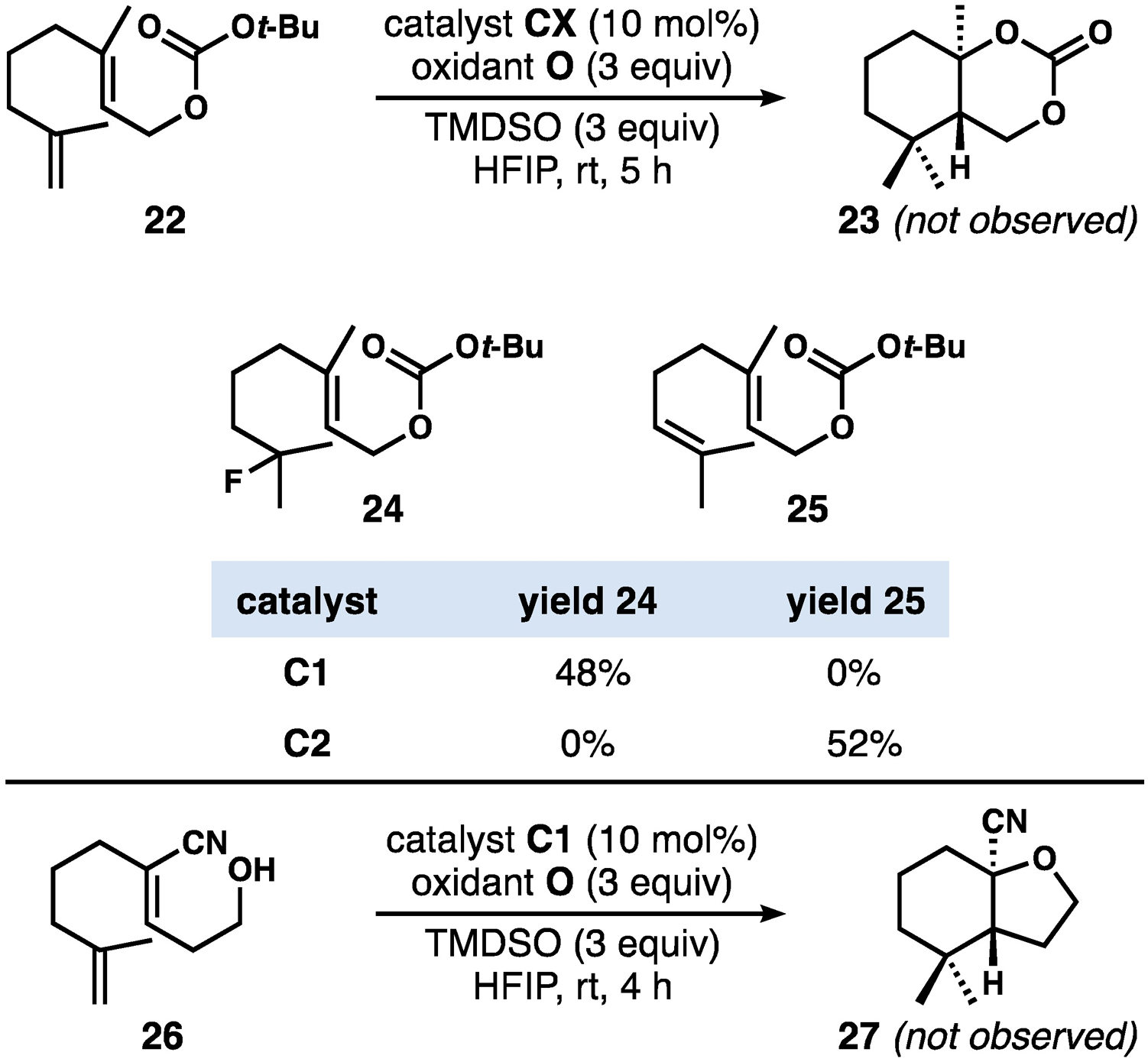
An attempt to probe the intermediacy of cationic intermediates instead supports radical-based reactivity (yields shown are isolated yields of purified compounds)
The bicyclizations with meta-anisole substrates 6a (nitrile) and 8a (methyl) provided products 6b and 8b, respectively, with a remarkably similar, slight preference for second ring closures at the para positions (1.7:1 para/ortho for 6b and 1.5:1 for 8b). While Rendler and MacMillan’s polycyclizations afforded predominantly the ortho regioisomer (2:1 ortho/para) and a radical process is strongly implicated,[3a] and cationic processes normally feature a moderate precedent for cyclization at the para position[30] (consistent with our results), we have trouble rationalizing how our two results could be so similar without the final bond construction proceeding via a radical mechanism in both cases. Simply put, with different mechanisms in the two cases, we would anticipate a different regiochemical outcome; moreover, a cationic mechanism for both would likely also provide different regioselectivities owing to the very different energies of the tertiary alkyl vs. tertiary α-nitrilo cations.
Reaction of a homologue of substrate 7a (extra isoprene unit, see SI) under our standard, optimized conditions failed in a tricyclization attempt—affording alkene isomerization and decomposition)—which is not easy to explain if the process features cationic intermediates, given that 7a itself cyclizes well. Such a result is easier to support with a radical cyclization mechanism, wherein polarity alternation is often required for high-yielding multi-bond-forming processes. For example, Rendler and MacMillan’s study showed that organocatalyzed radical polycyclizations benefit from the alternation of electron-rich and electron-deficient alkenes, leveraging unsaturated nitriles as key components of their substrates.[3a]
At this stage, the majority of our experiments provide support for mechanism C (Scheme 1), in which both C–C bond-forming reactions are radical in nature. We remained interested in further understanding the final stages of the reaction, in which an oxidation must occur. From the postulated cyclohexadienyl radical intermediate 12rad, both oxidation to the corresponding cation 12cat—by either the Co catalyst or the chemical oxidant—or back MHAT from 12rad to a Co(II) intermediate appear reasonable. To investigate, we synthesized pentadeuterated substrate 28 (see Scheme 4) from d6-benzene.[13] If back-MHAT from 12rad were involved, we would expect significant deuterium incorporation at the C18 or C19 methyl groups (or possibly at C3, if some alkene isomerization takes place) because of subsequent MDAT from Co–D. If oxidation to 12cat occurred, then the deuteron should be lost to base in solution, and there would be little expectation of reincorporation. The results of this key experiment showed the conversion of d5-28 into product 29 with >95% d4, strongly supporting an oxidation event followed by proton (deuteron) removal. The isolation of the overall reduction products indolines 6f and 6g are the only outlying data, but a slower deprotonation of the more stable cationic intermediate might allow for competitive ionic reduction by excess silane reagent. Interestingly, deuterated 14-E (14-E-d2) underwent cyclization in poor yield, but with substantial “reincorporation” of deuterium. This Shenvi-type cycloisomerization would be expected to generate intermediate cobalt deuterides for MDAT to the subsequently engaged substrate. Unexpectedly, in this particular case, we found most of the label appeared to be at the methylene rather than the methyl groups, which suggests alkene isomerization prior to cyclization.[13,31]
Scheme 4.
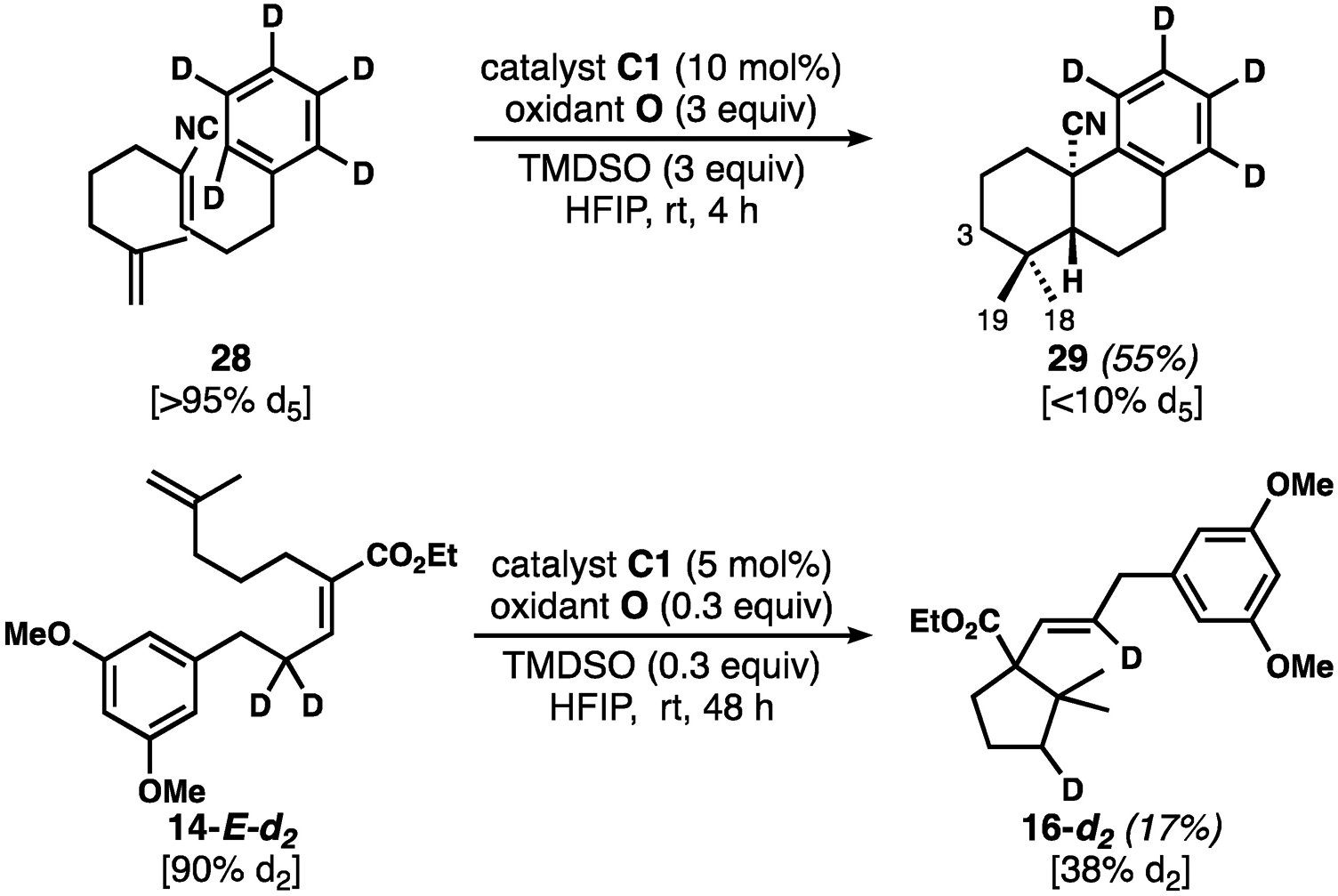
Deuteration experiments suggest oxidation followed by proton transfer, rather than back MHAT, as the final stages of the reactions.
Finally, the importance of HFIP as the solvent in these reactions is worth reiterating. Other common solvents, with the exception of acetone, did not lead to productive reactivity. In acetone, many side products were observed, and the overall reaction efficiency was lower. HFIP is known have powerful effects on cationic processes, but little is known about its ability to alter or improve the course of radical reactions.[32] We do not currently have a solid understanding of the role of this unusual solvent.
On the basis of the majority of our data, we tentatively propose that the two rings are forged via radical processes, followed by a final oxidation of the cyclohexadienyl radical (or equivalent) to the corresponding cation and subsequent proton removal. Overall, the similar behavior of nitrile- and alkyl-substituted systems under nearly identical reaction conditions remains the most interesting aspect of this bicyclization method.
Conclusions.
In summary, we have developed an oftentimes highly efficient HAT-inititated polyene cyclization for the stereocontrolled construction of terpenoid scaffolds. Although the method in its current form is limited to electron-rich terminating arenes, the intermediate alkene substituents can be either electron-withdrawing or donating, which is highly unusual. The reaction conditions are mild and tolerate a variety of functionalities, offering an attractive alternative to the few methods for protonative polyene cyclizations.[1] The experiments that we have performed to date suggest that the cyclization events proceed via radical intermediates, with a late-stage oxidation; however, some questions remain, underscoring the need for more refined experiments to improve our understanding of HAT-initiatied reactions that proceed under oxidative conditions. Finally, the ability to effect bicyclization reactions that directly deliver terpenoid-like scaffolds with C20 preoxidized opens up many opportunities for natural product synthesis;[33] these applications are currently ongoing in our laboratory.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health through NIH R01 grant GM-129264. D.V. was supported by an NSF Graduate Research Fellowship. We thank Dr. Joe Ziller for invaluable assistance with X-ray crystallographic structure determination[34] and Drs. Felix Grun and Vanessa Arredondo for assistance with mass spectrometry determination of isotopic labeling. We are most grateful for the many helpful comments and suggestions of a particularly conscientious reviewer, which led to a much improved paper.
References
- [1]. For an excellent review, see:; Yoder RA, Johnston JN, Chem Rev. 2005, 105, 4730–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. For representative pioneering achievements, see:; (a) Stork G, Burgstahler AW, J. Am. Chem. Soc 1955, 77, 5068–5077; [Google Scholar]; (b) Eschenmoser A, Ruzicka L, Jeger O, Arigoni D, Helv. Chim. Acta 1955, 38, 1890–1904; [Google Scholar]; (c) Johnson WS, Acc. Chem. Res 1968, 1, 1–8; [Google Scholar]; (d) van Tamelen EE, Willet J, Schwartz M, Nadeau R, J. Am. Chem. Soc 1966, 88, 5937–5938. [DOI] [PubMed] [Google Scholar]
- [3]. For selected examples of radical polycyclizations that mirror the biomimetic cationic reactions of the type described in this disclosure, see:; (a) Rendler S and MacMillan DWC, J. Am. Chem. Soc 2010, 132, 5027–5029; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barrero AF, Cuerva JM, Herrador MM, Valdivia MV, J. Org. Chem 2001, 66, 4074–4078; [DOI] [PubMed] [Google Scholar]; (c) Chen L, Gill GB, Pattenden G, Simonian H, J. Chem. Soc., Perkin Trans 1, 1996, 31–43; [Google Scholar]; (d) Zoretic PA, Fang H, Ribeiro AA, J. Org. Chem 1998, 63, 7213–7217; [DOI] [PubMed] [Google Scholar]; (e) Snider BB, Mohan RM, Kates SA, J. Org. Chem 1985, 50, 3659; [Google Scholar]; (f) Breslow R, Olin SS, Groves JT, Tetrahedron Lett. 1968, 9, 1837–1840. [Google Scholar]
- [4]. For selected examples of organometallic polycyclizations that mirror the biomimetic cationic reactions of the type described in this disclosure, see:; (a) Mullen CA, Gagné MR, J. Am. Chem. Soc 2007, 129, 11880–11881. [DOI] [PubMed] [Google Scholar]; (b) Schafroth MA, Sarlah D, Krautwald S, Carreira EM, J. Am. Chem. Soc 2012, 134, 20276–20278. [DOI] [PubMed] [Google Scholar]
- [5].Wang FP, Chen QH, Liu XY, Nat. Prod. Rep 1999, 16, 529–570. [DOI] [PubMed] [Google Scholar]
- [6].(a) Chen K, Shi Q, Fujioka T, Nakano T, Hu C-Q, Jin J-Q, Kilkuskie RE, Lee K-H, Bioorg. Med. Chem 1995, 3, 1345–1348; [DOI] [PubMed] [Google Scholar]; (b) Chen K, Shi Q, Fujioka T, Zhang DC, Hu C-Q, Jin J-Q, Kilkuskie RE, Lee KH, J. Nat. Prod 1992, 55, 88–92. [DOI] [PubMed] [Google Scholar]
- [7].Michalak SE, Nam S, Kwon DM, Horne DA, Vanderwal CD, J. Am. Chem. Soc 2019, 141, 9202–9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou S, Guo R, Yang P, Li A, J. Am. Chem. Soc 2018, 140, 9025–9029. [DOI] [PubMed] [Google Scholar]
- [9].Liu J, Ma D, Angew. Chem. Int. Ed 2018, 57, 6676–6680. [DOI] [PubMed] [Google Scholar]
- [10].Crossley SWM, Obradors C, Martinez RM, Shenvi Chem RA. Rev. 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].(a) Shigehisa H, Nishi E, Fujisawa M, Hiroya K, Org. Lett 2013, 15, 5158–5161; [DOI] [PubMed] [Google Scholar]; (b) Shigehisa H, Koseki N, Shimizu N, Fujisawa M, Niitsu M, Hiroya K, J. Am. Chem. Soc 2014, 136, 13534–13537; [DOI] [PubMed] [Google Scholar]; (c) Shigehisa H, Hayashi M, Ohkawa H, Suzuki T, Okayasu H, Mukai M, Yamazaki A, Kawai R, Kikuchi H, Satoh Y, Fukuyama A, Hiroya K, J. Am. Chem. Soc 2016, 138, 10597–10604; [DOI] [PubMed] [Google Scholar]; (d) Shigehisa H, Ano T, Honma H, Ebisawa K, Hiroya K, Org. Lett 2016, 18, 3622–3625; [DOI] [PubMed] [Google Scholar]; (e) Shigehisa H, Kikuchi H, Hiroya K, Chem. Pharm. Bull 2016, 64, 371–374. [DOI] [PubMed] [Google Scholar]
- [12]. Pronin and colleagues have recently used this catalytic system to convert allylic alcohols to either semi-pinacol or epoxide products, the latter with control of enantioselectivity. See:; (a) Touney EE, Foy NJ, Pronin SV, J. Am. Chem. Soc 2018, 140, 16982–16987; [DOI] [PubMed] [Google Scholar]; (b) Discolo CA, Touney EE, Pronin SV, J. Am. Chem. Soc 2019, 141, 17527–17532. [DOI] [PubMed] [Google Scholar]
- [13]. Please see the Supporting Information for details.
- [14].Obradors C, Martinez RM, Shenvi RA, J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Crossley SWM, Barabé F, Shenvi RA, J. Am. Chem. Soc 2014, 136, 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Crossley SWM, Martinez RM, Guevara-Zuluaga S, Shenvi RA, Org. Lett 2016, 18, 2620–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].(a) Lo JC, Yabe Y, Baran PS, J. Am. Chem. Soc 2014, 136, 1304–1307; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC, Gui J, Yabe Y, Pan C-M, Baran PS, Nature, 2014, 516, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Giese B, Angew. Chem. Int. Ed. Engl 1983, 22, 753–764. [Google Scholar]
- [19].(a) Deng H, Cao W, Liu R, Zhang Y, Liu B, Angew. Chem. Int. Ed 2017, 56, 5849–5852; [DOI] [PubMed] [Google Scholar]; (b) Cao W, Deng H, Sun Y, Liu B, Qin S, Chem. Eur. J 2018, 24, 9120–9129. [DOI] [PubMed] [Google Scholar]
- [20].Kametani T, Kondoh H, Tsubuki M, Honda T, J. Chem. Soc. Perkin 1 1990, 5–10. [Google Scholar]
- [21].González MA, Nat. Prod. Rep 2015, 32, 684–704. [DOI] [PubMed] [Google Scholar]
- [22].Fischedick JT, Standiford M, Johnson DA, Johnson JA, Bioorg. Med. Chem 2013, 21, 2618–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Substrates 7c–7g were used as an 89:11 mixture of the terminal alkene to the internal trisubstituted (geranyl-type) alkene. In control experiments, we found this alkene type to undergo bicyclization reactions under our standard conditions, but significantly more slowly and in depressed yield. Therefore, we would expect the yields of 8c and 8d to be higher than reported in Figure 3 if isomerically pure substrates were obtained and used in these reactions.
- [24].(a) Rosales V, Zambrano JL, Demuth M, J. Org. Chem 2002, 67, 1167–1170; [DOI] [PubMed] [Google Scholar]; (b) Ishibashi H, Ishihara K, Yamamoto H, J. Am. Chem. Soc 2004, 126, 11122–11123. [DOI] [PubMed] [Google Scholar]
- [25].Oswald JP, Woerpel KA, J. Org. Chem 2018, 83, 9067–9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Julia M, Acc. Chem. Res 1970, 4, 386–392. [Google Scholar]
- [27].Ma X, Herzon SB, Chem. Sci 2015, 6, 6250–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stolzenberg AM, Cao Y, J. Am. Chem. Soc, 2001, 123, 9078–9090. [DOI] [PubMed] [Google Scholar]; Shevick SL, Obradors C, Shenvi RA, J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Although unlikely, we note that some mesomeric stabilization of α-nitrilo cations is expected to partially offset the inductively destabilizing effect of this functional group. See:; Creary X, Chem. Rev 1991, 91, 1626–1678. [Google Scholar]
- [30]. For a relevant recent example of an epoxide-initiated cationic polycyclization, also carried out in HFIP (ortho/para = 1.6:1), see.; Tian Y, Xu X, Zhang L, Qu J, Org. Lett 2016, 18, 268–271. [DOI] [PubMed] [Google Scholar]
- [31]. We have investigated the reactivity of internal, “geranyl-type” trisubstituted alkenes as initiators in our reactions, and found them to be competent substrates; however, the reactions are slower and lower yielding, with the formation of a multitude of side products.
- [32].(a) Bégué J-P, Bonnet-Delpon D, Crousse B, Synlett 2004, 18–29; [Google Scholar]; (b) Shuklov IA, Dubrovina NV, Börner A, Synthesis 2007, 2925–2943; [Google Scholar]; (c) Colomer I, Chamberlain AER, Haughey MB, Donohoe TJ, Nat. Rev. Chem 2017, 1, 0088. [Google Scholar]
- [33]. For recent examples of MHAT-induced cyclization reactions in the synthesis of complex natural products, see:; (a) Ji Y, Xin Z, He H, Gao S, J. Am. Chem. Soc 2019, 141, 16208–16212; [DOI] [PubMed] [Google Scholar]; (b) Zhang B, Zheng W, Wang X, Sun D, Li C, Angew. Chem. Int. Ed 2016. 55, 10435–10438; [DOI] [PubMed] [Google Scholar]; ref. 19.
- [34]. The X-ray crystallographic structures of 6e and 6g have been deposited with CCDC numbers 1980218and 1980219, respectively.
Figure 3.
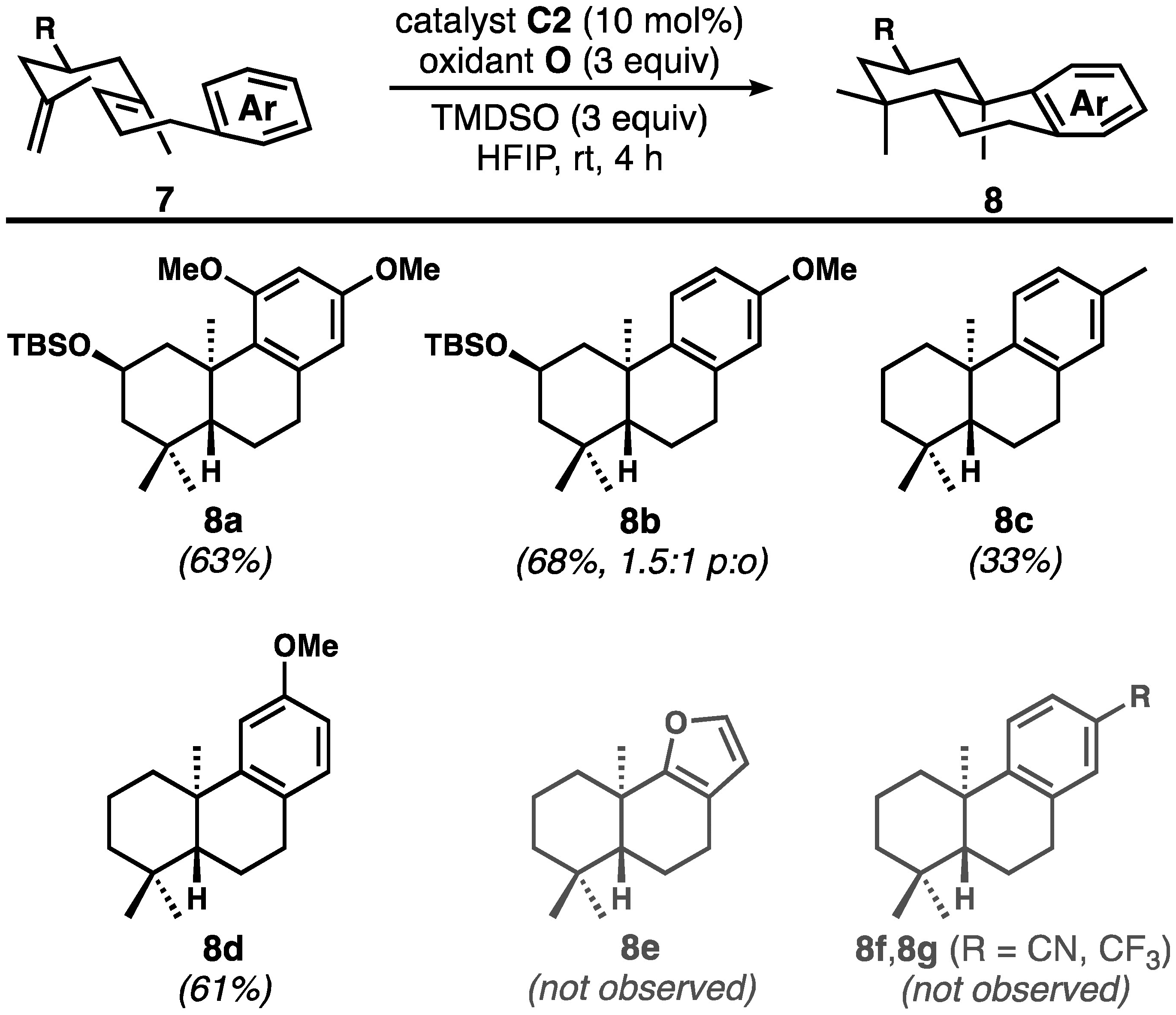
The reactivity of methyl-substituted substrates mirrors that of the corresponding nitrile-bearing compounds (yields shown are for isolated and purified compounds)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.