

Abstract
Folate supplementation in F0 mating rodents increases regeneration of injured spinal axons in vivo in 4 or more generations of progeny (F1-F4) in the absence of interval folate administration to the progeny. Transmission of the enhanced regeneration phenotype to untreated progeny parallels axonal growth in neuron culture after in vivo folate administration to the F0 ancestors alone, in correlation with differential patterns of genomic DNA methylation and RNA transcription in treated lineages. Enhanced axonal regeneration phenotypes are observed with diverse folate preparations and routes of administration, in outbred and inbred rodent strains, and in two rodent genera comprising rats and mice, and are reversed in F4-F5 progeny by pretreatment with DNA demethylating agents prior to phenotyping. Uniform transmission of the enhanced regeneration phenotype to progeny together with differential patterns of DNA methylation and RNA expression is consistent with a non-Mendelian mechanism. The capacity of an essential nutritional cofactor to induce a beneficial transgenerational phenotype in untreated offspring carries broad implications for the diagnosis, prevention, and treatment of inborn and acquired disorders.
Keywords: Transgenerational inheritance, central nervous system (CNS), spinal cord injury, axonal regeneration, DNA methylation, epigenetics, folic acid
Introduction
Parental diet and environmental exposures may influence the health of human offspring to the F2 generation and beyond with patterns of inheritance that are inconsistent with DNA sequence variation [1]. Because transmission of an altered trait from an exposed gestating dam (F0) to the F1 and F2 generations also comprises exposure to both the F1 embryo and to its F2 germ-line, evidence of transmission to the F3 generation is necessary to assure that transgenerational inheritance is independent of exposure other than to the F0 mother [2]. In males, exposure of the F1’s F2 germ-line does not occur, and therefore paternal transgenerational inheritance may be confirmed by presence of a trait in the F2 generation [3]. Accordingly, inheritance of impaired stress responses and anxiety [4,5], spermatogenetic capacity and infertility in males [6,2], as well as early puberty and decreased ovarian primordial follicle pool size in females [7] have been reported in up to 4 consecutive generations (i.e., F1-F4) after administration of toxins and endocrine disruptors confined to an F0 progenitor. Whereas manipulation of specific chromatin modifiers in C. elegans parents enhances longevity in progeny to the F3 generation [8], non-genomic, transgenerational transmission of a beneficial trait beyond the F2 generation in response to environmental stimuli has not been identified in mammalian species.
Folate promotes axonal regeneration in the spinal cord of adult rodents after sharp injury, and enhances functional recovery after blunt trauma [9]. In view of evidence that the beneficial effect of folic acid may be mediated at least in part through DNA methylation [10], and that altered folate metabolism may underlie methylation-related, transgenerational consequences during development [11,12], we hypothesized that central nervous system (CNS) repair mechanisms may be enhanced by non-genomic, transgenerational mechanisms after folate supplementation in a first parental generation alone. We report that substantial increments in spinal cord axon regeneration after sharp trauma are transmitted to serial generations of the progeny without interruption after folate supplementation during F0 gestation and weaning, and with no folate administration to intervening generations. Of particular note, inheritance of enhanced axonal regeneration in vivo is independent of folate preparation (folic acid vs. 5-methyltetrahydrofolate) and parallels enhanced in vitro axon growth, and with differential patterns of genomic DNA promoter methylation and differential patterns of RNA transcription.
Results
Intraperitoneal folate supplementation of outbred rats promotes in vivo regeneration of injured spinal axons in untreated progeny; the effect is reversed with both the histone deacetylase (HDAC) inhibitor trichostatin A (TSA), and the DNA methyltransferase (DNMT) inihibitor 5-aza-2’-deoxycytidine (5-azadCyD).
Intraperitoneal (IP) folate supplementation promotes axonal regeneration mediated by altered DNA methylation in the injured spinal cord of adult rodents, and the effect is biphasic in dose with an optimal response observed at 80μg/kg [9,10]. In order to determine whether the effect of folate is heritable, we administered 80μg/kg intraperitoneal (IP) doses of either folic acid or (6S)-5-methyltetrahydrofolate (methylfolate) to F0 mating rats starting 14 days before mating, and daily thereafter until the pups were weaned at approximately day 21. Methylfolate is the predominant form of folate in the circulation and in cellular metabolism. Unlike folic acid, methylfolate does not require enzymatic conversion for activation [13]. We conducted non-sibling mating of offspring to create F1-F5 (folic acid) or F1-F3 (methylfolate) generations with no additional intervening folate supplementation (Fig. 1A). We gave a second set of 3 mating pairs daily IP water (DDI) injections, and used them to generate a control lineage. Only F0 animals directly received either active compounds or vehicle alone IP. We otherwise treated progeny animals (F1-F4) identically with no IP supplementation. After mating, adult males and females (Fig. 1A) from the F1-F4 generations underwent sharp injury to the cervical spinal cord as previously described (Figs. 1B–D) [10]. The proportions of regenerated axons in F3 and/or F4 male animals descended from F0 progenitors given IP folic acid (Fig. 1E) or methylfolate (Fig. 1F) were 3- to 6-fold greater than in animals descended from F0 progenitors given IP vehicle alone. We observed no differences in axonal regeneration between F3 males and females (data available upon request). To determine whether the inherited regeneration phenotype is dependent on DNA methylation, we used 2 different inhibitors of DNA methylation: 1) F5 animals that descended from folic acid-treated F0 progenitors received 17 days of daily IP trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor that impairs DNA methylation [14]; 2) F3 animals descended from folic acid-treated F0 progenitors received 17 days of daily IP 5-aza-2’-deoxycytidine (5-azadCyD), a DNA methyltransferase (DNMT) inhibitor [15]. Both TSA-treated and 5-azadCyD-treated animals failed to regenerate spinal axons into peripheral nerve grafts in vivo (Figs. 2A,B).
Fig 1. Folate supplementation of Sprague-Dawley rat progenitors enhances in vivo regeneration of injured CNS axons into a peripheral nerve graft in untreated transgenerational progeny.
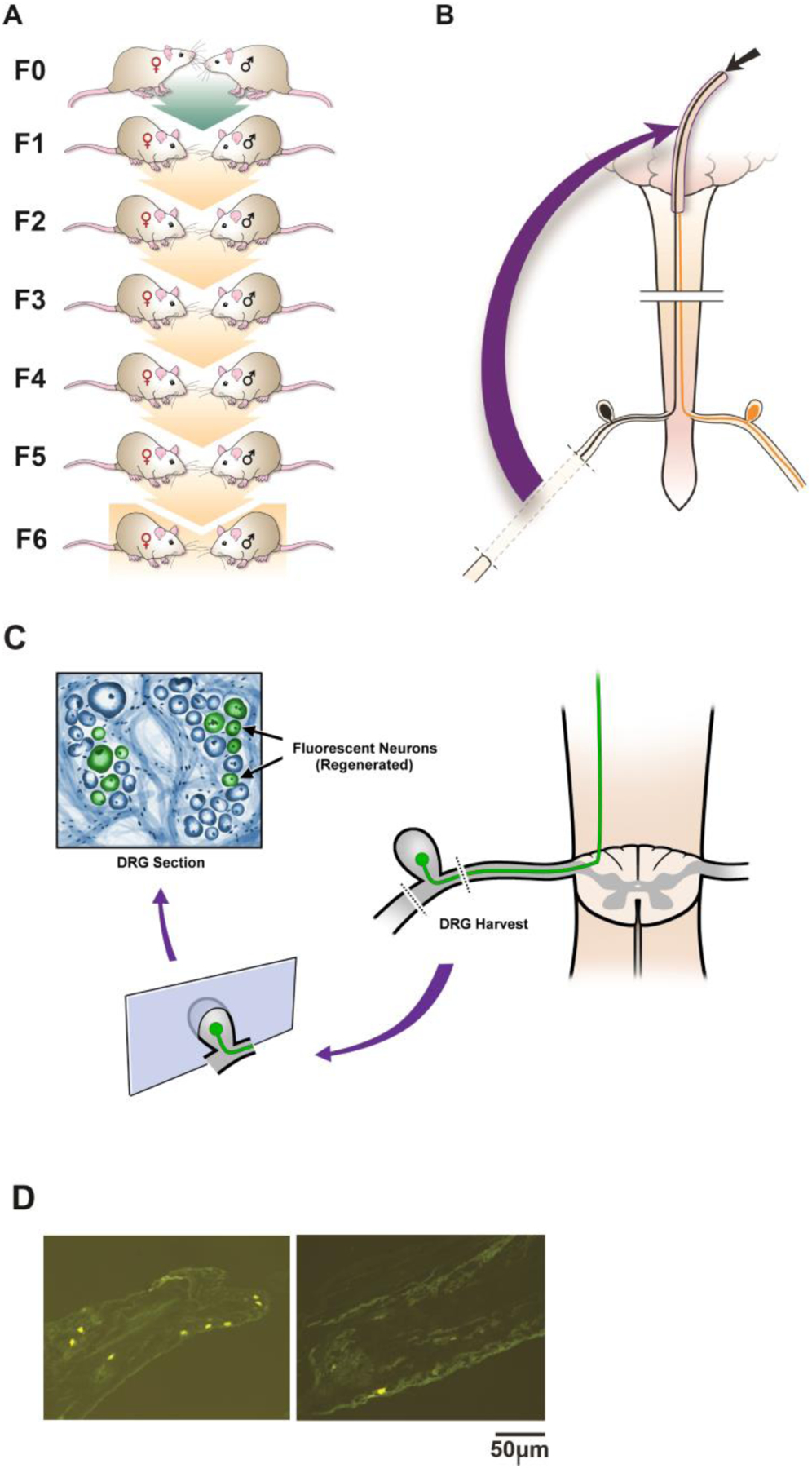
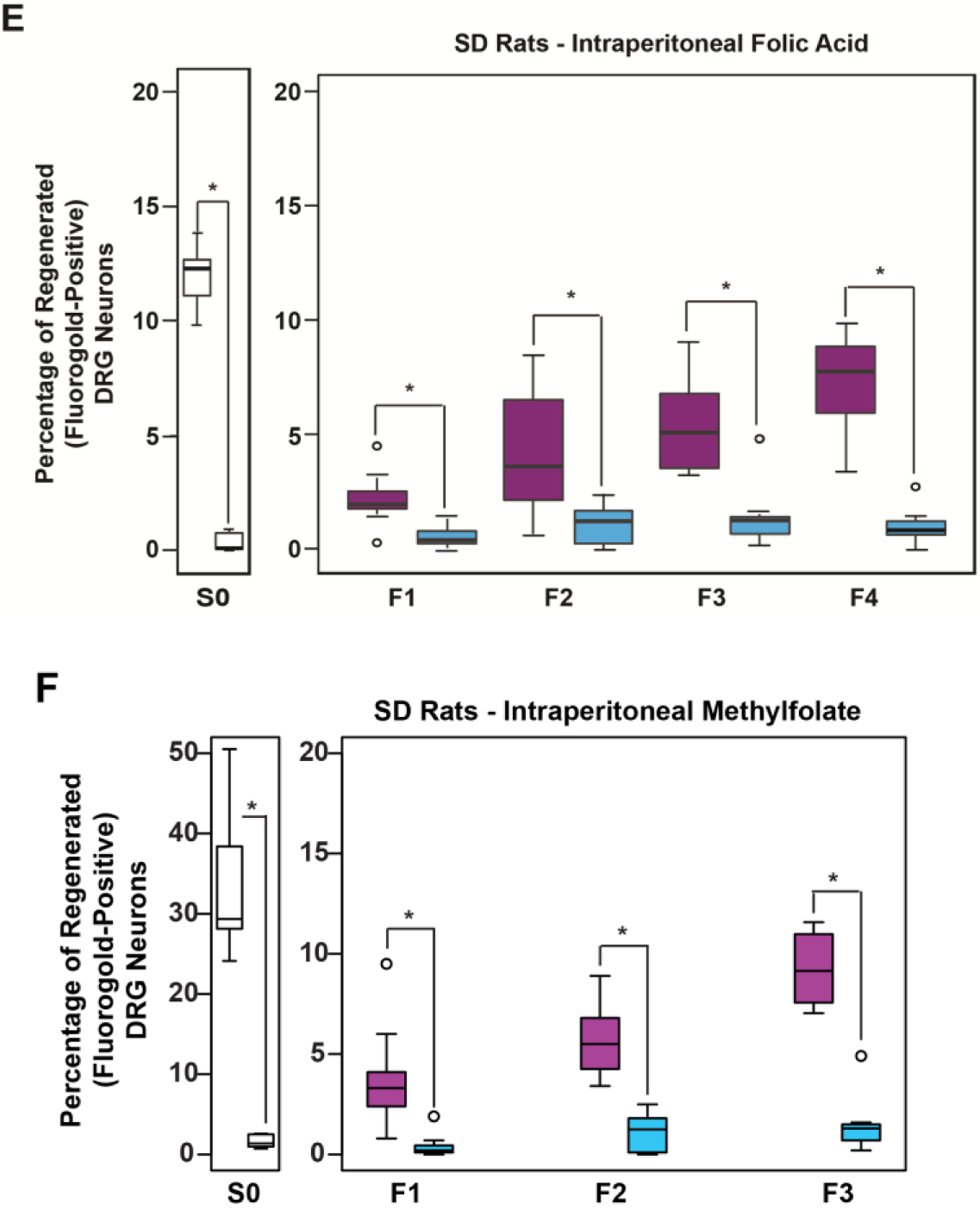
(A-C) Rat Spinal Cord Regeneration Model(SCRM): Experimental Design Schematic: (A) Mating pairs of animals (F0) were treated with folate or vehicle starting 2 weeks before breeding, and continuing in females until weaning, and in males until pregnancy was assured. Four to six generations of progeny (depending on intervention) were bred without treatment. (B) A sciatic nerve graft is implanted at the site of bilateral C3 dorsal column lesion (purple arrow). At 2 weeks, a fluorescent tracer is placed at the free end of the graft (black arrow), and (C,D) its uptake is detected 48 hours later in the lumbar DRG neuron cell bodies of axons that have extended into the graft ipsilateral to sciatic nerve harvest. The fluorescent tracer is taken up only by regenerated neurons. (E) Intraperitoneal (IP) folic acid supplementation of progenitors enhances spinal axon regeneration in untreated F1-F4 male progeny. Percentage of DRG neurons of the untreated offspring (F1-F4) of treated progenitors that have regenerated spinal axons into sciatic nerve grafts compared to DDI controls. Single generation control animals (S0) with direct exposure to folate supplementation exhibit approximately 12% regeneration. Animals whose progenitors (F0) were supplemented with folate regenerate injured spinal axons for 4 generations (n: (FA-purple, DDI-blue) = S0: 8, 8; F1: 12, 8; F2: 16, 10; F3: 10, 8; F4: 10, 10; *p<0.05). (F) Intraperitoneal methylfolate supplementation of progenitors enhances spinal axon regeneration in untreated F1-F3 progeny. Percentage of DRG neurons of the untreated offspring (F1-F3) of treated progenitors that have regenerated spinal axons into the sciatic nerve grafts compared to DDI controls (n (MF - purple, DDI- blue) = S0: 12, 7; F1: 14, 11; F2: 12, 10; F3: 8, 8; *p<0.05).
Figure 2. The folate-induced transgenerational phenotype is reversed with a histone deacetylase (HDAC) inhibitor and the DNA methyltransferase (DNMT) inihibitor 5-aza-2’-deoxycytidine (5-azadCyD).
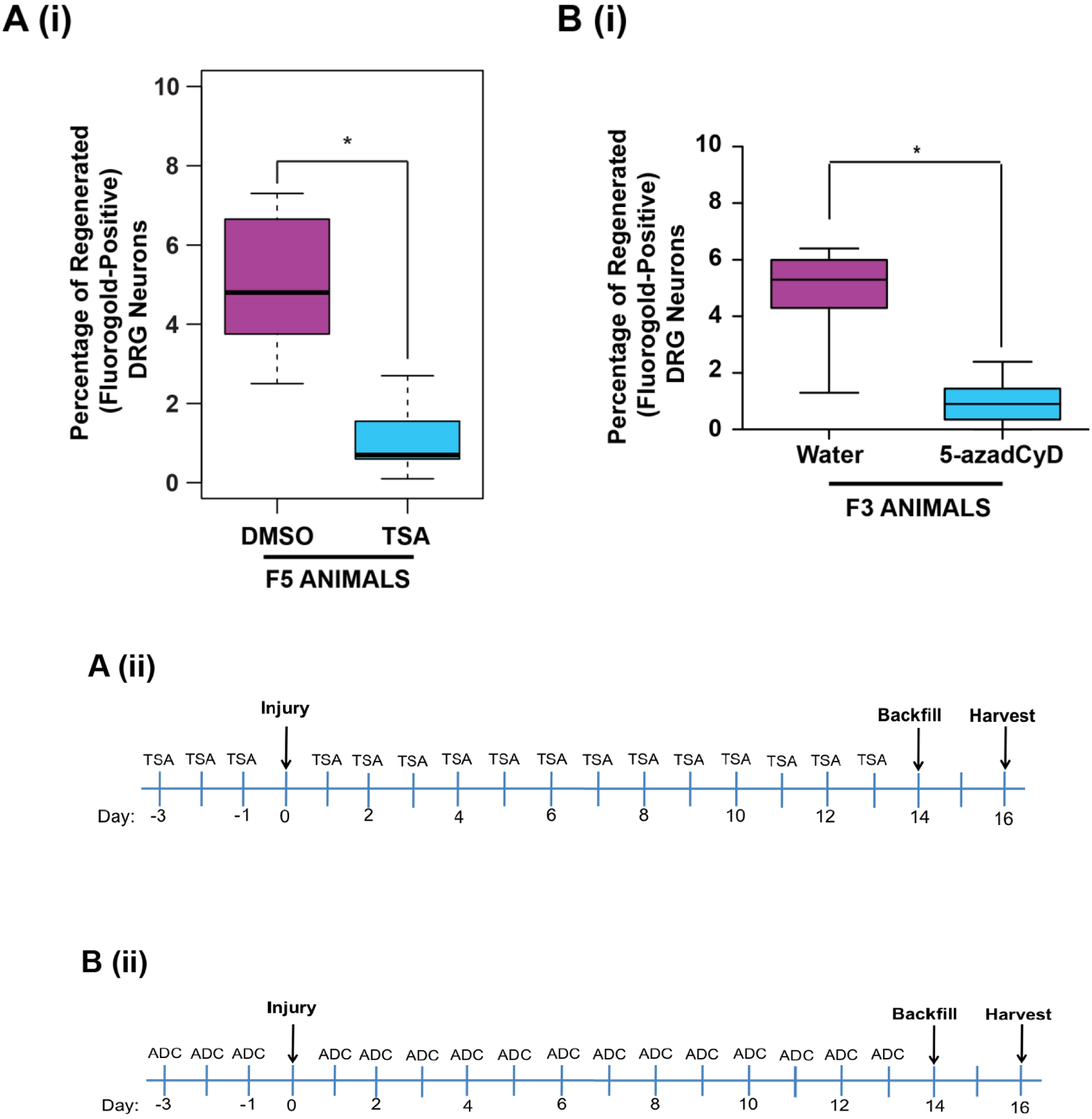
(A(i)) Intraperitoneal TSA treatment of F5 progeny from folic acid-treated F0 lineage with decreased axonal regeneration after spinal cord injury. Male F5 generation animals with F0 progenitors that received IP folic acid supplementation (80 μg/kg) were administered 15 days of IP histone deacetylase (HDAC) inhibitor trichostatin A (TSA; 0.5 mg in 33% DMSO) with SCRM surgery on day 3 (timeline – A(ii)). The graph shows the percentage of DRG neurons of the DMSO-only and TSA-DMSO F5 offspring of folate-supplemented progenitors that have regenerated spinal axons into the sciatic nerve grafts; n (DMSO-purple, TSA-blue) = 11, 11. (B(i)) Intraperitoneal 5-aza-2’-deoxycytidine treatment of F3 progeny from folic acid-treated F0 lineage with decreased axonal regeneration after spinal cord injury. Male F3 generation animals with F0 progenitors that received IP folic acid supplementation (80 μg/kg) were administered 15 days of DNMT inhibitor 5-aza-2’-deoxycytidine (0.1mg/kg in water) with SCRM surgery on day 3 (timeline – B(ii)). The graph shows the percentage of DRG neurons of the water-only and 5-azadCyD in water F3 offspring of folate-supplemented progenitors that have regenerated spinal axons into the sciatic nerve grafts; n (Water-purple, 5-AzadCyD-blue) = 11, 12. Y axis scales differ between the methylfolate S0 and transgenerational graphs. Boxes extend from the 25th to 75th percentiles, and whiskers from the minimum to the maximum, except for outliers indicated by a circle. Comparisons of differences within generations were made using the Wilcoxon rank-sum test, *p<0.05 (FA: folic acid, MF: methylfolate, ADC, 5-azadCyD: intraperitoneal 5-aza-2’-deoxycytidine; TSA: intraperitoneal trichostatin A; DMSO: dimethyl sulfoxide vehicle control for TSA; S0: unbred single generation control; F1-F4: generation number).
Oral methyl supplementation of outbred rats promotes in vivo regeneration of injured spinal axons in untreated progeny.
To test whether the transgenerational, pro-regenerative response to folate is reproducible with alternate routes of administration of folate, mating pairs (F0) of Sprague-Dawley outbred rats were fed a diet (NIH-31 7017-MS) supplemented with folic acid and other methyl donor compounds (choline, betaine, and vitamin B12) ad libitum, and compared the phenotypes of their unsupplemented F1-F6 progeny (Figs. 1A) with the phenotypes of the unsupplemented F1-F4 progeny of F0 rats fed an identical diet lacking methyl supplementation (NIH-31 7017). F1-F6 progeny (F1-F6) were maintained on a standard laboratory diet (Harlan Rodent Diet #8604). In the methyl-supplemented diet lineage, enhanced spinal cord regeneration after sharp traumatic injury is transmitted through 6 generations of untreated animals bred before phenotyping (Fig. 3A). Notably, the difference between folic acid treated progeny and control progeny increases with every subsequent generation, consistent with the “wash-in” phenomenon recognized in epigenetic inheritance [16].
Fig. 3. Folate-induced transgenerational enhancement of CNS axon regeneration is independent of route of administration, breeding protocol, and genus.
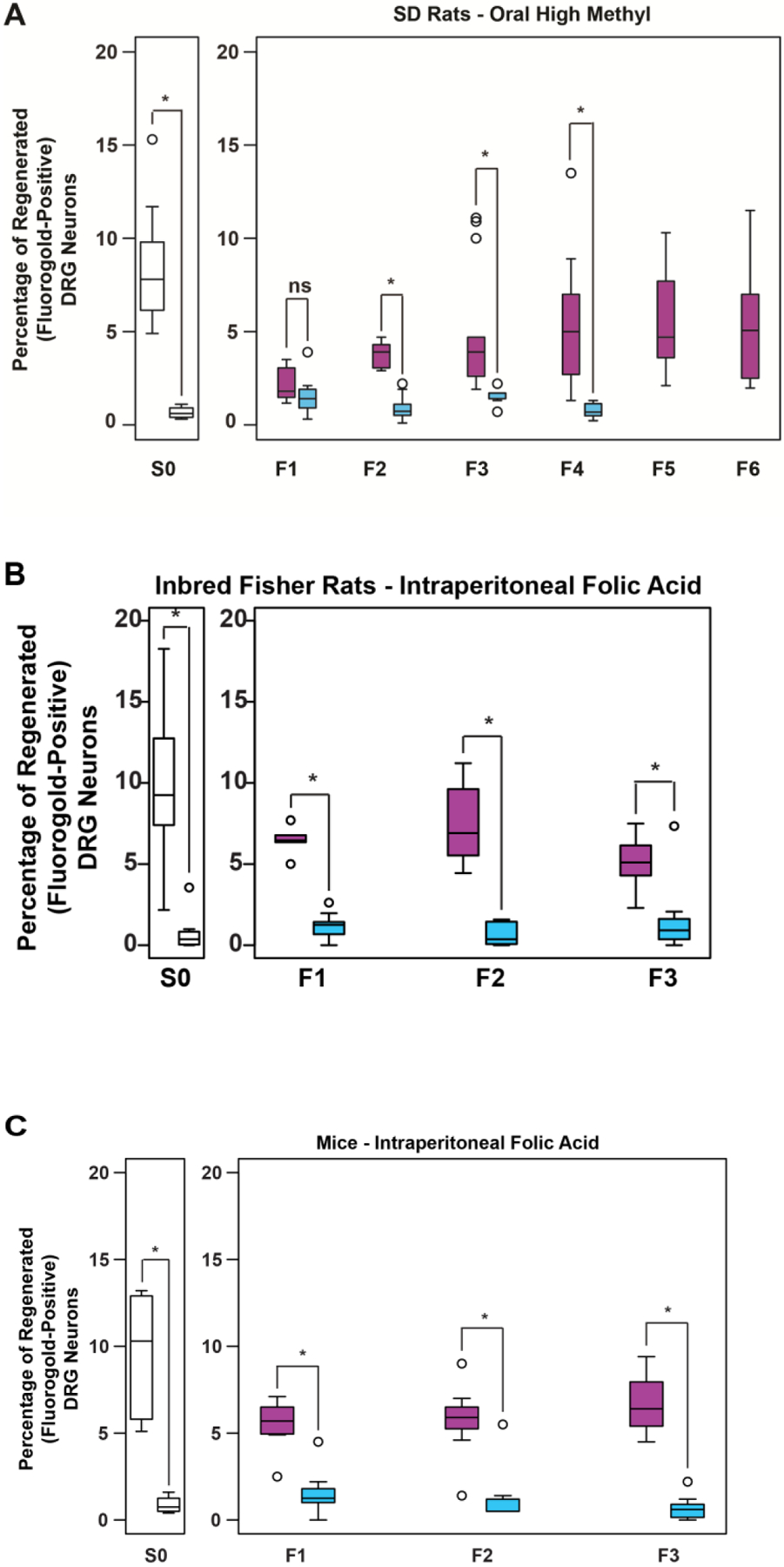
(A) Oral folate supplementation of progenitors enhances spinal axon regeneration in F1-F6 progeny on an unsupplemented diet. Graph shows the percentage of DRG neurons that regenerated into sciatic nerve grafts in S0 animals and the untreated offspring (F1-F6) of F0 progenitors with a methyl-supplemented diet, compared to an un-supplemented ‘normal’ diet (F1-F4). (n (high methyl-purple, normal-blue) = S0: 13, 6; F1: 7, 9; F2: 11, 9; F3: 14, 11; F4: 9, 10; F5: 10, - ; F6: 9, - ; the “normal diet” lineage was not bred beyond F4; *p<0.05; The high methyl diet includes supplemental folic acid and other methyl donor compounds comprising betaine, choline, and vitamin B12). (B) Folate supplementation of inbred Fisher rat progenitors enhances in vivo regeneration of injured spinal axons in untreated F1-F3 progeny. Percentage of DRG neurons of the S0 animals and the untreated offspring (F1-F3) of folate supplemented F0 progenitors that have regenerated spinal axons into the sciatic nerve grafts (n (FA-purple, DDI-blue) = S0: 12, 11; F1: 5, 11; F2: 8, 9; F3: 11, 11; *p<0.05). (C) Folate supplementation of mouse progenitors enhances in vivo regeneration of injured spinal axons in untreated F1-F3 progeny. Percentage of DRG neurons of S0 animals and the untreated offspring (F1-F3) of folate supplemented progenitors that have regenerated spinal axons into the sciatic nerve grafts (n (FA-purple, DDI-blue) = S0: 6, 4; F1: 7, 10; F2: 7, 9; F3: 7, 7; *p<0.05). Boxes extend from the 25th to 75th percentiles, and whiskers from the minimum to the maximum, except for outliers indicated by a circle. Comparisons of differences within generations were made using the Wilcoxon rank-sum test, *p<0.05 (S0: unbred single generation control).
Intraperitoneal folate supplementation of inbred rats promotes in vivo regeneration of injured spinal axons in untreated progeny.
To test whether transgenerational inheritance of enhanced axonal regeneration is an artifact of breeding in outbred strains, an identical experimental protocol in a lineage of inbred Fisher rats was conducted. Compared to the progeny of F0 ancestors treated with vehicle only, increased axonal regeneration to the F3 generation was observed in Fisher animals with F0 progenitors treated with IP folic acid (Fig. 3B).
Intraperitoneal folate supplementation of outbred mice promotes in vivo regeneration of injured spinal axons in untreated progeny.
To test whether the heritable, pro-regenerative response to folate observed in rats is confined to a single genus, identical experiments were performed in outbred CD-1 mice. F3 male mice with F0 ancestors exposed to folic acid exhibit 7-fold greater spinal axon regeneration compared to vehicle-only control mice (Fig. 3C).
In vivo intraperitoneal and oral folate supplementation of outbred rats promotes in vitro elongation of injured spinal axons in untreated progeny; the effect is reversed with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA).
To test whether the heritable axon growth phenotype is intrinsic to neurons [17] or, in the alternative, whether the phenotype arises from mediators extrinsic to neurons such as the glial scar, growth factors, and cell-cell interactions [18], we excised lumbar dorsal root ganglia (DRGs) post-mortem from F3 and F4 animals of the breeding lineages described in the in vivo experiments above after sharp injury to the spinal cord 3 days before excision. We dissociated the DRG neurons and placed them in a culture medium permissive for growth as previously described [19,10,20]. DRG neurons from F3 progeny descended from F0 progenitors exposed to IP folic acid grow markedly longer axons in vitro at 48 hours than corresponding DRG neurons from F3 progeny descended from control F0 progenitors (Fig. 4A,C), denoting that intrinsic neuronal mechanisms are responsible at least in part for the transgenerational, pro-regenerative effect that is independent of glial and other cellular factors such as vascular elements, dura, and circulating metabolites. DRG neurons from F4 progeny descended from F0 progenitors exposed to a high methyl diet grow markedly longer axons at 24 hours than DRG neurons from F4 progeny descended from control F0 progenitors (Fig. 4B,C). Dense growth and small sample sizes obscure detection of significant differences at 48 hours. Exposure of cultures of DRG neurons derived from F4 progeny of the folate-treated F0 lineage to TSA in the medium prior to culture initiation reverses the in vitro, pro-regenerative effects (Fig. 4D).
Fig 4. In vivo folate supplementation of F0 Sprague-Dawley rat progenitors enhances in vitro axonal elongation of cultured DRG neurons from untreated F3 and F4 progeny; the effect is reversed with an HDAC inhibitor.

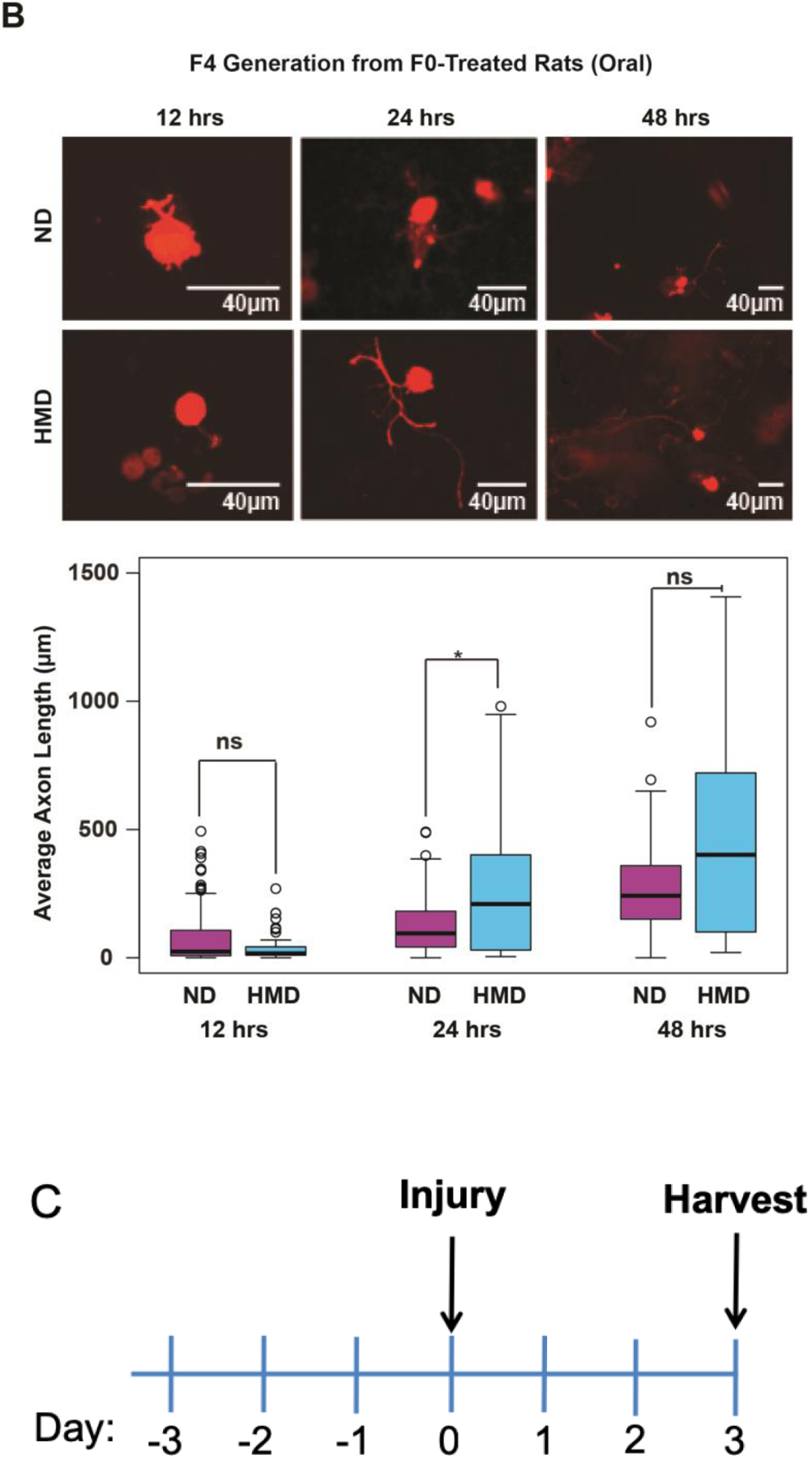
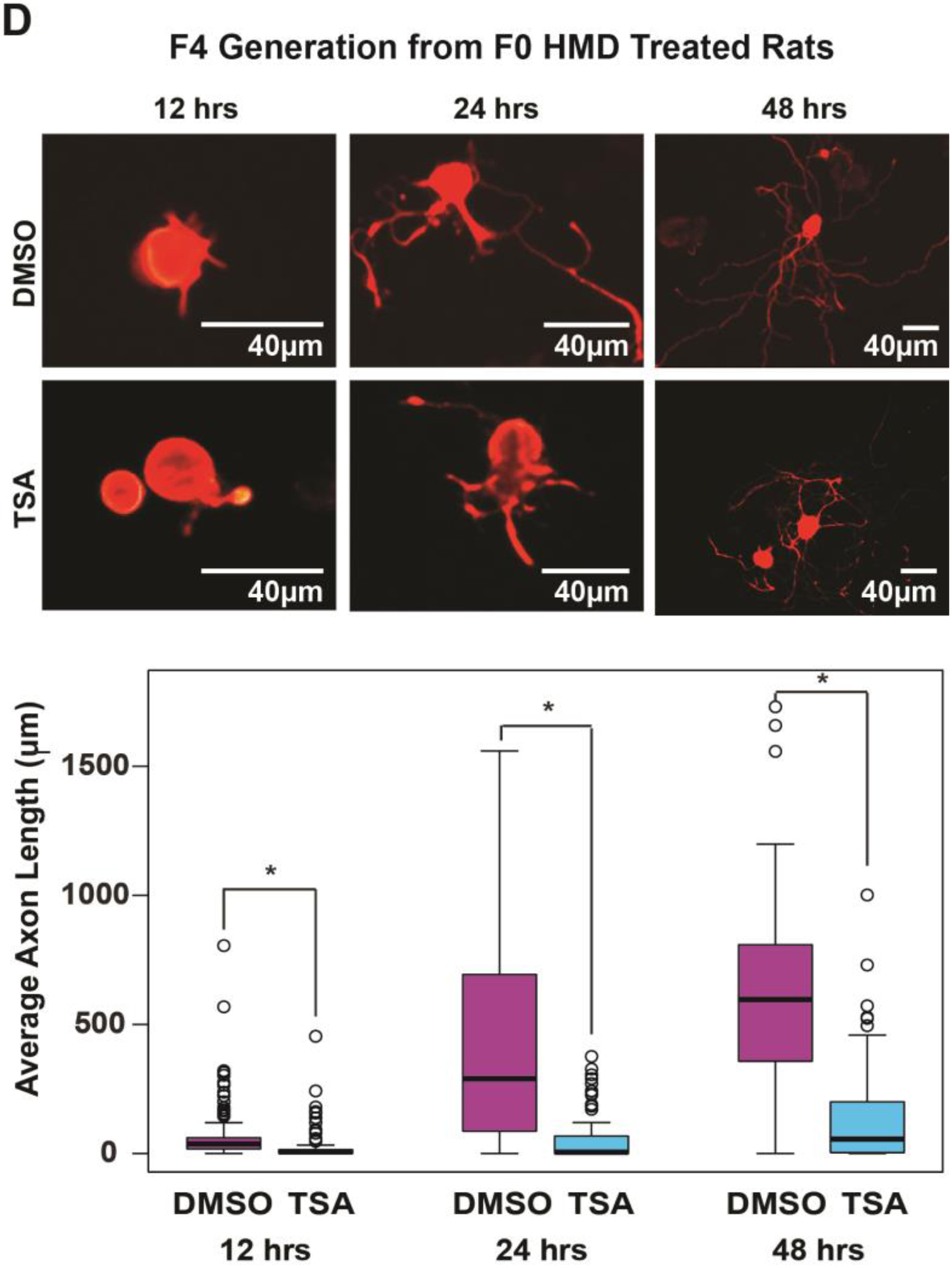
Twelve to 48 hours after a bilateral in vivo sharp injury to the dorsal columns of the spinal cord, 6 lumbar DRGs (L4-L6) containing the cell bodies of the injured axons were removed from each of 3 rats and pooled, dissociated, and placed in a neuronal culture medium that sustains growth. The cultured neurons were subsequently divided into 2 to 4 replicate pools, and each pool was separated into thirds for use at 12, 24, and 48-hour culture experiments, respectively. In each experiment, 4 images were selected at random from each of 4 quadrants as described in the text. The longest neurite was measured in all cell bodies identified. Average axon lengths were calculated for each of the 3 replicates. (A) F3 axon elongation in vitro after F0 IP folic acid in vivo: At 48 hours, DRGs obtained from F3 animals of F0 progenitors exposed to IP folic acid have longer average axon lengths compared to cells from progenitors that were treated with DDI alone (original magnifications 20X; see treatment timeline in Fig. 4C). Intervention: timepoint: number of replicates (number of cells examined in each replicate): Folic acid (blue): 12hr: n = 4 (56, 43, 72, 23); 24hr: n = 4 (21, 19, 10, 30); 48hr: n = 3 (43, 25, 36); DDI (purple): 12hr: n = 4 (33, 58, 15, 34); 24hr: n = 4 (2, 11, 3, 27); 48hr: n = 4 (64, 35, 27, 44); *p<0.05, ns – not significant. (B) F4 axon elongation in vitro after F0 oral methyl supplement in vivo: At 24 hours, DRGs obtained from F4 animals of F0 progenitors exposed to a high methyl diet have significantly larger average axon lengths compared to those whose F0 progenitors were exposed to DDI. Dense growth and small sample sizes obscure detection of significant differences at 48 hours; original magnifications 20X; see treatment timeline in Fig. 4C). Intervention: timepoint: number of replicates (number of cells examined in each replicate): high methyl diet (blue): 12hr: n = 4 (26, 16, 10, 8); 24hr: n = 4 (34, 20, 30, 44); 48hr: n = 2 (21, 9); normal diet (purple): 12hr: n = 3 (62, 62, 66); 24hr: n = 3 (76, 110, 42); 48hr: n = 2 (53, 70) ); *p<0.05, ns – not significant. (D) F4 axon elongation in vitro after F0 oral methyl supplement and F4 IP HDAC inhibitor in vivo: The effect of folic acid was reversed when TSA was added to the culture medium, resulting in shorter axons at 12, 24 and 48 hours (original magnifications 20X). Intervention: timepoint: number of replicates (number of cells examined in each replicate): F0 folic acid with F4 DMSO (purple): 12hr: n = 4 (49, 37, 29, 25); 24hr: n = 4 (15, 22, 28, 18); 48hr: n = 4 (20, 6, 20, 19); F0 folic acid with F4 TSA (blue): 12hr: n = 4 (25, 30, 20, 50); 24hr: n = 4 (32, 53, 16, 20); 48hr: n = 4 (26, 22, 28, 22). Boxes extend from the 25th to 75th percentiles, and whiskers from the minimum to the maximum, except for outliers are indicated by a circle. Comparisons of differences within generations were made using the Wilcoxon rank-sum test (FA: folic acid, DDI: Distilled de-ionized water control, ND: normal diet, HMD: high methyl diet, TSA: trichostatin A).
The greater proportion of spinal axon regeneration after injury in the progeny of folic-acid treated compared to untreated F0 progenitors is not caused by random selection of a founder effect, and is inconsistent with a Mendelian genetic model.
In theory, variations in regeneration proportions may arise from DNA sequence variations entering one or more founders and segregating in the experimental pedigrees. The presence of such genetic factors may induce dependencies between measurements, and thereby complicate assessment of the significance of empirical differences, and reduce the effective sample size from the number of phenotyped animals to a number closer to the number of founders (i.e.,~3 founding pairs per ~ n=10 member group). To test whether quantitative trait variation is affected by a segregating genetic factor, we combined results from 5 controlled experiments (i.e., the Oral high methyl, IP folic acid, IP methylfolate, Fisher inbred, and Mouse protocols each with untreated controls) using the van Elteren test, and rescaled the variance by a factor of 31/95, the number of independent pairs divided by the number of F3 animals, to conservatively account for the possible correlation within each F3 generation. Under the assumption that the F0 breeding pairs are independent, the rescaled Chi-square statistic is 58.7*31/95=19.2, with a corresponding p-value of 0.00001, indicating that the likelihood that the reported observations may be accounted for by a founder effect entering the experimental pedigrees is remote.
Second, we performed a likelihood ratio test using pedigree information and F1-F4 regeneration proportions from intraperitoneal FA treatment and matched controls. We developed a peeling algorithm to evaluate the null likelihood function for a single, dominantly-acting gene model [21], and we deployed linear regression to represent the likelihood for an alternative, epigenetic model, allowing generation and treatment status, but not genetic effects, to modulate the expected phenotype. Bootstrap simulation of the genetic model was used to assess the statistical significance of the observed likelihood ratio (details under Supplementary Materials 1). Again, we observed strong evidence that variations in the proportions of regenerating neurons are inconsistent with segregation of a genetic factor in the founders (p = 0.00005). Accordingly, experiments herein comprise sufficient sample size to disclose a statistically significant increased proportion of spinal axon regeneration after surgery in progeny of folic acid-treated progenitors compared to controls. The increased proportion of spinal axon regeneration is inconsistent with a simple Mendelian genetic model, as evidenced both by nonparametric and statistical-genetics-based approaches. Whether a more complex, non-Mendelian genetic mechanism could replicate the heritable phenotypic variation we report cannot be addressed by the present data.
Patterns of Differentially Methylated Regions (DMRs) of genomic DNA in F3 animals correlate with F0 folate-enhanced axonal regeneration.
To test whether the inherited, pro-regenerative phenotype is associated with heritable changes in DNA promoter methylation, we compared levels of methylated DNA immunoprecipitation (MeDIP) in F3 progeny of F0 ancestors exposed to IP folic acid or IP water, and in F3 progeny of F0 ancestors exposed to IP folic acid with TSA treatment on the identical schedule. F3 animals were not themselves exposed to folate supplementation. First, we sought to identify transgenerational changes in methylation levels by comparing F3 progeny of F0 ancestors exposed to IP folic acid to F3 progeny of F0 ancestors exposed to IP water. These comparisons identified 518 hyper-methylated (increases in DNA methylation) differentially methylated regions (DMRs), and 390 hypo-methylated (decreases in DNA methylation) DMRs associated with ancestral folic acid supplementation.
Next, to interrogate the array of transgenerational methylation abundances that may be reversible by TSA, we investigated five unique patterns of methylation in F3 progeny: 1) hyper-methylation derived from ancestral folate that is reversed with TSA (pattern 000.111.000), 2) hyper-methylation from ancestral folate that is not reversed with TSA (pattern 000.111.111), 3) hypo-methylation derived from ancestral folate that is reversed with TSA (pattern 111.000.111), 4) hypo-methylation derived from ancestral folate that is not reversed with TSA (pattern 111.000.000), and 5) methylation abundances not associated with ancestral folate that are lost with TSA (pattern 11.111.000). These methods identified a unique set of DMRs for each of the five patterns, specifically 229 DMRs, 289 DMRs, 278 DMRs, 112 DMRs, and 972 DMRs for each pattern, respectively (Table 2). Because folate is an integral component of the establishment of DNA methylation, we focused on genes that exhibited hyper-methylation for downstream analyses. Accordingly, Fig. 5A shows the relative position of hyper-methylated DMRs within each chromosome. Corresponding gene identifiers are provided in Fig. 5B. Nine genes were selected at random from a set of 34 genes (Supplementary Materials 2) to confirm the MeDIP array results using methylation-specific PCR (MS-PCR) [22,23]. Genes tested include those encoding myosin light chain-3 (Myl3), olfactory receptor 1401 (Olr1401), neuronal acetylcholine receptor subunit alpha-3 (Chrna3), erythropoietin receptor-1 (EpoR-1), mannan-binding lectin-associated serine protease-2 (Masp2), F-box protein l (Fbxl1), myelin basic protein (MBP), and retinoid X receptor (Rxrb). MS-PCR of this panel in samples derived from F3 generation animals confirmed the results of the MeDIP array analyses (Fig. 5C). These data demonstrate that a plurality of genes in the F3 offspring of folate-treated F0 progenitors comprise increased methylation that is reversed with TSA in turn.
Table 2. Summary of differential methylation patterns.
Following data extraction, normalization and peak selection, differentially methylated regions (DMRs) were identified by pattern generation and filtering. Each identified peak was classified into 5 patterns wherein the control, folic acid, and folic acid + TSA replicates are shown in the 13, 4–6, and 7–9 components of the pattern, respectively. The table classifies the DMRs with respect to CpG island and gene promoters.
| Pattern | 0.0.0.1.1.1.0.0.0 | 0.0.0.1.1.1.1.1.1 | 1.1.1.0.0.0.0.0.0 | 1.1.1.0.0.0.1.1.1 | 1.1.1.1.1.1.0.0.0 |
|---|---|---|---|---|---|
| Number of DMRs | 229 | 289 | 278 | 112 | 972 |
| Number of DMRs mapping to gene promoters | 180 | 237 | 210 | 96 | 828 |
| Number of genes with DMR | 89 | 72 | 149 | 30 | 306 |
| Number of genes with a highly significant DMR | 52 | 49 | 81 | 8 | 216 |
| Number of unique genes with a DMR in a CpG Island | 34 | 40 | 9 | 17 | 88 |
| Number of DMRs in non-genes CpG Islands | 22 | 12 | 10 | 9 | 70 |
Fig 5. F0 folate supplementation of outbred Sprague-Dawley rat progenitors results in DNA promoter methylation pattern alterations in untreated F3 progeny that parallel inheritance of the axonal regeneration phenotype.
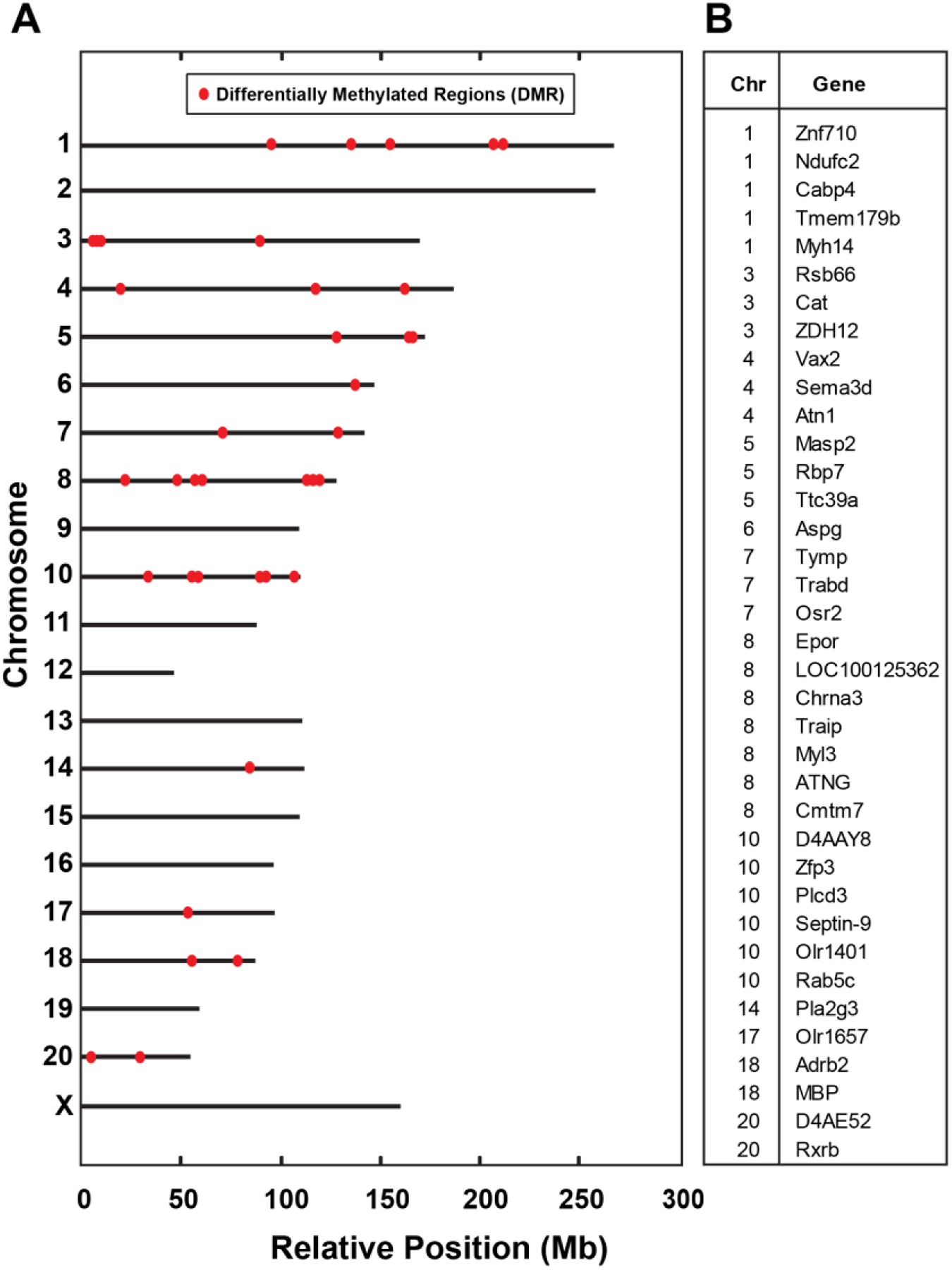
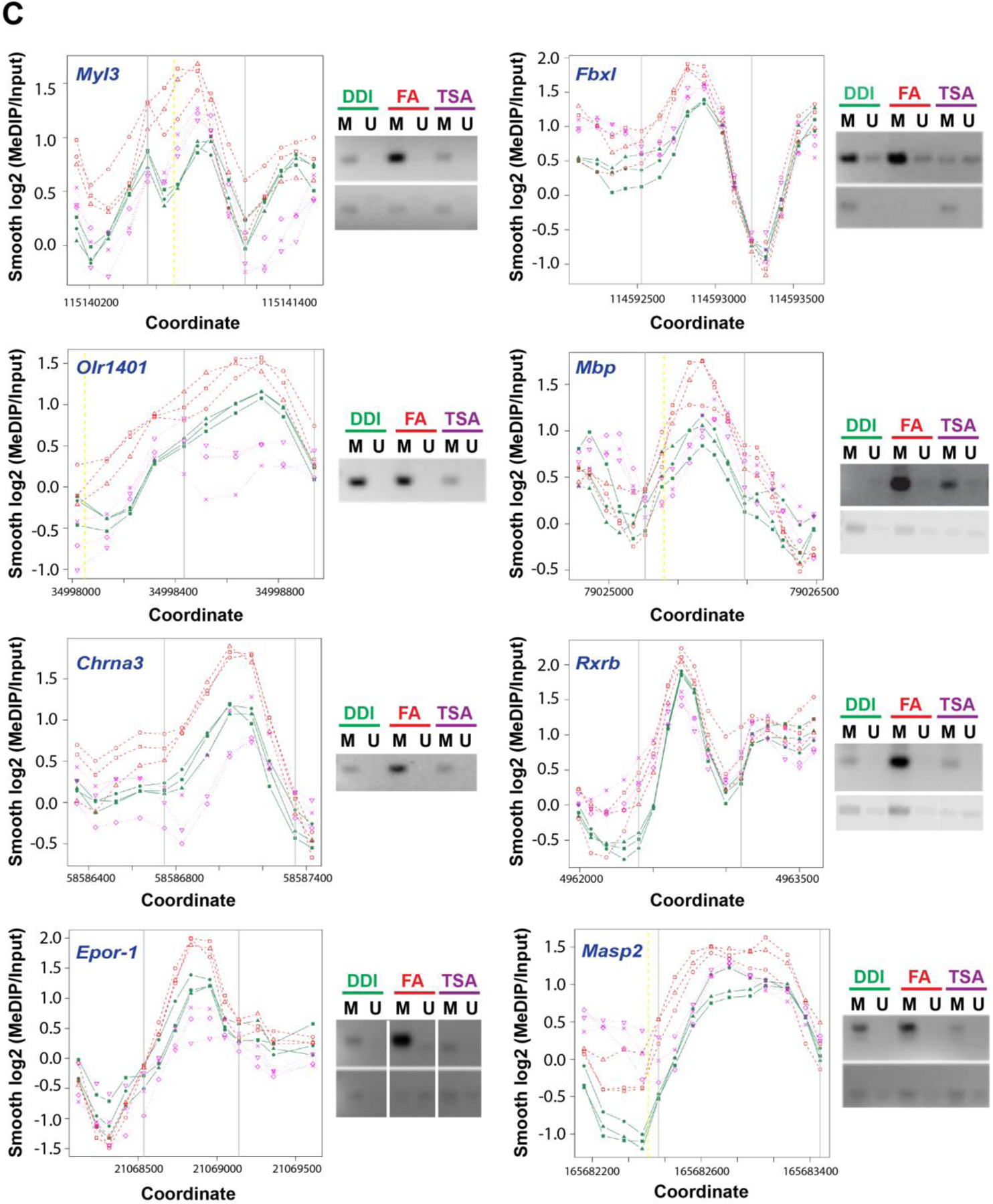
(A) Differentially methylated regions (DMRs) in response to IP folate in spinal cord tissue from F3 generation animals. Relative chromosomal positions of DMRs (X-axis: relative position (Mb); Y-axis: chromosome number). DNA promoter methylation array analysis was performed on spinal cord samples from F3 progeny of F0 progenitors supplemented with IP folic acid at 80μg/kg (FA) with and without TSA, compared to vehicle alone (DDI). F3 progeny of treated ancestors were not directly exposed to treatment. Red dots depict differentially methylated regions from pattern 000.111.000 across the genome. (B) Listing of genes with DMRs (also see Supplementary Materials 2). (C) Methylation-Specific PCR (MS-PCR) results validate methylation changes in selected genes from the MeDIP array results. Replicate level-smoothed log2(MEDIP/Input) data is displayed for each of 9 DMRs selected at random. Vertical gray bars mark the boundaries of the DMR, and the yellow dashed lines show transcription start sites (TSS) if they are in close proximity of the DMRs. Green indicates the DDI controls, Red indicates the folic acid-treated group, and Magenta indicates the folic acid and TSA-treated group. Methylation-specific primers (Table 3) were generated for the region of the DMR (between the gray lines) corresponding to widely separated regions between the 3 F3 (DDI, folic acid, folic acid + TSA dissolved in DMSO) shown in the methylation array plot (top panels, Full length gels in Supplementary Materials 3). Primers were generated for an experimental control from a region 5’ or 3’ to the DMR (bottom panels, Full-length gels in Supplementary Materials 3). Note Epor-1 includes a grouping of different parts of the same gel (delineated by white spaces). The MS-PCR was performed as in Methods, and the methylation levels are shown adjacent to graph (D1-D4) (FA: folic acid at 80 μg/kg intraperitoneal, DDI: Distilled de-ionized water control, TSA: trichostatin A, M: methylated, U: unmethylated, gene name in blue).
Ancestral folate results in stable transgenerational alterations in RNA transcription
We used direct, high throughput sequencing of cDNA (RNA-Seq) to test whether RNA expression in the spinal cords of F3 progeny of folate supplemented F0 ancestors differs from RNA expression in the spinal cords of F3 progeny of unsupplemented ancestors. A mean of 27.8 million paired end reads satisfied quality control standards and were aligned to the rat genome (Methods). Of 18,267 gene mRNA transcripts, 782 transcripts, associated to 752 unique genes, were dysregulated by ancestral folate (Figure 6A and 6B). Of these, the abundance of 395 genes were increased in the F3 progeny of folate supplemented ancestors compared to F3 progeny of unsupplemented F0 ancestors, while the mRNA abundance of 370 genes were decreased in F3 progeny of folate supplemented ancestors compared to F3 progeny of unsupplemented F0 ancestors. The abundance of 13 genes showed transcriptional variants in both directions (Supplementary Materials 4).
Fig. 6: Gene expression changes in F3 progeny are associated with ancestral folate supplementation.
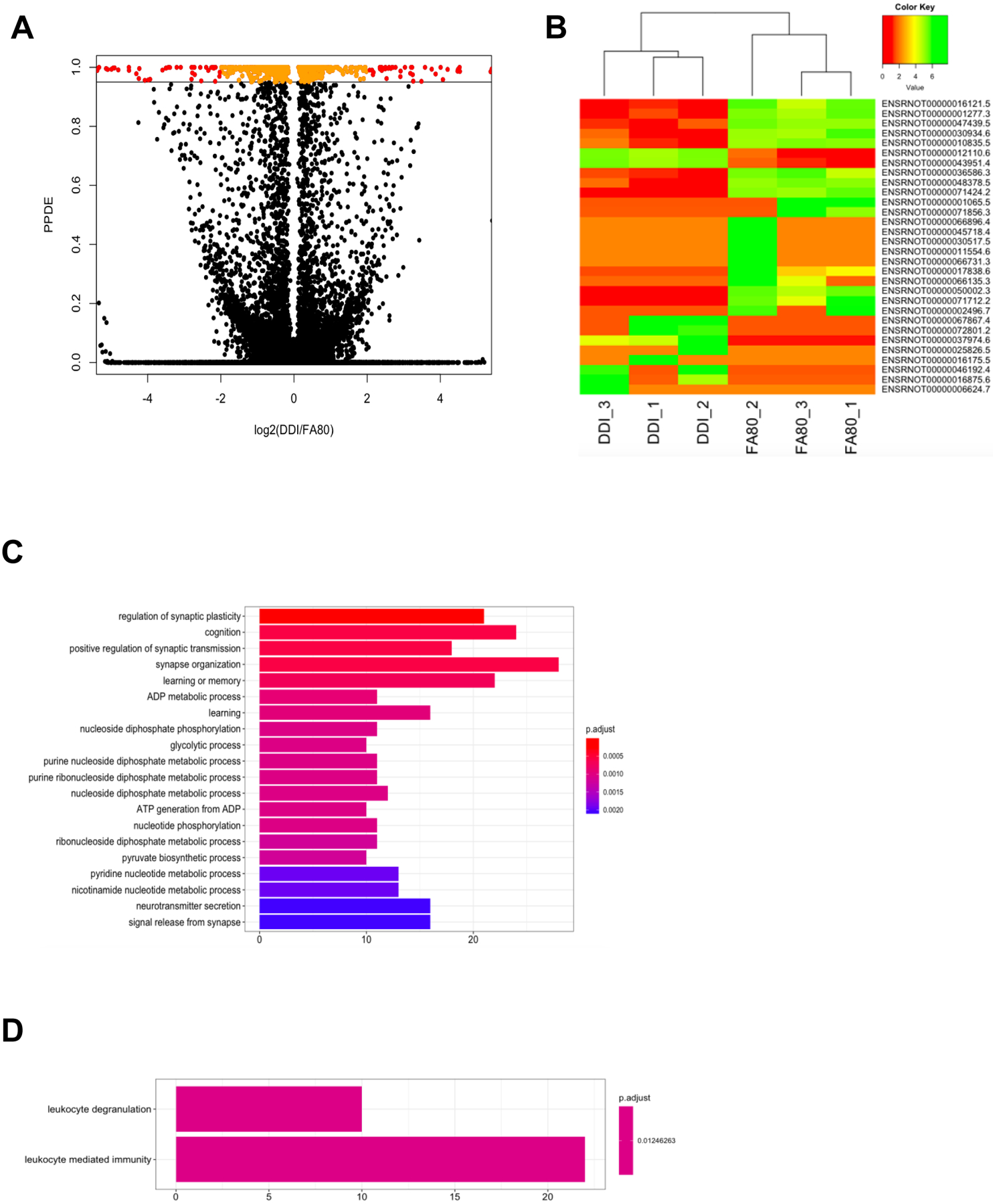
(A) A modified volcano plot depicts transcripts that are significantly dysregulated by ancestral folate. The –log2 value of the fold-change between DDI and FA80 is depicted on the x-axis while the posterior probability of differential expression (PPDE; analog of the adjusted P-value), as determined by EBSeq, is displayed on the y-axis. Transcripts with a PPDE >0.95 with a −log2(fold-change) < 2x are depicted in orange. Transcripts with a PPDE >0.95 with a –log2(fold-change) > 2x are depicted in red. Nonsignificant transcripts are shown in black. (B) A heat map of the top 30 dysregulated transcripts (y-axis) sequester FA80 samples from DDI samples (x-axis). Low expression of transcripts (log(read counts+1)) are depicted in red and range to high expression depicted in green. (C and D) The top gene ontology terms of biological processes for down-regulated genes (C) and up-regulated genes (D) are displayed. The number of dysregulated genes (x-axis) associated with the term (y-axis) are color-coordinated based on the adjusted P-value.
Examination of related biological functions using the gene ontologies of the 752 differentially expressed genes found significant ontological terms associated with synaptic-related processes, (e.g., regulation of synaptic plasticity), metabolic-related processes, (e.g., ATP generation from ADP), and immunological-related processes (e.g., such as leukocyte mediated immunity) (Figure 6C and 6D; Supplementary Materials 4). These data suggest that ancestral folate results in transgenerational alterations within the transcriptome, stably enhancing the CNS axonal regeneration.
DMRs associated with differentially expressed genes represent candidate functional DMRs that may have a direct role in gene regulation. An overlay of these data revealed 13 genes that were both differentially methylated and differentially expressed in F3 progeny of folate supplemented F0 ancestors. Among the six genes found to be hyper-methylated, two were up-regulated (Rxrb and RGD1565616), while four were down-regulated (Ant1, Chpf, Fam69b, and Galnt2). Among the seven genes found to be hypo-methylated, four were up-regulated (Cmtm6, Phb2, Unc13d, and MGC94199), while three were down-regulated (Nrcam, Arhgef7, and Eef1a2). Together, these data suggest a gene regulatory role for DNA methylation in axonal regeneration following ancestral folate supplementation.
Discussion
Neurons extend axons during growth, development, and regeneration after injury via transcriptionally-dependent mechanisms [24,20,17] that are susceptible to environmental and epigenetic influences [25–27]. Folate supplementation promotes axon regeneration after injury to CNS neurons, and alters genomic methylation in offspring [28]. Present data reveal that ancestral administration of folate augments axon growth in the spinal cord after sharp injury to at least the F4 generation without administration of folate to the intervening generations. A transgenerational rather than intergenerational effect is supported by transmission of the enhanced phenotype beyond the unexposed second (F2) generation [2]. Transmission of the phenotype after both synthetic folic acid and the naturally occurring 5-methyltetrahydrofolate indicates that the folate preparation is not a confounding factor. Enhanced axon regeneration after administration of distinct methyl donors points to participation of methylation-specific pathways [10]. Observation of folate-induced, transgenerational axon regeneration in both inbred and outbred rat lineages renders an artifact of breeding unlikely. Of particular note, observation of the pro-regenerative phenotype in both rats and mice establishes that the transgenerational inheritance is not genus-specific. In vitro culture data from cells harvested from animals with folic acid-supplemented ancestors indicate that the in vitro transgenerational phenotype is specific to neuronal rather than glial or other supportive cells. Transmission of the regenerative phenotype with uniform incidence of axon regeneration in succeeding generations diminishes the likelihood that inheritance is mediated by DNA sequence variation.
Antagonism of the in vivo phenotype with administration of the histone deacetylase (HDAC) inhibitor TSA [29,30] and the DNMT antagonist 5-azadCyD [15], and reversal of patterns of differentially methylated DNA promoter regions with TSA, indicates that patterns of DNA methylation are transmitted in tandem with in vivo and in vitro phenotypes to the F3 generation after ancestral folic acid supplementation. These observations are in keeping with global and gene-specific DNA methylation changes previously observed in rats directly exposed to folic acid [10], and correspond to differential transcription of genes that encode protein participants in axonal growth and regeneration, For example, the retinoid X receptor (Rxrb) and neuronal cell adhesion molecule (Nrcam) serve as transcription factors after spinal cord injury, initiating signaling in neurons, glia, and macrophages. Retinoic acid signaling increases during peripheral nerve regeneration to reduce glial scar formation while enhancing neurite outgrowth after spinal cord injury through cAMP, AKT, and other signaling pathways [31,32]. Nrcam expression promotes directional signal transduction during axonal cone growth [33], and is a critical component of dendritic spine remodeling of cortical neurons [34]. Nrcam is alternatively spliced after rat peripheral nerve injury [35], with alternative splicing as molecular contributor to the regulation of sciatic nerve regeneration. Accordingly, our data show that folate supplementation generates patterns of DNA methylation in the spinal cord that differ between F3 progeny of supplemented and unsupplemented F0 ancestors, and that differential patterns of DNA methylation correspond to differential patterns of RNA expression of genes known to participate in cellular pathways essential for axonal growth and regeneration.
These data have certain limitations, including: 1) A correlation observed between DNA methylation and the transgenerational axon regeneration phenotype does not preclude mechanisms of heritable transmission other than or in addition to DNA methylation, 2) DNA sequence variations may directly or indirectly affect other methylation pathways (e.g., mutations in histones, or in genes that encode folate and DNA methyltransferases) together with possible contributions from RNA and protein methylation, histone methylation and modifications, kinetics of chromatin conformation, non-coding RNAs, and other biological or environmental mechanisms yet to be identified [36]. 3) The gender of inheritance of the phenotype, and the critical interval during gestation most sensitive to supplementation, await ascertainment; and 4) Additional yet to be identified confounders in experimental design, handling, breeding, and stress mechanisms may introduce bias in animal experiments that address transgenerational inheritance, and must be sought and controlled as the field expands
To the present, reports of mammalian transgenerational inheritance of a phenotype in the absence of corresponding DNA sequence variation have employed models of deleterious effects after exposure to toxins and stressors [6,37,4,5], in addition to genetic disruption of folate metabolism [12]. To the contrary, we report that shared mechanisms may underlie transgenerational inheritance of an adaptive and potentially beneficial phenotype, i.e., enhanced axonal regeneration after spinal cord injury in two distinct genera. The central nervous system comprises tissues that are notoriously indolent in response to efforts to promote regeneration, and accordingly the present observations may be of particular clinical relevance. Moreover, the effects we observe arise in response to cofactors and essential nutrients that date to the origins of carbon-based life, not in response to exposure to compounds that have been newly introduced to the environment such as toxins, fungicides, and petrochemicals. The folate pathway fulfills a central role as the sole methyl donor in most mammalian tissues including the central nervous system [38]. Although DNA methylation inhibitors have long found utility in the suppression of cancer and other disorders of aberrant proliferation, our results suggest that methylation supplementation may provide transgenerational benefits when proliferation of targeted tissue is to be desired.
Evidence that pharmaceutical, environmental and nutritional exposures may have consequences that span generations has significant implications for maintenance of health and well-being. A more profound understanding of the biochemical mechanisms that give rise to adaptive, transgenerational phenotypes may broaden treatment options for inherited and acquired conditions, and frame the consequences of behavioral, dietary and medical choices beyond the life spans of single individuals. Implications of these data comprise a more profound understanding of tissue healing and repair, and deeper insights into how behavior, habits, diet, and therapy interact to affect our children and their progeny.
Methods
Animals and Surgical Procedures
Male and female Sprague-Dawley Outbred rats (SD) or ICR Outbred mice (CD-1) were used for the experiments. Fisher 344 inbred rats (F344/NHsd) were used in 1 experiment. All animals were obtained from Harlan Laboratories Inc. (Madison, WI), and housed in approved facilities at the University of Wisconsin-Madison staffed by licensed staff veterinarians. All surgical procedures were conducted with approval of the University of Wisconsin Research Animal Resources and Care Committee, and in accordance with published NIH guidelines.
SECTION I – IN VIVO AXONAL REGENERATION
Intraperitoneal Folic Acid, Methylfolate, and Control Groups
To establish a breeding lineage of rats with intraperitoneal (IP) folate-supplemented F0 progenitors, 250gm Sprague-Dawley rats were purchased from Harlan Laboratories for each experimental group: 1) Folic acid (lineage 1): 3 males and 3 females; DDI control: 2 males and 2 females; 2) Folic acid (lineage 2: 5-azadCyD group): 4 males and 4 females; 3) Methylfolate: 5 males and 5 females, DDI control: 3 males and 3 females; In a parallel experiment, inbred Fisher rats (Folic acid: 3 males and 3 females; DDI control: 3 males and 3 females) weighing 175gm to 225gm were used to determine whether the effect of folate is confined to inbred or outbred rat strains. Prior to breeding, all animals were weighed and given daily IP injections of 80 μg/kg of folic acid (APP Pharmaceuticals, Schaumberg, IL) or methylfolate ((6S)-5-methyltetrahydrofolic acid disodium salt, Sigma-Aldrich Co., St. Louis, MO), as a 5mg/mL solution diluted with double distilled H20 (DDI) to 0.125mg/mL, and injected daily in 20 μl volumes, based on the body weight obtained prior to the first injection. During pregnancy, the injections were made subcutaneously (SC) to avoid injury to the uterine contents. The animals were weighed before each injection. The injections started 14 days before the rats were mated in 3 separate cages. Injections in F0 males were continued daily until the pups were born. Injections in gravid F0 females were continued until pups were weaned at 21 days. Control F0 animals were injected with distilled de-ionized water (DDI) using an identical protocol. Both control and experimental animals were fed Harlan Rodent Diet #8604 (Table 1) throughout the experiment unless otherwise indicated. All breeding was performed in a non-sibling manner. In subsequent F1-F6 generations, females were used only for breeding. Male animals were used either for breeding or for spinal cord injury and measurement of the phenotypes, but not both. Only males were phenotyped in view of: 1) No difference in axonal regeneration after sharp injury is observed between females and males (data provided upon request); 2) conservation of females for further breeding; and 3) efficient use of resources with surgical phenotypes that cannot be repeatedly measured in single animals.
Table 1. Diet comparisons.
Constituents of the 3 diets used are provided. The 8604 diet is the laboratory baseline diet used in all animals other than for F0 animals treated with oral methyl supplementation fed NIH-31 7017-MS, and control F0 animals fed the identical diet lacking the supplementary methyl compounds (NIH-31 7017). The additional methyl nutrients are highlighted at the bottom of the 7017-MS column (grey shading). The 7017 and 7017-MS diets were fed only to the F0 progenitors. All progeny (F1-F6) were fed the laboratory baseline 8604 diet.
| Harlan Laboratories Diet | ||||
|---|---|---|---|---|
| Component | Unit | 8604 | 7017 | 7017MS |
| Macronutrients | ||||
| Crude Protein | % | 24.3 | 18 | 18 |
| Fat (ether extract) | % | 4.7 | 4.7 | 4.7 |
| Carbohydrate (available) | % | 40.2 | 46.5 | 46.5 |
| Crude Fiber | % | 4.0 | 4.0 | 4.0 |
| Natural Detergent Fiber | % | 12.4 | 13.6 | 13.6 |
| Ash | % | 7.4 | 6.2 | 6.2 |
| Energy Density | kcal/g | 3.0 | 3.0 | 3.0 |
| Calories from Protein | % | 32 | 24 | 24 |
| Calories from Fat | % | 14 | 14 | 14 |
| Calories from Carbohydrate | % | 54 | 62 | 62 |
| Minerals | ||||
| Calcium | % | 1.4 | 1.1 | 1.1 |
| Phosporus | % | 1.1 | 1.0 | 1.0 |
| Non-Phytate Phosporus | % | 0.7 | 0.7 | 0.7 |
| Sodium | % | 0.3 | 0.3 | 0.3 |
| Potassium | % | 1.0 | 0.6 | 0.6 |
| Chloride | % | 0.5 | 0.5 | 0.5 |
| Megnesium | % | 0.3 | 0.2 | 0.2 |
| Zinc | mg/kg | 80 | 47 | 47 |
| Manganese | mg/kg | 100 | 155 | 155 |
| Copper | mg/kg | 25 | 13 | 13 |
| Iodine | mg/kg | 2 | 2 | 2 |
| Iron | mg/kg | 300 | 270 | 270 |
| Selenium | mg/kg | 0.34 | 0.3 | 0.3 |
| Amino acids | ||||
| Aspartic Acid | % | 2.3 | 1.5 | 1.5 |
| Glutamic Acid | % | 4.1 | 3.2 | 3.2 |
| Alanine | % | 1.4 | 1.1 | 1.1 |
| Glycine | % | 1.3 | 1.0 | 1.0 |
| Threonine | % | 0.9 | 0.7 | 0.7 |
| Proline | % | 1.6 | 1.5 | 1.5 |
| Serine | % | 1.6 | 0.9 | 0.9 |
| Leucine | % | 1.9 | 1.4 | 1.4 |
| Isoleucine | % | 1.0 | 0.8 | 0.8 |
| Valine | % | 1.1 | 0.8 | 0.8 |
| Phenylalanine | % | 1.1 | 0.8 | 0.8 |
| Tyrosine | % | 0.9 | 0.7 | 0.7 |
| Methionine | % | 0.4 | 0.4 | 0.4 |
| Cystine | % | 0.4 | 0.3 | 0.3 |
| Lysine | % | 1.4 | 0.8 | 0.8 |
| Histidine | % | 0.6 | 0.4 | 0.4 |
| Arginine | % | 1.5 | 1.0 | 1.0 |
| Tryptophan | % | 0.3 | 0.2 | 0.2 |
| Vitamins | ||||
| Vitamin A | IU/g | 12.6 | 24.2 | 24.2 |
| Vitamin D3 | IU/g | 2.4 | 4.2 | 4.2 |
| Vitamin E | IU/kg | 120 | 41 | 41 |
| Vitamin K3 (menadione) | mg/kg | 40 | 22 | 22 |
| Vitamin B1 (thiamin) | mg/kg | 27 | 76 | 76 |
| Vitamin B2 (riboflavin) | mg/kg | 8 | 7 | 7 |
| Niacin (nicotinic acid) | mg/kg | 63 | 87 | 87 |
| Vitamin B6 (pyridoxine) | mg/kg | 13 | 9 | 9 |
| Pantothenic Acid | mg/kg | 21 | 39 | 39 |
| Vitamin B12 (cyanocobalamin) | mg/kg | 0.05 | 0.06 | 0.06 |
| Biotin | mg/kg | 0.38 | 0.3 | 0.3 |
| Folate | mg/kg | 3 | 2 | 2 |
| Choline | mg/kg | 2530 | 1890 | 1890 |
| Fatty Acids | ||||
| C16.0 Palmitic | % | 0.7 | 0.7 | 0.7 |
| C18.0 Stearic | % | 0.1 | 0.1 | 0.1 |
| C18.1ro9 Oleic | % | 0.9 | 1.0 | 1.0 |
| C18.2ro6 Linoleic | % | 1.9 | 1.9 | 1.9 |
| C18.3ro3 Linolenic | % | 0.2 | 0.2 | 0.2 |
| Total Saturated | % | 0.9 | 0.9 | 0.9 |
| Total Monounsaturated | % | 1.1 | 1.2 | 1.2 |
| Total Polyunsaturated | % | 2.1 | 2.1 | 2.1 |
| Other | ||||
| Cholesterol | mg/kg | 50 | 50 | 50 |
| Choline Chloride | g/kg | 0 | 0 | 5.76 |
| Betaine, anhydrous | g/kg | 0 | 0 | 5 |
| Folic acid | g/kg | 0 | 0 | 0.005 |
| Vitamin B12 (0.1% in mannitol) | g/kg | 0 | 0 | 0.5 |
Oral Supplemented Methyl and Control groups
To establish a breeding lineage of rats with orally treated F0 progenitors, 250gm Sprague-Dawley, male and female rats were purchased from Harlan Laboratories and were fed either of 2 experimental diets from Harlan Laboratories Inc. (Madison, WI): 1) Control rats received NIH-31 Open Formula Mouse/Rat Sterilizable Diet (7017), and 2) Supplemented methyl rats received the identical diet with added methyl supplements (7017-MS) comprising added choline, betaine, folic acid, and vitamin B12 (Table 1). The 7017 and 7017-MS diets were provided for 2 weeks before conception to the control (2 males and 2 females), and supplemented-methyl (3 males and 3 females) animals, respectively. The animals were then mated in 3 separate cages to generate 3 separate lineages for each experimental group. The mothers were maintained on either of the 2 experimental diets until the pups were weaned at 21 days, after which the pups were permitted unrestricted access to Harlan Rodent Diet #8604 (Harlan Laboratories, Madison, WI) (Table 1). The 7017 and 7017-MS experimental diets were the sole diets provided during pre-conception, gestation, and breast-feeding to the F0 parents. All subsequent breeding pairs, as well as all progeny in subsequent generations, consumed the Harlan Rodent #8604 diet throughout their lives. All breeding was performed in a non-sibling manner. The supplemented methyl lineage was maintained for 6 generations, and the untreated control lineage was maintained for 4 generations. In each generation, females were used only for breeding. Male animals were used either for breeding or for spinal cord injury and measurement of the phenotypes. Males were bred before phenotyping.
Spinal Cord Regeneration Animal Model (SCRM)
To quantify regeneration of injured spinal axons into a peripheral nerve graft, surgery for the SCRM was performed between 7 and 10 weeks of life in animals weighing 250 to 400gm as previously described [10]. In brief, adult male SD rats were anesthetized with ketamine (40–80mg/kg; Clipper Distributing Company LLC, St. Joseph, MO) and xylazine (5–10mg/kg; Bimeda-MTC Animal Health Inc., Le Sueur, MN) as previously described (Fig. 1B) [19,10,39]. The cervical spinal cord was exposed through a C3 laminectomy and the dura opened. With operating microscope guidance, a 0.5–1mm deep injury was made in both posterior columns with a sharp jeweler’s forceps or 25G needle. A sciatic nerve segment harvested from the left hindlimb was then implanted at the injury site and secured with 10–0 nylon sutures to the dura. The opposite (i.e., distal in situ) end of the graft was allowed to lie freely under the skin. Animals received buprenorphine (0.05 to 0.1 mg/kg, SQ) intraoperatively, and additional doses every 8 to 12 hours as needed for pain. Two weeks later, the wound was reopened under anesthesia as above, and a gelfoam (Johnson & Johnson Ethicon-SARL, Neuchatel, Switzerland) soaked in 5 μl of the retrograde tracer Fluoro-Gold™ (Fluorochrome, LLC, Denver, Co) was placed at the free end of the nerve graft. The fluorescent tracer tracks axons that have grown from the spinal cord into the graft, and is transported in a retrograde fashion to reach neuronal cell bodies in the dorsal root ganglia (DRG) (Fig. 1C) [39].
Tissue preparation and harvest:
Forty-eight hours thereafter animals were euthanized with 100mg/kg IP of Beuthanasia-D Special (a combination of pentobarbital sodium and phenytoin sodium; Schering-Plough Animal Health Corp., Union, NJ). The 4th and 5th lumbar (L4–5) DRGs were removed bilaterally, fixed in 4% paraformaldehyde at 4°C overnight , incubated in 30% sucrose for 3 hours at room temperature, and quick frozen in Optimal Cutting Temperature compound (OCT; Sakura Finetek, Torrance, CA). Twelve μm sections were cut with a cryostat, floated on pretreated glass slides (FisherFinest™, Fisher Scientific, Pittsburgh, PA), and frozen at −80°C.
Separation and Processing of Spinal Cord:
Following DRG removal, the spine was separated from the skull, and laminae were removed to expose the spinal cord. The intact spinal cord was divided into 4 equal parts that were stored separately at −80°C for biochemical and molecular analysis (see Methylation Studies below). The 1-cm section containing the cervical injury was used for the methylation studies.
Analysis:
DRG sections were scored for tracer uptake under an Axioscope 20 fluorescent microscope (Zeiss, Peabody, MA) using a gold filter (Fluorescence Zeiss Filter Set 02 For UV Shift Free, Excitation: G 365 nm, Dichroic: FT 395 nm, Emission: LP 420 nm) to detect Fluoro-Gold™. Each section was also examined with a red filter (Zeiss Filter Set 63 HE MRFP Shift Free, Excitation: BP 575/25 nm, Dichroic Mirror: FT 590 nm, Emission: BP 629/62 nm) to detect auto-fluorescence. Fluoro-Gold™-labeled cells were counted by 2 independent observers who were blinded to the treatment conditions. The percentage of labeled neuronal cells in each DRG was calculated, with the denominator comprising the average total number of neurons in rat DRG using laboratory normative data i.e., 3,040 cells on the side of sciatic nerve harvest. This value was previously derived using the assumption-based method of Abercombie [10], in which only cells with visible nucleoli are counted [40]. Harvesting the peripheral sciatic nerve axon of the ipsilateral DRG enhances growth of both the peripheral and central (i.e., dorsal column) components of DRG neurons [39]. The combination of peripheral nerve injury and IP folic acid supplementation may be additive or synergistic, thereby increasing the number of regenerating axons on the harvested side up to 10-fold [9]. SCRM results reported refer to the percentage of labeled neuronal cell bodies in DRGs ipsilateral to the site of the sciatic nerve harvest.
Trichostatin A (TSA) Rat Treatment in vivo
Male F3 generation (for methylation studies, Fig. 5A) and male F5 generation (for axon regeneration studies, Fig. 2A(i)) animals whose F0 progenitors received IP folic acid supplementation (80μg/kg) were treated with TSA (0.5mg/kg TSA in 33% DMSO (40–60 units; IP) (Selleck Chemicals, Houston) once daily for 17 days starting 3 days before SCRM surgery (Fig. 2A(ii)). Two weeks after surgery, Fluoro-Gold™ application, and DRG and spinal cord harvest and handling were performed as above. A control group of F3 and F5 animals whose F0 ancestors were similarly treated with IP folic acid received the same volume of DMSO vehicle without TSA on the identical schedule.
5-aza-2’-deoxycytidine (5-azadCyD) Rat Treatment in vivo
Male F3 generation (Fig. 2B(i)) animals whose F0 progenitors received IP folic acid supplementation (80μg/kg) were treated with 5-azadCyD (0.1mg/kg 5-azadCyD in water; IP) (Sigma-Aldrich Co., St. Louis, MO) once daily for 17 days starting 3 days before SCRM surgery (Fig. 2B(ii)). Two weeks after surgery, Fluoro-Gold™ application, and DRG and spinal cord harvest and handling were peformed as above. A control group of F3 animals whose F0 ancestors were similarly treated with IP folic acid received the same volume of water without 5-azadCyD on the identical schedule.
Mouse breeding and surgery
Twenty-eight gm to 40 gm CD-1 mice (Folic acid: 3 males and 3 female; DDI control: 3 males and 3 females) were purchased from Harlan Laboratories, Madison, WI. Prior to breeding, each animal was given daily IP injections of 80 μg/kg of folic acid (APP Pharmaceuticals, Schaumberg, IL), provided as 5mg/mL, diluted with DDI to 0.125mg/mL, and injected in 20 μl volumes. Mice were mated in 3 cages. Injections in the F0 males continued until pups were born. Injections of the pregnant F0 females were continued until pups were weaned. Control F0 animals (3 males and 3 females) were injected with IP DDI using an identical protocol. Both control and experimental animals were fed Harlan Rodent Diet #8604 throughout the experiment. All breeding was done in a non-sibling manner. In each succeeding generation (F1 to F3), females were used only for breeding, whereas male animals were used either for surgery or breeding. Males used for breeding did not undergo surgery.
Mice from the F1 to F3 generations were anesthetized with IP ketamine (100 mg/ml at 90–120mg/kg) and Xylazine (100mg/ml at 10 mg/kg). Additional 10–20 mg/kg doses of ketamine were administered as needed if, for example, the animal moved its extremities in response to pinching. Each animal underwent SCRM surgery as in the rat SCRM protocol described above. Two weeks later, the mice were anesthetized using the same technique, the wound was re-opened, and a gelfoam soaked in 5 μl of the retrograde tracer Fluoro-Gold™ (Fluorochrome, LLC, Denver, Co) was placed at the free end of the 1-cm long sciatic nerve graft. Tissue harvesting and sectioning were performed as in the rat protocol described above. The percentage of labeled DRG neurons in each animal was calculated, with the denominator comprising the average total number of cells in mouse DRG using laboratory normative data i.e., 1743 DRG neurons on the side of sciatic nerve injury, using the assumption-based method of Abercombie [10], in which only cells with visible nucleoli are counted [40].
Single Generation Controls (S0) vs. Lineage progenitors (F0)
To assure stability of laboratory standards after SCRM surgery, S0 animals (rats or mice) were treated directly with daily IP injections of 80μg/kg of folic acid, methylfolate, or DDI starting 3 days prior to surgery and continuing for a total of 17 days (i.e., the treatment dose and regimen that results in optimal axon regeneration in the model) [9,10], or oral supplementation with the supplemented methyl diets (7017 or 7017-MS) starting a week before injury, and continuing for a total of 3 weeks. S0 animals provide evidence of maintenance of SCRM results despite possible changes in drug stocks, media, operators, animal handling, and other variables over the months to years required to perform the present experiments. S0 animals treated with DDI show uniformly minimal capacity to regenerate axons, as previously shown [9,10]. Using S0 rats, animal age was investigated, and showed that within the 54–124 day range used (corresponding to 250–400gm weight range) in the present studies, age has no impact on axon regeneration (Data available upon request). F0 animals (actual lineage progenitors) were not phenotyped because of the variable number of days of exposure to test compounds and controls that is required by the unpredictable lengths of time needed for pregnancy to occur.
SECTION II – IN VITRO NEURITE OUTGROWTH ASSAYS
In vitro neuron cultures
To test the transgenerational effect of folate on axonal growth in neurons isolated from other cellular elements in vitro after in vivo supplementation, DRG neurons from animals with spinal cord injury were dissociated and grown in culture medium, as previously reported [19,10,20].
Spinal Cord Injury:
Adult male SD rats from the SCRM breeding lineages were subjected to spinal cord injury (but not sciatic nerve grafting) under ketamine/xylazine anesthesia (10:1). A C3 laminectomy and dural opening exposed the cervical spinal cord. A 0.5 – 1.0 mm-deep sharp transection injury was made across both posterior columns using jeweler’s forceps or 25G needle, as previously done.10
Dorsal Root Ganglia Cell Culture:
All experiments were performed in triplicate, with cells from 18 bilateral L4–6 DRGs harvested from 3 rats comprising each of 3 independent replicate pools for subsequent culture, as previously done.10 The right and left L4, L5, and L6 DRGs from 3 adult rats with a spinal cord injury identical to SCRM above (i.e. to a total of 18 DRGs) were removed 3 days after injury, and the rats were euthanized. The DRGs were placed in 70% ethanol, dissociated using micro-dissecting scissors, and transferred to cold media RPMI (Hyclone Lab, Logan, UT) and B-27 (Life Technology, Grand Island, NY). The DRG fragments were washed three times in PBS without Ca++ or Mg++, and placed in PBS with 100μl collagenase (35mg/mL; Sigma Chemical Company, St. Louis, MO) and 200μl dispase (10mg/mL; Gibco, Life Technologies Corporation, Grand Island, NY). After 20 minutes incubation at 37°C, 5% CO2, the cells were mixed 5–10 times with a Pasteur pipette, and 25 μl DNAse (10mg/mL) was added (Sigma-Aldrich, St. Louis, MO). Following a 20-minute incubation at 37°C, 5% CO2, the cells were washed twice with RPMI containing 5% FBS (GE Healthcare Life Sciences, Pittsburg, PA) by re-suspension and centrifugation. One hundred μl aliquots of cells with a target concentration of one DRG per coverslip were obtained from the pool, and plated on 12-mm diameter coverslips pre-coated overnight with Poly-D Lysine (Sigma-Aldrich, St. Louis, MO, Cat#7280) and laminin (Sigma-Aldrich, St. Louis, MO, Cat#2020). The cells were grown in a 37°C, 5% CO2 incubator, and coverslips were removed for scoring at 12, 24, and 48-hour intervals after plating.
Immunocytochemistry:
Cells were fixed with 4% paraformaldehyde for 30 minutes, and then washed 3 times in PBST (PBS + 0.01%Triton X). The coverslips were placed in block solution (1xPBS + 5%FBS + 0.2%Triton X (Sigma-Aldrich, St. Louis, MO) for 30 minutes. DRG neurons were stained using mouse anti-βIII tubulin antibody (Promega Co., Madison, WI), and Alexa Fluor 594 donkey anti-mouse secondary IgG antibody (Life Technologies, Carlsbad, CA), and placed on Fluoromount G medium (EM laboratories Inc., Electron Microscopy Sciences, Cat# 17984–25) for observation.
Analysis of Neurite Growth:
Quantitative analysis of neurite length was performed by an observer blinded to the experimental conditions. Images were acquired using Axiovision software (Zeiss, Peabody, MA) for an Axioscope 20 fluorescent microscope (Zeiss, Peabody, MA) at 20X magnification. Each coverslip was divided into 4 quadrants, and 4 images containing at least one complete cell each were selected at random in each quadrant by tracking the longest axon away from each cell body of each image to ensure that the entire axon length was measurable. ImageJ Software (imagej.nih.gov/ij/) was used to calibrate the measurement scale using the scale image taken at 20X magnification, and to archive the photomicrographs. The longest neurite from each cell body was measured, and average axon lengths were calculated for each of the 3 replicates.
Trichostatin A (TSA) treatment of neurons in culture
To examine the effect of TSA on axonal growth in culture, TSA was dissolved in DMSO and diluted in culture media (final concentration of 50nM in RPMI-1640 minus L-Glutamine (HiYClone Laboratories, Logan, Utah) supplemented with B27 (Life Technologies, Carlsbad, CA) prior to plating the DRG cells. Equal amounts of DMSO in culture media alone were used as controls (Fig. 4D). Immunohistochemistry and analysis of neurite growth were conducted as detailed above.
SECTION III – MOLECULAR ANALYSIS
Whole genome promoter methylation and RNA transcription analyses
Spinal cord tissue harvest:
To perform quantitative whole genome promoter methylation analysis using methyl-DNA immunoprecipitation (MeDIP) sequencing analysis (Array Star, Rockville, MD) [41], the injured 1-cm segment of cervical spinal cord was harvested from F3 SCRM animals whose F0 progenitors were treated with 80ug/kg IP folic acid with and without TSA treatment, and compared to F3 SCRM animals whose F0 progenitors were treated with IP DDI. All tissues were obtained from the animals used for axon regeneration experiments above. Spinal cord tissue from F3 generation animals was used to perform gene expression analysis (see below).
MeDIP Assay:
MeDIP assays (ArrayStar, Rockville, MD) were performed as previously described [42,41]. Genomic DNA was isolated by proteinase K treatment, phenol-chloroform extraction, ethanol precipitation and RNase digestion. Before carrying out MeDIP, genomic DNA was sonicated using a Bioruptor sonicator (Diagenode, Denville, NJ) at “Low” mode for 10 cycles of 30 seconds “ON” and 30 seconds “OFF” (with an ultrasound wave frequency of 20 kHz) to produce random fragments ranging from 200 to 1,000 bp. Five μg of fragmented DNA was used for a standard MeDIP assay, while 1 μg sheared DNA was used as input. DNA was denatured for 10 minutes at 94 °C and immunoprecipitated for 4 hours at 4 °C with 5 μg anti- 5-methycytidine (Diagenod, Denville, NJ) in a final volume of 500 μl IP buffer (0.5% NP40; 1.1% Triton X-100; 1.5 mM EDTA; 50 mM Tris-HCl; 150 mM NaCl). The mixture was then incubated with 50 μl of BiomagTM magnetic beads coupled anti-mouse IgG for 2 hours at 4 °C, and washed three times with 500 μl IP buffer. DNA was eluted at 65°C for 15 minutes in 200 μl elution buffer (1 mM Tris-HCl, pH 8.0; 0.5 mM EDTA, pH 8.0; 10% SDS). Methylated DNA was recovered by phenol-chloroform extraction followed by ethanol precipitation.
Real-time PCR of MeDIP samples:
To test the specificity of MeDIP, a known methylated gene locus (H19) was selected for real-time PCR using 1 μl of input DNA and immunoprecipitated methylated DNA. For real-time PCR, SYBR Green I nucleic acid gel stain (Molecular Probes, Thermo Fisher Scientific, Waltham, Massachusetts) and an ABI Prism 7900 System (Diagenode, Denville, NJ) were used. To evaluate the relative enrichment of target sequences after MeDIP, the ratios of signals in immunoprecipitated DNA vs. input DNA were calculated.
Analysis by microarrays:
Input and MeDIP fractions were differentially labeled with Cy3 and Cy5 and co-hybridized to NimbleGen RN34 CpG Island Plus RefSeq Promoter Microarray arrays as a two-color experiment (Roche NimbleGen, Inc., Madison, WI). The arrays were scanned with the Axon GenePix 4000B scanner, and analyzed using NimbleScan software and Bioconductor packages Ringo and limma. Promoters and CpG islands that gain methylation were defined as follows: sliding window width: 750 bp; mini probes per peak: 2; P-value minimum cutoff: 2; and maximum spacing between nearby probes within peak: 500 bp. Three replicates were generated for each of the DDI, folic acid, and TSA groups, resulting in a total of 9 arrays, with 3 animals per array. The arrays were configured in a 3×720k format design containing 15,790 CpG Islands annotated by UCSC (http://genome.ucsc.edu/), and 15,287 RefSeq promoter regions. Promoter regions spanned 3,880bp upstream to 970bp downstream of the transcription start sites (TSSs).
Data pre-processing:
Raw data were extracted as pair files by NimbleScan software (v2.5) with median-centering, quantile normalization, and linear smoothing by Bioconductor packages Ringo www.bioconductor.org/packages/release/bioc/html/Ringo.html, limmawww.bioconductor.org/packages/release/bioc/html/limma.html[43], and MEDMEwww.bioconductor.org/packages/release/bioc/html/MEDME.html. Normalization resulted in log base 2 ratios of the immunoprecipitated and total DNA for each sample, with Spearman correlations ranging from 0.86 to 0.91 between samples within each MeDIP group.
Peak calling at the replicate level:
The normalized log base 2 ratios within each array were smoothed using a simple moving average method with a window size of 3 probes. For each replicate, candidate peaks were generated with the Algorithm for Capturing Microarray Enrichment (ACME) (www.bioconductor.org/packages/release/bioc/html/ACME.html) [44] using the following parameter settings: window = 700; thresh = 0.95 (for do.aGFF.calc()); and thresh = 5e-3 (for findRegions()). Candidate peaks were filtered by requiring at least 2 consecutive probes within the peak to exceed the threshold used by the ACME.
Identifying differentially methylated regions (DMRs) by pattern generation and filtering:
Each identified peak was classified into one of the five patterns (0.0.0.1.1.1.0.0.0, 0.0.0.1.1.1.1.1.1, 1.1.1.0.0.0.0.0.0, 1.1.1.0.0.0.1.1.1, and 1.1.1.1.1.1.0.0.0) by evaluating overlap of peaks identified across each of the 3 replicates of the DDI, folic acid, and TSA groups, represented in the 1–3, 4–6, and 7–9 components of the pattern description, respectively (Supplementary Materials 2). The DMRs were further screened based on a limma differential enrichment analysis [43] at the probe level. At least 2 consecutive probes with a false discovery rate adjusted p-value [45] smaller than 0.1 were required within each DMR. In addition, fold changes within the DMRs consistent with the implied ordering of the patterns were required. For example, for the 0.0.0.1.1.1.0.0.0 pattern, we required that median of the average log base 2 ratios of the probes across replicates within the DMR region for the folic acid group was larger than those of both the control and the folic acid + TSA groups. This resulted in 52, 49, 81, 8, and 216 unique genes with a significant DMR in each of the 5 pattern groups. At least 2 consecutive probes with a false discovery rate adjusted p-value [45] smaller than 0.1 within each DMR were selected. Supplementary Materials 2 further classifies the DMRs with respect to CpG island and to gene annotations, and indicates that multiple genes have multiple DMRs.
RNA library preparation and sequencing
100 ng of total RNA from F3 generation spinal cord tissue was used for sequence library construction following instructions of the NuGen mRNA sample prep kit (cat# 0348). In brief, total RNA was copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNaseH. The cDNA fragments were end repaired, a single ‘A’ base was added, and then ligated to adapters. The products were gel purified and enriched by PCR to generate cDNA libraries. The size and concentration of library constructs were verified by a before sequencing on an Illumina HiSeq2500 sequencer (San Diego, CA). 100-cycle single-end sequencing was performed by the University of Wisconsin Biotechnology Center (Madison, WI, USA).
RNAseq processing and analysis
An average of 31.6 million paired-end reads were sequenced per sample. Quality was assessed for each pair-mate using FastQC. After reads were assured for quality, paired-end reads were aligned to the Rattus norvegicus genome (rn6) using RSEM v1.3.1 [46], which utilized STAR v2.7.0 [47]. A mean of 28.9 million reads satisfied quality filtering through STAR, and a mean of 27.8 million reads (~95.8%) were aligned for each sample, corresponding to 18,267 genes throughout the genome with mRNA expression. RNA transcription was quantified using RSEM that uses a forward probability of 0.0, followed by differential expression analysis that uses an empirical Bayes method and expectation maximization in the computation of normalized fold changes and to determine differential expression [48]. This method assigns a posterior probability on the status of expression per individual gene. To account for multiple comparisons and to reduce false positives, a false discovery rate (FDR) of 0.05 was used in the determination of differentially expressed genes (Supplementary Materials 4).
Methylation-Specific Polymerase Chain Reaction (MS-PCR)
To further validate DMRs resulting from MeDIP analyses, MS-PCR, a bisulfite conversion method that has been extensively used to validate the methylation status of specific genes, was used [22,23]. DMR sequences from selected chromosome coordinates identified in MeDIP analyses were obtained using UCSC (genome.ucsc.edu/.) Sequences were entered to the MethPrimer (www.urogene.org/methprimer/index1.html) algorithm to derive the methylation-specific primers (MSP) of the DMRs of each gene. Alogrithm-derived MSPs from a region of the DMR corresponding to highly separated regions between the 3 groups of F3 animals (DDI control, folic acid, and folic acid + TSA in DMSO) are provided in the methylation array (Fig. 5C). Primers were generated for a non-DMR experimental control from regions 5’ and 3’ to the DMR (Table 3). Non-DMR experimental controls were unavailable for a subset of the genes because specific primers were not identified by MethPrimer.
Table 3. Primer sequences used to confirm the methylation status of the DMRs of selected genes.
Ten genes were randomly selected from the list of genes in Fig. 5B to confirm array results. Methylation-specific primers were generated using MethPrimer (www.urogene.org/methprimer/index1.html) for DMR targets (between the gray lines in Figure 5C corresponding to the highly separated regions between the three F3 groups (DDI, folic acid, folic acid + TSA) shown in the methylation array plot. Primers were also synthesized for regions just upstream and downstream the DMR, thereby providing a non-DMR region as an experimental control (FA: folic acid at 80 μg/kg intraperitoneal, DMR: differentially methylated region, M: methylated, U: unmethylated).
| GENE | DMR | CONTROL | |
|---|---|---|---|
| Myl3 | M primer | (L) TTAAGAAGGACGAT GTTAAAATCGT (R) ACTCTACTTCCTTAAAACGTTCGAA |
(L) ATTTTGTACGGAGATAATTAGACGA (R) CCCTAATCAAAAACAAAAAACGTA |
| U Primer | (L) TTAAGAAGGATGATGTTAAAATTGT (R) ACTCTACTTCCTTAAAACATTCAAA |
(L) ATTTTGTATGGAGATAATTAGATGA (R) CCCTAATCAAAAACAAAAAACATA |
|
| Olr1401 | M primer | (L) ATTTTTTGATTTAGATAAGTTTCGT (R) CACAATAAAATAAAATACACACGTC |
No primers developed by MethPrimer |
| U Primer | (L) ATTTTTTGATTTAGATAAGTTTTGT (R) CACAATAAAATAAAATACACACATC |
||
| Charna3 | M primer | (L) TTATTTTATTATAATTCGGGGGTTC (R) CGTCGCGATAATTATTAACCG |
No primers developed by MethPrimer |
| U Primer | (L) ATTTTATTATAATTTGGGGGTTTGA (R) TTCATCACAATAATTATTAACCACC |
||
| Epor | M primer | (L) GGTTATATTTTAGGATTTTGTCGT (R) ATCTATACAATTATAAAACCCCGTA |
(L) ATTGATATATTTTAATGGTTTTCGG (R) TTTCTCTATTTTCTATAAATTACCGTT |
| U Primer | (L) TTTGGGTTATATTTTAGGATTTTGTTG (R) TCTATACAATTATAAAACCCCATA |
(L) ATTGATATATTTTAATGGTTTTTGG (R) TTATTTTTCTCTATTTTCTATAAATTACCA |
|
| Masp2 | M primer | (L) TTTTAATTTGGAATTTTTTTATCGT (R) TATACTCTCCTACCCACACAACGTA |
(L) TTTTTTATGTTTTTAATGTAGTCGT (R) AACCTAATAAACCAAAATCTCAACG |
| U Primer | (L) TTATTTATTTTAATTTGGAATTTTTTTATT (R) TATACTCTCCTACCCACACAACATA |
(L) TTTTTATGTTTTTAATGTAGTTGT (R) AAAACCTAATAAACCAAAATCTCAACA |
|
| Fbxl | M primer | (L) GGGTATTTTAGTAGGTGTGGTTTTC (R) CCACTATATAACATAAAACTAATCGCC |
(L) ATTATTTGAAGTTACGATGGGTATC (R) AACAAAAAAAACTCTATACAACGAA |
| U Primer | (L) GGGTATTTTAGTAGGTGTGGTTTTT (R) ACCTCCACTATATAACATAAAACTAATCAC |
(L) ATTTGAAGTTATGATGGGTATTGG (R) AAACAAAAAAAACTCTATACAACAAA |
|
| Mbp | M primer | (L) TTAGGGAATCGTTTTTATTTGATTC (R) TAACCAAATACTTAAATCCGTATCG |
(L) TATGTTTGGGGTTTTAGTTTATTTC (R) CGAACCATTATTTCACAACGAC |
| U Primer | (L) GGAATTGTTTTTATTTGATTTGT (R) CAAATACTTAAATCCATATCACT |
(L) GTTTGGGGTTTTAGTTTATTTTGT (R) CCAAACCATTATTTCACAACAAC |
|
| Rxrb | M primer | (L) TACGGAGATAGTTACGCGTACG (R) TCCGTATCCTACTCCAACGAA |
(L) GAGGGTTTGGGATTTTGATTAC (R) GAAAATAAAAAAACAACTTTCCCG |
| U Primer | (L) GTATGGAGATAGTTATGTGTATGG (R) TTTTTCCATATCCTACTCCAACAA |
(L) GGTTTGGGATTTTGATTATGA (R) CAAAAATAAAAAAACAACTTTCCCA |
M - Methylated, U – Unmethylated, L – Left, R – Right
DNA isolated from the injured spinal cord segments from each of the 3 groups of animals was bisulfite-converted using a Qiagen kit (Qiagen Sample and Assay Technologies Inc., Valencia, CA) according to the manufacturer’s instructions. Each sample in each of 3 replicates was obtained from a different animal in the IP folic acid group. The bisulfite-treated product was amplified using primers for the methylated and unmethylated regions (Table 3). Amplification products were then subjected to MSP kit assay from Qiagen according to manufacturer’s instructions to specifically characterize methylated DNA, followed by gel electrophoresis using 2% agarose (Ambion; Life Technologies Inc., Calrsbad, CA). The predicted product size was compared with the observed size to confirm validity of the assay. Spinal cord samples used for MS-PCR analyses belonged to different F3 animals from those used for MeDIP.
SECTION IV – STATISTICAL ANALYSES
Descriptive and inferential statistics were performed using R (version 2.15, R Core Team (2013). ISBN 3–900051-07–0, URL www.R-project.org/software). Figures show responses to test compounds by group and by time as box and whisker plots. Boxes extend from the 25th to 75th percentiles, and whiskers from the minimum to the maximum, except for outliers indicated by a circle. Comparisons of differences within generations were made using the Wilcoxon rank-sum test, and statistical significance was reported as p<0.05. Likelihood-based genetic and bootstrap methods are developed in order to account for relatedness of animals when assessing the statistical significance of observed treatment effects. Details are provided in Supplementary Materials 1.
Supplementary Material
Acknowledgments
Funding: This work was supported by the March of Dimes Gene Discovery and Translational Research Grant #6-FY14-435 (BJI), NICHD 1R01HD047516 (BJI), NIH 3R01HD047516-04S1 ARRA Supplement (BJI), NIAID U54AI117924 (MAN), American Association of Neurological Surgeons NREF Grant (NJP), the Department of Neurological Surgery R & D (BJI), and the Clinical and Translational Science Award (CTSA) program (SVM and BJI), through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
List of nonstandard abbreviations
- TSA
trichostatin A
- DMSO
dimethyl sulfoxide vehicle control for TSA
- 5-azadCyD
5-aza-2’-deoxycytidine
- DNMT
DNA methyltransferase
- FA
folic acid
- MF
methylfolate
- S0
unbred single generation control
- DDI
Distilled de-ionized water control
- DMR
differentially-methylated region
- HDAC
histone deacetylase
- DRG
dorsal root ganglion
- MeDIP
methylated DNA immunoprecipitation
- MS-PCR
methylation-specific PCR
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Data availability:
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Likelihood-based calculations performed using R language source code are available at www.biostat.wisc.edu/ñgroup/benny/. Additional datasets generated during and/or analyzed during the current study are available on Gene Expression Omnibus (GEO) upon request.
Ethical approval:
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Competing Interests:
The authors declare that they have no competing interests.
References
- 1.Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J (2006) Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 14 (2):159–166. doi:5201538 [pii] 10.1038/sj.ejhg.5201538 [DOI] [PubMed] [Google Scholar]
- 2.Skinner MK (2008) What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol 25 (1):2–6. doi:S0890–6238(07)00278-X [pii] 10.1016/j.reprotox.2007.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, Meissner A, Weng Z, Hofmann HA, Friedman N, Rando OJ (2010) Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143 (7):1084–1096. doi: 10.1016/j.cell.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews D, Gillette R, Scarpino SV, Manikkam M, Savenkova MI, Skinner MK (2012) Epigenetic transgenerational inheritance of altered stress responses. Proceedings of the National Academy of Sciences of the United States of America 109 (23):9143–9148. doi: 10.1073/pnas.1118514109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dias BG, Ressler KJ (2014) Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci 17 (1):89–96. doi: 10.1038/nn.3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science (New York, NY 308 (5727):1466–1469. doi: 10.1126/science.1108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nilsson E, Larsen G, Manikkam M, Guerrero-Bosagna C, Savenkova MI, Skinner MK (2012) Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PloS one 7 (5):e36129. doi: 10.1371/journal.pone.0036129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A (2011) Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479 (7373):365–371. doi: 10.1038/nature10572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iskandar BJ, Nelson A, Resnick D, Pate Skene JH, Gao P, Johnson C, Cook TD, Hariharan N (2004) Folic acid supplementation enhances repair of the adult central nervous system. Ann Neurol 56 (2):221–227 [DOI] [PubMed] [Google Scholar]
- 10.Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, Jarrard DF, Banerjee RV, Skene JH, Nelson A, Patel N, Gherasim C, Simon K, Cook TD, Hogan KJ (2010) Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest 120 (5):1603–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambrot R, Xu C, Saint-Phar S, Chountalos G, Cohen T, Paquet M, Suderman M, Hallett M, Kimmins S (2013) Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nature communications 4:2889. doi: 10.1038/ncomms3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padmanabhan N, Jia D, Geary-Joo C, Wu X, Ferguson-Smith AC, Fung E, Bieda MC, Snyder FF, Gravel RA, Cross JC, Watson ED (2013) Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 155 (1):81–93. doi: 10.1016/j.cell.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietrzik K, Bailey L, Shane B (2010) Folic acid and L-5-methyltetrahydrofolate: comparison of clinical pharmacokinetics and pharmacodynamics. Clinical pharmacokinetics 49 (8):535–548. doi: 10.2165/11532990-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 14.Szyf M (2009) Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol 49:243–263. doi: 10.1146/annurev-pharmtox-061008-103102 [DOI] [PubMed] [Google Scholar]
- 15.Christman JK (2002) 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21 (35):5483–5495. doi: 10.1038/sj.onc.1205699 [DOI] [PubMed] [Google Scholar]
- 16.Burggren WW (2015) Dynamics of epigenetic phenomena: intergenerational and intragenerational phenotype ‘washout’. J Exp Biol 218 (Pt 1):80–87. doi: 10.1242/jeb.107318 [DOI] [PubMed] [Google Scholar]
- 17.Sun F, He Z (2010) Neuronal intrinsic barriers for axon regeneration in the adult CNS. Current opinion in neurobiology 20 (4):510–518. doi: 10.1016/j.conb.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filbin MT (2003) Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nature reviews 4 (9):703–713. doi: 10.1038/nrn1195 [DOI] [PubMed] [Google Scholar]
- 19.Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH (2001) Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci 4 (1):38–43. doi: 10.1038/82881 [DOI] [PubMed] [Google Scholar]
- 20.Smith DS, Skene JH (1997) A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. Journal of Neuroscience 17 (2):646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson EA (2000) Statistical inferences from genetic data on pedigrees, vol 6 NSF-CBMS regional conference series in probability and statistics IMS, Beachwood, OH [Google Scholar]
- 22.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America 93 (18):9821–9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laird PW (2003) The power and the promise of DNA methylation markers. Nat Rev Cancer 3 (4):253–266. doi: 10.1038/nrc1045 [DOI] [PubMed] [Google Scholar]
- 24.Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI (2000) Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci 20 (12):4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finelli MJ, Wong JK, Zou H (2013) Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 33 (50):19664–19676. doi: 10.1523/JNEUROSCI.0589-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng YL, An R, Cassin J, Joseph J, Mi R, Wang C, Zhong C, Jin SG, Pfeifer GP, Bellacosa A, Dong X, Hoke A, He Z, Song H, Ming GL (2017) An Intrinsic Epigenetic Barrier for Functional Axon Regeneration. Neuron 94:337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yiu G, He Z (2006) Glial inhibition of CNS axon regeneration. Nature reviews 7 (8):617–627. doi: 10.1038/nrn1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li CC, Cropley JE, Cowley MJ, Preiss T, Martin DI, Suter CM (2011) A sustained dietary change increases epigenetic variation in isogenic mice. PLoS genetics 7 (4):e1001380. doi: 10.1371/journal.pgen.1001380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cervoni N, Szyf M (2001) Demethylase activity is directed by histone acetylation. The Journal of biological chemistry 276 (44):40778–40787. doi: 10.1074/jbc.M103921200 [DOI] [PubMed] [Google Scholar]
- 30.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7 (8):847–854. doi: 10.1038/nn1276 nn1276 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Maden M (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nature reviews 8 (10):755–765. doi: 10.1038/nrn2212 [DOI] [PubMed] [Google Scholar]
- 32.Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34 (6):885–893 [DOI] [PubMed] [Google Scholar]
- 33.Demyanenko GP, Mohan V, Zhang X, Brennaman LH, Dharbal KE, Tran TS, Manis PB, Maness PF (2014) Neural cell adhesion molecule NrCAM regulates Semaphorin 3F-induced dendritic spine remodeling. J Neurosci 34 (34):11274–11287. doi: 10.1523/JNEUROSCI.1774-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohan V, Wyatt EV, Gotthard I, Phend KD, Diestel S, Duncan BW, Weinberg RJ, Tripathy A, Maness PF (2018) Neurocan Inhibits Semaphorin 3F Induced Dendritic Spine Remodeling Through NrCAM in Cortical Neurons. Front Cell Neurosci 12:346. doi: 10.3389/fncel.2018.00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao S, Zhang S, Zhou Z, Shi X, Huang T, Feng W, Yao C, Gu X, Yu B (2018) Alternative RNA splicing associated with axon regeneration after rat peripheral nerve injury. Experimental neurology 308:80–89. doi: 10.1016/j.expneurol.2018.07.003 [DOI] [PubMed] [Google Scholar]
- 36.Zhang G, Pradhan S (2014) Mammalian epigenetic mechanisms. IUBMB Life 66 (4):240–256. doi: 10.1002/iub.1264 [DOI] [PubMed] [Google Scholar]
- 37.Blake GE, Watson ED (2016) Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr Opin Chem Biol 33:101–107. doi: 10.1016/j.cbpa.2016.06.008 [DOI] [PubMed] [Google Scholar]
- 38.Van den Veyver IB (2002) Genetic effects of methylation diets. Annual review of nutrition 22:255–282 [DOI] [PubMed] [Google Scholar]
- 39.Richardson PM, Issa VM (1984) Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309:791–793 [DOI] [PubMed] [Google Scholar]
- 40.Abercombie M (1946) Estimation of nuclear population from microtome sections. Anat Rec 94:239–247 [DOI] [PubMed] [Google Scholar]
- 41.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature genetics 37 (8):853–862. doi: 10.1038/ng1598 [DOI] [PubMed] [Google Scholar]
- 42.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nature genetics 39 (4):457–466. doi: 10.1038/ng1990 [DOI] [PubMed] [Google Scholar]
- 43.Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:Article3. doi: 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- 44.Scacheri PC, Crawford GE, Davis S (2006) Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol 411:270–282. doi: 10.1016/S0076-6879(06)11014-9 [DOI] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57 (1):289–300 [Google Scholar]
- 46.Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1):15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C (2013) EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29 (8):1035–1043. doi: 10.1093/bioinformatics/btt087 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.